
Submitted:
22 May 2023
Posted:
23 May 2023
You are already at the latest version
Abstract
Ligand-based virtual screening (LBVS) is a promising approach for rapid and low-cost screening of potentially bioactive molecules in the early stage of drug discovery. Compared with traditional similarity-based machine learning methods, deep learning frameworks for LBVS can more effectively extract high-order molecule structure representations from molecular fingerprints or structures. However, the 3D conformation of a molecule largely influences its bioactivity and physical properties, and has rarely been considered in previous deep learning-based LBVS methods. Moreover, the relative bioactivity benchmark dataset is still lacking. To address these issues, we introduce a novel end-to-end deep learning architecture trained from molecular conformers for LBVS. We first extracted molecule conformers from multiple public molecular bioactivity data and consolidated them into a large-scale bioactivity benchmark dataset, which totally includes millions of endpoints and molecules corresponding to 954 targets. Then, we devised a deep learning-based LBVS called EquiVS to learn molecule representations from conformers for bioactivity prediction. Specifically, graph convolutional network (GCN) and equivariant graph neural network (EGNN) are sequentially stacked to learn high-order molecule-level and conformer-level representations, followed by attention-based deep multiple-instance learning (MIL) to aggregate these representations and then predict the potential bioactivity for the query molecule on a given target. We conducted various experiments to validate the data quality of our benchmark dataset, and confirmed EquiVS achieved better performance compared with 10 traditional machine learning or deep learning-based LBVS methods. Further ablation studies demonstrate the significant contribution of molecular conformation for bioactivity prediction, as well as the reasonability and non-redundancy of deep learning architecture in EquiVS. Finally, a model interpretation case study on CDK2 shows the potential of EquiVS in optimal conformer discovery. The overall study shows that our proposed benchmark dataset and EquiVS method have promising prospects in virtual screening applications.
Keywords:
Virtual screening
; Bioactivity prediction
; Equivariant graph neural network
; Multiple instance learning
; Molecular conformation
; Benchmark dataset
1. Introduction
Virtual screening (VS) adopts computational methods to identify chemical candidates that may have binding bioactivities to a query target, which is widely used in the early stage of drug discovery1, 2. There are two main categories of VS: structure-based VS (SBVS) and ligand-based VS (LBVS). LBVS methods generally predict unknown bioactivities of new molecules based on known bioactivities of molecules. The commonly used methods of LBVS are pharmacophore mapping3, shape-based similarity4, fingerprint similarity5, and machine learning-based Quantitative Structure-Activity Relationship (QSAR) 6-8. The main assumption of these methods is that “structurally similar molecules have similar bioactivities for specific targets”9. Following the assumption, as there are good consistencies of structure differences and bioactivity differences, chemical knowledge (molecular fingerprints) and representations (molecular embeddings) extracted from known molecular structures can be used for bioactivity prediction.
With the massive development of public pharmaceutical databases and artificial intelligence technologies, data-driven deep learning frameworks have been widely used in the vast majority fields of drug discovery, such as de novo drug design10, 11, ADMET prediction12, 13, drug repositioning14, 15, and VS16, 17. Compared to traditional LBVS methods, deep learning-based methods map molecular representations into high-dimensional spaces, and implicitly identify molecular similarities and correlations to bioactivities based on high-order embedding, which can be regarded as a continuous way to discover bioactive groups better than those traditional and discrete ones. Recently, there are several studies constructing LBVS and bioactivity prediction models using deep learning. For instance, DeepScreening is a platform which uses deep neural networks trained from molecular fingerprints to predict molecular bioactivity values and categories18; GATNN is a graph neural network (GNN) framework extracts molecular fingerprints for similarity calculation in LBVS, which outperforms traditional fingerprints19; RealVS adopts a graph attention network (GAT) to learn molecule features from molecular graphs and optimizes multiple training loss with adversarial domain alignment and domain transferring for bioactivity prediction20. They experimentally proved that deep learning framework showed better structure representation ability than 2D structure-based molecular fingerprints.
However, the molecular 3D structures-molecular conformers, have seldom been considered in these studies. Previous studies have proven that there were certain relations between different molecular conformers and bioactivities21-23, and several traditional fingerprint-based QSAR methods have already adopted molecular three-dimensional features extracted from molecular conformers for LBVS24-27. Therefore, employing molecular conformation for molecular representation learning is a promising strategy for bioactivity prediction. However, the available bioactivity prediction benchmark dataset with multiple molecular conformers is still lacking, and the end-to-end deep learning architecture that effectively learns molecular representations from molecular conformers has not been designed and estimated.
To address the above challenges, we first constructed a large-scale molecular bioactivity benchmark dataset with over 3 million endpoints, including nearly 1 thousand targets, 1 million molecules, and 10 million calculated molecular conformers. Then, we proposed a new deep learning method called EquiVS to introduce molecular conformation information into LBVS, which effectively improved molecular bioactivity prediction. EquiVS was designed as an end-to-end architecture with graph convolutional network (GCN), equivariant graph neural network (EGNN), and deep multiple instance learning (MIL) layers, which can directly learn molecular high-order representations from the 2D topological level and 3D structural level. The model performance comparison results on large-scale benchmark datasets indicated that EquiVS outperformed multiple classical machine learning-based and graph neural network-based baseline methods. Furthermore, the ablation study emphasized that efficient representations and attention-based aggregations of multiple molecular conformers play an important role in the accurate bioactivity prediction in EquiVS. To enhance the interpretability of EquiVS in real LBVS scenarios, we introduced an attention-based mechanism in deep MIL for optimal molecular conformer discovery which was investigated by a case study.
2. Materials and Methods
2.1. Bioactivity data collecting, integrating and filtering
We introduced the data collection, integration, filtering, and preprocessing process of the molecular bioactivity benchmark dataset for drug virtual screening in detail. Figure 1 shows the overall process of the construction of the benchmark dataset, including (1) Collection and integration of multi-source drug bioactivity data; (2) Multilevel data filtering based on activity type, activity unit, molecular structure, and target; (3) Downstream processing, including molecular conformer generation and optimization, and molecular graph construction.
In the first steps, we collected large-scale molecular bioactivity data from five public databases, including ChEMBL28, PubChem29, BindingDB30, Probes & Drugs31, and IUPHAR/BPS32. We directly downloaded the integrated data of the above five databases from a previous study 33 to accelerate the data collection process. Brief descriptions of the introduced databases were listed in Table 1. The collected bioactivity data from 33 have nine items, including (1) Molecule ID corresponding to the source database; (2) Molecule name; (3) Molecule structure represented with SMILES (Simplified Molecular Input Line Entry Specification); (4) HGNC (HUGO Gene Nomenclature Committee) 34 named target ID; (5) Bioactivity type; (6) Bioassay type; (7) Bioactivity unit; (8) Bioactivity value; (9) Data source.
As for the data integration, in 33, the bioactivity data have been preprocessed, which first identified matched molecules from different sources and integrated their relevant bioactivity data, then labeling “1 structure”, “match”, “no structure”, or specific Tanimoto structure similarity based on Morgan fingerprints35 for each integrated data. Furthermore, the corresponding bioactivity values have been integrated too. Specifically, a set of buckets were set determined by negative decadic logarithm with a molar unit (e.g., a bucket with the range of 5 -logM to 6 -logM). Then, different bioactivity values to a matched molecule were classified into the corresponding buckets based on their ranges. The average value in each bucket was calculated. In our study, we selected the “1 structure” and “match” labeled bioactivity data to allow structural consistency, and the average values of the buckets with the highest frequency as the final bioactivity values.
In the second step, we proposed a multilevel data filtering strategy to identify high-quality bioactivity data. For bioactivity data, we selected endpoints with five widely used and studied activity types (IC50, Ki, Kd, EC50, and Potency) as candidates and filtered the last ones. For molecular structure, we adopted MolVS 36 for structure standardization, including (1) Normalization of functional groups to a consistent format; (2) Recombination of separated charges; (3) Breaking of bonds to metal atoms; (4) Competitive reionization to ensure strongest acids ionize first in partially ionize molecules; (5) Tautomer enumeration and canonicalization; (6) Neutralization of charges; (7) Standardization or removal of stereochemistry information; (8) Filtering of salt and solvent fragments; (9) Generation of fragment, isotope, charge, tautomer or stereochemistry insensitive parent structures; (10) Validations to identify molecules with unusual and potentially troublesome characteristics. Then, the mistake molecules which cannot be identified by RDkit package 37 were filtered. For biological targets, as the prediction performances of machine learning-based and deep learning-based LBVS methods highly rely on the quality and quantity of bioactivity data for specific targets, we only selected those targets with more than 300 bioactivity endpoints as candidates and filtered the corresponding bioactivity data of other targets. Finally, 3,888,765 bioactivity endpoints with 954 targets were left. Regarding bioactivity endpoints belonging to a specific target as a bioactivity sub-dataset, we gathered these 954 sub-datasets together and assembled them into a bioactivity benchmark dataset.
2.2. Molecular conformer and graph generating
Candidate bioactivity endpoints were determined by the above comprehensive process. However, these data contained only one-dimensional SMILES representations of molecular structures and lacked three-dimensional structural information. To address this issue, multiple molecular conformers were computationally generated and assembled with bioactivity endpoints to finish the bioactivity benchmark dataset construction.
