Submitted:
31 March 2023
Posted:
04 April 2023
Read the latest preprint version here
Abstract
The ability of 2D hybrid structures formed by boron, nitrogen and carbon atoms (h-BNCs) as potential substrates for surface enhanced Raman spectroscopy (SERS) detection of dioxin-like pollutants is theoretically analyzed. The strong confinement and high tunability of the electromagnetic response of the carbon nanostructures embedded within the h-BNC sheets point out that these hybrid structures could be promising for applications in optical spectroscopies, such as SERS. In this work, two model dioxin-like pollutants, TCDD and TCDF, and a model h-BNC surface composed by a carbon nanodisk of ninety-six atoms surrounded by a string of borazine rings, BNC96, are employed for the simulation of the adsorption complexes and the static and pre-resonance Raman spectra of the adsorbed molecules. A large affinity of BNC96 by these pollutants is reflected on the large interaction energies obtained for the most stable stacking complexes, with dispersion being the most important contribution to their stability. The large vibrational coupling of some active modes of TCDF and, specially, of TCDD makes the Raman static spectra display a ‘pure’ chemical enhancement of one order of magnitude. On the other hand, due to the strong electromagnetic response of BNC96, confined within the carbon nanodisk, pre-resonance Raman spectra obtained for TCDD and TCDF display large enhancement factors of 108 and 107, respectively. Promisingly, laser excitation wavelengths frequently used in SERS experiments also provoke significant Raman enhancements, around 104, for the TCDD and TCDF signals. Both the strong confinement of the electromagnetic response within the carbon domains and the high modulation of the resonance wavelengths within the visible and/or UV ranges in h-BNCs should lead to a higher sensitivity than graphene and white graphene parent structures, solving one of the main disadvantages of using 2D substrates for SERS applications.
Keywords:
Dioxins
; POPs
; h-BNC
; graphene
; white graphene
; SERS
; Raman
1. Introduction
Dioxins and dibenzofurans are persistent organic pollutants (POPs), the more toxic forms correspond to their tetrachlorinated derivatives, 2,3,7,8-tetrachloro-p-dibenzodioxin (TCDD) and 2,3,7,8-tetrachlorodibenzofuran (TCDF) [1,2]. These chemicals are sources of carcinogenic and mutagenic effects, producing serious damages to organs and tissues [3-7]. The risk posed by these substances to humans is twofold. On one hand, they may be taken by inhalation as they are expelled in gaseous form in combustion processes [8], both industrial and natural, or by reactions with anthracene in the incineration of waste or in the production of iron and steel [9]. On the other hand, they may enter the organism by the intake of animals or plants where they may be present in low concentration due to bioaccumulation [10-12]. Other examples of dioxins and dibenzofurans sources are the chlorine bleaching processes in the pulp and paper industry [13,14], sewage sludge and gardening compounds. Thus, many different processes can lead to the formation of dioxins, so that they are found throughout the world and pose a serious risk to health and environment.
Detection of dioxins and dibenzofurans is therefore a priority to prevent environment contamination and protect living organisms from their damages. The classical methods for detection of dioxin-like compounds are mainly classified as biological or chemical techniques [15]. Biological methods are based on in vivo or in vitro bioassays and protein binding assays [16-20], whereas chemical methods are based on gas chromatography and mass spectrometry [21-25]. More recently, different kind of chemical sensors [26-28] and biosensors [29-32] have been successfully developed to detect dioxin-like pollutants. Sensor detection offers a series of advantages over biological assays and chemical analysis, such as the possibility for on-site application and lower costs. Among all the sensors developed and tested for the detection of dioxin-like pollutants, those based on surface enhanced Raman scattering (SERS) deserve special attention [28,33-44]. SERS is a very sensitive spectroscopic technique based on the Raman scattering enhancement of molecules adsorbed on surfaces, which act as enhancer platforms by means of two different resonant mechanisms, electromagnetic and chemical [45].
Among the multiple applications of SERS based detection, those related to disease diagnosis [46-49] and environmental analysis [37,44,50-54] are particularly interesting. For the latter, very important advances have been made in the last decade. Therefore, SERS technique has been pointed out as one of the most promising alternatives for POPs detection [28,33-44], mainly due to the very low detection limits obtained [45], limits unreachable with traditional detection procedures based on bioassays or chromatography techniques. Another advantage of spectroscopic techniques in general and SERS in particular is the lower costs compared to bioassays and gas chromatography [15]; costs that have been reduced gradually by improving the fabrication techniques of SERS substrates or replacing the traditional gold and silver nanoparticles by less expensive nanostructured materials [55-59].
Particularly interesting for SERS applications are the 2D materials [59-64]. They may provide some advantages over metal-based 3D materials, such as: higher inertness, due to the non-covalent nature exhibited, in general, by their interactions with the analyte; lower production costs, due to the reduction of the material amount employed; and a reduced signal-to-noise ratio, due to their smoother surfaces. Graphene and white graphene (h-BN) are good examples of 2D SERS substrates that may operate both independently or incorporated to hybrid structures [60,62,65-72]. In the case of hybrid structures, they exert a twofold effect: on one hand, wrapping metal nanoparticles with few-layer graphene or h-BN protect them from oxidation and other chemical degradation processes; on the other hand, they increase the Raman enhancement factors with respect to the metal nanoparticles alone, which come with a higher sensitivity. Raman enhancements of adsorbed molecules on pristine graphene and h-BN were also reported. In the particular case of graphene, it has given rise to a specific branch of SERS, known as graphene enhanced Raman scattering (GERS) [67]. The problem is, however, the magnitude of the enhancements reached are significantly lower than those obtained by metallic plasmonic materials. Thus, the possible advantages of using graphene or h-BN as SERS platforms are overcome by the extremely high sensitivity displayed by noble metal nanoparticles, which allows reaching detection limits down to single-molecule level [73].
The relatively small Raman enhancement factors obtained with graphene compared to metal nanoparticles or hybrid substrates stem from the lack of plasmonic activity within the visible and UV regions. It is well-known that plasmon emission in graphene occurs at terahertz frequencies. Thus, the mechanism responsible of the Raman enhancement in GERS has been associated with charge transfer excitations between the surface and the adsorbed molecule [67], in combination with vibrational coupling between molecule and surface atoms as suggested by theoretical simulations [74-76]. A strong optical response of the 2D substrate in the visible or UV regions, where the laser sources in SERS experiments operate, would be desirable in order to increase the Raman enhancement factors of the adsorbed molecules. This is the case of other finite carbon structures related to graphene, such as graphene nanoribbons, nanodisks or capped nanotubes. Theoretical simulations have shown that, at near resonance conditions, electronic excitation modes of a graphene nanodisk can hugely enhance the Raman activity of molecules adsorbed on its surface [75]. In order to exploit the optical activity at the visible or UV range of these graphene related structures for SERS detection, they must be immobilized within another substrate, either by adsorption or by integration within another 2D material. An example of the latter is the synthesis of hybrid h-BN/graphene structures (h-BNCs), where carbon domains of different sizes and morphologies are embedded within a h-BN sheet [77]. Recent experimental and theoretical works on h-BNCs have focused, respectively, on the development of reliable techniques for a controllable synthesis and the study of their optical response properties [78-80]. Thus, previous studies have shown the high tunability of the band gap in h-BNC structures, which is directly related to the localization/isolation of -electron systems within the carbon domains. This localization gives rise to a strong confinement of their optical response, which, for a given carbon domain, only requires two surrounding BN strings [81]. Thus, the optical response can be widely modulated by changing the size and the shape of these carbon domains [82-85].
In this work, the suitability of h-BNCs for SERS detection of dioxin-like pollutants has been investigated using quantum chemical simulations. A high affinity of these molecules by graphene and h-BN sheets, a required condition for SERS substrates, was previously found both in gas phase and water solution [86]. Their affinity by a hybrid h-BNC surface formed by a graphene nanodisk of ninety-six carbon atoms embedded within a h-BN sheet has been also investigated in this work. Subsequently, Raman spectra of TCDD and TCDF attached to this h-BNC model have been simulated. The whole system has been subjected to electromagnetic radiations of wavelengths coincident with laser sources frequently employed in SERS experiments. Herein, it is shown that suitable laser sources approaching the resonance wavelength of the optically active electronic excitation modes of carbon domains in h-BNCs induce Raman enhancement factors much larger than those previously reported for graphene and h-BN sheets.
2. Materials and Methods
2.1 Interaction Energy Decomposition Analysis
The total interaction energy between the molecule (M) and the model surface (S) can be obtained using the supermolecule approach,
The interaction energy of eqn (1) can be further partitioned into components with different physical origin, as shown in eqn (2),
It must be noticed that the dispersion energy of eqn (2) includes, at Kohn-Sham Density Functional Theory (KS-DFT) level, the part implicitly accounted for by the DFT functional, obtained from the interaction energy decomposition scheme, and the empirical dispersion correction added (see the next section for details about the functional employed in this work).
2.2. Simulation of Raman Spectra
The Raman intensity may be represented by the differential Raman scattering cross section. Within the harmonic approximation, the differential Raman scattering for a given vibrational mode k is expressed as,
where and are isotropic and anisotropic invariants of the Raman tensor, . The Raman tensor indicates how the electric polarizability of a molecular system, , oscillates along with a given vibrational mode. It is obtained from the derivative of the electric polarizability tensor, , with respect to the coordinate of the vibrational mode. Since vibrational modes are constructed by combination of atomic displacements, the Raman tensor may be calculated using eqn (5) [91],
The form of eqn (5) allows for the splitting of the Raman tensor into molecule and surface contributions in SERS simulations [74,75]. Thus, the first summation of eqn (5) can be split into two summations: one running over the atoms belonging to the molecule and another one over those belonging to the surface.
Using this fragmentation of the Raman tensor, the Raman activity of gets also partitioned into a sum of molecule (M), surface (S) and intermolecular (MS) contributions, the latter arising from crossed products of the components and in eqn (6).
The reader may find further details about derivation of eqn (7) elsewhere [75].
Eqn (7) provides a way to analyze the effect of molecule-surface vibrational coupling on the Raman scattering enhancement of adsorbed molecules. This has been an overlooked effect in the SERS literature, but nevertheless it was shown to play a very important role in carbon based substrates such as graphene [74-76], although residual in metals such as silver [92]. Thus, the Raman activity for a given molecular mode in the molecule-surface complex can be decomposed into the ‘pure’ molecular contribution and the surface terms using eqn (7). This allows representing hypothetical spectra for the molecule and the surface separately using their corresponding contributions to the Raman activities. The superposition of these hypothetical spectra gives rise to the Raman spectrum.
2.3. Real Space Representation of Electric Polarizabilities
Real-space visualization of the linear optical response of the surfaces allows characterizing the regions where this response is larger. Localization of a strong optical response within the carbon nanoislands of h-BNCs is well-known and was recently exploited to simulate the Raman enhancement of a single water molecule adsorbed on a pyrene nanoisland embedded in a h-BN sheet [81]. However, the magnitude of the Raman enhancement depends on a physical property in particular, the frequency-dependent polarizability, , which is expected to increase drastically when the system is subjected to an electromagnetic perturbation that matches (resonance) or is close to (pre-resonance) an electronic excitation. In this case, the excitation may involve the electrons of the molecule, those of the surface or a charge-transfer process between both. Therefore, the real-space representation of would be a valuable tool here, providing information about the atoms or bonding regions where the Raman enhancement is expected to be larger.
