Submitted:
28 April 2023
Posted:
04 May 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
This review brings together the current knowledge regarding the risk factors, and the clinical, radiologic and histological features of both post-COVID-19 interstitial pul-monary fibrosis (PCPF) and Idiopathic Pulmonary Fibrosis (IPF) with a particular fo-cus on describing the similarities and the disparities between the fibrotic changes in these two diseases. It is important to highlight the common points of PCPF and IPF to observe if they are some targetable changes to improve patient outcomes. The litera-ture review was performed using numerous databases to identify relevant articles published in English through October 2022. This review would help clinicians, pathologists and researchers to make an accurate diagnosis, which can be useful in identifying the group of patients who can be selected for antifibrotic therapies, and future therapeutic perspectives.
Keywords:
Covid-19
; idiopathic pulmonary fibrosis
; interstitial lung disease
; pulmonary fibrosis
1. Introduction
The novel, „severe acute respiratory syndrome coronavirus 2” (SARS-CoV-2) is the cause of the coronavirus disease (COVID-19), which has become a global pandemic causing millions of deaths. According to the currently available data, SARS-CoV-2 can affect various organs in the body, leading to acute organ damage and long-term sequelae [1,2]. Prospective data regarding the long-term changes of SARS-CoV-2 infection are just beginning to emerge up to three years after the start of the pandemic. While clinical studies regarding the safety and effectiveness of antiviral agents and vaccines are ongoing, chronic pulmonary consequences of COVID-19 have become steadily more recognized and are of increasing concern.
Although pulmonary fibrosis has been observed in varying degrees of evolution in patients with SARS-CoV-2 infection, its mechanism has not been sufficiently studied and elucidated.
Various mechanisms of lung injury have been described in COVID-19, both viral and immune-mediated. Pulmonary fibrosis can be either caused by chronic inflammation or can be an idiopathic, genetic or age-related fibroproliferative process. It is a known fact that pulmonary fibrosis is a sequel of ARDS and the radiological anomalies are of little clinical significance and have diminished after lung ventilation. The data show that 40% of patients with Covid-19 develop ARDS and only 20% of them have severe outcomes [3]. The prevalence of post-COVID-19 fibrosis will be visible in time, but early analysis suggests that more than a third of the recovered patients develop fibrotic anomalies. One study showed that 4% of patients with a disease duration of more than a week, 24% of patients with a duration between 1 and 3 weeks and 61% of patients with a disease duration greater than 3 weeks develop fibrosis [4].
Retrospective studies have shown that over 90% of hospitalized patients have presented persistent modifications of the lungs at the moment of discharge from the hospital [5,6], while the majority of patients with mild forms of COVID-19 had ground glass opacities which disappeared in three weeks after discharge [7]. In a series of severe COVID-19 patients who had recovered and who were followed up for one year after hospital discharge, nearly 25% of them had persistent radiologic abnormalities with features characteristic of pulmonary fibrosis including reticular opacities, septal thickening, and traction bronchiectasis [5].
Interstitial lung diseases are a heterogeneous group of parenchymal lung diseases characterized by varying degrees of inflammation and fibrosis. Lung fibrosis might be a possible complication of COVID-19 pneumonia leading to irreversible lung dysfunction. Although pulmonary fibrosis is not a common complication in other viral pneumonia, lung sequelae are found in a large number of COVID-19 patients [8]. Pulmonary fibrosis occurs when the normal ability to repair the tissue is affected by abnormal regeneration of the alveolar epithelium, with fibroblast persistence and excessive collagen deposition, which will lead to abnormal lung architecture [9]. Different potential contributing risk factors for persistent fibrotic lung changes in COVID-19 patients were proposed, such as age over 40 years old, tachycardia at admission, hospitalization duration over 16 days, acute respiratory distress syndrome (ARDS), noninvasive mechanical ventilation and computer tomography (CT) score over 17 at the initial evaluation [10]. Due to the tremendous number of COVID-19 cases and the severity of illness in many patients, there is a crucial need to consider the potential long-term implications of this disease. The features of post-COVID-19 interstitial pulmonary fibrosis (PCPF) and whether it will be permanent or progressive, as in other fibrotic lung diseases such as Idiopathic Pulmonary Fibrosis (IPF) are needed to be established by further studies.
Idiopathic pulmonary fibrosis (IPF) is a diffuse, fibrosing interstitial pneumonia, of unknown origin, having a chronic progressive evolution and a very poor prognosis, with a mean survival of 2.5–5 years after definite diagnosis. Although it is considered a rare disease, IPF represents around 17-37% of the cases of interstitial lung disease [11]. It affects primarily older adults, has exclusively pulmonary involvement, and is defined by the histopathologic and/or radiologic pattern of usual interstitial pneumonia (UIP). The histological pattern of UIP is characterized by marked parenchymal fibrosis with or without a „honeycombing” pattern with a predominantly sub-pleural and para-septal distribution, presence of fibroblast foci, traction bronchiectasis and remodelling of the alveoli [12].
Idiopathic pulmonary fibrosis is a lung lesion characterised by chronic, progressive scarring of the lungs and the pathological hallmark of usual interstitial pneumonia, and which diagnosis requires specialist expertise. Regarding etiopathogenic, current theories hypothesize that the initiating factor is alveolar epithelial cell injury. In this sense, it is important to exclude another interstitial lung disease. The lesion consists of excessive proliferation of fibroblasts and myofibroblasts and excessive deposition of disorganized collagen and extracellular matrix, with resulting distortion of the normal lung architecture, with or without honeycomb cyst formation [13]. Identifying and managing associated diseases, such as obstructive sleep apnea, pulmonary hypertension, and emphysema is also an important feature of monitoring patients.
2. Materials and Methods
This literature review was performed using the following databases: MEDLINE, Clarivate, PubMed, Scopus, Google Scholar and Science Direct, to identify relevant articles published in English through October 2022. Search items included: severe acute respiratory syndrome coronavirus 2, COVID-19, post-COVID-19 pulmonary fibrosis, and idiopathic pulmonary fibrosis. Review articles, case reports and case series were also included, especially due to the lack of RCTs. The search resulted in 2,567 total articles. The titles and abstracts were independently reviewed by the authors to be included in this review. The search was supplemented by reviewing reference lists of included studies and related review papers.
3. Risk factors
The recognition of the potential risk factors for post-COVID-19 pulmonary fibrosis is a main goal for improving the clinical course of these patients. It has been demonstrated that severe forms of COVID-19 have several shared major risk factors with IPF [14]. Although the risk factors have not yet been completely defined, some studies from different countries showed that patients at higher risk for PCPF are elders, males, smokers and have underlying conditions, such as diabetes and cardiovascular and lung diseases [15,16,17,18]. Furthermore, there were identified different features related to the severity of the acute phase of the disease which were associated with a higher risk of developing PCPF on CT scan in the follow-up, such as the presence of dyspnea, prolonged hospital stay, intensive care unit (ICU) admission, use of high-flow oxygen support, the need for intubation and mechanical ventilation, the severity of the acute infection and development of ARDS [15,19,20]. It was observed that mechanical ventilation can determine ventilator-induced lung injury and release of proinflammatory modulators, increasing the risk of pulmonary fibrosis and mortality [21]. Opposite to the severe forms of COVID-19, patients with mild-to-moderate forms of the disease had no fibrotic abnormalities on the CT scan follow-up [22].
As the global COVID-19 pandemic has progressed, a range of studies has identified different markers and biomarkers that may predict the development of PCPF [17,19]. In retrospective studies from China, higher levels of serum lactic dehydrogenase and inflammatory biomarkers, including C-reactive protein (CRP) and interleukin-6 (IL-6), and D-dimer levels, were found in the subgroups of patients who presented changes compatible with pulmonary fibrosis on follow-up CT scans [17,18]. Other laboratory markers identified to be associated with a higher risk of developing PCPF are represented by the decrease in the lymphocyte count [23] and lower plasma levels of interferon-γ (IFN-γ) [24].
Additionally, host genetic predisposition was proposed as a risk factor for severe courses of COVID-19 [25]. The first genome-wide association studies (GWAS) identified the 3p21.31 gene cluster (rs11385942) associated with severe forms of COVID-19 and respiratory failure and confirmed a potential involvement of the ABO blood group system [26]. The COVID-19 Host Genetics Initiative (HGI) meta-analysis also confirmed the 3p21.31 locus as significant, with two signals within the locus, one associated with severity (rs10490770), the other with infection susceptibility (lead variant rs2271616) [27]. CXCR6 was proposed as a causal gene for the severe disease (lead variant rs10490770). CXCR6 recruits CD8-resident memory T-cells in the respiratory tract to combat respiratory pathogens [28].
Different studies reported that several genes are associated with IPF predisposition, including the genes that encode the surfactant and proteins A and C (SFTPC, SFTPA1, SFTPA2), genes associated with telomerase dysfunction (TERT, TERC, DKC1, RTEL1, PARN), genes affecting the integrity of the epithelial barrier (DSK), and genes affecting host defence (MUC5B, TOLLIP [29,30]. An association between MUC5Brs35705950 and hospitalization due to COVID-19 was reported in the most recent meta-analysis by the HGI [31]. The study of Fadista J et al. (2021). showed that genetic variants associated with IPF did not predispose to an increased risk of severe COVID-19 [14]. As mucins are involved in the first defence against pathogens in the airways and play an essential role in mucociliary clearance, high expression may protect against SARS-CoV-2 infection. Nevertheless, this study was driven by a single abnormal variant at the MUC5B locus, which had an apparent protective effect on the severity of COVID-19. Removal of this outlier demonstrated that, collectively, the remaining variants associated with increased risk of IPF were also associated with increased risk of severe COVID-19.
Regarding the risk factors for the development of IPF, there is increasing evidence to support the role played by the intrinsic risk factors (e.g., advanced age, male sex, genetics, lung microbiome), comorbidities (e.g., diabetes mellitus, gastroesophageal reflux, obstructive sleep apnea, herpes virus infection), and extrinsic risk factors (e.g., smoking, air pollution, environmental exposures). These risk factors, part of them common with those predisposing to PCPF, may independently increase susceptibility for IPF or work in a synergistic mode to contribute to a higher risk for disease development [32] or be associated with shorter survival rates [33]. The incidence and prevalence of IPF increase with age, being higher in adults 65 years or older. The mechanisms include the alteration of the proliferation/apoptosis ratio [4], decrease alveolar stability and reduced differentiation capacity which promote fibrosis [35].
Around 70% of all IPF patients are male [36], males also being 1.3 times more subjected to PCPF than females [37]. A possible explanation is given by studies performed on animals, which have shown that female hormones have a role in protection against pulmonary fibrosis [38,39]. Another cause may be that men are more exposed to tobacco smoking and occupational exposures [36]. Smoking history increases the risk of developing IPF by 60% [40], and, also, the risk of progression to severe forms of COVID-19 by 1.3 times [4]. Smoker patients with COVID-19 are 2.4 times more likely to need ICU admission and mechanical ventilation or die compared to nonsmokers [41,42]. Studies demonstrated that cigarette smoking determines endoplasmic reticulum stress, production of TGFB (transforming growth factor beta) [43] which mediates fibrosis, increased epithelial permeability, production of reactive oxidative species, and alteration of tissue regeneration, inducing lung micro-injuries [44]. The environmental occupational factors associated with IPF are metal dust, wood dust, stone, sand, and farming substances [45].
Studies have shown that microbial pathogens like Streptococcus and Staphylococcus are associated with the development and progression of IPF [46,47], while COVID-19 patients with bacterial infections were more frequently admitted to the ICU and needed invasive ventilation [48]. A recent meta-analysis [49] showed a prevalence of bacterial co-infections of 3.5% in COVID-19 patients admitted to the ICU, in most of the cases being hospital-acquired infections with Gram-negative germs (e.g., Acinetobacter baumanii) [50].
