
Submitted:
15 February 2023
Posted:
17 February 2023
You are already at the latest version
Abstract
HAX1 is a multifunctional protein involved in the antagonism of apoptosis in cellular response to oxidative stress. In the present study we identified HAX1 as novel binding partner for Che-1/AATF, a pro-survival factor which plays a crucial role in fundamental processes, including response to multiple stresses and apoptosis. HAX1 and Che-1 proteins show extensive colocalization in mitochondria and we demonstrated that their association is strengthened after oxidative stress stimuli. Interestingly, in MCF-7 cells, resembling luminal estrogen receptor (ER) positive breast cancer, we found that Che-1 depletion correlates with decreased HAX1 mRNA and protein levels, and this event is not significantly affected by oxidative stress induction. Furthermore, we observed an enhancement of the previously reported interaction between HAX1 and estrogen receptor alpha (ERα) upon H2O2 treatment. These results indicate the two anti-apoptotic proteins HAX1 and Che-1 as coordinated players in cellular response to oxidative stress with a potential role in estrogen sensitive breast cancer cells.
Keywords:
HAX1
; Che-1/AATF
; apoptosis
; mitochondria
; oxidative stress
; breast cancer
; ERα
1. Introduction
HAX1 (HCLS1-associated protein X-1) is an anti-apoptotic protein ubiquitously expressed in various tissues and tumors [1,2]. Mutations in the human HAX1 gene are responsible for autosomal recessive severe congenital neutropenia (SCN or Kostmann disease) and neurological abnormalities, mainly resulting from defective mitochondrial control of apoptosis [3,4]. HAX1 is involved in many cellular processes and functions, including regulation of apoptosis, cell proliferation [5-7], cell adhesion and migration [8-11], endocytosis [10], autophagy [12], ribosome biogenesis [13] and mRNA metabolism [13-16]. HAX1 is localized in mitochondria, endoplasmic reticulum, nucleus, plasma membrane/on the leading edge of lamellipodia and cytoplasmic vesicles/granules, as P-bodies [17]. It is described mainly as mitochondrial protein and is critical in maintaining the inner mitochondrial membrane potential [3]. HAX1 participates in the regulation of apoptosis through attenuation of the damaged signals from mitochondria and endoplasmic reticulum [18-26]. In the antioxidative stress response HAX1 exerts its anti-apoptotic role through different pathways not fully clarified. It can affect the Akt1/MDM2/p53 axis and the expression of p21, Bax and p53 proteins to participate in cell apoptosis control [27]. Moreover, it has been recently reported that HAX1 in response to oxidative or genotoxic stress activates the non-receptor tyrosine kinase c-Abl, thereby promoting c-Abl-mediated activation of catalase and glutathione peroxidase resulting in reactive oxygen species (ROS) clearance [28]. Deregulation of the expression levels and subcellular distribution of HAX1 is associated with the development and progression of severe diseases, including cancer and psoriasis [29]. HAX1 overexpression was reported in several cancers [5,10,30-32], including breast cancer and its expression analysis in correlation to metastasis revealed its significant prognostic value for luminal estrogen receptor positive (ER+) breast cancer metastasis [2,7,10,33]. It has been reported that HAX1 forms a complex with prohibitin 2 (PHB2) that in the presence of Estrogen receptor alpha (ERα) and estradiol, translocates to the nucleus, suggesting a link between HAX1 and estrogen-receptor signaling [17,34]. Moreover, a possible involvement of HAX1 in estrogen signaling is also supported by microarray studies, which classified HAX1 as estrogen-responsive [35,36]. Of note, HAX1 was shown to directly bind ERα that represents a pro-metastatic factor [7,37].
Pathologic overexpression of HAX1 was observed also in psoriasis, a severe inflammatory disease characterized by increased proliferation and dysfunctional apoptosis of keratinocytes [38,39]. Interestingly, we recently reported that HAX1 interacts with the psoriasis candidate Coiled-Coil alpha-Helical Rod 1 (CCHCR1) protein [14]. Considering the ability of HAX1 to bind RNA, we identified pools of mRNAs associated with both HAX1 and CCHCR1, including Vimentin, Cortactin and CAMP/LL37 mRNAs, whose genes are all deregulated in psoriasis. HAX1 may influence fate and stability of specific transcripts via protein/protein interaction [14]. Due to HAX1 protein intrinsically disordered structure a large number of HAX1 protein targets are weakly bound, making difficult to complete HAX1 interactome [17].
In the present study, HAX1 was determined to be a novel binding partner of Apoptosis Antagonizing Transcription Factor (AATF)/Che-1 protein. Che-1 is an RNA polymerase (RNA Pol) I and II interacting protein highly conserved during evolution [40,41]. It is involved in many fundamental cellular processes, such as transcriptional regulation, ribosome biogenesis, cell-cycle and apoptosis control, cellular response to DNA damage and stress, RNA binding and inflammatory response activation [42-46]. At the cellular level, Che-1 predominantly shows a nuclear and nucleolar localization [41]. However, Che-1 protein distribution has been also reported in centrosomes, focal adhesion, Golgi apparatus, and mitochondria [47-50]. Che-1 protects cells from multiple stress stimuli such as DNA damage, endoplasmic reticulum (ER) stress, hypoxia, or glucose deprivation by inducing cell cycle arrest, autophagy, or apoptosis inhibition. The critical role of Che-1 in the modulation of apoptosis can be played either by activation of antiapoptotic pathways or by inhibition of pro-apoptotic pathways [42]. The interaction of Che-1 with different proteins such as DAP-like kinase (Dlk), prostate apoptosis response 4 (Par-4), and neurotrophin receptor-interacting MAGE homolog (NRAGE/MAGED1) resulted in the inhibition of apoptosis [43,51-53]. In addition, Che-1 shows a protective role in human kidney proximal tubule cells, where this protein has been observed to counteract apoptotic cell death following induced-renal injury by preserving mitochondrial function and reducing oxidative damage [54]. Over the last years, several pieces of evidence indicate that Che-1 serves as a critical regulator in various cancers and promotes tumorigenesis by protecting cancer cells from apoptosis induction, favoring cell proliferation, or promoting cell survival by autophagy [44,55-58]. In breast cancer Che-1 is upregulated and its gene silencing leads to stimulation of apoptotic cell death via upregulation of proapoptotic genes and downregulation of ERα, a key factor in breast cancer development [42,59]. Of importance, Bruno and colleagues have recently shown that Che-1 depletion leads to downregulation of HAX1 in Multiple Myeloma [44].
In this study, we show that HAX1 interacts with Che-1 and that Che-1 depletion correlates with downregulation of HAX1 in MCF-7 cells. HAX1 and Che-1 proteins colocalize mainly in mitochondria and notably we found that their association is strengthened after oxidative stress stimuli in MCF-7 cells. Interestingly, we observed an enhancement also of the previously reported interaction between HAX1 and ERα, upon H2O2 treatment [37]. These results suggest that the two anti-apoptotic proteins HAX1 and Che-1 cooperate in cellular response to oxidative stress, likely in association with ERα in estrogen sensitive contexts, as ER positive breast cancer subtypes.
2. Materials and Methods
2.1. Yeast two-hybrid selection
The cDNA region encoding the C-terminal part of human Che-1 (aa 470 to 558) was cloned into the pGBKT7 vector (Takara Bio Inc., Kusatsu, Shiga, Japan) in frame with the Gal4 DNA binding domain (pGBKT7-Che-1-C) and used as bait for yeast two-hybrid screening as previously described [51]. Bait interaction specificity was further analyzed cloning in pGBKT7 vector both the complete open reading frame of human Che-1 (pGBKT7-Che-1) and the Che-1 portion from aa 1 to aa 470 (pGBKT7- Che-1ΔC).
2.2. Cell cultures and treatments
HeLa human cervical cancer cells (ATCC, CCL-2) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS), L-glutamine and penicillin/streptomycin (Thermo Fisher Scientific, Inc., Waltham, MA, USA). MCF-7 human breast cancer cells (ATCC, HTB-22) were grown in DMEM supplemented with 10% heat-inactivated FBS, L-glutamine and penicillin/streptomycin. All cell cultures were maintained at 37°C in a humidified atmosphere of 5% CO2. Mycoplasma contamination was periodically checked by polymerase chain reaction (PCR) analysis.
Hydrogen peroxide (H2O2) was purchased from Merck & Co Inc. (Rahway, NJ USA). Oxidative stress was induced with daily fresh H2O2 solution prepared from stock solution 30 % (w/w) in H2O at concentration and time indicated.