Herein, intrinsic polarizabilities, first introduced by Van Alsenoy et al [93], were employed to represent in real-space and to understand the Raman enhancement in the different complexes and at different excitation wavelengths. The intrinsic polarizabilities avoid the well-known problem of the origin dependence of distributed polarizabilities, which hinders any partitioning of the total polarizability into atomic polarizabilities or its real-space representation. Thus, Otero et al implemented a general scheme to obtain maps of intrinsic polarizability density distributions by means of a sum over a set of atoms [94]:
In order to compare the changes provoked by two interacting systems (the model surface and the molecules considered in this work), one can define an interacting intrinsic polarizability () only replacing the electron density, , by a BSSE-corrected deformation density [76],
2.4. Computational Details
Geometry optimizations and vibrational analysis of the molecule-surface complexes, were carried out using Gaussian16 [96]. Optimized geometries and ground state energies and electron densities for the EDA were obtained at the DFT level with the M06-2X functional in combination with the 6-311G(d,p) basis set. This is an appropriate functional for the study of aromatic systems such as those considered in this work. The EDA analysis was performed with the EDA-NCI program [97].
Time-dependent density functional theory (TDDFT), as implemented in Gaussian16, was employed to characterize the electronic transitions with larger oscillator strengths in the complexes. Afterwards, using an own Fortran utility, the transition density (TD) and the ground to excited state density difference (DD) were generated for each optically active mode from the excitation and deexcitation coefficients obtained after the solution of the Casida’s equation. Plots of TDs and DDs were drawn with Gaussview 6.0 [98]. The excitation wavelengths of these transitions were then selected to calculate frequency-dependent polarizabilities at pre-resonance conditions using the coupled perturbed Kohn-Sham theory (CPDFT), and the corresponding Raman tensors for the vibrational modes of the adsorbed molecules. This information was transferred to another Fortran utility written to compute the total Raman activities and the molecule and surface components according to eqns (6) and (7). Besides simulations at pre-resonance conditions, excitation wavelengths of laser sources usually employed in SERS experiments and close to the electronic transition wavelengths (488 and 532 nm) were also considered. Owing to the large computational cost of CPDFT calculations, the basis set employed in this case was reduced to a hybrid basis including 6-31G(d,p) for the molecule and the internal C96 structure and 3-21G for the external boron, nitrogen and hydrogen atoms in the h-BNC surface model. The geometries of the complexes were reoptimized with the new basis set in order to perform the vibrational analysis at energy minima. In order to check the reduction of the basis set does not affect significantly the electronic spectra of the complexes and, therefore, the rest of the analysis, TDDFT excitation energies and oscillator strengths were compared with those obtained with the 6-311G(d,p) basis set.
Raman spectra were represented with the Gabedit [99] software using Lorentzian lineshapes with a half-height width of 5 cm-1. To simplify the analysis, the Raman spectra represented in this work for the complexes include only the signals that correspond to vibrations within the TCDD and TCDF molecules. In order to identify these vibrations, the atomic weights for the different vibrations were calculated and only those vibrations where the weight of the molecule was larger than 50% were considered. Afterwards, the vibration modes that give rise to the most intense peaks in the Raman spectra of the complexes were matched with those of the isolated molecules by comparing their symmetry features and atomic displacements.
Intrinsic polarizabilities were calculated using CPDFT and the POLACUBE program [94]. The real-space representation of these polarizabilities were done with the Chemcraft visualization software [100]. Fragment polarizabilities were also calculated to compare the polarizability of the isolated molecules with that of the adsorbed molecules. In order to avoid the coordinates’ origin dependence and the basis set dependence of fragment polarizabilities, the calculations were done with the geometry, coordinates’ origin and basis set of the complex even for the isolated molecules, the latter with the help of the counterpoise method [101] (more details about the calculation of fragment polarizabilities may be found elsewhere [74]).
3. Results
3.1 Energetic Stability of the Complexes
The complexes analyzed are depicted in Figure 1. They correspond to the stable conformations of TCDD and TCDF molecules adsorbed on a h-BNC model surface composed by a carbon nanodisk of ninety-six carbon atoms (C96) surrounded by a string of borazine rings. This model of h-BNC will be denoted as BNC96 and present a lower symmetry (D3h) than the C96 carbon nanodisk (D6h). In previous studies, where the interaction energies of aromatic molecules with carbon nanodisks of increasing size were compared [88,102], the C96 carbon structure was shown to provide interaction energies close to those found between an infinite graphene sheet and molecules of similar size to TCDD and TCDF. It must be also noted that the BNC96 structure was fully relaxed during the calculations of the adsorption complexes, giving rise to a significant bending of its structure.
The interaction energy and its different components are shown in Table 1 for the complexes shown in Figure 1. For the sake of comparison, the same data obtained previously for complexes formed by TCDD and TCDF adsorbed on a “pure” C96 carbon nanodisk are included in Table 1 [86]. As it can be observed from these data, the external borazine rings have a little influence on the interaction of the pollutants with the internal carbon nanodisk. Thus, neither the total interaction energy nor its components display significant differences between C96 and BNC96. The most important contribution to the formation of these complexes is by far the dispersion energy. Accordingly, the pollutants and the surfaces adopt stacking conformations with a molecule–surface distance inversely proportional to the magnitude of the interaction (3.2 and 3.3 Å for TCDD and TCDF, respectively). Moreover, the slightly lower interaction energy of the complexes formed with BNC96 with respect to C96 might be related to the bent structure adopted by the former, providing a more favourable balance between first order energies (electrostatic and Pauli) since the polarization energy (induction + dispersion) is practically the same in both surfaces.
3.2 Static Raman Spectra
Raman spectra obtained at static conditions for the isolated molecules and for the molecules adsorbed on BNC96 are shown in Figure 2. The spectrum of the isolated TCDD is dominated by five peaks at 1302, 1323, 1554, 1661 and 1679 cm-1. Vibrations associated with these peaks correspond to ring vibrations parallel to the molecular plane. It can be observed that these frequencies are slightly shifted in the BNC96-TCDD complex. A deeper analysis of these vibrational modes in the complex denotes an important contribution of the surface atoms, with weights of 57% and 34% for the most intense peaks at 1302 and 1679 cm-1, respectively. Due to this large vibrational coupling with the surface, the Raman activity of these modes is enhanced in the complex, reaching an enhancement factor of 10 for the signal at 1679 cm-1, even when the resonance mechanisms (electromagnetic and charge transfer) are omitted.
The spectrum of the isolated TCDF is dominated by four peaks at 1327, 1350, 1528 and 1727 cm-1, which correspond to ring vibrations similar to those observed in TCDD. Due to the partial loss of symmetry in TCDF (C2v) with respect to the centrosymmetric TCDD (D2h), the Raman activities of the active modes in the isolated molecule are larger than in TCDD, and consequently the Raman intensities in the spectrum. Interestingly, the Raman activities of all the active modes, except one (1685 cm-1), lessen when TCDF is adsorbed on BNC96. It can be observed in Figure 2 that the peak at 1685 cm-1, which shows a low intensity in the spectrum for the isolated molecule, is largely enhanced in the spectrum of the complex. It can be also extracted from these spectra that the intense peak in the complex correspond to the superposition of two vibrational modes with a scarce activity in the isolated molecule. The reason of the significant Raman enhancement of these modes in the complex is, as in the case of TCDD, the large vibrational coupling with the surface, with contributions of 62% and 42% of the surface atoms, respectively. As mentioned above, the Raman activity of the rest of modes decreases in the complex with respect to the isolated molecule, in these cases the vibrational coupling is much lower with contributions from the surface atoms that in no case exceed 5%.
In previous works, the static Raman spectra of aromatic molecules was found to be consistent with the electric polarizability changes experienced upon stacking interaction with a carbon surface [74,75]. Therefore, leaving out the two modes with a strong vibrational coupling with the surface, the decrease of the Raman activity in TCDF should stem from a decrease in the polarizability of the molecule in the complex. In order to confirm that, the polarizability tensor and the isotropic polarizability of the molecules isolated and adsorbed on BNC96 have been calculated and compared in Table 2.
The results in Table 2 confirm that the important decrease of the Raman activity of TCDF upon adsorption on BNC96 can be mainly associated with a decrease in its polarizability along the in-plane directions ( and ). These are the relevant components of the polarizability due to the in-plane character of the modes (ring vibrations) observed in the spectrum. In addition, one can see that despite the increase of the polarizability along the perpendicular direction (), the isotropic polarizability is still significantly smaller in the complex. A nice pictorial view of this result is provided by Figure 3 with the representation of the Hirshfeld-based total and interacting intrinsic polarizabilities of TCDD and TCDF. Considering first the isolated molecules (Figures 3a and 3c), it is obvious the most polarizable regions, represented by orange lobes, are systematically centered on chlorine atoms due to several factors: they occupy peripheral positions and are singly bonded to one atom uniquely, in contrast with the carbon atoms; their electron density, coming presumably from electron lone pairs, can be more expanded and accessible to the effect of an electric field; and, finally, they are larger than the rest of atoms according to the atomic radius scale. The second most polarizable regions are centered on oxygen atoms, since they also accumulate an important fraction of electron density at lone pairs, as chlorine atoms, but are considerably smaller. Less polarizable regions are centered on carbon atoms, due to absence of electron lone pairs, their smaller atomic size and the fact that they are normally positively charged, in contrast to more electronegative atoms as oxygen and chlorine. The plots also reflect polarizability density distributed uniformly along the rings, characteristic of multicenter electron delocalization in aromatic systems. Regarding the molecules interacting with BNC96 (Figures 3b and 3d), the most polarizable regions previously commented (corresponding to lone pairs of chlorine and oxygen atoms) are systematically the regions with larger reduction of the polarizability (in gray) in contrast with bonded regions, such as C−Cl, C−O or C−C. Curiously, this effect is highly localized, since multicenter electron delocalization seems to be affected negatively in view of the gray lobes found inside the rings. Therefore, the interaction with the surface can locally boost the aforementioned bonds to the detriment of reducing lone pairs and the multicenter electron delocalization of the molecule.
Summarizing, the results obtained here for TCDD and TCDF reflect the molecule-surface vibrational coupling is a key factor to interpret the static Raman spectra, provoking a significant Raman enhancement in those vibrational modes with a large contribution of the surface atoms. In contrast, the strong stacking interaction with the surface leads to a general decrease of the polarizability of the adsorbed molecules, which is reflected on a reduction of the Raman activity as long as the molecule-surface vibrational coupling is small, as already observed in previous works for different carbon allotropes [74,75].
