Submitted:
08 May 2023
Posted:
15 May 2023
You are already at the latest version
Abstract
Abstract
Coenzyme Q10 (CoQ10) has a number of vital functions in all cells, both mitochondrial and extra-mitochondrial. In addition to its key role in mitochondrial oxidative phosphorylation, CoQ10 serves as a lipid soluble antioxidant, plays an important role in fatty acid beta-oxidation and pyrimidine and lysosomal metabolism, as well as directly mediating the expression of a number of genes, including those involved in inflammation. Because of the multiplicity of roles in cell function, it is not surprising that deficiency of CoQ10 has been implicated in the pathogenesis of a wide range of disorders. CoQ10 deficiency is broadly divided into primary and secondary types. Primary CoQ10 deficiency results from mutations in genes involved in the CoQ10 biosynthetic pathway. In man, at least 10 genes are required for the biosynthesis of functional CoQ10, a mutation in any one of which can result in a deficit in CoQ10 status. Patients may respond well to oral CoQ10 supplementation, although the condition must be recognised sufficiently early, before irreversible tissue damage has occurred. In this article, we have reviewed clinical studies (up to March 2023) relating to the identification of these deficiencies, and the therapeutic outcomes of CoQ10 supplementation.
Keywords:
coenzyme Q10
; oxidative stress
; primary deficiency
; blood brain barrier
; mitochondrial
; supplementation
1. Introduction
Coenzyme Q10 (CoQ10) is usually described as a vitamin-like substance, although it is endogenously synthesised within most cell types. CoQ10 has a number of functions of vital importance to normal cell function; these include i) its key role in cellular energy supply/ATP synthesis via mitochondrial oxidative phosphorylation (Figure 1) ; ii) its role as a major endogenously synthesised lipid soluble antioxidant, protecting cellular/sub-cellular organelle membranes from free radical induced oxidative damage; (iii) its role in the metabolism of lysosomes, sulphides, amino acids, pyrimidine and cholesterol; (iv) its role in directly mediating the expression of more than one hundred genes, including those involved in the inflammatory process [1]. Because of the multiplicity of roles in cell function, it is not surprising that deficiency of CoQ10 has been implicated in the pathogenesis of a wide range of disorders. CoQ10 deficiency is broadly divided into primary and secondary types. Primary CoQ10 deficiency results from mutations in genes involved in the CoQ10 biosynthetic pathway. In man, at least 10 genes are required for the biosynthesis of functional CoQ10, a mutation in any one of which can result in a deficit in CoQ10 status [2]. Primary CoQ10 deficiency has been estimated to affect approximately 120,000 patients worldwide [3]. In general, mitochondrial disorders are not treatable; the exception is primary CoQ10 deficiency, where patients may respond well to oral CoQ10 supplementation. However, the condition must be recognised sufficiently early, since once damage to critical organs such as the kidney or the nervous system is established, only minimal recovery is possible. In this article, we have reviewed clinical studies (up to March 2023), on a case by case basis, relating to the identification of these deficiencies, and the therapeutic outcomes of CoQ10 supplementation.
2. Biosynthesis of CoQ10
The biosynthesis of CoQ10 is a complex multi-step process which takes place in various sub-cellular locations [4]. The polyisoprenoid tail is synthesised (via polyprenyl diphosphate synthase) in the cytosol via the mevalonate pathway, with attachment to the benzoquinone ring (originating from tyrosine and its metabolite 4-hydroxybenzoate) taking place within mitochondria [4]. The ring structure is then further modified via hydroxylation, methylation and decarboxylation by a set of enzymes grouped in a complex [4]. At least 10 genes are required for the biosynthesis of functional CoQ10, a mutation in any one of which can result in a deficit in CoQ10 status [2]. Much of the data relating to CoQ10 biosynthesis was initially obtained from studies in yeast, with deficiencies corresponding to the above genes denoted as CoQ1 to CoQ11 (numbering refers to date order of identification) [5]. The biosynthesis of CoQ10 in yeast and man has subsequently been shown to be highly conserved [6]. With regard to the corresponding enzymatic/protein gene products, COQ1 (heterotetrameric decaprenyl diphosphate synthase, comprising PDSS1 and PDSS2) is involved in the synthesis of the polyisoprenoid chain [7] and COQ2 in the condensation of the isoprenoid chain with the benzoquinone ring [8]. COQ3, COQ5, COQ6 and COQ7 are involved in concomitant methylation, decarboxylation, hydroxylation and deamination reactions [9-12]. COQ4 is involved in the stabilisation of the CoQ10 synthetic complex [13]. COQ8A is necessary for phosphorylation of COQs 3, 5 and 7 [14]. The COQ9 lipid-binding protein is necessary for stabilisation of COQ7 [15], and COQ10A/COQ10B direct the localisation of CoQ10 within the mitochondrial membrane [16]. In humans, mutations in 10 of these genes have been identified to date, as described below: the corresponding gene products respectively are PDSS1 (phenyl diphosphate synthase subunit 1), PDSS2 (decaprenyl diphosphate synthase subunit 2), COQ2 (para-hydroxybenzoate-polyprenyl transferase), COQ4 (multienzyme complex organisation enzyme), COQ5 (methyltransferase), COQ6 (monooxygenase), COQ7 (DMG hydroxylase ADCK3 (renamed COQ8A, protein kinase), ADCK4 (renamed COQ8B, protein kinase), COQ9 (lipid-binding protein), and COQ10A/B (CoQ10 chaperone proteins).
3. Assessment of primary CoQ10 deficiency
Primary CoQ10 deficiency can be identified via both biochemical and genetic analysis. Biochemically, the determination of CoQ10 is not usually included as part of routine analysis by hospital pathology laboratories. When CoQ10 levels are measured, this is typically carried out using plasma samples, with an approximate reference range of 0.5 to 1.7 µM [17]. However for identification of primary CoQ10 deficiency, the tissues of choice are skeletal muscle biopsies or skin fibroblasts, since primary CoQ10 deficiency in tissues may not be manifest in plasma. Blood mononuclear cells have been suggested as an appropriate low invasive means of assessing endogenous CoQ10 status, and these can be isolated from 5 ml of EDTA blood [18]. Furthermore, in view of the increasing association of primary CoQ10 deficiency with kidney dysfunction, the determination of urinary CoQ10 has been suggested a reliable and non-invasive method of assessing urinary tract CoQ10 status [19].The most common analytical techniques used to assess CoQ10 status are based on high-pressure liquid chromatography (HPLC) with either ultraviolet (HPLC-UV) or electrochemical (HPLC-ED) detection. The demonstration of reduced biochemical activities of respiratory chain complexes, in particular, complexes I+III and II+III is also of relevance, since the activity of these enzyme complexes are dependent upon endogenous CoQ10 status. For genetic analysis, mutations are identified using EDTA blood samples for whole-exome sequencing and Sanger sequencing section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
4. Clinical studies relating to COQ mutations
In the following sections, clinical studies relating to each of the COQ genes 1-10 have been listed on a case by case basis in chronological order; each clinical study has been briefly summarised to include the number of patients, age of onset, symptoms, lethality, variant type and effect of CoQ10 supplementation (where attempted). Mutations (where known) in the various COQ genes are shown in parentheses for the DNA nucleotide change and corresponding protein change respectively.
COQ1: The COQ1 gene (also known as PDSS1/PDSS2) encodes subunits 1 and 2 of the enzyme hexaprenyl pyrophosphate synthetase, which catalyses the first step in CoQ10 biosynthesis i.e. synthesis of the polyisoprenoid tail. The PDSS1 gene is located on chromosome 10 and comprises 14 exons; PDSS2 is located on chromosome 6 and comprises 12 exons. Mutations in PDSS1 reportedly result in steroid resistant nephrotic syndrome, encephalopathy, and optic nerve atrophy. To date a total of 6 patients with PDSS1 mutations have been identified. These include two siblings with infantile onset neurosensorial deafness and optic atrophy, resulting from mutation ([c.924T>G] [p.Asp308Glu]) in the PDSS1 gene [20]; a more severe birth onset phenotype with kidney failure and death at 16 months, resulting from PDSS1 mutations ([c.661C>T] [p.Arg221Ter] and [c661-662inST] [pArg221Leufs16]) [21]; and two sisters (aged 6 and 14 years) with sensorineural deafness and optic atrophy, resulting from a PDSS1 mutation ([c.735G>T] [p.Gln245His]) [22]. An infant with mitochondrial encephalopathy, pulmonary hypertension, and chronic distal phalangeal erythema who subsequently died aged 3 years was shown to have mutations ([c.716 T > G] [p.Val239Gly] and [c.1183C > T] [p.Arg395*]) in the PDSS1 gene [23]. Only one of the above patients was subject to CoQ10 supplementation, where CoQ10 supplementation (15mg/kg/day) was ineffective in altering disease progression [23].
CoQ10 deficiency (COQ10D3) resulting from mutations in the PDSS2 gene are associated particularly with neonatal/infantile onset renal disease, with variable neurological involvement. To date a total of 7 patients with PDSS2 mutations have been identified. The first clinical report relating to mutation of the PDSS2 gene was by Lopez et al [24], who described an infant with nephrotic syndrome and encephalopathy, who subsequently died at 8 months. The PDSS2 mutation in this patient involved a C→T transition at nucleotide 964, changing amino acid 322 from glutamine to a stop codon, and a C→T transition at nucleotide 1145, changing amino acid 382 from serine to leucine. Subsequent cases included a fatal PDSS2 mutation ([c485A>G] [pHis162Arg]) in a 7-month-old male infant with nephrotic syndrome, encephalomyopathy, cardiomyopathy, deafness and retinitis pigmentosa [25], and four patients from 2 families with a less severe phenotype (cerebral palsy, ataxia) resulting from a ([c1145C>T] [pSer382Leu]) PDSS2 mutation described by Sadowski et al [26] , and by Rahman et al [27] (mutation not specified) respectively. Supplementation with CoQ10 was attempted only in the patient described by Lopez et al [24], where administration of CoQ10 (50mg/day) had no effect on disease progression.
