Submitted:
21 April 2023
Posted:
23 April 2023
You are already at the latest version
Abstract
The makeup of human microbiota has been linked to a number of autoimmune disorders. Recent developments in whole metagenome sequencing and 16S rRNA sequencing technology have considerably aided research into the microbiome and its relationship to disease. Due to the inherent high dimensionality and complexity of data generated by high-throughput platforms, conventional bioinformatics techniques could only provide an inadequate explanation for the most relevant changes and seldom provide correct predictions. Machine learning, on the other hand, is a subset of artificial intelligence applications that enable the untangling of high-dimensional systems and intricate knots in correlation by learning complex patterns and improving automatically from training data without being explicitly programmed. Machine learning is increasingly being utilized to research the influence of microbes on the onset of illness and other clinical features since computer power has increased dramatically in the last few decades. In this review paper, we focused on emerging methodological approaches of supervised machine learning algorithms for identification of autoimmune disorders utilizing metagenomics data, as well as the potential benefits and limitations of machine learning models in clinical applications.
Keywords:
Gut microbiome
; Machine learning
; Deep learning
; Metagenomics
; Autoimmune diseases
; Biomarkers discovery
; Diagnostic models
1.0. Introduction
Clinically autoimmune diseases are defined as scenarios in which the immune system gets triggered by healthy cells of the body instead of diseased cells or foreign particles. Autoimmune diseases occur due to genetic susceptibility, environmental triggers, and auto reactivity toward healthy cells or cell products [1]. The human body system maintains a high level of vigilance against autoreactive immune cells. Central and peripheral control carries out the vigilance. During the formation in the thymus, autoreactive lymphocytes are negatively selected and removed in the thymic medulla due to central tolerance. After maturity, the lymphocytes that enter the bloodstream undergo peripheral tolerance, where autoreactive cells are negatively selected and removed [2]. Despite a strong check system, some autoimmune cells survive and can cause allergic reactions or inflammation. An autoimmune disease can occur by chance, but several factors increase the possibility of the disease. For example, the microbiome and epigenome have been explored for their role in triggering autoimmune responses [3]. Studies have indicated that autoimmune disease pathogenesis is highly associated with gut dysbiosis, a phenomenon of microbiota imbalance [3], suggest that microbiome changes result in epigenetic changes that ultimately trigger autoimmunity. The microbiome is highly sensitive to environmental triggers and diet. For example, in the case of inflammatory bowel disease, an autoimmune disease, the microbiome undergoes a shift in terms of population and causes inflammation. Sometimes, the microbiome can also come in contact with the damaged lining of intestines, which can also cause an infection or inflammation [4]. Similarly, imbalances in liver microbiota result in autoimmune diseases like primary sclerosing cholangitis (PSC), primary biliary cholangitis (PBC), and an autoimmune hepatitis (AH) [5]. Liver microbiota is also known to interact with its intestinal counterparts. As the microbiome and host interactions are still poorly understood, the field of metagenomics becomes extremely important in diagnosing and treating autoimmune diseases. Recent approaches like metagenomics, meta-transcriptomics, and high-throughput sequencing have made it possible to diagnose autoimmune disorders as well understanding the role of microbiomes in development of autoimmune disorder [6].
1.1. Current Limitation in Diagnosis of Autoimmune Diseases
Due to the heterogeneity of onset and progression, diagnosis and prognosis for autoimmune diseases are unpredictable. The diagnosis and prognosis of autoimmune diseases remain uncertain because of the complexity of symptoms and progression of the disease. Unfortunately, studies have indicated that diagnosing chronic autoimmune diseases like systemic lupus erythematosus (SLE), multiple sclerosis (MS), and rheumatoid arthritis (RA) may still be challenging and depend on a specific set of criteria [7,8], To make a definitive diagnosis, several criteria must be satisfied, such as clinical symptoms, functional outcomes, and biochemical and imaging evidence [9,10]. Misdiagnosis and delayed diagnosis of such disorders are quite typical when using imprecise and insensitive criteria [11]. Typically time required from the emergence of symptoms to the confirmation of the autoimmune diseases reported as two years [12]. Patients can thereby miss the appropriate timeframe required for disease intervention. Numerous improvements have been achieved in the detection and treatment of autoimmune disorders during the last decade. To improve scientists' ability for the early detection of autoimmune disorders, several novel molecular or immunological biomarkers have been identified [13,14,15]. However, due to the extremely heterogeneous nature of these disorders and the inadequate understanding of their pathogenesis, the outcomes remained unsatisfactory [16]. Therefore, more work remains to be done to ensure accurate and timely identification of auto-immune illnesses.
Numerous research studies have been conducted with the aim of enhancing the accuracy of disease diagnosis and prognosis, encompassing various ailments such as cancer, among other diseases using diverse number of data including metagenomics [17,18]. Figure 1 presents a concise overview of the sequential process involved in developing a diagnostic model for diseases. The utilization of machine learning techniques has the potential to expedite the identification of autoimmune diseases by leveraging metagenomics data analysis. In this review paper we present an overview of the latest methodologies that integrate machine learning and metagenomics to detect autoimmune diseases, which may serve as a viable diagnostic model for autoimmune diseases. In the final part, we also discusses the limitations and possible drawbacks of developing a machine learning-based diagnostic model.
1.2. Metagenomics and Machine Learning for Microbial Analysis
The total number of microbes in the human body is a controversial topic but it is widely agreed among researchers that abundance of microbes in human body are at least equal to the number of body cells [19]. These microbes play an important role in many bodily functions, including people's health and mood [20]. Numerous microorganism species exhibit symbiotic relationships with host cells, while a certain number of species function as pathogens or opportunists that exploit a weakened immune system. Therefore, investigating these microorganisms is crucial for comprehending their impact on human health and the onset of illnesses [21]. The in-vitro culture methodology is a conventional approach utilized for the examination of microorganisms within a particular setting. However, it should be noted that this particular approach may not be universally applicable to all microorganisms, and thus, it may encounter limitations when confronted with unfamiliar microorganisms or those inhabiting complex environments. Metagenomics sequencing techniques can be employed to access microbial organisms’ analysis that are non-culturable in laboratory settings owing to the difficulties in reproducing intricate environments, such as human tissue. The metagenomics process involves the extraction and analysis of genetic material from microbial sources through high-throughput sequencing techniques [22]. There are two main methods utilized for sequencing microbial DNA, namely whole genome sequencing (WGS), which is also referred to as Shotgun metagenomics sequencing or untargeted sequencing, and Amplicon sequencing, also known as targeted sequencing [23]. Figure 2 demonstrates the key steps involved in targeted and untargeted microbial sequencing method and machine learning for analyses of patterns from metagenomics data. The Amplicon sequencing approach utilizing 16s rRNA and 18s rRNA has been extensively employed in microbial studies in previous years. However, the Shotgun sequencing method has gained popularity in recent times due to a significant reduction in sequencing costs and its ability to provide information on previously unidentified species of microbial organisms. Advantages and disadvantages of Amplicon and Shotgun sequencing methods are mentioned in Table 1.
The utilization of amplicon sequencing and shotgun sequencing techniques has ushered in a new era of extensive data analysis, enabling researchers to explore the intricate interactions between microorganisms and the human body, as well as their involvement in the onset and management of various diseases. For instance, the Human Microbiome Project (HMP) by the National Institutes of Health (NIH) [24] assessed the human body's microbiota and generated over 35 billion reads using 16S rRNA MG data, utilizing 690 samples from various body sites. In addition to the HMP study, two additional projects, the American Gut Project [25] and the Human Intestinal Tract [26], have greatly expanded the knowledge of the composition and function of the human microbiome with generation of huge amount of data. To research the microbiome's unique nature, composition, function, and heterogeneity, advanced analytics tools for collecting and interpreting microbial data are still necessary [27]. The establishment of reliable methodologies for scrutinizing microbial data is of utmost importance in comprehending the interplay between the host and microbiome, thereby facilitating the identification of diseases and the formulation of therapeutic interventions aimed at enhancing individual well-being [28].
Machine learning is a subfield of artificial intelligence that involves the development of algorithms capable of learning from past data and generating predictions based on trained data. In contemporary times, machine learning has become a prevalent method for analyzing patterns of diseases utilize clinical and omics data due to its capacity to detect patterns from high dimensional datasets through a variety of algorithms [29,30,31,32]. The discipline of machine learning is commonly classified into four discrete subdomains, including supervised learning, unsupervised learning, semi-supervised learning, and reinforcement learning. Supervised learning pertains to datasets that have been labeled, whereas unsupervised learning involves clustering of data samples with unknown labels based on similarities or dissimilarities. Semi-supervised learning, on the other hand, involves the utilization of both labeled and unlabeled data. The integration of machine learning with metagenomics data holds promise for uncovering the fundamental etiology of diseases and advancing the development of targeted diagnostic and therapeutic modalities. This can be achieved through various machine learning methodologies, such as dimensionality reduction techniques to identify significant biomarkers and the stratification of cohorts based on selected biomarkers [33].
2.0. Biomarker Discovery with Machine Learning Approaches
Biomarkers are defined as measurable indicators of a biological state, encompassing both normal physiological processes and pathological conditions, as well as responses to specific stimuli such as treatment or exposure to hazardous environments. Several approaches can be utilized to obtain biomarkers. Meanwhile utilization of contemporary high-throughput technologies, coupled with diverse statistical and machine learning methodologies, has considerably amplified the identification of biomarkers. While high throughput methodologies present various advantages for biomarker detection, they are not free to constraints. A limitation that arises in this context involves the generation of data featuring a high number of dimensions. Consequently, it becomes crucial to devise advance tools to attain a more profound comprehension of the biological implications and mitigate the risk of inaccurate identification of biomarkers.
2.1. High-Dimensional Data Analysis Using Machine Learning and Conventional Statistical Methods
High-throughput methodologies generate omics datasets that typically comprise a multitude of features, spanning from several hundred to several thousand. This results in a high ratio of features to samples. For example, a single sample of microarray data produces more than fifty thousand features, representing the gene expression of an individual. The proteomic data consists of ten thousand features that are employed to represent proteins abundance in a comparable fashion. The analysis of large datasets in biological research is becoming increasingly difficult due to the presence of irrelevant and insignificant information. The aforementioned outcomes lead to escalated computational expenses and a heightened susceptibility to overfitting while building diagnostic models [34]. Additionally, there exists a constraint in discerning significant features for distinguishing or categorizing among various groups, such as healthy individuals versus those with a disease.
Dimension reduction is a machine learning technique that aims to reduce the dimensionality of data by eliminating redundant and unnecessary features while retaining significant information. This approach can facilitate the development of reliable and interpretable machine learning models [35]. Dimension reduction is commonly classified into two primary branches, namely supervised machine learning based dimension reduction and unsupervised machine learning based dimension reduction. Supervised learning ML-based dimension reduction can be subdivided into two categories: one that employs conventional statistical methods and the other that utilizes machine learning methodologies. The present review exclusively focuses on dimensionality reduction methods utilizing supervised ML-based dimension reduction. However, the exploration of unsupervised ML-based dimension reduction falls beyond the scope of the present review and may be further examined in a succinct and comprehensive review article [36].
The literature has extensively documented four dimension reduction strategies that are based on supervised machine learning: filter technique, wrapper, embedding, and hybrid feature selection methods. Filter technique employs conventional statistical approaches, while wrapper, embedded, and hybrid feature selection methods utilize ML-algorithmic approaches. Figure 3 shows the types of supervised based dimension reduction techniques along with their variants.