2.3. Transfection with siRNA and DNA constructs
Transfections were carried out using Lipofectin® Transfection Reagent and PLUS™ reagent or Lipofectamine 3000 reagents cells according to the manufacturer’s instructions (Thermo Fisher Scientific Inc.). Cells were analyzed 48-72 h after transfection, and the efficiency of transfection and silencing was determined by RT-qPCR, immunoblotting, or immunofluorescence. siRNA-mediated interference experiments were performed using Stealth siRNA oligonucleotides (siAATF cat. n. 1299003 - HSS120157/HSS120158/HSS120159 and siControl cat. n. 12935300) (Thermo Fisher Scientific Inc.) for Che-1 and siSMART pool oligonucleotides (siHAX1 cat. M012168-01 or siControl cat. 001810-10) (Dharmacon Inc, Carlo Erba Reagents Srl, Cornaredo, MI, Italy) for HAX1.
2.4. Immunofluorescence
For endogenous proteins, cells were fixed in 3:7 methanol-acetone for 20 min at –20°C, air dried and then preincubated in SM buffer (PBS containing 0.05% saponin and 5% BSA (Merck & Co Inc., Rahway, NJ, USA)) for 30 min at room temperature. Cells were then incubated overnight at 4°C with the appropriate primary antibodies diluted in SM buffer. Cells were washed three times with PBS and incubated with secondary antibody for 45 min in SM buffer at room temperature. Cells transiently transfected with the indicated constructs were fixed 24 h after transfection with 4% paraformaldehyde solution for 10 min at room temperature and then permeabilized with 0.2% IgepalCA 630 (Merck & Co Inc.) for 10 min at room temperature. To label mitochondria, cells were incubated with 250 nM of MitoTracker® Red CMXRos M7512 (Thermo Fisher Scientific, Inc.) according to the manufacturer’s instructions before fixation. Slides were mounted with ProLong™ Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific, Inc.). Slides were examined by conventional epifluorescence microscopy (Olympus BX51). Images were captured using a digital camera SPOT RT3 and merged using the IAS2000 software.
2.5. Cell lysis and sub-cellular fractionation
Whole-cell extracts were prepared using the Lysis buffer (50 mM Tris-HCl pH 7.5, 250 mM NaCl, 5 mM EDTA pH 8, 50 mM NaF, 0.1 mM Na3VO4, 0.1% Triton X-100, 5% glycerol) supplemented with a proteinase inhibitor cocktail (Merck & Co Inc.). Lysates were centrifuged at 12000 g for 10 min at 4 °C and supernatants collected. The mitochondrial enriched fraction was obtained by the Qproteome Mitochondria Isolation Kit (QIAGEN Hilden, Germany) according to the manufacturer's instructions. The heavy membrane (HM) enriched fraction was obtained as previously described [50].
2.6. Co-immunoprecipitation and Western blotting
After protein extraction as described above, co-immunoprecipitations were performed. An equal amount of proteins (either whole-cell extract or mitochondria/heavy membrane enriched cell extract), was diluted in a 2:1 ratio between Lysis Buffer and Dilution buffer (50 mM Tris-HCl pH 7.5, 50 mM NaCl, 50 mM NaF, 0.1 mM Na3VO4, 15% glycerol). Then, the diluted cell extract was pre-cleared with protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). Immunoprecipitation assays were performed overnight at 4◦C with 2/4 μg of indicated antibodies, previously bound to A/G PLUS- Agarose beads. A normal mouse IgG or a “no-antibody” immunoprecipitation were performed as a negative control. Protein-bound beads were recovered by centrifugation and washed three times with the Wash buffer (1 Lysis buffer: 1 Dilution buffer) and once in PBS buffer. Immunoprecipitated proteins were eluted from beads heating samples at 70 °C in LDS Sample Buffer for 10 min, followed by electrophoresis on NuPAGE ® Bis-Tris Gel System (Thermo Fisher Scientific, Inc.). Western blotting analysis of protein samples was carried out by standard procedures and signals were visualized by chemiluminescence ECL™ Select Western Blotting Detection Reagent (Cytiva, Marlborough, MA, USA) according to the manufacturer’s instructions.
2.7. Cell viability assay
Cell viability was determined by MTT reduction assay. MCF-7 cells were plated at a density of 8 × 103 cells/well in octuplicate on 96-well plate and transfected the day after with the indicated constructs. 24 h after transfection cells were exposed to H2O2 at various concentrations (0 control, 0.25 mM, 0.5 mM, 1 mM) for 16 h. The culture medium was discarded, and cells were incubated with MTT (0.5 mg/mL in PBS; 100 µL/well) (Thermo Fisher Scientific, Inc.) at 37 °C for 4 h. Then, MTT was removed and 100 µL of DMSO was added to each well to dissolve the purple formazan crystals. Lastly, 570 nm absorbance values against 620 nm absorbance were calculated from the automated microplate reader (Bio-Rad Laboratories, Inc. Hercules, California, United States).
2.8. Padlock assay
Phosphorylation of the padlock probe and Padlock assays were performed as previously described [14,60]. The Padlock probe sequences are presented in Supplementary Table S1.
2.9. RNA extraction, retrotranscription and Quantitative real-time PCR (qPCR)
RNA was isolated using TRIzol® reagent (Thermo Fisher Scientific, Inc.) and was then reverse transcribed using a HighCapacity cDNA Reverse Transcription kit (Thermo Fisher Scientific, Inc.). Quantitative real-time PCR assays were performed using the Fast SYBR™ Green Master Mix (Thermo Fisher Scientific, Inc.) in a StepOnePlus™ Real-Time PCR System according to the manufacturer’s protocol. GAPDH was used for the normalization of mRNA and relative expression was calculated using the comparative Ct methods (2− ΔΔCt). Primers are presented in Supplementary Table S1.
2.10. Antibodies
The following antibodies were used: anti-Che-1 rat polyclonal antibody [50] for immunofluorescence (1:20 dilution); anti-Che-1 rabbit polyclonal antibody [41] for immunoprecipitation, western blotting (1:1000 dilution) and immunofluorescence (1:300 dilution); anti-AATF mouse monoclonal antibody (Merck & Co Inc.) for immunoprecipitation and western blotting (1:250 dilution); anti-HAX1 rabbit polyclonal antibody (Proteintech Group, Rosemont, IL, USA) for immunoprecipitation, western blotting (1:200 dilution) and immunofluorescence (1:20 dilution); anti-HAX1 mouse monoclonal antibody (BD Bioscences, Franklin Lakes, New Jersey, USA) for western blotting (1:250 dilution); anti-EGFP mouse monoclonal antibody (Takara Bio USA, Inc.) for western blotting (1:2000 dilution); anti-Cytochrome c purified mouse antibody (BD Bioscences) for western blotting (1:500 dilution); anti-Tom20 rabbit polyclonal antibody (cat. no. sc-11415 Santa Cruz Biotechnology) for western blotting (1:500 dilution); anti-myc mouse monoclonal antibody 9E10 clone hybridoma-conditioned medium for immunoprecipitation and western blotting (1:15 dilution); anti-α-tubulin mouse monoclonal antibody, anti-α-tubulin rabbit polyclonal antibody (Merck & Co Inc) for western blotting (1:10000 dilution); anti-Estrogen Receptor alpha mouse monoclonal antibody (cat. no. sc-8002 Santa Cruz Biotechnology) for western blotting (1:200 dilution); anti-Nucleolin mouse monoclonal antibody (Medical & Biological Laboratories Co., Ltd., Nagoya, Japan) for immunofluorescence (1:50 dilution); Alexa-Fluor-488 or Alexa-Fluor-594-conjugated secondary antibodies (Thermo Fisher Scientific, Inc.) for immunofluorescence (1:200 dilution); the secondary antibodies conjugated to horseradish peroxidase (Jackson ImmunoResearch Laboratories West Grove, Pennsylvania, USA) for western blotting (1:5000 dilution).
2.11. Statistics
All experiments were repeated at least 3 times. Data are presented as means ± SD, when indicated. Two-tailed Student’s t-tests with Benjamini–Hochberg correction were performed to compare one parameter between two groups. Statistical significance is indicated by asterisks as follows: *P<0.05, **P<0.01, ***P<0.001. Microscopy images shown in the paper correspond to the most representative experiments.
3. Results
3.1. HAX1 interacts with Che-1
To detect candidate proteins that interact with Che-1/AATF, yeast two-hybrid (Y2H) assays were performed. A cDNA fragment encoding the C-terminal 88 amino acid residues of Che-1 (Che-1-C) (Figure 1A), highly conserved among eucaryotes [61], was fused in-frame with the yeast Gal4 DNA binding domain in the vector pGBKT7 and used to screen a two-hybrid cDNA library prepared from human adult brain [51]. Several clones were isolated and the clone encoding for the HCLS1 Associated Protein X-1 (HAX1) was selected for further studies. The specificity of the Che-1/HAX1 interaction was confirmed in a two-hybrid assay co-transforming HAX1 with either Che-1-C or PGBKT7 constructs containing full-length Che-1 or Che-1 lacking the C-terminal region (Che-1-ΔC) (Figure 1A). Both Che-1 and Che-1-C bound HAX1 whereases Che-1- ΔC did not interact with HAX1 (Figure 1A).