4. Clinical aspects
The clinical manifestations of PCPF and IPF are mostly similar, with both diseases sharing numerous symptoms, including dyspnea, dry cough, fatigue, chest pain and weight loss, which are related to decreased life quality [51,52]. It should be mentioned that there is a paucity of data for the clinical course of PCPF, but this gap will be filled by the results of the appropriate prospective studies. An Italian study conducted by Carfi A assessed persistent symptoms in COVID-19 patients who were discharged from the hospital, with 60.3 day mean period of assessment after the initial onset of COVID-19 symptoms, showing that at the time of evaluation, 55% of the patients had 3 or more COVID-19 related symptoms, 32% had 1 or 2 symptoms, and only 12.6% were completely free of any symptoms, with 44.1% of patients reporting worsened quality of life. The most common persistent symptoms beyond discharge were fatigue (53.1%), dyspnea (43.4%), joint pain (27.3%) and chest pain (21.7%) [53]. Another prospective cohort study of 76 severe COVID-19 patients requiring supplemental oxygen found a positive correlation between the presence of radiographic abnormalities (fibrotic-like patterns) 4 months after hospitalization and decreased lung function, cough and frailty [54]. The study conducted by Kamal M showed that almost 90% of the 287 included patients suffered from several symptoms and diseases after recovery from COVID-19. Most individuals suffered from fatigue (72.8%), anxiety (38%), joint pain (31.4%), continuous headache (28.9%), chest pain (28.9%), dementia (28.6%), depression (28.6%) and dyspnea (28.2%), while 2.4% of recovered patients have newly been diagnosed with diabetes [55]. A positive correlation between the severity of the initial disease and the severity of post-COVID-19 manifestations was noted. It was also observed that many of those manifestations were related to the central nervous system, including continuous headache, anxiety, depression, and obsessive-compulsive disorder.
Psychological morbidities such as anxiety and depression were also reported in IPF patients [56]. Anxiety and depression are known to be strongly associated with health-related quality of life [57]. For IPF patients and, also, for subjects recovering from COVID-19, the need for continuous counselling is very important for the early detection of warning signs of developing serious manifestations and for maintaining good adherence to the medications.
A case-control study performed on the UK population in 2017 has shown that there is a strong correlation between dyspnea and cough as debut symptoms in IPF, appearing up to 4 years before the diagnosis. [58].
Obstructive sleep apnea is also highly associated with IPF, with patients reporting snoring, insomnia, daytime sleepiness, and witnessed apneas. Gastro-oesophagal reflux can be present in 35% up to 100% of patients with IPF. These patients can be asymptomatic or can present different digestive symptoms, such as regurgitation, belching, dysphagia, dysphonia, and chest pain. Cough can be associated with about 28% of cases of gastro-oesophagal reflux [59].
The physical findings of IPF patients are much better documented in the literature data compared with patients with COVID-19. A study analyzing patients with PCPF 6 months after the acute episode has shown the presence of pathological auscultation sounds in 4% to 12% of the patients, represented by velcro crackles and wheezing [60].
Fine crackles, usually in the lower posterior parts of the lung are usually reported in IPF patients while clubbing fingers are found in 30-50% of the cases, being correlated with smooth muscle proliferation in the areas of fibrosis observed in lung biopsy. Body mass index has also been associated with survival in IPF patients. [61].
5. Pulmonary function tests
Pulmonary function tests (PFTs) indicate a restrictive pattern and an altered lung diffusion capacity for carbon monoxide (DLCO) in the vast majority of cases of patients with IPF and patients who have recovered after severe forms of COVID-19 [62,63]. In a recently published meta-analysis, which included 380 post-COVID-19 patients, the authors found impaired DLCO, restrictive pattern and obstructive pattern in 39%, 15%, and 7% of subjects, respectively [63]. A high prevalence of decreased DLCO (66%) was found in patients with severe forms of COVID-19, especially those who had high inflammatory markers, being more likely to develop pulmonary fibrosis [18]. The British Thoracic Society (BTS) guide suggests the evaluation of patients with severe forms of COVID-19 should be done with full PFTs until 12 weeks after hospital discharge [64]. According to the findings of Cherrez-Ojeda I, the impairment in PFTs appears to persist well beyond this timeframe [65]. A prospective observational study which analyzed the evolution of functional and radiological features between 3 to 6 months after hospital discharge in critical COVID-19 survivors reported the persistence of functional abnormalities such as impairments in total lung capacity (TLC) (41% and 33%) and DLCO (88% and 80%) at the end of monitoring period [66]. Significant improvements were observed only in forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC) and distance covered during the 6-min walking test (6MWT).
Acute phase severity markers such as the presence of previous conditions (arterial hypertension and diabetes), invasive mechanical ventilation (IMV) and prone positioning during the ICU stay were associated with a low level of DLCO and its non-improvement during the 3rd and 6th months of follow-up. The decrease in DLCO could be the result of interstitial or pulmonary vascular abnormalities caused by critical COVID-19. The authors observed that despite the partial radiological resolution, gas-blood exchange abnormalities persisted at 6 months follow-up, which could suggest the initial establishment of an irreversible, chronic lung disorder.
Although the restrictive pattern is consistent also in IPF, in one study 25% of the patients had normal TLC, and more than 50% had normal FVC. As the restriction’s severity increases, FEV1/FVC increases and DLco decreases. The lung function decline is a mortality predictor in IPF [67].
6. Radiologic aspects
In the early stages of COVID-19, the most common radiological findings are bilateral „ground glass opacities” (GGOs) and consolidations, predominantly in the lower lobes, posterior and peripheral. Other aspects that may be observed include a „crazy-paving” pattern, nodular opacities, halo sign, reversed halo sign, pleural effusions, cavitation and lymph node enlargement [15,70,71]. The extent of the lesions varies significantly, they can be patchy or diffuse, and a predominance of central and upper distribution may also be present. Different pulmonary CT abnormalities may be found even in patients without respiratory symptoms.
The lesions present in the acute phase of the disease may progress to fibrotic abnormalities such as interlobular septal thickening and traction bronchiectasis, especially in survivors of critical forms of COVID-19 [72]. However, the data regarding the long-term evolution of pulmonary changes in these patients are scarce. Liu D reported that between three to four weeks after the acute COVID-19 pneumonia, a transitory extension of the GGOs occurs, with the decreasing of density, an aspect described as “tinted sign”, accompanied by the distortion of the broncho-vascular bundle [7]. Most patients with mild or moderate pneumonia have complete resolution of the imagistic lesions in the first month after the episode, the first lesions that are resolving being GGOs. Other lesions like subpleural bands and bronchial dilations are resolving slowly, the severity of the initial clinical manifestations being the most common determinant of the resolution time.
In terms of imaging evaluation of lung parenchyma, there has been developed a score for the degree of lung affection, based on dividing the lung into five lung lobes; each lobe affection was visually scored on a scale of 0–5, with 0 indicating no involvement, 1 indicating less than 5% involvement, 2 indicating 5–25% involvement, 3 indicating 26–49% involvement, 4 indicating 50–75% involvement, and 5 indicating more than 75% involvement. The total CT score represents the sum of the individual lobar scores and ranged from 0 to 25 [73].
Recent studies evaluated long-term longitudinal changes in chest CT findings in COVID-19 survivors. Yin X showed that GGOs continued to be present on CT images in 30% of the subjects more than 6 months after discharge [74]. At the same time, reticulation was observed in 46% of cases, more commonly after severe forms of the disease, with a longer duration of hospitalization. In a 1-year follow-up study, the CT scans were normal in 16% of cases, stable in 19%, and with a reduction in extension of the lesion in 65% of the patients, abnormalities consisting in reticular bands opacities persisting at 1-year follow-up, while the extent of GGOs was being substantially reduced [75]. The long-term persistence of CT abnormalities after COVID-19 was correlated with the severity of the acute phase of the disease, the high inflammatory state, and the need for more intensive ventilator requirements.
According to the CT scoring method proposed by Camiciottoli, the degree of pulmonary fibrosis was evaluated by chest CT images in ground-glass opacity, linear opacity, interlobular septal thickening, reticulation, honeycombing or bronchiectasis features [76]. In the study of Jia-Ni Zou, approximately 80% of the 284 COVID-19 patients had pulmonary fibrosis at discharge, more pronounced in patients with severe disease compared to those with mild/moderate form (73.8%).
It was observed that PCPF changes appear in the areas where there were previously GGOs during COVID-19 pneumonia [6]. As a result, the CT distribution of the fibrotic changes will be predominantly bilateral, peripheral and in the lower lobes. The same distribution of CT abnormalities is also described in IPF cases, in which usual interstitial pneumonia (UIP) represents the hallmark radiologic CT pattern (Figure 1).
Regarding the CT findings, the main difference between IPF and PCPF is represented by the presence of honeycombing, which is essential for the definite diagnosis of IPF, but it is rarely observed in PCPF, currently, just a few cases are being reported [77]. Another chest CT feature that differentiates those two diseases consist of the large extent of the GGOs in PCPF, while in IPF is commonly absent or minimum, or may be present in case of exacerbations. The evolution of pulmonary manifestation on CT scan is very well documented in IPF, the data from the literature showing an irreversible, progressive course [78,79], while despite the scarce data regarding the evolution of post-COVID-19 CT abnormalities, recent studies found an improvement, with a 10-40% decrease in the extent of CT lesions and no progression at 1-year follow-up CT [74,75]. Prospective studies from large cohorts undergoing longer monitoring are likely to further clarify the evolution of PCPF on CT scans.
In 2011 the clinical practice guidelines for the diagnosis of IPF from the American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association (ATS/ERS/JRS/ALAT) proposed the concept of multidisciplinary diagnosis for IPF in patients without surgical lung biopsy (SLB) if the patient had UIP pattern on HRCT and clinical suggestive presentation, and recommend that HRCT features of IPF should be referred to as UIP, possible UIP, or inconsistent with UIP. The imagistic UIP pattern is accurate for the presence of histopathological UIP patterns.
UIP pattern consists of reticular opacities, honeycombing, traction bronchiectasis and bronchiolectasis, which may be observed with the concurrent presence of fine reticulation and GGOs. Honeycombing is a defining feature of UIP and its presence is mandatory for a definite diagnosis of UIP to be made. It can be associated with or without peripheral traction bronchiectasis or bronchiolectasis (Figure 2a,b). The presence on the HRCT of imaging features of UIP except for the presence of honeycombing defines the “possible UIP” pattern, in these case lung biopsy is necessary for the positive diagnosis. The presence of other abnormalities, such as extensive GGOs, micronodules, pleural abnormalities, non-honeycombing cysts, consolidation areas, or the predominance of peribronchovascular or perilymphatic distribution is characteristic of inconsistent with the UIP pattern. However, differentiating honeycombing from traction bronchiectasis and emphysema can be difficult, honeycombing has a thicker wall, and subpleural distribution, parallel with the chest’s wall, while emphysema is seen as cystic airspaces, with thin walls, located further away from the chest wall [80].
7. Histopathologic characterization
The histopathological data is mostly based on autopsy findings. In contrast to other viral infections such as H1N1, when the death occurred within a few days after the symptomatic debut, the patients infected with SARS-CoV-2 died within a mean duration of three weeks. An explanation may be offered by the more progressive lung injury caused by SARS-CoV-2 that favours the repair with extracellular markers deposition. However, histopathological studies in prolonged or in post-COVID-19 patients are very limited. The autopsy studies show that during the course of the disease, type I and type II pneumocytes are infected by the SARS-CoV-2 virus, resulting in a cytopathic effect, pneumocyte desquamation, accumulation of fibrinoid material in alveolar spaces and numerous inflammatory cells in the lungs, macrophages, lymphocytes and neutrophils [81].