2.1.1. Filter Method
The filter technique selects a subset of features based on statistical criteria such as, Pearson correlation, mutual information, chi-square, analysis of variance (ANOVA) etc. To deal with curse of dimensionality, statistically significant features are extracted to provide a subset of features used to train a classifier model [37]. Filter techniques are broadly classified into two categories. Univariate approaches assess the significance of each attribute to the group separately, whereas multivariate methods assess a subset of features. Even though the filters technique may successfully reduce data dimensionality while requiring less processing time, there are several limitations that must be solved. In the univariate approach, for example, the problem of collinearity arises when each independent variable is evaluated individually to establish a link with the dependent variable, but the fact that independent variables are connected to each other is ignored [38]. To overcome collinearity difficulties, multivariate filter techniques can be used to remove features indicating correlation with other features [40]; however, these approaches have limitations when it comes to considering interactions between independent features. On the other hand, more recent advanced approaches of filter method have demonstrated the ability to find interactions between features and eliminate redundant features [39,40].
Relief family-based algorithms (RBA) represent a different kind of filter method utilized to address the challenge of the curse of dimensionality. The approach of RBAs does not operate as a comprehensive exploration of feature interactions and statistical associations between independent and dependent variables. Instead, it prioritizes features based on their significance within each class and evaluates a feature's importance by its ability to differentiate between both classes. [41].
2.1.2. Wrapper Method
In contrast to the filter approach, wrapper method rather depending on statistical score makes use of classifier algorithms to select subset of features; as a result, ultimately selects best subset that fares better accuracy in the classification of groups. For the wrapper method, exhaustive search is computationally impractical with big dimensional data for all the feature combinations in space; therefore, other approaches such as heuristic search and sequential search method utilized. Heuristic method includes Genetic algorithm approach [42] and ant colony search optimization [43] whereas sequential search utilizes two methods including sequential forward selection (SFS) and sequential backward selection (SBS) to generate a subset of features [44]. Wrapper methods are applicable to various classifier methodologies, enabling the assessment of features with the use of diverse classifiers. Wrapper methods assess the significance of features by evaluating their impact on the model's performance. The evaluation criteria frequently employed in heuristic and sequential search methods comprise the accuracy score and the area under the curve. However recursive feature elimination method, which is another type of wrapper approach, uses feature weight (i.e Gini impurity score) to select subset of features.
Wrapper methods possess a primary advantage over filter methods in that they inherently consider feature dependencies, including interactions and redundancies, when selecting the appropriate feature subset. However, it should be noted that wrapper methods require more computational resources compared to filter and embedded methods due to the extensive computations involved in designing and evaluating feature subsets [45]. The utilization of the wrapper approach is a prevalent technique for reducing dimensions in the development of diagnostic models. Nonetheless, the approach of utilizing wrapper techniques presents several limitations. Specifically, the wrapper approach relies on the selection of features according to specific classifier models, and it cannot be assured that the chosen features will yield optimal performance when implemented with alternative classifiers. Wrapper approaches encounter a notable challenge in the form of overfitting, as the selection of features is contingent upon particular data and cannot be ensured to remain optimal when confronted with additional data that exhibits variance.
2.1.3. Embedded Method
Embedded feature selection method carries out feature selection and model building simultaneously. Classifier model adjusts its internal settings and chooses the proper weights/importance given to each feature to generate the best classification accuracy during the training phase. Consequently, with an embedded method, finding the ideal feature subset and building the model are merged in a single phase.
There are several algorithms work as embedded feature selection algorithms such as decision-trees based models including random forest, gradient boosting trees which give weight/importance to feature by mean decrease impurity (MDI) [46] and regularization model such as logistic regression and its variant (lasso and elastic net) determine importance of features with penalization or shrinkage of coefficients that do not enhance classification accuracy of models or meaningfully interact with models [47]. The output of the embedded technique includes feature rankings determined by factors that make significant contributions to classification model accuracy.
2.1.4. Hybrid Search
The hybrid approach encompasses the integration of both filter and wrapper techniques. The filter method is initially employed to perform dimensionality reduction on data, followed by the application of the wrapper method on a subset of features that have been selected by the filter method. Hybrid methods are known to possess filter and wrapper characteristics, which enable them to select features that exhibit high accuracy from the wrapper approach and high efficacy from the filter method. Numerous methodologies have been proposed in recent times to accomplish hybrid feature selection, such as hybrid genetic algorithm [48], fuzz random forest for feature selection [49], hybrid ant colony optimization [50]. Table 2 contains the dimension reduction algorithm types along with advantages and disadvantages.
3.0. Supervised Learning Algorithms
3.1. Linear Regression
Linear Regression is a simple statistical method used for predictive analysis, when outcome class in data exist in continuous values rather than binary outcomes [51,52]. The linear regression algorithm predicts possible outcomes by establishing a linear relationship between an independent variable and a dependent variable. The dependent variable must be continuous, but the independent variables can be binary continuous or categorical. There are two types of linear regression 1) Simple linear regression 2) Multiple linear regression. A simple linear regression model consists of one independent variable and one dependent variable. In contrast, multiple linear regression model use more than one independent variable to predict outcome of dependent variable.
The equation for a simple linear regression is:
Whereas, Y is dependent variable or outcome, X is independent variable or predictor, i is intercept (value of Y when X = 0) and c = coefficient of X (slope line).
In order to construct the best-fitted regression line, cost function (the difference of error between the actual and predicted value of outcome variable) must be at a minimum. The cost function of linear regression is most commonly estimated by the root mean squared error (RMSE) method. It is pivotal to adjust the values of i and c to obtain a minimum cost function. So, the model uses gradient descent to reduce the RMSE by adjusting i and c values and constructing the best-fit regression line.
3.2. Logistic Regression
Due to straightforward mathematical structure, logistic regression is one of the most popular algorithms for classification tasks. Basically, LR predicts the likelihood that an event will occur. For example, whether or not obesity leads to autoimmune disorder? Logistic regression is classified into three types. 1) Binary-When there are only two possible outcomes, such as in the preceding example, obesity leads to autoimmune disorders? Is it yes or not? 2) Multinomial- When there are multiple possible outcomes, such as whether obesity leads to diabetes, IBD or RA? 3) Ordinal- When the outcome variables are ordered, for example, is obesity associated with organ specific- or systemic specific- autoimmune disorder? Logistic regression employs the sigmoid or logit function to compute probabilities. In the case of binary classification, the logit function used which is simply an S-shaped curve (sigmoid curve) that transform dependent variables into value of 0 and 1 [53]. To fit best logistic regression, a few assumptions must be met: the dependent or outcome variable must be categorical or dichotomous. Multi-collinearity between independent variables must be minimal or zero [54]. To train the logistic regression model, a relatively large sample size is needed.
3.3. Naïve Bayes
The Naïve Bayes (Nb) is a simple probabilistic ML classifier based on the Naive Bayes theorem that expressed as,
Where, P(A) = Probability of occurrence of event A, P(B) = Probability of occurrence of event B, P(A|B) = Probability of occurrence of event A with conditional event B, & P(B|A) = Probability of occurrence of event B with conditional event A
Naïve Bayes assumes that predictor variables are independent of the outcome variables that is, the presence/absence of one variable has no influence on the presence/absence of another variable. Thus, it is referred to as the term “naïve”. Since the crux of the Bayesian method is to estimate the means and differences of each variable in the input data, NB requires a small training set to solve the classification task [55]. As NB implementation is easy with no complicated hyper parameters tuning, thus it can handle large sample size. Additionally, during model training it is possible to incorporate new variables that can enhance the probability followed by classification improvement. Thus, this simple classifier method can efficiently handle high dimensional data for binary or multiclass classification.
3.4. Random Forest
The Random Forest algorithm is widely utilized in the field of machine learning for both classification and regression tasks involving data with a high number of dimensions. The Random Forest algorithm is comprised of a collection of decision trees that are combined through an ensemble method [56]. In Random Forest (RF), each tree is trained on a specific subset of features from the training data and subsequently generates predictions for unseen samples, referred to as out-of-bag samples (OOBs). For new sample predictions, random forest method uses a number of different decision trees trained on data and averages their outputs and decides the outcome based on the prediction with the most votes. As random forest chooses feature’s subset randomly, it reduces the correlation between decision trees [57]. This is also a distinctive characteristic of RF forest that discriminates RF to decision trees, as RF selects subset of features whereas decision trees tend to include all features. Random forest can handle both regression and classification tasks with advantage of robustness with outliers and missing value data, thus the most widely used classifier found in literature [58]. Despite random forests flexibility in parameters adjustment, hyper parameter tuning of certain parameters needed to be set prior to model training. Including, number of trees, node size and number of features subset.
3.5. Support Vector Machine
Support vector machine was introduced by Vladimir Vapnik [59] which, due to its benefit of resilience in noisy data and outliers, is among the widely used algorithms in omics domain. Support vector machine strive to locate the hyperplane (decision boundary) that most effectively establishes a separation between data points belonging to distinct classes or groups. Support vectors are data points placed near decision borders that are used to find the optimal hyperplane. Margin is utilized in SVM to optimize the distance between hyperplanes of each class. There are 2 types of margins: 1) Hard margin 2) Soft margin. Hard margins are suitable when two classes (support vectors) are clearly separated, whereas soft margin allows SVM to loosen strict boundaries and misclassify certain data points so that other data points can be properly classified [60]. As data points frequently overlap in metagenomics data, soft margins are the ideal choice in this situation but increment in soft margin leads to overfitting of model hence optimal number of soft margin adjustment should be determined. SVM was originally developed as a linear classifier, but subsequently, the Kernel function was added to address non-linear classification issues [60]. In non-linear tasks, kernels introduce additional dimensions to data points, where non-linear data can be separated linearly [61]. There are several kernels for SVM model including Polynomial kernel, Radial basis function kernel, sigmoidal kernel yet there is no definitive method for selecting the optimal kernel; rather, this choice is determined by the nature of the data as well as the classification or regression problem at hand. However, RBF is frequently utilized in omics research [62].
3.6. Artificial Neural Network
The biological interactions that exist between neurons in the organic brain inspired the development of artificial neural network algorithm. Artificial neurons, like actual brain neurons, are the fundamental unit of an ANN and follow three different and simple sets of rules: multiplication, summation, and activation [63]. In the first multiplication step, neurons are weighted by multiplying the input value (protein expression level, metabolites, and abundances of micro-organisms in metagenomics case) by individual weights. The following stage of the artificial neuron's model contains a sum function that sums all of the weighted inputs and introduces a bias term (a value that change the result of the activation function toward a negative or positive threshold) and passes the output to an activation function that sum up previously weighted inputs and bias to determine whether or not a specific neuron should be activate. To address complex issues, the activation function simply changes linear input into nonlinear [64]. There are several well-known activation functions for non-linear transformations, including sigmoid activation function, ReLU (Rectified linear unit) activation function, and Tanh (hyperbolic tangent activation function) [64].
Artificial neurons in a neural network are frequently organized in a multi-layer structure, such as a simple feed forward neural network with an input layer, a hidden layer, and an output layer. Data with features are introduced at the input layer of a simple feed forward neural network. This layer contains no mathematical calculation performance. Following that, the input data is transferred to a hidden layer, which is in charge of performing all computations such as adding weights, bias, and activation functions, before the outcome layer determines the class of given samples. Hidden layer can vary according to topology of neural network model and tasks. Neural network models can have one or more hidden layers as well hundreds of hidden layers in the case of deep learning models [65].
3.7. Deep Learning
Deep learning is an area of machine learning that has gained notoriety for its ability to handle challenging real-world problems, such as defeating top human players in the ‘’Go’’ game [66]. While both deep learning and conventional neural network such as feedforward use a neural network structure with multiple layers of neurons to learn patterns, the key distinction between the two is the complexity and large number of hidden layers that deep learning employs [67]. By contrast, feedforward neural networks have fewer hidden layers and are constrained in their ability to build sophisticated representations of the data. Using deep learning techniques, state-of-the-art results have been achieved in a number of different areas, including picture classification, speech recognition, and natural language processing. However, the incomprehensibility of deep learning models—often called "black box" models—restricts their applicability in many morally fraught contexts, such as in clinical settings [68]. Figure 4 shows supervised learning algorithms used for linear and non-linear tasks.