With the intent to define HAX1/Che-1 interaction in mammalian cells we performed a series of immunofluorescence and co-immunoprecipitation assays in two different tumoral cell lines: HeLa and MCF-7 cells. In Figure 1B, immunofluorescence assays showed consistent partial colocalization of HAX1 and Che-1 proteins with a major site in mitochondria in both HeLa and MCF-7 cell lines. To confirm the interaction between the two proteins in this district, mitochondrial enriched lysate obtained from HeLa cells was used in co-immunoprecipitation experiments. A clear interaction between HAX1 and Che-1 proteins is shown in Figure 1C. In addition, we demonstrated HAX1/Che-1 interaction in MCF-7 cells performing co-immunoprecipitation experiments using heavy membrane cellular fraction (HM), highly enriched in mitochondria (Figure 1D).
3.2. Characterization of HAX1/Che-1 interaction and dominant negative effect of HAX1 deletion construct
Next, we intended to determine the protein regions of HAX1 (isoform 001) (Figure 2A) crucial for the interaction with Che-1. To this end, HeLa cells were transiently co-transfected with myc-Che-1 full-length and EGFP-HAX1 or EGFP-HAX1 deletion constructs (Figure 2B). Then, whole cell extracts were immunoprecipitated with anti-myc antibody and analyzed by western blot. Figure 2B and Figure 2C show that the construct expressing the C terminal portion of HAX1 (179-279 aa) displays the highest binding affinity toward Che-1 protein, with two crucial interacting regions working in synergy, one region spanning from amino acids 179 to 190 and the other between amino acids 234 and 279. In parallel the same HAX1 mutants were characterized by immunofluorescence, and the two complementary mutants HAX1N-190 (1 aa-190 aa), mimicking HAX1-Kostmann mutant protein, and HAX1C-89 (190 aa-279 aa) together with HAX1C-55, do not maintain strictly mitochondrial localization (Supplementary Figure S1A). Conversely, as shown in figure 1D the HAX1C-100 mutant with a high binding affinity to Che-1 maintains mainly mitochondrial localization. The high affinity of HAX1C-100 mutant to Che-1 binding, prompted us to investigate whether HAX1C-100 could play any role(s) in interfering with HAX1/Che-1 protein interaction. As shown in Figure 2E, increased expression of HAX1C-100 resulted in a marked decrease of EGFP-HAX1 full length co-immunoprecipitated with myc-Che-1. This result indicates that HAX1C-100 mutant acts as a dominant negative by inhibiting HAX1/Che-1 interaction. To elucidate HAX1C-100 mutant dominant negative properties upon oxidative stress, we measured cellular viability in MCF-7 cells transfected with EGFP (control), EGFP-HAX1 and EGFP-HAX1C-100 respectively, treated with increasing concentrations of H2O2 for 16 hr. Cell viability following H2O2 treatment was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. As shown in Figure 2F cells transfected with EGFP-HAX1, compared to cells transfected with EGFP, display in response to oxidative stress a clear increase in viability related to the anti-apoptotic role of HAX1, as expected. The viability of cells transfected with EGFP-HAX1C-100 goes in the opposite trend. We suppose that HAX1C-100 acts as effective dominant negative mutant by competing with the interaction of endogenous Che-1 and HAX1 proteins and counteracting their anti-apoptotic effect [28,42,43].
3.3. The association of HAX1 and Che-1 proteins is strengthened by oxidative stimuli
Then we examined HAX1 and Che-1 endogenous proteins both individually and in association under oxidative stress conditions. First, we compared subcellular localization of HAX1 and Che-1 proteins before and after H2O2 treatment (Figure 3A and Supplementary Figure S2A). In HeLa cells, in the absence of stress condition, HAX1 localized primarily within the mitochondria and Che-1 in nucleus/nucleolus and mitochondria. Upon H2O2 treatment, we observed a clear alteration of the subcellular localization of both HAX1 and Che-1. Consistent with previous finding that reported HAX1 to be a nucleocytoplasmic shuttling protein after specific cellular stress [62], we detected a certain HAX1 nuclear accumulation upon H2O2 treatment (Figure 3A).
Concomitantly, we observed a statistically significant loss of nucleolar Che-1 staining. The intensity of Che-1 fluorescence signal in the nucleolus was decreased in both HeLa and MCF-7 cells (Figure 3A and Supplementary Figure S2A and S2B). Co-immunostaining with nucleolin, a major component of nucleoli, underlined an independent mislocalization of Che-1 and nucleolin upon H2O2 treatment (Supplementary Figure S3A). We then quantified by ImageJ analysis the colocalization between HAX1 and Che-1 before and after H2O2 treatment. As shown in Figure 3B a reinforced colocalization between HAX1 and Che-1 was observed in both cytosol and nucleus after H2O2 treatment. In agreement with these findings, by co-immunoprecipitation in MCF-7 cells, it was detected an enhanced interaction between HAX1 and Che-1 proteins upon H2O2 stimulation (Figure 3C and 3D). These results underline the importance of HAX1/Che-1 interaction in cellular response to oxidative stress. Modulation of oxidative stress status in the cells is a risk factor associated with breast cancer. Previous reports have demonstrated that HAX1 is overexpressed in breast cancer and represents an independent risk factor for metastasis. Importantly, HAX1 was previously shown to directly bind ERα, a protein involved in breast cancer initiation and progression via an oxidative stress-mediated pathway [7,34,37,63,64]. We confirmed in MCF-7 cells the interaction of HAX1 with ERα by co-immunoprecipitation experiments. Remarkably, we observed that the HAX1/ERα association is also strengthened by oxidative stimuli (Figure 3D).
3.4. Knockdown of Che-1 induces downregulation and altered cellular distribution of HAX1
To further clarify the significance of HAX1/Che-1 interaction in steady state condition and upon oxidative stress, we performed RNAi-mediated knockdown studies with or without oxidative stress induction. Taking into account that HAX1 and Che-1 proteins are key regulators of apoptosis, MCF-7 cells were transiently transfected with either Che-1 or HAX1 siRNAs and then treated or not with H2O2. Quantitative real time RT-PCR (qPCR) analysis showed that Che-1 knockdown specifically reduces the HAX1 mRNA levels of about 50 % in steady state condition, and this reduction is slightly less when Che-1 silencing is combined to H2O2 treatment (Figure 4A and Supplementary Figure S4A). The effect of Che-1 knockdown on HAX1 mRNA levels in steady state condition was also confirmed by padlock experiments that allowed sensitive detection of HAX1 endogenous mRNAs (Figure 4B).
Next, we evaluated the effects on HAX1 protein levels following Che-1 silencing in MCF-7 cells, treated or not with H2O2. As shown in Figure 4C, western blot analysis revealed that Che-1 knockdown induces a decrease of HAX1 protein levels coherent with the HAX1 mRNA decrease in untreated cells, with a slight further decrease upon H2O2 treatment (Figure 4C and Supplementary Figure S4B). The effect of Che-1 knockdown on HAX1 protein level/localization was explored by immunofluorescence experiments following Che-1 silencing, in MCF-7 untreated and treated with H2O2. Figure 4 D shows upon Che-1 silencing a general decrease of HAX1 protein staining coupled with an increased HAX1 presence in nuclear discrete dots. This phenomenon in our experimental conditions appears unaffected by oxidative stress induction. All together these data indicate that Che-1 depletion induces downregulation and altered cellular distribution of HAX1. In contrast, HAX1 silencing in our experimental conditions induces only a slight decrease of Che-1 mRNA levels both in untreated and H2O2 treated MCF-7 cells (Supplementary Figure S5A). Accordingly, Che-1 protein levels analyzed in steady state condition appear not significantly altered upon HAX1 knockdown (Supplementary Figure S5B).
These results suggest a direct involvement of Che-1 in the regulation of HAX1 regardless of oxidative stress induction.