Infected type II pneumocytes contain numerous autophagosomes, ultrastructural characterized by double membranes and the presence of organelles in the cytoplasm, also containing viral aggregates, that can be present in tracheal epithelial cells and within the extracellular mucus in the tracheal lumen. The existence of virus particles can be demonstrated by immunohistochemical staining using monoclonal antibodies against the SARS-CoV-2 nucleocapsid protein [82]. All these changes represent the spectrum of diffuse alveolar damage (DAD), which is the characteristic of severe COVID-19 in the acute or exudative phase, with numerous reactive pneumocytes, lung haemorrhage, fibrin deposits in the alveolar spaces, interstitial oedema, hyaline membrane, giant cell formation and bronchiolitis obliterans [83,84,85,86]. Thrombotic events in pulmonary arteries may also occur in this phase.
The guideline panel updated the diagnostic criteria for idiopathic lung fibrosis. Previously, defined patterns of usual interstitial pneumonia (UIP) were refined to patterns of UIP, probable UIP, indeterminate for UIP, and alternate diagnosis. For patients with newly detected interstitial lung disease (ILD) who have a high-resolution CT scan pattern of probable UIP, indeterminate for UIP, or an alternative diagnosis, conditional recommendations were made for performing BAL and surgical lung biopsy [87]. The study performed by Hanley B. et al. (2020) on 10 autopsies, reported DAD presence in all the cases, as well as lymphocyte inflammation, predominantly CD4+ positive T cells, along with macrophages and scattered plasma cells. CD56-positive natural killer cells were rarely found. Chronic bronchiolitis was also a common finding (Figure 4b). Thromboembolism was a frequent finding in small and medium-sized vessels, without any sign of deep venous thrombosis at the external examination [88].
The 2018 ATS/ERS/JRS/ALAT guideline classifies the histopathological findings in UIP, probable UIP, and indeterminate UIP. IPF is macroscopically characterized by fibrosis situated in the interior part of the lobes, pre dominantly subpleural, this pattern being called honeycombing. IPF is microscopically characterized by a UIP pattern, consisting of patchy dense fibrosis, with architectural distortion, predominantly in the periphery of the lobule and paraseptal, with a normal central portion of the lobule, as a result, the patterns will evolve from chronic to acute to absent from the periphery to the centum of the lobule. The honeycombing aspect can be seen subpleural as dense fibrosis surrounding bronchial epithelium which lines irregular airspaces. In the centrum of the lobule important inflammation and fibrosis is absent. Fibroblast foci can be seen in the area between normal regions of the lobule and fibrotic lesions, arranged parallel with the alveolar foci, on a basophilic myxoid ground (Figure 3a,b).
A probable UIP pattern is characterized by the presence of some histological UIP features, to an extent that precludes the definitive diagnosis and the absence of features of an alternative diagnosis, or the presence of honeycombing alone. Indeterminate UIP is characterized by fibrosis with or without architectural distortion suggesting UIP secondary to other causes or non-suggestive for UIP, or the presence of some features of UIP along with features of an alternative diagnosis. An alternative diagnosis is suggested by features of other histological patterns suggestive of other diseases at biopsy, such as airway-centred lesions, granulomas, interstitial inflammation without fibrosis, chronic fibrous pleuritis, and hyaline membranes [87].
The histopathological guidelines for UIP have been changed in 2021 and require advanced fibrosis with distortion of the architecture, beginning at the periphery of the lobule, going to centrilobular regions, with a sharp demarcation with the fibrotic and non-fibrotic areas, at this level often being encountered fibroblast foci as evidence of active injury, often situated at the interface between fibrotic and non-fibrotic areas, without features characteristic for an alternative diagnosis.
UIP from IPF is characterized by a paucity of inflammation, and ununiform affection of lung parenchyma. In acute exacerbations of UIP ALI pattern can be seen, predominantly in the regions without chronic fibrosis, while the peripheric fibrotic lesions are unmodified. In the centrum of the lobule, we can see modifications of DAD, such as type II pneumocyte hyperplasia, diffuse alveolar septal thickening caused by oedema, accumulation of airspace fibrin or nonspecific changes such as thrombi within small pulmonary arteries, distal airway squamous metaplasia, and the presence of hyaline membranes [80].
In acute exacerbation of IPF, we can find DAD along with UIP features, so DAD is a histopathological feature present in IPF and COVID-19. DAD can be caused by numerous factors such as infections, shock, sepsis, connective tissue disorders or disseminated intravascular coagulation. When the aetiology is unidentified it is called acute interstitial pneumonia, which was previously referred to as Hamman-Rich syndrome [89]. DAD is a result of acute lung injury (ALI) determined by direct or indirect causes, or in the case of ARDS requiring mechanical ventilation. ARDS is defined as acute hypoxemia with a ratio of partial pressure of arterial oxygen to the fraction of inspired oxygen (PaO2:FiO2) of a maximum of 200 mmHg, while ALI, which is less severe, refers to acute hypoxemia with a PaO2:FiO2 ratio of 300 mg Hg [90]. Other histopathologic findings in the case of ARDS and ALI are acute eosinophilic pneumonia (AEP) and acute fibrinous and organizing pneumonia (AFOP) [91].
DAD has two phases: the acute phase in the first week after lung injury, followed by the organizing or proliferative phase [92]. Two days after the lung injury, hyaline membranes are developed, while thrombi can also be seen as a result of the alteration of the coagulation, without any underlying thromboembolic disorder [93]. The organizing phase is defined by cellular fibroblastic proliferation, type 2 pneumocyte hyperplasia, squamous metaplasia, and residual fibrin rest. In this phase, the hyaline membranes become integrated into the alveolar septa and cannot be seen anymore [91]. In autopsies, other modifications such as polypoid plugs, alterations of the basal membrane and thickening of the alveolar wall were also present [84,94,95]. After this phase, DAD can be resolved gradually or can evolve into interstitial fibrosis, with most of the survivors presenting lung function alteration [96].
After the organizing stage, the disease may progress to fibrosis (fibrosing stage) (Figure 4a,b), with the excessive extracellular matrix, dense collagen deposition and diffuse thickening of alveolar walls, resulting in an architectural disorder, similar to other cellular and fibrous interstitial pneumonia patterns [94,96,97]. In a series of 30 minimal invasive autopsies, the most common finding was organizing DAD (70%), acute DAD (40%) and/or fibrosing patterns. Fibrosing DAD may be involved in the development of post-Covid-19 pulmonary fibrosis in long term. There is still limited data about the pathology of prolonged disease [94].
The histological patterns observed in organizing pneumonia are represented by fibroblasts and myofibroblasts that fill the alveolar space and ducts as an inflammatory response to the virus. This mechanism may have a favourable evolution towards resorption, or may chronically evolve to excessive deposition of collagen, and thus progress to pulmonary fibrosis [98]. In explanted lungs from patients with lung transplants due to COVID-19, the main pathologic characteristic was extensive pulmonary fibrosis, acute interstitial pneumonia or organizing pneumonia, micro-thrombosis, alveolar haemorrhage and acute bronchopneumonia from superimposed bacterial infection [85,99].
The morphologic changes of the lung in COVID-19 can be grouped into three main patterns: 1. epithelial reactive changes and DAD; 2. vascular with microvascular damage, microthrombi and acute fibrinous and organizing pneumonia and 3. fibrotic, with evidence of interstitial fibrosis [100]. The epithelial and vascular changes appear alone, simultaneously, or consecutively in all stages. Buja described in some cases the association of DAD with microvascular involvement and proposed three stages of lung injury: early infection stage, pulmonary stage and severe hyper-inflammation stage. In the first stage, the morphologic aspects are of interstitial pneumonia with DAD, in some cases associated with micro-thrombosis, peripheral lung haemorrhage and in severe cases pulmonary thromboembolism [101]. In COVID-19, interstitial inflammatory infiltrate is reduced (Figure 5a,b), unlike typical interstitial pneumonia.
Another study by Ackerman highlights the presence of thrombosis (Figure 6a,b), pulmonary vascular endothelitis and angiogenetic alterations in patients with COVID-19 [102], while Burel’s study showed the loss of pericytes, the cells responsible for micro-vessel integrity, which may be the trigger of the micro-vasculopathy [103]. Ackerman’s study has described three angiogenetic features of COVID-19, the first being severe endothelial injury associated with the destruction of the endothelial cell membranes, the second being disseminated vascular thrombosis in the lungs, associated with microangiopathy and capillaries occlusion, while the third feature described new vessel growth in the lungs through angiogenesis [102]. One study has shown an increased number of angiotensin-converting enzyme 2 (ACE2)- positive cells in the lung, in the case of COVID-19 patients. The presence of SARS-CoV-2 within the endothelial cells suggests that perivascular inflammation and the direct effects of the virus determine endothelial injury [104].
Some authors suggest two patterns of fatal COVID-19, with different clinical courses: one with high viral load, high cytokine expression in the lung but a limited morphologic expression, and a second one with low viral load and cytokine expression but a large number of immune cells (including CD8 + T lymphocytes and macrophages), that correlates with the presence of DAD [105].
The proliferative/organizing phase of DAD shows type II pneumocyte hyperplasia, reactive pneumocytes (Figure 7a), alveolar wall thickening, and myofibroblast proliferation (Figure 7b). A case report of an 80-year-old woman autopsy, with eradicated COVID-19, showed severe reactive and inflammatory changes in all the lung samples. The architecture was destroyed in larger areas with fibrinous organization and collagenized fibrosis. Widespread angiogenesis was seen as well as focal bleeding. Local moderate chronic inflammation dominated by lymphocytes was present. The fibrosis had a honeycomb-like pattern with enlarged airspaces and bronchial metaplasia in some areas. A small subpleural area showed alveoli with hyaline membranes representing the acute stage of lung injury, as is seen in acute DAD.
7. Therapeutic perspectives
Acute exacerbations of IPF could be triggered by viral respiratory tract infections, having a bad prognosis and high mortality rate [106]. Pirfenidone (a pyridine) and nintedanib (a tyrosine kinase inhibitor) are antifibrotic drugs that reduce lung function decline by 50% and improve life expectancy by 2-5 years [107]. Neither of these drugs has an immunosuppressive effect so they should not be stopped in case of infections.
Starting from April 2020 these drugs are available exclusively in oral form, as a result, it is not possible to administrate them in case of mechanically ventilated and intubated patients such as patients with severe forms of COVID-19. A form of pirfenidone with inhalator administration is under evaluation for COVID-19 patients. Pirfenidone should not be administrated in patients with a glomerular filtration rate of less than 30 mL/min per 1·73 m2. Patients with severe COVID-19 are at high risk to develop renal dysfunction, so pirfenidone should be carefully considered in these patients.
Pirfenidone and nintedanib can determine hepatotoxicity, while liver function test alterations are commonly found in COVID-19, especially in patients with severe forms, so in this case, temporary disruption of the antifibrotic treatment might be necessary [108].
Nintedanib has an increased risk of bleeding in case of the concomitant administration with full-dose of anticoagulant. COVID-19 patients have an increased risk of acute pulmonary embolism, and anticoagulant therapy is necessary in these cases. The use of antifibrotic therapy in COVID-19 might be useful in the case of patients with poor prognosis and high risk to develop pulmonary fibrosis and ALI [109].