In this study, we used the RDKit package to generate and optimize molecular conformers. Specifically, hydrogen atoms were first added to the molecular structure to simulate the real molecular geometric conformation. Then, ETKDG (Experimental-Torsion basic Knowledge Distance Geometry) was used for conformation generation. ETKDG is a knowledge-based conformer generation method that combines distance geometry and torsion angle preferences proposed by Riniker et al38. Additionally, since a molecule could have multiple three-dimensional conformers at different chemical environments, we used ETKDG methods with different random initializations to generate 10 conformers for each molecule to achieve sufficient sampling of molecule three-dimensional structures. To better approximate the actual geometric conformations of molecules, we optimized the generated molecular conformers using MMFF94 force field, which is developed by Merck that uses multiple empirical parameters, including atomic types, charges, bond lengths, bond angles, torsional angles, van der Waals force, etc., to represent the potential energy and optimize the molecular conformation to finally obtain approximately optimal low-energy conformers39.
Here, we aligned the three-dimensional coordinates of different conformers of each molecule to normalize coordinate information. All hydrogen atoms were removed from the molecular structures to reduce the computing complexity and training time of LBVS methods. Finally, we assembled all conformers to corresponding molecule SDF structure files to finish the construction of the large-scale bioactivity benchmark dataset.
To introduce the molecular conformer information to our GNN-based EquiVS method, we constructed each molecular graph with an adjacency matrix, a node feature matrix , an edge feature matrix , and a coordinate feature matrix , where an example of the representations of molecules in our study was shown in Figure 2.
Given a molecule, its graph can be represented as:
where denotes the set of atoms, and denotes the set of bonds. The topological structure of can be represented as an adjacency matrix . Given an atom node , its neighbor atom node , and its neighbor node set , the adjacency relation can be represented as:
As for feature matrices in the molecular graph, we first calculated atom physiochemical properties as with the dimension of 74, including (1) one-hot encoding of atomic elements; (2) one-hot encoding of atomic degrees; (3) one-hot encoding of the number of implicit hydrogens; (4) one-hot encoding of the formal charges; (5) one-hot encoding of the number of radical electrons; (6) one-hot encoding of the atom hybridizations; (7) one-hot encoding of the aromatics; (8) one-hot encoding of the number of total hydrogens. Then, the chemical bond properties were adopted as with the dimension of 12, including (1) one-hot encoding of bond types; (2) conjugation; (3) ring; (4) one-hot encoding of the stereo configuration. Finally, we collected the atomic spatial coordinates in each conformer and concatenated them as with a dimension of 30. Through the above processes, the molecular three-dimensional structural information was represented in molecule graphs, which can be identified in EquiVS.
2.3. Bioactivity prediction model construction
In this study, we proposed an EGNN and deep MIL-based virtual screening method, EquiVS, to facilitate the representation learning of current LBVS methods via utilizing molecular conformations. The architecture of EquiVS (Figure 3) comprises two core modules: the graph representation learning module and the deep multiple instance learning module. We introduced the computing flow of these modules in detail.
Graph representation learning module. We considered learning molecule high-order representations from both two-dimensional topological level and three-dimensional structural level. Therefore, a stepwise molecular graph learning strategy with skip connection was designed to learn and aggregate the molecular representations. Specifically, a GCN layer is first used to learn graph topological representations , which can be represented as:
where , are learned node features and initial node features, respectively. is a trainable parameter matrix. Considering the message passing process in GCN, given a target node and one of its neighbor nodes , the message from is:
where is a linear transformation function. Then, GCN aggregates the message and the input feature to finish node feature updating:
where is a linear transformation function. Then, a readout function is further adopted to aggregate node feature to graph feature :
where is Sigmoid activation function, and are trainable parameter matrices. Then, EGNN layers were used to learn structural representations for each conformer. Given conformers for a specific molecule, by dividing its coordinate feature matrix into matrices, the overall graph can be represented as:
where is a given molecular conformer. Taking -th EGNN layer as an example, the molecule representations of each is learned through:
where , , and are node feature, edge feature, and coordinate feature in -th EGNN layer, respectively. Considering the message passing process in , given a target node and one of its neighbor nodes , the message from is:
where is a linear transformation function. Meanwhile, the coordinate features are also updated in EGNN:
where is the number of neighbor nodes minus 1, and is a linear transformation function. Finally, EGNN aggregates the message and the input feature to finish node feature updating:
where is a linear transformation function After the node features are updates through the above message passing process, similarly, a readout function is used to generate graph feature or :
Finally, as the “over-smoothing” and “vanishing gradient” widely exist in deep graph neural network models, which could significantly harm the model performances, we built skip connections between different EGNN layers to allow direct gradient propagation to the shallow layers by simply concatenating the graph feature output of each layer as the final graph representations. For conformer , its final graph feature can be formulated as:
Deep multiple instance learning module. After EquiVS captures the high-order representations for molecular conformers, the aggregation process of these conformer representations to generate comprehensive molecular representations, and effective bioactivity prediction through both conformer-level and molecule-level should be elaborately considered and designed. Regarding this, we introduced MIL theory into bioactivity prediction and designed our deep multiple instance learning module with an interpretable attention mechanism. Based on MIL theory, a molecule can be regarded as a “bag”, and its multiple conformers are “instances”. The bioactivity value is only labeled at the molecule level, but the conformer-level bioactivities still remain unknown. Therefore, the training object of MIL is to accurately predict the molecular bioactivity value, while identifying the conformer which fits this bioactivity value best, simultaneously. Regarding this, EquiVS first dynamically aggregates the conformer instance representations with an attention mechanism. The attention score of can be formulated as:
where is a Tanh activation function. , , and are trainable parameter matrices. The attention score can be further converted to normalized attention coefficient :
The attention coefficient represents the importance weight of a conformer. Hence, the conformer instance representations can be aggregated to acquire molecule representation based on the attention coefficients:
Then, EquiVS predicts the bioactivity value from both conformer-level and molecule-level. For conformer-level prediction, a multilayer perceptron (MLP) is used for bioactivity prediction:
where is a ReLU activation function. , , , and are trainable parameter matrices. For molecule-level prediction, another MLP is constructed:
where is a ReLU activation function. , , , and are trainable parameter matrices. Based on the above processes, EquiVS achieves molecular three-dimensional structure-based bioactivity prediction.
2.4. Training optimization
Inspired by loss-based deep multiple instance learning 40, we adopted both conformer-level prediction and molecule-level prediction to calculate model loss and update model parameters. Meanwhile, we also differentiated the conformer predictions by attention coefficients, thus alleviating the impact of noisy conformers for model training. Specifically, we introduced mean square error (MSE) as the optimization function. Given samples as the batched data, the conformer-level prediction loss can be formulated as:
where is the attention coefficient for -th conformers in -th molecule. and are the true label for -th molecule and predicted label for -th conformers in -th molecule, respectively. The molecule-level prediction loss can be formulated as:
where is the molecule-level predicted label for -th molecule. The final loss function can be represented as:
where is a contribution factor to determine the importance of and . In this study, we set as 0.5.
The overall EquiVS computing flow was shown in Algorithm 1. Furthermore, we used Adam optimizer for training optimization and added a dropout layer after each GNN layer and MLP layer to alleviate overfitting.

2.5. Model interpretation
As described in Deep multiple instance learning module, EquiVS employs the attention mechanism to aggregate the conformer representations, which could also be used for conformer-level model interpretation. Given a bioactivity endpoint with a molecule and multiple conformers, we ranked the importance of different conformers based on the attention coefficients to discover the optimal conformer which matches the current bioactivity value best.
2.6. Settings
For model hyper-parameter settings, we set the learning rate as 0.001, dropout rate as 0.05, the number of epochs as 200, batch size as 128, hidden feature dimensions as 128, and the number of EGNN layers as 2 to construct and train EquiVS on our proposed bioactivity prediction benchmark dataset.
For baseline settings, we adopted 2 molecular fingerprints (ECFP435 and MACCS41), 3 machine learning methods (Linear regression LR, gradient boosting decision tree GBDT42, and extreme gradient boosting decision tree XGB43), and 4 GNN methods (GCN44, GAT45, AttentiveFP46, and Weave47) to construct baseline models for model performance comparisons.
For experimental settings, we randomly split the benchmark dataset into training set, validating set, and testing set with a ratio of 8:1:1. EquiVS and all baseline methods were trained on the training set, adjusting hyper-parameters on the validating set, and evaluated on the testing set. We adopted the coefficient of determination (R2), MSE, and mean absolute error (MAE) to assess the performances of bioactivity prediction. These metrics can be formulated as:
where denotes the sample size. , , and are true label, predicted label, and overall average label.
3. Results
3.1. Benchmark dataset quality analysis
We examined our constructed bioactivity benchmark dataset quality through visualization analysis. Figure 4A showed the distribution of major bioactivity types in our raw data. It is suggested that our adopted five bioactivity types (pIC50, pPotency, pKi, pKd, and pEC50) occupy the majority of the data (87.56%). Figure 4B showed the overlap between different source databases, indicating that there are only 12.4% of bioactivity endpoints that are duplicated, and the majority of the raw endpoints are unique (e.g., 5.13 million unique data from Probe&Drugs and 3.88 million unique data from ChEMBL, with a proportion of 83.6% among overall endpoints). Therefore, these non-redundant bioactivity endpoints from different sources should be integrated to expand the data scale and promote bioactivity prediction.
