
Submitted:
14 April 2023
Posted:
17 April 2023
You are already at the latest version
Abstract
The exploitation of the metallacarboranes’ potential in various fields of research and practical ap-plications requires the availability of convenient and versatile methods for their functionalization with various functional moieties and/or linkers of different types and lengths. Herein we report a study on cobalt bis(1,2-dicarbollide) functionalization at 8,8’-boron atoms with different het-ero-bifunctional moieties containing protected hydroxyl function allowing further modification after deprotection. Moreover, an approach to the synthesis of three and four functionalized metallacarboranes, at boron and carbon atoms simultaneously, via additional functionalization at carbon to obtain derivatives carrying three or four rationally oriented and distinct reactive surfaces, is described
Keywords:
metallacarboranes
; cobalt bis(1
; 2-dicarbollide)
; oligofunctionalization
; alkylation
; stereochemistry
1. Introduction
Boron clusters, a class of polyhedral caged compounds is playing an increasing role in the development of a broad range of technologies including material science [1,2] and medicinal chemistry [3,4,5]. They find also applications as labels of nucleic acid fragments [6,7,8]. Conjugates of DNA-oligonucleotides and functionalized boron clusters have been proposed recently as building blocks for the construction of new nanomaterials for biomedical applications [9,10].
The ability of boron clusters type of dicarbaborate anion (nido-7,8-C2B9H11) to coordinate a wide spectrum of metal ions, such as Fe, Co, Cr, Ta, Mo, W, V, Nb, and form metal complexes, metallacarboranes, additionally extends the range of their prospective applications [11,12,13,14]. Among them, the widely used metallocarborane is cobalt bis(1,2-dicarbolide), a sandwich of two [C2B9H11]2- (dicarbollide) units with a cobalt ion in the center of the complex structure.
The broad technological potential of metallacarboranes requires access to a diverse array of functionalities (reactive functional groups, alkyl chains, spacers or polymers of various lengths, pharmacologically active species, etc). Furthermore, it could be necessary that more than one of this kind of functionalities will coexist in the same metallacarborane structure to have hetero-functionalized, tailor-made metallacarborane derivatives. The possibility to arrange these functionalities in a specific spatial orientation with respect to the topology of the 3D cluster core makes it possible to achieve the desired covalent modification, following atomic-level precision and allowing control of size and surface composition. Last but not least, highly relevant for molecular design is the opportunity to space the metallacarborane cage from the introduced functional group and the possibility of their further modifications and/or extensions.
The successful use of functionalized 1,2-dicarbadodecaborane as a core unit in the synthesis of building blocks containing a boron cluster and DNA for the construction of functional nanoparticles carrying therapeutic nucleic acids [9,10] prompted us to extend this technology towards metallacarboranes. Due to the different shape of metallacarboranes, it may be possible to obtain nanoparticles with a different topology than in the case of 1,2-dicarbadodecaborane, which in turn can affect their biological properties. The ability of boron clusters to function as membrane carriers for a broad range of cargo molecules, facilitating the therapeutic nucleic acid cellular uptake, would be an additional advantage of metallacarborane-containing DNA nanoparticles [15,16,17,18].
2. Results
The results of a study on oligofunctionalization of the cobalt bis(1,2-dicarbollide) at boron atoms 8 and 8’ and 1,1’ and/or 2,2’ at carbon atoms with extendable ligands are described. Ligands such as hydroxyalkyls, bearing protected hydroxy function separated from the metallacarborane core by spacers were used. They offer the possibility to maximize the distance of functional moieties from each other and the cluster core, to avoid the influence of the metallacarborane (“metallacarborane effect”) [19] on the chemical properties of these moieties. Analogous, but unprotected and substituted at cage carbon atoms alkylhydroxy derivatives of cobalt bis(1,2-dicarbollide) were described by Grüner and colleagues [20,21].
Moreover, the derivatives were designed to test the applicability of two commonly employed in the chemical synthesis of DNA hydroxyl protecting groups, trityl and alkylsilyl protections, to allow the chemo-selection in subsequent chemical manipulation. These results are complemented by studies on further functionalization of the metallocarborane substituted on the 8,8' boron atoms by substitution on the 1,1' and 2,2’ carbon atoms of the carborane ligands.
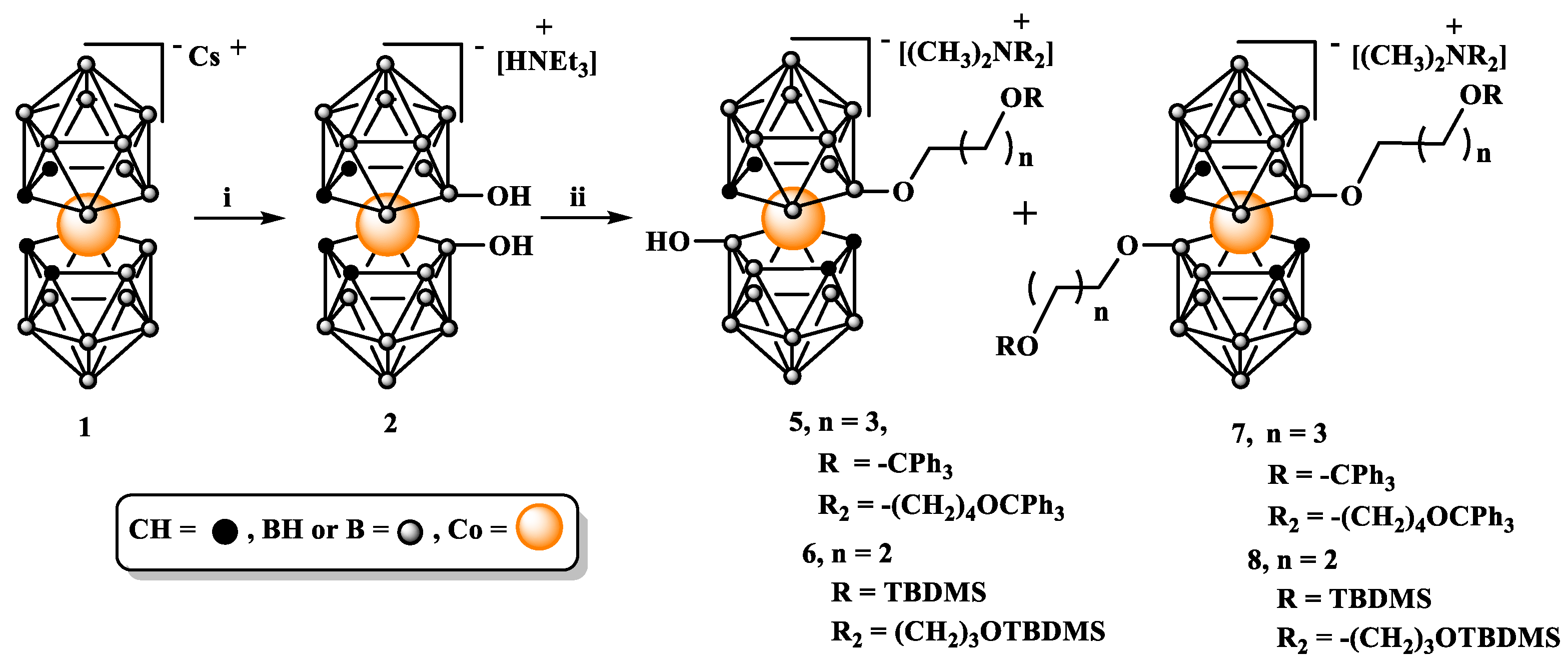
2.1. Functionalization at 8 and 8’ boron atoms via direct alkylation of hydroxy groups in 8,8’-dihydroxy-bis(1,2-dicarbollide)-3-cobalt(1-)ate (2).
The functionalization procedure starts with converting cobalt bis(1,2-dicarbollide) (1) into easily available 8,8’-dihydroxy-bis(1,2-dicarbollido)-3-cobalt(1-)ate (2) in the reaction with 80% aqueous sulphuric acid [22]. The substitution reaction proceeds selectively at the 8 and 8’ boron atoms, which are the ones with maximum electron density [23].
As alkylating agents for compound 2, 4-(trityloxy)butyl-4-methylbenzenesulfonate (3) [24] or (3-bromopropoxy)-tert-butyldimethylsilane (4) differing in leaving groups (tosyl or bromine) and hydroxyl group protection (trityl, -CPh3 or tert-butyldimethylsilyl, TBDMS) were used, NaH was used as a base activating hydroxyl groups in 2 (Scheme 1).
For the reactions involving both the tosylate 3 and the bromide 4, even if excess of NaH was used, the complete alkylation of both hydroxyl groups was not achieved after 24 h, yielding a mixture of mono-substitution products 5 or 6 (minor products), and bis-substitution products 7 or 8 (major products). Both, mono- and bis alkylated products are easily separable by column chromatography on silica gel using a gradient of methanol or acetonitrile in chloroform as eluting solvent system.
The final yield of trityl protected 7 after purification was found to vary considerably, ranging from 30 to 60 %, although the yield of conversion detected in the crude reaction mixture was high with bis-functionalized derivative as the major product formed. This variability in recovery of the tritylated products 7 and 8 can be ascribed to the relative instability of the trityl protection in 5 and 7. A reason for this instability could be the known Lewis acidity of the boron cluster cage [25] and the acid lability of trityl protection. Interestingly, it seems that one of the factors influencing this effect may be a distance of the trityl protection from the metallocarborane core, because in compounds 15, 16, and 21, with a longer linker, the instability of the trityl groups do not appear to be a problem.
Compounds 6 and 8 containing silyl protection are reasonably stable anions, which can be purified and separated by column chromatography on silica gel. TBDMS protecting groups can be quantitatively removed from terminal hydroxyls by treatment with 2.5 equivalents of TBAF in THF at room temperature as demonstrated for 8 (Scheme 2). This treatment lead to a counterion exchange in 9 as demonstrated by NMR analysis.
As indicated by 1H NMR (Supplementary Information, Fig. S2, S7, S12) alkylated compounds 5-8 are isolated in the form of tetraalkylammonium (CH3)2NR2 salt where R2 is -(CH2)4OCPh3 or -(CH2)3OSi(CH3)2C(CH3)3 (Scheme 1). An unexpected structure of tetraalkylammonium counterions is most probably due formation in situ sodium dimethylamine as a result of a side reaction involving the reduction of DMF used as a solvent by NaH and its reaction with the alkylating agent [26]. The modest nucleophilicity of the hydroxyls from one side and the erosion of reagents because of this competing reaction with solvent from the other side, account for the use of an excess of reagents to achieve good yields. These undesirable properties of the pair of NaH and DMF are also responsible for the incomplete alkylation of both hydroxyl groups in 2 mentioned above under the condition used, despite other advantages of DMF solvent.
Next, we attempted to functionalize carbon atoms bisubstituted on boron atoms compound 8 (Scheme 3). The reaction was carried out in anhydrous THF at temperature -70 °C to room temperature following a usual procedure. First, to 8 solution in THF nBNuLi in hexane and then ethylene oxide solution in THF were added. After 24 h, the reaction was quenched providing after standard workup, a mono-substitution product 10 formed in the minority and bis-substitution product 11 formed as the major one. Although moderate amounts of the target compounds can be obtained using this approach, synthetic yields are generally not high.
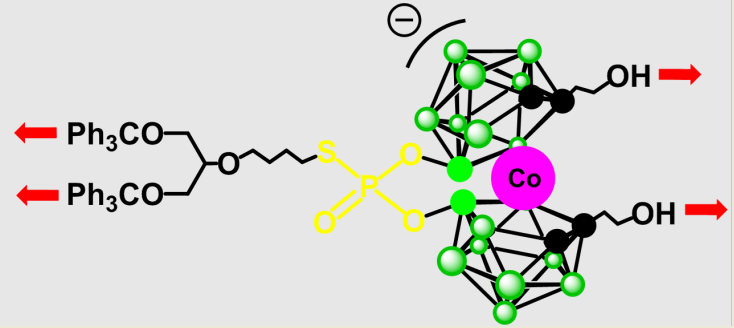
We hypothesize that the reasons for the limited susceptibility of compounds type 8 to functionalization on carbon atoms may include the electron-donating effect of boron cluster ligands, steric hindrance due to the presence of large substituents at the 8 and 8’ positions hampering the electrophile's access to the activated carbon atoms, formation of intramolecular hydrogen bonds between oxygen atoms of substituents at 8,8’ position and acidic carborane C-H groups (Figure 1) analogously to C-H⋅⋅⋅X-B hydrogen bonds [27,28], and rotations of 1,2-dicarbollide ligands around an axis decreasing susceptibility of C-H groups to activation and alkylation. Consequently, we decided to test a different approach based on derivatives of cobalt bis(1,2-dicarbollide) with arrested rotation containing phosphorothioate bridging moiety, 13.
2.2. Functionalization at boron atoms 8 and 8’ via S-alkylation of 8,8'-O,O-[cobalt bis(1,2-dicarbollide)]phosphorothioate (13)
2.2.1. Synthesis of 8,8’-bridged 8,8'-O,O-[cobalt bis(1,2-dicarbollide)]phosphorothioate (13)
The target compound 13 was obtained in a simple, two-step procedure. In the first step the 8,8’-dihydroxy-derivative 2 was converted into 8,8’-bridged 8,8'-O,O-[cobalt bis(1,2-dicarbollide)] H-phosphonate acid ester (12) in the reaction with tris(1H-imidazol-1-yl)phosphine [29] and in situ hydrolysis of the resultant imidazolide.