SARS-CoV-2 spike protein has an Arg-Gly-Asp integrin-binding domain raising the possibility that inhibitory integrins or galectins therapies might be useful as COVID-19 treatment. Some drugs in development target molecules from TGF-β pathways, such as those against αvβ6 integrin (BG0001, PLN-74809) and galectins (TD139). Studies on mice have shown that those who did not have αvβ6 integrin or had treatment with an αvβ6 blocking antibody had increased protection against viral infections [110]. In another study performed on mice, galectin Gal-3 determined reduced pulmonary inflammation and protection against TGF-β-induced lung injury and fibrosis [111].
mTOR is a target in IPF, two recent studies showed that mTOR might be an anti-SARS-CoV-2 target, with rapamycin being taken into consideration in COVID-19 patients [112].
Pentraxins are response proteins of the acute phase, with a role in immunity and inflammation. One of them, PRM-151 an analogue of SAP (PTX2) has shown good results in IPF trials. SAP determines suppression of JNK family signalling, JNK1 inhibitor preventing fibrosis 55 and inhibiting ALI [113].
C21 role (an agonist of AT2R) is studied for COVID-19 and has clinical trial applicability in IPF, having anti-inflammatory properties. ACE2 receptors are the main SAR-CoV-2 receptors. A study has shown that in patients who took AT1R blockers before hospitalization the risk of severe COVID-19 was significantly decreased [114].
Treamid or bisamide derivative of dicarboxylic acid (BDDA) is an experimental drug with promising results used in animals with pulmonary fibrosis, that inhibits the production and deposition of collagen, being in trial for use in cases of IPF and post-COVID-19 fibrosis [115].
LYT-100 (deupirfenidon) is an N-aryl-pyridinone derivative, an analogue of pirfenidone, which has an antifibrotic effect and is in trial for usage COVID-19 and IPF [116].
Corticosteroids can be used in COVID-19 and IPF exacerbation. Long-term use of corticoid therapy might have a role in reducing the risk or the severity of PCPF, in rats with IPF having the role of slowing down the progression of the fibrosis [117].
8. Conclusions
There are numerous common features between PCPF and IPF regarding the clinical aspect, risk factors, pulmonary function tests, imagistic and histopathological aspects, as well as some notable differences. As clinical aspects both pathologies have common symptoms such as dyspnoea and cough; functionally restriction syndrome and decreasing of DLco can be observed; radiologically in PCPF, we can find signs of fibrosis in both cases, in IPF the aspect being defined as UIP. Regarding histopathology, findings of lung fibrosis in COVID-19 are limited, PCPF causing significant, irreversible consequences that affect patients’ quality of life after SARS-CoV-2 infection. DAD appears in PCPF, but it can appear also in an IPF exacerbation, alongside UIP features.
This review would help clinicians, pathologists and researchers to understand the mechanisms of fibrosis, and make a diagnosis as accurate as possible, which can be useful in identifying the group of patients who can be selected for antifibrotic therapies, and future therapeutic perspectives.
9. Patents
Author Contributions
Conceptualization, R.E.C. and A.P.F; methodology R.E.C, M.D., M.E., G.I.B., A.A.N.; software, I.S.G; validation, M.D., M.E., T.E.D. and A.P.F.; formal analysis, R.E.C and O.C.A.; investigation, S.I.G and M.D.; resources, A.V; data curation, R.E.C.; writing—original draft preparation, R.E.C and A.P.F; writing—review and editing, M.D., G.I.B., A.A.N. and O.C.A.; visualization, T.E.D; supervision, A.P.F; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgements
There is no funding or administrative support provided to the authors.
Conflicts of Interest
All the authors declare no conflict of interest.
References
- Bourgonje, A.R.; Abdulle, A.E; Timens, W.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol, 2020, 251, 228–48. [Google Scholar] [CrossRef] [PubMed]
- Shi L, Z.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 2020, 8, 420–422. [Google Scholar]
- Ioannidis, J.P.A. Infection fatality rate of COVID-19 inferred from seroprevalence data. Bull World Health Organ 2021, 99, 19–33F. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.K.; Sharma, P.; Kumar, R. Post covid 19 pulmonary fibrosis. Is it real threat? Indian J. Tuberc. 2020, 68, 330–333. [Google Scholar] [CrossRef]
- Han, X.; Cao, Y.; Jiang, N.; et al. Novel Coronavirus Disease 2019 (COVID-19) Pneumonia Progression Course in 17 Discharged Patients: Comparison of Clinical and Thin-Section Computed Tomography Features During Recovery. Clin Infect Dis 2020, 71, 723–731. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, C.; Hu, Y.; Li, C.; Ren, Q.; Zhang, X.; Shi, H.; Zhou, M. Temporal Changes of CT Findings in 90 Patients with COVID-19 Pneumonia: A Longitudinal Study. Radiology 2020, 296, E55–E64. [Google Scholar] [CrossRef] [PubMed]
- Liu, D. , Zhang W. ; Pan F.; et al. The pulmonary sequelae in discharged patients with COVID-19: a short-term observational study. Respir Res 2020, 21, 125. [Google Scholar]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Lechowicz, K.; Drozdzal, S.; Machaj, F.; Rosik, J.; Szosta, B.; Zegan-Baranska, M.; Biernawska, J.; Dabrowski, W.; Rotter, I.; Kotfis, K. COVID-19: The potential treatment of pulmonary fibrosis associated with SARS-CoV-2 infection. J. Clin. Med 2020, 9, 1917. [Google Scholar] [CrossRef]
- Han, X.; Fan, Y. ; Alwalid O:; Li N. ; Jia X.; Yuan M.; et al. Six-month Follow-up Chest CT Findings after Severe COVID-19 Pneumonia. Radiology 2021, 299, E177–E186. [Google Scholar]
- Idiopathic pulmonary fibrosis. National Heart, Lung, and Blood Institute website, 2019.
- Kawabata, Y. Pathology of IPF. In Idiopathic Pulmonary Fibrosis; Nakamura, H., Aoshiba, K., Eds.; Publisher: Springer, Tokyo, 2016. [Google Scholar] [CrossRef]
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef]
- Fadista, J.; Kraven, L.M.; Karjalainen, J.; Andrews, S.J.; Geller, F.; Baillie, J.K.; Wain, L.V.; Jenkins, R.; Feenstra, B. Shared genetic etiology between idiopathic pulmonary fibrosis and COVID-19 severity. EBioMedicine 2021, 65, 103277–103277. [Google Scholar] [CrossRef]
- Marvisi, M.; Ferrozzi, F.; Balzarini, L.; Mancini, C.; Ramponi, S.; Uccelli, M. First report on clinical and radiological features of COVID-19 pneumonitis in a Caucasian population: Factors predicting fibrotic evolution. Int. J. Infect. Dis. 2020, 99, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm. Med. 2020, 2020, 6175964. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Wu, Q.; Chen, Z.; Xiong, Z.; Wang, K.; Tian, J.; Zhang, S. The potential indicators for pulmonary fibrosis in survivors of severe COVID-19. J. Infect 2020. [CrossRef]
- Yu, M.; Liu, Y.; Xu, D.; Zhang, R.; Lan, L.; Xu, H. Prediction of the development of pulmonary fibrosis using serial thin-section CT and clinical features in patients discharged after treatment for COVID-19 pneumonia. Korean J. Radiol 2020, 21, 746–755. [Google Scholar] [CrossRef]
- Vasarmidi, E.; Tsitoura, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K.M. Pulmonary fibrosis in the aftermath of the Covid-19 era (Review). Exp. Ther. Med. 2020, 20, 2557–2560. [Google Scholar] [CrossRef] [PubMed]
- Guler, S.A.; Ebner, L.; Beigelman, C.; et al. Pulmonary function and radiological features four months after COVID-19: first results from the national prospective observational Swiss COVID-19 lung study. Eur Respir J 2021, 2003690. in press. [CrossRef]
- Grasselli, G.; Pesenti, A.; Cecconi, M. Critical Care Utilization for the COVID-19 Outbreak in Lombardy, Italy: Early experience and forecast during an emergency response. JAMA 2020, 323, 1545–1546. [Google Scholar] [CrossRef]
- Rogliani, P.; Calzetta, L.; Coppola, A.; et al. Are there pulmonary sequelae in patients recovering from COVID-19? Respir Res 2020, 21, 286. [Google Scholar] [CrossRef]
- Ding, M.; Zhang, Q.; Li, Q.; et al. Correlation analysis of the severity and clinical prognosis of 32 cases of patients with COVID-19. Respir Med 2020, 167, 105981. [Google Scholar] [CrossRef]
- Hu, Z.J.; Xu, J.; Yin, J.M.; et al. Lower circulating interferon-gamma is a risk factor for lung fibrosis in COVID-19 patients. Front Immunol 2020, 11, 585647. [Google Scholar] [CrossRef] [PubMed]
- Velavan, T.P.; Pallerla, S.R.; Rüter, J.; Augustin, Y.; Kremsner, P.G.; Krishna, S.; Meyer, C.G. Host genetic factors determining COVID-19 susceptibility and severity. EBioMedicine 2021, 72, 103629. [Google Scholar] [CrossRef] [PubMed]
- Severe Covid-19 GWAS Group. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Covid-19 Host Genetics Initiative Mapping the human genetic architecture of COVID-19. Nature, 2021.