Meanwhile, Figure 4C visualized the bioactivity value distribution among different source data. We set 1μM(6 -logM) as the threshold to classify active/inactive molecules, and the corresponding specific distribution results were listed in Table 2. The above results showed that the standard deviations of bioactivity values (within-group differences) among 5 source databases are close. The majority of bioactivity endpoints in ChEMBL and Probes&Drugs are inactive, while the proportions of active bioactivity endpoints in PubChem, BindingDB, and IUPHAR/BPS are larger than those of inactive ones. Considering between-group differences, the average bioactivity value differences between different source databases are acceptable (differences between average values are less than 1 -logM among ChEMBL, PubChem, BindingDB, and IUPHAR/BPS, which contains 99.2% of the overall endpoints). The above findings proved the reasonability of data integration from those source databases to expand the bioactivity benchmark data scale.
As for the selection of candidate targets, Figure 4D showed the distribution of bioactivity data scale of different targets in our benchmark dataset. It suggested that a considerable number of targets are with insufficient bioactivity endpoints (less than 100). Therefore, setting 300 as the data scale can help to filter the targets on which are more possible to construct low-capacity LBVS models, thus improving the quality of the benchmark dataset and the reliability of LBVS models for specific targets.
Hence, eight physiochemical and structural properties were calculated for all molecules in the benchmark dataset to explore the distributions of Linpski rules 48 and Quantitative Estimate of Drug-likeness (QED) 49. These properties include molecular weights (MW), number of hydrogen bond donors (HBD), number of hydrogen bond acceptors (HBA), AlogP, number of rotatable bonds (ROTB), polar surface areas (PSA), number of aromatic rings (AROM), number of alert structures (ALERTS). The distributions of these properties in different source databases are shown in Figure 5. According to the results, approximately 83.03% of molecules’ MWs are lower than 500 Dalton (Da), with an average MW of 405; 97.89% of molecules have lower than 10 HBDs; 97.89% of molecules have lower than 5 HBAs; 82.66% of molecules’ AlogP are lower than 5, with an average AlogP of 3.49; 94.46% of molecules’ have lower than 10 ROTBs; 81.05% of molecules’ PSA obey the QED rule; 89.43% of molecules have lower than 4 and larger than 1 AROMs; 76.27% of molecules’ have lower than 1 ALERTS. Therefore, our benchmark roughly fulfills the requirements of Linpski and QED rules for drug-like molecules in HBD, HBA, ROTB, and AROM properties. However, about 20% of collected molecules are out-of-bag of Linpski and QED rules in MW, AlogP, PSA, and ALERTS properties. As all of these properties should take into account to comprehensively estimate the drug-likeness, and a study has observed many recently approved drugs excess the MW range in these rules50, we could conclude that our benchmark overall includes enough chemical space to discover drug-like candidates by LBVS methods.
3.2. Model performance comparison
We then trained EquiVS and 10 baseline models on our organized bioactivity benchmark dataset to compare the overall performances of these LBVS bioactivity prediction models, which were listed in Table 3. The results showed that EquiVS outperformed other baseline methods and achieved optimal performances on 3 metrics. Especially on MSE, the relative improvement of EquiVS compared with the suboptimal method (GBDT_ECFP) is 13.33%, while the improvements are even larger when compared to other deep learning-based methods. Meanwhile, EquiVS also performed stabler than these deep learning methods, gaining lower standard variations. The above results indicated that EquiVS achieves competitive performances in bioactivity prediction tasks, which could be used as a promising LBVS method to discover potentially active molecules for specific targets.
Furthermore, as the qualities and confidences of bioactivity endpoints varied in different bioactivity sub-dataset, there are a proportion of bioactivity sub-datasets that failed to be used to train bioactivity prediction models with sufficient performances. Therefore, it is necessary to exclude low-quality bioactivity sub-datasets corresponding to infeasible targets and focus on those feasible ones which have sufficient bioactivity data with fewer noises and better consistencies. Considering this, we set as the threshold and filtered 1,702 (82.98%) feasible targets and their bioactivity sub-datasets. The performances of EquiVS and 10 baseline methods on such feasible targets were shown in Figure 6. The results suggested that EquiVS showed a more significant superiority on good-quality targets. Compared to methods with suboptimal performances (R2: GBDT_ECFP, MSE: GBDT_ECFP, MAE: GAT), the relative improvements of R2, MSE, and MAE of EquiVS reached 3.60%, 33.01%, and 5.45%, respectively. Also, the performances of EquiVS are generally stabler than other deep learning methods.
The overall model performance comparison experiments emphasized that our designed EquiVS method can give relatively accurate predictions on molecular bioactivities rather than widely used machine learning and deep learning-based baseline methods. Especially in the scenario of bioactivity prediction with good-quality training data, the superiority of EquiVS is highlighted.
3.3. Ablation study
After the effectiveness of EquiVS on large-scale bioactivity prediction has been proven, we then explored the potential mechanisms and core modules that play important roles in the molecular representation learning and model performances of EquiVS through an ablation study. Specifically, we designed five variant models, and each of them deleted or replaced a core module in EquiVS and left other modules. These variant models are as follows:
- EquiVS-Single: EquiVS architecture which adopts one conformer for each molecule for structural representation learning.
- EquiVS-w/o GCN: EquiVS architecture which replaces the GCN layer with a simple linear layer.
- EquiVS-w/o Skip: EquiVS architecture which deletes the skip connection between different EGNN layers.
- EquiVS-w/o AA: EquiVS architecture which replaces the attention-based conformer representation aggregation process with a simple sum calculation.
- EquiVS-w/o IP: EquiVS architecture which deletes the conformer-level predictor and the corresponding conformer-level prediction loss.
After these variants were defined, considering the running time and computing complexity, we randomly selected 50 targets and their bioactivity sub-datasets and trained EquiVS and these variants on them for performance comparisons. The data splitting settings are the same as those in model performance comparison experiments. The performances of EquiVS and its 5 variants were shown in Figure 7. The results show that EquiVS achieved optimal performances on all metrics. In addition, compared EquiVS with each of the variants, we can summarize the following five observations:
- Compared to EquiVS-Single: EquiVS gained relative improvements of 7.79% on R2, 35.14% on MSE, and 27.94% on MAE. The results indicated that multiple sampling of molecular conformers can enhance molecular representation learning, and it is easier than a single conformer to obtain the right conformer which matches the bioactivity endpoint best, thus “diluting” the potential molecular three-dimensional structural noises caused by unsupervised conformer generation methods, which is also supported by a previous study27.
- Compared to EquiVS-w/o GCN: EquiVS gained relative improvements of 5.28% on R2, 28.81% on MSE, and 23.44% on MAE. The results proved that topological molecular representations learned by GCN are better than initial molecular features. Meanwhile, the effectiveness of combining two-dimensional topological level features and three-dimensional structural level features for comprehensive molecular representations.
- Compared to EquiVS-w/o Skip: EquiVS gained relative improvements of 5.80% on R2, 26.96% on MSE, and 21.22% on MAE. The results implied that the “over-smoothing” phenomenon caused by stacking multiple EGNN layers could decrease the model performance. The use of the skip connection process can alleviate the negative impact to some degree.
- Compared to EquiVS-w/o AA: EquiVS gained relative improvements of 90.67% on R2, 80.84% on MSE, and 63.70% on MAE. Such vastly different performance results indicated that although the fusion of multiple molecular conformer coordinates can supplement molecular three-dimensional structural information, some sampled conformers do not match the current bioactivity endpoint. Therefore, massive structural noises will be introduced and model performances will drastically decrease unless an elaborate strategy is adopted to differentiate and identify different importance for multiple molecular conformers. In contrast, EquiVS weighted aggregates different conformer representations using the attention mechanism to focus on the high-confident conformers thus benefiting the molecular representation learning and model performances.
- Compared to EquiVS-w/o IP: EquiVS gained relative improvements of 5.41% on R2, 27.27% on MSE, and 20.97% on MAE. The results indicated that conformer-level prediction and the corresponding attention-weighted conformer-level loss could effectively assist model training and improve model performances.
3.4. Case study: optimal molecular conformer discovery
In EquiVS, the attention mechanism is adopted to calculate the attention coefficients of multiple molecular conformers and weighted aggregate the conformer representations. As the attention coefficients represent the importance of the molecule conformers to specific bioactivities, they can be used to interpret which conformer matches the current bioactivity and the actual molecule crystal structure best when there is a ligand-protein binding. To investigate the potential of EquiVS in optimal molecular conformer discovery, we took human cyclin-dependent kinase 2 (CDK2) as a candidate target and its bioactivity sub-dataset to visualize all attention coefficients (Figure 10A). The results showed that quite a few attention coefficients generated by EquiVS are close to 0, indicating that there are noises in some calculated molecular conformers. Representing the molecule structures with single conformers or not differentiating the reliabilities of multiple conformers could lead to a negative impact on the correctness and effectiveness of molecular structural representations. Combining the result that a proportion of attention coefficients are significantly larger than the average (0.10), the overall distribution visualization indicated that EquiVS can recognize and distinguish different molecular conformers to reduce the influence of structurally unreasonable conformers on the bioactivity predictions.