4. Discussion
Although HAX1 protein is implicated in many cellular processes, its molecular mechanisms of action remain elusive. We have previously shown that HAX1 interacts with the psoriasis candidate Coiled-Coil alpha-Helical Rod 1 (CCHCR1) protein and it may influence fate and stability of specific transcripts via protein/protein interaction [14]. In the present study, we provide new insights into the biological role of the HAX1 protein, by showing that HAX1 is a new Che-1/AATF interaction partner and notably their interaction is strengthened after oxidative stress stimuli. We have shown that HAX1 binds to the C-terminal region of Che-1 and colocalize with Che-1 mainly at mitochondrial level. Previous studies indicated that HAX1 interacts weakly with a wide spectrum of proteins and its interactome tends to be cell-specific [17]. We revealed the presence of HAX1 and Che-1 in the same immune-complex in heavy membrane/mitochondria enriched fractions in two different human cell lines, HeLa derived from cervical carcinoma and MCF-7 derived from breast cancer. To understand more fully the nature of the HAX1/Che-1 interaction, we defined the regions of HAX1 protein critical for Che-1 binding, using HAX1 deletion constructs. Significantly, HAX1 C-terminal region spanning the last 100aa (HAX1C-100), including the transmembrane domain with a conserved α-helix motif crucial for protein-protein interaction [17,28], is responsible for Che-1 interaction. The HAX1C-100 construct maintains mainly mitochondrial localization and it shows the highest binding affinity to Che-1. Importantly, HAX1C-100 acts as effective dominant negative mutant by competing with the interaction of endogenous Che-1 and HAX1 proteins and counteracting their anti-apoptotic effect [28,42,43]. Since HAX1 is involved in the antagonism of apoptotic processes in cellular response to oxidative stress [12,17,28] and Che-1 as well participates in cellular response to different types of stresses [42-45,52,55,65,66], we examined whether HAX1 and Che-1 individually and in association were regulated under oxidative stress conditions. We observed an altered subcellular localization of both HAX1 and Che-1 proteins upon H2O2 treatment. In accordance with previous studies, we detected an evident HAX1 nuclear accumulation upon H2O2 treatment [62]. We also reported a significant decrease of Che-1 nucleolus-staining in both HeLa and MCF-7 cells. The Che-1 shuttling between the nucleolus and nucleoplasm in response to oxidative stress conditions is in accordance with the recent evidence that Che-1 is essential in ribosome biogenesis and with the emerging role of nucleoli in cellular stress response [40,45,46,67-69]. Anyway, we observed and independent mislocalization of Che-1 and nucleolin upon H2O2 treatment, indicating for Che-1 an individual fate, that is not merely a consequence of stress-related rearrangement of nucleolar compartment. Furthermore, we showed that HAX1/Che-1 colocalization is significantly reinforced after H2O2 treatment in the whole cell as well as in both cytosol and nucleus. Significantly, the increased percentage of colocalization between the two proteins correlates with their strengthened interaction upon oxidative stress induction. These results underline the importance of HAX1/Che-1 interaction in cellular response to oxidative stress. Oxidative stress plays an important role in both initiation and progression of breast cancer. Both HAX1 and Che-1 are crucial targets in the field of cancer research on account of their involvement in regulation of apoptosis and cell survival. Importantly HAX1 regulates cell migration, a key process in carcinogenesis and metastasis [9]. Of note, both HAX1 and Che-1 have been shown by other research groups to directly bind ERα, a protein involved in breast cancer initiation and progression via an oxidative stress-mediated pathway [7,34,37,63,64,70,71]. We report for the first time that the interaction of HAX1 with ERα is strengthened by oxidative stimuli. Moreover, we demonstrated that Che-1 depletion correlates with HAX1 downregulation and altered cellular distribution, regardless of oxidative stress. In this perspective, HAX1 could contribute to antiapoptotic activity exerted by Che-1, acting as Che-1 downstream gene. Our findings on HAX1/Che-1 interaction provide insights into the molecular mechanisms underlying their antiapoptotic activity in both physiological and pathological conditions. Importantly, HAX1, Che-1 and ERα have been independently shown to be RNA binding proteins [13,45,72], this common ability allows to speculate that at least part of their functions can be exerted through the control of specific RNA metabolism. It is possible to hypothesize that these three proteins may bind and regulate single transcripts or group of transcripts in shared pathways. Further studies are needed to define the potential coordinated activity of HAX1, Che-1 and ERα in cellular stress response pathways involved in breast cancer progression, pointing to the use of these molecules as biomarkers and effective therapeutic targets.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: Subcellular distribution of EGFP-HAX1 and its deletion constructs; Figure S2: Subcellular distribution and colocalization of HAX1 and Che-1 upon H2O2 treatment; Figure S3: Subcellular distribution and colocalization of Che-1 and nucleolin upon H2O2 treatment; Figure S4: Che-1 mRNA and protein levels upon Che-1 siRNA in MCF-7 untreated and treated with H2O2; Figure S5: HAX1 and Che-1 mRNA and protein levels upon HAX1 siRNA in MCF-7; Table S1: Oligos used in the present study.
Author Contributions
C.P. (Cinzia Pisani): investigation, conceptualization, formal analysis, writing—original draft preparation; A.O: investigation, methodology, data curation; F.G.: methodology, formal analysis, data curation; S.I: methodology, review and editing; R.D.A.: methodology; M. F., A.C. and M.d.V.: resources, review and editing; M.G.D.C.: investigation, writing—review and editing; C.P. (Claudio Passananti) and N.C.: conceptualization, investigation, supervision, funding acquisition, writing—original draft preparation. All authors have read and agreed the present version of the manuscript.
Funding
This research was supported by MUR PNRR PE13 INF-ACT grant.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We thank Michele Sansone for his constant support in imaging acquisition.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Suzuki, Y.; Demoliere, C.; Kitamura, D.; Takeshita, H.; Deuschle, U.; Watanabe, T. HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J Immunol 1997, 158, 2736–2744. [Google Scholar] [CrossRef] [PubMed]
- Trebinska, A.; Rembiszewska, A.; Ciosek, K.; Ptaszynski, K.; Rowinski, S.; Kupryjanczyk, J.; Siedlecki, J.A.; Grzybowska, E.A. HAX-1 overexpression, splicing and cellular localization in tumors. BMC Cancer 2010, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Murgia, M.; Linder, M.I.; Mizoguchi, Y.; Wang, C.; Łyszkiewicz, M.; Ziȩtara, N.; Liu, Y.; Frenz, S.; Sciuccati, G.; et al. HAX1-dependent control of mitochondrial proteostasis governs neutrophil granulocyte differentiation. J Clin Invest 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Klein, C. Kostmann's Disease and HCLS1-Associated Protein X-1 (HAX1). J Clin Immunol 2017, 37, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, T.; You, B.; Shan, Y.; Shi, S.; Cao, X.; Qian, L. Expression and Function of HAX-1 in Human Cutaneous Squamous Cell Carcinoma. J Cancer 2015, 6, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhong, Y.; Meng, L.; Chen, Y.; Liu, Y.; Wu, A.; Li, X.; Wang, M. HAX1 enhances the survival and metastasis of non-small cell lung cancer through the AKT/mTOR and MDM2/p53 signaling pathway. Thorac Cancer 2020, 11, 3155–3167. [Google Scholar] [CrossRef] [PubMed]
- Trebinska-Stryjewska, A.; Szafron, L.; Rembiszewska, A.; Wakula, M.; Tabor, S.; Sienkiewicz, R.; Owczarek, J.; Balcerak, A.; Felisiak-Golabek, A.; Grzybowska, E.A. Cytoplasmic HAX1 Is an Independent Risk Factor for Breast Cancer Metastasis. J Oncol 2019, 2019, 6375025. [Google Scholar] [CrossRef] [PubMed]