- Wein, A.N.; McMaster, S.R.; Takamura, S.; Dunbar, P.R.; Cartwright, E.K.; Hayward, S.L.; McManus, D.T.; Shimaoka, T.; Ueha, S.; Tsukui, T.; et al. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med. 2019, 216, 2748–2762. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S.; Loyd, J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1717–1727. [Google Scholar] [CrossRef]
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. 2017, 4, 154. [Google Scholar] [CrossRef]
- van Moorsel, C.H.M.; van der Vis, J.J.; Duckworth, A.; Scotton, C.J.; Benschop, C.; Ellinghaus, D.; Ruven, H.J.T.; Quanjel, M.J.R.; Grutters, J.C. The MUC5B Promoter Polymorphism Associates With Severe COVID-19 in the European Population. Front. Med. 2021, 8, 668024. [Google Scholar] [CrossRef]
- Zaman, T.; Lee, J.S. Risk Factors for the Development of Idiopathic Pulmonary Fibrosis: a Review. Curr. Pulmonol. Rep. 2018, 7, 118–125. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Gottardi, C.J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Finding Similarities within Differences. Am. J. Respir. Cell Mol. Biol. 2019, 61, 667–668. [Google Scholar] [CrossRef]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef]
- Leung, J.; Cho, Y.; Lockey, R.F.; Kolliputi, N. The Role of Aging in Idiopathic Pulmonary Fibrosis. Lung 2015, 193, 605–610. [Google Scholar] [CrossRef]
- Jo, H.E.; Glaspole, I.; Grainge, C.; Goh, N.; Hopkins, P.M.; Moodley, Y.; Reynolds, P.N.; Chapman, S.; Walters, E.H.; Zappala, C.; et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur. Respir. J. 2017, 49, 1601592. [Google Scholar] [CrossRef]
- Ali, R.M.M.; Ghonimy, M.B.I. Post-COVID-19 pneumonia lung fibrosis: a worrisome sequelae in surviving patients. Egypt. J. Radiol. Nucl. Med. 2021, 52, 101. [Google Scholar] [CrossRef]
- Voltz, J.W.; Card, J.W.; Carey, M.A.; DeGraff, L.M.; Ferguson, C.D.; Flake, G.P.; Bonner, J.C.; Korach, K.S.; Zeldin, D.C. Male Sex Hormones Exacerbate Lung Function Impairment after Bleomycin-Induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 45–52. [Google Scholar] [CrossRef]
- Lekgabe, E.D.; Royce, S.G.; Hewitson, T.D.; Tang, M.L.K.; Zhao, C.; Moore, X.L.; Tregear, G.W.; Bathgate, R.A.D.; Du, X.-J.; Samuel, C.S. The Effects of Relaxin and Estrogen Deficiency on Collagen Deposition and Hypertrophy of Nonreproductive Organs. Endocrinology 2006, 147, 5575–5583. [Google Scholar] [CrossRef]
- Baumgartner, K.B.; Samet, J.M.; A Stidley, C.; Colby, T.V.; A Waldron, J. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef]
- Liu, W.; Tao, Z.W.; Wang, L.; et al. Analysis of factors associated with disease outcomes in hospitalized patients with 2019 novel coronavirus disease. Chinese Med J 2020, 133, 1032e1038. [Google Scholar] [CrossRef]
- Vardavas, C.I.; Nikitara, K. COVID-19 and smoking: A systematic review of the evidence. Tob. Induc. Dis. 2020, 18, 20. [Google Scholar] [CrossRef]
- Jensen, K.; Nizamutdinov, D.; Guerrier, M.; Afroze, S.; Dostal, D.; Glaser, S. General mechanisms of nicotine-induced fibrogenesis. FASEB J. 2012, 26, 4778–4787. [Google Scholar] [CrossRef]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The epithelium in idiopathic pulmonary fibrosis: breaking the barrier. Front. Pharmacol. 2014, 4. [Google Scholar] [CrossRef]
- Taskar, V.S.; Coultas, D.B. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006, 3, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.G.; Mallia, P.; Russell, K.E.; Russell, A.-M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef]
- Han, M.K.; Zhou, Y.; Murray, S.; Tayob, N.; Noth, I.; Lama, V.N.; Moore, B.B.; White, E.S.; Flaherty, K.R.; Huffnagle, G.B.; et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: An analysis of the COMET study. Lancet Respir. Med. 2014, 2, 548–556. [Google Scholar] [CrossRef]
- Nasir, N.; Rehman, F.; Omair, S.F. Risk factors for bacterial infections in patients with moderate to severe COVID-19: A case-control study. J. Med Virol. 2021, 93, 4564–4569. [Google Scholar] [CrossRef]
- Langford, B.J.; So, M.; Raybardhan, S.; Leung, V.; Westwood, D.; MacFadden, D.R.; Soucy, J.-P.R.; Daneman, N. Bacterial co-infection and secondary infection in patients with COVID-19: a living rapid review and meta-analysis. Clin. Microbiol. Infect. 2020, 26, 1622–1629. [Google Scholar] [CrossRef]
- Sharifipour, E.; Shams, S.; Esmkhani, M.; Khodadadi, J.; Fotouhi-Ardakani, R.; Koohpaei, A.; Doosti, Z.; Golzari, S.E. Evaluation of bacterial co-infections of the respiratory tract in COVID-19 patients admitted to ICU. BMC Infect. Dis. 2020, 20, 1–7. [Google Scholar] [CrossRef]
- Alhiyari, M.A.; Ata, F.; Alghizzawi, M.I.; I Bilal, A.B.; Abdulhadi, A.S.; Yousaf, Z. Post COVID-19 fibrosis, an emerging complicationof SARS-CoV-2 infection. IDCases 2020, 23, e01041. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef]
- Carfi, A.; Bernabei, R.; Landi, F. For the Gemelli against COVID-19 post-acute care study group. Persistent symptoms in patients after acute COVID-19. J Am Med Assoc 2020, 324, 603–605. [Google Scholar]
- McGroder, C.F.; Zhang, D.; A Choudhury, M.; Salvatore, M.M.; D’Souza, B.M.; A Hoffman, E.; Wei, Y.; Baldwin, M.R.; Garcia, C.K. Pulmonary fibrosis 4 months after COVID-19 is associated with severity of illness and blood leucocyte telomere length. Thorax 2021, 76, 1242–1245. [Google Scholar] [CrossRef]
- Kamal, M.; Omirah, M.A.; Hussein, A.; Saeed, H. Assessment and characterisation of post-COVID-19 manifestations. Int. J. Clin. Pr. 2020, 75. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.M.; Patel, S.; E Barker, R.; George, P.; Maddocks, M.M.; Cullinan, P.; Maher, T.M.; Man, W.D.-C. Anxiety and depression in idiopathic pulmonary fibrosis (IPF): prevalence and clinical correlates. ERS International Congress 2017 abstracts. LOCATION OF CONFERENCE, COUNTRYDATE OF CONFERENCE; p. PA848.
- Lechtzin, N.; Hilliard, M.E.; Horton, M.R. Validation of the Cough Quality-of-Life Questionnaire in Patients With Idiopathic Pulmonary Fibrosis. Chest 2013, 143, 1745–1749. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Katsura, H.; Sawabe, M.; Kida, K. A Clinical Study of Idiopathic Pulmonary Fibrosis Based on Autopsy Studies in Elderly Patients. Intern. Med. 2003, 42, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Carvajalino, S.; Reigada, C.; Johnson, M.J.; Dzingina, M.; Bajwah, S. Symptom prevalence of patients with fibrotic interstitial lung disease: a systematic literature review. BMC Pulm. Med. 2018, 18, 78. [Google Scholar] [CrossRef]
- Faverio, P.; Luppi, F.; Rebora, P.; Busnelli, S.; Stainer, A.; Catalano, M.; Parachini, L.; Monzani, A.; Galimberti, S.; Bini, F.; et al. Six-Month Pulmonary Impairment after Severe COVID-19: A Prospective, Multicentre Follow-Up Study. Respiration 2021, 100, 1078–1087. [Google Scholar] [CrossRef]
- Kanematsu, T.; Kitaichi, M.; Nishimura, K.; Nagai, S.; Izumi, T. Clubbing of the Fingers and Smooth-Muscle Proliferation in Fibrotic Changes in the Lung in Patients With Idiopathic Pulmonary Fibrosis. Chest 1994, 105, 339–342. [Google Scholar] [CrossRef]
- Bonella, F. , di Marco F., Spagnolo P. Pulmonary Function Tests in Idiopathic Pulmonary Fibrosis. In: Idiopathic Pulmonary Fibrosis. Respiratory Medicine, Meyer K., Nathan S. Eds.; Humana Press, Cham, 2019. [CrossRef]
- Torres-Castro, R.; Vasconcello-Castillo, L.; Alsina-Restoy, X.; Solis-Navarro, L.; Burgos, F.; Puppo, H.; Vilaró, J. Respiratory function in patients post-infection by COVID-19: a systematic review and meta-analysis. Pulmonology 2020, 27, 328–337. [Google Scholar] [CrossRef]
- British Thoracic Society. British Thoracic Society Guidance on Respiratory Follow-Up of Patients with a Clinico-Radiological Diagnosis of COVID-19 Pneumonia [Internet]. 2020. Available from: https://www.brit-thoracic.org.uk/document-library/quality-improvement/covid-19/resp-follow-up-guidance-post-covid-pneumonia/.
- Cherrez-Ojeda, I.; Robles-Velasco, K.; Osorio, M.F.; Cottin, V.; Centeno, J.V.; Felix, M. Follow-up of two cases of suspected interstitial lung disease following severe COVID-19 infection shows persistent changes in imaging and lung function. Clin. Case Rep. 2021, 9, e04918. [Google Scholar] [CrossRef]
- Cabo-Gambin, R.; Benítez, I.D.; Carmona, P.; Santiesteve, S.; Mínguez, O.; Vaca, R.; Moncusí-Moix, A.; Gort-Paniello, C.; García-Hidalgo, M.C.; de Gonzalo-Calvo, D.; et al. Three to Six Months Evolution of Pulmonary Function and Radiological Features in Critical COVID-19 Patients: A Prospective Cohort. 58, 62. [CrossRef]
- Fidler, L.; Shapera, S.; Mittoo, S.; Marras, T.K. Diagnostic Disparity of Previous and Revised American Thoracic Society Guidelines for Idiopathic Pulmonary Fibrosis. Can. Respir. J. 2015, 22, 86–90. [Google Scholar] [CrossRef]
- Collard, H.R.; King, T.E., Jr.; Bartelson, B.B.; Vourlekis, J.S.; Schwarz, M.I.; Brown, K.K. Changes in Clinical and Physiologic Variables Predict Survival in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 538–542. [Google Scholar] [CrossRef]
- Nathan, S.D.; Shlobin, O.A.; Weir, N.; Ahmad, S.; Kaldjob, J.M.; Battle, E.; Sheridan, M.J.; du Bois, R.M. Long-term Course and Prognosis of Idiopathic Pulmonary Fibrosis in the New Millennium. Chest 2011, 140, 221–229. [Google Scholar] [CrossRef]
- Tanni, S.E.; Fabro, A.T.; de Albuquerque, A.; Ferreira, E.V.M.; Verrastro, C.G.Y.; Sawamura, M.V.Y.; Ribeiro, S.M.; Baldi, B.G. Pulmonary fibrosis secondary to COVID-19: a narrative review. Expert Rev. Respir. Med. 2021, 15, 791–803. [Google Scholar] [CrossRef]
- Atabati, E.; Dehghani-Samani, A.; Mortazavimoghaddam, S.G. Association of COVID-19 and other viral infections with interstitial lung diseases, pulmonary fibrosis, and pulmonary hypertension: A narrative review. Can. J. Respir. Ther. 2020, 56, 70–78. [Google Scholar] [CrossRef]
- Wei, J.; Yang, H.; Lei, P.; Fan, B.; Qiu, Y.; Zeng, B.; Yu, P.; Lv, J.; Jian, Y.; Wan, C. Analysis of thin-section CT in patients with coronavirus disease (COVID-19) after hospital discharge. J. X-Ray Sci. Technol. 2020, 28, 383–389. [Google Scholar] [CrossRef]
- Francone, M.; Iafrate, F.; Masci, G.M.; Coco, S.; Cilia, F.; Manganaro, L.; Panebianco, V.; Andreoli, C.; Colaiacomo, M.C.; Zingaropoli, M.A.; et al. Chest CT score in COVID-19 patients: correlation with disease severity and short-term prognosis. Eur. Radiol. 2020, 30, 6808–6817. [Google Scholar] [CrossRef]