Moreover, we selected “Nc1ccc(-c2cc(Nc3ccc(S(N)(=O)=O)cc3)[nH] n2)cc1” as a case active molecule (bioactivity value: 9.0 -logM), visualized its conformers, predicted the bioactivity values by the conformer-level predictor in EquiVS, generated the binding score and pose by Autodock Vina51, to investigate the consistency of attention coefficients, predicted bioactivities, and vina scores. The binding pose of CDK2 and the case molecule was shown in Figure 8B, showing that molecular docking successfully discover two binding sites for the case molecule. Then, the molecular conformers, attention coefficients, predicted bioactivities, and vina scores were shown in Figure 8C. It is inspiring to discover the observation that there is kind of consistencies and correlations between attention coefficients and the accuracy of predicted bioactivities and vina scores. That is, the conformers with higher attention coefficients tend to get more accurate bioactivity predictions and higher vina scores. For instance, conformer 4 with the second biggest attention coefficient achieved the second most accurate prediction and Vina score. Also, the prediction accuracies and vina scores of those with attention coefficients below the average (conformer 2, 3, 6, 7, and 8) are much lower than the else.
The overall results suggested that the attention-based conformer-level model interpretability of EquiVS can differentiate molecular conformers with varied structure reliabilities and discover the optimal conformers for specific bioactivity endpoints and targets.
Conclusion
In this study, we investigate the role of molecular conformation in LBVS and bioactivity prediction scenarios. A large-scale bioactivity prediction benchmark dataset is proposed to assemble the requirement of molecular conformation to bioactivity endpoints, which is collected from multiple public pharmaceutical databases and contains thousands of targets and millions of bioactivity endpoints, molecules, and molecule conformers. Then, an EGNN and deep MIL-based LBVS method is designed for bioactivity prediction, which is called EquiVS. Compared to other widely-used ML-based and GNN-based methods, EquiVS achieved notable improvements on our large-scale benchmark dataset. Combining the ablation analysis, the performance results prove employing molecular conformation could enhance molecular representation learning and further contribute to better bioactivity prediction with elaborate neural network architecture design and reasonable feature extraction and aggregation. To promote the practical application of EquiVS, two case studies are designed to explore the effectiveness of conformer-level interpretation in EquiVS. The overall results reveal a promising prospect of molecular conformation as well as our proposed benchmark dataset and EquiVS method in bioactivity prediction and LBVS.
It should also be emphasized that there are several major limitations in our study. First, from the data quality control aspect, some of the sub-datasets and targets in our integrated benchmark dataset should be filtered as all tested LBVS methods could not give reliable predictions on them, but they are still retained in the current version; Second, from the model training aspect, a predictable optimization is to pre-train EGNN-based models using molecular conformation and finetune them for the downstream bioactivity prediction52-54. Also, improving the GNN model training period with chemical domain knowledge insights is another promising strategy55, 56; Third, from the architecture design aspect, more advanced GNN backbones which take 3D graphs as input should be adopted for conformer representation learning, such as SchNet57, GemNet58, PaiNN59, and etc.
As for future research, we will focus on developing practical tools to enhance the accessibility of EquiVS, and employing molecular conformation from massive unlabeled molecules to pre-train and optimize EquiVS models with more robustness and generality.
Author Contributions
Y.G, J.L, H.K, B.Z, and S.Z conceptualized the study; Y.G collected and curated the dataset, designed the model, and wrote the manuscript; J.L provide funding support; S.Z revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Chinese Academy of Medical Sciences (Grant No. 2021-I2M-1–056), Fundamental Research Funds for the Central Universities (Grant No. 3332022144).
Data Availability Statement
The source codes of EquiVS and molecular conformer generation process and the benchmark dataset are available at https://github.com/gu-yaowen/EquiVS.
Acknowledgments
The authors would like to thank all anonymous reviewers for their constructive advice.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bajorath, J. Selected concepts and investigations in compound classification, molecular descriptor analysis, and virtual screening. Journal of chemical information and computer sciences 2001, 41, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hernandez, C.; Fernández, A.; Serratosa, F. Ligand-Based Virtual Screening Using Graph Edit Distance as Molecular Similarity Measure. Journal of chemical information and modeling 2019, 59, 1410–1421. [Google Scholar] [CrossRef]
- Sun, H. Pharmacophore-based virtual screening. Current medicinal chemistry 2008, 15, 1018–1024. [Google Scholar] [CrossRef]
- Kirchmair, J.; Distinto, S.; Markt, P.; Schuster, D.; Spitzer, G.M.; Liedl, K.R.; Wolber, G. How to optimize shape-based virtual screening: choosing the right query and including chemical information. Journal of chemical information and modeling 2009, 49, 678–692. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods (San Diego, Calif.) 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Kong, W.; Wang, W.; An, J. Prediction of 5-hydroxytryptamine transporter inhibitors based on machine learning. Computational Biology and Chemistry 2020, 87, 107303. [Google Scholar] [CrossRef]
- Kong, W.; Tu, X.; Huang, W.; Yang, Y.; Xie, Z.; Huang, Z. Prediction and optimization of NaV1. 7 sodium channel inhibitors based on machine learning and simulated annealing. Journal of Chemical Information and Modeling 2020, 60, 2739–2753. [Google Scholar] [CrossRef]
- Kong, W.; Huang, W.; Peng, C.; Zhang, B.; Duan, G.; Ma, W.; Huang, Z. Multiple machine learning methods aided virtual screening of NaV1. 5 inhibitors. Journal of Cellular and Molecular Medicine 2023, 27, 266–276. [Google Scholar] [CrossRef]
- Johnson, M.A.; Maggiora, G.M. Concepts and applications of molecular similarity; Wiley: 1990.
- Wang, M.; Wang, Z.; Sun, H.; Wang, J.; Shen, C.; Weng, G.; Chai, X.; Li, H.; Cao, D.; Hou, T. Deep learning approaches for de novo drug design: An overview. Current Opinion in Structural Biology 2022, 72, 135–144. [Google Scholar] [CrossRef]
- Li, Y.; Hu, J.; Wang, Y.; Zhou, J.; Zhang, L.; Liu, Z. DeepScaffold: a comprehensive tool for scaffold-based de novo drug discovery using deep learning. Journal of chemical information and modeling 2019, 60, 77–91. [Google Scholar] [CrossRef]
- Gu Yaowen, Z.B. , Zheng Si, Yang Fengchun, Li Jiao. Predicting Drug ADMET Properties Based on Graph Attention Network. Data Analysis and Knowledge Discovery 2021, 5, 76–85. [Google Scholar] [CrossRef]
- Yang, L.; Jin, C.; Yang, G.; Bing, Z.; Huang, L.; Niu, Y.; Yang, L. Transformer-based deep learning method for optimizing ADMET properties of lead compounds. Physical Chemistry Chemical Physics 2023. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zheng, S.; Yin, Q.; Jiang, R.; Li, J. REDDA: Integrating multiple biological relations to heterogeneous graph neural network for drug-disease association prediction. Computers in biology and medicine 2022, 150, 106127. [Google Scholar] [CrossRef]
- Gu, Y.; Zheng, S.; Zhang, B.; Kang, H.; Li, J. MilGNet: A Multi-instance Learning-based Heterogeneous Graph Network for Drug repositioning. Proceedings of 2022 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); pp. 430–437.