3.3 Raman Spectra Under Pre-resonance Conditions and Laser Wavelengths
The characterization of the electronic transitions from the ground to the different excited states in the adsorption complexes is the first step to simulate pre-resonance Raman spectra. Subsequently, excitation wavelengths near those corresponding to the most intense transitions are included in the calculation of frequency-dependent polarizabilities and the corresponding Raman activities. In this case, the electronic absorption spectra for the isolated BNC96 structure and for the complexes of Figure 1 were simulated using the results obtained from TDDFT calculations (see Figure S1 in the Supplementary Material). The spectrum of BNC96 shows a very intense band around 513.6 nm, corresponding to two degenerate electronic transitions, x-polarized and y-polarized, respectively, with oscillator strengths of 2.560. These transitions are significantly stronger than those found for C96, where the oscillator strength was found to be 1.864 [74,75]. In addition, the absorption band in BNC96 is red-shifted with respect to C96 (457 nm) [74,75], making this band closer to the most employed laser source in SERS experiments (532 nm). In the complexes, this band is even more red-shifted to 518.8 nm and 518.0 nm for BNC96-TCDD and BCN96-TCDF, respectively, and corresponds, due to the loss of symmetry with respect to the isolated surface, to two quasi-degenerate electronic transitions with oscillator strengths of 2.368 and 2.313 for BNC96-TCDD and 2.364 and 2.286 for BNC96-TCDF.
The analysis of the DDs for the electronic transitions engaged in these bands allows discarding a significant charge transfer between surface and molecule, confirming that the excitations involve mainly electrons of the surface. The DDs obtained for the x-polarized electronic excitation in C96-TCDD and BNC96-TCDD complexes are represented in Figure 4. Differences between the DD distribution in C96 and BNC96 are related to the different symmetry of the surface models, D6h and D3h, respectively. As it can be observed, the optical response is mainly located on the carbon nanodisk with a residual contribution from the borazine rings. This confinement of the optical response within the carbon structures in h-BNCs was previously observed and investigated at theoretical level [81,84]. Thus, the number of BN strings necessary to confine the optical response in a phenyl ring embedded within a h-BNC nanodisk of D3h symmetry was found to be only two (a single string of borazine rings) [81], so that enlarging our surface model BNC96 with more strings of borazine rings is not expected to modify significantly its optical response. The y-polarized excitation in BNC96-TCDD and excitations in BNC96-TCDF complex provide identical information and can be seen in Figure S2 of the Supplementary Material.
On the other hand, the TDs for the same electronic excitations represented in Figure 4 are shown in Figure 5. Notice that the distribution of the TD reflects the polarization of the electronic mode (see the differences between x-polarized and y-polarized excitations in Figure S3 of the Supplementary Material). The plots of Figure 5 denote a highly polarized electronic mode strongly localized within the carbon structure. This high polarization is a characteristic feature of collective electronic excitations such as those involved in molecular plasmons, where a large transition dipole moment spans the entire system. On the other hand, a weak local polarization within each individual ring at the molecule-surface interacting region can be also observed. This local polarization opposes the global polarization of the mode. Furthermore, TD plots in the BNC96 complexes also reflect the low participation of the borazine rings and the molecule in the optical response, confirming its local confinement within the carbon nanodisk.
As aforementioned above, the dominant ground to excited state electronic transitions in BNC96-TCDD and BNC96-TCDF complexes are centered at 518.8 nm and 518.0 nm, respectively, involving electrons localized on the carbon nanodisk. These wavelengths might be used as reference to simulate the Raman spectra under pre-resonance conditions. However, the computational cost of these calculations forces the use of a lower basis set, as mentioned in the computational details section. TDDFT calculations show that the dominant ground to excited state electronic transitions are slightly blue-shifted with the lower basis set to 514.0 nm and 514.2 nm for BNC96-TCDD and BNC96-TCDF complexes, respectively. The oscillator strengths for these excitations are very similar to those obtained with the larger basis set. In order to avoid numerical problems when solving the CPDFT equations at resonance conditions, a detuning of ~2 nm in the excitation wavelength with respect to the resonance wavelength was applied to simulate pre-resonance Raman spectra [103]. In addition, Raman spectra were also simulated using, as electromagnetic perturbations, typical laser wavelengths (488 nm and 532 nm) employed in SERS experiments. These are the most interesting spectra, as they give us a more realistic prediction of the expected Raman enhancement under experimental conditions.
The spectra obtained under incident wavelengths blue-shifted by 2 nm with respect to resonance are shown in Figure 6. These spectra are represented using the same scale for the Raman activity as in the static spectra, only changing the multiplicative factors indicated next to the axis. Comparing the factors of pre-resonance spectra with those of the static spectra, it can be observed that the Raman enhancement in TCDD and TCDF complexes reaches 108 and 107, respectively. Practically the same enhancements are observed under incident wavelengths red-shifted 2 nm with respect to resonance (see Figure S6).
Since the electromagnetic response is mainly located on the surface, those vibrational modes from the molecules with the largest vibrational couplings with the surface atoms are expected to display the largest Raman enhancements. It can be observed that, effectively, the largest Raman enhancements are found for signals which were already enhanced in the static spectra: for TCDD at 1302 and 1679 cm-1 and for TCDF at 1685 cm-1, which are precisely those vibrational modes with the highest vibrational coupling with the surface. On the other hand, noticeable enhancements are also found at 1248 cm-1 and 1350 cm-1 for BNC96-TCDF, which correspond to signals associated with vibrational modes with a small vibrational coupling with the surface, with a weight of the surface atoms of 1.4% and 0.4%, respectively).
Raman spectra obtained under an excitation wavelength of 532 nm are shown in Figure 7. The spectra obtained under an excitation wavelength of 488 nm are shown in Figure S7 of the Supplementary Material. Both lead to the same conclusions, so that only the results obtained with 532 nm will be discussed here. In this case, the hypothetical spectra obtained by neglecting the contribution to the Raman activity of the surface atoms or the contribution of the molecule are also shown. They provide a pictorial view of the effect of the molecule-surface vibrational coupling on the Raman spectra of the pollutants.
There are clear similarities between the pre-resonance Raman spectra discussed above and those shown in Figure 7. Thus, the shape of the spectra is quite similar with the same enhanced peaks. Since the laser wavelength is significantly red-shifted with respect to resonance (around 18 nm) the Raman enhancement decreases with respect to pre-resonance spectra, although it is still large, with enhancements around 104 for both molecules, being slightly larger in TCDD. As it can be extracted from the molecule-surface decomposition of the Raman spectrum of BNC96-TCDD, the largest enhancement comes from the surface contribution. The enhancement associated with the molecule lies in a lower scale, so that no signals can be observed in Figure 7c. On the contrary, in the BNC96-TCDF spectrum (Figure 7b) both the molecule and the surface contributions lie in the same scale and depending on the vibrational mode the enhancement is mainly due to one or the other (see Figures 7d and 7f). For instance, the contribution of the molecule is clearly dominant at 1727 cm-1. This is due to the fact that, in this complex, the molecule-surface vibrational coupling of some modes is significantly lower than in the TCDD complex. Anyway, the enhancement of the most intense peak of the spectrum, located at 1685 cm-1, is mainly due to the contribution of the surface atoms (Figure 7f). As mentioned in the previous section, this signal is already enhanced in the static spectrum because of the large vibrational coupling with the surface atoms of the modes associated with it.
4. Conclusions
In this work, the ability of 2D hybrid h-BNC structures as substrates for the Raman-enhancement of dioxins and dibenzofurans has been theoretically investigated. First, the interaction energies obtained for the stacking complexes formed between two model compounds, TCDD and TCDF, and a model h-BNC surface, BNC96, show the large affinity of the substrates by these families of persistent organic pollutants. The most important contribution to the energetic stabilization of these complexes is the dispersion interaction, as previously found for pure carbon 2D structures.
Under static conditions, the Raman activity in the BNC96-TCDF complex displays a general decrease as a consequence of the decrease experienced by the polarizability of the molecule upon the adsorption by the surface. The only exception is the peak located at 1685 cm-1, which is associated with vibrational modes of the molecule with a large vibrational coupling with the surface atoms. On the other hand, the Raman activity in the BNC96-TCDD complex is dominated by the large vibrational coupling between the molecule and the surface atoms observed for the most active modes of TCDD, which overpass the general decrease of its polarizability, giving rise to a general enhancement of the Raman spectrum.
Transition electron densities and ground to excited state density differences obtained for the most active electronic modes of the complexes reflect that these modes correspond to collective electronic excitations characteristic of molecular plasmons, strongly confined within the carbon nanodisk of the BNC96 structure. Under excitation wavelengths close to resonance with these active electronic modes, large Raman enhancement factors of 108 and 107 are obtained for some vibrational modes of TCDD and TCDF, respectively. Under laser excitation wavelengths frequently used in SERS experiments, significant Raman enhancement factors around 104 are also obtained, being slightly larger in TCDD. Due to the large vibrational coupling with the surface atoms in TCDD, its Raman spectrum displays a general enhancement mainly due to the contribution of the surface. In TCDF, some active vibrational modes display a small vibrational coupling with the surface, so that the signals associated with these modes also display an important contribution of the molecule to the Raman enhancement.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Main electronic adsorption band obtained for BCN96-TCDD and BCN96-TCDF complexes (solid lines) compared to that obtained for BCN96 (dashed lines). The excitation wavelengths employed as reference for the calculation of pre-resonance Raman spectra are indicated by vertical lines in red; Figure S2: Ground to excited state density differences (DDs) in BNC96-TCDD and BNC96-TCDF (right) and C96-TCDD and C96-TCDF (left) complexes. Isosurface value is 4·10-4; Figure S3: Transition densities (TDs) in BNC96-TCDD and BNC96-TCDF (right) and C96-TCDD and C96-TCDF (left) complexes. Isosurface value is 1·10-4; Figure S4: Simulated static Raman spectra of TCDD (left) and TCDF (right) adsorbed on BNC96 (first row) and hypothetical spectra obtained from the contributions to the Raman activity of the molecule (second row); Figure S5: Simulated pre-resonance Raman spectra of TCDD (left) and TCDF (right) adsorbed on BNC96 obtained, respectively, with excitation wavelengths of 512.0 and 512.2 nm (first row) and hypothetical spectra obtained from the contributions to the Raman activity of the molecule (second row); Figure S6: Simulated pre-resonance Raman spectra of TCDD (left) and TCDF (right) adsorbed on BNC96 obtained, respectively, with excitation wavelengths of 516.0 and 516.2 nm (first row) and hypothetical spectra obtained from the contributions to the Raman activity of the molecule (second row); Figure S7: Simulated Raman spectra of TCDD (left) and TCDF (right) adsorbed on BNC96 obtained with an excitation wavelength of 488 nm (first row) and hypothetical spectra obtained from the contributions to the Raman activity of the molecule (second row) and the surface (third row).