- Yin, X.; Xi, X.; Min, X.; et al. Long-term chest CT follow-up in COVID-19 Survivors: 102-361 days after onset. Ann Transl Med 2021, 9, 1231. [Google Scholar] [CrossRef]
- Vijayakumar, B.; Tonkin, J.; Devaraj, A.; Philip, K.E.J.; Orton, C.M.; Desai, S.R.; Shah, P.L. CT Lung Abnormalities after COVID-19 at 3 Months and 1 Year after Hospital Discharge. Radiology 2022, 303, 444–454. [Google Scholar] [CrossRef]
- Camiciottoli, G.; Orlandi, I.; Bartolucci, M.; Meoni, E.; Nacci, F.; Diciotti, S.; Barcaroli, C.; Conforti, M.L.; Pistolesi, M.; Matucci-Cerinic, M.; et al. Lung CT Densitometry in Systemic Sclerosis. 131. [CrossRef]
- Combet, M.; Pavot, A.; Savale, L.; Humbert, M.; Monnet, X. Rapid onset honeycombing fibrosis in spontaneously breathing patient with COVID-19. Eur. Respir. J. 2020, 56, 2001808. [Google Scholar] [CrossRef]
- Balestro, E.; Cocconcelli, E.; Giraudo, C.; Polverosi, R.; Biondini, D.; Lacedonia, D.; Bazzan, E.; Mazzai, L.; Rizzon, G.; Lococo, S.; et al. High-Resolution CT Change over Time in Patients with Idiopathic Pulmonary Fibrosis on Antifibrotic Treatment. J. Clin. Med. 2019, 8, 1469. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Elicker, B.M.; Hartman, T.E.; Ryerson, C.J.; Vittinghoff, E.; Ryu, J.H.; Lee, J.S.; Jones, K.D.; Richeldi, L.; King, T.E.; et al. Idiopathic pulmonary fibrosis: CT and risk of death. . 2014, 273, 570–9. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Adachi, T.; Chong, J.-M.; Nakajima, N.; Sano, M.; Yamazaki, J.; Miyamoto, I.; Nishioka, H.; Akita, H.; Sato, Y.; Kataoka, M.; et al. Clinicopathologic and Immunohistochemical Findings from Autopsy of Patient with COVID-19, Japan. Emerg. Infect. Dis. 2020, 26, 2157–2161. [Google Scholar] [CrossRef]
- Deshmukh, V.; Motwani, R.; Kumar, A.; Kumari, C.; Raza, K. Histopathological observations in COVID-19: a systematic review. J. Clin. Pathol. 2020, 74, 76–83. [Google Scholar] [CrossRef]
- Martines, R.B.; Ritter, J.M.; Matkovic, E.; et al. COVID-19 pathology working group. Pathology and pathogenesis of SARS-CoV-2 associated with fatal coronavirus disease, United States. Emerg Infect Dis 2020, 26, 2005–2015. [Google Scholar]
- Sauter, J.L.; Baine, M.K.; Butnor, K.J.; Buonocore, D.J.; Chang, J.C.; A Jungbluth, A.; Szabolcs, M.J.; Morjaria, S.; Mount, S.L.; Rekhtman, N.; et al. Insights into pathogenesis of fatal COVID-19 pneumonia from histopathology with immunohistochemical and viral RNA studies. Histopathology 2020, 77, 915–925. [Google Scholar] [CrossRef]
- Chen, J.Y.; Qiao, K.; Liu, F.; et al. Lung transplantation as therapeutic option in acute respiratory distress syndrome for coronavirus disease 2019-related pulmonary fibrosis. Chin Med J (Engl) 2020, 133, 1390–1396. [Google Scholar] [CrossRef]
- Tian, S.; Xiong, Y.; Liu, H.; et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol 2020, 33, 1007–1014. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Hanley, B.; Naresh, K.N.; Roufosse, C.; Nicholson, A.G.; Weir, J.; Cooke, G.S.; Thursz, M.; Manousou, P.; Corbett, R.; Goldin, R.; et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: a post-mortem study. Lancet Microbe 2020, 1, e245–e253. [Google Scholar] [CrossRef] [PubMed]
- Parambil, J.G.; Myers, J.L.; Aubry, M.-C.; Ryu, J.H. Causes and Prognosis of Diffuse Alveolar Damage Diagnosed on Surgical Lung Biopsy. Chest 2007, 132, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; Legall, J.R.; Morris, A.; Spragg, R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994, 149, 818–824. [Google Scholar] [CrossRef]
- Beasley, M.B.; Franks, T.J.; Galvin, J.R.; Gochuico, B.; Travis, W.D. Acute fibrinous and organizing pneumonia: a histological pattern of lung injury and possible variant of diffuse alveolar damage. . 2002, 126, 1064–70. [Google Scholar] [CrossRef]
- Tomashefski, J.F., Jr. PULMONARY PATHOLOGY OF ACUTE RESPIRATORY DISTRESS SYNDROME. Clin. Chest Med. 2000, 21, 435–466. [Google Scholar] [CrossRef]
- Sapru, A.; Wiemels, J.L.; Witte, J.S.; Ware, L.B.; Matthay, M.A. Acute lung injury and the coagulation pathway: potential role of gene polymorphisms in the protein C and fibrinolytic pathways. Intensiv. Care Med. 2006, 32, 1293–1303. [Google Scholar] [CrossRef]
- Li, Y.; Wu, J.; Wang, S.; Li, X.; Zhou, J.; Huang, B.; Luo, D.; Cao, Q.; Chen, Y.; Chen, S.; et al. Progression to fibrosing diffuse alveolar damage in a series of 30 minimally invasive autopsies with COVID-19 pneumonia in Wuhan, China. Histopathology 2020, 78, 542–555. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, P.; Wei, Y.; et al. Histopathologic Changes and SARS-CoV-2 immunostaining in the lung of a patient with COVID-19. Ann Intern Med 2020, 172, 629–632. [Google Scholar] [CrossRef]
- Kory, P.; Jp, K. SARS-CoV-2 organizing pneumonia: has there been a widespread failure to identify and treat this prevalent condition in COVID-19? BMJ Open Respir Res 2020, 7, e000724. [Google Scholar] [CrossRef]
- Kommoss, F.K.; Schwab, C.; Tavernar, L.; Schreck, J.; Wagner, W.L.; Merle, U.; Jonigk, D.; Schirmacher, P.; Longerich, T. The Pathology of Severe COVID-19-Related Lung Damage. Dtsch. Aerzteblatt Online 2020, 117, 500. [Google Scholar] [CrossRef]
- Copin, M.-C.; Parmentier, E.; Duburcq, T.; Poissy, J.; Mathieu, D. ; The Lille COVID-19 ICU and Anatomopathology Group Time to consider histologic pattern of lung injury to treat critically ill patients with COVID-19 infection. Intensiv. Care Med. 2020, 46, 1124–1126. [Google Scholar] [CrossRef]
- Bharat, A.; Querrey, M.; Markov, N.S.; Kim, S.; Kurihara, C.; Garza-Castillon, R.; Manerikar, A.; Shilatifard, A.; Tomic, R.; Politanska, Y.; et al. Lung transplantation for patients with severe COVID-19. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- John, A.E.; Joseph, C.; Jenkins, G.; Tatler, A.L. COVID-19 and pulmonary fibrosis: A potential role for lung epithelial cells and fibroblasts. Immunol. Rev. 2021, 302, 228–240. [Google Scholar] [CrossRef]
- Buja, L.M.; Wolf, D.; Zhao, B. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc Pathol 2020, 48, 107233. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Burel-Vandenbos, F.; Cardot-Leccia, N.; Passeron, T. Pulmonary vascular pathology in Covid-19. N Engl J Med 2020, 383, 886–887. [Google Scholar] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef] [PubMed]
- Nienhold, R.; Ciani, Y.; Koelzer, V.H.; Tzankov, A.; Haslbauer, J.D.; Menter, T.; Schwab, N.; Henkel, M.; Frank, A.; Zsikla, V.; et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med 2016, 194, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Nathan, S.D.; Hill, C.; Marshall, J.; Dejonckheere, F.; Thuresson, P.-O.; Maher, T.M. Predicting Life Expectancy for Pirfenidone in Idiopathic Pulmonary Fibrosis. J. Manag. Care Spéc. Pharm. 2017, 23, S17–S24. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Jolly, L.; Stavrou, A.; Vanderstoken, G. Influenza promotes collagen deposition via αvβ6 integrin-mediated transforming growth factor β activation. J Biol Chem 2014, 289, 35246–35263. [Google Scholar] [CrossRef]
- MacKinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of Transforming Growth Factor-β1–driven Lung Fibrosis by Galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; van den Blink, B.; Hamblin, M.J. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: a randomized clinical trial. JAMA 2018, 319, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, F.; Xu, J.; Yang, P.; Qin, Y.; Cao, M.; Wang, Z.; Li, X.; Zhang, S.; Ye, L.; et al. Anti-hypertensive Angiotensin II receptor blockers associated to mitigation of disease severity in elderly COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Skurikhin, E.; Nebolsin, V.; Widera, D.; Ermakova, N.; Pershina, O.; Pakhomova, A.; Krupin, V.; Pan, E.; Zhukova, M.; Novikov, F.; et al. Antifibrotic and Regenerative Effects of Treamid in Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 8380. [Google Scholar] [CrossRef]
- A Phase 2 Randomized, Double-Blind, Placebo-Controlled Trial and Open Label Extension to Evaluate the Safety and Efficacy of Deupirfenidone (LYT-100) in Post-Acute COVID-19 Respiratory Disease. PureTech; Boston, MA, USA: 2020. Identifier NCT04652518.
- Bazdyrev, E.; Rusina, P.; Panova, M.; Novikov, F.; Grishagin, I.; Nebolsin, V. Lung Fibrosis after COVID-19: Treatment Prospects. Pharmaceuticals 2021, 14, 807. [Google Scholar] [CrossRef]
Figure 1.
By courtesy of Dr. Oana Cristina Arghir, who provided images of post COVID-19 pulmonary fibrosis, from Pneumology Hospital of Constanta, Romania. Thoracic CT scan of a patient with PCF, 1 year after the acute episode, showing subpleural and peribronchovascular reticular opacities, traction bronchiectasis and GGO bilateral, and fibrotic lines predominantly in the upper lobes, in a 77 year-old male, with history of severe form of COVID-19 in October 2021 and PCPF in October 2022.
Figure 1.
By courtesy of Dr. Oana Cristina Arghir, who provided images of post COVID-19 pulmonary fibrosis, from Pneumology Hospital of Constanta, Romania. Thoracic CT scan of a patient with PCF, 1 year after the acute episode, showing subpleural and peribronchovascular reticular opacities, traction bronchiectasis and GGO bilateral, and fibrotic lines predominantly in the upper lobes, in a 77 year-old male, with history of severe form of COVID-19 in October 2021 and PCPF in October 2022.

Figure 2.
By courtesy of Dr. Ariadna Petronela Fildan, who provided images of IPF, from Pneumology Hospital of Constanta, Romania. Thoracic CT scan of a patient with IPF, showing honeycombing and reticular opacities with the basal and subpleural distribution in a 73 year-old male, smoker, with exposure to environmental noxes, with idiopathic pulmonary fibrosis, who died in 2017, 3 years after the diagnosis.
Figure 2.
By courtesy of Dr. Ariadna Petronela Fildan, who provided images of IPF, from Pneumology Hospital of Constanta, Romania. Thoracic CT scan of a patient with IPF, showing honeycombing and reticular opacities with the basal and subpleural distribution in a 73 year-old male, smoker, with exposure to environmental noxes, with idiopathic pulmonary fibrosis, who died in 2017, 3 years after the diagnosis.
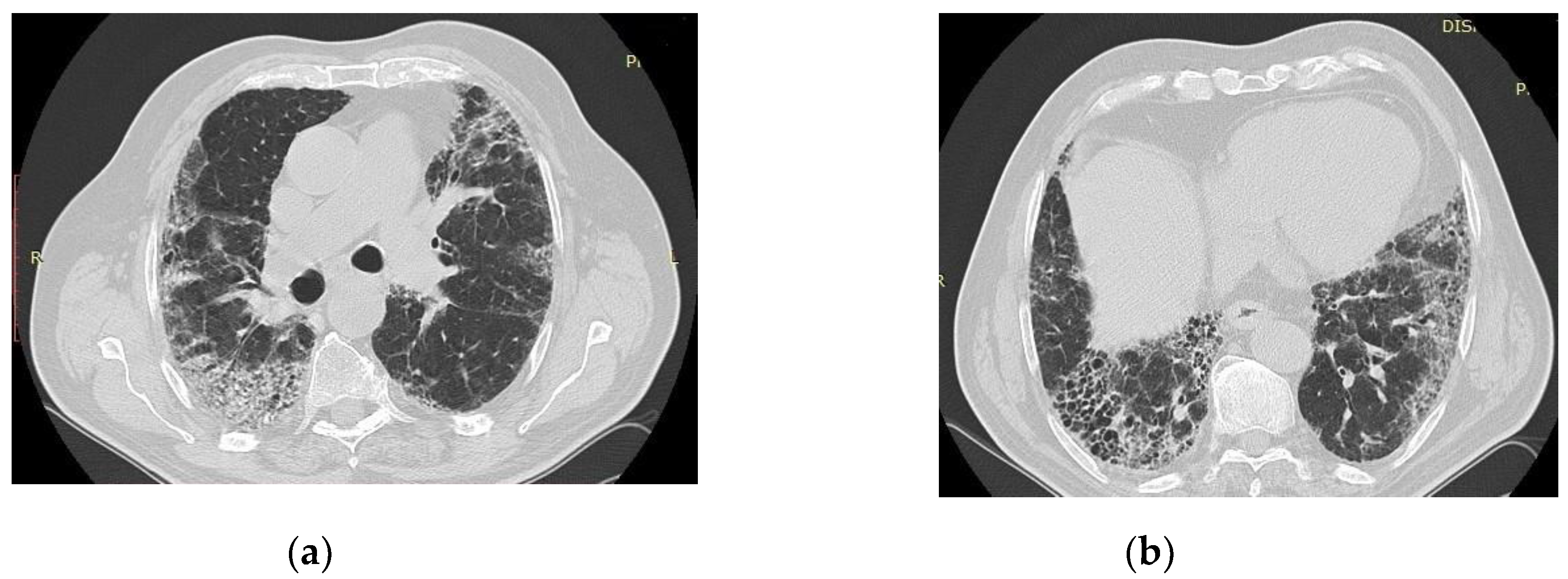
Figure 3.