4.0. Validation Strategies and Performance Metrics for Machine Learning Models
4.1. Data for Training and Validation of Models
The accurate assessment of machine learning model performance and generalization is contingent upon the availability of reliable validation data. Therefore, it is necessary to evaluate the performance of a machine learning model on independent data that is not accessible during the model training phase. One frequently employed approach for producing precise validation data involves the implementation of a basic holdout validation technique. This technique entails partitioning the data into two distinct sets: one set for training models and another set for evaluating models. Nonetheless, the utilization of a small number of samples renders this approach unfeasible[69] thus, another validation approach called “K-fold cross-validation” is popular with small amount of data to train and validate ML models [70]. The implementation of K-fold cross-validation requires the division of the dataset into K subsets or folds, with K being a variable determined by the user. The model is trained using the initial K-1 folds and subsequently evaluated on the remaining K-subsets. This procedure is done K times, with each fold representing the validation set exactly once. Then, the performance of the model is determined by averaging all K simulations. K-fold validation has limits with class imbalance data and can lead to over optimism outcomes. Instead, stratified K-fold cross-validation should be employed, which divides samples equally by class or groups and ensures that each fold has a balanced representation of all class labels in training and validation set. However, research study with small data using cross validation demonstrated that K-fold CV can lead to biased results [71].
Nested cross-validation is a variant of cross-validation that employs multiple layers of cross-validation to assess the efficacy of a machine learning model. The nested cross-validation technique involves two layers, where the outer layer is utilized to assess the model's performance by partitioning the data into training and testing sets. On the other hand, the inner layer is employed for the purpose of model selection and hyper parameter tuning. The inner layer of the model is utilized to train on a subset of the training data, while its efficacy is assessed through cross-validation. This facilitates the identification of the most suitable model and hyperparameters, along with an assessment of their ability to generalize. Subsequent to the inner layer's outcomes, the model and hyper parameters are modified for the ensuing outer layer iteration. The assessment of the model's performance is conducted by utilizing the optimal hyper parameters, followed by an evaluation of an independent data set in the outer layer of cross-validation. The exclusion of test data exposure during the hyper parameter tuning phase results in a more precise evaluation of the model's generalization performance. In small sample sizes, nested cross-validation is comparatively more robust against overfitting than k-fold cross-validation. [71].
4.2. Performance Estimation of Machine Learning Models
The evaluation of a model's performance in supervised machine learning involves the application of various metrics. There exist multiple techniques for assessing models in the context of classification and regression tasks. The model's classification performance is evaluated through various metrics such as accuracy, sensitivity, specificity, recall, F1 score, and area under curve (AUC). In regression tasks, it is preferable to utilize R2 (correlation) score, adjusted R2 score, MSE (mean squared error), and RMSE (root mean squared error).
5.0. Application of Machine Learning and Metagenomics in Autoimmune Diseases
5.1. Machine Learning in Inflammatory Bowel Diseases (IBD) Diagnosis
Several research have been conducted to investigate autoimmune disease using metagenomics and machine learning technologies, such as Forbes et al. used 16S rRNA sequencing data to compare the association of gut microbiome association with different autoimmune diseases including ulcerative colitis, Crohn's disease, rheumatoid arthritis, and multiple sclerosis. Gut microbiome from healthy subjects were also examined. Sequencing data of was clustered into OTUs and Naïve bayes classifier was used to classify different taxonomic features. Furthermore, the RF model was used to classify overall OTUs as well as OTUs at genus level and attained a highest AUC in between 93% to 99%. This analysis found significant differences in microbial abundance in different cohorts. Relative abundance of Actinomyces, Clostridium III, Eggerthella, Faecalicoccus, and Streptococcus were found higher all diseases group than healthy individuals [72]. Similarly, to identify IBD-related microbial biomarkers, 16S rRNA sequencing data were amplified into ASVs by using the multi-taxonomic assignment (MTA) method. The fecal microbiome from a geographically distinct cohort of 654 healthy subjects, as well as that from 274 CD and 175 UC patients, underwent taxonomic and functional analysis. The principal component analysis (PCA) and principal coordinate analysis (PCoA) methods were used to estimate metabolic and taxonomic features of IBD patients, respectively. Study revealed that each patient from different geographical regions had different microbial compositions. The acquired taxonomic genus-level features and metabolic features were used to assess the ability of the microbiome to predict clinical subtypes of IBD by using a random forest model. dBacteroides, Bifidobacterium, and Blautia were the most predictive genera with five metabolic phenotypes [73]. In order to build a predictive model that can differentiate IBD patients from healthy subjects, Manandhar et al. analyzed gut microbiomes of 729 IBD patients and 700 non-IBD samples using 16S rRNA metagenomics data and five supervised machine learning based algorithms. To find out the relative abundance, metagenomics data underwent OTU clustering. In order to construct a predictive model to classify IBD patients from non-IBD subjects, ML models were trained with 50 taxonomic markers and RF achieved AUC of 80% on test dataset. The individuals with IBD and those without IBD showed significant differences in the intestinal microbiota. These taxonomic features were either overexpressed or under expressed in IBD and non-IBD samples. For instances, Acinetobacter, Alistipes, Paraprevotella, Phascolarctobacterium, Pseudomonas, and Stenotrophomonas found more abundant in non-IBD and relative abundance of Akkermansia, Bifidobacterium, Blautia, Coprococcus, Dialister, Fusobacterium, Lachnospira, Morganella, Oscillospira and Ruminococcus were observed in IBD. Along with this, researchers also identified 117 taxonomic biomarkers that were significantly differentiated IBD subtypes into CD and UC [74]. Several health determinants, such as diet, lifestyle, and environmental factors, influenced the composition of the gut microbiome at disease onset. To assess the compositional differences of the gut microbiome in the development of IBD, Clooney et al. analyzed 16S rRNA metagenomic sequences of IBD patients (228 UC and 303 CD) and 161 healthy subjects from two different geographical regions. 16S rRNA sequencing data were clustered into OTUs, and 200 taxonomic species underwent machine learning analysis. Analysis revealed that 27 species in UC and 35 species in CD were considerably high compared to healthy controls. Eggerthella lenta, Holdemania filiformis, and Clostridium innocuum were found higher in UC patients, and abundances of E. lenta and Ruminococcus gnavus were found higher in CD patients than in healthy subjects. Gradient boosted trees and hierarchical clustering was applied for machine learning based classification. Results showed that ML models could classify IBD and control samples as well as distinguished active and inactive states of CD and UC with highest AUC of 88% and 91% for IBD vs HC and inactive vs active IBD respectively [75]. Similarly, a work published by Liñares-Blanco et al. analyzed relative microbial abundance at taxonomic genus level to distinguish IBD subtypes. 16 rRNA amplicons were sequenced from ulcerative colitis (UC) and Crohn’s disease (CD) patients. After the initial pre-processing of raw OTU counts, to obtain the most influential features that discriminate IBD subtypes, four feature selection techniques were applied on taxonomic profiles. This study identified that following representative genus including Rhodoplanes, Streptococcus, Xenorhabdus, Janthinobacterium, Propionivibrio/Limnohabitans and representative phyla including, Bacteroidetes, Firmicutes and Proteobacteria with potential to classify IBD subtypes with highest AUC by using random forest algorithm [76].
5.2. Machine Learning in Type 1 Diabetes (T1D) Diagnosis
Researchers examined the association of gut microbiomes with the development of T1D in children. For this purpose, 16S metagenomics data obtained from 7 different polymorphic regions from 31 children diagnosed with T1D and 25 healthy children were analyzed via random forest and l1 l2 regularization-based machine learning. The samples identified 1606 OTUs in the T1D group and 1552 OTUs in control group with significant difference in composition of gut microbiome in both groups. The relative abundances of B. stercoris, B. fragilis, B. intestinalis, B. bifidum, Gammaproteobacteria, Holdemania, and Synergistetes species were higher, while B. vulgatus, Deltaproteobacteria, Parasutterella, Lactobacillus, and Turicibacter species were lower in children with T1D compared to healthy children. Furthermore, diverse microbial flora was found in healthy subjects than T1D subjects. Upon analysis, researchers concluded that machine learning methods along with taxonomic relative abundance analysis identified Bacteroidetes stercoris species and phylum Synergistetes as a significant taxonomic signature of T1D [77]. Similar to this, taxonomic genus level profiles of 124 newborns were acquired from a public dataset for the diagnosis of pediatrics T1D. Using the random forest classification approach, investigators were able to effectively identify 45 genera with the ability to predict T1D status with an area under the ROC Curve of 91.5%. Prevotella, Anaerotruncus, Scherichia, Eubacterium and Odorib were the high abundant taxonomic genera of T1D [78]. In another study, research conducted on the gut microbiomes of a cohort of 33 infants divided into three groups, including four cases of type 1diabetes (T1D), seven seroconverted infants, who were not clinically diagnosed with T1D but positive for at least two autoantibodies and twenty-two non-seroconverted infants.16S RNA sequencing metagenomics profiles clustered into different taxonomic profiles and analyzed. Researchers were capable of identifying 25 biomarkers at the taxonomic species level with the knack of predicting T1D with an AUC of 98.7% by using the random forest classification algorithm. Bacteroides vulgatus and Prevotella copri were found enriched in T1D patients. Subsequently, based on those specific species-level metagenomics biomarkers, RF model was able to predict seroconverted patients with an AUC of 99%. As a result, the authors concluded that the ML model could stratify the T1D cohort accurately and that the acquired metagenomics markers were strongly associated with the onset of T1D [79].
5.3. Machine Learning in Other Autoimmune Diseases Diagnosis
To identify disease related microbial biomarkers, shotgun metagenomics were sequenced from 123 rheumatoid arthritis (RA), 130 liver cirrhosis, 170 type 2 diabetes (T2D) patients and 383 healthy controls. This analysis successfully identified overall 594 taxonomic biomarkers including, 257 biomarkers of rheumatoid arthritis, 220 of liver cirrhosis and 117 of T2D. DOF003_GL0053139 identified as a topmost marker which is belong to the genus Clostridium and phylum Firmicutes relatively found with high abundance in RA then the other phenotypes whereas, 469590.BSCG_05503 was enriched in liver cirrhosis patients. Moreover, research showed that none of the three phenotypes had shared biomarkers. Wu, Honglong, et al. in their experiment used seven different machine learning algorithms and mRMR was used for feature selection. The selected features from mRMR were evaluated by using 7 different ML classifiers and the most predictive algorithm was a logistic regression with a ROC of 94% [80].