COQ2: The COQ2 gene is located on chromosome 4 and comprises 7 exons. COQ2 encodes the enzyme p-hydroxybenzoate-polyprenyl transferase, which mediates the conjugation of the benzoquinone ring with the decaprenyl side chain in CoQ10 biosynthesis. COQ2 mutations (causing CoQ10 deficiency COQ10D1) typically manifest as encephalopathy and nephropathy of variable severity. The first report of primary CoQ10 deficiency resulting from a COQ2 mutation ([c890A>G] [pTyr297Cys]) was by Quinzii et al [28], two siblings with infantile nephropathy. Diomedi-Camassei et al [29] described two patients with COQ2 mutations, an infant with fatal encephalopathy and nephropathy who died at 6 months ([c.437G > A] [p437G > A]), and an 18 month old infant with non-fatal nephropathy ([c590G>A] [pArg197His] and [c683A>G] [pAsn228Ser]). Supplementation with CoQ10 (30mg/kg/day) resulted in clinical stabilisation of the latter patient. A fatal neonatal case of nephropathy resulting from a COQ2 mutation ([c1047delT] [pAsn351Ilefs15]) was reported by Mollet et al [20]. Other fatal neonatal or infantile cases resulting from COQ2 mutations included a two month old infant with myoclonic epilepsy and hypertrophic cardiomyopathy, resulting from a [c.326G > A] [p.Ser109Asn] COQ2 mutation, who subsequently died at 5 months despite supplementation with CoQ10 (5mg/kg/day) [30], and a fatal case of a newborn with multi-organ failure resulting from a [c.545T>G] [p.Met182Arg] COQ2 mutation [31]. Eroglu et al [32] identified 4 infants from two families with the [c.437G→A] [p.Ser146Asn]) COQ2 mutation; in two of these patients CoQ10 supplementation normalised renal function but did not improve neurological symptoms. Xu et al [33] described a case of infantile nephrotic syndrome in a 14 month old Chinese boy, resulting from COQ2 mutations ([c.518G>A] and [c.973A>G]); the patient’s renal function was improved following CoQ10 supplementation (30mg/kg/day). Starr et al [34] reported 3 children (aged 2-10 years) with nephrotic syndrome resulting from COQ2 mutations (2 missense (p.Thr325Ala and p.Thr294Ile), and one frameshift (c.176dupT), in two of whom renal function was restored following CoQ10 supplementation (30mg/kg/day). Wu et al [35] reported a 6 month old girl presenting with nephropathy with fatal outcome, resulting from a COQ2 mutation ([c.832 T > C] [p. Cys278Arg]). Abdelhakim et al [36] described three siblings from a Jewish family presenting with nephropathy and retinopathy, resulting from two COQ2 mutations ([c.288dupC] [p.(Ala97Argfs*56] and [c.376C > G] [p.(Arg126Gly]); supplementation with CoQ10 (30mg/kg/day for 6 months) prevented further visual deterioration in these patients. Li et al [37] identified COQ2 mutations [c.1058A > G] [p.Y353C] and [c.973A > G], [p.T325A]) in two Chinese infants with nephropathy; proteinuria was reduced following CoQ10 supplementation (30mg/kg/day). Rosado-Santos et al [38] described death in a newborn with severe fetal growth restriction, abnormal kidney function and lissencephaly resulting from COQ2 mutations ([c.590G>A] [p.(Arg197His] and [c.827del p.(Gly276Valfs*20]). Stallworth et al [39] described an eight-year-old child with nephropathy requiring renal transplantation who subsequently developed progressive cone-rod dystrophy and optic atrophy, resulting from the COQ2 variants [c683A>G] [pAsn228Ser] and [c518G>A] [parg173His]. In total, 39 COQ2 variants in 63 patients (including previously unreported cases) have been identified to date by Drovandi et al [40]; patients carried at least one missense variant, the most common of which was [c683A>G] [pAsn178Ser]. Supplementation with CoQ10 resulted in the partial or complete remission of proteinuria in more than half of the patients in the above study [41]. COQ2 variants have been associated with increased risk of developing multisystem atrophy or Parkinson’s disease [42].
COQ3: The COQ3 gene is located on chromosome 6 and comprises 9 exons. The COQ3 gene encodes a methyltransferase enzyme that catalyses both the O-methylation steps in CoQ10 biosynthesis. To date there have been no clinical studies reported in the medical literature relating to COQ3 mutations.
COQ4: The COQ4 gene is located on chromosome 9 and comprises 7 exons. The COQ4 gene encodes a multienzyme complex organisation/stabilisation enzyme localized to the matrix side of the mitochondrial inner membrane , which when deficient results in CoQ10 Deficiency-7 (COQ10D7), causing childhood onset neurodegeneration. As with other COQ mutations, the severity of disease depends on the location of the mutation. Mutations in in exons 1-4 are associated with less life-threating presentations, late onset, responsiveness to CoQ10 therapy, and a relatively long lifespan. In contrast, pathogenic mutations in exons 5-7 are associated with early onset, unresponsiveness to CoQ10 therapy, and early death. The first patient with primary CoQ10 deficiency resulting from mutation of the COQ4 gene was reported by Salviati et al [43], a three year old with neurological symptoms. Oral CoQ10 supplementation (30mg/kg/day) resulted in a significant improvement of neuromuscular symptoms.
COQ4 mutations resulting in neurological deterioration were described in 5 patients ([c718C>T] [pArg240Cys]; [c433C>G] [pArg145Gly]; [c190C>T] [pPro64Ser]; [c155T>C] [pLeu52Ser]) by Brea-Calvo et al [44], and 6 patients ([202G>C] [pAsp68His]; [c245T>A] [pLeu82Gln]; [c473G>A] pArg158GLN]; [c718C>T] [pArg240Cys]) by Chung et al [45]; onset was prenatal or perinatal, with early fatal outcome in the neonatal period or early infancy. Romero-Moya et al [46] reported a 4 year old with mental retardation and lethal rhabdomyolysis with a COQ4 mutation ([c.483G > C](E161D)). Sondheimer et al [47] described an infant with seizures and cardiomyopathy dying in early infancy, resulting from a deletion ([c.23_33del11] [p.Val8AlafsX19]), and two missense mutations ([c.331G>T] [p.Asp111Tyr] and [c.356C>T] [p.Pro119Leu]) in the COQ4 gene. Bosch et al [48] identified a patient with childhood onset spinocerebellar ataxia with stroke-like episodes, (with a different phenotype from the lethal infantile presentation described previously), resulting from mutation ([c.230C > T] [(p.Thr77Ile]) in the COQ4 gene. Supplementation with CoQ10 (1000mg/day) failed to prevent further stroke-like episodes.
Caglayan et al [49] described two siblings with childhood-onset, slowly progressive ataxia resulting from a COQ4 mutation (exon2:c.G164T:p.G55V). Supplementation with CoQ10 (200mg/day for 1 month) resulted in significant improvement of neurological symptoms in the more severely affected sibling. Ling et al [50] identified a COQ4 mutation (c.370G > A) in 3 Asian patients presenting with infantile encephalopathy or cardiomyopathy. Lu et al [51] found the same COQ4 mutation (c.370 G > A (p.G124S) in a Chinese patient with Leigh syndrome (age of onset at 2 months). Supplementation with CoQ10 allowed the patient to maintain a relatively stable health status. Yu et al [52] reported on a cohort of 11 Chinese patients from 9 unrelated families with the [c.370G>A] [p.Gly124Ser]) COQ4 mutation, 5 of whom had classical neonatal-onset encephalo-cardiomyopathy, with the others having infantile onset with more heterogeneous clinical presentations. Supplementation with CoQ10 (15-40mg/kg/day) was attempted in 7 of these 11 patients; seizure control was improved in two of these patients, while no benefit was noted in 5 patients who subsequently died. Chen et al [53] described a 5 month old patient with epileptic seizures resulting from a c.370G>A mutation in the COQ4 gene. Mero et al [54] reported two patients each with two COQ4 mutations ([c.577C>T] [p.Pro193Ser] and [c.718C>T] [p.Arg240Cys]), and [c.284G>A] [p.Gly95Asp] and [c.305G>A] [p.Arg102], respectively), presenting with motor impairment and ataxia.
In summary, some 28 COQ4 variants have been identified to date, comprising a total of 44 patients; three phenotypes have been described, an early-onset phenotype with neonatal brain anomalies and epileptic encephalopathy, an intermediate phenotype with distinct stroke-like lesions, and a moderate phenotype with non-specific brain pathology and a stable disease course [55]. Patients With COQ4 Variants in exons 1–4 i(.e. amino acid changes in the N-terminus of COQ4), and patients with the East Asian-specific c.370G > A COQ4 variant are generally responsive to supplementation with CoQ10, whereas patients with COQ4 variants in exons 5-7 (amino acid changes in the C-terminus of COQ4) are generally unresponsive.
COQ5: The COQ5 gene is located on chromosome 12 and contains 8 exons. The COQ5 gene encodes the enzyme responsible for the C-methyltransferase step in CoQ10 biosynthesis (i.e. conversion of 2 methoxy-6-polyprenyl-1,4-benzoquinol to 2 methoxy-5-methyl-6-polyprenyl-1,4 benzoquinol). To date, there has been only one clinical study reported relating to mutation of the COQ5 gene. Malicdan et al [56] reported 3 female siblings from a Middle East family, presenting in early childhood with seizures, cerebellar ataxia and cognitive disability, resulting from biallelic duplication of 9590bp in the COQ5 gene. Supplementation with CoQ10 (15mg/kg/day for 6 months) resulted in a significant improvement in their respective ICARS (International Cooperative Ataxia Rating Scale) scores.