Author Contributions
Conceptualization, M.M. and N.R-B..; methodology, M.M., N.R.-B. and N.O.; software, M.M. and N.O..; validation, R.A., M.M. and N.R.-B.; formal analysis, R.A., N.O., M.M. and N.R.-B.; investigation, R.A., M.M. and N.R.-B.; data curation, R.A.; writing—original draft preparation, R.A., N.O., M.M. and N.R.-B.; writing—review and editing, R.A., N.O., M.M. and N.R.-B.; supervision, M.M. and N.R.-B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Xunta de Galicia, grant number GRC2019/24.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy constraints.
Acknowledgments
N.R-B. thanks Xunta de Galicia for a postdoctoral grant. N.O. thanks University of Vigo for a posdoctoral grant within the "Retención de Talento Investigador da Universidade de Vigo 2020" program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Van Den Berg, M.; Birnbaum, L.; Bosveld, A. T. C.; Brunström, B.; Cook, P.; Feeley, M.; Giesy, J. P.; Hanberg, A.; Hasegawa, R.; Kennedy, S. W.; Kubiak, T.; Larsen, J. C.; Van Leeuwen, F. X. R.; Liem, A. K. D.; Nolt, C.; Peterson, R. E.; Poellinger, L.; Safe, S.; Schrenk, D.; Tillitt, D.; Tysklind, M.; Younes, M.; Wærn, F.; Zacharewski, T. Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environ. Health Perspect. 1998, 106, 775. [CrossRef]
- Van den Berg, M.; Birnbaum, L.; Denison, M.; De Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Hakansson, H.; Hanberg, A.; Haws, L.; Rose, M.; Safe, S.; Schrenk, D.; Tohyama, C.; Tritscher, A.; Tuomisto, J.; Tysklind, M.; Walker, N.; Peterson, R. E. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 2006, 93, 223. [CrossRef]
- World Health Organization, Polychlorinated dibenzo-para-dioxins and dibenzofurans. Environ. Health Criteria. 1989, 88, 1.
- Polychlorinated Dibenzo-para-dioxins and Polychlorinated Dibenzofurans. IARC Monogr. Eval. Carcinog. Risks Hum. Suppl. 1997, 69, 1.
- Schecter, A. (Ed), Dioxin and Health, Plenum Press. Springer. New York, USA 1994.
- Steenland, K.; Bertazzi, P.; Baccarelli, A.; Kogevinas, M.; Dioxin revisited: developments since the 1997 IARC classification of dioxin as a human carcinogen. Environ. Health Perspect. 2004, 112, 1265. [CrossRef]
- Biswas, G.; Srinivasan, S.; Anandatheerthavarada, H. K.; Avadhani, N. G. Dioxin-mediated tumor progression through activation of mitochondria-to-nucleus stress signaling. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 186. [CrossRef]
- Wielgosiński, G. The Reduction of Dioxin Emissions from the Processes of Heat and Power Generation, J. Air & Waste Manage. Assoc. 2011, 61, 511.
- Rappe, C. Dioxins, patterns and source identification, Fresenius´ J. Anal. Chem. 1994, 348, 63. [CrossRef]
- Merrill, M. L.; Emond, C.; Kimi, M. J.; Antignaci, J.-P.; Bizeci, B. L.; Birnbaum, K. C. L. S.; Barouki, R. Toxicological function of adipose tissue: focus on persistent organic pollutants. Environ. Health Perspect. 2013, 121, 162. [CrossRef]
- Regnier, S. M.; Sargis, R. M. Adipocytes under assault: environmental disruption of adipose physiology. Biochim. Biophys. Acta, Mol. Basis Dis. 2014, 1842, 520. [CrossRef]
- Schrenk, D.; Chopra, M. Dioxins and Polychlorinated Biphenyls in Foods. In Chemical Contaminants and Residues in Food, Woodhead Publishing Series in Food Science, Technology and Nutrition, D. Schrenk, A. Cartus (eds), Elsevier. Cambridge, USA 2017.
- Beck, H.; Droß, A.; Eckart, K.; Mathar, W.; Wittkowski, R. PCDDs, PCDFs and related compounds in paper products. Chemosphere 1989, 19, 655. [CrossRef]
- Wiberg, K.; Lundström, K.; Glas, B.; Rappe, C. PCDDs and PCDFs in consumers’ paper products. Chemosphere 1989, 19, 735. [CrossRef]
- Sany, S. B. T.; Narimani, L.; Soltanian, F. K.; Hashim, R.; Rezayi, M.; Karlend, D. J.; Mahmud, H. N. M. E. An overview of detection techniques for monitoring dioxin-like compounds: latest technique trends and their applications. RSC Adv. 2016, 6, 55415. [CrossRef]
- Schecter, A.; Birnbaum, L.; Ryan, J. J.; Constable, J. D. Dioxins: an overview. Environ. Res. 2006, 101, 419. [CrossRef]
- Behnisch, P. A.; Hosoe, K.; Sakai, S.-i. Bioanalytical screening methods for dioxins and dioxin-like compounds - a review of bioassay/biomarker technology. Environ. Int. 2001, 27, 413. [CrossRef]
- Torres, M. A.; Barros, M. P.; Campos, S. C. G.; Pinto, E.; Rajamani, S.; Sayre, R. T.; Colepicolo, P. Biochemical biomarkers in algae and marine pollution: a review. Ecotoxicol. Environ. Saf. 2008, 71, 1. [CrossRef]
- Eichbaum, K.; Brinkmann, M.; Buchinger, S.; Reifferscheid, G.; Hecker, M.; Giesy, J. P.; Engwall, M.; van Bavel, B.; Hollert, H. In vitro bioassays for detecting dioxin-like activity - Application potentials and limits of detection, a review. Sci. Total Environ. 2014, 487, 37. [CrossRef]
- Beníšek, M.; Kukučka, P.; Giulio Mariani, G.; Suurkuusk, G.; Gawlik, B. M.; Locoro, G.; Giesy, J. P.; Bláha, L. Dioxins and dioxin-like compounds in composts and digestates from European countries as determined by the in vitro bioassay and chemical analysis. Chemosphere 2015, 122, 168. [CrossRef]
- Du, B.; Liu, A.; Huang, Y. Uncertainty evaluation of the determination of toxic equivalent quantity of polychlorinated dibenzo-p-dioxins and dibenzofurans in soil by isotope dilution high resolution gas chromatography and high resolution mass spectrometry. Chin. J. Chromatogr. 2014, 32, 967. [CrossRef]
- L'Homme, B.; Scholl, G.; Eppe, G.; Focant, J. F. Validation of a gas chromatography-triple quadrupole mass spectrometry method for confirmatory analysis of dioxins and dioxin-like polychlorobiphenyls in feed following new EU Regulation 709/2014. J. Chromatogr. A 2015, 1376, 149. [CrossRef]
- Baughman, R.; Meselson, M. An analytical method for detecting TCDD (dioxin): levels of TCDD in samples from Vietnam. Environ. Health Perspect. 1973, 5, 27. [CrossRef]
- Schecter, A.; Tiernan, T. Occupational exposure to polychlorinated dioxins, polychlorinated furans, polychlorinated biphenyls, and biphenylenes after an electrical panel and transformer accident in an office building in Binghamton. Environ. Health Perspect. 1985, 60, 305. [CrossRef]
- Schecter, A.; Ryan, J. J.; Päpke, O.; Ball, M.; Lis, A. Elevated dioxin levels in the blood of male and female Russian workers with and without chloracne 25 years after phenoxyherbicide exposure: The UFA “Khimprom” incident. Chemosphere 1993, 27, 253. [CrossRef]
- Wei, Y.; Kong, L.-T.; Yang, R.; Wang, L.; Liu, J.-H.; Huang, X.-J. Electrochemical impedance determination of polychlorinated biphenyl using a pyrenecyclodextrin-decorated single-walled carbon nanotube hybrid. Chem. Commun. 2011, 47, 5340. [CrossRef]
- Wang, M.; Meng, G.; Huang, Q.; Li, M.; Lia, Z.; Tang, C. Fluorescence detection of trace PCB101 based on PITC immobilized on porous AAO membrane. Analyst 2011, 136, 278. [CrossRef]
- Zhu, C.; Meng, G.; Huang, Q. Vertically aligned Ag nanoplate-assembled film as a sensitive and reproducible SERS substrate for the detection of PCB-77. J. Hazard. Mater. 2012, 211, 389. [CrossRef]
- Silva, E.; Mascini, M.; Centi, S.; Turner, A. P. F. Detection of polychlorinated biphenyls (PCBs) in milk using a disposable immunomagnetic electrochemical sensor. Anal. Lett. 2007, 40, 1371. [CrossRef]
- Kurosawa, S.; Aizawa, H.; Park, J.-W. Quartz crystal microbalance immunosensor for highly sensitive 2,3,7,8-tetrachlorodibenzo-p-dioxin detection in fly ash from municipal solid waste incinerators. Analyst 2005, 130, 1495. [CrossRef]
- Chobtang, J.; De Boer, I. J. M.; Hoogenboom, R. L. A. P.; Haasnoot, W.; Kijlstra, A.; Meerburg, B. G. The need and potential of biosensors to detect dioxins and dioxin-like polychlorinated biphenyls along the milk, eggs and meat food chain. Sensors 2011, 11, 11692. [CrossRef]
- Centi, S.; Silva, E.; Laschi, S.; Palchetti, I.; Mascini, M. Polychlorinated biphenyls (PCBs) detection in milk samples by an electrochemical magnetoimmunosensor (EMI) coupled to solid-phase extraction (SPE) and disposable low-density arrays. Anal. Chim. Acta 2007, 594, 9. [CrossRef]
- Wang, S.; Sun, B.; Feng, J.; An, F.; Li, N.; Wang, H.; Tian, M. Development of affinity between target analytes and substrates in surface enhanced Raman spectroscopy for environmental pollutant detection. Analytical Methods 2020, 12, 5657. [CrossRef]
- Fang, X.; Song, Y.; Huang, Y.; Yang, G.; Han, C.; Li, H.; Qu, L. Two-dimensional MXene modified AgNRs as a surface-enhanced Raman scattering substrate for sensitive determination of polychlorinated biphenyls. Analyst 2020, 145, 7421. [CrossRef]
- Patrizi, B.; De Cumis, M.S.; Viciani, S.; D’Amato, F. Dioxin and related compound detection: Perspectives for optical monitoring. Int. J. Mol. Sci. 2019, 20, 2671. [CrossRef]
- Rindzevicius, T.; Barten, J.; Vorobiev, M.; Schmidt, M.S.; Castillo, J. J.; Boisen, A. Detection of surface-linked polychlorinated biphenyls using surface-enhanced Raman scattering spectroscopy. Vib. Spectrosc. 2017, 90, 1. [CrossRef]
- Potara, M.; Farcau, C.; Botiz, I.; Astilean, S. Detection of Environmental Pollutants by Surface-Enhanced Raman Spectroscopy, In RSC Detection Science, 2017, pp. 479.