By courtesy of Dr. Angela Ștefania Mărgescu, who provided images of IPF from Pneumology Institute “Marius Nasta” Bucharest, Romania. (a) In this image is evident the spatial variability, with normal lung parenchyma mixed with fibrotic areas (fibroblastic focus); HE, 40x. (b) Interstitial fibrosis with architectural distortion and cystic changes in the pulmonary parenchyma; HEx40, in a 62 –year-old women with marked pulmonary fibrosis.
Figure 3.
By courtesy of Dr. Angela Ștefania Mărgescu, who provided images of IPF from Pneumology Institute “Marius Nasta” Bucharest, Romania. (a) In this image is evident the spatial variability, with normal lung parenchyma mixed with fibrotic areas (fibroblastic focus); HE, 40x. (b) Interstitial fibrosis with architectural distortion and cystic changes in the pulmonary parenchyma; HEx40, in a 62 –year-old women with marked pulmonary fibrosis.
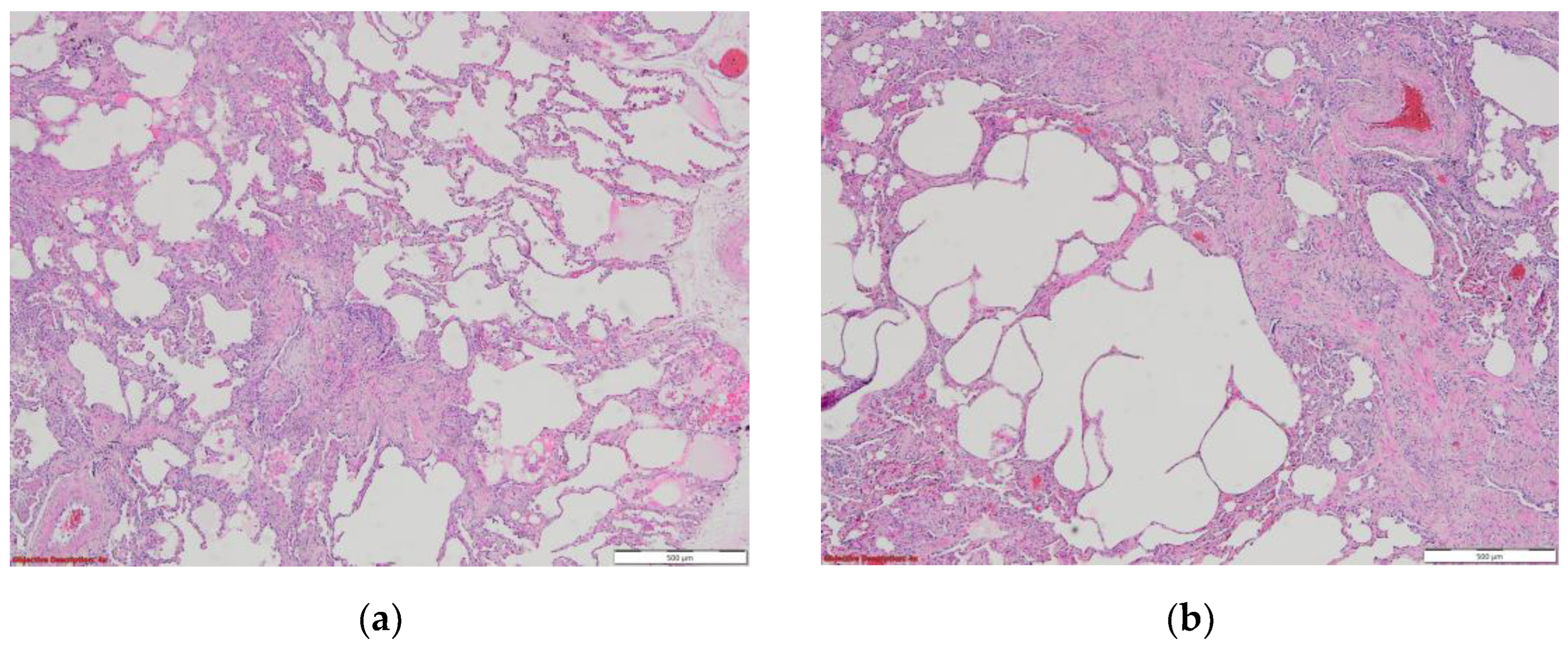
Figure 4.
By courtesy of Dr. Mariana Deacu, who provided images of post COVID-19 pulmonary fibrosis, from “St. Andrei ‘’Emergency County Hospital of Constanta, Romania (a) Macroscopic and (b) microscopic aspect of the lung in a 55-years-old male patient patient with COVID-19 interstitial pneumonia in the fibrosing stage, showing architectural disorder caused by excessive extracellular matrix, dense collagen deposition and diffuse thickening of alveolar walls. HE x40.
Figure 4.
By courtesy of Dr. Mariana Deacu, who provided images of post COVID-19 pulmonary fibrosis, from “St. Andrei ‘’Emergency County Hospital of Constanta, Romania (a) Macroscopic and (b) microscopic aspect of the lung in a 55-years-old male patient patient with COVID-19 interstitial pneumonia in the fibrosing stage, showing architectural disorder caused by excessive extracellular matrix, dense collagen deposition and diffuse thickening of alveolar walls. HE x40.
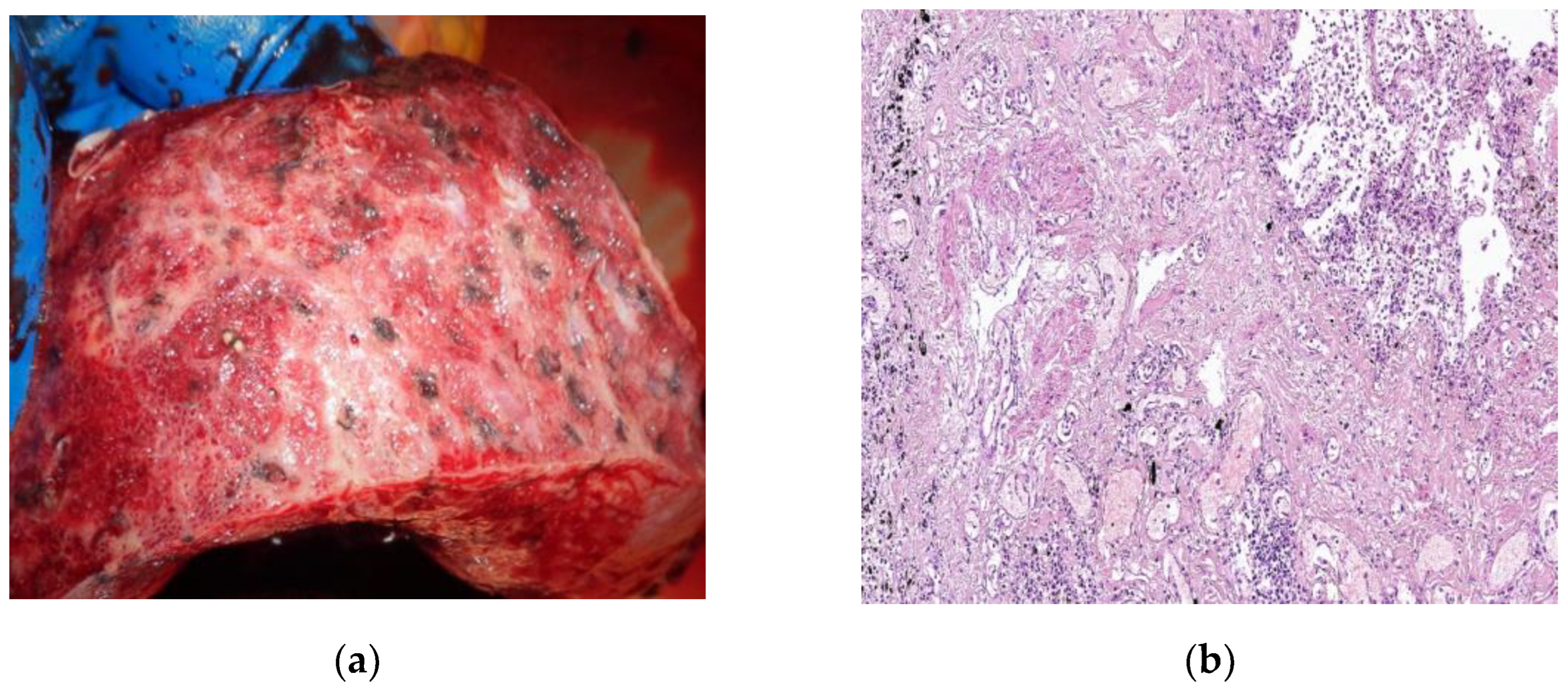
Figure 5.
By courtesy of Dr. Mariana Deacu, who provided images of post COVID-19 pulmonary fibrosis, from “St. Andrei ‘’Emergency County Hospital of Constanta, Romania (a) Interstitial inflammatory infiltrate reduced seen in a microscopic view of the lung, in a 61 –years old patient with COVID-19. HE x100 (b) Chronic bronchiolitis is seen in a microscopic view of the lung, in a patient with COVID-19. HE x100.
Figure 5.
By courtesy of Dr. Mariana Deacu, who provided images of post COVID-19 pulmonary fibrosis, from “St. Andrei ‘’Emergency County Hospital of Constanta, Romania (a) Interstitial inflammatory infiltrate reduced seen in a microscopic view of the lung, in a 61 –years old patient with COVID-19. HE x100 (b) Chronic bronchiolitis is seen in a microscopic view of the lung, in a patient with COVID-19. HE x100.