COQ6: The COQ6 gene is located on chromosome 14 and comprises 14 exons. The protein encoded by this gene is a flavin-dependent monooxygenase localised to the matrix side of the inner mitochondrial membrane, which is responsible for the C5-hydroxylation of the quinone ring during CoQ10 synthesis. Mutations in this gene result in primary CoQ10 deficiency-6 (COQ10D6), an autosomal recessive disorder which typically manifests as progressive infantile-onset steroid-resistant nephrotic syndrome resulting in end-stage renal failure, together with sensorineural deafness; individuals may also be susceptible to development of Scwannomatosis a form of neurofibromatosis characterised by formation of benign tumours in the nervous system. The COQ6 mutation results in damaging effects in renal podocytes by inducing cell apoptosis, increasing cellular oxidative stress, and destroying the cytoskeleton. Whereas most forms of monogenic nephrotic syndrome in children do not respond to treatment, the identification of children with nephrotic syndrome resulting from defective CoQ10 biosynthesis is vital, since such individuals may respond to CoQ10 supplementation. Primary CoQ10 deficiency due to COQ6 mutations should be considered in children presenting with both steroid resistant nephrotic syndrome and sensorineural hearing loss.
Heeringa et al [57] first identified 6 different COQ6 mutations ([c484C>T] [pArg162]; [c564G>A] [pTrp188]; [c763G>A] [pGly255Arg]; [c1058C>A pAla353Asp]; [ c1341G>A] [pTrp447]; [c12383delG] [pGln461fs478])in 13 individuals from 7 families by homozygosity mapping. Each mutation was linked to early-onset (1-2 years) steroid resistant nephrotic syndrome and sensorineural deafness, with some patients responding favourably (reduced proteinuria, improved hearing) to oral CoQ10 supplementation (50mg twice daily). However such patients may not benefit from CoQ10 therapy when severe renal and neurological damage is established, so an early and accurate diagnosis of COQ10D6 and simultaneous CoQ10 intervention are critical in improving prognosis.
Sadowski et al [26] described 6 patients with infancy/childhood onset of nephropathy resulting from COQ6 mutations ([c1058C>A] [pAla353Asp]; [c1154A>C] [pAsp385Ala]; [c1235A>G] [pTyr412Cys]). Park et al [58] identified COQ6 mutations ([c189 191 del GAA] [pLys64del]; [c686A>C] [pGln229Pro]; [c782C>T] [pPro26Leu]) in a series of 6 children (age of onset 15-47 months), all of whom progressed to end stage renal disease requiring transplantation. Li et al [59] diagnosed a child at 1 year with a mutation ([c.1078C > T] [p.Arg360Trp]]) in the COQ6 gene; proteinuria was subsequently completely resolved following supplementation with CoQ10 (30mg/kg per day for 3 months). Cao et al [60] described a case of infantile nephrotic syndrome resulting from a COQ6 mutation ([c1078C>T] [pArg360Trp]); supplementation with CoQ10 (30mg/kg/day) over a period of 3 months restored normal renal function. Song et al [61] reported a 16 year old presenting with proteinuria, and subsequently identified as having a COQ6 mutation ([c.G41A] [p.W14X]). Supplementation with CoQ10 over a 6 month period reduced the 24 hour urinary protein by approximately 50%.
Stanczyk et al [62] described a 4 year old presenting with steroid resistant glomerulopathy with COQ6 mutations ([c.1078C > T] [p.Arg360Trp]]; [c.804delC] [pLeu269Trpfs13]). Supplementation with CoQ10 (30mg/kg per day for one month resulted in complete symptomatic remission. Yildirim et al [63] reported on a 7-year-old girl diagnosed with steroid-resistant nephrotic syndrome and sensorineural deafness, requiring haemodialysis and subsequent renal transplantation (mutation [c.1058C > A] [rs397514479] in exon 9 of COQ6).
Perrin et al [64] described COQ6 mutation in a patient with renal disease (requiring transplantation) and deafness, who also developed loss of vision. In this case, supplementation with the CoQ10 analogue idebenone, which has been used for the treatment of other types of optic neuropathy, improved visual impairment without affecting deafness. Wang et al [65] identified two Chinese siblings with COQ10D6 who presented with severe metabolic acidosis, proteinuria, hypoalbuminemia, growth retardation, and muscle hypotonia and died in early infancy (mutations : [c.249C > G] [p.Tyr83Ter] in exon 2 and [c.1381C > T] [p.Gln461Ter] in exon 12 of COQ6). A 19 month old patient with cardiomyopathy was found to have the COQ6 variant [c763G>A] [p.Gly255Arg]; the patient died from cardiorespiratory failure before CoQ10 supplementation could be started [66]. Nam et al [67] identified 12 children from 11 unrelated Korean families resulting from COQ6 mutations (c.189_191delGAA (p.Lys64del), c.484C>T (p.Arg162∗), c.686A>C (p.Gln229Pro), and c.782C>T (p.Pro261Leu). The effects of CoQ10 supplementation (30 mg/kg) on hearing loss was further investigated in 7 of these children (mean age 7.2 years at start of CoQ10 treatment). In terms of prevention of hearing loss, approximately 50% of patients responded well to treatment with CoQ10; those with the c.686A>C variant responded poorly to CoQ10 supplementation, but those with c.189_191delGAA or c.782C>T variants responded well.
The total number of patients identified with COQ6 related primary CoQ10 deficiency is currently 45, with 14 of these responding to CoQ10 supplementation (where attempted).
COQ7: The COQ7 gene is located on chromosome 16 and comprises 11 exons. The COQ7 gene encodes the enzyme 5-demethoxyubiquinone hydroxylase, which catalyzes the hydroxylation of converting demethoxyubiquinone to 5-hydroxy-ubiquinone. Mutations in the COQ7 gene result in primary CoQ10 deficiency-8 (COQ10D8). To date four clinical cases relating to mutations in the COQ7 gene have been reported. The first case was described by Freyer et al [68], a 9-year-old Syrian boy with progressive encephalo-neuro-nephro-cardiopathy resulting from a missense mutation ([c.422T>A] [p.Val141Glu]) in exon 4 of the COQ7 gene. CoQ10 levels were severely decreased in the skeletal muscle and fibroblasts of the patient. The authors demonstrated that the coenzyme Q analogue 2,4-dihydroxybenzoic acid was able to specifically bypass the COQ7 deficiency, increase cellular coenzyme Q levels and rescue the biochemical defect in patient fibroblasts. The second case was reported by Wang et al [69], a 6-year-old girl presenting with spasticity and bilateral sensorineural hearing loss resulting from two mutations ([c.308C>T[ [p. Thr103Met] and [c.332T>C] [p.Leu111Pro]) in the COQ7 gene. Only a moderate decrease in CoQ10 levels were found in cells from this patient. The third case was reported by Kwong et al [70], a Chinese boy presenting with encephalo-myo-nephro-cardiopathy, with a fatal outcome at one year of age, resulting from frameshift [c.599_600delinsTAATGCATC] [p.(Lys200Ilefs*56] and missense substitution [c.319C>T] [p.(Arg107Trp]) in the COQ7 gene. This patient showed a poor response to supplementation with CoQ10 (20mg/kg/day). For the fourth case, Wang et al [71] identified a 4 year old Turkish boy with a mutation ([c.161G>A] [p.Arg54Gln]) in exon 2 of the COQ7 gene, presenting with hypotonia, difficulty walking, motor developmental delay, ataxia, and spasticity. The patient’s skin fibroblasts showed a 45% reduction in CoQ10 level with concomitant reduction in mitochondrial respiratory capacity, which could be rescued using a CoQ10/ caspofungin formulation.