Scheme 4.
Synthesis of H-phosphonate (12) and thiophosphate (13) esters of 8,8’-dihydroxy-bis(1,2-dicarbollido)-3-cobalt(1-)ate (2); i. PCl3, imidazole, Et3N in THF; ii. H2O, iii. S8, DBU in MeOH. DBU = 1,8-diazabicyclo(5.4.0)undec-7-en.
Scheme 4.
Synthesis of H-phosphonate (12) and thiophosphate (13) esters of 8,8’-dihydroxy-bis(1,2-dicarbollido)-3-cobalt(1-)ate (2); i. PCl3, imidazole, Et3N in THF; ii. H2O, iii. S8, DBU in MeOH. DBU = 1,8-diazabicyclo(5.4.0)undec-7-en.
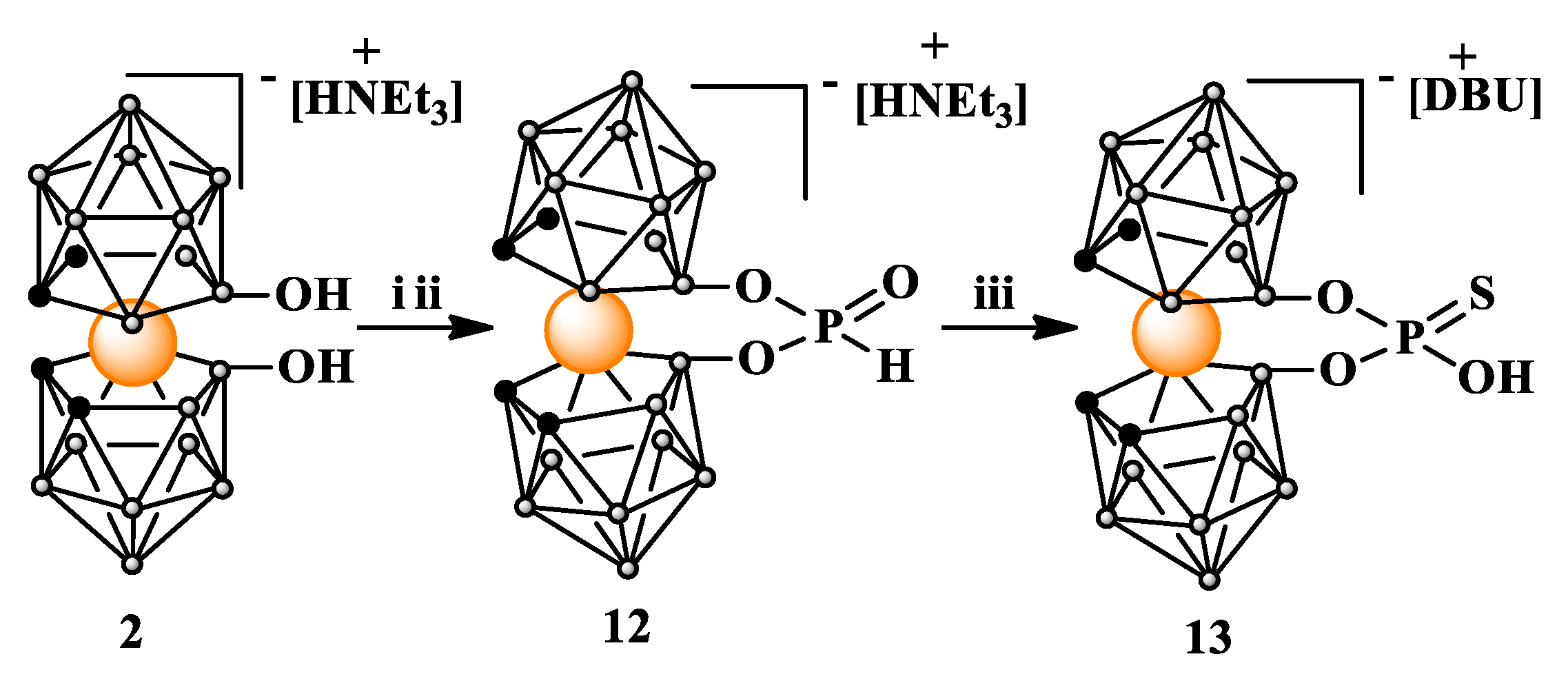
In the second step H-phosphonate acid ester 12 was dissolved in anhydrous methanol then elemental sulfur S8 was added. To the resultant suspension strong organic base, 1,8-diazabicyclo(5.4.0)undec-7-en (DBU) was added and the reaction was left for 96 hours at room temperature with stirring. After evaporation of the methanol, the residue containing crude product was purified by silica gel column chromatography using a gradient of acetonitrile in chloroform as eluting solvent system.
2.2.2. S-alkylation of 8,8’-bridged 8,8'-O,O-[cobalt bis(1,2-dicarbollide)]phosphorothioate (13) with linear and branched alkylating agents.
Sulfur is a larger atom than oxygen, making its electrons more polarizable and sulfur more nucleophilic. Alkylation of the sulfur atom of phosphorothioates is a viable method for the synthesis of their S-alkylated derivatives. This methodology takes an advantage of the excellent nucleophilic properties of sulfur and is commonly used in organophosphorus chemistry [30].
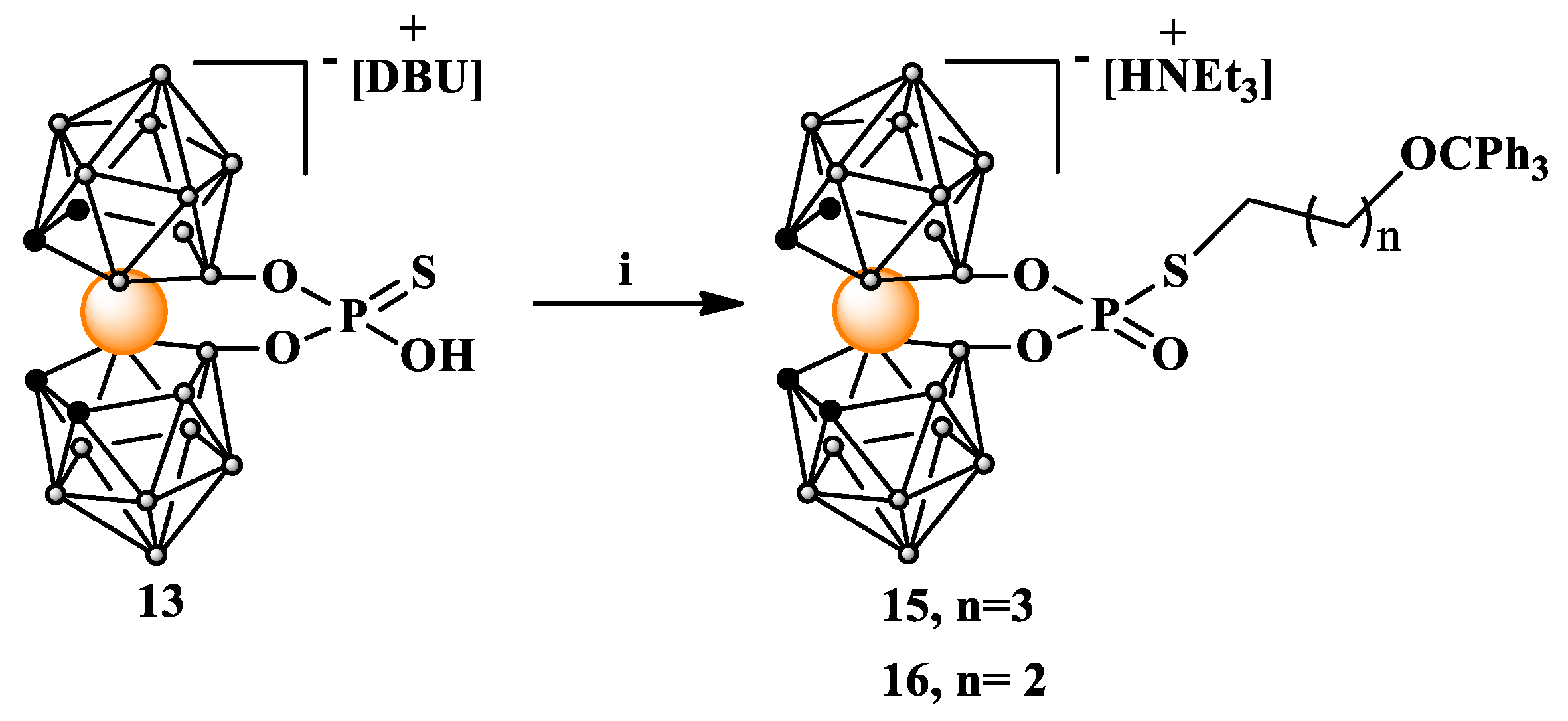
Using these advantageous properties of phosphorothioates, we attached both linear 13, 14, or branched 20 ligands containing hydroxyl functions protected by a trityl group to the metallocarborane derivative 13. The alkylation reaction proceeds smoothly and yields of isolated alkylated products 15, 16, and 21 were high (Scheme 5).
It is worth noting that derivatives of known 8,8’-bridged 8,8'-O,O-[cobalt bis(1,2-dicarbollide)]phosphate [22], a counterpart of 13 containing an 8,8’-O,O-phosphate bridge instead of a phosphorothioate one, have not been described so far. The expected low nucleophilicity of the phosphorus center resulting from the metallocarborane effect [19] and the low nucleophilicity of oxygen atoms can be one of the reasons for this.
Then study on the functionalization of 13 carbon atoms was undertaken. For this purpose, a method for the synthesis of a branched alkylating agent 20, in this case, a glycerol derivative, was first developed. In the first step bis(trityloxy)propan-2-ol (18) was synthesized according to the literature method [31]. Next 18 was reacted with 1,4-bis(p-toluenesulfonyloxy)butane (19) providing a branched alkylating agent with elongated linker 20.
Scheme 6.
Synthesis of branched alkylation agent 4-(1,3-bis(trityloxy)propan-2-yloxy)butyl-4-methylbenzenesulfonate (20), its use to alkylate of 8,8'-O,O-[cobalt bis(1,2-dicarbollide)]phosphorothioate (13) and functionalization of 21 on carbon atoms: i. ClCPh3 (trityl chloride), pyridine; ii. NaH, DMF, 1,4-bis-(4-methylbenzenesulfonate)butane (19); iii. NaH, 20 in DMF; iv. nBuLi, THF; ethylene oxide; DBU = 1,8-diazabicyclo(5.4.0)undec-7-en.
Scheme 6.
Synthesis of branched alkylation agent 4-(1,3-bis(trityloxy)propan-2-yloxy)butyl-4-methylbenzenesulfonate (20), its use to alkylate of 8,8'-O,O-[cobalt bis(1,2-dicarbollide)]phosphorothioate (13) and functionalization of 21 on carbon atoms: i. ClCPh3 (trityl chloride), pyridine; ii. NaH, DMF, 1,4-bis-(4-methylbenzenesulfonate)butane (19); iii. NaH, 20 in DMF; iv. nBuLi, THF; ethylene oxide; DBU = 1,8-diazabicyclo(5.4.0)undec-7-en.