Likewise, Bang, Sohyun, et al. investigated 696 samples of intestinal microbiome obtained from 6 different diseases including multiple sclerosis (MS), juvenile idiopathic arthritis (JIA), colorectal cancer (CRC), acquired immune deficiency syndrome (AIDS), myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), and stroke as well as healthy controls. 16S rRNA amplicons were sequenced and clustered into different OTUs. In this analysis author considered two feature selection algorithms, forward feature selection (FS) and backward feature elimination (BE), along with four efficient multi-classifier algorithms, including K nearest neighbor (KNN), support vector machine (SVM), Logit Boost and logistic model tree (LMT). Logit Boost was the most predictive model in terms of accuracy. Predictive models were trained using taxonomic markers and identified 17 genera commonly. This analysis concluded that the identified taxonomic genus markers were capable to distinguished different diseases, which conferred the potential of ML and metagenomics to multi-class classification which aid in the diseases diagnosis and identification of disease related taxonomic markers. For instance, PSBM3 from the family of Erysipelotrichaceae were identified as candidate biomarker that potentially distinguished different diseases [81]. Recent work by Volkova and Ruggles reanalyzed 42 papers connecting gut microbiota with 12 autoimmune diseases, including multiple sclerosis (MS), inflammatory bowel disease (IBD), rheumatoid arthritis (RA), and general autoimmune disease. In this study, 16s RNA sequencing and shotgun metagenomics data were used with the following four machine learning methods: support vector with radial kernel (SVM-RBF), ridge regression, random forest, and XGBoost. RFE was used for feature selection. ML was utilized in order to classify autoimmune diseases in contrast to controls and compare each condition on an individual basis. The overall experiment revealed that both XGBoost and RF out-performed other classifiers [82]. Another research was conducted on the gut microbiomes of 162 patients divided into 3 groups including 62 healthy, 36 mild Graves’ disease and 64 severe Graves’ disease subjects. Metagenome assembled genomes (MAGs), metagenome annotated genes (MAG) with metabolic function, and metabolite profiles underwent taxonomic and functional investigation. Researchers were able to successfully identify a set of overall 32 biomarkers, including 4 microbiological species, 19 MAGs, six related genes, and 3 SNPs, with ability to predict Graves’s diseases status with area under curve score of 88% by using the random forest classification method. Among these 32 biomarkers, five MAGs markers under the family Erysipelotrichaceae and the genera Coprobacillus, Streptococcus and Rothia were found enriched in all Grave disease patients [83]. Vinod K., et al. examined the intestinal flora that is linked with the minimum clinically important improvement (MCII) in RA patients. The author considered multiple linear regression models and deep learning neural networks to identify the most influential microbial signatures associated with MCII. In this experiment shotgun metagenomics sequencing data was examined for disease classification. Neural network was applied to classify RA patients into MCHII positive and MCHII negative and it demonstrated good accuracy of 90% in identifying individuals in predicting which patients might acquire MCII. Along taxonomic investigation they also measured alpha and beta diversity and revealed that MCII patients possessed intestinal microbiome with high alpha diversity. Researchers also efficiently acknowledged Bacteroidaceae as the most representative family, Bacteroidales and Clostridiales as an order and Firmicutes and Bacteroidetes as a phylum [84]. Table 3 presents studies on autoimmune diseases using metagenomics data with supervised machine learning algorithms.
6.0. Challenges and Risks Inherent in Developing ML-Based Diagnosis Models
6.1. Explanation Matters
Artificial intelligence (AI) methods may be useful in deciphering intricate disease patterns, but the reasoning behind the algorithms' decisions is not always obvious, earning them the "black box" label. Deep learning is top of the list in black box models where calculations done in hundreds of hidden layers while training and predicting a certain condition which are beyond human understanding [85], followed by random forest where prediction is determined by hundreds of trees and non-linear SVM models where several dimensions introduced to data for classification. Increasing model complexity reduces model interpretability. Therefore, it is possible that black box models do not fulfill the high standards of accountability, transparency, and dependability required in medical decisions [86]. It has been fiercely argued whether or not AI models are truly unintelligible, with examples of models achieving great accuracy using means that are of little help in predictive analysis.
Explainable Artificial Intelligence (XAI) on the other hand, seeks to train AI systems to explain their predictions and actions in a way that humans can understand. The goal of XAI models is to create AI models capable of explaining their logic and consequences to humans [87]. Given the complexity and sometimes inexplicable outcomes of conventional AI models such as deep learning, XAI models are gaining traction. XAI models would lead to higher acceptance of AI in traditionally skeptical professions such as medicine, finance, and law. Some examples of XAI approaches include feature significance analysis, saliency maps [88], decision trees [89], and simplistic models such as linear and logistic regression [90] are already being widely used to build interpretable models. Nevertheless, the accuracy of XAI models might be restricted when complicated modeling is required rather than simple models based on traditional statistical and mathematical methodologies [91]. Other recently developed tools such as SHAPLY based on game theory [92] and LIME (Local Interpretable Model-agnostic Explanations) [93] are widely used to interpret the decision of complex non-linear ML models.
6.2. Pitfalls to Avoid in Development of Diagnostic Models
6.2.1. Data Collection and Data Representation for Machine Learning Models
The development of machine learning-based diagnostic models often neglects several crucial factors, such as the significance of commencing with data collection. Prior to developing machine learning models, it is recommended to conduct exploratory analysis, which may include assessing the proportion of missing values and evaluating the consistency of the sample. The occurrence commonly referred to as "garbage in, garbage out" transpires when a model is supplied with imprecise or deficient data during its training phase, resulting in a flawed model and erroneous deductions. An additional factor to take into account is that the majority of machine learning models are trained on data encompass particular races or geographic regions, potentially leading to prejudiced outcomes for other demographic groups [94]. To address this issue, it is recommended to collect diverse data.
6.2.2. Imputations of Missing Values
Metagenomics data, similar to other omics data, frequently exhibit missing values due to the intrinsic imprecision of biological processes or diverse experimental factors. The absence of certain values not only diminishes the dependability of statistical evaluations, but also amplifies the probability of detecting false biomarkers. [95]. There are numerous ways available for researchers to address the issue of missing values; nevertheless, incorrect implementation of imputing missing values can possibly lead to overfitting and biased results [96], for example imputation conducted prior to partitioning data into training and testing sets for constructing a machine learning model may result in the leakage of information from the testing set into the training set. The reason for this is that the imputed values are derived from information encompassing all samples within the dataset. This involves the imputation of missing values through the utilization of the mean or median of all samples present in the data. The integration of information from the test sets into the training set can lead to an overly optimistic assessment of the model's performance on unseen data.
6.2.3. Feature Selection and Performance Metrics Selection
As previously stated, the feature selection procedure is crucial in the development of machine learning models because it facilitates understanding of the models' decision-making process while also improving understanding of biological significance. Therefore, it is imperative to execute the task with great care. For example, applying a feature selection method to the complete dataset in order to obtain a subset of features and subsequently constructing a ML model using the chosen features may lead to overfitting. This is due to the likelihood of information leakage from the test set, since the features will be selected based on population estimates for the entire data set [97].
The appropriate utilization of accurate performance metrics for the purpose of characterizing outcomes is another crucial aspect of ML based diagnostic model research that is sometime overlooked. In the context of constructing models for imbalanced data classification, relying solely on the accuracy score of the model is inadequate. It is necessary to consider additional metrics such as Precision, Recall, F1, and area under the curve (AUC) to accurately evaluate the effectiveness of the model for each class in data set. In the context of data analysis involving regression tasks, some researchers exclusively exhibit the R-squared metric of multiple regression models as a means of demonstrating the associations in term of correlation between the independent and dependent variables. Nonetheless, it is important to note that this methodology may produce inaccurate results since the R-squared metric does not provide evidence as to whether the introduction of a new variable significantly improves the predictive ability of the model. Therefore, it is necessary to incorporate the adjusted R-squared alongside the R-squared metric, as it accounts for changes in significance that arise from the introduction of independent variables into the model.
7.0. Conclusion
Machine learning and metagenomics present a potent approach for the diagnosis of autoimmune diseases, especially in terms of early detection and the categorization of cohorts for tailored treatments. Recent research has indicated that the application of machine learning techniques to the analysis of metagenomics data has the potential to effectively diagnose autoimmune disorders with a high degree of accuracy. Nonetheless significant number of research has been conducted on a specific demographic or ethnicity, it remains imperative to gather additional data from diverse sources to authenticate models and the statistical reliability of identified biomarkers. Similarly, machine learning domain presents several issues that necessitate further investigation, particularly with regard to its acceptance in the medical field. These issues include the transparency and interpretability of models, as well as the reproducibility of biological findings. These considerations are of utmost importance in clinical settings and represent the primary limitation of utilizing machine learning algorithms in medical applications..
Author Contributions
Conceptualization, S.K.; methodology, S.K; writing—original draft preparation, S.K and I.Z; writing—review and editing, S.K., I.Z., U.H., M.P., S.B., U.R., and O.H. All authors have read and agreed to the published version of the manuscript.
Funding
None.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Davidson, A.; Diamond, B. Autoimmune diseases. N Engl J Med 2001, 345, 340–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, F. S.; Gershwin, M. E. Human autoimmune diseases: a comprehensive update. J Intern Med 2015, 278, 369–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Sun, L.; Zhang, X. Integration of microbiome and epigenome to decipher the pathogenesis of autoimmune diseases. J Autoimmun 2017, 83, 31–42. [Google Scholar] [CrossRef]
- Hacilar, H.; Nalbantoglu, O. U.; Bakir-Güngör, B. Machine Learning Analysis of Inflammatory Bowel Disease-Associated Metagenomics Dataset. 2018 3rd International Conference on Computer Science and Engineering (UBMK), 2018; pp. 434–438. [Google Scholar]
- Biewenga, M.; Farina Sarasqueta, A.; Tushuizen, M. E.; de Jonge-Muller, E. S. M.; van Hoek, B.; Trouw, L. A. The role of complement activation in autoimmune liver disease. Autoimmun Rev 2020, 19, 102534. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ran, Y.; Zhang, H.; Wang, B.; Zhou, L. The Microbiome in Autoimmune Liver Diseases: Metagenomic and Metabolomic Changes. Frontiers in Physiology 2021, 12. [Google Scholar] [CrossRef]
- Narváez, J. Lupus erythematosus 2020. Medicina Clínica (English Edition) 2020, 155, 494–501. [Google Scholar] [CrossRef]
- Onuora, S. Rheumatoid arthritis: Methotrexate and bridging glucocorticoids in early RA. Nat Rev Rheumatol 2014, 10, 698. [Google Scholar] [CrossRef]