COQ8A: The COQ8A gene is located on chromosome 1 and comprises 18 exons. The COQ8A gene, also known as ADCK3 or CABC1, encodes an atypical protein kinase. Patients with mutations in this gene typically develop childhood onset cerebellar atrophy and ataxia, and have decreased CoQ10 content in their muscle, fibroblast, and lymphoblast cells. Mollet et al [72] described 4 children (aged 18 months to 3 years) from 3 unrelated families with seizures and cerebellar atrophy resulting from various COQ8A mutations ([c1655G>A] [Glu551Lys]; [p 815G>T] [Gly272Val]; [c636C>T] [Arg213Trp]; [c815G>A] [Gly272Asp]). Supplementation with CoQ10 (10mg/kg/day) did not result in clinical improvement in these patients. Lagier-Tourenne et al [73] identified 6 patients with childhood onset cerebellar atrophy and ataxia resulting from COQ8A mutations ([c500521del22insTTG] [pGln167Leufs36]; [c993C>T] [pLys314Gln360del]; c17501752delACC] [pThr584del]; [c16456G>A] [pGly549Ser]; [c1541A>G] [pTyr514Cys]; [c139+2T>C] [pAsp420Trpfs40]); no CoQ10 supplementation was attempted in any of these patients. Blumkin et al [74] reported two sisters with COQ8A mutations ([c805C>G] [pPro602Arg] and [c1750 1752 delACC] [pThr584del]) with variable phenotype; one sister had childhood onset severe progressive ataxia which was partially resolved following CoQ10 supplermentation (20mg/kg/day), whilst the second sister had only mild dysarthria. Hikmat et al [75] reported 3 Norwegian patients with childhood onset cerebellar ataxia and epilepsy, with stroke-like episodes, resulting from COQ8A mutations ([c895C>T] [pArg229Trp]; [c1732T>G] [pPhe578Val]). Two children with cerebellar atrophy and ataxia resulting from COQ8A mutations ([c803T>C] [pLeu227Pro] and [c1506+1G>A] [pVal503Metfs21]) were described by Jacobsen et al [76]; supplementation with CoQ10 (20mg/kg/day) over a 12 month period resulted in an improvement in ataxia and mental capacity in both siblings. Chang et al [77] carried out a literature review of 22 cases with COQ8A mutations for which CoQ10 supplementation had been attempted; 11 patients showed clinical benefit, with ataxia being the symptom showing greatest response. On this basis, the authors recommended a CoQ10 dosage regime of at least 15mg/kg/day for 6 months for patients with COQ8A related ataxia. Nair et al [78] detailed a 5 year old girl with severe intellectual disability and ataxia resulting from a COQ8A mutation ([c1534C>T] [pArg512Trp]). Schirinzi et al [79] described an improvement in motor performance following CoQ10 supplementation (15mg/kg/day) over a period of 6-12 months in 4 children with ataxia. Uccella et al [80] reported a child with early onset ataxia resulting from a COQ8 mutations (c.901C>T;c.589-3C>G), where CoQ10 supplementation slowed down disease progression. Paprocka et al [81] described a 22-month-old girl with cerebellar ataxia and developmental regression resulting from a COQ8A mutation ([c.811C>T][p.Arg271Cys] who showed improved communication and growth following supplementation with CoQ10 (300mg/day for 3 months). Degerliyurt et al [82] reported on a 16 year old patient with ataxia, cerebellar atrophy and cardiomyopathy resulting from a COQ8A mutation ([c901 C > T] [p.Arg301Trp]); the authors noted that supplementation with CoQ10 was started at too late a stage in disease to prevent the death of the patient. The most comprehensive study on patients with COQ8A mutations was carried out by Traschutz et al [83], who assessed 64 patients (including 39 previously unreported) from 51 families from 16 different countries. This work incorporates results from previous studies by Horvath et al [84], Mignot et al [85], and Mallaret et al [86]. Cerebellar ataxia was evident in all patients, with a mean age of onset of 7 years. A total of 44 COQ8A variants were identified in these patients, comprising of 26 missense and 18 loss of function (10 frameshift, 4 stop, 3 canonical splice, and 1 in-frame deletion), distributed across the entire COQ8A gene. Out of a total of 30 patients supplemented with CoQ10 (mean daily dose of 11mg/kg/day), approximately half of these patients were classed as responders to treatment.
COQ8B: The COQ8B gene is located on chromosome 19 and comprises 16 exons. The COQ8B gene (also known as ADCK4) similarly encodes an atypical protein kinase, variants in which result in steroid resistant nephrotic syndrome with variable neurological involvement (presenting mainly in childhood or adolescence). To date 99 patients with COQ8B mutations have been identified. Ashraf et al [87] reported a patient with nephropathy resulting from a homozygous COQ8B frameshift mutation, who subsequently had partial remission following CoQ10 supplementation. Korkmaz et al [88] identified 26 adolescent patients with nephropathy resulting from COQ8B mutations ([c293T>G] [pLeu98Arg]; [c332C>T] [[pArg178Trp]; [c929C>T [pPro301Leu]; [c748G>A] [pAsp250Asn]; [c1199dupA] [pHis400Glnfs11]; [c1339dupG] [pGlu447Glyfs10]). Two patients in the early stage of renal damage showed improved proteinuria following CoQ10 supplementation (15mg/kg/day) over a 6 week period. Atmaca et al [89] reported significant improvement in proteinuria in 8 patients with COQ8B-related nephropathy, following CoQ10 supplementation. Feng et al [90] identified 2 children with proteinuria resulting from a COQ8B mutation, one of whom showed a good response to CoQ10 therapy. Song et al [91] identified 20 Chinese children from 17 families with renal disease resulting from COQ8B mutations, with the c.737G>A (p.S246N) and c.748G>A (p.D250H) variants being most prevalent. In a trial group of 5 patients, supplementation with CoQ10 (15-30 mg/kg/day) reduced proteinuria.
COQ9: The COQ9 gene is located on chromosome 16 and comprises 9 rexons. The COQ9 gene encodes a lipid binding protein that is responsible, via its interaction with other COQ encoded enzymes (particularly COQ7), for stabilising the COQ10 biosynthetic complex. Mutations in this gene (responsible for COQ10D5) have been reported in 7 patients to date. Duncan et al [92] reported an infant with multisytem disease, including intractable seizures, global developmental delay, hypertrophic cardiomyopathy, and renal tubular dysfunction, who died at 2 years of age despite supplementation with CoQ10 (up to 300mg/day). This patient was found to have the COQ9 mutation [c.730C>T] [p.Arg244]. Danhauser et al [93] identified an infant of Turkish origin with fatal neonatal lactic acidosis and encephalopathy, resulting from a COQ9 mutation ([c.521+1del] [p.Ser127-Arg202del]). Smith et al [94] described 4 siblings with multisystem disease, two of whom died soon after birth with the COQ9 variants [c711+3G.A] [pAla203Asp237del] and [c521+2T>C] [pSer127Arg202del]. Olgac et al [95] described a 9 month old girl of Pakistani origin presenting with microcephaly and seizures, resulting from a COQ9 mutation ([c.384delG] [Gly129Valfs*17]). There was no improvement in neurological symptoms following supplementation with CoQ10 (5-50mg/kg/day).
COQ10A and COQ10B: The COQ10A gene is located on chromosome 12 and comprises 6 exons; COQ10B is located on chromosome 2 and comprises 6 exons. The COQ10A and COQ10B genes encode lipid binding proteins which act as molecular chaperones, directing CoQ10 molecules to their final placement within membranes. COQ10A and COQ10B are expressed in all tissues, although COQ10A is predominantly expressed in heart and skeletal muscle. Polymorphisms in COQ10A or COQ10B have been implicated in predisposing patients to statin-associated myopathy [96]. To date there have been no clinical studies published relating to COQ10A or COQ10B variants.
5. Conclusions
Primary CoQ10 deficiency resulting from COQ gene mutations is associated with a heterogeneous spectrum of clinical phenotypes; however from the data presented above (summarised in Table 1), the following generalisations may be made: 1) the disorder typically has a neonatal, infantile or childhood age of onset; 2) patients typically present with neurological dysfunction (encephalopathy, psychomotor delay, cerebellar atrophy/ataxia, optic atrophy), nephropathy, cardiomyopathy, or any combination thereof; 3) the outcome for patients is typically either serious disability or fatality. To date, some 300 patients with primary CoQ10 deficiency have been identified, in approximately 100 of whom CoQ10 supplementation was attempted, as described in the present review. Some of the above syndromes can be rescued by oral supplementation with CoQ10, particularly when diagnosed sufficiently early; however the response to therapy depends on which COQ gene has been affected, and the particular location of the mutation within each of the respective genes. Steroid resistant nephrotic syndrome resulting from COQ mutations appears to be particularly amenable to treatment via CoQ10 supplementation. For example Drovandi et al [41] assessed the longer term efficacy of supplemental CoQ10 in a series of 40 patients (less than 18 years) with chronic kidney disease (stage 1-4) compared to an untreated cohort matched by genotype, age, kidney function, and proteinuria. Supplementation with CoQ10 supplementation resulted in a substantial and significant sustained reduction of proteinuria (by 88%) after 12 months. Complete remission of proteinuria was more frequently observed in patients with renal disease resulting from COQ6 mutations. Supplementation with CoQ10 resulted in significantly better preservation of kidney function (5-year kidney failure-free survival 62% vs. 19%), together with improvements in neurological manifestations and general health. The authors of this study consider that patients with this type of primary CoQ10 deficiency should receive early and life-long supplementation with CoQ10 to decrease kidney disease progression and prevent further damage to other organs.
In addition to the above, the response to therapy can depend on the dose and duration of supplementation, as well as the formulation of the supplement; formulations which have not been subject to CoQ10 crystal dispersion may have their bioavailability reduced by 75% [97]. There is currently no consensus as to the dosage of CoQ10 for treatment of primary deficiency disorders, although a dose of 15-30mg/kg/day has been used in a number of studies. In patients who have died, lack of response to CoQ10 supplementation results when irreversible tissue damage has already occurred prior to diagnosis; in such patients CoQ10 deficiency in cultured fibroblasts can typically be rescued following CoQ10 supplementation. In neonatal fatalities, there is a rationale for CoQ10 supplementation of at-risk mothers during pregnancy, in order to reduce tissue damage during fetal development. It is also of note that the correction of CoQ10 deficiency in cultured fibroblasts or yeast models, but not in patients with neurological symptoms, may indicate insufficient uptake of exogenous CoQ10 across the blood-brain barrier in these patients. Finally, there is a need for a method of determining CoQ10 during newborn screening to enable the identification and treatment of primary CoQ10 deficient patients prior to the maturation of the blood brain barrier.
Author Contributions
Both authors contributed equally to all aspects of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not relevant
Informed Consent Statement
Not relevant
Conflicts of Interest
Dr Mantle is medical adviser to Pharma Nord (UK) Ltd.
References
- Crane, FL. Biochemical functions of coenzyme Q10. Journal of the American College of Nutrition 2001, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L. , Trevisson E., Doimo M., Navas P. Primary Coenzyme Q10 Deficiency. In: Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Mirzaa G., Amemiya A., editors. GeneReviews((R)) University of Washington; Seattle, WA, USA: 2017.