- Kimber, T.B.; Chen, Y.; Volkamer, A. Deep Learning in Virtual Screening: Recent Applications and Developments. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Yaowen, G.; Si, Z.; Fengchun, Y.; Jiao, L. GNN-MTB: An Anti-Mycobacterium Drug Virtual Screening Model Based on Graph Neural Network. Data Analysis and Knowledge Discovery 2023, 6, 93–102. [Google Scholar]
- Liu, Z.; Du, J.; Fang, J.; Yin, Y.; Xu, G.; Xie, L. DeepScreening: a deep learning-based screening web server for accelerating drug discovery. Database 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, L.; Popovic, M.; Tijanic, N.; Rakocevic, G.; Kalinic, M. Improved scaffold hopping in ligand-based virtual screening using neural representation learning. Journal of Chemical Information and Modeling 2020, 60, 4629–4639. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Hu, H.; Yang, Z.; Xu, H.; Wu, J. Realvs: toward enhancing the precision of top hits in ligand-based virtual screening of drug leads from large compound databases. Journal of Chemical Information and Modeling 2021, 61, 4924–4939. [Google Scholar] [CrossRef]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: a conformational search method for efficient generation of bioactive conformers. Journal of chemical information and modeling 2010, 50, 534–546. [Google Scholar] [CrossRef]
- Méndez-Lucio, O.; Ahmad, M.; del Rio-Chanona, E.A.; Wegner, J.K. A geometric deep learning approach to predict binding conformations of bioactive molecules. Nature Machine Intelligence 2021, 3, 1033–1039. [Google Scholar] [CrossRef]
- Sauer, W.H.; Schwarz, M.K. Molecular shape diversity of combinatorial libraries: a prerequisite for broad bioactivity. Journal of chemical information and computer sciences 2003, 43, 987–1003. [Google Scholar] [CrossRef]
- Hu, G.; Kuang, G.; Xiao, W.; Li, W.; Liu, G.; Tang, Y. Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. Journal of chemical information and modeling 2012, 52, 1103–1113. [Google Scholar] [CrossRef]
- Shang, J.; Dai, X.; Li, Y.; Pistolozzi, M.; Wang, L. HybridSim-VS: a web server for large-scale ligand-based virtual screening using hybrid similarity recognition techniques. Bioinformatics (Oxford, England) 2017, 33, 3480–3481. [Google Scholar] [CrossRef]
- Riniker, S.; Landrum, G.A. Open-source platform to benchmark fingerprints for ligand-based virtual screening. Journal of cheminformatics 2013, 5, 26. [Google Scholar] [CrossRef]
- Zankov, D.V.; Matveieva, M.; Nikonenko, A.V.; Nugmanov, R.I.; Baskin, I.I.; Varnek, A.; Polishchuk, P.; Madzhidov, T.I. QSAR modeling based on conformation ensembles using a multi-instance learning approach. Journal of Chemical Information and Modeling 2021, 61, 4913–4923. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: towards direct deposition of bioassay data. Nucleic acids research 2019, 47, D930–d940. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: new data content and improved web interfaces. Nucleic acids research 2021, 49, D1388–d1395. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic acids research 2016, 44, D1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Škuta, C.; Southan, C.; Bartůněk, P. Will the chemical probes please stand up? RSC medicinal chemistry 2021, 12, 1428–1441. [Google Scholar] [CrossRef]
- Harding, S.D.; Armstrong, J.F.; Faccenda, E.; Southan, C.; Alexander, S.P.H.; Davenport, A.P.; Pawson, A.J.; Spedding, M.; Davies, J.A. The IUPHAR/BPS guide to PHARMACOLOGY in 2022: curating pharmacology for COVID-19, malaria and antibacterials. Nucleic acids research 2022, 50, D1282–d1294. [Google Scholar] [CrossRef]
- Isigkeit, L.; Chaikuad, A.; Merk, D. A Consensus Compound/Bioactivity Dataset for Data-Driven Drug Design and Chemogenomics. Molecules (Basel, Switzerland) 2022, 27. [Google Scholar] [CrossRef]
- Tweedie, S.; Braschi, B.; Gray, K.; Jones, T.E.M.; Seal, R.L.; Yates, B.; Bruford, E.A. Genenames.org: the HGNC and VGNC resources in 2021. Nucleic acids research 2021, 49, D939–d946. [Google Scholar] [CrossRef]
- Morgan, H.L. The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. Journal of chemical documentation 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Swain, M. MolVS: molecule validation and standardization. Web Page 2018. [Google Scholar]
- Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. Greg Landrum 2013, 8. [Google Scholar]
- Riniker, S.; Landrum, G.A. Better informed distance geometry: using what we know to improve conformation generation. Journal of chemical information and modeling 2015, 55, 2562–2574. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. Journal of computational chemistry 1999, 20, 730–748. [Google Scholar] [CrossRef]
- Shi, X.; Xing, F.; Xie, Y.; Zhang, Z.; Cui, L.; Yang, L. Loss-based attention for deep multiple instance learning. In Proceedings of Proceedings of the AAAI conference on artificial intelligence; pp. 5742–5749.
- Polton, D. Installation and operational experiences with MACCS (Molecular Access System). Online Review 1982, 6, 235–242. [Google Scholar] [CrossRef]
- Drucker, H.; Cortes, C. Boosting decision trees. Advances in neural information processing systems 1995, 8. [Google Scholar]
- Chen, T.; He, T.; Benesty, M.; Khotilovich, V.; Tang, Y.; Cho, H.; Chen, K.; Mitchell, R.; Cano, I.; Zhou, T. Xgboost: extreme gradient boosting. R package version 0.4-2 2015, 1, 1–4. [Google Scholar]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. Graph attention networks. arXiv 2017, arXiv:1710.10903. [Google Scholar]
- Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H.; et al. Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism. Journal of medicinal chemistry 2020, 63, 8749–8760. [Google Scholar] [CrossRef]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: moving beyond fingerprints. Journal of computer-aided molecular design 2016, 30, 595–608. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nature chemistry 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Shultz, M.D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. Journal of medicinal chemistry 2019, 62, 1701–1714. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2. 0: New docking methods, expanded force field, and python bindings. Journal of chemical information and modeling 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Liu, S.; Wang, H.; Liu, W.; Lasenby, J.; Guo, H.; Tang, J. Pre-training molecular graph representation with 3d geometry. arXiv 2021, arXiv:2110.07728. [Google Scholar]
- Stärk, H.; Beaini, D.; Corso, G.; Tossou, P.; Dallago, C.; Günnemann, S.; Liò, P. 3d infomax improves gnns for molecular property prediction. Proceedings of International Conference on Machine Learning; pp. 20479–20502.
- Jiao, R.; Han, J.; Huang, W.; Rong, Y.; Liu, Y. 3D equivariant molecular graph pretraining. arXiv 2022, arXiv:2207.08824. [Google Scholar]
- Gu, Y.; Zheng, S.; Li, J. CurrMG: A Curriculum Learning Approach for Graph Based Molecular Property Prediction. Proceedings of 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 9-12 Dec. 2021; pp. 2686–2693. [Google Scholar]
- Gu, Y.; Zheng, S.; Xu, Z.; Yin, Q.; Li, L.; Li, J. An efficient curriculum learning-based strategy for molecular graph learning. Briefings in bioinformatics 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Schütt, K.T.; Sauceda, H.E.; Kindermans, P.-J.; Tkatchenko, A.; Müller, K.-R. Schnet–a deep learning architecture for molecules and materials. The Journal of Chemical Physics 2018, 148, 241722. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Becker, F.; Günnemann, S. Gemnet: Universal directional graph neural networks for molecules. Advances in Neural Information Processing Systems 2021, 34, 6790–6802. [Google Scholar]
- Schütt, K.; Unke, O.; Gastegger, M. Equivariant message passing for the prediction of tensorial properties and molecular spectra. Proceedings of International Conference on Machine Learning; 2021; pp. 9377–9388. [Google Scholar]
Figure 1.
Bioactivity data collecting, integrating, filtering and processing workflow.
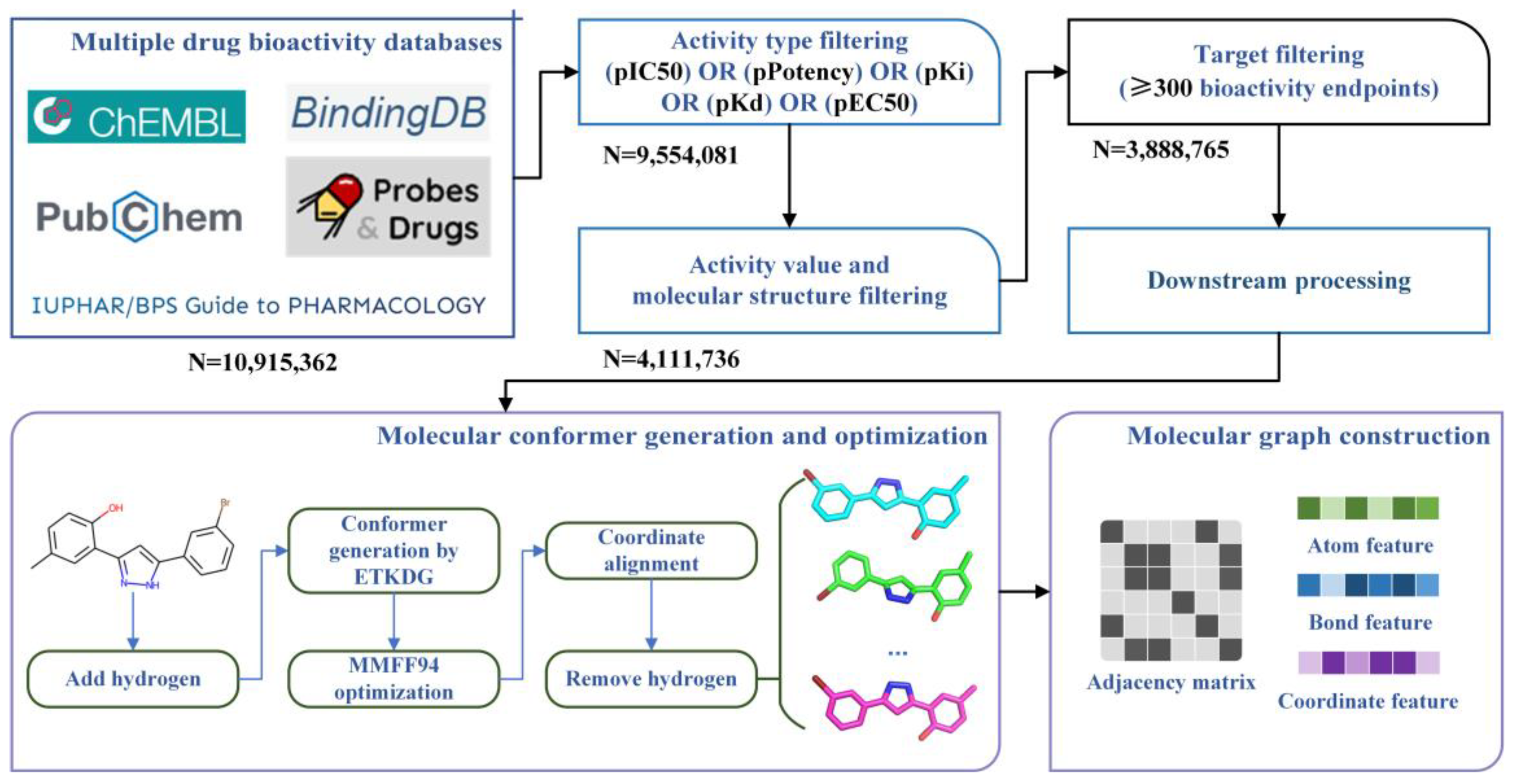
Figure 2.