- Radhika, V.; Onesime, D.; Ha, J.H.; Dhanasekaran, N. Galpha13 stimulates cell migration through cortactin-interacting protein Hax-1. J Biol Chem 2004, 279, 49406–49413. [Google Scholar] [CrossRef]
- Balcerak, A.; Trebinska-Stryjewska, A.; Wakula, M.; Chmielarczyk, M.; Smietanka, U.; Rubel, T.; Konopinski, R.; Macech-Klicka, E.; Zub, R.; Grzybowska, E.A. HAX1 impact on collective cell migration, cell adhesion, and cell shape is linked to the regulation of actomyosin contractility. Mol Biol Cell 2019, 30, 3024–3036. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Keppler, M.D.; Jazayeri, M.; Thomas, G.J.; Parsons, M.; Violette, S.; Weinreb, P.; Hart, I.R.; Marshall, J.F. HS1-associated protein X-1 regulates carcinoma cell migration and invasion via clathrin-mediated endocytosis of integrin alphavbeta6. Cancer Res 2007, 67, 5275–5284. [Google Scholar] [CrossRef]
- Hu, Y.L.; Feng, Y.; Ma, P.; Wang, F.; Huang, H.; Guo, Y.B.; Li, P.; Mao, Q.S.; Xue, W.J. HAX-1 promotes the migration and invasion of hepatocellular carcinoma cells through the induction of epithelial-mesenchymal transition via the NF-κB pathway. Exp Cell Res 2019, 381, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Cai, W.F.; Wang, L.; Liu, G.S.; Paul, C.; Jiang, L.; Wang, B.; Gao, X.; Wang, Y.; Wu, S.Z. Identification of the Functional Autophagy-Regulatory Domain in HCLS1-Associated Protein X-1 That Resists Against Oxidative Stress. DNA Cell Biol 2018, 37, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Balcerak, A.; Macech-Klicka, E.; Wakula, M.; Tomecki, R.; Goryca, K.; Rydzanicz, M.; Chmielarczyk, M.; Szostakowska-Rodzos, M.; Wisniewska, M.; Lyczek, F.; et al. The RNA-Binding Landscape of HAX1 Protein Indicates Its Involvement in Translation and Ribosome Assembly. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Pisani, C.; Onori, A.; Gabanella, F.; Di Certo, M.G.; Passananti, C.; Corbi, N. Identification of protein/mRNA network involving the PSORS1 locus gene CCHCR1 and the PSORS4 locus gene HAX1. Exp Cell Res 2021, 399, 112471. [Google Scholar] [CrossRef]
- Al-Maghrebi, M.; Brule, H.; Padkina, M.; Allen, C.; Holmes, W.M.; Zehner, Z.E. The 3' untranslated region of human vimentin mRNA interacts with protein complexes containing eEF-1gamma and HAX-1. Nucleic Acids Res 2002, 30, 5017–5028. [Google Scholar] [CrossRef] [PubMed]
- Sarnowska, E.; Grzybowska, E.A.; Sobczak, K.; Konopinski, R.; Wilczynska, A.; Szwarc, M.; Sarnowski, T.J.; Krzyzosiak, W.J.; Siedlecki, J.A. Hairpin structure within the 3'UTR of DNA polymerase beta mRNA acts as a post-transcriptional regulatory element and interacts with Hax-1. Nucleic Acids Res 2007, 35, 5499–5510. [Google Scholar] [CrossRef]
- Wakula, M.; Balcerak, A.; Rubel, T.; Chmielarczyk, M.; Konopinski, R.; Lyczek, F.; Grzybowska, E. The interactome of multifunctional HAX1 protein suggests its role in the regulation of energy metabolism, de-aggregation, cytoskeleton organization and RNA-processing. Biosci Rep 2020. [Google Scholar] [CrossRef] [PubMed]
- Yap, S.V.; Vafiadaki, E.; Strong, J.; Kontrogianni-Konstantopoulos, A. HAX-1: a multifaceted antiapoptotic protein localizing in the mitochondria and the sarcoplasmic reticulum of striated muscle cells. J Mol Cell Cardiol 2010, 48, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.B.; Deng, X.; Wei, Y. Hematopoietic Substrate-1-Associated Protein X-1 Regulates the Proliferation and Apoptosis of Endothelial Progenitor Cells Through Akt Pathway Modulation. Stem Cells 2018, 36, 406–419. [Google Scholar] [CrossRef]
- Szwarc, M.; Sarnowska, E.; Grzybowska, E.A. [HAX-1 protein: multifunctional factor involved in apoptosis, cell migration, endocytosis and mRNA transport]. Postepy Biochem 2007, 53, 218–227. [Google Scholar]
- Yan, J.; Ma, C.; Cheng, J.; Li, Z.; Liu, C. HAX-1 inhibits apoptosis in prostate cancer through the suppression of caspase-9 activation. Oncol Rep 2015, 34, 2776–2781. [Google Scholar] [CrossRef]
- Chao, J.R.; Parganas, E.; Boyd, K.; Hong, C.Y.; Opferman, J.T.; Ihle, J.N. Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature 2008, 452, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, Y.S.; Liu, Z.; Bodyak, N.; Rigor, D.; Bisping, E.; Pu, W.T.; Kang, P.M. Overexpression of HAX-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circ Res 2006, 99, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Kirshenbaum, L.A. HAX-1 represses postmitochondrial caspase-9 activation and cell death during hypoxia-reoxygenation. Circ Res 2006, 99, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Li, W.B.; Feng, J.; Geng, S.M.; Zhang, P.Y.; Yan, X.N.; Hu, G.; Zhang, C.Q.; Shi, B.J. Induction of apoptosis by Hax-1 siRNA in melanoma cells. Cell Biol Int 2009, 33, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, Z.; Li, X.; Wang, G.; Liu, W.; Dong, S.; Cai, S.; Tao, D.; Yan, Q.; Wang, J.; et al. hSav1 interacts with HAX1 and attenuates its anti-apoptotic effects in MCF-7 breast cancer cells. Int J Mol Med 2011, 28, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Song, L.; Zhao, W.; Wei, Y.; Guo, X.B. HAX-1 Protects Glioblastoma Cells from Apoptosis through the Akt1 Pathway. Front Cell Neurosci 2017, 11, 420. [Google Scholar] [CrossRef]
- Dong, Q.; Li, D.; Zhao, H.; Zhang, X.; Liu, Y.; Hu, Y.; Yao, Y.; Zhu, L.; Wang, G.F.; Liu, H.; et al. Anti-apoptotic HAX-1 suppresses cell apoptosis by promoting c-Abl kinase-involved ROS clearance. Cell Death Dis 2022, 13, 298. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Grzybowska, E. HAX-1: a multifunctional protein with emerging roles in human disease. Biochim Biophys Acta 2009, 1790, 1139–1148. [Google Scholar] [CrossRef]
- Li, X.; Jiang, J.; Yang, R.; Xu, X.; Hu, F.; Liu, A.; Tao, D.; Leng, Y.; Hu, J.; Gong, J.; et al. Expression of HAX-1 in colorectal cancer and its role in cancer cell growth. Mol Med Rep 2015, 12, 4071–4078. [Google Scholar] [CrossRef]
- Huang, F.; Wu, X.; Wei, M.; Guo, H.; Li, H.; Shao, Z.; Wu, Y.; Pu, J. miR-654-5p Targets HAX-1 to Regulate the Malignancy Behaviors of Colorectal Cancer Cells. Biomed Res Int 2020, 2020, 4914707. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Cai, Z.Q.; He, R.M.; Wang, X.C.; Cong, L.L.; Qiu, F.H. Knockdown of Long Noncoding RNA 01124 Inhibits the Malignant Behaviors of Colon Cancer Cells via miR-654-5p/HAX-1. Evid Based Complement Alternat Med 2022, 2022, 1092107. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Ni, Q. Expression of HAX1 and Ki-67 in breast cancer and its correlations with patient's clinicopathological characteristics and prognosis. Int J Clin Exp Med 2015, 8, 20904–20910. [Google Scholar] [PubMed]
- Kasashima, K.; Ohta, E.; Kagawa, Y.; Endo, H. Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J Biol Chem 2006, 281, 36401–36410. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, V.R.; Kates, R.E.; Kantelhardt, E.; Vetter, M.; Wuerstlein, R.; Fischer, T.; Schmitt, M.; Jaenicke, F.; Untch, M.; Thomssen, C.; et al. Health economic impact of risk group selection according to ASCO-recommended biomarkers uPA/PAI-1 in node-negative primary breast cancer. Breast Cancer Res Treat 2013, 138, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.P.; Zhang, M.; Le, T.P.; Wu, P.; Lainé, M.; Greene, G.L. RAC3 is a pro-migratory co-activator of ERα. Oncogene 2011, 30, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Yap, S.V.; Koontz, J.M.; Kontrogianni-Konstantopoulos, A. HAX-1: a family of apoptotic regulators in health and disease. J Cell Physiol 2011, 226, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- Mirmohammadsadegh, A.; Tartler, U.; Michel, G.; Baer, A.; Walz, M.; Wolf, R.; Ruzicka, T.; Hengge, U.R. HAX-1, identified by differential display reverse transcription polymerase chain reaction, is overexpressed in lesional psoriasis. J Invest Dermatol 2003, 120, 1045–1051. [Google Scholar] [CrossRef]
- Sorino, C.; Catena, V.; Bruno, T.; De Nicola, F.; Scalera, S.; Bossi, G.; Fabretti, F.; Mano, M.; De Smaele, E.; Fanciulli, M.; Iezzi, S. Che-1/AATF binds to RNA polymerase I machinery and sustains ribosomal RNA gene transcription. Nucleic Acids Res 2020, 48, 5891–5906. [Google Scholar] [CrossRef]