- Hughes BG, Harrison PM, Hekimi S. Estimating the occurrence of primary ubiquinone deficiency by analysis of large-scale sequencing data. Sci Rep. 2017, 7, 17744. [Google Scholar] [CrossRef]
- Stefely JA, Pagliarini DJ. Biochemistry of mitochondrial coenzyme Q biosynthesis. Trends Biochem Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef] [PubMed]
- Allan CM, Awad AM, Johnson JS, Shirasaki DI, Wang C, Blaby-Haas CE, Merchant SS, Loo JA, Clarke CF. Identification of Coq11, a new coenzyme Q biosynthetic protein in the CoQ-synthome in Saccharomyces cerevisiae. J Biol Chem. 2015, 290, 7517–7534. [Google Scholar] [CrossRef] [PubMed]
- Awad AM, Bradley MC, Fernández-Del-Río L, Nag A, Tsui HS, Clarke CF. Coenzyme Q 10 deficiencies: pathways in yeast and humans. Essays Biochem. 2018, 62, 361–376. [CrossRef]
- Yen HC, Yeh WY, Lee SH, Feng YH, Yang SL. Characterization of human mitochondrial PDSS and COQ proteins and their roles in maintaining coenzyme Q10 levels and each other's stability. Biochim Biophys Acta Bioenerg. 2020, 1861, 148192. [Google Scholar] [CrossRef]
- Forsgren M, Attersand A, Lake S, Grünler J, Swiezewska E, Dallner G, Climent I. Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem J. 2004, 382, 519–526. [Google Scholar] [CrossRef]
- Jonassen T, Clarke CF. Isolation and functional expression of human COQ3, a gene encoding a methyltransferase required for ubiquinone biosynthesis J Biol Chem. 2000, 275, 12381–12387. [CrossRef]
- Nguyen TP, Casarin A, Desbats MA, Doimo M, Trevisson E, Santos-Ocaña C, Navas P, Clarke CF, Salviati L. Molecular characterization of the human COQ5 C-methyltransferase in coenzyme Q10 biosynthesis. Biochim Biophys Acta. 2014, 1841, 1628–1638. [Google Scholar] [CrossRef]
- Ozeir M, Pelosi L, Ismail A, Mellot-Draznieks C, Fontecave M, Pierrel F. Coq6 is responsible for the C4-deamination reaction in coenzyme Q biosynthesis in Saccharomyces cerevisiae. J Biol Chem. 2015, 290, 24140–24151. [Google Scholar] [CrossRef]
- Marbois BN, Clarke CF. The COQ7 gene encodes a protein in saccharomyces cerevisiae necessary for ubiquinone biosynthesis. J Biol Chem. 1996, 271, 2995–3004. [Google Scholar] [CrossRef] [PubMed]
- Belogrudov GI, Lee PT, Jonassen T, Hsu AY, Gin P, Clarke CF. Yeast COQ4 encodes a mitochondrial protein required for coenzyme Q synthesis. Arch Biochem Biophys. 2001, 392, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Chang A, Ruiz-Lopez M, Slow E, Tarnopolsky M, Lang AE, Munhoz RP. ADCK3-related Coenzyme Q10 Deficiency: A potentially treatable genetic disease. Mov Disord Clin Pract. 2018, 5, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Lohman DC, Forouhar F, Beebe ET, Stefely MS, Minogue CE, Ulbrich A, et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc Natl Acad Sci U S A. 2014, 111, E4697–705. [Google Scholar] [CrossRef]
- 16. Tsui HS, Pham NVB, Amer BR, Bradley MC, Gosschalk JE, Gallagher-Jones M, Ibarra H, Clubb RT, Blaby-Haas CE, Clarke CF. Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function. 60, 2019; 60. [CrossRef]
- Yubero, D. , Montero R., Artuch R., Land J.M., Heales S.J., Hargreaves I.P. Biochemical diagnosis of coenzyme q10 deficiency. Mol. Syndromol. 2014, 5, 147–155. [Google Scholar] [CrossRef]
- Duncan AJ, Heales SJ, Mills K, Eaton S, Land JM, Hargreaves IP. Determination of coenzyme Q10 status in blood mononuclear cells, skeletal muscle, and plasma by HPLC with di-propoxy-coenzyme Q10 as an internal standard. Clin Chem. 2005, 51, 2380–2382. [Google Scholar] [CrossRef]
- Yubero D, Montero R, Ramos M, Neergheen V, Navas P, Artuch R, Hargreaves I. Determination of urinary coenzyme Q10 by HPLC with electrochemical detection: Reference values for a paediatric population. Biofactors. 2015, 41, 424–430. [Google Scholar] [CrossRef]
- Mollet J, Giurgea I, Schlemmer D, Dallner G, Chretien D, Delahodde A, Bacq D, de Lonlay P, Munnich A, Rötig A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest. 2007, 117, 765–772. [Google Scholar] [CrossRef]
- Vasta V, Merritt JL 2nd, Saneto RP, Hahn SH. Next-generation sequencing for mitochondrial diseases: a wide diagnostic spectrum. Pediatr Int. 2012, 54, 585–601. [Google Scholar] [CrossRef]
- Nardecchia F, De Giorgi A, Palombo F, Fiorini C, De Negri AM, Carelli V, Caporali L, Leuzzi V. Missense PDSS1 mutations in CoenzymeQ10 synthesis cause optic atrophy and sensorineural deafness. Ann Clin Transl Neurol. 2021, 8, 247–251. [Google Scholar] [CrossRef]
- Bellusci M, García-Silva MT, Martínez de Aragón A, Martín MA. Distal phalangeal erythema in an infant with biallelic PDSS1 mutations: Expanding the phenotype of primary Coenzyme Q 10 deficiency. JIMD Rep. 2021, 62, 3–5. [Google Scholar] [CrossRef] [PubMed]
- López LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, Dimauro S, Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Iványi B, Rácz GZ, Gál P, Brinyiczki K, Bódi I, Kalmár T, Maróti Z, Bereczki C. Diffuse mesangial sclerosis in a PDSS2 mutation-induced coenzyme Q10 deficiency. Pediatr Nephrol. 2018, 33, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015, 26, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Rahman S, Clarke C, Hirano M. 176th ENMC international workshop on the diagnosis and treatment of CoQ10 disorders. Neuromusc Disord. 2011, 22, 76–86. [Google Scholar]
- Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, Dimauro S, Hirano M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef]
- Scalais E, Chafai R, Van Coster R, Bindl L, Nuttin C, Panagiotaraki C, et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur J Paediatr Neurol. 2013, 17, 625–630. [Google Scholar] [CrossRef]
- Desbats MA, Vetro A, Limongelli I, Lunardi G, Casarin A, Doimo M, et al. Primary coenzyme Q10 deficiency presenting as fatal neonatal multiorgan failure. Eur J Hum Genet. 2015, 23, 1254–1258. [Google Scholar] [CrossRef]
- Eroglu FK, Ozaltin F, Gönç N, Nalçacıoğlu H, Özçakar ZB, Yalnızoğlu D, Güçer Ş, Orhan D, Eminoğlu FT, Göçmen R, Alikaşifoğlu A, Topaloğlu R, Düzova A. Response to Early Coenzyme Q10 Supplementation Is not Sustained in CoQ10 Deficiency Caused by CoQ2 Mutation. Pediatr Neurol. 2018, 88, 71–74. [Google Scholar] [CrossRef]
- Xu K, Mao XY, Yao Y, Cheng H, Zhang XJ. Clinical analysis of one infantile nephrotic syndrome caused by COQ2 gene mutation and literature review. Zhonghua Er Ke Za Zhi. 2018, 56, 662–666. [Google Scholar] [CrossRef]
- Starr MC, Chang IJ, Finn LS, Sun A, Larson AA, Goebel J, Hanevold C, Thies J, Van Hove JLK, Hingorani SR, Lam C. COQ2 nephropathy: a treatable cause of nephrotic syndrome in children. Pediatr Nephrol. 2018, 33, 1257–1261. [Google Scholar] [CrossRef] [PubMed]
- Wu X, Wang W, Liu Y, Chen W, Zhao L. A steroid-resistant nephrotic syndrome in an infant resulting from a consanguineous marriage with COQ2 and ARSB gene mutations: a case report. BMC Med Genet. 2019, 20, 165. [Google Scholar] [CrossRef]
- 36. Abdelhakim AH, Dharmadhikari AV, Ragi SD, de Carvalho JRL Jr, Xu CL, Thomas AL, Buchovecky CM, Mansukhani MM, Naini AB, Liao J, Jobanputra V, Maumenee IH, Tsang SH. Orphanet J Rare Dis. 2020 Nov 13;15, 320. https://doi.org/10.1186/s13023-020-01600-8.Compound heterozygous inheritance of two novel COQ2 variants results in familial coenzyme Q deficiency. Orphanet J Rare Dis.
- Li M, Yue Z, Lin H, Wang H, Chen H, Sun L. COQ2 mutation associated isolated nephropathy in two siblings from a Chinese pedigree. Ren Fail. 2021, 43, 97–101. [CrossRef]
- Rosado Santos R, Rodrigues M, Loureiro T. Prenatal diagnosis of lissencephaly associated with biallelic pathologic variants in the COQ2 gene. Prenatal diagnosis of lissencephaly associated with biallelic pathologic variants in the COQ2 gene. Acta Med Port. 2022. [CrossRef]
- 39. Stallworth JY, Blair DR, Slavotinek A, Moore AT, Duncan JL, de Alba Campomanes AG. Retinopathy and optic atrophy in a case of COQ2-related primary coenzyme Q10 deficiency. Ophthalmic Genet. [CrossRef]
- Drovandi S, Lipska-Ziętkiewicz BS, Ozaltin F, Emma F, Gulhan B, Boyer O, et al. Variation of the clinical spectrum and genotype-phenotype associations in Coenzyme Q10 deficiency associated glomerulopathy. Kidney Int. 2022, 102, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Drovandi S, Lipska-Ziętkiewicz BS, Ozaltin F, Emma F, Gulhan B, Boyer O, et al. Oral Coenzyme Q10 supplementation leads to better preservation of kidney function in steroid-resistant nephrotic syndrome due to primary Coenzyme Q10 deficiency. Kidney Int. 2022, 102, 604–612. [Google Scholar] [CrossRef]
- Mantle, D. Turton N, Hargreaves I. Multiple system atrophy: role of CoQ10. J Clin Med Res.