Diagram of constructing a molecular graph from the query molecule and its conformers.
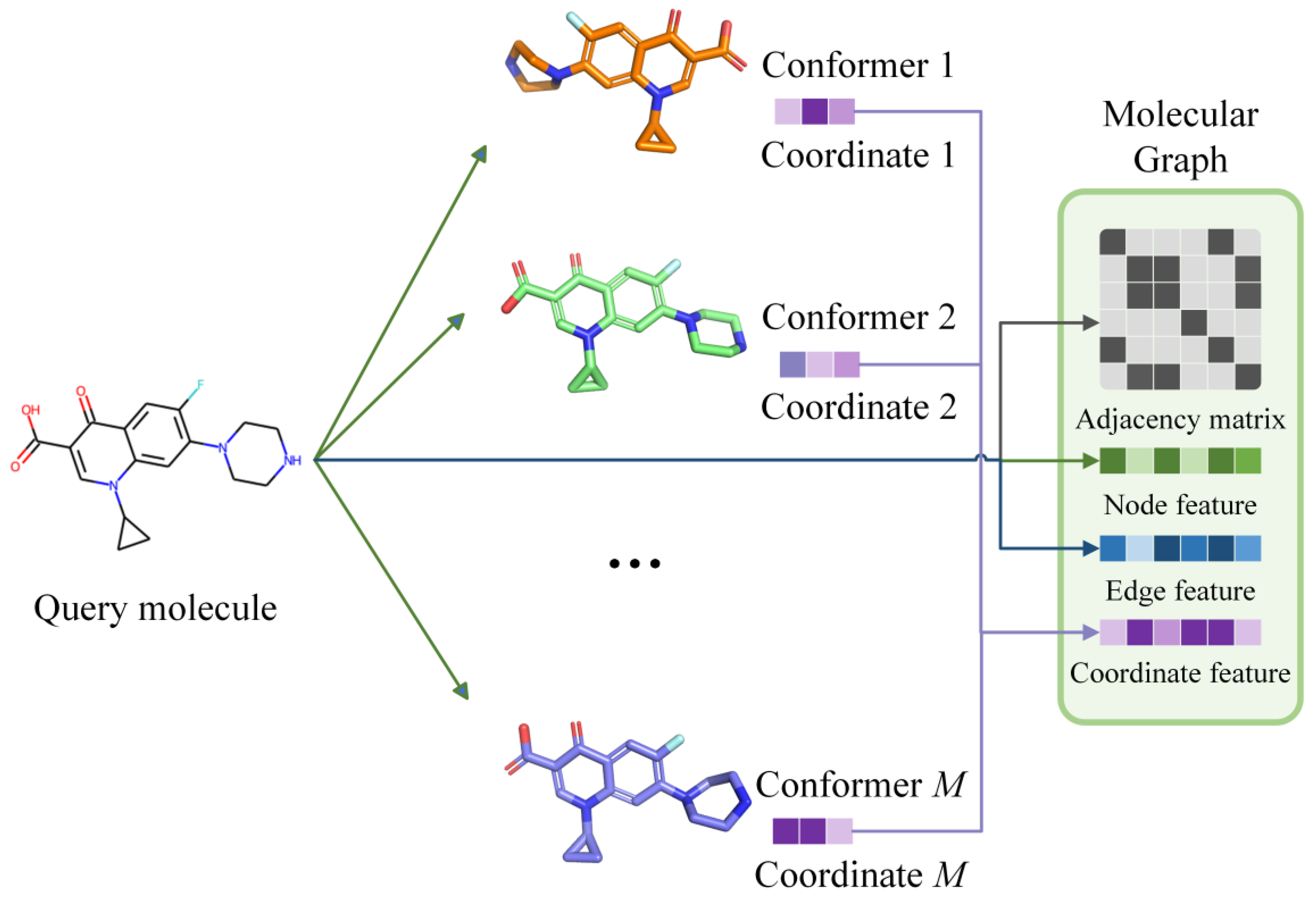
Figure 3.
The architecture of EquiVS.
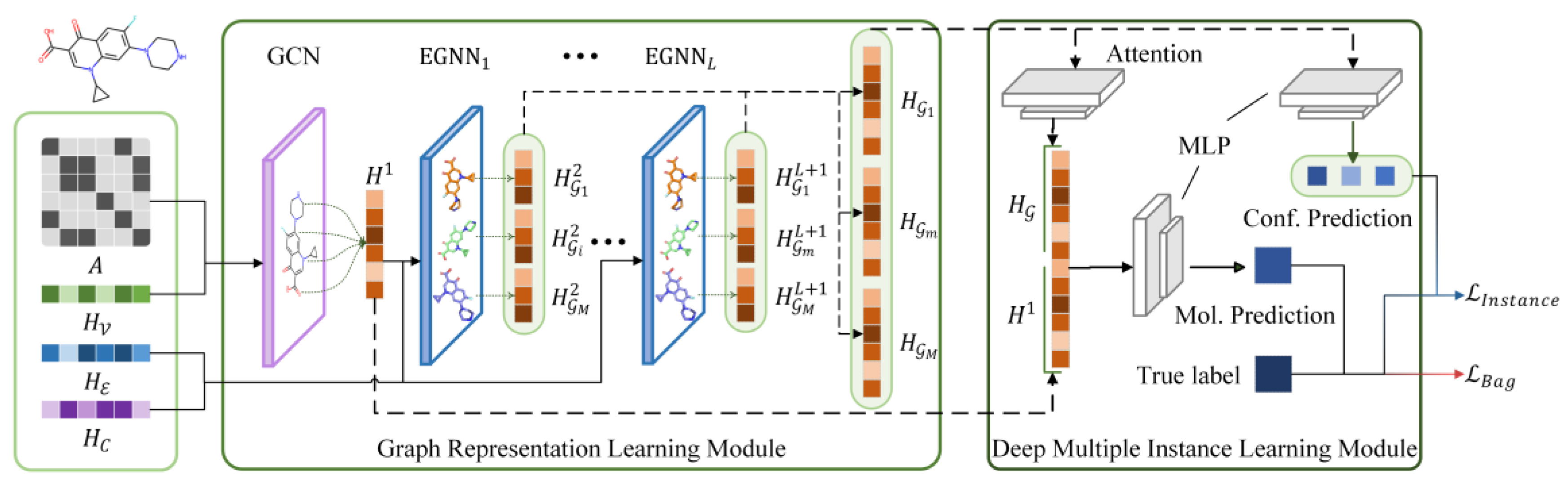
Figure 4.
Distribution visualization of raw bioactivity endpoints. A. Pie plot of bioactivity type proportion; B. Venn plot of data overlap in different source databases; C. Distribution plot of bioactivity value in different source databases; D. Distribution of data scale of different targets.
Figure 4.
Distribution visualization of raw bioactivity endpoints. A. Pie plot of bioactivity type proportion; B. Venn plot of data overlap in different source databases; C. Distribution plot of bioactivity value in different source databases; D. Distribution of data scale of different targets.