- Fanciulli, M.; Bruno, T.; Di Padova, M.; De Angelis, R.; Iezzi, S.; Iacobini, C.; Floridi, A.; Passananti, C. Identification of a novel partner of RNA polymerase II subunit 11, Che-1, which interacts with and affects the growth suppression function of Rb. Faseb j 2000, 14, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, A.N.; Suresh, D.; Mirshahi, F.; Santhekadur, P.K.; Sanyal, A.J.; Kumar, D.P. Emerging roles of AATF: Checkpoint signaling and beyond. J Cell Physiol 2021, 236, 3383–3395. [Google Scholar] [CrossRef] [PubMed]
- Iezzi, S.; Fanciulli, M. Discovering Che-1/AATF: a new attractive target for cancer therapy. Front Genet 2015, 6, 141. [Google Scholar] [CrossRef]
- Bruno, T.; Corleone, G.; Catena, V.; Cortile, C.; De Nicola, F.; Fabretti, F.; Gumenyuk, S.; Pisani, F.; Mengarelli, A.; Passananti, C.; et al. AATF/Che-1 localizes to paraspeckles and suppresses R-loops accumulation and interferon activation in Multiple Myeloma. Embo j 2022, e109711. [Google Scholar] [CrossRef]
- Kaiser, R.W.J.; Erber, J.; Höpker, K.; Fabretti, F.; Müller, R.U. AATF/Che-1-An RNA Binding Protein at the Nexus of DNA Damage Response and Ribosome Biogenesis. Front Oncol 2020, 10, 919. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.W.J.; Ignarski, M.; Van Nostrand, E.L.; Frese, C.K.; Jain, M.; Cukoski, S.; Heinen, H.; Schaechter, M.; Seufert, L.; Bunte, K.; et al. A protein-RNA interaction atlas of the ribosome biogenesis factor AATF. Sci Rep 2019, 9, 11071. [Google Scholar] [CrossRef]
- Barbato, C.; Corbi, N.; Canu, N.; Fanciulli, M.; Serafino, A.; Ciotti, M.; Libri, V.; Bruno, T.; Amadoro, G.; De Angelis, R.; et al. Rb binding protein Che-1 interacts with Tau in cerebellar granule neurons. Modulation during neuronal apoptosis. Mol Cell Neurosci 2003, 24, 1038–1050. [Google Scholar] [CrossRef]
- Sorino, C.; Bruno, T.; Desantis, A.; Di Certo, M.G.; Iezzi, S.; De Nicola, F.; Catena, V.; Floridi, A.; Chessa, L.; Passananti, C.; et al. Centrosomal Che-1 protein is involved in the regulation of mitosis and DNA damage response by mediating pericentrin (PCNT)-dependent Chk1 protein localization. J Biol Chem 2013, 288, 23348–23357. [Google Scholar] [CrossRef]
- Thomas, T.; Voss, A.K.; Petrou, P.; Gruss, P. The murine gene, Traube, is essential for the growth of preimplantation embryos. Dev Biol 2000, 227, 324–342. [Google Scholar] [CrossRef]
- Pisani, C.; Onori, A.; Gabanella, F.; Delle Monache, F.; Borreca, A.; Ammassari-Teule, M.; Fanciulli, M.; Di Certo, M.G.; Passananti, C.; Corbi, N. eEF1Bγ binds the Che-1 and TP53 gene promoters and their transcripts. J Exp Clin Cancer Res 2016, 35, 146. [Google Scholar] [CrossRef]
- Di Certo, M.G.; Corbi, N.; Bruno, T.; Iezzi, S.; De Nicola, F.; Desantis, A.; Ciotti, M.T.; Mattei, E.; Floridi, A.; Fanciulli, M.; et al. NRAGE associates with the anti-apoptotic factor Che-1 and regulates its degradation to induce cell death. In J Cell Sci; England, 2007; Volume 120, pp. 1852–1858.
- Xie, J.; Guo, Q. AATF protects neural cells against oxidative damage induced by amyloid beta-peptide. In Neurobiol Dis; United States, 2004; Volume 16, pp. 150–157.
- Page, G.; Lödige, I.; Kögel, D.; Scheidtmann, K.H. AATF, a novel transcription factor that interacts with Dlk/ZIP kinase and interferes with apoptosis. FEBS Lett 1999, 462, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Guo, Q. Apoptosis antagonizing transcription factor protects renal tubule cells against oxidative damage and apoptosis induced by ischemia-reperfusion. J Am Soc Nephrol 2006, 17, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Desantis, A.; Bruno, T.; Catena, V.; De Nicola, F.; Goeman, F.; Iezzi, S.; Sorino, C.; Ponzoni, M.; Bossi, G.; Federico, V.; et al. Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. Embo j 2015, 34, 1214–1230. [Google Scholar] [CrossRef] [PubMed]
- Bruno, T.; Desantis, A.; Bossi, G.; Di Agostino, S.; Sorino, C.; De Nicola, F.; Iezzi, S.; Franchitto, A.; Benassi, B.; Galanti, S.; et al. Che-1 promotes tumor cell survival by sustaining mutant p53 transcription and inhibiting DNA damage response activation. Cancer Cell 2010, 18, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Desantis, A.; Bruno, T.; Catena, V.; De Nicola, F.; Goeman, F.; Iezzi, S.; Sorino, C.; Gentileschi, M.P.; Germoni, S.; Monteleone, V.; et al. Che-1 modulates the decision between cell cycle arrest and apoptosis by its binding to p53. Cell Death Dis 2015, 6, e1764. [Google Scholar] [CrossRef] [PubMed]
- Folgiero, V.; Sorino, C.; Pallocca, M.; De Nicola, F.; Goeman, F.; Bertaina, V.; Strocchio, L.; Romania, P.; Pitisci, A.; Iezzi, S.; et al. Che-1 is targeted by c-Myc to sustain proliferation in pre-B-cell acute lymphoblastic leukemia. EMBO Rep 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M. Apoptosis-antagonizing transcription factor (AATF) gene silencing: role in induction of apoptosis and down-regulation of estrogen receptor in breast cancer cells. Biotechnol Lett 2013, 35, 1561–1570. [Google Scholar] [CrossRef]
- Gabanella, F.; Onori, A.; Ralli, M.; Greco, A.; Passananti, C.; Di Certo, M.G. SMN protein promotes membrane compartmentalization of ribosomal protein S6 transcript in human fibroblasts. Sci Rep 2020, 10, 19000. [Google Scholar] [CrossRef] [PubMed]
- Lindfors, K.; Halttunen, T.; Huotari, P.; Nupponen, N.; Vihinen, M.; Visakorpi, T.; Mäki, M.; Kainulainen, H. Identification of novel transcription factor-like gene from human intestinal cells. In Biochem Biophys Res Commun; 2000 Academic Press.: United States, 2000; Volume 276, pp. 660–666. [Google Scholar]
- Grzybowska, E.A.; Zayat, V.; Konopinski, R.; Trebinska, A.; Szwarc, M.; Sarnowska, E.; Macech, E.; Korczynski, J.; Knapp, A.; Siedlecki, J.A. HAX-1 is a nucleocytoplasmic shuttling protein with a possible role in mRNA processing. Febs j 2013, 280, 256–272. [Google Scholar] [CrossRef]
- Mobley, J.A.; Brueggemeier, R.W. Estrogen receptor-mediated regulation of oxidative stress and DNA damage in breast cancer. Carcinogenesis 2004, 25, 3–9. [Google Scholar] [CrossRef]
- Nazmeen, A.; Maiti, S. Redox Regulation of Estrogen Signaling in Human Breast Cancer.
- Wang, D.; Chen, T.Y.; Liu, F.J. Che-1 attenuates hypoxia/reoxygenation-induced cardiomyocyte apoptosis by upregulation of Nrf2 signaling. In Eur Rev Med Pharmacol Sci; Italy, 2018; Volume 22, pp. 1084–1093.
- Ishigaki, S.; Fonseca, S.G.; Oslowski, C.M.; Jurczyk, A.; Shearstone, J.R.; Zhu, L.J.; Permutt, M.A.; Greiner, D.L.; Bortell, R.; Urano, F. AATF mediates an antiapoptotic effect of the unfolded protein response through transcriptional regulation of AKT1. Cell Death Differ 2010, 17, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Yang, J.; Yi, J. Nucleolar Stress: hallmarks, sensing mechanism and diseases. Cell Stress 2018, 2, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Sapio, R.T.; Burns, C.J.; Pestov, D.G. Effects of Hydrogen Peroxide Stress on the Nucleolar Redox Environment and Pre-rRNA Maturation. Front Mol Biosci 2021, 8, 678488. [Google Scholar] [CrossRef] [PubMed]
- Shcherbik, N.; Pestov, D.G. The Impact of Oxidative Stress on Ribosomes: From Injury to Regulation. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Nassa, G.; Giurato, G.; Salvati, A.; Gigantino, V.; Pecoraro, G.; Lamberti, J.; Rizzo, F.; Nyman, T.A.; Tarallo, R.; Weisz, A. The RNA-mediated estrogen receptor α interactome of hormone-dependent human breast cancer cell nuclei. Sci Data 2019, 6, 173. [Google Scholar] [CrossRef] [PubMed]
- Leister, P.; Burgdorf, S.; Scheidtmann, K.H. Apoptosis antagonizing transcription factor AATF is a novel coactivator of nuclear hormone receptors. Signal Transduction 2003, 3, 17–25. [Google Scholar] [CrossRef]
- Xu, Y.; Huangyang, P.; Wang, Y.; Xue, L.; Devericks, E.; Nguyen, H.G.; Yu, X.; Oses-Prieto, J.A.; Burlingame, A.L.; Miglani, S.; Goodarzi, H.; Ruggero, D. ERα is an RNA-binding protein sustaining tumor cell survival and drug resistance. Cell 2021, 184, 5215–5229.e5217. [Google Scholar] [CrossRef]
Figure 1.