- Salviati L, Trevisson E, Rodriguez Hernandez MA, Casarin A, Pertegato V, Doimo M, et al. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. Med Genet. 2012, 49, 187–191. [Google Scholar] [CrossRef]
- Brea-Calvo G, Haack TB, Karall D, Ohtake A, Invernizzi F, Carrozzo R, et al. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am J Hum Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef]
- Chung WK, Martin K, Jalas C, Braddock SR, Juusola J, Monaghan KG, et al. Mutations in COQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J Med Genet. 2015, 52, 627–635. [Google Scholar] [CrossRef]
- Romero-Moya D, Castaño J, Santos-Ocaña C, Navas P, Menendez P. Generation, genome edition and characterization of iPSC lines from a patient with coenzyme Q 10 deficiency harboring a heterozygous mutation in COQ4 gene. Stem Cell Res. 2017, 24, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Sondheimer N, Hewson S, Cameron JM, Somers GR, Broadbent JD, Ziosi M, Quinzii CM, Naini AB. Novel recessive mutations in COQ4 cause severe infantile cardiomyopathy and encephalopathy associated with CoQ 10 deficiency. Mol Genet Metab Rep. 2017, 12, 23–27. [Google Scholar] [CrossRef]
- Bosch AM, Kamsteeg EJ, Rodenburg RJ, van Deutekom AW, Buis DR, Engelen M, Cobben JM. Coenzyme Q10 deficiency due to a COQ4 gene defect causes childhood-onset spinocerebellar ataxia and stroke-like episodes. Mol Genet Metab Rep. 2018, 17, 19–21. [Google Scholar] [CrossRef]
- Caglayan AO, Gumus H, Sandford E, Kubisiak TL, Ma Q, Ozel AB, Per H, Li JZ, Shakkottai VG, Burmeister M. COQ4 Mutation Leads to Childhood-Onset Ataxia Improved by CoQ10 Administration. Cerebellum. 2019, 18, 665–669. [Google Scholar] [CrossRef] [PubMed]
- 50. Ling TK, Law CY, Yan KW, Fong NC, Wong KC, Lee KL, Chu WC, Brea-Calvo G, Lam CW. Clinical whole-exome sequencing reveals a common pathogenic variant in patients with CoQ 10 deficiency: An underdiagnosed cause of mitochondriopathy. Clin Chim Acta. [CrossRef]
- Lu M, Zhou Y, Wang Z, Xia Z, Ren J, Guo Q. Clinical phenotype, in silico and biomedical analyses, and intervention for an East Asian population-specific c.370G>A (p.G124S) COQ4 mutation in a Chinese family with CoQ10 deficiency-associated Leigh syndrome. J Hum Genet. 2019, 64, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Yu MH, Tsang MH, Lai S, Ho MS, Tse DML, Willis B, et al. Primary coenzyme Q10 deficiency-7: expanded phenotypic spectrum and a founder mutation in southern Chinese. NPJ Genom Med. 2019, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen XR, Xu JP, Yao YH. Zhonghua Er Ke Za Zhi. Primary coenzyme Q10 deficiency-7: a case report and literature review. 2020, 58, 928–932. [CrossRef]
- Mero S, Salviati L, Leuzzi V, Rubegni A, Calderan C, Nardecchia F, et al. New pathogenic variants in COQ4 cause ataxia and neurodevelopmental disorder without detectable CoQ 10 deficiency in muscle or skin fibroblasts. J Neurol. 2021, 268, 3381–3389. [Google Scholar] [CrossRef]
- Laugwitz L, Seibt A, Herebian D, Peralta S, Kienzle I, Buchert R, et al. Human COQ4 deficiency: delineating the clinical, metabolic and neuroimaging phenotypes. J Med Genet. 2022, 59, 878–887. [Google Scholar] [CrossRef]
- Malicdan MCV, Vilboux T, Ben-Zeev B, Guo J, Eliyahu A, Pode-Shakked B, et al. A novel inborn error of the coenzyme Q10 biosynthesis pathway: cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum Mutat. 2018, 39, 69–79. [Google Scholar] [CrossRef]
- Heeringa SF, Chernin G, Chaki M, Zhou W, Sloan AJ, Ji Z, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. Clin Invest. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Park E, Ahn YH, Kang HG, Yoo KH, Won NH, Lee KB, Moon KC, Seong MW, Gwon TR, Park SS, Cheong HI. COQ6 Mutations in Children With Steroid-Resistant Focal Segmental Glomerulosclerosis and Sensorineural Hearing Loss. Am J Kidney Dis. 2017, 70, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Li GM, Cao Q, Shen Q, Sun L, Zhai YH, Liu HM, An Y, Xu H. Gene mutation analysis in 12 Chinese children with congenital nephrotic syndrome. BMC Nephrol. 2018, 29, 382. [Google Scholar] [CrossRef]
- Cao Q, Li GM, Xu H, Shen Q, Sun L, Fang XY, Liu HM, Guo W, Zhai YH, Wu BB. Coenzyme Q(10) treatment for one child with COQ6 gene mutation induced nephrotic syndrome and literature review. Zhonghua Er Ke Za Zhi. 2017, 55, 135–138. [Google Scholar] [CrossRef]
- Song CC, Hong Q, Geng XD, Wang X, Wang SQ, Cui SY, Guo MD, Li O, Cai GY, Chen XM, Wu D. New Mutation of Coenzyme Q 10 Monooxygenase 6 Causing Podocyte Injury in a Focal Segmental Glomerulosclerosis Patient. Chin Med J (Engl). 2018, 131, 2666–2675. [Google Scholar] [CrossRef] [PubMed]
- Stańczyk M, Bałasz-Chmielewska I, Lipska-Ziętkiewicz B, Tkaczyk M. CoQ10-related sustained remission of proteinuria in a child with COQ6 glomerulopathy-a case report. Pediatr Nephrol. 2018, 33, 2383–2387. [Google Scholar] [CrossRef] [PubMed]
- Yuruk Yildirim Z, Toksoy G, Uyguner O, Nayir A, Yavuz S, Altunoglu U, et al. Primary coenzyme Q10 Deficiency-6 (COQ10D6): Two siblings with variable expressivity of the renal phenotype. Eur J Med Genet. 2020, 63, 103621. [Google Scholar] [CrossRef]
- 64. Justine Perrin R, Rousset-Rouvière C, Garaix F, Cano A, Conrath J, Boyer O, Tsimaratos M. COQ6 mutation in patients with nephrotic syndrome, sensorineural deafness, and optic atrophy. JIMD Rep. [CrossRef]
- Wang N, Zheng Y, Zhang L, Tian X, Fang Y, Qi M, Du J, Chen S, Chen S, Li J, Shen B, Wang L. A Family Segregating Lethal Primary Coenzyme Q10 Deficiency Due to Two Novel COQ6 Variants. Front Genet. 2022, 12, 811833. [Google Scholar] [CrossRef]
- Leeuwen L, Lubout CMA, Nijenhuis HP, Meiners LC, Vos YJ, Herkert JC. Expanding the clinical spectrum of primary coenzyme Q10 deficiency type 6: The first case with cardiomyopathy. Clin Genet. 2022, 102, 350–351. [Google Scholar] [CrossRef]
- Nam DW, Park SS, Lee SM, Suh MW, Park MK, Song JJ, Choi BY, Lee JH, Oh SH, Moon KC, Ahn YH, Kang HG, Cheong HI, Kim JH, Lee SY. Effects of CoQ10 replacement therapy on the audiological characteristics of pediatric patients with COQ6 variants. Biomed Res Int. 2022, 2022, 5250254. [Google Scholar] [CrossRef]
- Freyer C, Stranneheim H, Naess K, Mourier A, Felser A, Maffezzini C, et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J Med Genet. 2015, 52, 779–783. 52. [CrossRef]