- Psarras, A.; Emery, P.; Vital, E. M. Type I interferon-mediated autoimmune diseases: pathogenesis, diagnosis and targeted therapy. Rheumatology (Oxford) 2017, 56, 1662–1675. [Google Scholar] [CrossRef]
- Ghorbani, F.; Abbaszadeh, H.; Mehdizadeh, A.; Ebrahimi-Warkiani, M.; Rashidi, M. R.; Yousefi, M. Biosensors and nanobiosensors for rapid detection of autoimmune diseases: a review. Mikrochim Acta 2019, 186, 838. [Google Scholar] [CrossRef]
- Solomon, A. J. Diagnosis, Differential Diagnosis, and Misdiagnosis of Multiple Sclerosis. Continuum (Minneap Minn) 2019, 25, 611–635. [Google Scholar] [CrossRef]
- Lazar, S.; Kahlenberg, J. M. Systemic Lupus Erythematosus: New Diagnostic and Therapeutic Approaches. Annual Review of Medicine 2023, 74, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, M.; Sintes, J.; Lányi, Á.; Engel, P. CD84 cell surface signaling molecule: An emerging biomarker and target for cancer and autoimmune disorders. Clin Immunol 2019, 204, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Rönnblom, L.; Leonard, D. Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med 2019, 6, e000270. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, R.; Puxeddu, I.; Pratesi, F.; Migliorini, P. New biomarkers in SLE: from bench to bedside. Rheumatology (Oxford) 2020, 59 (Suppl5), v12–v18. [Google Scholar] [CrossRef] [PubMed]
- Ziemssen, T.; Akgün, K.; Brück, W. Molecular biomarkers in multiple sclerosis. Journal of Neuroinflammation 2019, 16, 272. [Google Scholar] [CrossRef] [PubMed]
- Casimiro-Soriguer, C. S.; Loucera, C.; Peña-Chilet, M.; Dopazo, J. Towards a metagenomics machine learning interpretable model for understanding the transition from adenoma to colorectal cancer. Sci Rep 2022, 12, 450. [Google Scholar] [CrossRef] [PubMed]
- Pei, Q.; Luo, Y.; Chen, Y.; Li, J.; Xie, D.; Ye, T. Artificial intelligence in clinical applications for lung cancer: diagnosis, treatment and prognosis. Clin Chem Lab Med 2022, 60, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Huang, T.-T.; Lai, J.-B.; Du, Y.-L.; Xu, Y.; Ruan, L.-M.; Hu, S.-H. Current Understanding of Gut Microbiota in Mood Disorders: An Update of Human Studies. Frontiers in Genetics 2019, 10. [Google Scholar] [CrossRef]
- [21] Dey, P.; Ray Chaudhuri, S. The opportunistic nature of gut commensal microbiota. Crit Rev Microbiol 2022, 1–25. [Google Scholar]
- Streit, W. R.; Schmitz, R. A. Metagenomics--the key to the uncultured microbes. Curr Opin Microbiol 2004, 7, 492–8. [Google Scholar] [CrossRef] [PubMed]
- Brumfield, K. D.; Huq, A.; Colwell, R. R.; Olds, J. L.; Leddy, M. B. Microbial resolution of whole genome shotgun and 16S amplicon metagenomic sequencing using publicly available NEON data. PLoS One 2020, 15, e0228899. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P. J.; Ley, R. E.; Hamady, M.; Fraser-Liggett, C. M.; Knight, R.; Gordon, J. I. The human microbiome project. Nature 2007, 449, 804–10. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Hyde, E.; Debelius, J. W.; Morton, J. T.; Gonzalez, A.; Ackermann, G.; Aksenov, A. A.; Behsaz, B.; Brennan, C.; Chen, Y.; DeRight Goldasich, L.; Dorrestein, P. C.; Dunn, R. R.; Fahimipour, A. K.; Gaffney, J.; Gilbert, J. A.; Gogul, G.; Green, J. L.; Hugenholtz, P.; Humphrey, G.; Huttenhower, C.; Jackson, M. A.; Janssen, S.; Jeste, D. V.; Jiang, L.; Kelley, S. T.; Knights, D.; Kosciolek, T.; Ladau, J.; Leach, J.; Marotz, C.; Meleshko, D.; Melnik, A. V.; Metcalf, J. L.; Mohimani, H.; Montassier, E.; Navas-Molina, J.; Nguyen, T. T.; Peddada, S.; Pevzner, P.; Pollard, K. S.; Rahnavard, G.; Robbins-Pianka, A.; Sangwan, N.; Shorenstein, J.; Smarr, L.; Song, S. J.; Spector, T.; Swafford, A. D.; Thackray, V. G.; Thompson, L. R.; Tripathi, A.; Vázquez-Baeza, Y.; Vrbanac, A.; Wischmeyer, P.; Wolfe, E.; Zhu, Q.; Knight, R. American Gut: an Open Platform for Citizen Science Microbiome Research. mSystems 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K. S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; Mende, D. R.; Li, J.; Xu, J.; Li, S.; Li, D.; Cao, J.; Wang, B.; Liang, H.; Zheng, H.; Xie, Y.; Tap, J.; Lepage, P.; Bertalan, M.; Batto, J. M.; Hansen, T.; Le Paslier, D.; Linneberg, A.; Nielsen, H. B.; Pelletier, E.; Renault, P.; Sicheritz-Ponten, T.; Turner, K.; Zhu, H.; Yu, C.; Li, S.; Jian, M.; Zhou, Y.; Li, Y.; Zhang, X.; Li, S.; Qin, N.; Yang, H.; Wang, J.; Brunak, S.; Doré, J.; Guarner, F.; Kristiansen, K.; Pedersen, O.; Parkhill, J.; Weissenbach, J.; Bork, P.; Ehrlich, S. D.; Wang, J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Lugli, GA.; Ventura, M. A breath of fresh air in microbiome science: shallow shotgun metagenomics for a reliable disentangling of microbial ecosystems. Microbiome. Res. Rep. 2022, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N. A.; Ziemski, M.; Robeson, M. S., 2nd; Kaehler, B. D. Measuring the microbiome: Best practices for developing and benchmarking microbiomics methods. Comput Struct Biotechnol J 2020, 18, 4048–4062. [Google Scholar] [CrossRef]
- Barberis, E.; Khoso, S.; Sica, A.; Falasca, M.; Gennari, A.; Dondero, F.; Afantitis, A.; Manfredi, M. Precision Medicine Approaches with Metabolomics and Artificial Intelligence. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Kwon, Y. W.; Jo, H.-S.; Bae, S.; Seo, Y.; Song, P.; Song, M.; Yoon, J. H. Application of Proteomics in Cancer: Recent Trends and Approaches for Biomarkers Discovery. Frontiers in Medicine 2021, 8. [Google Scholar] [CrossRef]
- Barberis, E.; Amede, E.; Khoso, S.; Castello, L.; Sainaghi, P. P.; Bellan, M.; Balbo, P. E.; Patti, G.; Brustia, D.; Giordano, M.; Rolla, R.; Chiocchetti, A.; Romani, G.; Manfredi, M.; Vaschetto, R. Metabolomics Diagnosis of COVID-19 from Exhaled Breath Condensate Metabolites [Online], 2021.
- Avanzo, M.; Stancanello, J.; Pirrone, G.; Sartor, G. Radiomics and deep learning in lung cancer. Strahlenther Onkol 2020, 196, 879–887. [Google Scholar] [CrossRef]
- Mreyoud, Y.; Song, M.; Lim, J.; Ahn, T.-H. MegaD: Deep Learning for Rapid and Accurate Disease Status Prediction of Metagenomic Samples Life [Online], 2022.
- Jia, W.; Sun, M.; Lian, J.; Hou, S. Feature dimensionality reduction: a review. Complex & Intelligent Systems 2022, 8, 2663–2693. [Google Scholar]
- Cantini, L.; Zakeri, P.; Hernandez, C.; Naldi, A.; Thieffry, D.; Remy, E.; Baudot, A. Benchmarking joint multi-omics dimensionality reduction approaches for the study of cancer. Nat Commun 2021, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Solorio-Fernández, S.; Carrasco-Ochoa, J. A.; Martínez-Trinidad, J. F. A review of unsupervised feature selection methods. Artif. Intell. Rev. 2020, 53, 907–948. [Google Scholar] [CrossRef]
- Hopf, K.; Reifenrath, S. Filter Methods for Feature Selection in Supervised Machine Learning Applications - Review and Benchmark. arXiv 2021, arXiv:2111.12140. [Google Scholar]
- Rajab, M.; Wang, D. Practical Challenges and Recommendations of Filter Methods for Feature Selection. Journal of Information & Knowledge Management 2020, 19, 2040019. [Google Scholar]
- Wang, L.; Jiang, S.; Jiang, S. A feature selection method via analysis of relevance, redundancy, and interaction. Expert Syst. Appl. 2021, 183 (C), 11. [Google Scholar] [CrossRef]
- Anitha, M. A.; Sherly, K. K. In A Novel Forward Filter Feature Selection Algorithm Based on Maximum Dual Interaction and Maximum Feature Relevance(MDIMFR) for Machine Learning. 2021 International Conference on Advances in Computing and Communications (ICACC), 21-23 Oct. 2021; pp. 1–7. [Google Scholar]
- Urbanowicz, R. J.; Meeker, M.; La Cava, W.; Olson, R. S.; Moore, J. H. Relief-based feature selection: Introduction and review. Journal of Biomedical Informatics 2018, 85, 189–203. [Google Scholar] [CrossRef]
- Yang, J.; Honavar, V. G. Feature Subset Selection Using a Genetic Algorithm. IEEE Intelligent Systems 1998, 13, 44–49. [Google Scholar] [CrossRef]
- Forsati, R.; Moayedikia, A.; Jensen, R.; Shamsfard, M.; Meybodi, M. R. Enriched ant colony optimization and its application in feature selection. Neurocomputing 2014, 142, 354–371. [Google Scholar] [CrossRef]
- Xiong, M.; Fang, X.; Zhao, J. Biomarker identification by feature wrappers. Genome Res 2001, 11, 1878–87. [Google Scholar] [CrossRef]
- Chandrashekar, G.; Sahin, F. A survey on feature selection methods. Computers & Electrical Engineering 2014, 40, 16–28. [Google Scholar]
- Liu, H.; Zhou, M.; Liu, Q. An Embedded Feature Selection Method for Imbalanced Data Classification. IEEE/CAA Journal of Automatica Sinica 2019, 6, 703. [Google Scholar] [CrossRef]
- Okser, S.; Pahikkala, T.; Airola, A.; Salakoski, T.; Ripatti, S.; Aittokallio, T. Regularized machine learning in the genetic prediction of complex traits. PLoS Genet 2014, 10, e1004754. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.-S.; Lee, J.-S.; Moon, B.-R. Hybrid Genetic Algorithms for Feature Selection. IEEE transactions on pattern analysis and machine intelligence 2004, 26, 1424–37. [Google Scholar] [PubMed]
- Cadenas, J.; Garrido, M.; Martínez-España, R. Feature subset selection Filter–Wrapper based on low quality data. Expert Systems with Applications 2013, 40, 6241–6252. [Google Scholar] [CrossRef]
- Ali, S.; Shahzad, W. A FEATURE SUBSET SELECTION METHOD BASED ON CONDITIONAL MUTUAL INFORMATION AND ANT COLONY OPTIMIZATION. International Journal of Computer Applications 2012.
- Stanton, J. M. Galton, Pearson, and the Peas: A Brief History of Linear Regression for Statistics Instructors. Journal of Statistics Education 2001, 9. [Google Scholar] [CrossRef]
- Schneider, A.; Hommel, G.; Blettner, M. Linear regression analysis: part 14 of a series on evaluation of scientific publications. Dtsch Arztebl Int 2010, 107, 776–82. [Google Scholar]
- Yoo, C.; Ramirez, L.; Liuzzi, J. Big Data Analysis Using Modern Statistical and Machine Learning Methods in Medicine. International neurourology journal 2014, 18, 50–7. [Google Scholar] [CrossRef]
- Prabhat, A.; Khullar, V. Sentiment classification on big data using Naïve bayes and logistic regression. 2017 International Conference on Computer Communication and Informatics (ICCCI), 2017; pp. 1–5. [Google Scholar]
- Vijayarani, D. S.; Dhayanand, M. S. In Liver Disease Prediction using SVM and Naïve Bayes Algorithms, 2015.
- Breiman, L. Random Forests. Machine Learning 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Ibrahim, M. Reducing correlation of random forest–based learning-to-rank algorithms using subsample size. Computational Intelligence 2019, 35, 774–798. [Google Scholar] [CrossRef]
- Touw, W. G.; Bayjanov, J. R.; Overmars, L.; Backus, L.; Boekhorst, J.; Wels, M.; van Hijum, S. A. F. T. Data mining in the Life Sciences with Random Forest: a walk in the park or lost in the jungle? Briefings in Bioinformatics 2013, 14, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-vector networks. Machine Learning 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Cristianini, N.; Shawe-Taylor, J. An Introduction to Support Vector Machines and Other Kernel-based Learning Methods; Cambridge University Press: Cambridge, 2000. [Google Scholar]
- Huang, S.; Cai, N.; Pacheco, P. P.; Narrandes, S.; Wang, Y.; Xu, W. Applications of Support Vector Machine (SVM) Learning in Cancer Genomics. Cancer Genomics Proteomics 2018, 15, 41–51. [Google Scholar] [PubMed]
- Debik, J.; Sangermani, M.; Wang, F.; Madssen, T. S.; Giskeødegård, G. F. Multivariate analysis of NMR-based metabolomic data. NMR Biomed 2022, 35, e4638. [Google Scholar] [CrossRef] [PubMed]
- Andrej, K.; Janez, B. t.; Andrej, K. Introduction to the Artificial Neural Networks. In Artificial Neural Networks; Kenji, S., Ed.; IntechOpen: Rijeka, 2011; p. Ch. 1. [Google Scholar]
- Parhi, R.; Nowak, R. The Role of Neural Network Activation Functions. IEEE Signal Processing Letters 2020, 27, 1779–1783. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. In Deep Residual Learning for Image Recognition. 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), 27-30 June 2016; pp. 770–778. [Google Scholar]
- Silver, D.; Huang, A.; Maddison, C. J.; Guez, A.; Sifre, L.; van den Driessche, G.; Schrittwieser, J.; Antonoglou, I.; Panneershelvam, V.; Lanctot, M.; Dieleman, S.; Grewe, D.; Nham, J.; Kalchbrenner, N.; Sutskever, I.; Lillicrap, T.; Leach, M.; Kavukcuoglu, K.; Graepel, T.; Hassabis, D. Mastering the game of Go with deep neural networks and tree search. Nature 2016, 529, 484–489. [Google Scholar] [CrossRef]
- Mohamed, E. A.; Rashed, E. A.; Gaber, T.; Karam, O. Deep learning model for fully automated breast cancer detection system from thermograms. PLoS One 2022, 17, e0262349. [Google Scholar] [CrossRef]
- Yang, G.; Ye, Q.; Xia, J. Unbox the black-box for the medical explainable AI via multi-modal and multi-centre data fusion: A mini-review, two showcases and beyond. Information Fusion 2022, 77, 29–52. [Google Scholar] [CrossRef]
- Eertink, J. J.; Heymans, M. W.; Zwezerijnen, G. J. C.; Zijlstra, J. M.; de Vet, H. C. W.; Boellaard, R. External validation: a simulation study to compare cross-validation versus holdout or external testing to assess the performance of clinical prediction models using PET data from DLBCL patients. EJNMMI Res 2022, 12, 58. [Google Scholar] [CrossRef]
- Raschka, S. Model Evaluation, Model Selection, and Algorithm Selection in Machine Learning. 2018.