- Hou, M.; Huang, Y.; Ma, L.; Zhang, Z. Sensitivity and Reusability of SiO2 NRs@ Au NPs SERS Substrate in Trace Monochlorobiphenyl Detection. Nanoscale Res. Lett. 2015, 10, 444. [CrossRef]
- Chen, S.N.; Li, X.; Han, S.; Liu, J. H.; Zhao, Y.Y. Synthesis of surface-imprinted Ag nanoplates for detecting organic pollutants in water environments based on surface enhanced Raman scattering. RSC Adv. 2015, 5, 99914. [CrossRef]
- Jing, L.; Shi, Y.-E.; Cui, J.; Zhang, X.; Zhan, J. Hydrophobic gold nanostructures via electrochemical deposition for sensitive SERS detection of persistent toxic substances. RSC Adv. 2015, 5, 13443. [CrossRef]
- Lu, Y.; Yao, G.; Sun, K.; Huang, Q. β-Cyclodextrin coated SiO2@Au@Ag core-shell nanoparticles for SERS detection of PCBs. Phys. Chem. Chem. Phys. 2014, 17, 21149. [CrossRef]
- Bao, Z.Y.; Liu, X.; Chen, Y.; Wu, Y.; Chan, H. L. W.; Dai, J.; Lei, D. Y. Quantitative SERS detection of low-concentration aromatic polychlorinated biphenyl-77 and 2,4,6-trinitrotoluene. J. Hazard. Mater. 2014, 280, 706. [CrossRef]
- Chen, B.; Meng, G.; Zhou, F.; Huang, Q.; Zhu, C.; Hu, X.; Kong, M. Ordered arrays of Au-nanobowls loaded with Ag-nanoparticles as effective SERS substrates for rapid detection of PCBs. Nanotechnology, 2014, 25, 145605. [CrossRef]
- Li, D.-W.; Zhai, W.-L.; Li, Y.-T.; Long, Y.-T. Recent progress in surface enhanced Raman spectroscopy for the detection of environmental pollutants. Microchim. Acta 2014, 181, 23. [CrossRef]
- Xia Han, X.; Rodriguez, R. S.; Haynes, C. L.; Ozaki, Y.; Zhao, B. Surface-enhanced Raman spectroscopy. Nat. Rev. Methods Primers 2021, 1, 87. [CrossRef]
- Tripp, R.A.; Dluhy, R.A.; Zhao, Y. Novel nanostructures for SERS biosensing, Nano Today 2008, 3, 31.
- Hudson, S. D.; Chumanov, G. Bioanalytical applications of SERS (surface-enhanced Raman spectroscopy), Anal Bioanal. Chem. 2009, 394, 679.
- El-Ansary, A.; Faddah, L. M. Nanoparticles as biochemical sensors, Nanotechnol. Sci. Appl. 2010, 3, 65.
- Sharma, B.; Frontiera, R. R.; Henry, A.-I.; Ringe, E.; Van Duyne, R. P. SERS: Materials, applications, and the future, Materials Today 2012, 15, 16.
- Halvorson, R. A.; Vikesland, P. J. Surface-Enhanced Raman Spectroscopy (SERS) for Environmental Analyses, Environ. Sci. Technol. 2010, 44, 7749. [CrossRef]
- Ong, T. T. X.; Blanch, E. W.; Jones, O. A. H. Surface Enhanced Raman Spectroscopy in environmental analysis, monitoring and assessment, Sci. Total Environ. 2020, 720, 137601. [CrossRef]
- Terry, L. R.; Sanders, S.; Potoff, R. H.; Kruel, J. W.; Jain, M.; Guo, H. Applications of surface-enhanced Raman spectroscopy in environmental detection, Anal. Sci. Adv. 2022, 3, 113.
- Bodelón, G.; Pastoriza-Santos, I. Recent Progress in Surface-Enhanced Raman Scattering for the Detection of Chemical Contaminants in Water, Front. Chem. 2020, 8, 478.
- Guo, H.; He, L.; Xing, B. Applications of surface-enhanced Raman spectroscopy in the analysis of nanoparticles in the environment, Environ. Sci.: Nano 2017, 4, 2093.
- Liu, X.; Guo, J.; Li, Y.; Wang, B.; Yang, S.; Chen, W.; Wu, X.; Guo, J.; Ma, X. SERS substrate fabrication for biochemical sensing: towards point-of-care diagnostics, J. Mat. Chem. B 2021, 9, 8378. [CrossRef]
- Xie, X.; Pu, H.; Sun, D.-W. Recent advances in nanofabrication techniques for SERS substrates and their applications in food safety analysis, Crit. Rev. Food Sci. Nutr. 2018, 58, 2800. [CrossRef]
- Ouyang, L.; Ren, W.; Zhu, L.; Irudayaraj, J. Prosperity to challenges: recent approaches in SERS substrate fabrication, Rev. Anal. Chem. 2016, 36, 20160027. [CrossRef]
- Ge, K.; Hu, Y.; Li, G. Recent Progress on Solid Substrates for Surface-Enhanced Raman Spectroscopy Analysis, Biosensors 2022, 12, 941.
- Guselnikova, O.; Lim, H.; Kim, H.-J.; Kim, S. H.; Gorbunova, A.; Eguchi, M.; Postnikov, P.; Nakanishi, T.; Asahi, T.; Na, J.; Yamauch, Y. New Trends in Nanoarchitectured SERS Substrates: Nanospaces, 2D Materials, and Organic Heterostructures, Small 2022, 18, 2107182.
- Chen, M.; Liu, D.; Du, X.; Lo, K. H.; Wang, S.; Zhou, B.; Pan, H. 2D materials: Excellent substrates for surface-enhanced Raman scattering (SERS) in chemical sensing and biosensing, Trends Anal. Chem. 2020, 130, 115983.
- Jebakumari, K. A. E.; Murugasenapathi, N. K.; Palanisamy, T. Engineered Two-Dimensional Nanostructures as SERS Substrates for Biomolecule Sensing: A Review, Biosensors 2023, 13, 102.
- Liu, Y.; Qin, Z.; Deng, J.; Zhou, J.; Jia, X.; Wang, G.; Luo, F. The Advanced Applications of 2D Materials in SERS, Chemosensors 2022, 10, 455.
- Luo, W.; Xiong, W.; Han, Y.; Yan, X.; Mai, L. Application of two-dimensional layered materials in surface-enhanced Raman spectroscopy (SERS), Phys. Chem. Chem. Phys. 2022, 24, 26398. [CrossRef]
- Kannan, P. K.; Shankar, P.; Blackman, C.; Chung, C.-H. Recent Advances in 2D Inorganic Nanomaterials for SERS Sensing, Adv. Mater. 2019, 31, 1803432.
- Zhang, N.; Tong, L.; Zahng, J. Graphene-Based Enhanced Raman Scattering toward Analytical Applications, Chem. Mater. 2016, 28, 6426.
- Silver, A.; Kitadai, H.; Liu, H.; Granzier-Nakajima, T.; Terrones, M.; Ling, X.; Huang, S. Chemical and Bio Sensing Using Graphene-Enhanced Raman Spectroscopy, Nanomaterials 2019, 9, 516.
- Xu, W.; Mao, N.; Zhang, J. Graphene: a platform for surface-enhanced Raman spectroscopy, Small 2013, 9, 1206.
- Cai, Q.; Mateti, S.; Watanabe, K.; Taniguchi, T.; Huang, S.; Chen, Y.; Li, L. H. Boron Nitride Nanosheet-Veiled Gold Nanoparticles for Surface-Enhanced Raman Scattering, ACS Appl. Mater. Interfaces 2016, 8, 15630. [CrossRef]
- Kim, G; Kim, M.; Hyun, C.; Hong, S.; Ma, K. Y.; Shin, H. S.; Lim, H. Hexagonal Boron Nitride/Au Substrate for Manipulating Surface Plasmon and Enhancing Capability of Surface-Enhanced Raman Spectroscopy, ACS Nano 2016, 10, 11156.
- Kim, N.-Y.; Leem, Y.-C.; Hong, S.-H.; Park, J.-H.; Yim, S.-Y. Ultrasensitive and Stable Plasmonic Surface-Enhanced Raman Scattering Substrates Covered with Atomically Thin Monolayers: Effect of the Insulating Property, ACS Appl. Mater. Interfaces 2019, 11, 6363. [CrossRef]
- Chugh, D.; Jagadish, C.; Tan, H. Large-Area Hexagonal Boron Nitride for Surface Enhanced Raman Spectroscopy, Adv. Mater. Technol. 2019, 4, 1900220.
- Basu, N.; Satya Bharathi, M. S.; Sharma, M.; Yadav, K.; Parmar, A. S.; Soma, V. R.; Lahiri, J. Large Area Few-Layer Hexagonal Boron Nitride as a Raman Enhancement Material, Nanomaterials 2021, 11, 622.
- Nie, S. M.; Emory, S. R. Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering, Science 1997, 275, 1102.
- Ramos-Berdullas, N.; López-Carballeira, D.; Pérez-Juste, I.; Mandado, M. On the mechanism responsible of Raman enhancement on carbon allotropes surfaces: the role of molecule-surface vibrational coupling in SERS, J. Raman Spectrosc. 2015, 46, 1205. [CrossRef]
- López-Carballeira, D.; Ramos-Berdullas, N.; Pérez-Juste, I.; Mandado, M. Can single graphene nanodisks be used as Raman enhancement platforms?, RSC Adv. 2016, 6, 71397. [CrossRef]
- Ramos-Berdullas, N.; Otero, N.; Mandado, M. Graphene Molecules as Platforms for SERS Detection: A Future Perspective, In Handbook of Graphene: Biosensors and Advanced Sensors, Palys, B. (ed.), Vol 6, pp 431, John Wiley & Sons Inc, Hoboken, 2019.
- Ci, L.; Song, L.; Jin, C.; Jariwala, D.; Wu, D.; Li, Y.; Srivastava, A.; Wang, Z.F.; Storr, K.; Balicas, L.; Liu, F.; Ajayan, P. M. Atomic layers of hybridized boron nitride and graphene domains, Nat. Mater. 2010, 10, 430.
- Gong, Y.; Shi, G.; Zhang, Z.; Zhou, W.; Jung, J.; Gao, W.; Ma, L.; Yang, Y.; Yang, S.; You, G.; Vajtai, R.; Xu, Q.; MacDonald, A. H.; Yakobson, B. I.; Lou, J.; Liu, Z.; Ajayan, P. M. Direct chemical conversion of graphene to boron- and nitrogen- and carbon-containing atomic layers, Nat. Commun. 2014, 5, 3193.
- Lorenzo-García, M.M.; Bonifazi, D. Renaissance of an Old Topic: From Borazines to BN-doped Nanographenes, Chimia 2017, 71, 550.