- Wang Y, Smith C, Parboosingh JS, Khan A, Innes M, Hekimi S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J Cell Mol Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Kwong AK, Chiu AT, Tsang MH, Lun KS, Rodenburg RJT, Smeitink J, Chung BH, Fung CW. A fatal case of COQ7-associated primary coenzyme Q 10 deficiency. JIMD Rep. 2019, 47, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Wang Y, Gumus E, Hekimi S. Mol Genet Metab Rep. A novel COQ7 mutation causing primarily neuromuscular pathology and its treatment options. Mol Genet Metab Rep. 2022, 31, 100877. [Google Scholar] [CrossRef]
- Mollet J, Delahodde A, Serre V, Chretien D, Schlemmer D, Lombes A, Boddaert N, Desguerre I, de Lonlay P, de Baulny HO, Munnich A, Rötig A. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008, 82, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne C, Tazir M, López LC, Quinzii CM, Assoum M, Drouot N, et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008, 82, 661–672. [Google Scholar] [CrossRef]
- Blumkin L, Leshinsky-Silver E, Zerem A, Yosovich K, Lerman-Sagie T, Lev D. Heterozygous Mutations in the ADCK3 Gene in Siblings with Cerebellar Atrophy and Extreme Phenotypic Variability. JIMD Rep. 2014, 12, 103–107. [CrossRef]
- Hikmat O, Tzoulis C, Knappskog PM, Johansson S, Boman H, Sztromwasser P, Lien E, Brodtkorb E, Ghezzi D, Bindoff LA. ADCK3 mutations with epilepsy, stroke-like episodes and ataxia: a POLG mimic? Eur J Neurol. 2016, 23, 1188–1194. [Google Scholar] [CrossRef]
- Jacobsen JC, Whitford W, Swan B, Taylor J, Love DR, Hill R, Molyneux S, George PM, Mackay R, Robertson SP, Snell RG, Lehnert K. Compound Heterozygous Inheritance of Mutations in Coenzyme Q8A Results in Autosomal Recessive Cerebellar Ataxia and Coenzyme Q 10 Deficiency in a Female Sib-Pair. JIMD Rep. 2018, 42, 31–36. [Google Scholar] [CrossRef]
- Chang A, Ruiz-Lopez M, Slow E, Tarnopolsky M, Lang AE, Munhoz RP. ADCK3-related Coenzyme Q10 Deficiency: A Potentially Treatable Genetic Disease. Mov Disord Clin Pract. 2018, 5, 635–639. [Google Scholar] [CrossRef]
- Nair P, Lama M, El-Hayek S, Abou Sleymane G, Stora S, Obeid M, Al-Ali MT, Delague V, Mégarbané A. COQ8A and MED25 Mutations in a Child with Intellectual Disability, Microcephaly, Seizures, and Spastic Ataxia: Synergistic Effect of Digenic Variants? Mol Syndromol. 2019, 9, 319–323. [CrossRef]
- Schirinzi T, Favetta M, Romano A, Sancesario A, Summa S, Minosse S, Zanni G, Castelli E, Bertini E, Petrarca M, Vasco G. One-year outcome of coenzyme Q10 supplementation in ADCK3 ataxia (ARCA2). Cerebellum Ataxias. 2019, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- 80. Uccella S, Pisciotta L, Severino M, Bertini E, Giacomini T, Zanni G, Prato G, De Grandis E, Nobili L, Mancardi MM. Photoparoxysmal response in ADCK3 autosomal recessive ataxia: a case report and literature review. Epileptic Disord. [CrossRef]
- Paprocka J, Nowak M, Chuchra P, Śmigiel R. COQ8A-Ataxia as a manifestation of primary Coenzyme Q deficiency. Metabolites. 2022, 12, 955. [CrossRef]
- 82. Değerliyurt A, Gülleroğlu NB, Kibar Gül AE. Primary CoQ10 deficiency with a severe phenotype due to the c.901 C > T (p.R301W) mutation in the COQ8A gene. Int J Neurosci. [CrossRef]
- Traschütz A, Schirinzi T, Laugwitz L, Murray NH, Bingman CA, Reich S, et al. Clinico-Genetic, Imaging and Molecular Delineation of COQ8A-Ataxia: A Multicenter Study of 59 Patients. Ann Neurol. 2020, 88, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Horvath R, Czermin B, Gulati S, Demuth S, Houge G, Pyle A, et al. J Neurol Neurosurg Psychiatry. 2012, 83, 174–178. [CrossRef]
- 85. Mignot C, Apartis E, Durr A, Marques Lourenço C, Charles P, et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J Rare Dis. [CrossRef]
- Mallaret M, Renaud M, Redin C, Drouot N, Muller J, Severac F, et al. Validation of a clinical practice-based algorithm for the diagnosis of autosomal recessive cerebellar ataxias based on NGS identified cases. J Neurol. 2016, 263, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- 87. Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 5189. [CrossRef]
- Korkmaz E, Lipska-Ziętkiewicz BS, Boyer O, Gribouval O, Fourrage C, Tabatabaei M, et al. ADCK4-Associated Glomerulopathy Causes Adolescence-Onset FSGS. J Am Soc Nephrol. 2016, 27, 63–68. [Google Scholar] [CrossRef]
- Atmaca M, Gülhan B, Atayar E, Bayazıt AK, Candan C, Arıcı M, Topaloğlu R, Özaltın F. Long-term follow-up results of patients with ADCK4 mutations who have been diagnosed in the asymptomatic period: effects of early initiation of CoQ10 supplementation. Turk J Pediatr. 2019, 61, 657–663. [Google Scholar] [CrossRef]
- Feng C, Wang Q, Wang J, Liu F, Shen H, Fu H, Mao J. Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal disease and ADCK4 mutation: Case reports and literature review. Medicine (Baltimore). 2017, 96, e8880. [Google Scholar] [CrossRef]
- Song X, Fang X, Tang X, Cao Q, Zhai Y, Chen J, et al. COQ8B nephropathy: Early detection and optimal treatment. Mol Genet Genomic Med. 2020, 8, e1360. [Google Scholar] [CrossRef]
- 92. Duncan AJ, Bitner-Glindzicz M, Meunier B, Costello H, Hargreaves IP, López LC, et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet. [CrossRef]
- Danhauser K, Herebian D, Haack TB, Rodenburg RJ, Strom TM, Meitinger T, et al. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur J Hum Genet. 2016, 24, 450–454. [Google Scholar] [CrossRef]
- Smith AC, Ito Y, Ahmed A, Schwartzentruber JA, Beaulieu CL, Aberg E, et al. A family segregating lethal neonatal coenzyme Q 10 deficiency caused by mutations in COQ9. J Inherit Metab Dis. 2018, 41, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Olgac A, Öztoprak Ü, Kasapkara ÇS, Kılıç M, Yüksel D, Derinkuyu EB, et al. A rare case of primary coenzyme Q10 deficiency due to COQ9 mutation. J Pediatr Endocrinol Metab. 2020, 33, 165–170. [Google Scholar] [CrossRef] [PubMed]
- 96. Vrablik M, Zlatohlavek L, Stulc T, Adamkova V, Prusikova M, Schwarzova L, Hubacek JA, Ceska R. Statin-associated myopathy: from genetic predisposition to clinical management. Physiol Res. [CrossRef]
- López-Lluch G, Del Pozo-Cruz J, Sánchez-Cuesta A, Cortés-Rodríguez AB, Navas P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition. 2019, 57, 133–140. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram illustrating the key role of CoQ10 in cellular energy generation where CoQ10 acts an electron carrier in the mitochondrial respiratory chain (MRC). The MRC Enzyme complexes I-V and the electron carrier, cytochrome c (C).
Figure 1.
Diagram illustrating the key role of CoQ10 in cellular energy generation where CoQ10 acts an electron carrier in the mitochondrial respiratory chain (MRC). The MRC Enzyme complexes I-V and the electron carrier, cytochrome c (C).
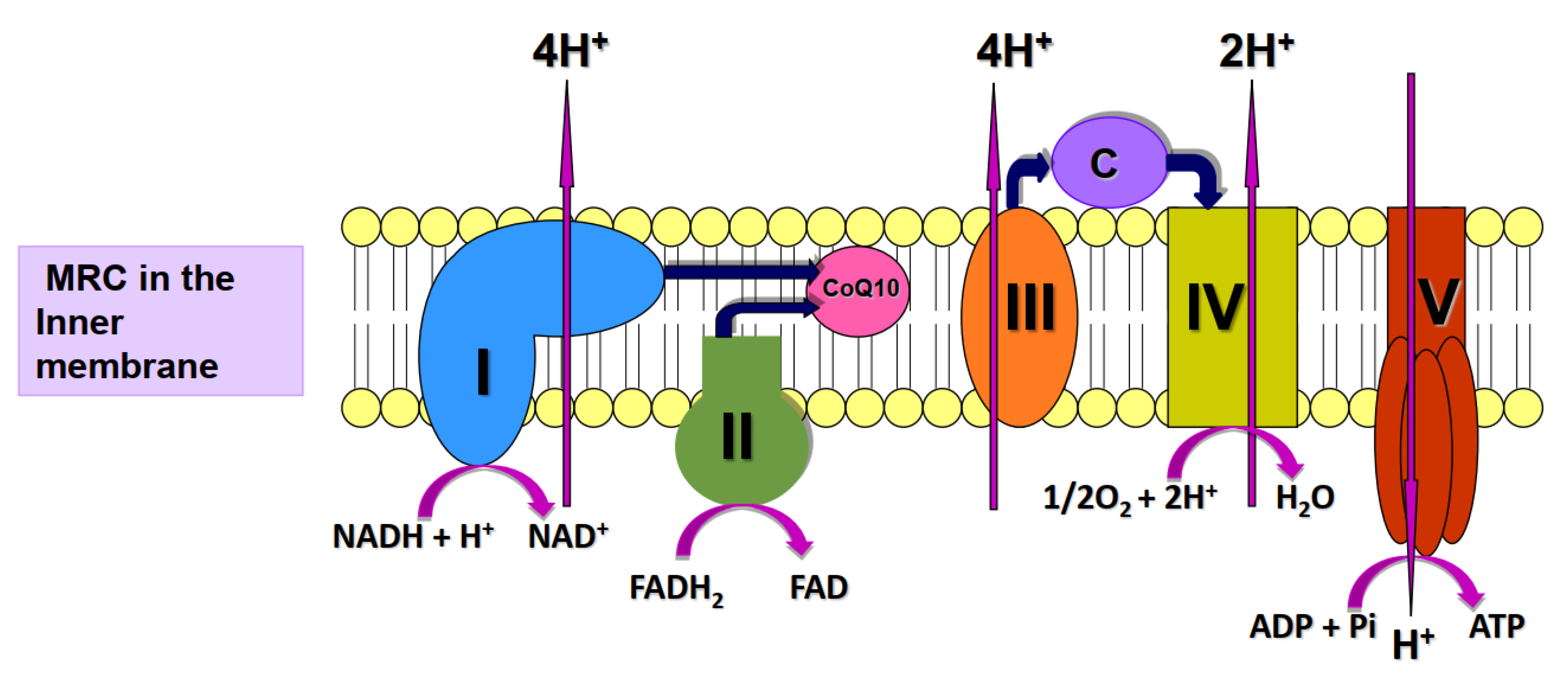

Table 1.