- Vabalas, A.; Gowen, E.; Poliakoff, E.; Casson, A. J. Machine learning algorithm validation with a limited sample size. PLoS One 2019, 14, e0224365. [Google Scholar] [CrossRef]
- Forbes, J. D.; Chen, C.-y.; Knox, N. C.; Marrie, R.-A.; El-Gabalawy, H.; de Kievit, T.; Alfa, M.; Bernstein, C. N.; Van Domselaar, G. A comparative study of the gut microbiota in immune-mediated inflammatory diseases—does a common dysbiosis exist? Microbiome 2018, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Iablokov, S. N.; Klimenko, N. S.; Efimova, D. A.; Shashkova, T.; Novichkov, P. S.; Rodionov, D. A.; Tyakht, A. V. Metabolic Phenotypes as Potential Biomarkers for Linking Gut Microbiome With Inflammatory Bowel Diseases. Front Mol Biosci 2020, 7, 603740. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, I.; Alimadadi, A.; Aryal, S.; Munroe, P. B.; Joe, B.; Cheng, X. Gut microbiome-based supervised machine learning for clinical diagnosis of inflammatory bowel diseases. Am J Physiol Gastrointest Liver Physiol 2021, 320, G328–g337. [Google Scholar] [CrossRef] [PubMed]
- Clooney, A. G.; Eckenberger, J.; Laserna-Mendieta, E.; Sexton, K. A.; Bernstein, M. T.; Vagianos, K.; Sargent, M.; Ryan, F. J.; Moran, C.; Sheehan, D.; Sleator, R. D.; Targownik, L. E.; Bernstein, C. N.; Shanahan, F.; Claesson, M. J. Ranking microbiome variance in inflammatory bowel disease: a large longitudinal intercontinental study. Gut 2021, 70, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Liñares-Blanco, J.; Fernandez-Lozano, C.; Seoane, J. A.; López-Campos, G. Machine Learning Based Microbiome Signature to Predict Inflammatory Bowel Disease Subtypes. Frontiers in Microbiology 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Biassoni, R.; Di Marco, E.; Squillario, M.; Barla, A.; Piccolo, G.; Ugolotti, E.; Gatti, C.; Minuto, N.; Patti, G.; Maghnie, M.; d'Annunzio, G. Gut Microbiota in T1DM-Onset Pediatric Patients: Machine-Learning Algorithms to Classify Microorganisms as Disease Linked. J Clin Endocrinol Metab 2020, 105. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Edreira, D.; Liñares-Blanco, J.; Fernandez-Lozano, C. Identification of Prevotella, Anaerotruncus and Eubacterium Genera by Machine Learning Analysis of Metagenomic Profiles for Stratification of Patients Affected by Type I Diabetes. Proceedings 2020, 54, 50. [Google Scholar]
- Fernández-Edreira, D.; Liñares-Blanco, J.; Fernandez-Lozano, C. Machine Learning analysis of the human infant gut microbiome identifies influential species in type 1 diabetes. Expert Systems with Applications 2021, 185, 115648. [Google Scholar] [CrossRef]
- Wu, H.; Cai, L.; Li, D.; Wang, X.; Zhao, S.; Zou, F.; Zhou, K. Metagenomics Biomarkers Selected for Prediction of Three Different Diseases in Chinese Population. Biomed Res Int 2018, 2018, 2936257. [Google Scholar] [CrossRef]
- Bang, S.; Yoo, D.; Kim, S. J.; Jhang, S.; Cho, S.; Kim, H. Establishment and evaluation of prediction model for multiple disease classification based on gut microbial data. Sci Rep 2019, 9, 10189. [Google Scholar] [CrossRef] [PubMed]
- Volkova, A.; Ruggles, K. V. Predictive Metagenomic Analysis of Autoimmune Disease Identifies Robust Autoimmunity and Disease Specific Microbial Signatures. Frontiers in Microbiology 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Hou, Q.; Huang, S.; Ou, Q.; Huo, D.; Vázquez-Baeza, Y.; Cen, C.; Cantu, V.; Estaki, M.; Chang, H.; Belda-Ferre, P.; Kim, H.-C.; Chen, K.; Knight, R.; Zhang, J. Compositional and genetic alterations in Graves’ disease gut microbiome reveal specific diagnostic biomarkers. The ISME Journal 2021, 15, 3399–3411. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V. K.; Cunningham, K. Y.; Hur, B.; Bakshi, U.; Huang, H.; Warrington, K. J.; Taneja, V.; Myasoedova, E.; Davis, J. M.; Sung, J. Gut microbial determinants of clinically important improvement in patients with rheumatoid arthritis. Genome Medicine 2021, 13, 149. [Google Scholar] [CrossRef] [PubMed]
- Tjoa, E.; Guan, C. A Survey on Explainable Artificial Intelligence (XAI): Toward Medical XAI. IEEE Trans Neural Netw Learn Syst 2021, 32, 4793–4813. [Google Scholar] [CrossRef]
- Gilvary, C.; Madhukar, N.; Elkhader, J.; Elemento, O. The Missing Pieces of Artificial Intelligence in Medicine. Trends Pharmacol Sci 2019, 40, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Vilone, G.; Longo, L. Explainable Artificial Intelligence: a Systematic Review. 2020.
- Lu, X.; Tolmachev, A.; Yamamoto, T.; Takeuchi, K.; Okajima, S.; Takebayashi, T.; Maruhashi, K.; Kashima, H. Crowdsourcing Evaluation of Saliency-based XAI Methods. arXiv 2021, arXiv:2107.00456. [Google Scholar]
- Estivill-Castro, V.; Gilmore, E.; Hexel, R. Constructing Explainable Classifiers from the Start—Enabling Human-in-the Loop Machine Learning Information [Online], 2022.
- Guleria, P.; Naga Srinivasu, P.; Ahmed, S.; Almusallam, N.; Alarfaj, F. K. XAI Framework for Cardiovascular Disease Prediction Using Classification Techniques Electronics [Online], 2022.
- Xu, F.; Uszkoreit, H.; Du, Y.; Fan, W.; Zhao, D.; Zhu, J. In Explainable AI: A Brief Survey on History, Research Areas, Approaches and Challenges, Natural Language Processing and Chinese Computing, Cham, 2019//; Tang, J., Kan, M.-Y., Zhao, D., Li, S., Zan, H., Eds.; Springer International Publishing: Cham, 2019; pp. 563–574. [Google Scholar]
- Lundberg, S.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. 2017.
- Ribeiro, M. T.; Singh, S.; Guestrin, C. "Why Should I Trust You?": Explaining the Predictions of Any Classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; Association for Computing Machinery: San Francisco, California, USA, 2016; pp. 1135–1144. [Google Scholar]
- Ntoutsi, E.; Fafalios, P.; Gadiraju, U.; Iosifidis, V.; Nejdl, W.; Vidal, M. E.; Ruggieri, S.; Turini, F.; Papadopoulos, S.; Krasanakis, E.; Kompatsiaris, I.; Kinder-Kurlanda, K.; Wagner, C.; Karimi, F.; Fernandez, M.; Alani, H.; Berendt, B.; Kruegel, T.; Heinze, C.; Staab, S. Bias in data-driven artificial intelligence systems—An introductory survey. WIREs Data Mining and Knowledge Discovery 2020, 10. [Google Scholar] [CrossRef]
- Ou, F. S.; Michiels, S.; Shyr, Y.; Adjei, A. A.; Oberg, A. L. Biomarker Discovery and Validation: Statistical Considerations. J Thorac Oncol 2021, 16, 537–545. [Google Scholar] [CrossRef]
- Harrell, F. E., Jr.; Lee, K. L.; Mark, D. B. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med 1996, 15, 361–87. [Google Scholar] [CrossRef]
- Demircioğlu, A. Measuring the bias of incorrect application of feature selection when using cross-validation in radiomics. Insights Imaging 2021, 12, 172. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Steps involved in development of diagnostic model using machine learning for diseases using high throughput (metagenomics) data.
Figure 1.
Steps involved in development of diagnostic model using machine learning for diseases using high throughput (metagenomics) data.
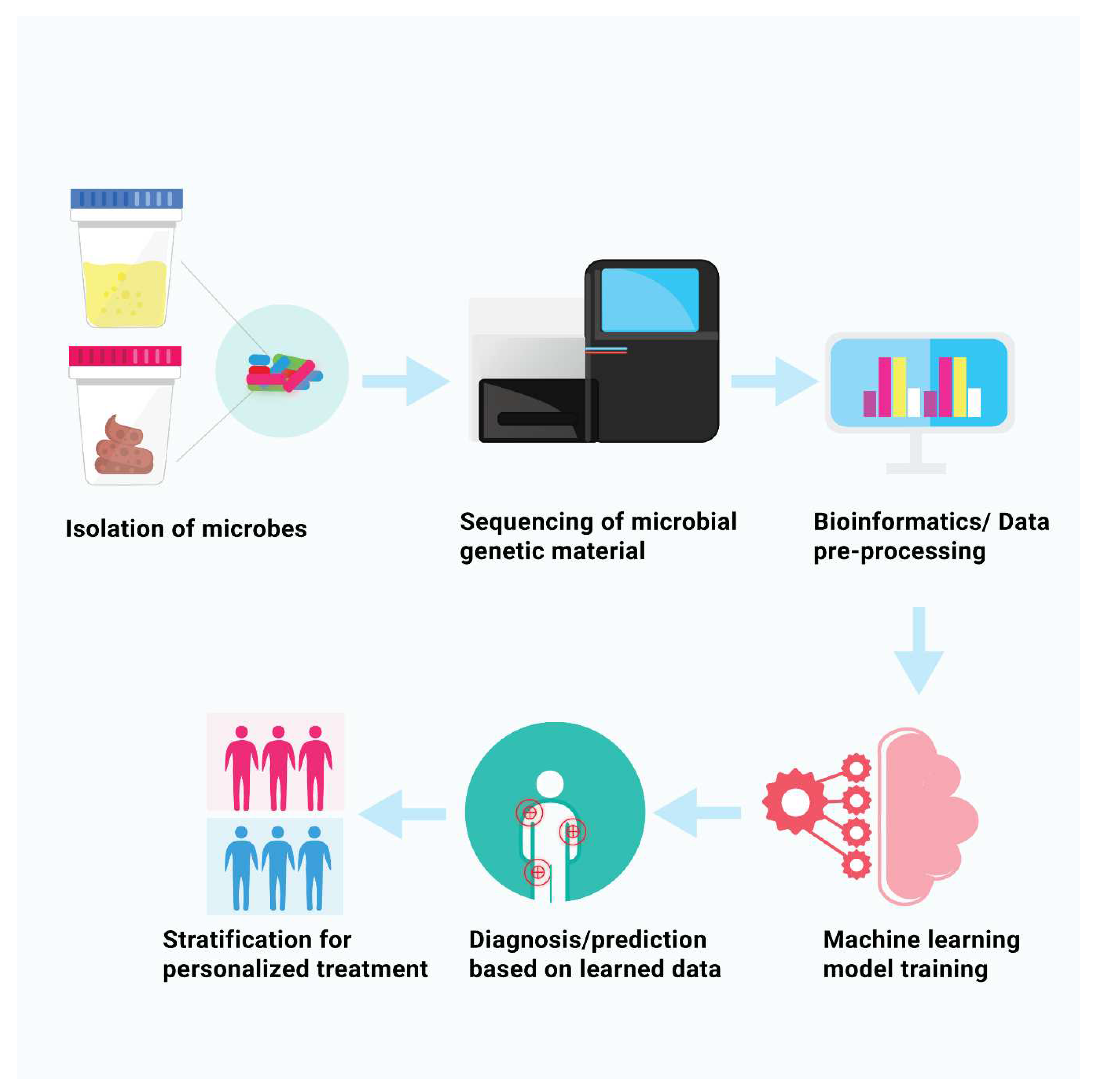
Figure 2.