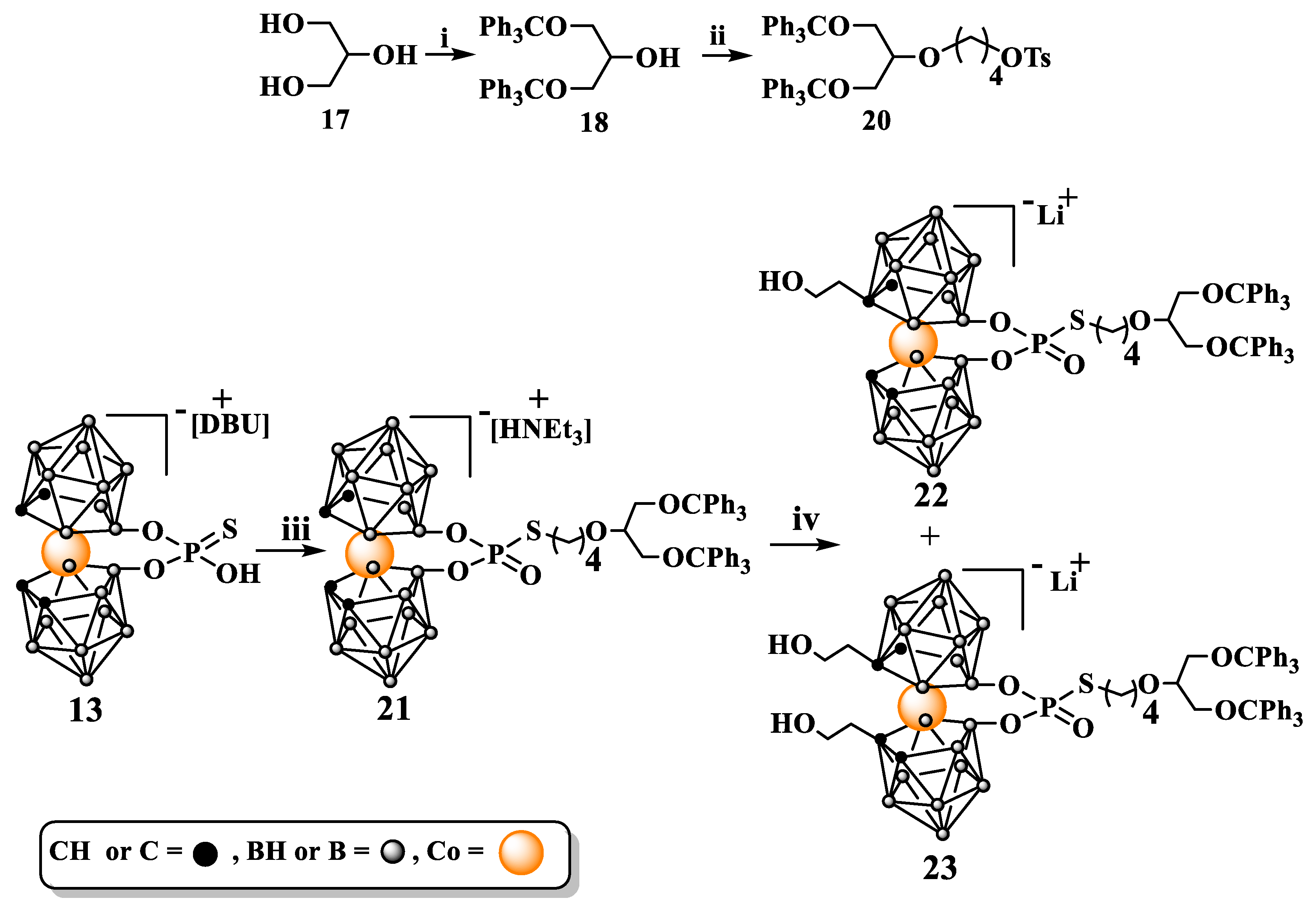
Functionalization of 21 at carbon atoms was achieved via activation of cage C-H groups with nBuLi and then treatment with ethylene oxide. As expected carrying out the synthesis of 22 and 23 required overcoming the low susceptibility of the intermediate 21 towards the substitution on carbon atoms. However, mono- and disubstituted derivatives 22 and 23 were obtained with yields enabling their full characterization and use for further chemical manipulations. Easy availability and high yields of synthesis of intermediates 13 and 21 allow for easy scale-up of the 22 and 23 synthesis if needed.
3. Discussion
Description in the literature of metallacarborane building blocks with hydroxyl or other functional groups separated from the cluster cage by an alkyl spacer seems limited. In this contribution, we report on the development of the methods for functionalization of cobalt bis(1,2-dicarbollide) (1) with hetero bifunctional derivatives of silyl- or trityl-protected alcohols attached directly to boron atoms, and trityl protected branched alcohols attached to the metallacarborane through a phosphorothioate bridge. An approach to the synthesis of oligofunctionalized metallacarboranes via carbon deprotonation through nBuLi to obtain derivatives carrying substituents at both, boron and carbon atoms is also described.
After a long period of discussion, the aromatic, three-dimensional (3D) nature of boron clusters is now widely accepted [32,33]. As in the case of aromatic organic molecules, the attachment of a substituent to one of the atoms of a 3D aromatic boron cluster system changes the distribution of electron density of the entire molecule and affects the properties of other reactive centers. This makes the oligofunctionalization of metallacarboranes, especially simultaneously on boron and carbon atoms, a particular challenge. On this, an complicated stereochemistry of substituted metallacarboranes having a source in the presence of various types of chirality in the same molecule is superimposed.
One of the practical manifestations of an effect of synchronized changes in electron density within the whole metallacarborane molecule on properties of reactive centers is the preference for the formation of disubstituted derivatives on boron atoms 8 and 8' and carbon atoms 1,2 and 1',2'. This is illustrated by the preferential formation of disubstituted derivatives compared to monosubstituted ones in the case of 7 vs. 5, 8 vs. 6, 11 vs. 10, or 23 vs. 22. This is clearly due to activation of a second nucleophilic center such as B-OH or C-H groups activated by a strong base, after a previous substitution at the first center. However, it does not change the fact that the overall alkylation efficiency of C-H groups in derivatives of 1 already substituted on 8 and 8" boron atoms is low.
For example, monoalkylation derivatives of 8,8'-dihydroxy-bis(1,2-dicarbollide)-3-cobalt(1-)ate (2) and bisalkylation derivatives are isolated in the ratio 1:12 for 5 and 7 and 1:4 for 6 and 7. Mono-substituted and bis-substituted at carbon atoms derivatives of bis-substituted at boron atoms compound 8 are isolated in the ratio 1:3 for 10 and 11. The same trend, though less pronounced is observed for boron and carbon functionalized derivatives 22 and 23 with arrested rotation where the ratio of mono-substitution on carbon bis-substitution is 1:1.2.
Another property significantly influencing the chemistry of oligofunctionalized metallacarboranes is their stereochemistry. The phenomenon of boron cluster chirality was recognized early [34,35,36], however, this did not initially arouse much interest. The increasing use of boron clusters in the field of new materials, nanotechnology, and medical chemistry makes the stereochemistry of boron clusters increasingly important.
Perversely, though the most symmetric species in nature is the B12H12- ion, relatively minor changes in the boron cluster structure might render the basic framework dissymmetric enough to provoke chirality [37,38]. This is particularly evident in the case of oligofunctionalized metallacarboranes. The derivatives type 22 and 23 are an extreme examples containing various sources of chirality such as chiral center at phosphorus atom, axial and planar chirality due to bending of the metallacarborane molecule and the existence of a number of isomers due to substitution at carbon atoms in addition to boron substitution.
Comparison of 11B-NMR spectra for compounds 13, 15, 16, 21, 22, and 23 with arrested rotation as well as their 31P-NMR spectra clearly shows changes in the number of isomers of individual products depending on substitution. Compounds 13, 15, 16, and 21 show a singlet at δ about 23 ppm attributed to 2B(8,8’) in the 11B-NMR spectrum and a singlet at 48.62 ppm for 13 and at about 16 ppm for compounds 15, 16, 21 in the 31P-NMR spectrum which is consistent with the relative symmetry of the complexes.
On the contrary, 11B- and 31P-NMR spectra of mono- and bisalkylated at carbon atoms derivatives 22 and 23 show dramatic change in symmetry due to many possible combinations of substitution on carbon atoms as well as due to the formation of a center of chirality on the phosphorus atom in monosubstituted 22 and in some isomers of bissubstituted 23. In consequence, four signals at 25.37, 24.65, 23.69, 22.93 ppm in integral intensity ratio 2:2:1:1 corresponding to B8,8’ for 22 and five signals at 25.35, 24.40, 23.64, 22.80, 22.23 ppm in approximate integral intensity ratio 1:2:1.5:1:1 for 23 are observed. A similar effect in 31P-NMR spectra with seven signals at 15.10, 14.94, 14.57, 14.42, 14.11, 13.74, 13.49 ppm for 22 and five signals at 14.99, 14.39, 14.16, 14.01, 13.38 ppm for 23 reflecting asymmetry of these compounds and formations of isomers is seen.
The sensitivity of standard chromatographic techniques is insufficient to distinguish these isomers and allows only the separation of mono- and disubstituted derivatives on carbon atoms, already substituted on boron atoms as in case 10, 11, and 22, 23. A complete or partial separation of the isomers is probably possible by HPLC, however, for further practical applications of these derivatives, it is not necessary at the present stage.
4. Materials and Methods
4.1. Materials
All solvents were purchased in the highest available quality required for the specific application. The reactions requiring anhydrous conditions were carried under argon using anhydrous solvents treated with activated molecular sieves for at least 24 h. Molecular sieves (4Å and 3Å) were purchased by Alfa Aesar and heat activated under vacuum before use. Triethylamine, trityl chloride, toluene-sulfonyl chloride, NaHCO3, Na2SO4, 1,4-butanediol, P2O5, sodium hydride, n-butyllithium (1.6 M solution in hexane), (3-bromopropoxy)-tert-butyldimethylsilane (4) and ethylene oxide (2.9-3.1 M solution in THF) were purchased from Sigma Aldrich. Cobalt bis(1,2-dicarbollide) was purchased from Katchem Ltd., Czech Republic.
4.2. Methods
Chromatography. A DIONEX Ultramate 3000 HPLC system equipped with a photodiode array detector (fixed wavelength 210, 270, 310, 330 nm) was used to check purity of products. The method consisted in a gradient elution from 20% to 90% aqueous acetonitrile trough a Thermo Scientific Hypersil Gold (5μm particle size) reverse phase column at 25°C. HPLC data were acquired and processed by Chromaleon 6.8 software. Chromatography for purification of products was performed on a 230–400 mesh silica gel (Sigma Aldrich) filled glass column. Analytical TLC was performed on F254 silica gel plates purchased by Sigma Aldrich. Compounds were visualized using UV light (254 nm) and or by staining with 0.05 %w/v palladium chloride solution in MeOH/HCl.
NMR spectroscopy. 1H, 11B, 11B{1H}, 13C{1H}, 31P, 31P{1H} NMR spectras were recorded with a Bruker Advance III 600 MHz spectrometer.
UV-Vis spectrophotometry and line fitting analysis. Measurements were performed with a Jasco V-750 UV-spectrophotometer at room temperature in acetonitrile.
MS and FT-IR MALDI-TOF MS spectras were recorded with a Voyager–Elite mass spectrometer (PerSeptive Biosystems) with 3-hydroxypicolinic acid (HPA) as the matrix. ESI MS mass spectra were recorded using Agilent 6546 LC/Q-TOF (Santa Clara, Kalifornia, United States). Negative ions were detected. Infrared absorption spectra were recorded using a Nicolet 6700 FT-IR spectrometer (Thermo Scientific) equipped with a Smart orbit diamond Attenuated Total Reflectance (ATR) accessory. Samples to be analyzed were placed on a diamond ATR element in the solid form or by casting from CH2Cl2 solution. Data were acquired and processed by Omnic 8.1 software.
8,8’-dihydroxy-bis(1,2-dicarbollido)-3-cobalt(1-)ate HNEt3 (2) was synthesized according to the procedure reported by Plešek et al. [22].
4-Trityloxybutyl 4-methylbenzenesulfonate (3) was obtained as described [39].
1,4-bis-(4-methylbenzenesulfonate)butane (19) was synthesized according to the modified literature procedure [40].
Synthesis of 3,3’-Co{[(8-O(CH2)4OCPh3]-1,2-C2B9H10}(8’-OH-1,2-C2B9H10) (CH3)2N[(CH2)4OPh3]2 (5) and 3,3’-Co[(8-O(CH2)4OCPh3-1,2-C2B9H10)]2 (CH3)2N[(CH2)4OPh3]2 (7). Compound 2 (50 mg, 0.10 mmol) was dissolved in 0.5 mL of anhydrous dimethoxyethane (DME) and added to 60% NaH in mineral oil dispersion (4.4 mg, 0.10 mmol NaH) under argon atmosphere. The reaction mixture was stirred for 2 h at room temperature. After this time the solvent was evaporated under reduced pressure and the resultant solid residue was dissolved in 0.5 mL of anhydrous dimethylformamide (DMF) and added to a second aliquot of 60% NaH in mineral oil dispersion (26.2 mg, 0.65 mmol NaH). After 2 h at room temperature the mixture was dropped into a solution of 3 (318.8 mg, 0.65 mmol) in 0.5 mL of anhydrous DMF and then the reaction mixture was heated at 80 ˚C (oil bath temperature) for 24 h. The post-reaction mixture was concentrated then 3 mL of H2O was added. The resulting precipitate was washed with several aliquots of H2O. Then the solid was dissolved in 5 mL of CH2Cl2, and the solution was dried with anhydrous Na2SO4, filtered and concentrated. The repeated column chromatography on silica gel using a gradient of CH3OH in CH3Cl (0 to 20%) as eluting solvent system gave 5 and 7 as the first and second band respectively. Fractions containing compound 7 were collected, evaporated to dryness then crystallized from hexane, providing 96% pure product as determined by HPLC. Monosubstituted compound 5 was obtained as a red oil.
(5): Yield: ca 5 %; TLC (CHCl3 : MeOH 4:1); Rf: 0.53; MALDI-MS (m/z): 670.5 (calc. for C27B18H44O3Co1: 670.17). Due to obtaining too small quantities of this product, it was not further analyzed by NMR.