- Herrera-Reinoza, N.; dos Santos, A. C.; de Lima, L. H.; Landers, R.; de Siervo, A. Atomically Precise Bottom-Up Synthesis of h-BNC: Graphene Doped with h-BN Nanoclusters, Chem. Mat. 2021, 33, 2871. [CrossRef]
- Mandado, M.; Ramos-Berdullas, N. Confinement on the Optical Response in h-BNCs: Towards Highly Efficient SERS-active 2D Substrates, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2022, 266, 120451. [CrossRef]
- Karamanis, P.; Otero, N.; Pouchan, C. Unleashing the quadratic nonlinear optical responses of graphene by confining white-graphene (h-BN) sections in its framework, J. Am. Chem. Soc. 2014, 113, 7464. [CrossRef]
- Karamanis, P.; Otero, N.; Pouchan, C. Electric property variations in nanosized hexagonal boron nitride/graphene hybrids, J. Phys. Chem. C 2015, 119, 11872. [CrossRef]
- Otero, N.; El-Kelany, K.E.; Pouchan, C.; Rérat, M.; Karamanis, P. Establishing the pivotal role of local aromaticity in the electronic properties of boron-nitride graphene lateral hybrids, Phys. Chem. Chem. Phys. 2016, 18, 25315. [CrossRef]
- Bevilacqua, A. C.; Köhler, M. H.; Azevedo, S.; Baierle, R. J. Stability, and optical and electronic properties of ultrathin h-BNC, Phys. Chem. Chem. Phys. 2017, 19, 5629. [CrossRef]
- Alvarado, R.; Ramos-Berdullas, N.; Mandado, M. On the adsorption affinity of graphene and white graphene sheets by dioxin-like pollutants, Int. J. Quantum Chem. 2021, 121, e26591. [CrossRef]
- Mandado, M.; Hermida-Ramón, J. M. Electron Density Based Partitioning Scheme of Interaction Energies, J. Chem. Theo. Comput. 2011, 7, 633. [CrossRef]
- Ramos-Berdullas, N.; Pérez-Juste, I.; C. Van Alsenoy, C.; Mandado, M. Theoretical study of the adsorption of aromatic units on carbon allotropes including explicit (empirical) DFT dispersion corrections and implicitly dispersion-corrected functionals: the pyridine case, Phys. Chem. Chem. Phys. 2015, 17, 575. [PubMed]
- Cárdenas, G.; Pérez-Barcia, A.; Mandado, M.; Nogueira, J. J. Characterization of cisplatin/membrane interactions by QM/MM energy decomposition analysis, Phys. Chem. Chem. Phys. 2021, 23, 20533. [CrossRef]
- Pérez-Barcia, A.; Cárdenas, G.; Nogueira, J. J.; Mandado, M. Effect of the QM Size, Basis Set, and Polarization on QM/MM Interaction Energy Decomposition Analysis, J. Chem. Inf. Model. 2023, 63, 882. [CrossRef]
- Le Ru, E.; Etchegoin, P. Principles of Surface Enhanced Raman Spectroscopy and related plasmonic effects, pp. 14-20, Elsevier, Amsterdam, 2009.
- Ramos-Berdullas, N.; López-Carballeira, D.; Mandado, M.; Pérez-Juste, I. Revisiting the mechanism and the influence of the excitation wavelength on the surface-enhanced Raman scattering of the pyridine–Ag20 system, Theo. Chem. Acc. 2015, 134, 60. [CrossRef]
- Krishtal, A.; Senet, P.; Yang, M.; Van Alsenoy, C. A Hirshfeld partitioning of polarizabilities of water clusters, J. Chem. Phys. 2006, 125, 034312. [CrossRef]
- Otero, N.; Van Alsenoy, C.; Pouchan, C.; Karamanis, P. Hirshfeld-based intrinsic polarizability density representations as a tool to analyze molecular polarizability, J. Comput. Chem. 2015, 36, 1831. [CrossRef]
- Geldof, D.; Krishtal, A.; Blockhuys, F.; Van Alsenoy, C. An Extension of the Hirshfeld Method to Open Shell Systems Using Fractional Occupations, J. Chem. Theo. Comput. 2011, 7, 1328. [CrossRef]
- Gaussian 16, Revision C.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian, Inc., Wallingford CT, 2016.
- Mandado, M.; Van Alsenoy, C. EDA-NCI: A program to perform energy decomposition analysis of non-covalent interactions, available at: https://github.com/marcos-mandado/EDA-NCI.
- GaussView, Version 6, Dennington, Roy; Keith, Todd A.; Millam, John M. Semichem Inc., Shawnee Mission, KS, 2016.
- Allouche, R. A. Gabedit - A graphical user interface for computational chemistry softwares, J. Comput. Chem. 2011, 32, 174. [CrossRef]
- Chemcraft - graphical software for visualization of quantum chemistry computations, available at: https://www.chemcraftprog.com.
- Boys, S. F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys. 1970, 19, 553.
- López-Carballeira, D.; Ramos-Berdullas, N.; Pérez-Juste, I.; Cagide-Fajín, J. L.; Cordeiro, M. N. D. S., Mandado, M. A computational study of the interaction of graphene structures with biomolecular units, Phys. Chem. Chem. Phys. 2016, 18, 15312.
- Norman, P.; Bishop, D. M.; Jensen, H. J. A.; Oddershede, J. Near-resonant absorption in the time-dependent self-consistent field and multiconfigurational self-consistent field approximations, J. Chem. Phys. 2001, 115, 10323. [CrossRef]
Figure 1.
Optimized structures of the adsorption complexes BNC96-TCDD (left) and BNC96-TCDF (right).
Figure 1.
Optimized structures of the adsorption complexes BNC96-TCDD (left) and BNC96-TCDF (right).
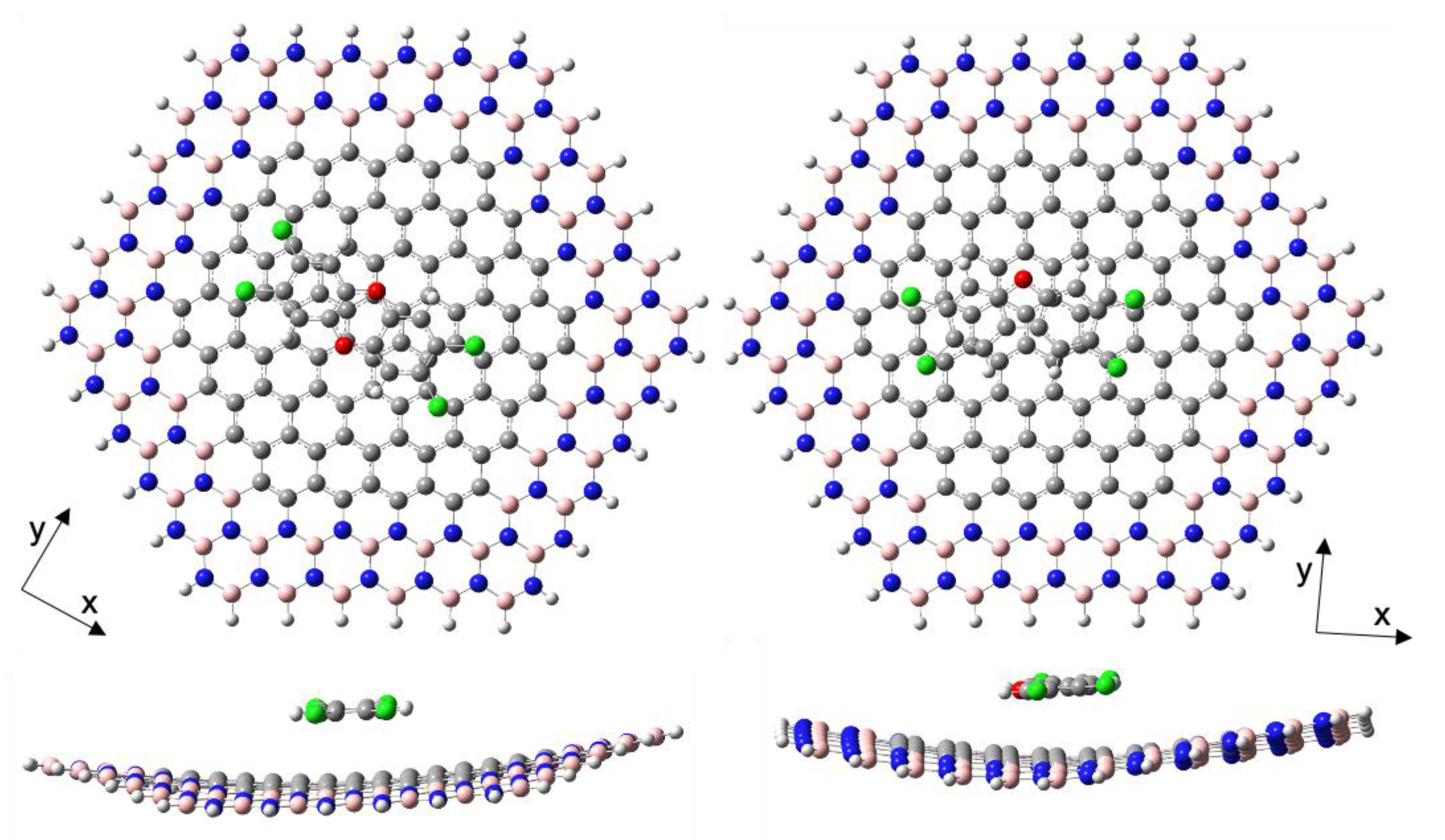
Figure 2.
Simulated static Raman spectra of TCDD (a) and TCDF (b) adsorbed on BNC96 (blue) confronted with the spectra of the corresponding isolated molecules (orange).
Figure 2.
Simulated static Raman spectra of TCDD (a) and TCDF (b) adsorbed on BNC96 (blue) confronted with the spectra of the corresponding isolated molecules (orange).
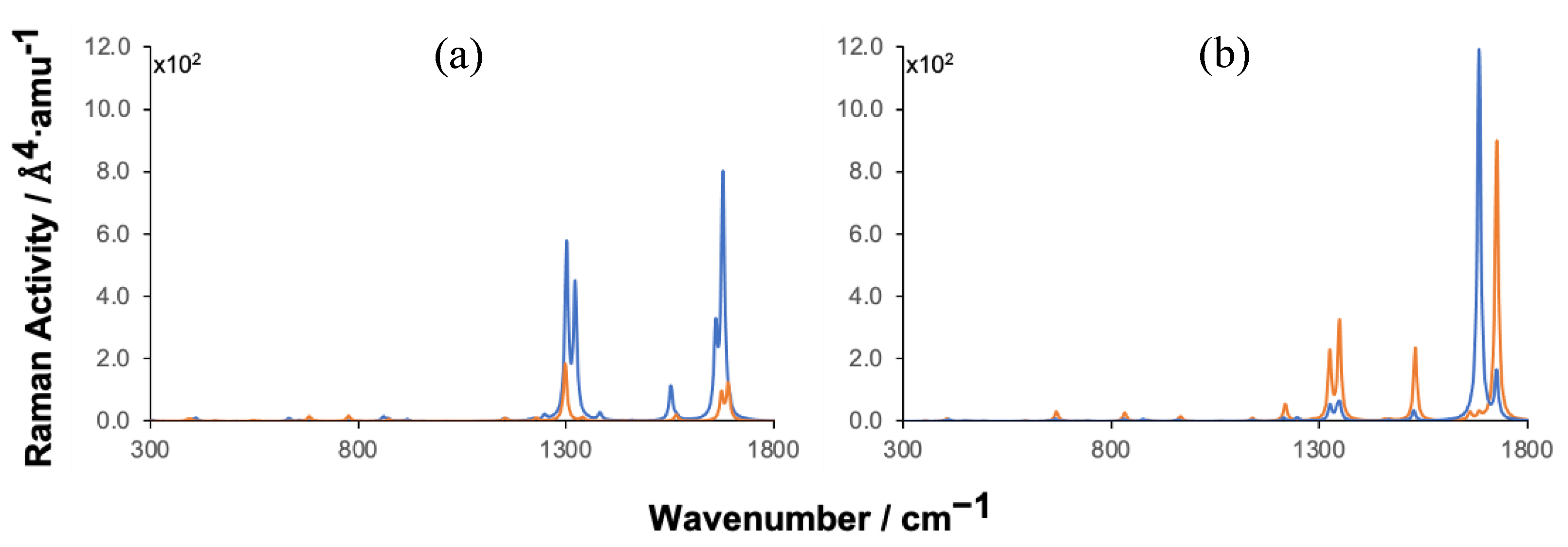
Figure 3.