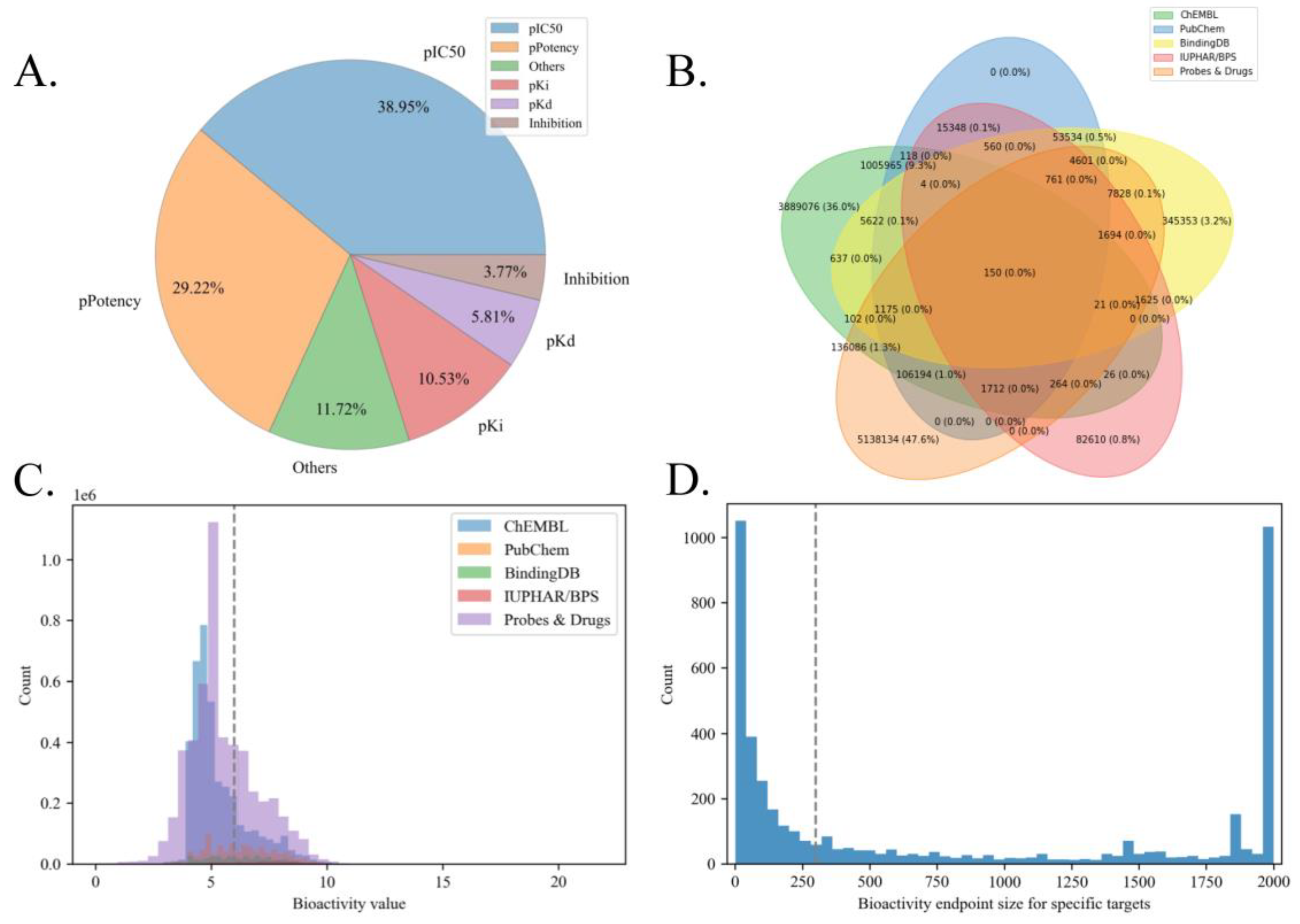
Figure 5.
QED property distributions of benchmark dataset. A. Distribution of MW; B. Distribution of Num. HBD; C. Distribution of Num. HBA; D. Distribution of AlogP; E. Distribution of Num. ROTB; F. Distribution of PSA; G. Distribution of Num. AROM; H. Distribution of Num. ALERTS.
Figure 5.
QED property distributions of benchmark dataset. A. Distribution of MW; B. Distribution of Num. HBD; C. Distribution of Num. HBA; D. Distribution of AlogP; E. Distribution of Num. ROTB; F. Distribution of PSA; G. Distribution of Num. AROM; H. Distribution of Num. ALERTS.
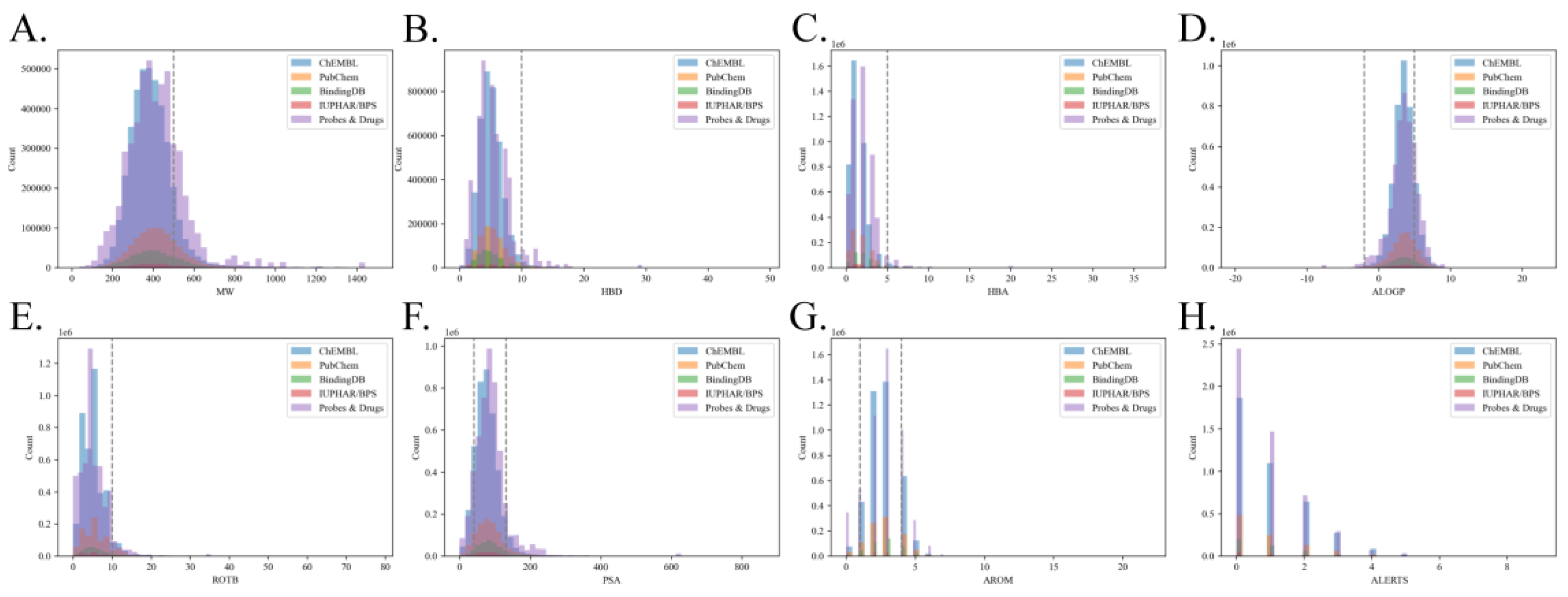
Figure 6.
Model performances (R2, MSE, and MAE) of EquiVS and 10 baseline methods on the bioactivity benchmark dataset of feasible targets.
Figure 6.
Model performances (R2, MSE, and MAE) of EquiVS and 10 baseline methods on the bioactivity benchmark dataset of feasible targets.
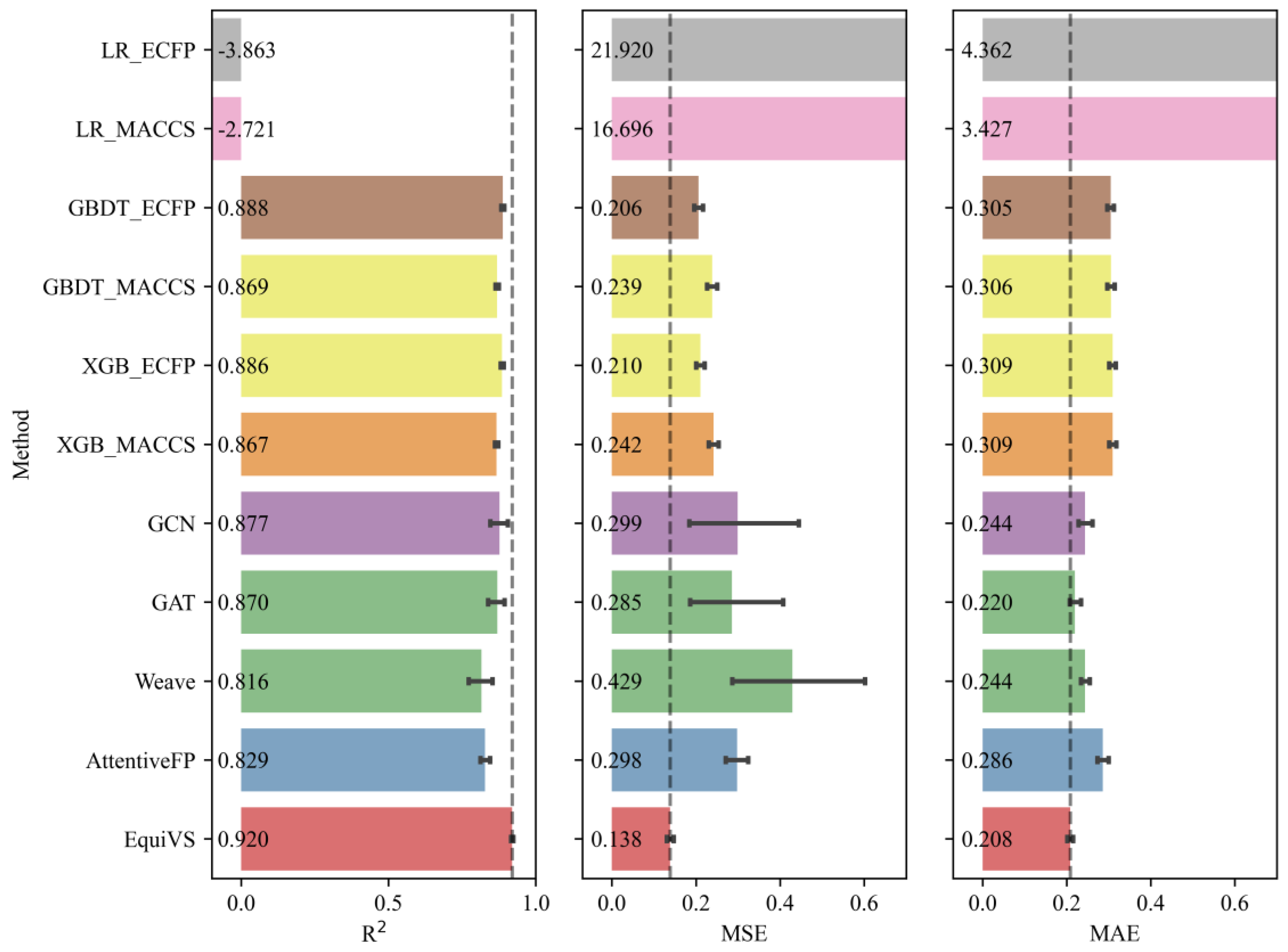
Figure 7.