(7): Yield: 62% (68.2 mg); TLC (CHCl3 : MeOH 4:1); Rf 0.64; 1H NMR (500 MHz, CD3CN) δ: 7.43 (m, 24H, Harom), 7.32 (m, 24H, Harom), 7.25 (m, 12H, Harom), 4.18 (s, 4H, CHcarborane), 3.31 (t, J = 6.2 Hz, 4H, BOCH2CH2CH2CH2OTr), 3.07 (m, 8H, CH2OTr), 2.99 (t, J = 6.4 Hz, 4H, NCH2CH2CH2CH2OTr), 2.86 (s, 6H, N(CH3)2), 1.68 (m, 4H, NCH2CH2CH2CH2OTr), 1.60 (m, 8H, CH2CH2OTr), 1.48 (m, 4H, BOCH2CH2CH2CH2OTr); 13C{1H} NMR (126 MHz, CD3CN) δ: 145.53, 145.18 (12C, aromatictrityl); 129.44, 129.41 (24C, aromatictrityl); 129.37, 128.83 (24C, aromatictrityl); 128.21 (12C, aromatictrityl); 87.43, 87.06 (4C, C(Ph)3), 69.43 (2C, BOCH2CH2CH2CH2OTr), 64,30 (2C, BOCH2CH2CH2CH2OTr), 63,30 (2C, NCH2CH2CH2CH2OTr), 62.33 (2C, NCH2CH2CH2CH2OTr), 51.56 (4C, CHcarb), 30.39, 29.68 (4C, CH2CH2OTr), 27.51, 27.13 (2C, BOCH2CH2CH2CH2OTr), 20.29 (NCH2CH2CH2CH2OTr); 11B{1H} NMR (160 MHz, CD3CN) δ: 20.63 (s, 2B, B8,8’), -3.56 (s, 2B, B10,10’), -7.52 (s, 4B, B4,4’,7,7’), -9.03 (s, 4B, B9,9’,12,12’), -20.53 (s 4B, B5,5’,11,11’), -28.38 (s, 2B, B6,6’) 11B NMR (160 MHz, CD3CN) δ: 20.66 (s, 2B, B8,8’), -3.59 (d, 2B, B10,10’), -8.22 (m, 8B, B9,9’,12,12’,4,4’,7,7’), -20.48 (d, 2B, B5,5’,11,11’), -28.09 (d, 2B, B6,6’); FT-IR (cm-1): 3083.53, 3055.16, 3029.62 (ν C-H aromatic, (C-Hcarb); 2927.18 (ν C-Hasym, CH2); 2867.43 (ν C-H, CH2O, ν C-Hsym, CH2); 2543.23 (ν B-H); 1596.06, 1488.72, 1447.49 (ν C=C); 1152.23; 745.10, 693.37 (δ C-H aromatic); 703.89 (ν B-B); MALDI-MS (m/z): found 984.2 (calc. for C50B18H66O4Co1: 984.59).
Synthesis of 3,3’-Co[8-O(CH2)3OTBDMS]-1,2-C2B9H10)(8’-OH-1,2-C2B9H10) (CH3)2N[(CH2)3OTBDMS)]2 (6) and 3,3’-Co[8-O(CH2)3OTBDMS-1,2-C2B9H10)]2 (CH3)2N[(CH2)3OTBDMS)]2 (8). Compound 2 (300 mg, 0.65 mmoles) was dissolved in 3 mL of anhydrous DME under argon atmosphere and added to 60% NaH in mineral oil dispersion (26.2 mg, 0.65 mmol NaH). The reaction mixture was stirred for 2h at room temperature. After this time the solvent was evaporated and the solid residue was dissolved in 3 mL of anhydrous DMF and added to a second portion of 60% NaH in mineral oil dispersion (157 mg, 3.93 mmol NaH). After 2 h at room temperature the mixture was heated at 60 ˚C (oil bath temperature) and 911 μL of (3-bromopropoxy)(tert-butyl)dimethylsilane (4) (3.93 mmol) was added dropwise. After 22 h, additional quantity of 4 (600 μL, 2.59 mmol) was added and the mixture was stirred for next 24 h at 60 ˚C. A white solid was formed. The post-reaction mixture was then filtered through syringe filter (5 μm, PTFE, Carl Roth) and DMF was evaporated under reduced pressure. The solid residue was treated with 2 mL of CHCl3, filtered, the filtrate was concentrated and loaded on a silica gel column for separation of products. 100% CHCl3 first and then 5% and 10% CH3CN in CHCl3 were used as eluting solvent system. Compound 8 was isolated as first fraction and was obtained in the form of orange crystals after solvent evaporation, its purity was above 95% as determined by HPLC. The second fraction containing 6 was obtained as red oil after solvent evaporation. Both products, 6 and 8 were isolated as N,N-bis[(3-(tert-butyldimethylsilyloxypropyl)]-N,N-dimethyl ammonium salts [26].
6: Yield: 10%; TLC (CHCl3 : CH3CN 3:1) Rf 0.53; 1H NMR (600 MHz, CD3CN) δ: 6.05 (s, 1H, OH), 3.73 (t, J = 6.5 Hz, 2H, BOCH2CH2CH2OSi), 3.69 (m, 4H, NCH2CH2CH2OSi), 3.65 (m, 2H, BOCH2CH2CH2OSi), 3.56 (s, 2H, CHcarb), 3.45 (s, 2H, CHcarb), 3.27 (m, 4H, NCH2CH2CH2OSi), 2.97 (s, 6H, N(CH3)2), 1.88 (m, 4H, NCH2CH2CH2OSi), 1.74 (m, J = 6.4 Hz, 2H, BOCH2CH2CH2OSi), 0.90 (s, 9H, BOCH2CH2CH2OSi(CH3)2C(CH3)3), 0.89 (s, 18H, NCH2CH2CH2OSi(CH3)2C(CH3)3), 0.07 (s, 6H, BOCH2CH2CH2OSi(CH3)2C(CH3)3), 0.05 (s, 12H, NCH2CH2CH2OSi(CH3)2C(CH3)3); 13C{1H} NMR (126 MHz, CD3CN) δ: 67.05 (1C, BOCH2CH2CH2OSi), 62.56 (2C, NCH2CH2CH2OSi), 61.23 (1C, BOCH2CH2CH2OSi), 60.17 (2C, NCH2CH2CH2OSi), 52.03 (2C, N(CH3)2), 46.23 (2C, CHcarb), 45.51 (2C, CHcarb), 35.92 (1C, BOCH2CH2CH2OSi), 26.45 (2C, NCH2CH2CH2OSi), 26.27 (6C, OSi(CH3)2C(CH3)3), 26.11 (3C, OSi(CH3)2C(CH3)3), 18.81 (1C, OSi(CH3)2C(CH3)3), 18.72 (2C, OSi(CH3)2C(CH3)3), -5.10 (2C, OSi(CH3)2C(CH3)3), -5.40 (4C, OSi(CH3)2C(CH3)3) 11B{1H} NMR (193 MHz, CD3CN) δ: 27.16 (s, 1B, B8’), 25.10 (s, 1B, B8), -5.02 to -9.09 (10B, overlapped B9,9’,10,10’12,12’,4,4’,7,7’), -20.10 to -20.69 (4B, B5,5’,11,11’), -29.18 to -30.16 (2B, B6,6’); 11B NMR (160 MHz, CD3CN) δ: 27.16 (s, 1B, B8’), 25.09 (s, 1B, B8), -3.22 to -9.40 (10B, overlapped B9,9’,10,10’12,12’,4,4’,7,7) -19.72 to -21.11 (4B, B5,5’,11,11’), -28.81 to -30.49 (2B, B6,6’), FT-IR (cm-1): 3278.64, 2952.00, 2927.58, 2855.64, 2539.99, 1470.65, 1387.53, 1360.69, 1252.66, 1156.76, 1093.25, 1005.54, 972.16, 938.48, 880.24, 832.61, 774.98, 718.29, 661.26; ESI- MS (m/z): found 530.39, 472.35 [M-tButyl]- (calc. for C13B18H43Si1O3Co1: 529.09)
8: Yield: 80%; TLC (CHCl3 : CH3CN 3:1) Rf 0.70; 1H NMR (500 MHz, CD3CN) δ: 4.18 (s, 4H, CHcarb), 3.69 (m, 4H, BOCH2CH2CH2OSi), 3.64 (t, J = 6.4 Hz, 4H, NCH2CH2CH2OSi), 3.38 (t, J = 6.0 Hz, 4H, BOCH2CH2CH2OSi), 3.27 (m, 4H, NCH2CH2CH2OSi), 2.97 (s, 6H, N(CH3)2), 1.88 (m, 4H, NCH2CH2CH2OSi), 1.58 (m, J = 6.2 Hz, 4H, BOCH2CH2CH2OSi), 0.90 (s, 18H, OSi(CH3)2C(CH3)3), 0.88 (s, 18H, OSi(CH3)2C(CH3)3), 0.07 (s, 12H, OSi(CH3)2C(CH3)3), 0.04 (s, 12H, OSi(CH3)2C(CH3)3); 13C NMR (126 MHz, CD3CN) δ: 66.18 (2C, BOCH2CH2CH2OSi), 62.54 (2C, NCH2CH2CH2OSi), 60.95 (2C, BOCH2CH2CH2OSi), 60.17 (2C, NCH2CH2CH2OSi), 52.06 (2C, N(CH3)2), 51.60 (4C, CHcarb), 35.97 (2C, BOCH2CH2CH2OSi), 26.46 (2C, NCH2CH2CH2OSi), 26.28 (6C, OSi(CH3)2C(CH3)3), 26.12 (6C, OSi(CH3)2C(CH3)3 18.83 (2C, OSi(CH3)2C(CH3)3), 18.72 (2C, OSi(CH3)2C(CH3)3), -5.09 (4C, OSi(CH3)2C(CH3)3), -5.39 (4C, OSi(CH3)2C(CH3)3) 11B{1H} NMR (160 MHz, CD3CN) δ: 20.61 (s, 2B, B8,8’), -3.61 (s, 2B, B10,10’), -7.45 (s, 4B, B4,4’,7,7’), -9.03 (s, 4B, B9,9’,12,12’), -20.51 (s, 4B, B5,5’,11,11’), -28.38 (s, 2B, B6,6’) 11B NMR (160 MHz, CD3CN) δ: 20.76 (s, 2B, B8,8’), -3.59 (d, 2B, B10,10’), -7.18 to -9.37 (m, 8B, overlapped B4,4’,7,7’,9,9’,12,12’), -20.48 (d, 4B, B5,5’,11,11’), -28.02 (d, 2B, B6,6’); FT-IR (cm-1): 3048.6 (C-Hcarb); 2949.4 (ν C-Hasym, CH3); 2927.4 (ν C-Hasym, CH2), 2891.6, 2884.2 (ν C-Hsym CH3); 2856.2 (ν C-H, CH2O, ν C-Hsym, CH2); 2605.2, 2550.6 (ν B-H); 1470.8 (δ C-Hsym, CH2), 1436.0; 1386.2; 1359.5; 1251.2 (Si-CH3), 1161.9 (Si-O-C); 1093.1 (ν Si-O); 1019.3; 1006.3; 975.2; 955.3; 942.7; 881.; 831.8, 772.4 (ν Si-C); 710.9; 661.0; MALDI-MS (m/z): found 700.5 (calc. for C22B18H62CoO4Si2 700.43).
Deprotection of compound 8 to obtain [3,3’-Co(8-O(CH2)3OH-1,2-C2B9H10)2] TBA (9). Compound 8 (20 mg, 0.027 mmol) was dissolved in 0.2 mL of tetrahydrofuran (THF). To the obtained solution 69 μL of terabutylammonium fluoride (TBAF) (1M solution in THF, 0.069 mmol) was added and the reaction mixture was stirred at room temperature overnight. Next, solvent was evaporated under reduced pressure and the resulting oil was dissolved in CHCl3 and applied to the silica gel column prepared in the same solvent. Elution in a gradient 1 to 20% CH3CN in CHCl3 provided 96 % pure 9 (determined by HPLC) as TBA salt. Yield: 95%; TLC (CHCl3 : CH3CN 3:1) Rf 0.47; 1H NMR (500 MHz, CD3CN) δ: 4.17 (s, 4H, CHcarb), 3.52 (t, , J = 6.3 Hz, 4H, BOCH2CH2CH2OH), 3.43 (t, J = 6.0 Hz, 4H, BOCH2CH2CH2OH), 3.10 – 3.03 (m, 8H, NCH2CH2CH2CH3), 2.58 (s, 2H, OH), 1.59 (m, J = 8.2, 3.7 Hz, 12H, BOCH2CH2CH2OH, NCH2CH2CH2CH3), 1.41 – 1.28 (m, 8H, NCH2CH2CH2CH3), 0.96 (t, J = 7.4, 12H, NCH2CH2CH2CH3); 13C NMR (126 MHz, CD3CN) δ: 67.47 (2C, BOCH2CH2CH2OH), 60.63 (2C, BOCH2CH2CH2OH), 59.23 (4C, NCH2CH2CH2CH3), 51.39 (4C, CHcarborane), 35.54 (2C, BOCH2CH2CH2OH), 24.21 (4C, NCH2CH2CH2CH3), 20.25 (4C, NCH2CH2CH2CH3), 13.72 (4C, NCH2CH2CH2CH3); 11B{1H} NMR (160 MHz, CD3CN) δ: 20.95 (s, 2B, B8,8’), -3.49 (s, 2B, B10,10’), -7.71 (s, 4B B4,4’,7,7’), -8.85 (s, 4B, B9,9’,12,12’), -20.44 (s, 4B, B5,5’,11,11’), -28.14 (s, 2B, B6,6’); 11B NMR (160 MHz, CD3CN) δ: 20.97 (s, 2B, B8,8’), -3.48 (d, 2B, B10,10’), -7.18 to -9.26 (m, 8B, B4,4’7,7’9,9’,12,12’ ), -20.42 (d, 4B, B5,5’,11,11’), -28.19 (d, 2B, B6,6’); FT- IR (cm-1): 3588.76 (ν O-H); 3450.79 (ν N+-R); 3052.83 (C-Hcarb); 2962.10 (ν C-Hasym, CH2); 3932.36, 2873.70 (ν C-H, CH2O, ν C-Hsym, CH2); 2529.19 (ν B-H); 1467.75 (ν N+-R), 1380.63; 1161.25 (HO-C); 1105.54; 1062.05; 1007.15; 969.00; 945.10; 920.29; 876.90; 789.11; 735.69; 696.07; 664.83; MALDI-MS (m/z): found 472.8 (calc. for C10B18H34O4Co1 471.90).