Front views of isotropic polarizability density distribution maps of TCDD and TCDF molecules isolated, (a) and (c), and adsorbed on BNC96, (b) and (d). For isolated molecules the map represents the total intrinsic polarizability (eqn (8)) whereas for the adsorbed molecules the map represents the interacting intrinsic polarizability (eqn (9)). Isosurface values for total and interacting intrinsic polarizabilities are ±0.03 and ±0.01, respectively. Orange and blue regions correspond to positive values of the isosurface, whereas purple and gray represent the negative ones. We have removed the polarizability density distributions from the host surface to simplify the visualization.
Figure 3.
Front views of isotropic polarizability density distribution maps of TCDD and TCDF molecules isolated, (a) and (c), and adsorbed on BNC96, (b) and (d). For isolated molecules the map represents the total intrinsic polarizability (eqn (8)) whereas for the adsorbed molecules the map represents the interacting intrinsic polarizability (eqn (9)). Isosurface values for total and interacting intrinsic polarizabilities are ±0.03 and ±0.01, respectively. Orange and blue regions correspond to positive values of the isosurface, whereas purple and gray represent the negative ones. We have removed the polarizability density distributions from the host surface to simplify the visualization.
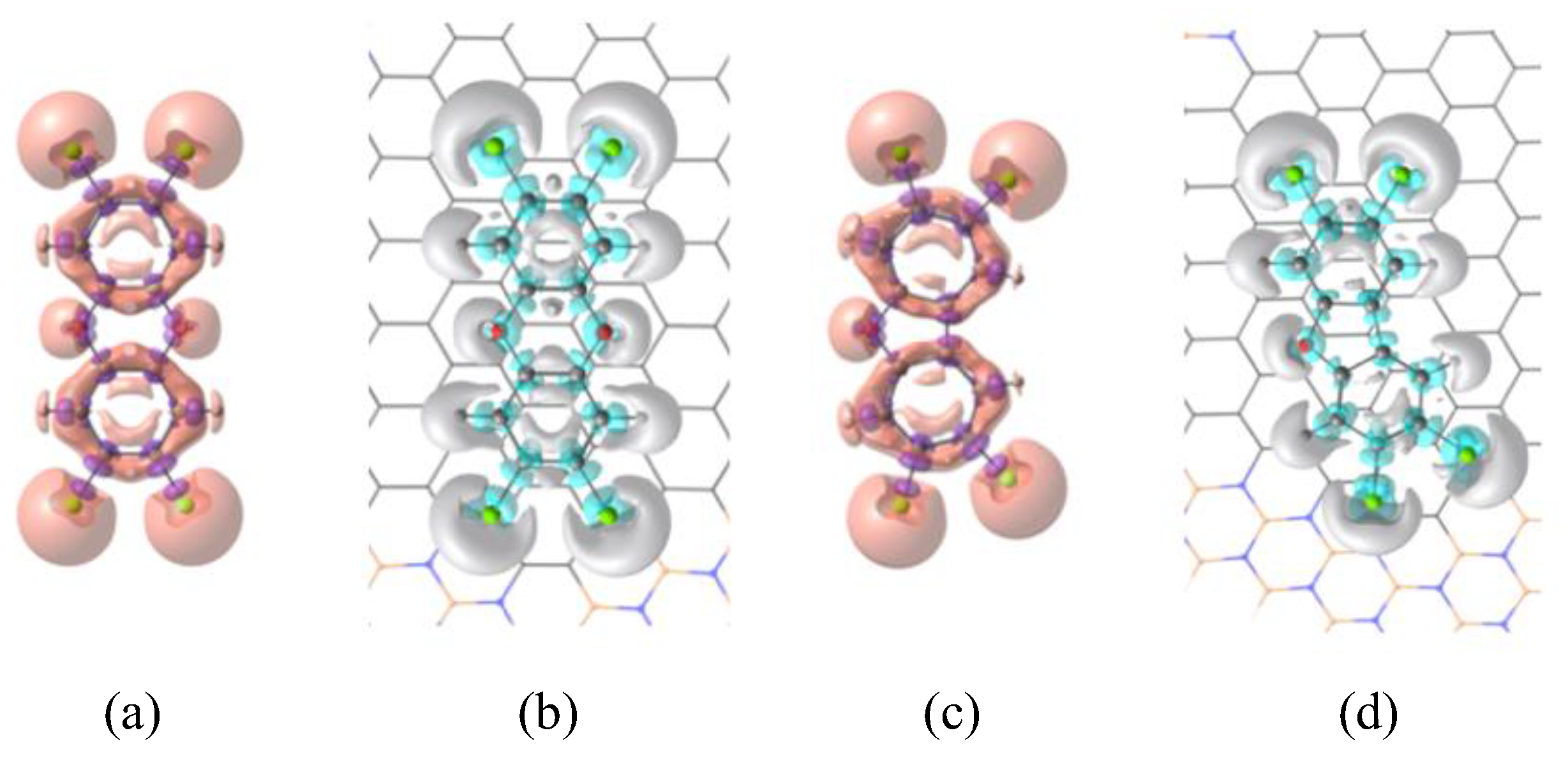
Figure 4.
Ground to excited state density differences (DDs) in BNC96-TCDD (right) and C96-TCDD (left) complexes. Isosurface value is 4·10-4.
Figure 4.
Ground to excited state density differences (DDs) in BNC96-TCDD (right) and C96-TCDD (left) complexes. Isosurface value is 4·10-4.
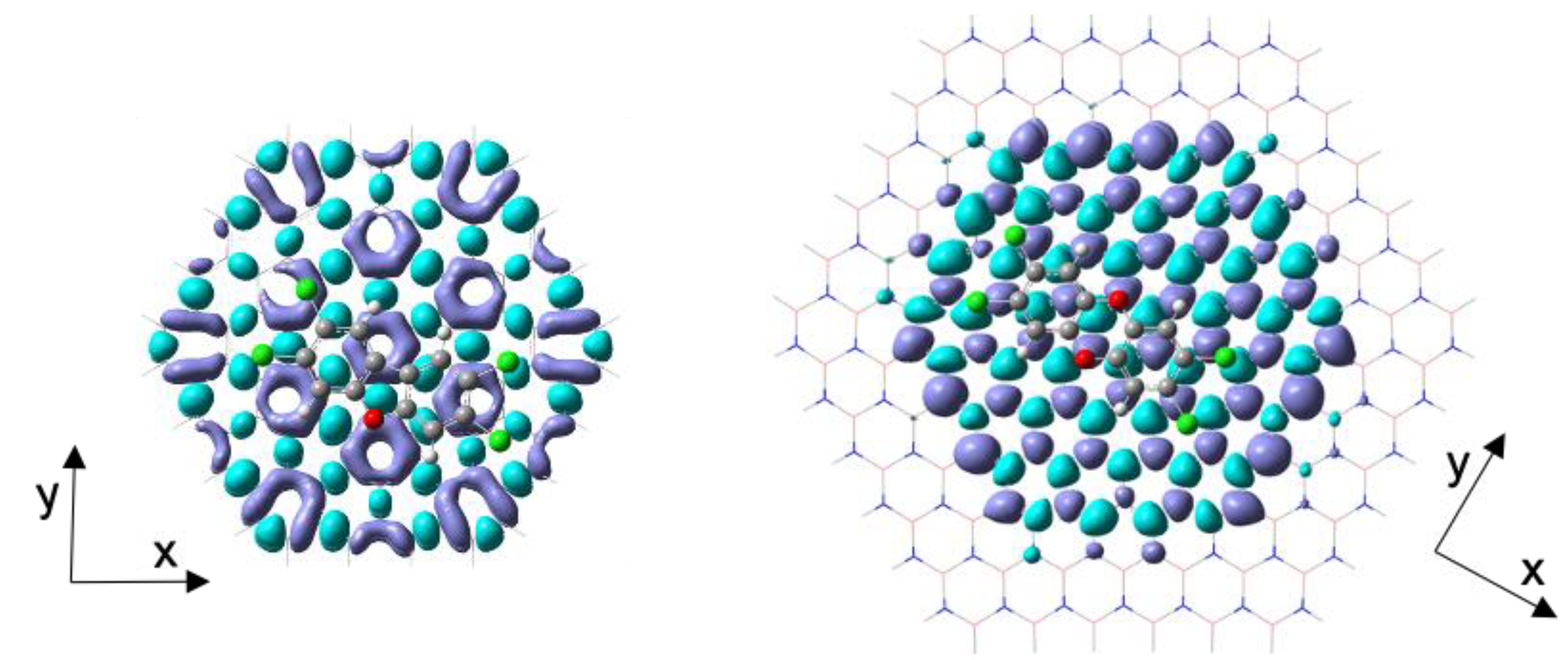
Figure 5.
Transition densities (TDs) in BNC96-TCDD (right) and C96-TCDD (left) complexes. Isosurface value is 1·10-4.
Figure 5.
Transition densities (TDs) in BNC96-TCDD (right) and C96-TCDD (left) complexes. Isosurface value is 1·10-4.