Model performances (R2, MSE, and MAE) of EquiVS and 5 variant models on a selective set of bioactivity benchmark dataset.
Figure 7.
Model performances (R2, MSE, and MAE) of EquiVS and 5 variant models on a selective set of bioactivity benchmark dataset.

Figure 8.
An attention visualization case for optimal conformer discovery. A. Distribution of attention coefficients for conformers in CDK2 sub-dataset; B. Docking pose of CDK2 complexed with the case molecule; C. 10 conformers of the case molecule with bioactivity predictions, attention coefficients, and vina scores.
Figure 8.
An attention visualization case for optimal conformer discovery. A. Distribution of attention coefficients for conformers in CDK2 sub-dataset; B. Docking pose of CDK2 complexed with the case molecule; C. 10 conformers of the case molecule with bioactivity predictions, attention coefficients, and vina scores.
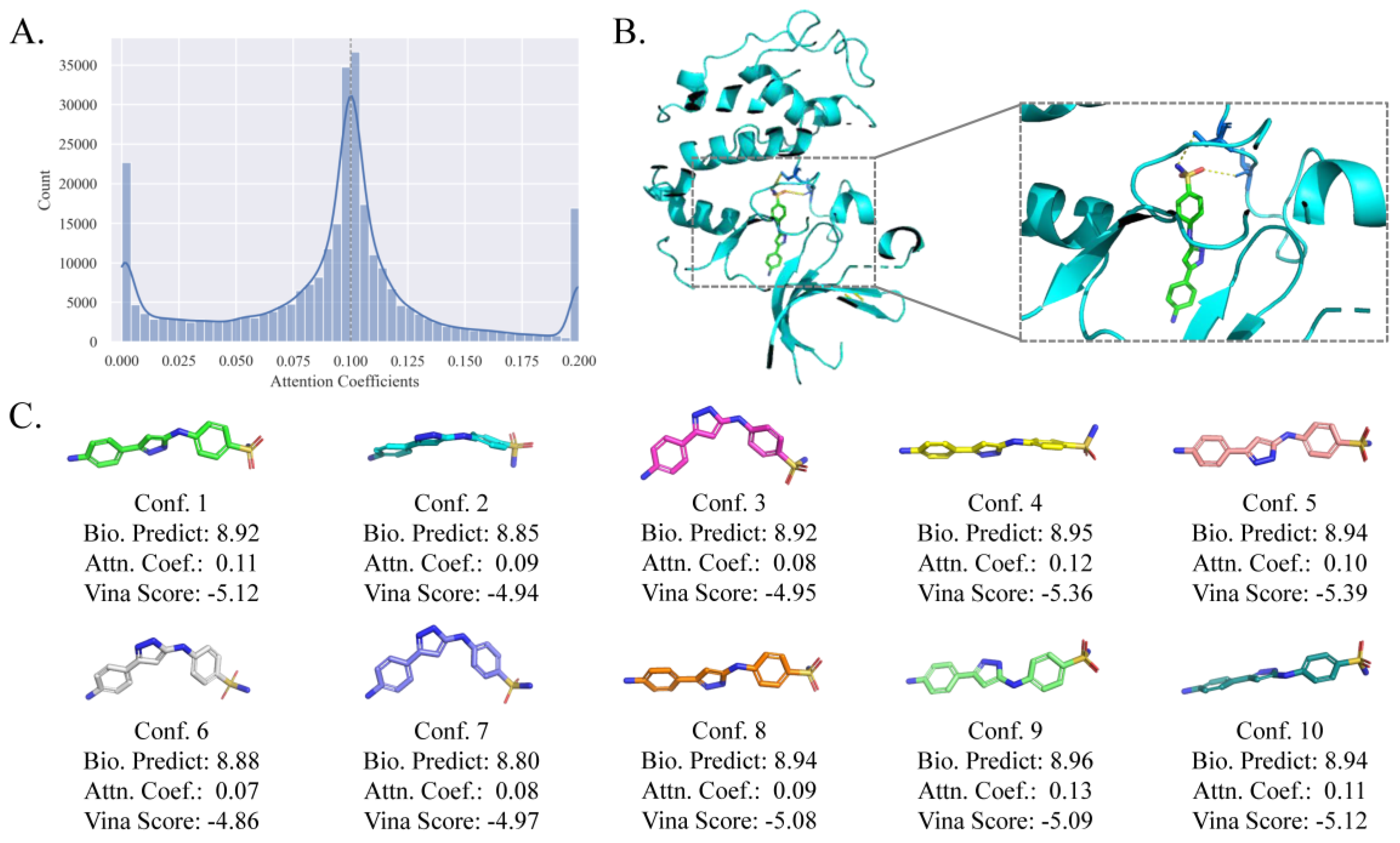

Table 1.
Detailed information about five databases.
| Database | Description | Version | Num. Molecule |
|---|---|---|---|
| ChEMBL | Contains the bioactivity data of more than 2.1 million experimentally determined drug-like molecules. | 28 | 1,131,947 |
| PubChem | Contains the bioactivity data and physiochemical properties of more than 1.1 million molecules. | 11.01.21 | 444,152 |
| BindingDB | Contains binding affinity data of approximately 26,000 drug-like molecules to specific biological targets. | 25.02.21 | 26,856 |
| Probes&Drugs | Contains manually collected biological target and bioactivity data of pharmacologically active compounds. | 2021.1 | 34,211 |
| IUPHAR/BPS | Contains bioactivity data, target and signaling pathway information of approximately 29,000 compounds from 30 public and commercial libraries. | 02b_2021 | 7,371 |
Table 2.
Descriptive statistical results of bioactivity endpoints in different source databases.
| Bioactivity | Statistics | ChEMBL | PubChem | BindingDB | IUPHAR/BPS | Probes&Drugs |
|---|---|---|---|---|---|---|
| Value | Avg. | 5.33 | 6.29 | 6.17 | 7.31 | 5.52 |
| Std. | 1.25 | 1.52 | 1.58 | 1.48 | 1.48 | |
| Min. | 0.10 | 0.10 | 0.10 | 0.80 | 0.10 | |
| Max. | 15.90 | 13.00 | 12.20 | 18.00 | 21.80 | |
| Category | Num. Active | 939,036 | 540,384 | 228,907 | 78,483 | 1,656,557 |
| PCT. Active | 23.56% | 56.72% | 54.9% | 81.42% | 32.93% | |
| Num. Inactive | 3,047,145 | 412,288 | 188,067 | 17,915 | 3,373,950 | |
| PCT. Inactive | 76.44% | 43.28% | 45.1% | 18.58% | 67.07% |
Footnote: Avg.: Average; Std.: Standard Deviation; Num.: Number of; PCT.: Percentile of.
Table 3.
Model performances of EquiVS and 10 baseline methods on bioactivity benchmark dataset.
| Type | Method | R2 | MSE | MAE |
|---|---|---|---|---|
| Machine Learning |
LR_ECFP | -4.543±5.443 | 24.844±24.904 | 4.960±4.956 |
| LR_MACCS | -3.438±5.302 | 19.853±24.339 | 4.063±4.798 | |
| GBDT_ECFP | 0.831±0.189 | 0.240±0.256 | 0.325±0.176 | |
| GBDT_MACCS | 0.810±0.202 | 0.273±0.286 | 0.328±0.190 | |
| XGB_ECFP | 0.830±0.188 | 0.243±0.254 | 0.329±0.175 | |
| XGB_MACCS | 0.808±0.201 | 0.276±0.285 | 0.331±0.189 | |
| Deep Learning |
GCN | 0.768±0.960 | 0.509±3.868 | 0.280±0.412 |
| GAT | 0.760±0.838 | 0.413±2.962 | 0.259±0.282 | |
| Weave | 0.705±1.056 | 0.601±4.051 | 0.286±0.297 | |
| AttentiveFP | 0.746±0.390 | 0.359±0.587 | 0.330±0.300 | |
| EquiVS | 0.833±0.243 | 0.208±0.282 | 0.257±0.189 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