Synthesis of 3,3’-Co{[8-O(CH2)3OTBDMS-1-(CH2)2OH]-1,2-C2B9H9)}[8’-O(CH2)3OTBDMS-1’,2’-C2B9H10)]- (10) and 3,3’-Co[(8-O(CH2)3OTBDMS-1-(CH2)2OH-1,2-C2B9H9)]2- (11). Compound 8 (50 mg, 0.04 mmol) was dried by co-evaporation with anhydrous benzene and then kept under vacuum over P2O5 overnight. Next it was dissolved in anhydrous DME (1 mL) and the solution was cooled in CO2/isopropanol cooling bath. After 15 min. n-BuLi (43 µL, 1.6 M solution in hexane, 1.5 eq) was added and the reaction mixture was stirred for 10 min. After that time, cooling bath was removed and the mixture was stirred for next 10 min. Then the reaction mixture was cooled again in cooling bath and another portion of n-BuLi (43 µL) was added. After 15 min, ethylene oxide (60 µL, 2.9-3.1 M solution in THF, 4.5 eq) was added and the reaction was left overnight in cooling bath. Next CH2Cl2 (3 mL) was added to the reaction mixture, the reaction was quenched by addition of water and the organic solution was washed three times with 5 mL portions of water. Organic layer was separated and dried over MgSO4 then solvents were evaporated. Crude product was purified and mono- and bis-substituted products separated by silica gel column chromatography using a gradient of MeOH in CH2Cl2 from 0 to 3% of MeOH.
10 Yield: 4,7 mg (9%) TLC (silica gel on Al, 8% MeOH/CH2Cl2): Rf: 0.28, ESI-MS (m/z): found 744.55, (calc. for C24B18H66O5Si2Co1 744.48). Due to obtaining too small quantities of this product, it was not further analyzed by NMR.
11 Yield: 15 mg (27%) TLC (silica gel on Al, 8% MeOH/CH2Cl2): Rf: 0.16 1H NMR (500 MHz, CD3CN) δ: 4.32-4.08 (s, 2H, diastereoizomeric CHcarborane), 3.77 to 3.52 (m, 16H, overlapped NCH2CH2CH2O, NCH2CH2CH2O, BOCH2CH2CH2O, BOCH2CH2CH2O), 3.52 to 3.39 (m, 4H, HOCH2CH2Ccarb), 3.39 to 3.31 (m, 4H, HOCH2CH2Ccarb), 3.03 (s, 6H, N(CH3)2), 1.74 (m, 4H, NCH2CH2CH2O), 1.65 (m, 4H, BOCH2CH2CH2O) 0.90 (s, 18H, NCH2CH2CH2OSi(CH3)2C(CH3)3), 0.88 (s, 18H, BOCH2CH2CH2OSi(CH3)2C(CH3)3), 0.07 (s, 12H, BOCH2CH2CH2OSi(CH3)2C(CH3)3), 0.05 (s, 12H, NCH2CH2CH2OSi(CH3)2C(CH3)3,13C{1H} NMR (126 MHz, CD3CN) δ: 67.39 (2C, BOCH2CH2CH2O), 66.93 (1C, CHcarborane), 64.82 (1C, CHcarborane), 64.10 (2C, NCH2CH2CH2OSi), 62.28 (2C, BOCH2CH2CH2O), 61.00 (2C, HOCH2CH2Ccarb), 57.17 (2C, NCH2CH2CH2O), 56.24 (2C, N(CH3)2), 53.22 (2C, Ccarborane), 45.17 (2C, HOCH2CH2Ccarb) 36.89 (2C, NCH2CH2CH2O), 27.28 (2C, BOCH2CH2CH2O), 26.98 (6C, OSi(CH3)2C(CH3)3) 26.77 (6C, OSi(CH3)2C(CH3)3), 19.53 (2C, OSi(CH3)2C(CH3)3), 19.39 (2C, OSi(CH3)2C(CH3)3), -4.38 (4C, OSi(CH3)2C(CH3)3), -4.73 (4C, OSi(CH3)2C(CH3)3). 11B{1H} NMR (120 MHz, CD3CN) δ: 29.65, 25.25, 24.28, 23.33, 21.98 (in ratio: 3:1.5:1:1:10), 31.27 to 19.66 (m, overlapped diastereoizomeric B8,8’), -2.63 to -13.75 (m, overlapped diastereoizomeric, B10,10’,9,9’,12,12’,4,4’,7,7’), -14.09 to -21.65 (m, overlapped diastereoizomeric B5,5’,11,11’), -22.11 to -29.38 (m, overlapped diastereoizomeric B6,6’) 11B NMR (120 MHz, CD3CN) δ: 30.85-20.54 (m, overlapped diastereoizomeric B8,8’), -2.16 to -13.03 (m, overlapped diastereoizomeric, B10,10’,9,9’,12,12’,4,4’,7,7’), -13.91 to -21.45 (m, overlapped diastereoizomeric, B5,5’,11,11’), -21.52 to -27.10 (m, overlapped diastereoizomeric, B6,6’), ESI-MS (m/z): found 788.58 (calc. for C26B18H70O6Si2Co1 788.53)
Synthesis of 8,8’-bridged [8,8'-O2P(O)H-3,3'-Co(1,2-C2B9H10)2] HNEt3 H-phosphonate (12). Imidazole (0.51 g, 7.5 mmol) was dissolved in minimum ammount of anhydrous acetonitrile. The solvent was evaporated under reduced pressure and the procedure was repeated twice. After drying under vacuum for 1.5 h imidazole was redissolved in 16 mL of anhydrous THF and the solution was cooled down at -70 °C in a dry ice/isopropanol bath under argon atmosphere. PCl3 (210 µL, 2.40 mmol) was added dropwise followed by Et3N (1 mL, 7.17 mmol) mixed with 1 mL of anhydrous THF. The whole was stirred at -70 °C for 15 min. and then solution of 8,8’-dihydroxy-bis(1,2-dicarbollido)-3-cobalt(1-)ate HNEt3 (2) (320 mg, 0,69 mmoles) in 13 mL of THF was added dropwisely. After further 30 min., the reaction mixture was removed from cooling bath and allowed to warm up to room temperature. After another 1 h the reaction was quenched with 30 mL of water and extracted four times with 40 mL of diethyl ether (Et2O). The combined ether extracts were dried with MgSO4 and the solvent was evaporated. The resultant solid crude product was dryed under vacuum then was purified by silica gel (230-400 mesh) column chromatography using CH3CN : CHCl3 1:4 as eluting solvent system. Yeld: 91%; TLC (CH3CN : CHCl3 1:2): Rf 0.35; 1H NMR (500 MHz, CD3CN) δ: 7.47,6.07 (1H, P-H), 3.70 (s, 4H, CHcarborane), 3.10 (m, 6H, NCH2CH3), 1.24 (t, 9H, NCH2CH3); 13C{1H} NMR (125 MHz, CD3CN) δ: 47.96 (4C, Ccarb), 47.74 (3C, NCH2CH3), 9.24 (3C, NCH2CH3); 11B{1H} NMR (120 MHz, CD3CN) δ: 23.02 (s, 2B, B8,8’), -2.83 (s, 2B, B10,10’), -5.70 (s, 4B, B9,9’,12,12’), -7.94 (s, 2B, B4,4’), -8.74 (s, 2B, B7,7’), -18.89 (s, 4B, B5,5’,11,11’), -27.85 (s, 2B, B6,6’); 11B NMR (125 MHz, CD3CN) δ: 23.02 (s, 2B, B8,8’), -2.83 (d, 2B, B10,10’), -5.70 (d, 4B, B9,9’,12,12’), -8,36 (t, 4B, B4,4’,7,7’), -18,91 (d, 4B, B5,5’,11,11’), -27.84 (d, 2B, B6,6’); 31P{1H} NMR (202 MHz, CD3CN) δ: -3.01 (s, P-H); 31P NMR (202 MHz, CD3CN) δ: -3,00 (d, P-H); ATR-IR (cm-1): 3621, 3029, 2993, 2544, 1609, 1474, 1446, 1393, 1218, 1152, 1137, 1094, 1025, 993, 981, 920, 903, 871, 849, 787, 743, 691, 666. UV-Vis λmax (nm): 297, 445. ESI-MS (m/z): found 402,24 (calc. for C4H21O3B18P1Co1: 401,71).
Synthesis of 8,8’-bridged [8,8'-O2P(O)SH-3,3'-Co(1,2-C2B9H10)2] HDBU phosphorothioate (13). H-phosphonate acid ester 12 (130 mg, 0,26 mmol) was dissolved in anhydrous MeOH (6,5 mL). The solution was added under argon atmosphere to S8 (85 mg, 2.6 mmol). 1,8-Diazabicyclo(5.4.0)undec-7-en (DBU) (160 µL, 1.05 mmoles) was then added and the mixture was stirred for 96 h at room temperature. After this time solvent was evaporated under reduced pressure. The crude product was dissolved in CH3CN then purified by silica gel column chromatography using a gradient of CH3CN : CHCl3 from 1:4 to 1:1 as eluting solvent system. Finally, the product 13 was eluted from the column using 100% MeOH as eluent. Yield: 70%; TLC (CH3CN : CHCl3 2:1) Rf: 0.5; 1H NMR (500 MHz, CD3CN) δ: 9.14 (s, 1H, NH), 3.58 (s, 4H, CHcarborane), 3.50 (m, 2H, NHCH2CH2), 3.44 (t, J = 5.9 Hz, 2H, CH2NCH2CH2CH2NH), 3.31 (s, 2H, CH2NCH2CH2CH2NH), 2.69 (dd, J = 6.6, 3.5 Hz, 2H, NHCCH2), 1.97 (dd, J = 6.6, 2H, CH2NCH2CH2CH2NH), 1.72 (m, 4H, NHCCH2CH2CH2), 1.65 (dt, J = 14.9, 5.2 Hz, 2H, NHCCH2CH2CH2CH2); 13C{1H} NMR (125 MHz, CD3CN) δ: 166.94 (NHCCH2), 54.98 (CH2NCH2CH2CH2NH), 49.26 (CH2NCH2CH2CH2NH), 46.76 (Ccarb), 46.63 (Ccarb), 39.04 (NHCH2), 33.42 (NHCCH2), 29.47 (NHCCH2CH2CH2), 27.05 (NHCCH2CH2CH2CH2), 24.52 (NHCCH2CH2CH2), 19.96 (CH2NCH2CH2CH2NH); 11B{1H} NMR (120 MHz, CD3CN) δ: 23.46 (s, 2B, B8,8’), -3.54 (s, 2B, B10,10’), -5.72 (s, 4B, B9,9’,12,12’), -8.73 (s, 4B, B4,4’,7,7’), -19.49 (s, 4B, B5,5’,11,11’), -28.25 (s, 2B, B6,6’); 11B NMR (120 MHz, CD3CN) δ: 23.46 (s, 2B, B8,8’), -3.50 (d, 2B, B10,10’), -5.74 (d, 4B, B9,9’,12,12’), -8.71 (d, 4B, B4,4’,7,7’), -19.53 (d, 4B, B5,5’,11,11’), -28.34 (d, 2B, B6,6’), 31P{1H}NMR (202 MHz, CD3CN) δ: 48.63 (s); 31P NMR (202 MHz, CD3CN) δ: 48.62 (s); ATR-IR (cm-1): 3383 (ν OH), 3223 (ν NH), 3091 (ν NH), 3026, 2926 (ν CH), 2856(ν CH), 2799 (ν CH), 2545 (ν BH), 1725, 1640, 1607, 1465, 1444, 1363, 1321, 1292, 1205, 1157, 1103, 1076, 978, 936, 910, 887, 836, 747, 690. UV-Vis λmax (nm) 215, 296, 450. ESI- MS (m/z): found: 434.20 (calc. for C4H21O3B18P1S1Co1 433.78).