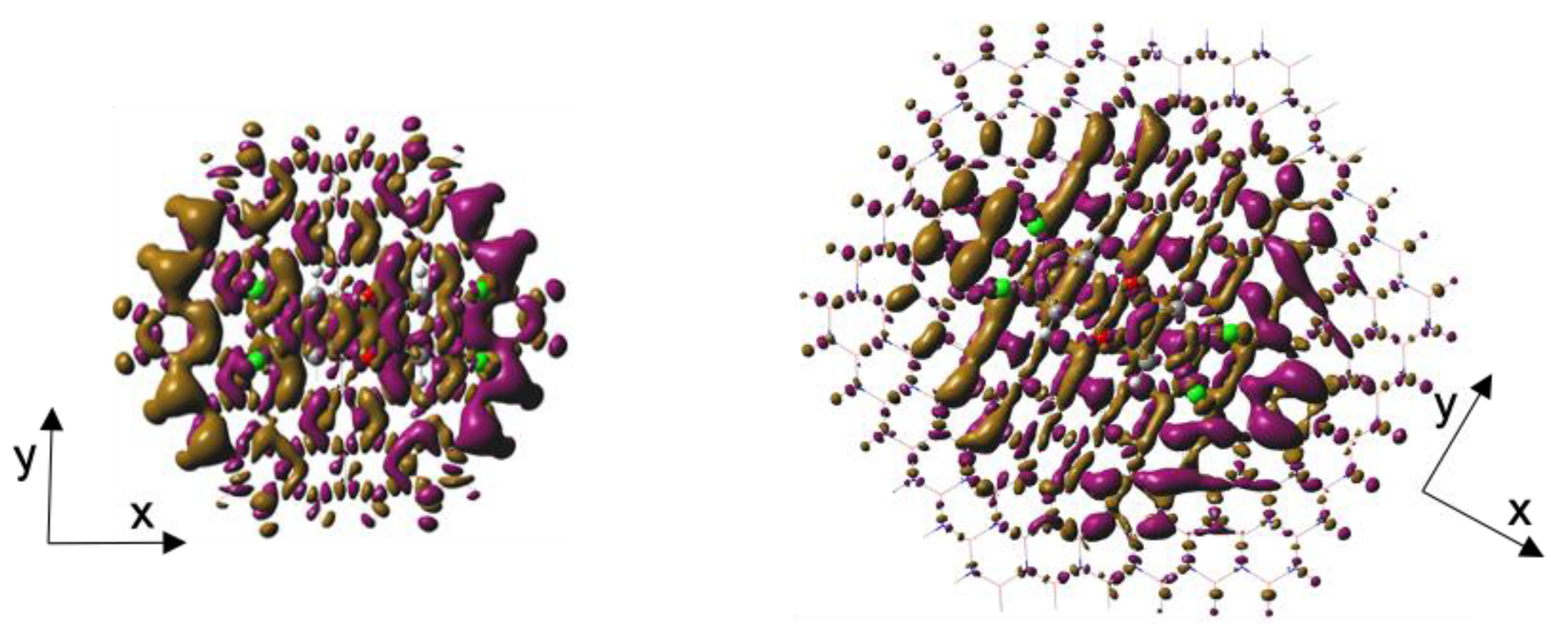
Figure 6.
Simulated Raman spectra of TCDD (a) and TCDF (b) adsorbed on BNC96 obtained with an excitation wavelength of 512.0 nm and 512.2 nm, respectively.
Figure 6.
Simulated Raman spectra of TCDD (a) and TCDF (b) adsorbed on BNC96 obtained with an excitation wavelength of 512.0 nm and 512.2 nm, respectively.
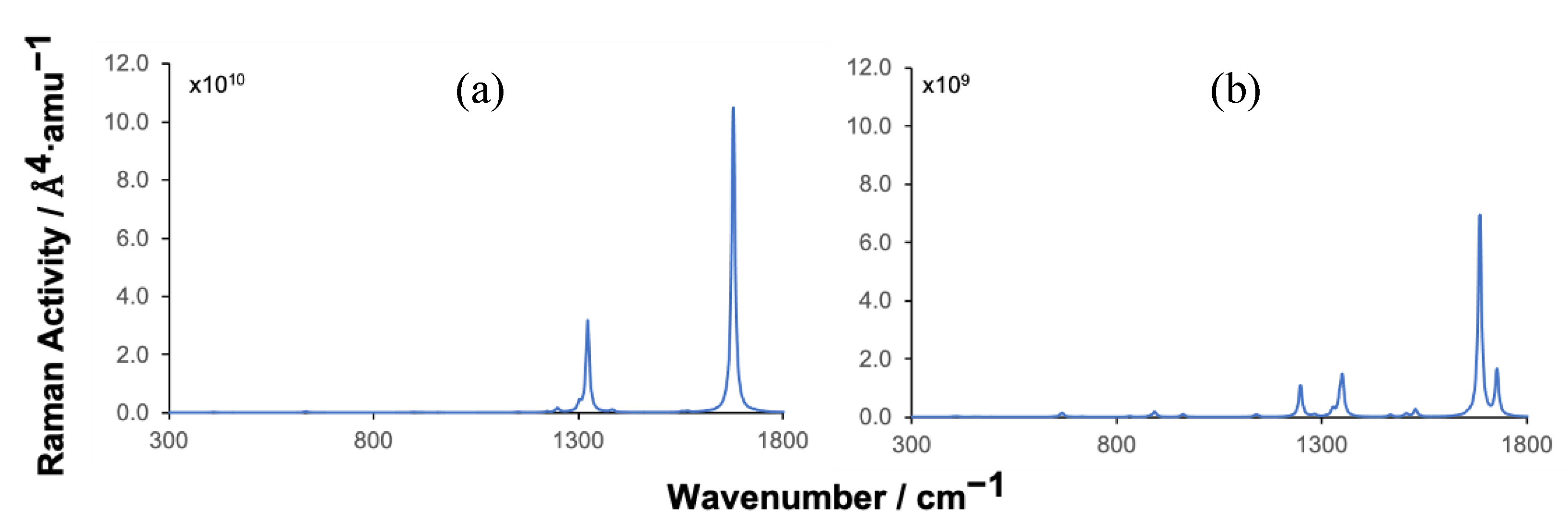
Figure 7.
Simulated Raman spectra of TCDD (a, c and e) and TCDF (b, d and f) adsorbed on BNC96 (a and b) and hypothetical spectra obtained from the contributions of molecule (c and d) and surface (e and f) obtained with an excitation wavelength of 532 nm.
Figure 7.
Simulated Raman spectra of TCDD (a, c and e) and TCDF (b, d and f) adsorbed on BNC96 (a and b) and hypothetical spectra obtained from the contributions of molecule (c and d) and surface (e and f) obtained with an excitation wavelength of 532 nm.
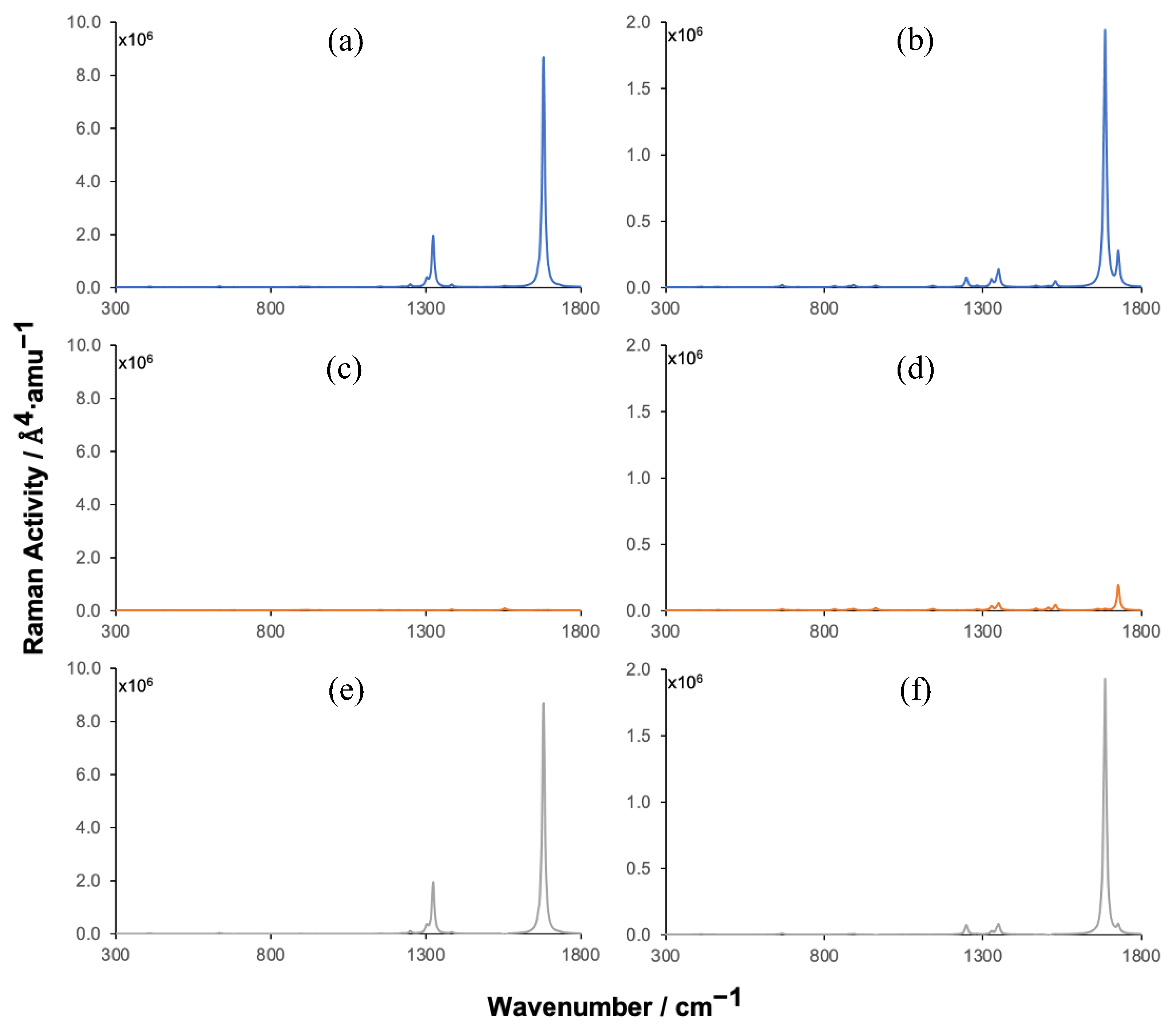

Table 1.
Interaction energy and its components for the complexes formed by the surface finite models of C96 and BNC96 with TCDD and TCDF. All data are given in kcal mol−1.
Table 1.
Interaction energy and its components for the complexes formed by the surface finite models of C96 and BNC96 with TCDD and TCDF. All data are given in kcal mol−1.
| TCDD | TCDF | ||||
| C96 | BNC96 | C96 | BNC96 | ||
| -21.3 | -21.1 | -19.6 | -19.0 | ||
| 52.5 | 51.2 | 48.7 | 46.9 | ||
| -5.0 | -5.1 | -4.2 | -4.3 | ||
| -59.5 | -59.7 | -56.2 | -55.9 | ||
| -33.4 | -34.7 | -31.4 | -32.3 | ||
Table 2.
Polarizability tensor diagonal components and isotropic polarizability (in au) for the isolated molecules and adsorbed on BNC96.
Table 2.
Polarizability tensor diagonal components and isotropic polarizability (in au) for the isolated molecules and adsorbed on BNC96.
| TCDD | TCDF | ||||
| Isolated | BNC96 | Isolated | BNC96 | ||
| 302.20 | 182.42 | 310.05 | 172.82 | ||
| 174.38 | 87.55 | 171.89 | 75.30 | ||
| 82.91 | 129.73 | 80.23 | 139.99 | ||
| 186.50 | 133.23 | 187.39 | 129.37 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