Human Che-1 interacts with HAX1. (A): Che-1 C-terminal deletion mutants and full-length proteins were used as the bait for the yeast two-hybrid screening (left panel). Yeast transformed with the indicated constructs were plated onto SD media lacking leucine and tryptophan (-LW) to verify the expression of both bait (W+) and prey (L+) plasmids, or onto media lacking leucine, tryptophan, histidine and adenine (-LWHA) to examine the interaction between bait and prey proteins (right panel). (B): Dual-label indirect immunofluorescence performed in HeLa cells (up) and MCF-7 cells (bottom) with the anti-Che-1 rat polyclonal antibody and the anti-HAX1 rabbit polyclonal antibody to visualize the immunolocalization of endogenous HAX1 (green) and Che-1 (red). Extensive co-localization (yellow) between HAX1 and Che-1 is visualized by the merged-colour image. Nuclei were stained with DAPI (blue). Scale bar, 50 μm. (C): Mitochondria-enriched lysate (MITO) from HeLa cells was immunoprecipitated with rabbit polyclonal anti-Che-1 antibody or with control IgG. Immunoprecipitated samples were analyzed by western blot using the indicated antibodies. (D): MCF-7 cells extract enriched in heavy membrane fraction (HM) was immunoprecipitated using anti-Che-1 rabbit polyclonal antibody or with no antibodies (no-Ab). Immunoprecipitation and co-immunoprecipitation were analyzed by western blot using the indicated antibodies.
Figure 1.
Human Che-1 interacts with HAX1. (A): Che-1 C-terminal deletion mutants and full-length proteins were used as the bait for the yeast two-hybrid screening (left panel). Yeast transformed with the indicated constructs were plated onto SD media lacking leucine and tryptophan (-LW) to verify the expression of both bait (W+) and prey (L+) plasmids, or onto media lacking leucine, tryptophan, histidine and adenine (-LWHA) to examine the interaction between bait and prey proteins (right panel). (B): Dual-label indirect immunofluorescence performed in HeLa cells (up) and MCF-7 cells (bottom) with the anti-Che-1 rat polyclonal antibody and the anti-HAX1 rabbit polyclonal antibody to visualize the immunolocalization of endogenous HAX1 (green) and Che-1 (red). Extensive co-localization (yellow) between HAX1 and Che-1 is visualized by the merged-colour image. Nuclei were stained with DAPI (blue). Scale bar, 50 μm. (C): Mitochondria-enriched lysate (MITO) from HeLa cells was immunoprecipitated with rabbit polyclonal anti-Che-1 antibody or with control IgG. Immunoprecipitated samples were analyzed by western blot using the indicated antibodies. (D): MCF-7 cells extract enriched in heavy membrane fraction (HM) was immunoprecipitated using anti-Che-1 rabbit polyclonal antibody or with no antibodies (no-Ab). Immunoprecipitation and co-immunoprecipitation were analyzed by western blot using the indicated antibodies.
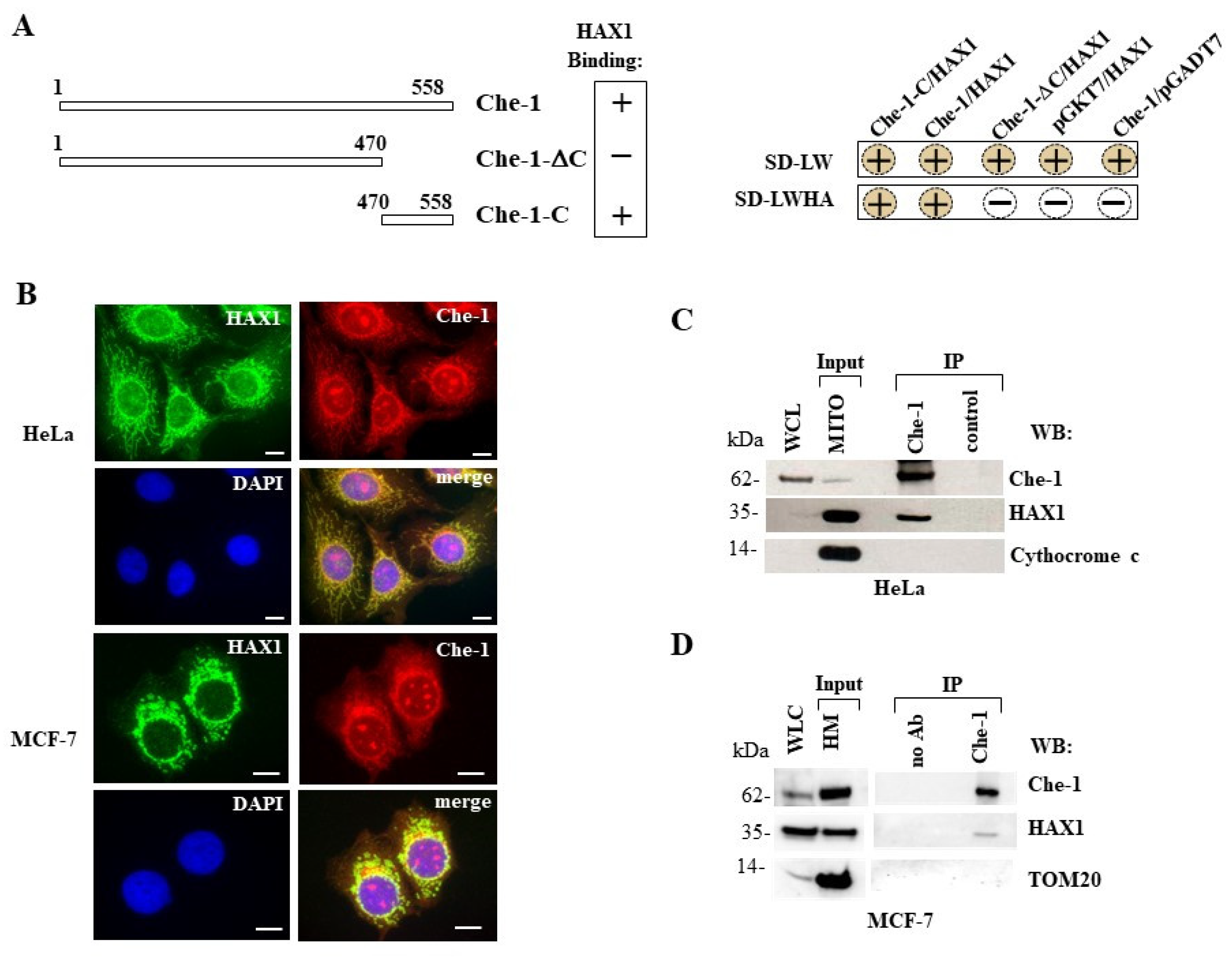
Figure 2.
Characterization of HAX1 regions that bind Che-1 protein and effect of HAX1C-100 dominant negative. (A): Domain map of HAX1 protein. Acid box with the EE motif, BH1 and BH2 (Bcl-2 protein homology domains); HD1 and HD2 (HAX1 domains conserved among species); PEST domain and TMD (transmembrane-like domain) were indicated. (B): Schematic representation of EGFP-HAX1 derived deletion mutants and their ability to bind Che-1 protein. (C): Whole-cell extracts of HeLa cells transiently transfected with myc-Che-1 and EGFP-HAX1 or its deletion constructs were immunoprecipitated with anti-myc monoclonal antibody and analyzed by western blot using anti-EGFP monoclonal antibody (right panel). The total cell lysates (Input) were immunoblotted to verify the correct expression of the transfected molecules (left panel). The asterisks indicate the main bands. (D): HeLa cells transfected with EGFP-HAX1C-100 were stained with mitochondrion-selective dye MitoTracker (red). Nuclei were stained with DAPI (blue). Scale bar, 50 μm. (E): Competition between HAX1C-100 and HAX1 for binding to Che-1. The total protein extracts of HeLa cells transfected with myc-Che-1, EGFP-HAX1 and increasing amounts of EGFP-HAX1C-100 were immunoprecipitated with anti-myc monoclonal antibody and analyzed by western blot using anti-EGFP monoclonal antibody. Arrows indicate HAX1 and HAX1C-100 bands. (F): Cell viability following H2O2-induced oxidative stress. MCF-7 cells were transfected with EGFP, EGFP-HAX1, and EGFP-HAX1C-100 respectively and 24h later treated with increasing concentrations of H2O2 (0.25mM, 0.5 mM, 1 mM) for 16 hr. The viability curve was performed by MTT assay. Data are reported as the mean ± SD of at least four independent experiments (*p≤0.05; **p≤0.01; ***p≤0.001).
Figure 2.