Isolation and fragmentation of genetic material to produce raw readings is the first stage in the microbial sequencing process, which is used to identify microbes. (A) Amplicon technology begins with the extraction of genetic material and then amplifies DNA. Clean amplicon reads can be generated by first using amplicon sequencing libraries to add barcodes to the ends of raw amplicons, followed by the removal of low-quality amplicons and chimera sequencing. Then, depending on sequence similarity or dissimilarity, the sequenced reads are projected to Operational Taxonomic Units (OTUs) or the Amplicon sequence variant (ASV) technique, and the obtained OTUs are given a taxonomic profile by comparing them to reference databases. (B) To prepare the sequencing library in shotgun metagenomics, DNA is first extracted, then fragmented. Quality inspection of the raw reads is the first stage since clean data is essential for Shotgun sequencing analysis. In the cleaning stage, low-quality reads, host DNA sequences, primers, and adapter contaminations are removed from the raw sequencing data. After this, shotgun analysis can be performed with reference-based or assembly-based techniques. Clean readings are mapped using the reference-based technique to curated databases of genomic sequencing to get Taxonomic classification. In contrast, the assembly-based approach groups comparable sequences into contigs, which are then clustered into OTUs or contig bins (binning). The following phase, refinement, entails the discovery of possible microbial genes and the elimination of redundant genes. (C) Table data with taxonomic profile can be projected to machine learning for building diagnostic model and biomarker selection where data split into training and test set and model builds on using training data and evaluated on test data.
Figure 2.
Isolation and fragmentation of genetic material to produce raw readings is the first stage in the microbial sequencing process, which is used to identify microbes. (A) Amplicon technology begins with the extraction of genetic material and then amplifies DNA. Clean amplicon reads can be generated by first using amplicon sequencing libraries to add barcodes to the ends of raw amplicons, followed by the removal of low-quality amplicons and chimera sequencing. Then, depending on sequence similarity or dissimilarity, the sequenced reads are projected to Operational Taxonomic Units (OTUs) or the Amplicon sequence variant (ASV) technique, and the obtained OTUs are given a taxonomic profile by comparing them to reference databases. (B) To prepare the sequencing library in shotgun metagenomics, DNA is first extracted, then fragmented. Quality inspection of the raw reads is the first stage since clean data is essential for Shotgun sequencing analysis. In the cleaning stage, low-quality reads, host DNA sequences, primers, and adapter contaminations are removed from the raw sequencing data. After this, shotgun analysis can be performed with reference-based or assembly-based techniques. Clean readings are mapped using the reference-based technique to curated databases of genomic sequencing to get Taxonomic classification. In contrast, the assembly-based approach groups comparable sequences into contigs, which are then clustered into OTUs or contig bins (binning). The following phase, refinement, entails the discovery of possible microbial genes and the elimination of redundant genes. (C) Table data with taxonomic profile can be projected to machine learning for building diagnostic model and biomarker selection where data split into training and test set and model builds on using training data and evaluated on test data.
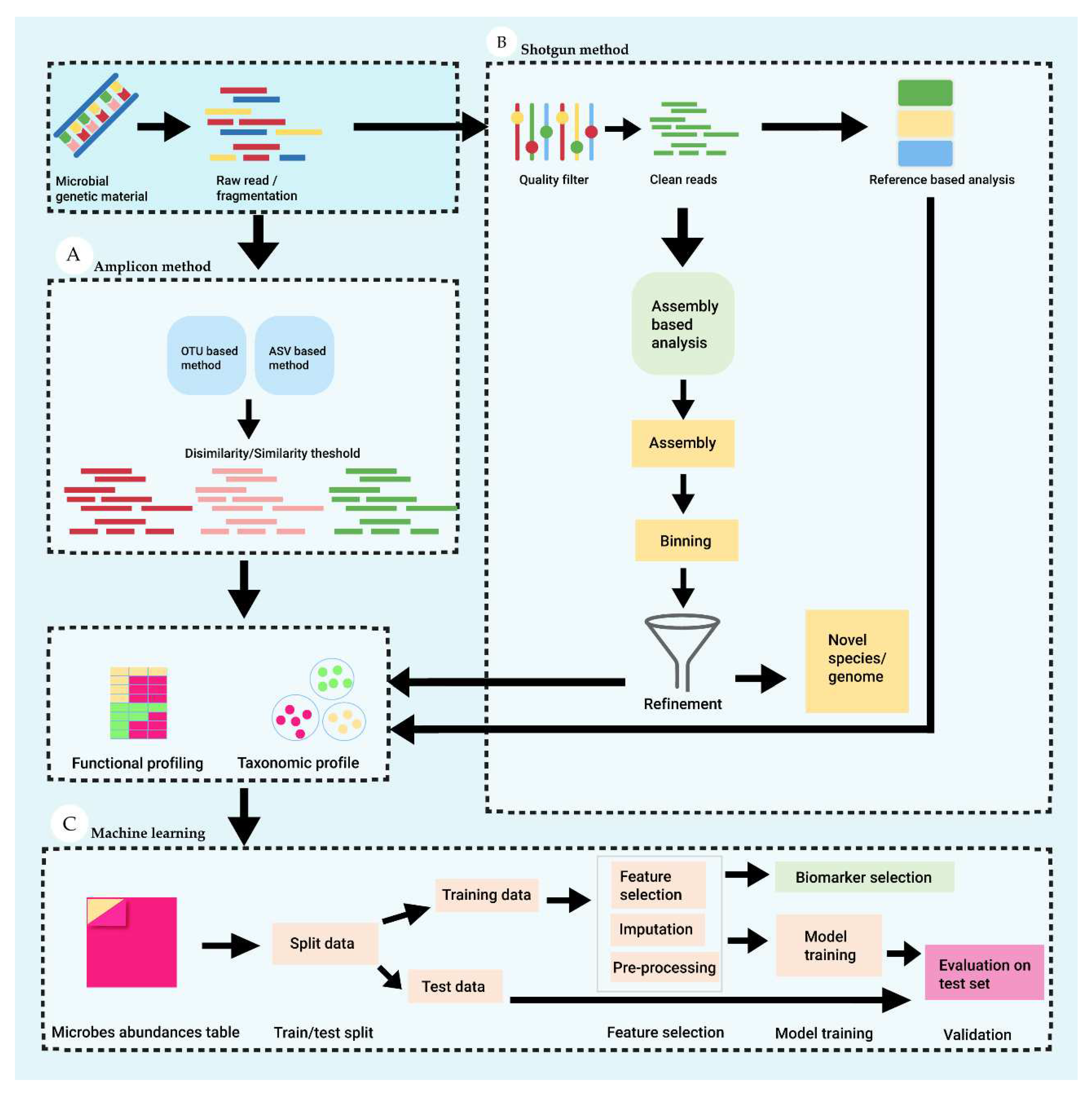
Figure 3.
Types of supervised learning-based dimension reduction techniques.
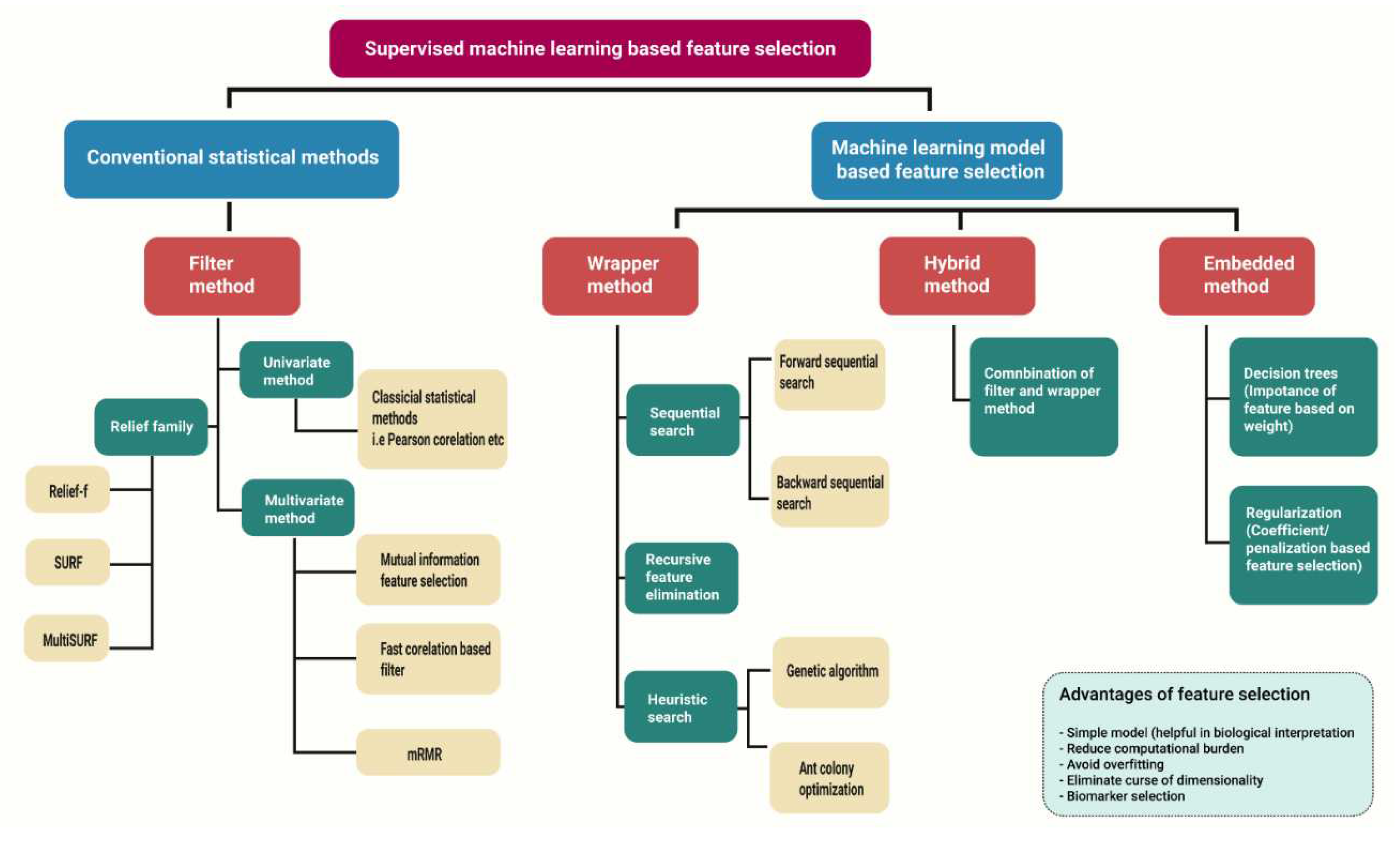
Figure 4.