Synthesis of 8,8’-bridged [8,8'-O2P(O)S(CH2)nOCPh3-3,3'-Co(1,2-C2B9H10)2] HNEt3 S-alkylated phosphorothioates 15 and 16. [8,8'-O2P(O)SH-3,3'-Co(1,2-C2B9H10)2] HDBU (13) (13 mg, 0.02 mmol) was dissolved in acetone (0.520 mL) then Et3N (65 µL, 0.46 mmol) was added under stirring at room temperature. The mixture was heated to 60 °C in an oil bath and then the alkylating agent 3 or 14 (0.04 mmol, dissolved in 130 µL of CH2Cl2) was added. The reaction mixture was kept overnight at 60 °C with stirring, then was cooled down to room temperature and solvents were evaporated under reduced pressure. The residue was dispersed in CH2Cl2, filtered and the solution was loaded on silica gel column prepared in CH2Cl2. The chromatography was performed using a gradient of MeOH in CH2Cl2 from 0 to 3% MeOH. 15, Yield: 90%; TLC (silica gel on Al, CH3CN : CHCl3 1:2): Rf: 0.71; 1H NMR (500 MHz, CD3CN): δ 7.41 (d, J = 7.5 Hz, 6H, Harom), 7.31 (t, J = 7.6 Hz, 6H, Harom), 7.23 (t, J = 7.2 Hz, 3H, Harom), 3.67 (s, 4H, CHcarborane), 3.07 (q, J = 7.3 Hz, 6H, NCH2CH3), 3.00 (t, J = 6.0 Hz, 2H SCH2CH2CH2CH2OTr), 2.78 (dt, 2H, SCH2CH2CH2CH2OTr), 1.72 (m, J = 7.1 Hz, 2H SCH2CH2CH2CH2OTr ), 1.64 (m, J = 6.9 Hz, 2H, SCH2CH2CH2CH2Otr), 1.21 (t, J = 7.3 Hz, 9H, NCH2CH3); 13C{1H} NMR (126 MHz, CD3CN): δ 145.55 (3C, aromatictrityl), 129.54 (6C, aromatictrityl), 128.87 (6C, aromatictrityl), 128.01 (3C, aromatictrityl), 87.30 (1C, OC(Ph)3), 63.90 (1C, SCH2CH2CH2CH2OTr), 47.76 (overlapped 3C, HNCH2CH3, 4C, CHcarborane), 31.20 (1C, SCH2CH2CH2CH2OTr), 29.75 (1C, SCH2CH2CH2CH2OTr), 28.84 (1C, SCH2CH2CH2CH2OTr), 9.29 (1C, NCH2CH3) 11B{1H} NMR (160 MHz, CD3CN): δ 23.08 (s, 2B, B8,8’), -2.86 (s, 2B, B10,10’), -5.54 (s, 4B, B9,9’,12,12’), -8.36 (s, 4B, B4,4’,7,7’), -18.87 (s, 4B, B5,5’,11,11’), -27.70 (s, 2B, B6,6’); 11B NMR (160 MHz, CD3CN): δ 23.06 (s, 2B, B8,8’), -2.87 (d, 2B, B10,10’), -5.57 (d, 4B, B9,9’,12,12’), -8.16 (d, 4B, B4,4’,7,7’), -18.92 (d, 4B, B5,5’,11,11’), -27.63 (d, 2B, B6,6’); 31P NMR{1H} (202 MHz, CD3CN): δ 16.72 (s), 11.37 (s);31P NMR (202 MHz, CD3CN): δ 16.72 (t), 11.37 (t); ATR-IR (cm-1) 3032 (ν CH aromatic), 2987 (ν CH aliphatic), 2925 (ν CH aliphatic), 2851 (ν CH aliphatic), 2681, 2566 (ν BH), 1727, 1595, 1474, 1447, 1392, 1264, 1200, 1134, 1103, 1068, 1032, 1016, 980, 941, 917, 901, 849, 735 (aromatic CH bending), 705(aromatic CH bending). UV-Vis λmax (nm) 196, 299, 440. ESI-MS (m/z): found: 748.37 (calc. for C27B18H43O4P1S1Co1: 748.24).
16, Yield: 86%; TLC (silica gel on Al, CH3CN : CHCl3 1:2) Rf: 0.69; 1H NMR (500 MHz, CD3CN) δ: 7.43 (m, 6H, Harom), 7.33 (m, 6H, Harom), 7.25 (m, 3H, Harom), 3.67 (s, 4H, CHcarborane), 3.10 (t, J = 6.0 Hz, 2H SCH2CH2CH2OTr), 3.08 (q, J = 7.3 Hz, 6H, NCH2CH3), 2.97 (dt, J = 14.9, 7.3 Hz, 2H, PSCH2CH2CH2OTr), 2.58 (s, 1H, CH3OH), 1.27 (t, J = 5.4 Hz, 2H PSCH2CH2CH2OTr ), 1.22 (t, J = 7.3 Hz, 9H, NCH2CH3), 1.18 (s, 3H, CH3OH); 13C{1H} NMR (126 MHz, CD3CN) δ: 145.27 (3C, aromatictrityl), 129.45 (6C, aromatictrityl), 128.78 (6C, aromatictrityl), 127.94 (3C, aromatictrityl), 87.25 (OC(Ph)3), 62.71 (PSCH2CH2CH2OTr), 55.13 (4C, CHcarb), 47.64 (HNCH2CH3), 47.59, 32.14, 31.98 (d, J = 6.1), 30.29, 29.66, 28.41 (d, J = 3.8), 9.17 (HNCH2CH3); 11B{1H} NMR (160 MHz, CD3CN): δ (ppm) 23.08 (s, 2B, B8,8’), -2.86 (s, 2B, B10,10’), -5.53 (s, 4B, B9,9’,12,12’), -8.36 (s, 4B, B4,4’,7,7’), -18.87 (s, 4B, B5,5’,11,11’), -27.70 (s, 2B, B6,6’); 11B NMR (160 MHz, CD3CN): δ 23.07 (s, 2B, B8,8’), -2.92 (d, 2B, B10,10’), -5.60 (d, 4B, B9,9’,12,12’), -8.38 (d, 4B, B4,4’,7,7’), -18.98 (d, 4B, B5,5’,7,7’), -27.88 (d, 2B, B6,6’); 31P{1H} NMR (202 MHz, CD3CN) δ: 16.54 (s); 31P NMR (202 MHz, CD3CN) δ: 16.54 (t); ATR-IR (cm-1) 3032 (ν CH aromatic), 2987(ν CH aliphatic), 2925 (ν CH aliphatic), 2851 (ν CH aliphatic), 2684, 2567 (ν BH), 1699, 1596, 1474, 1447, 1392, 1265, 1200, 1135, 1104, 1066, 1032, 1016, 980, 941, 917, 902, 849, 735 (aromatic CH bending), 705(aromatic CH bending). UV-Vis λmax (nm) 194, 299, 436. ESI-MS (m/z): 734.35 (calc. for C26B18H41O4P1S1Co1 734.17).
Synthesis of 4-[1,3-bis(trityloxy)propan-2-yl-oxy]butyl-4-methylbenzenesulfonate (20). The reaction was performed under argon atmosphere under anhydrous conditions. 1,3-Bis(trityloxy)propan-2-ol (18) (2.35 g, 4.07 mmol) was dissolved in 18 mL of anhydrous DMF then NaH60% (195 mg, 4.87 mmol) was added. After stirring for 15 min 1,4-bis(p-toluenesulfonyloxy)butane [40] (4.23 g, 10.63 mmol), dissolved in 18 mL of DMF was added. The reaction mixture was stirred for another 2 h at room temperature then was cooled in ice bath then an excess of NaH was centrifuged. The supernatant was poured into cooled 40 mL of phosphate buffer. The mixture was extracted with AcOEt (4x 100 mL). Organic extracts were combined, washed with H2O and dried over MgSO4. Solvents were evaporated under reduced pressure. The crude product was purified by silica gel column chromatography using a gradient of AcOEt in hexane from 0% to 10% as eluting solvent system. Yield: 27%, 1H NMR (600 MHz, CDCl3) δ: 7.78 (d, 2H, Harom ), 7.41 (d, 12H, Harom), 7.27 (m, 20H, Harom), 4.04 (t, 2H, OCH2CH2CH2CH2OSO2), 3.55 (p, 1H, TrOCH2CHOCH2OTr), 3.48 (t, 2H, OCH2CH2CH2CH2OSO2), 3.23 (ddd, 4H, TrOCH2CH), 2.43 (s, 3H, CH3tosyl), 1.75 (m, 2H, OCH2CH2CH2CH2OSO2), 1,58 (m, 2H, OCH2CH2CH2CH2OSO2). 13C{1H} NMR (125 MHz, CD3CN) δ: 144.62 (1C, aromatictosyl), 144.05 (6C, aromatictrityl), 133.19 (1C, aromatictosyl), 129.81 (2C, aromatictosyl,), 128.73 (12C, aromatictrityl,), 127.88 (2C, aromatictosyl), 127.76 (12C, aromatictrityl), 126.91 (6C, aromatictrityl), 86.52 (2C, OC(Ph)3), 78.59 (1C, TrOCH2CHOCH2OTr), 70.51 (1C, OCH2CH2CH2CH2OSO2-), 69.47 (1C, OCH2CH2CH2CH2OSO2-), 63.39 (2C, TrOCH2CH), 26.10 (1C, OCH2CH2CH2CH2OSO2), 25.86 (1C, OCH2CH2CH2CH2OSO2), 21.64 (1C, CH3tosyl); ATR-IR (cm-1): 3054, 3018, 2946, 2929, 2869, 1978, 1732, 1596, 1488, 1447, 1352, 1304, 1218, 1187, 1172, 1122, 1094, 1065, 1029, 992, 950, 922, 841, 811, 768, 745, 699; UV-Vis λmax (nm): 198, 229, 260; ESI-MS (m/z): found 825.32 [M+Na]-, 841.29 [M+K]-, (calc. for C52H50O6S1 803.01).