Summary of clinical publications relating to primary CoQ10 deficiency.
| Author | Gene | Condition | Number of patients | CoQ10 dose and Comment |
|---|---|---|---|---|
| Mollet et al [20] |
COQ1 (PDSS1) |
Infantile neurosensorial deafness and optic atrophy | 2 | |
| Vasta et al [21] | Kidney failure and death at 16 months | 1 | ||
| Nardecchia et al [22] | Sensorineural deafness and optic atrophy (ages 6 and 14 years) | 2 | ||
| Bellusci et al [23] | Numerous morbidities; death aged 3 years | 1 | 15 mg/Kg/day No effect on disease progression | |
| Lopez et al [24] |
COQ1 (PDSS2) |
Nephrotic syndrome & encephalopathy; died at 8 months | 1 | 50 mg/day. No effect on disease progression |
| Ivanyi et al [25] | Numerous morbidities; death at 7 months | 1 | ||
| Sadowski et al [26] | Less severe morbidities | 2 _________ 2 |
||
| Rahman et al [27] | Infantile onset encephalopathy | |||
| Quinzii et al [28] | COQ2 | Infantile nephropathy | 2 | |
| Diomedi-Camassei et al [29] | Encephalopathy and nephropathy in first patient who died at 6 months. Non-fatal nephropathy in second patient. | 2 | 30 mg/Kg/day clinical stabilisation in second patient | |
| Mollet et al [20] | Fatal neonatal nephropathy | 1 | ||
| Scalais et al [30] | Myoclonic epilepsy and hypertrophic cardiomyopathy; died at 5 months | 1 | 5 mg/Kg/day did not prevent fatality | |
| Desbats et al [31] | Newborn multi-organ failure and death | 1 | ||
| Eroglu et al [32] | Infantile renal and neurological symptoms | 4 | Supplement normalised renal function in 2 patients | |
| Xu et al [33] | Infantile nephrotic syndrome | 1 | 30 mg/Kg/day improved renal function | |
| Starr et al [34] | Nephrotic syndrome (patients 2-10 years old) | 3 | 30mg/Kg/day restored renal function in 2 patients | |
| Wu et al [35] | Fatal neuropathy at 6 months | 1 | ||
| Abdelhakim et al [36] | Nephropathy and retinopathy | 3 | Visual deterioration prevented after 30 mg/Kg/day CoQ10 for 6 months | |
| Li et al [37] |
Infantile nephropathy | 2 | Proteinuria reduced after 30 mg/Kg/day CoQ10 | |
| Rosado-Santos et al [38] |
Newborn fatality with severe fetal growth restriction | 1 | ||
| Stallworth et al [39] | 8 year old with nephropathy and optic atrophy | 1 | ||
| Drovandi et al [40, 41] |
Review articles to include many of the above studies relating to COQ2, plus some previously unreported cases. | 63 | Partial or complete remission of proteinuria in more than half of patients after CoQ10 | |
| No reports identified |
COQ3 | |||
| Salviati et al [43] | COQ4 | 3 year old with neuromuscular symptoms | 1 | Significant symptomatic improvement after 30mg/kg/day CoQ10 |
| Brea-Calvo et al [44] | Neurological deterioration with neonatal or early infancy fatality | 5 | ||
| Chung et al [45] | Neurological deterioration and early deaths | 6 | ||
| Romero-Moya et al [46] | Mental retardation and lethal rhabdomyolysis aged 4 | 1 | ||
| Sondheimer et al [47] | Seizures, cardiomyopathy and death in infancy | 1 | ||
| Bosch et al [48] | Infantile spinocerebellar ataxia and stroke-like episodes | 1 | No improvement after 1000 mg/day CoQ10 | |
| Caglayan et al [49] | Childhood onset slow progressive ataxia | 2 | One sibling improved after 200 mg/day CoQ10 for 1 month | |
| Ling et al [50] | Infantile encephalopathy or cardiomyopathy | 3 | ||
| Lu et al [51] | 2 month old with Leigh syndrome | 1 | Patient stabilised after CoQ10 | |
| Yu et al [52] | Numerous morbidities with neonatal or infantile onset | 11 | Seizure control improved in 2 patients after 15-40 mg/Kg/day CoQ10; no benefit in 5 patients who subsequently died | |
| Chen et al [53] | 5 month old with epileptic seizures | 1 | ||
| Mero et al [54] | Motor impairment and ataxia | 2 | ||
| Malicdan et al [56] | COQ5 | Seizures, ataxia and cognitive disability in early childhood | 3 | Symptomatic improvement in all cases after 15 mg/Kg/day CoQ10 for 6 months |
| Heeringa et al [57] | COQ6 | Early onset (1-2 years) nephrotic and sensory syndromes | 13 | Symptomatic improvement in some patients with 100mg/day CoQ10 |
| Sadowski et al [26] | Infancy/early childhood onset nephropathy | 6 | ||
| Park et al [58] | Renal disease (onset 15-47 months) requiring transplant | 6 | ||
| Li et al [59] | One year old with proteinuria | 1 | Proteinuria completely resolved after 30 mg/day CoQ10 for 3 months | |
| Cao et al [60] | Infantile nephrotic syndrome | 1 | Normal renal function restored after 30 mg/Kg/day CoQ10 for 3 months | |
| Song et al [61] | Proteinuria in 16 year old | 1 | 50% reduction in proteinuria after CoQ10 | |
| Stanczyk et al [62] | Glomerulopathy ion 4 year old | 1 | Complete symptomatic remission after 30 mg/Kg/day CoQ10 for 1 month | |
| Yildirim et al [63] | Nephrotic syndrome and sensorineural deafness in 7 year old | 1 | ||
| Perrin et al [64] | Renal disease, deafness and optic neuropathy | 1 | CoQ10 analogue, idebenone, improved vision | |
| Wang et al [65] | Numerous morbidities: died in infancy | 2 | ||
| Leeuwen et al [66] | 19 month old with fatal cardiomyopathy | 1 | ||
| Nam et al [67] | Patients (<18 years) with nephropathy and deafness | 12 | Hearing loss in some patients responded well after 30mg/kg CoQ10 | |
| Freyer et al [68] | COQ7 | Numerous morbidities oin 9 year old; muscle CoQ10 levels severely decreased | 1 | |
| Wang et al [69] | Spasticity and sensorineural hearing loss in 6 yesar old; moderate decrease in CoQ10 levels | 1 | ||
| Kwong et al [70] | Fatal morbidities at 1 year | 1 | Poor response to treatment after 10 mg/Kg/day CoQ10 | |
| Wang et al [71] | 4 year old with numerous morbidities: 45% reduction in fibroblast CoQ10 levels | 1 | ||
| Mollet et al [72] |
COQ8A | Seizures and cerebellar atrophy (ages 18-36 months) | 4 | Not improved after 15 mg/Kg/day CoQ10 |
| Lagier-Tourenne et al [73] | Childhood onset cerebellar atrophy and ataxia | 6 | ||
| Blumkin et al [74] |
Ataxia and mild dysarthria | 2 | Ataxia partially resolved after 20 mg/Kg/day CoQ10 | |
| Hikmat et al [75] | Childhood onset cerebellar ataxia and epilepsy | 3 | ||
| Jacobsen et al [76] | Childhood onset cerebellar atrophy and ataxia | 2 | Improvement in ataxia and mental capacity after 20mg/kg/day CoQ10 | |
| Nair et al [78] |
5 year old with intellectual disability and ataxia | 1 | Improved motor performance following 15 mg/kg/day CoQ10 | |
| Schirinzi et al [79] | Ataxia in patients aged 4-12 months | 4 | ||
| Uccella et al [80] |
Childhood onset ataxia | 1 | Disease progression slowed after CoQ10 | |
| Paprocka et al [81] | Cerebellar ataxia and developmental regression in 22 month old | 1 | Improved communi-cation and growth after 300 mg/day CoQ10 | |
| Degerliyurt et al [82] | 16 year old with ataxia, cerebellar atrophy and cardiomyopathy | 1 | treatment with CoQ10 started at too late a stage to prevent death of the patient. | |
| Traschutz et al 2020 | Review incorporating some previously published data; cerebellar ataxia in all patients with mean onset age 7 years | 64 | 50% of patients responded after mean dose of 11 mg/Kg/day CoQ10 | |
| Ashraf et al [87] | COQ8B | Nephropathy | 1 | Partial remission after CoQ10 |
| Korkmaz et al [88] | Adolescent nephropathy | 26 | Improved proteinuria after 15 mg/kg/day CoQ10 in 2 patients | |
| Atmaca et al [89] | Nephropathy | 8 | Improved proteinuria after CoQ10 | |
| Feng et al [90] | Patients aged 9 months and 11 years with proteinuria | 2 | Younger subject ahowed good response after 15 mg/kg/day CoQ10 | |
| Song et al [91] | Patients aged 1-18 years with renal disease | 20 | Reduced proteinuria in trial group of 5 subjects after 15-30 mg/Kg/day CoQ10 | |
| Duncan et al [92] | COQ9 | Multiple morbidities; died aged 2 years | 1 | Up to 300mg/day CoQ10 did not prevent fatality |
| Danhauser et al [93] | Fatal neonatal lactic acidosis and encephalopathy | 1 | ||
| Smith et al [94] | Multisysystem disease in 4 siblings, 2 of whom died soon after birth | 4 | ||
| Olgac et al [95] | Microcephaly and seizures in 9 month old | 1 | No improvement after 5-50 mg/Kg/day CoQ10 | |
| No reports identified |
COQ 10A and 10B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