Supervised machine learning algorithms for linear and non-linear task.
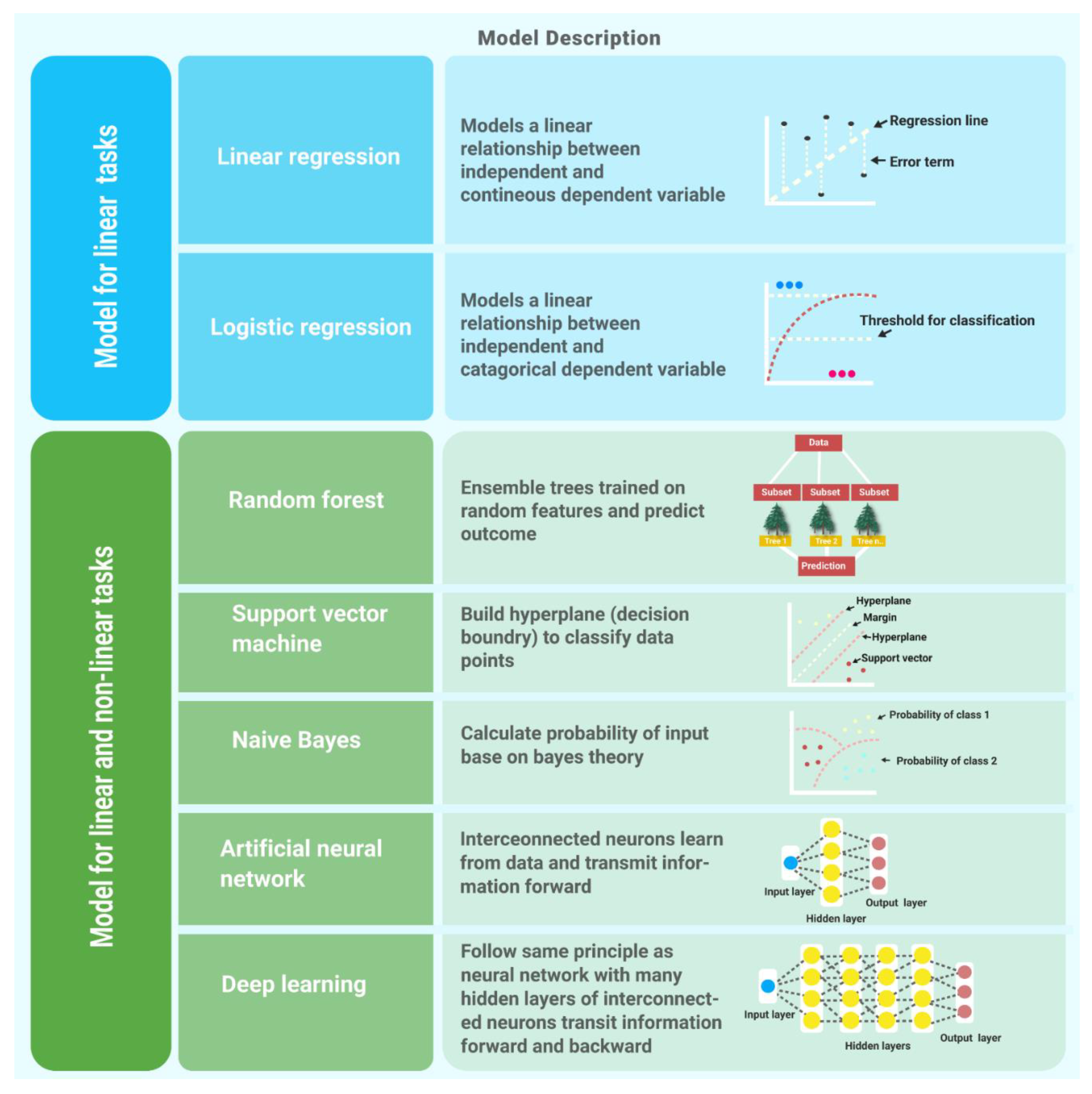

Table 1.
Advantages and disadvantages of amplicon and shotgun microbial sequencing methods.
| Methods | Advantages | Disadvantages |
|---|---|---|
| Amplicon sequencings | Great depth | Uneven amplification |
| More precise | Focus on specific fragment of genes (rRNA/ITS sequencing) | |
| Sequence a specific region (16s-, or 18s- rRNA) | Low enough resolution for species/subspecies identification-OTUs instead | |
| ITS and entire operon introduce more info | Not assess microbe’s function directly | |
| Shotgun sequencing | Theoretically sequence 100% of specimen | Rarely sequence everything of specimen |
| Greater resolution to genetic content | Sequence host/contaminant DNA | |
| Assess functional profiling | Produce very complex dataset | |
| Identify novel organisms/genes/genomic features | Costly | |
| Sequence host/contaminant DNA |
Table 2.
Dimension reduction techniques with advantages and disadvantages along with subtypes.
| Feature Selection Methods |
Subtype | Advantage | Disadvantage | Examples |
|---|---|---|---|---|
| Filter method | Univariate | Computational inexpensive High efficacy Scalable Independent of any classifier |
Multi-collinearity Lack of interaction of feature with classifier |
Fisher’s exact test |
| χ2 test, | ||||
| Information gain | ||||
| Euclidean distance | ||||
| Mann-Whitney U test | ||||
| Multivariate | Feature dependencies Independent of any classifier High efficacy |
Computational expensive in comparison with univariate Lack of interaction of features with classifier |
Minimal-redundancy-maximal-relevance (mRMR) | |
| Fast correlation-based filter (FCBF) | ||||
| Mutual information feature selection (MIFS) | ||||
| Conditional mutual information maximization (CMIM) | ||||
| Relief based family |
Can handle non-linear relationships. Computational efficient Interaction between variables |
Sensitivity to the choice of distance metric Sensitive to parameter tuning. Limited to nearest neighbors |
Relief-f | |
| MultiSURF | ||||
| SURF | ||||
| Wrapper method | Heuristically search approach. | Less prone to local optima | High risk of overfitting Computational expensive |
Genetic algorithm |
| Ant colony optimization | ||||
| Sequential search method | High performance in contrast to filter method Feature dependencies Feature interaction with classifiers |
High risk of overfitting Computational expensive More likely to stuck at local optima |
Sequential forward selection (SFS) | |
| Sequential backward selection (SBS) | ||||
| Recursive feature elimination | More robust against stuck in local optima than sequential methods. Can handle noisy data. Can remove multiple features with less accuracy |
Sensitive to hyperparameters Limited interpretability |
Recursive feature elimination with random forest | |
| Recursive feature elimination with SVM | ||||
| Recursive feature elimination with logistic regression | ||||
| Embedded method | Penalization/shrinkage-based feature selection. | More robust against stuck in local optima than sequential methods. Can handle noisy data. Can remove multiple features with less accuracy. More effective than wrapper method in handling noisy data and handling inter-correlation between variables. Automatic feature selection |
Sensitive to hyperparameters Limited interpretability Difficult to select optimal regularization strength or penalization type. Not robust again non-linear relationship between variable and class |
Lasso |
| Elastic net | ||||
| Weight-based feature selection | Robust against handling complex relationship between dependent and independent variable More interpretable than penalization-based models |
Sensitive to hyper-parameters Sensitive to non-linear relationship |
Decision tree | |
| Random forest | ||||
| Naïve Bayes | ||||
| Hybrid method | Filter and wrapper combination | Efficient in accuracy than filter method Less complexity in comparison to wrapper Robust for high dimensional data |
Classifier dependent Inherit wrapper techniques complexities (overfitting) |
Hybrid genetic algorithm |
| Hybrid ant colony | ||||
| Fuzz random forest |
Table 3.
Study of autoimmune diseases using metagenomics and supervised machine learning approaches.
Table 3.
Study of autoimmune diseases using metagenomics and supervised machine learning approaches.
| Disease | Input | Aim of the Study | Feature Selection Algorithm | Classifier | References |
|---|---|---|---|---|---|
| Different autoimmune diseases such as ulcerative colitis, Crohn's disease, rheumatoid arthritis and multiple sclerosis. | 16s RNA sequencing | To compare the gut microbiome associated with different autoimmune diseases such as ulcerative colitis, Crohn's disease, rheumatoid arthritis and multiple sclerosis. | Feature importance determined by Gini score using random forest | Random Forest | [72] |
| IBD | 16s RNA sequencing | To identify IBD-related microbial biomarkers | Principal component analysis (PCA) and principal coordinate analysis (PCoA) | Random Forest | [73] |
| IBD & Subtypes | 16s RNA sequencing | To discriminate IBD from non-IBD and distinguish IBD subtypes as well | Linear discriminant analysis | Random Forest, SVM radial kernel, NNET, EN | [74] |
| IBD | 16S rRNA sequencing | Assess the compositional differences of the gut microbiome associated with lifestyle, diet and environmental factors in the development of IBD | Feature importance determined by Gradient boosted trees t | Gradient boosted trees | [75] |
| Inflammatory bowel diseases subtypes | 16S rRNA sequencing | To distinguish IBD subtypes: ulcerative colitis (UC) from Crohn’s disease (CD). | Kruskal-Wallis Tests, Fast Correlation Based Filter for Feature Selection (FCBF), Linear decomposition model, Differential abundance | Random Forest, Generalized linear model (glmnet) | [76] |
| T1D | 16S rRNA sequencing | To determine the association of gut microbes in T1D onset in children | Embedded feature selection with random forest | Random forest | [77] |
| T1D | 16S rRNA sequencing | Identified a potential genus for T1D Classification | - | Glmnet, Random Forest | [78] |
| T1D | 16s RNA sequencing | Categorise risk factors associated with the development of type 1 diabetes in infants. | Wilcoxon test | SVM, Random Forest, Glmnet | [79] |
| rheumatoid arthritis, liver cirrhosis and type 2 diabetes | Shotgun metagenomics | To identify biomarkers for three different diseases by multiclass classification | mRMR. | Random forest, SVM, logistic regression, KNN, gradient boosting decision tree (GBDT), AdaBoost, and stochastic gradient descent (SGD) were employed in this study. | [80] |
| Colorectal cancer (CRC), Multiple sclerosis (MS), Juvenile idiopathic arthritis (JIA) | 16s RNA sequencing | To distinguish gut microbiome from six different diseases using multiclass classification | Forward feature selection, Backward feature elimination | KNN, SVM, Logistic model tree, Logiboost | [81] |
| Inflammatory bowel disease, Multiple sclerosis, Rheumatoid arthritis, and general autoimmune diseases. | Shotgun sequence and 16s rRNA sequence | Compare and classify each autoimmune diseases by using machine learning algorithms | RFE used for feature selection in the study | XGBoost, Random Forest, SVM Radial, Ridge regression were employed for classification | [82] |
| Grave's disease | Shotgun metagenomic sequence | To investigate the relation between gut microbiota and Grave diseases with four layers data derived from shotgun metagenomics. | Wilcoxon rank-sum test, two-tailed | Random Forest | [83] |
| Rheumatoid arthritis | Shotgun metagenomic sequencing | To examine the gut microbiomes that are linked with the minimum clinically important improvement (MCII) in RA patients. | Multiple regression model | Deep neural network, Random Forest, SVM & Logistic regression | [84] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