Synthesis of 8,8’-bridged {8,8'-O2P(O)S[(CH2)4OCH(CH2OCPh3)2]-3,3'-Co(1,2-C2B9H10)2} HNEt3 (21). [8,8'-O2P(O)SH-3,3'-Co(1,2-C2B9H10)2] HDBU (13) (440 mg, 0.75 mmol) was dissolved in 20 mL of anhydrous acetone then to the resultant solution anhydrous Et3N (2.15 mL, 15.42 mmol) was added under stirring at room temperature. The mixture was heated to 60 °C then 4-[1,3-bis(trityloxy)propan-2-yl-oxy]butyl-4-methylbenzenesulfonate (20) (901 mg, 1.12 mmol) dissolved in 20 mL of anhydrous AcOEt was added dropwise. After stirring overnight at 60 °C the mixture was cooled down and solvents evaporated under reduced pressure. The residue was dispersed in CH2Cl2, filtered and the filtrate was loaded to the silica gel column prepared in CH2Cl2. The chromatography was performed using a gradient of CH3OH in CH2Cl2 from 0 to 3% of CH3OH. Yield: 560 mg 64% TLC (silica gel on Al, CH3CN:CHCl3 1:2), Rf = 0.60, 1H NMR (600 MHz, CD3CN) δ: 7.41 (d, 12H, Haromatic), 7.31 (m, 18H, Haromatic), 3.70 (s, 4H, CHcarborane), 3.60 (p, 1H, TrOCH2CHOCH2OTr), 3.45 (t, 2H, PSCH2CH2CH2CH2O), 3.19 (ddd, 4H, CHOCH2OTr), 3,10 (q, 6H, HNCH2CH3), 2.85 (dt, 2H, PSCH2CH2CH2CH2O), 1.71 (m, 2H, PSCH2CH2CH2CH2O), 1.61 (m, 2H, PSCH2CH2CH2CH2O), 1.24 (t, 9H, NCH2CH3) 13C{1H} NMR (125 MHz, CD3CN) δ: 144.73 (6C, aromatictrityl), 129.10 (12C, aromatictrityl), 128.41 (12C, aromatictrityl), 127.60 (6C, aromatictrityl), 86.89 (2C, OC(Ph)3), 78.47 (1C, TrOCH2CHOCH2OTr), 69.92 (1C, PSCH2CH2CH2CH2O), 63.66 (2C, CHOCH2OTr), 47.28 (3C, HNCH2CH3), 47.18 (4C, CHcarborane), 31.89 (1C, PSCH2CH2CH2CH2O), 22.93 (1C, PSCH2CH2CH2CH2O), 13.96 (1C, PSCH2CH2CH2CH2O), 8.80 (3C, HNCH2CH3) 11B{1H} NMR (120 MHz, CD3CN) δ: 23.02 (s, 2B, B8,8’), -3.04 (s, 2B, B10,10’), -5.67 (s, 4B, B9,9’,12,12’), -8.55 (s, 4B, B4,4’,7,7’), -19.07 (s, 4B, B5,5’,11,11’), -27.99 (s, 2B, B6,6’). 11B NMR (120 MHz, CD3CN) δ: 23.02 (s, 2B, B8,8’), -2.98 (d, 2B, B10,10’), -5.64 (d, 4B, B9,9’,12,12’), -8.19 (d, 4B, B4,4’,7,7’), -19.05 (d, 4B, B5,5’,11,11’), -27.99 (d, 2B, B6,6’). 31P{1H}NMR (202 MHz, CD3CN) δ (ppm) 16.49 (s); 31P NMR (202 MHz, CD3CN) δ: 16.48(t); ATR-IR (cm-1): 3031, 2929, 2870, 2564, 2564, 2161, 1978, 1644, 1595, 1489, 1447, 1322, 1202, 1134, 1096, 1031, 940, 899, 848, 763, 747, 697; UV-Vis λmax (nm): 196, 233, 299, 453; MS (ESI) (m/z): found 1064.52 (calc. for C49B18H63O6P1S1Co1 1064.59)
Synthesis of 8,8’-bridged {8,8'-O2P(O)S[(CH2)4OCH(CH2OCPh3)2]-3,3'-Co[1-(CH2)2OH-1,2-C2B9H10)](1’,2’-C2B9H10)} HNEt3 (22) and {8,8'-O2P(O)S[(CH2)4OCH(CH2OCPh3)2]-3,3'-Co[1-(CH2)2OH-1,2-C2B9H10)] [1’-(CH2)2OH-1’,2’-C2B9H10)]} HNEt3 (23).Compound 21 (175 mg, 0.15 mmol) was dried by co-evaporation with anhydrous benzene and then kept under vacuum over P2O5 overnight. Next it was dissolved in anhydrous DME (3 mL) and the solution was cooled in CO2/isopropanol cooling bath. After 15 min. n-BuLi (140 µL, 1.6 M solution in hexane, 1.5 eq) was added and the reaction mixture was stirred for 10 min. After that time, cooling bath was removed and the mixture was stirred for next 10 min. Then the reaction mixture was cooled again in cooling bath and another portion of n-BuLi (140 µL) was added. After 15 min, ethylene oxide (200 µL, 2.9-3.1 M solution in THF)) was added and the reaction was left overnight in cooling bath. Next CH2Cl2 (5 mL) was added to the reaction mixture, the reaction was quenched by addition of water and then the organic solution was washed three times with 5 mL portions of water. Organic layer was separated and dried over MgSO4 then solvents were evaporated. Crude product was purified and mono- and bis-substituted products separated by silica gel column chromatography using a gradient of MeOH in CH2Cl2 from 0 to 3% of MeOH. 22, Yield: 17 mg (10 %); TLC (silica gel on Al, 8% MeOH/CH2Cl2): Rf: 0.13; 1H NMR (500 MHz, CD3CN): δ (ppm) 7.41 (d, 12H, Haromatic), 7.31 (m, 18H, Haromatic), 3.90 (s, CHcarborane), 3.78 to 3.54 (m, overlapped, CHcarborane, HOCH2CH2Ccarb, TrOCH2CHOCH2OTr), 3.44 (t, 2H, PSCH2CH2CH2CH2O-), 3.19 (ddd, 4H, CHOCH2OTr), 3.02 to 2.64 (m, overlapped, PSCH2CH2CH2CH2O-, HOCH2CH2Ccarb), 1.65 (m, 4H, overlapped, PSCH2CH2CH2CH2O);11B{1H} NMR (120 MHz, CD3CN) δ (ppm) 25.37, 24.65, 23.69, 22.93 (in ratio 2:2:1:1) 26.6 to 21.37 (m, overlapped diastereoizomeric B8,8’), 0.52 to -11.61 (m, overlapped diastereoizomeric, B10,10’,9,9’,12,12’,4,4’,7,7’), -12.05 to -11.61 (d, overlapped diastereoizomeric B5,5’,11,11’), -21.45 to -28.25 (s, overlapped diastereoizomeric B6,6’); 11B NMR (120 MHz, CD3CN) δ (ppm) 27.17 to 21.57 (m, overlapped diastereoizomeric B8,8’), 2.85 to -11.77 (m, overlapped diastereoizomeric, B10,10’,9,9’,12,12’,4,4’,7,7’), -11.82 to -21.00 (d, overlapped diastereoizomeric B5,5’,11,11’), -21.10 to -29.92 (s, overlapped diastereoizomeric B6,6’); 31P{1H}NMR (202 MHz, CD3CN) δ: 14.94, 14.58, 14.43, 14.12, 13.49 (in ratio: 4:1:2:1.5:15); 31P NMR (202 MHz, CD3CN) δ: 14.94(t), 14.42 (t), 14.12 (t), 13.49 (t); ATR-IR (cm-1): 3630, 3370, 3057, 3031, 2925, 2869, 2565, 2161, 1979, 1596, 1489, 1448, 1255, 1202, 1128, 1077, 1032, 985, 898, 871, 763, 746, 699; ESI-MS (m/z): found: 1108.54 m/z (calc. for C51B18H67O7P1S1Co1 1108.64).
23, Yield: 21 mg (12%); TLC (silica gel on Al, 8% MeOH/CH2Cl2): Rf: 0.27; 1H NMR (500 MHz, CD3CN) δ: 7.40 (d, 12H, Haromatic), 7.31 (m, 18H, Haromatic), 3.84 to 3.51 (m, overlapped, CHcarborane, HOCH2CH2-, TrOCH2CHOCH2OTr,), 3.44 (t, 2H, PSCH2CH2CH2CH2O-), 3.18 (ddd, 4H, CHOCH2OTr), 3.12 to 2.66 (m, overlapped, PSCH2CH2CH2CH2O, HOCH2CH2Ccarborane,), 1.65 to 1.55 (m, 4H, overllaped, PSCH2CH2CH2CH2O);11B{1H} NMR (120 MHz, CD3CN) δ: 25.36, 24.41, 23.65, 22.80, 22.23 (in ratio 1:2:1.5:1:1) 26.59 to 20.45 (m, overlapped diastereoizomeric B8,8’), 2.75 to -12.40 (m, overlapped diastereoizomeric, B10,10’,9,9’,12,12’,4,4’,7,7’), -12.34 to -24.64 (m overlapped diastereoizomeric B5,5’,11,11’6,6’); 11B NMR (120 MHz, CD3CN) δ: 26.65 to 20.21 (m, overlapped diastereoizomeric B8,8’), 2.32 to -12.88 (m, overlapped diastereoizomeric, B10,10’,9,9’,12,12’,4,4’,7,7’), -12.84 to -25.84 (m overlapped diastereoizomeric B5,5’,11,11’6,6’;); 31P{1H}NMR (202 MHz, CD3CN) δ: 15.00, 14.16, 14.02, 13.38 (in ratio 5:1:3:1); 31P NMR (202 MHz, CD3CN) δ: 15.99(t), 14.40 (s), 14.01 (t), 13.38 (t); ATR-IR (cm-1): 3566, 3357, 3056, 3027, 2920, 2889, 2857, 2565, 2166, 1596, 1489, 1448, 1291, 1255, 1201, 1120, 1078, 1032, 1001, 889, 871, 764, 746, 699 ESI-MS (m/z): found: 1152.57 (calc. for C53B18H71O8P1S1Co 1152.69).
5. Conclusions
Although derivatives of metallocarboranes containing various, simple substituents attached to the boron or carbon atoms of the complex carboranyl ligands are abundant, they usually do not allow further chemical transformations. One of the notable exceptions are the adducts of some metallacarboranes and cyclic ethers. In this work, we propose methods that attempt to, at least partially, fill this gap by using extendable ligands
The exploitation of the icosahedral metallacarborane’s immense potential in various fields of chemistry and technology requires the availability of convenient and versatile methods for their modification with various functional moieties and/or linkers of various type and length. Herein we report a convenient approach to introduce extendible arms on 8,8’-dihydroxy cobalt bis(1,2-dicarbollide). The approach can be used to introduce different hetero-bifunctional electrophiles containing protected hydroxyl function allowing further modification.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, MS, 1H-, 13C-, 11B-NMR spectra of compounds 6-13, 15,16, 20-23; 31P-NMR spectra of compounds 12, 13, 15, 16, 21-23, FT-IR spectra of compounds 12, 13, 15, 16, 22, 23.
Author Contributions
Conceptualization, investigation, data curation, writing—original draft preparation, KS; conceptualization, investigation, data curation, writing—original draft preparation, CS; conceptualization, validation, formal analysis, investigation, data curation, writing—review and editing, supervision, project administration, funding acquisition, ZL.
Funding
This work was supported in part by the National Science Center, Poland, grant 2015/16/W/ST5/00413.
Acknowledgments
The electrospray ionization (ESI) mass spectra for 10-13, 15, 16, 21-23, were recorded on Agilent 6546 LC/Q-TOF at the National Library of Chemical Compounds (NLCC) established within the project POL-OPENSCREEN financed by the Ministry of Science and Higher Education (decision no. DIR/WK/2018/06 of October 24, 2018). The authors thank Ms. Dorota Borowiecka and Ms. Agata Kraj of NLCC for recording the mass spectra.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the new compounds synthesized in this work are available from the authors.
References
- Dash, B.P.; Satapathy, R.; Maguireband, J.A.; Hosmane, N.S. Polyhedral boron clusters in materials science. New J. Chem. 2011, 35, 1955–1972. [CrossRef]
- Nuñez, R.; Romero, I.; Teixidor, F.; Viñas, C. Icosahedral boron clusters: A perfect tool for the enhancement of polymer features. Chem. Rev. 2016, 45, 5137–5434. [CrossRef]
- Leśnikowski, Z.J. Challenges and Opportunities for the Application of Boron Clusters in Drug Design. J. Med. Chem. 2016, 59, 7738-7758. [CrossRef]
- Messner, K.; Vuong, B.; Tranmer, G.K. The Boron Advantage: The Evolution and Diversification of Boron’s Applications in Medicinal Chemistry. Pharmaceuticals. 2022, 15, 264. [CrossRef]
- Marfavi, A.; Kavianpour, P.; Rendina, L.M. Carboranes in drug discovery, chemical biology and molecular imaging. Nat Rev Chem. 2022, 6, 486-504. [CrossRef]