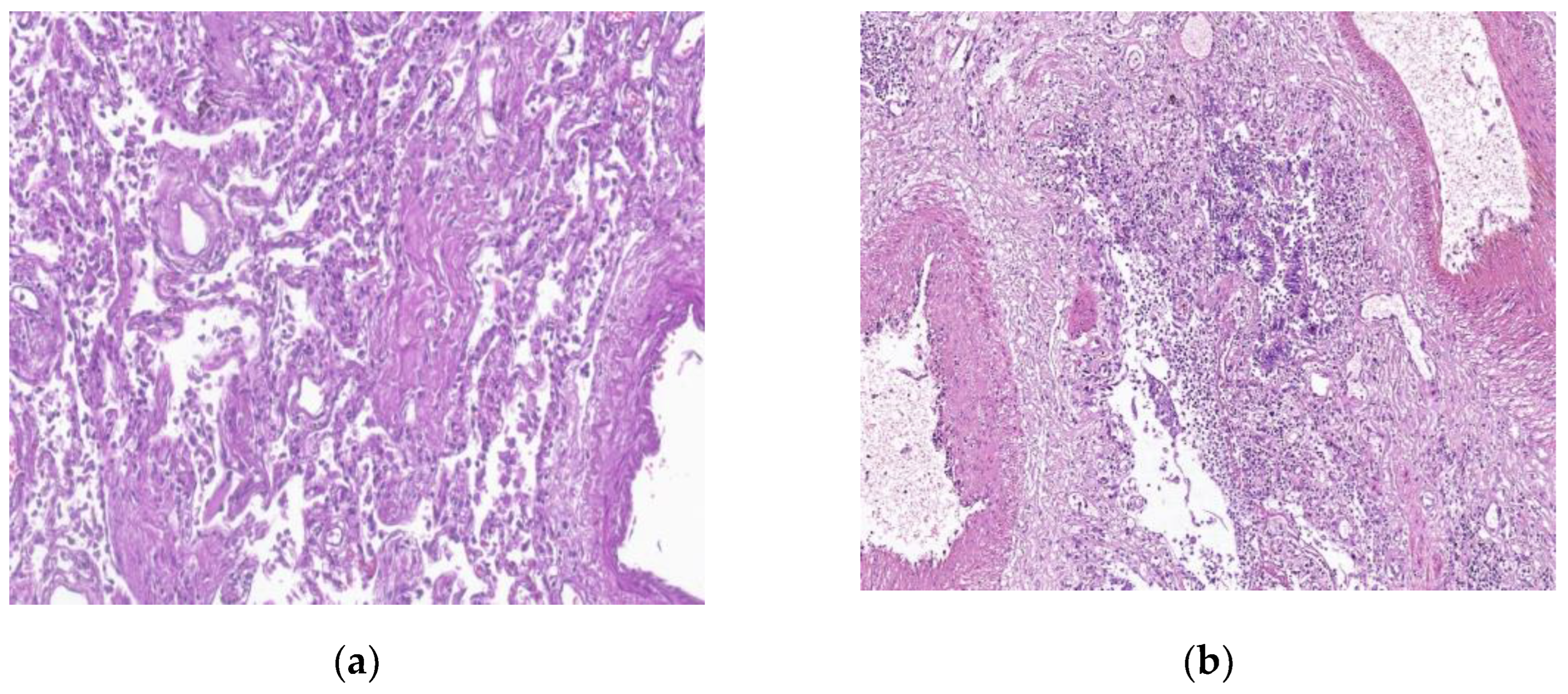
Figure 6.
By courtesy of Dr. Mariana Deacu, who provided images of post COVID-19 pulmonary fibrosis, from “St. Andrei ‘’Emergency County Hospital of Constanta, Romania (a) Macroscopic and (b) microscopic aspects of the lung in a 62-years-old patient showing thrombosis, pulmonary vascular endothelitis and angiogenetic alterations. HE x40.
Figure 6.
By courtesy of Dr. Mariana Deacu, who provided images of post COVID-19 pulmonary fibrosis, from “St. Andrei ‘’Emergency County Hospital of Constanta, Romania (a) Macroscopic and (b) microscopic aspects of the lung in a 62-years-old patient showing thrombosis, pulmonary vascular endothelitis and angiogenetic alterations. HE x40.
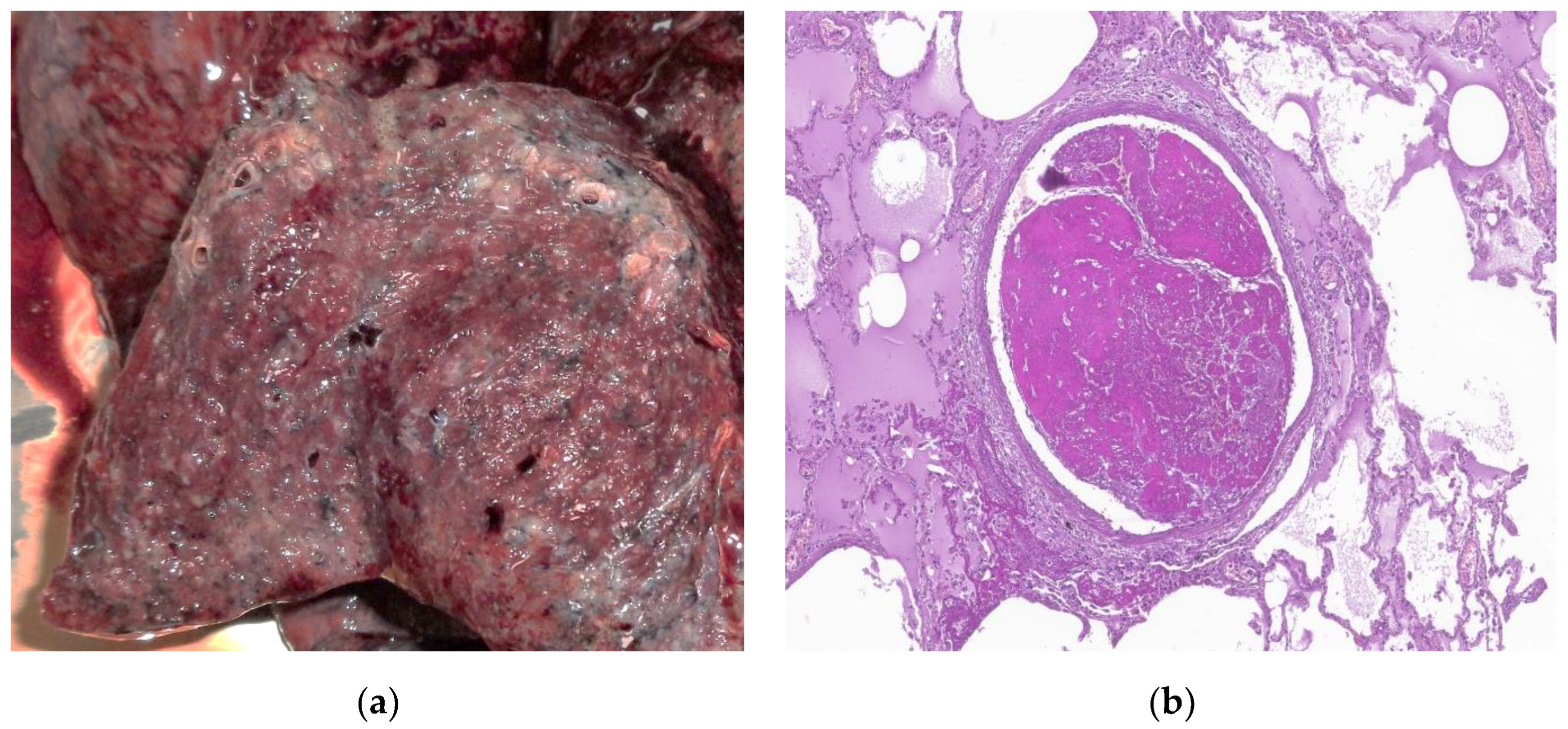
Figure 7.
By courtesy of Dr. Mariana Deacu, who provided images of post COVID-19 pulmonary fibrosis, from “St. Andrei ‘’Emergency County Hospital of Constanta, Romania Microscopic view of the lung in a 68-years-old patient, showing: (a) type II pneumocyte hyperplasia and reactive pneumocytes (b) alveolar wall thickening, and myofibroblast proliferation. HE x100.
Figure 7.
By courtesy of Dr. Mariana Deacu, who provided images of post COVID-19 pulmonary fibrosis, from “St. Andrei ‘’Emergency County Hospital of Constanta, Romania Microscopic view of the lung in a 68-years-old patient, showing: (a) type II pneumocyte hyperplasia and reactive pneumocytes (b) alveolar wall thickening, and myofibroblast proliferation. HE x100.
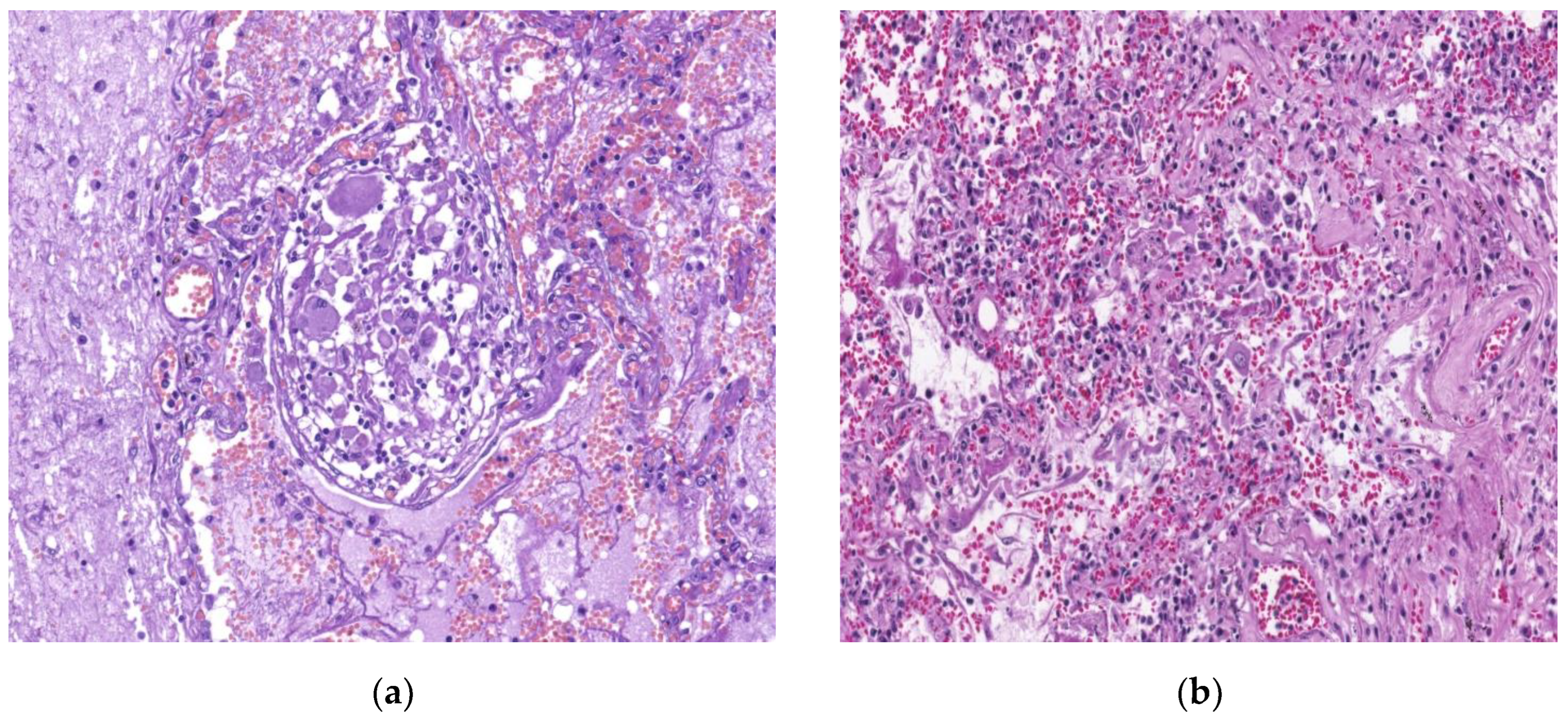
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