- Olejniczak, A.B.; Sut, A.; Wróblewski, A.E.; Leśnikowski, Z.J. Infrared spectroscopy of nucleoside and DNA-oligonucleotide conjugates labeled with carborane or metallacarborane group. Vibr. Spectrosc. 2005, 39, 177-185. [CrossRef]
- Olejniczak, A.B.; Lesnikowski, Z.J. Boron Clusters as Redox Labels for Nucleosides and Nucleic Acids. In Handbook of Boron Science, Hosmane N., Eagling R., Eds.; World Scientific: New Jersey, USA, 2018; Volume 4, pp. 1-13. [CrossRef]
- Kodr, D.; Yenice, C.P.; Simonova, A.; Saftić, D.P.; Pohl, R.; Sýkorová, V.; Ortiz, M.; Havran, L.; Fojta, M.; Lesnikowski, Z.J.; O'Sullivan C.K.; Hocek M. Carborane- or Metallacarborane-Linked Nucleotides for Redox Labelling. Orthogonal Multipotential Coding of all Four DNA Bases for Electrochemical Analysis and Sequencing. J. Am. Chem. Soc. 2021, 143, 7124-7134. [CrossRef]
- Janczak, S.; Olejniczak, A.; Balabańska, S.; Chmielewski, M.K.; Lupu, M.; Viñas, C.; Lesnikowski Z.J. The boron clusters as a platform for new materials: Synthesis of functionalized o-carborane (C2B10H12) derivatives incorporating DNA fragments. Chem. Eur. J. 2015, 21, 15118-15122. [CrossRef]
- Kaniowski, D.; Ebenryter-Olbinska, K.; Kulik, K.; Janczak, S.; Maciaszek, A.; Bednarska-Szczepaniak, K.; Nawrot, B.; Lesnikowski, Z.J. Boron Clusters as a Platform for New Materials: Composites of Nucleic Acids and Oligofunctionalized Carboranes (C2B10H12) and their Assembly into Functional Nanoparticles. Nanoscale. 2020, 12, 103–114. [CrossRef]
- Sivaev, I.B. Ferrocene and transition metal bis(dicarbollides) as platform for design of rotatory molecular switches. Molecules. 2017, 22, 2201. [CrossRef]
- Dash, B.P.; Satapathy, R.; Swain, B.R.; Mahanta, C.S.; Jena, B.B.; Hosmane, N.S. Cobalt bis(dicarbollide) anion and its derivatives. J. Oranomet. Chem. 2017, 849-850, 170-194. [CrossRef]
- Fink K.; Boratyński J.; Paprocka M.; Goszczyński T.M. Metallacarboranes as a tool for enhancing the activity of therapeutic peptides. Ann. NY Acad. Sci. 2019, 1457, 128-141. [CrossRef]
- Gozzi, M.; Schwarze, B.; Hey-Hawkins, E. Preparing (metalla)carboranes for nanomedicine. ChemMedChem. 2021, 16, 1533-1565. [CrossRef]
- Barba-bon, A.; Salluce, G.; Lostalé-Seijo, I.; Assaf, K.I.; Henning, A.; Montenegro, J.; Nau, W.M. Boron clusters as broadband membrane carriers. Nature 2022, 603, 637-642. [CrossRef]
- Verdiá-Báguena, C.; Alcaraz, A.; Aguilella, V.M.; Cioran, A.M.; Tachikawa, S.; Nakamura, H.; Teixidor, F.; Viñas, C. Amphiphilic COSAN and I2-COSAN crossing synthetic lipid membranes: Planar bilayers and liposomes. Chem. Commun. 2014, 50, 6700-6703. [CrossRef]
- Tarŕes, M.; Canetta, E.; Paul, E.; Forbes, J.; Azzouni, K.; Viñas, C.; Teixidor, F.; Harwood A.J. Biological interaction with living cells of COSAN-based synthetic vesicles. Sci. Rep. 2015, 5, 7804. [CrossRef]
- Avdeeva, V.V.; Garaev, T.M.; Malinina, E.A.; K. Zhizhin K.Yu.; Kuznetsov, N.T.; Physiologically Active Compounds Based on Membranotropic Cage Carriers–Derivatives of Adamantane and Polyhedral Boron Clusters, Russ. J. Inorg. Chem. 2022, 67, 28-47. [CrossRef]
- Sardo, C.; Janczak, S.; Leśnikowski, Z.J. Unusual resistance of cobalt bis dicarbollide phosphate and phosphorothioate bridged esters towards alkaline hydrolysis: The “metallacarborane effect”. J. Organomet. Chem. 2019, 896, 70-76. [CrossRef]
- Grüner, B.; Švec, P.; Šícha, V.; Padělkova, Z. Direct and facile synthesis of carbon substituted alkylhydroxy derivatives of cobalt bis(1,2-dicarbollide), versatile building blocks for synthetic purposes. Dalton Trans. 2012, 41, 7498–7512. [CrossRef]
- Nekvinda, J.; Švehla, J.; Císařovâ, I.; Grüner, B. Chemistry of cobalt bis(1,2-dicarbollide) ion; the synthesis of carbon substituted alkylamino derivatives from hydroxyalkyl derivatives via methylsulfonyl or p-toluenesulfonyl esters. J. Organomet. Chem. 2015, 798, 112-120. [CrossRef]
- Plešek, J.; Grüner, B.; Báča, J.; Fusek, J. Syntheses of the B(8)-hydroxy- and B(8,8′)-dihydroxy-derivatives of the bis(1,2-dicarbollido)-3-cobalt(1-)ate ion by its reductive acetoxylation and hydroxylation: Molecular structure of [8,8′-μ-CH3C(O)2 < (1,2-C2B9 H10)2-3-Co]0 zwitterion determined by X-ray diffraction analysis. J. Organomet. Chem. 2002, 649, 181-190. [CrossRef]
- Sivaev, I.; Bregadze, V.I. Chemistry of cobalt bis(dicarbollides). A Review. Collect. Czech. Chem. Commun. 1999, 64, 783-805. [CrossRef]
- van den Berg, R. J. B. H. N.; Korevaar, C.G.N.; Herman, S.; Overkleeft, H.S.; van der Marel, G.A; van Boom, J.H. Effective, High-Yielding, and Stereospecific Total Synthesis of d-erythro-(2R,3S)-Sphingosine from d-ribo-(2S,3S,4R)-Phytosphingosine. J. Org. Chem. 2004, 69, 5699–5704. [CrossRef]
- Sivaev, I.B.; Bregadze, V.I., Lewis acidity of boron compounds. Coord. Chem. Rev. 2014, 270-271, 75-88. [CrossRef]
- Hesek, D.; Lee, M.; Noll, B.C.; Fisher, J.F.; Mobashery, S. Complications from Dual Roles of Sodium Hydride as a Base and as a Reducing Agent. J. Org. Chem. 2009, 74, 2567–2570. [CrossRef]
- Planas, J.G.; Teixidor, F.; Viñas, C.; Light, M.E.; Hursthouse, M.B. Self-Assembly of Halogenated Cobaltacarborane Compounds: Boron-Assisted C-H⋅⋅⋅X-B Hydrogen Bonds? Chem. Eur. J. 2007, 2493-2502. [CrossRef]
- Stogniy, M.Y.; Suponitsky, K.Y.; Chizhov, A.O.; Sivaev, I.B.; Bregadze, V.I. Synthesis of 8-alkoxy and 8,8’-dialkoxy derivatives of cobaltbis(dicarbollide), J. Organomet. Chem. 2018, 865, 138-144. [CrossRef]
- Stawinski, J.; Strömberg, R. Di- and Oligonucleotide Synthesis using H-phosphonate chemistry. In Methods in Molecular Biology, Oligonucleotide Synthesis: Methods and Applications. Herdewijn P., Eds.; Humana Press Inc.: Totowa, New Jersey, USA, 2008; Volume 288, pp. 81-100.
- Jones, D. J.; O'Leary, E.M.; O'Sullivan, T. P. Synthesis and application of phosphonothioates, phosphonodithioates, phosphorothioates, phosphinothioates and related compounds. Tetrahedron Lett. 2018, 59, 4279-4292. [CrossRef]
- Hronowski, L.J.J.; Szarek, W.A.; Hay, G.W.; Krebs, A.; Depew, W.T. Synthesis and characterization of 1-O-β-lactosyl-(R,S)-glycerols and 1,3-di-O-β-lactosylglycerol. Carbohydr. Res. 1989, 190, 203-218. [CrossRef]
- King, B.R. Three-Dimensional Aromaticity in Polyhedral Boranes and Related Molecules. Chem. Rev. 2001, 101, 1119–1152. [CrossRef]
- Poater, J.; Viñas, C.; Bennour I.; Escayola S.; Solà M.; Teixidor F. Too Persistent to Give Up: Aromaticity in Boron Clusters Survives Radical Structural Changes. J. Am. Chem. Soc. 2020, 142, 9396–9407. [CrossRef]
- Fulcrand-El Kattan, G.; Lesnikowski, Z.J.; Yao, S.; Tanious, F.; Wilson, W.D.; Shinazi, R.F. Carboranyl Oligonucleotides. 2. Synthesis and Physicochemical Properties of Dodecathymidylate Containing 5-(o-Carboranyl-1-yl)-2'-O-Deoxyuridine. J. Am. Chem. Soc. 1994, 116, 7494-7501. [CrossRef]
- Lesnikowski, Z.J.; Shi, J.; Schinazi, R.F. Nucleic acids and nucleosides containing carboranes. J. Organomet. Chem. 1999, 581, 156-169. [CrossRef]
- Plešek, J. The age of chiral deltahedral borane derivatives. Inorg. Chim. Acta. 1999, 289, 45-50. [CrossRef]
- Grűner, B.; Plzàk, Z. High-performance liquid chromatographic separations of boron cluster compounds. J. Chromatogr. 1997, 789, 497-517. [CrossRef]
- Horàkovà, H.; Grűner, B.; Vespalec, R. Emerging Subject for Chiral Separation Science: Cluster Boron Compounds. Chirality 2011, 23, 307-319. [CrossRef]
- Van Swieten, P.F.; Rene Maehr, R.; van den Nieuwendijk, A.M.C.H.; Kessler, B.M.; Reich, M.; Wong, C-S.; Kalbacher, H.; Leeuwenburgh, M.A.; Driessen, C.; van der Marel, G.A.; Ploegh, H.L.; Overkleefta, H.S. Development of an isotope-coded activity-based probe for the quantitative profiling of cysteine proteases. Bioor. Chem. Med. Lett. 2004, 14, 3131-3134. [CrossRef]
- Martin, A.E.; Ford, T.M.; Bulkowski, J.E. Synthesis of selectively protected tri- and hexaamine macrocycles. J. Org. Chem. 1982, 47, 3, 412–415. [CrossRef]
Scheme 1.
Alkylation of 8,8’-dihydroxy-bis(1,2-dicarbollide)-3-cobalt(1-)ate (2) with Ts(CH2)4OCPh3 (3) or Br(CH2)3OTBDMS (4) as electrophiles. i. H2SO4 80% 140 °C; ii. NaH, 3 or 4, 80 °C or 60 °C, DMF; TBDMS = -Si(CH3)2C(CH3)3.
Scheme 1.
Alkylation of 8,8’-dihydroxy-bis(1,2-dicarbollide)-3-cobalt(1-)ate (2) with Ts(CH2)4OCPh3 (3) or Br(CH2)3OTBDMS (4) as electrophiles. i. H2SO4 80% 140 °C; ii. NaH, 3 or 4, 80 °C or 60 °C, DMF; TBDMS = -Si(CH3)2C(CH3)3.
Scheme 2.
Deprotection of TBDMS protected 8. i. 2.5 eq TBAF in THF. TBDMS = -Si(CH3)2C(CH3)3, TBA = N[(CH2)3CH3]4.
Scheme 2.
Deprotection of TBDMS protected 8. i. 2.5 eq TBAF in THF. TBDMS = -Si(CH3)2C(CH3)3, TBA = N[(CH2)3CH3]4.
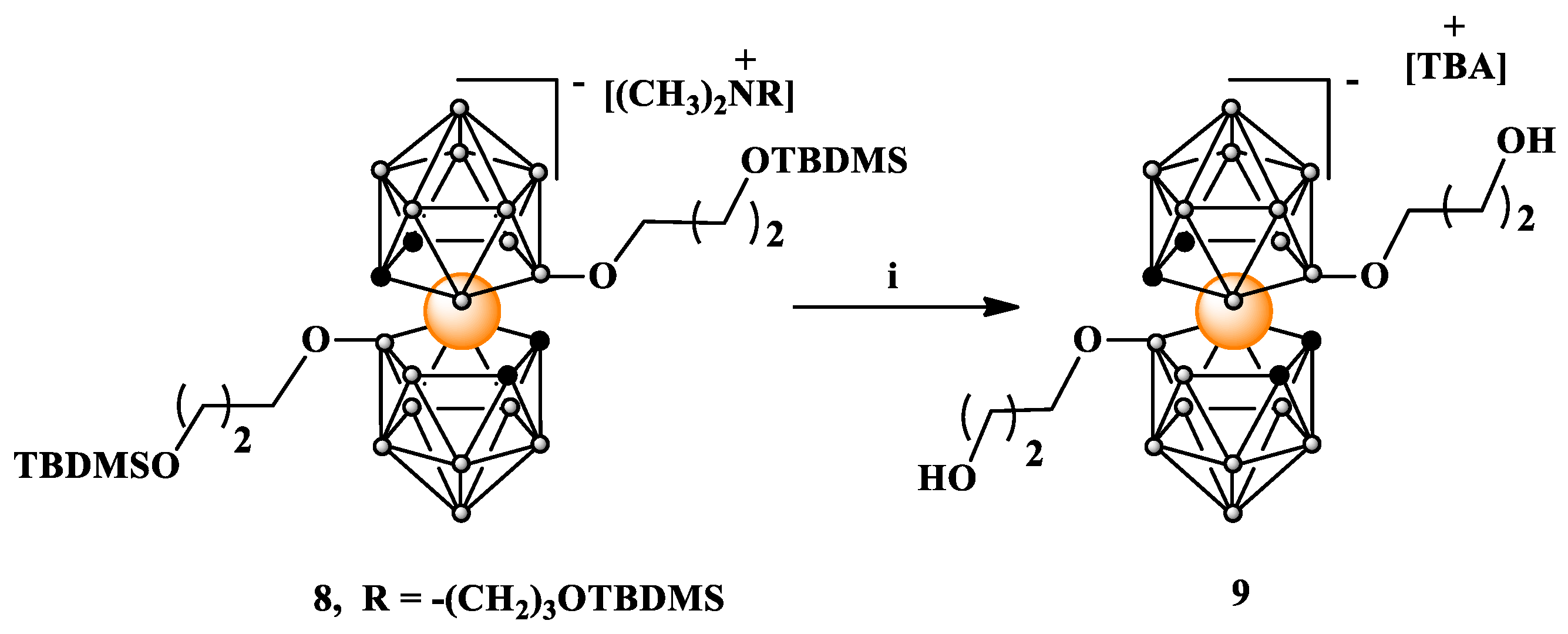
Scheme 3.
Substitution on carbon atoms in 8,8’-alkoxy functionalized metallacarborane 8. i. nBuLi, THF; ii. ethylene oxide. TBDMS = -Si(CH3)2C(CH3)3.
Scheme 3.
Substitution on carbon atoms in 8,8’-alkoxy functionalized metallacarborane 8. i. nBuLi, THF; ii. ethylene oxide. TBDMS = -Si(CH3)2C(CH3)3.
Figure 1.
Intramolecular hydrogen bonds in halogenated cobalt bis(1,2-dicarbollide) (A) and hypothetical intramolecular interactions in 8,8’-bisalkylated 8 (B).
Figure 1.
Intramolecular hydrogen bonds in halogenated cobalt bis(1,2-dicarbollide) (A) and hypothetical intramolecular interactions in 8,8’-bisalkylated 8 (B).

Scheme 5.
Alkylation of 8,8'-O,O-[cobalt bis(1,2-dicarbollide)]phosphorothioate (13) with linear alkylating agent. i. Ts(CH2)4OCPh3 (3) or Br(CH2)3OCPh3 (14), acetone, NEt3, 60 oC, 24 h. DBU = 1,8-diazabicyclo(5.4.0)undec-7-en.
Scheme 5.
Alkylation of 8,8'-O,O-[cobalt bis(1,2-dicarbollide)]phosphorothioate (13) with linear alkylating agent. i. Ts(CH2)4OCPh3 (3) or Br(CH2)3OCPh3 (14), acetone, NEt3, 60 oC, 24 h. DBU = 1,8-diazabicyclo(5.4.0)undec-7-en.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