Characterization of HAX1 regions that bind Che-1 protein and effect of HAX1C-100 dominant negative. (A): Domain map of HAX1 protein. Acid box with the EE motif, BH1 and BH2 (Bcl-2 protein homology domains); HD1 and HD2 (HAX1 domains conserved among species); PEST domain and TMD (transmembrane-like domain) were indicated. (B): Schematic representation of EGFP-HAX1 derived deletion mutants and their ability to bind Che-1 protein. (C): Whole-cell extracts of HeLa cells transiently transfected with myc-Che-1 and EGFP-HAX1 or its deletion constructs were immunoprecipitated with anti-myc monoclonal antibody and analyzed by western blot using anti-EGFP monoclonal antibody (right panel). The total cell lysates (Input) were immunoblotted to verify the correct expression of the transfected molecules (left panel). The asterisks indicate the main bands. (D): HeLa cells transfected with EGFP-HAX1C-100 were stained with mitochondrion-selective dye MitoTracker (red). Nuclei were stained with DAPI (blue). Scale bar, 50 μm. (E): Competition between HAX1C-100 and HAX1 for binding to Che-1. The total protein extracts of HeLa cells transfected with myc-Che-1, EGFP-HAX1 and increasing amounts of EGFP-HAX1C-100 were immunoprecipitated with anti-myc monoclonal antibody and analyzed by western blot using anti-EGFP monoclonal antibody. Arrows indicate HAX1 and HAX1C-100 bands. (F): Cell viability following H2O2-induced oxidative stress. MCF-7 cells were transfected with EGFP, EGFP-HAX1, and EGFP-HAX1C-100 respectively and 24h later treated with increasing concentrations of H2O2 (0.25mM, 0.5 mM, 1 mM) for 16 hr. The viability curve was performed by MTT assay. Data are reported as the mean ± SD of at least four independent experiments (*p≤0.05; **p≤0.01; ***p≤0.001).
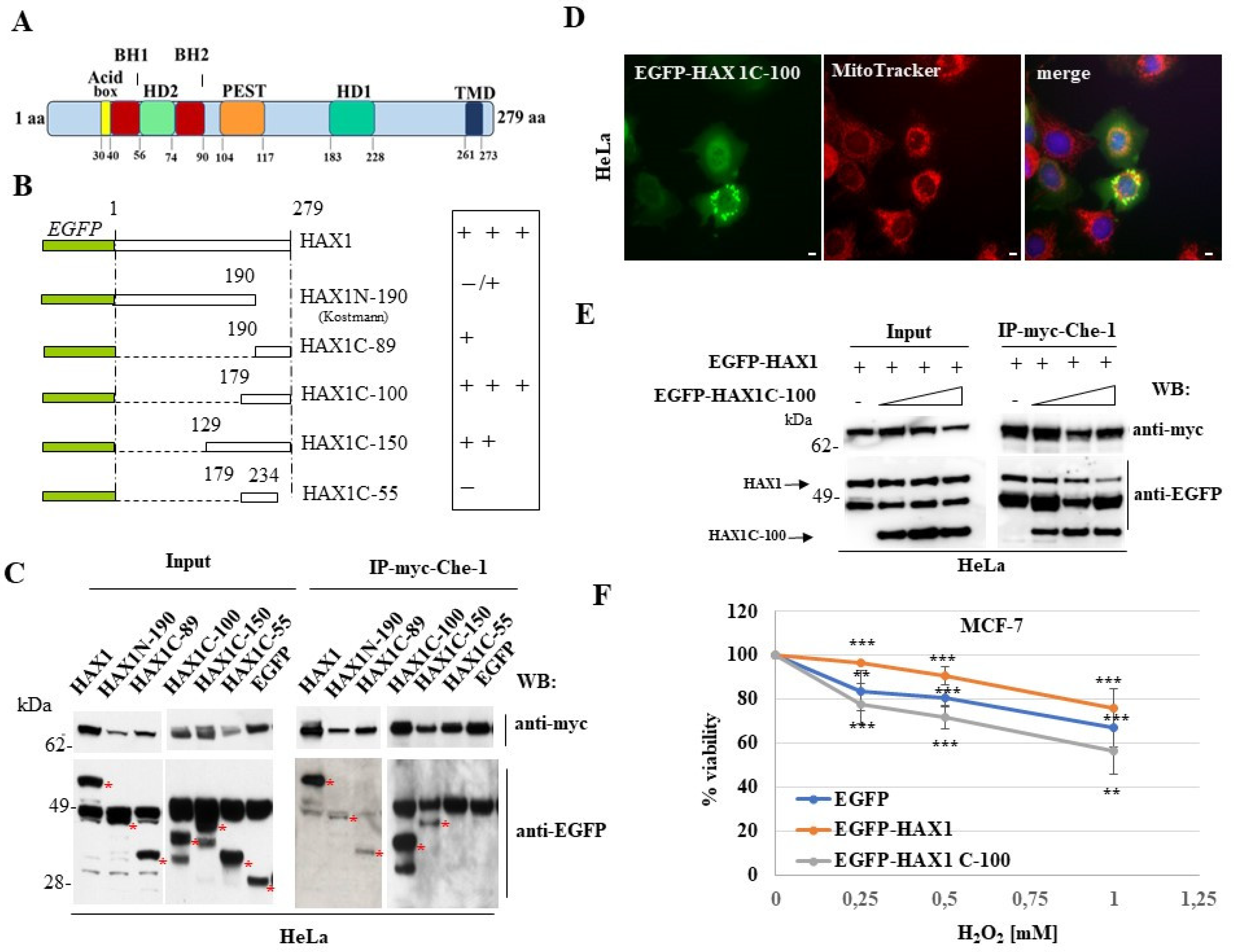
Figure 3.
The association of HAX1 and Che-1 proteins is strengthened by oxidative stimuli. (A): Dual-label indirect immunofluorescence with anti-HAX1 rabbit polyclonal antibody (green) and the anti-Che-1 rat polyclonal antibody (red) in HeLa cells untreated (NT) or treated with H2O2 (1 mM, 3 h). Nuclei are stained with DAPI (blue). Scale bar, 50 μm (left panel). Pearson correlation coefficient of relative nuclear localization of HAX1 and Che-1 in cells untreated or treated with H2O2 (1 mM, 3 h) shown in the histogram was calculated by ImageJ JACoP. At least 15 fields by three different experiments were analyzed, and the data were shown as mean±S.D (*p≤0.05; **p≤0.01; ***p≤0.001, Student’s t test) (right panel). (B): Histograms represent the Pearson correlation coefficient of HAX1/Che-1 total colocalization ratio (left panel) or HAX1/Che-1 colocalization ratio in cytosol and nuclei (right panel) in cells untreated or treated with H2O2 (1 mM, 3 h), calculated by ImageJ JACoP. At least 15 fields by three different experiments were analyzed, and the data were shown as mean±S.D. (*p≤0.05; **p≤0.01; ***p≤0.001, Student’s t test). (C): Whole cell lysates prepared from MCF-7 cells untreated or treated with H2O2 (1 mM, 3 h) were immunoprecipitated with anti-Che-1 monoclonal antibody and analyzed by western blotting. Used antibodies are indicated. (D): Whole cell lysates prepared from MCF-7 cells untreated or treated with H2O2 (1 mM, 3 h) were immunoprecipitated with anti-HAX1 polyclonal antibody and analyzed by western blotting. Used antibodies are indicated.
Figure 3.
The association of HAX1 and Che-1 proteins is strengthened by oxidative stimuli. (A): Dual-label indirect immunofluorescence with anti-HAX1 rabbit polyclonal antibody (green) and the anti-Che-1 rat polyclonal antibody (red) in HeLa cells untreated (NT) or treated with H2O2 (1 mM, 3 h). Nuclei are stained with DAPI (blue). Scale bar, 50 μm (left panel). Pearson correlation coefficient of relative nuclear localization of HAX1 and Che-1 in cells untreated or treated with H2O2 (1 mM, 3 h) shown in the histogram was calculated by ImageJ JACoP. At least 15 fields by three different experiments were analyzed, and the data were shown as mean±S.D (*p≤0.05; **p≤0.01; ***p≤0.001, Student’s t test) (right panel). (B): Histograms represent the Pearson correlation coefficient of HAX1/Che-1 total colocalization ratio (left panel) or HAX1/Che-1 colocalization ratio in cytosol and nuclei (right panel) in cells untreated or treated with H2O2 (1 mM, 3 h), calculated by ImageJ JACoP. At least 15 fields by three different experiments were analyzed, and the data were shown as mean±S.D. (*p≤0.05; **p≤0.01; ***p≤0.001, Student’s t test). (C): Whole cell lysates prepared from MCF-7 cells untreated or treated with H2O2 (1 mM, 3 h) were immunoprecipitated with anti-Che-1 monoclonal antibody and analyzed by western blotting. Used antibodies are indicated. (D): Whole cell lysates prepared from MCF-7 cells untreated or treated with H2O2 (1 mM, 3 h) were immunoprecipitated with anti-HAX1 polyclonal antibody and analyzed by western blotting. Used antibodies are indicated.
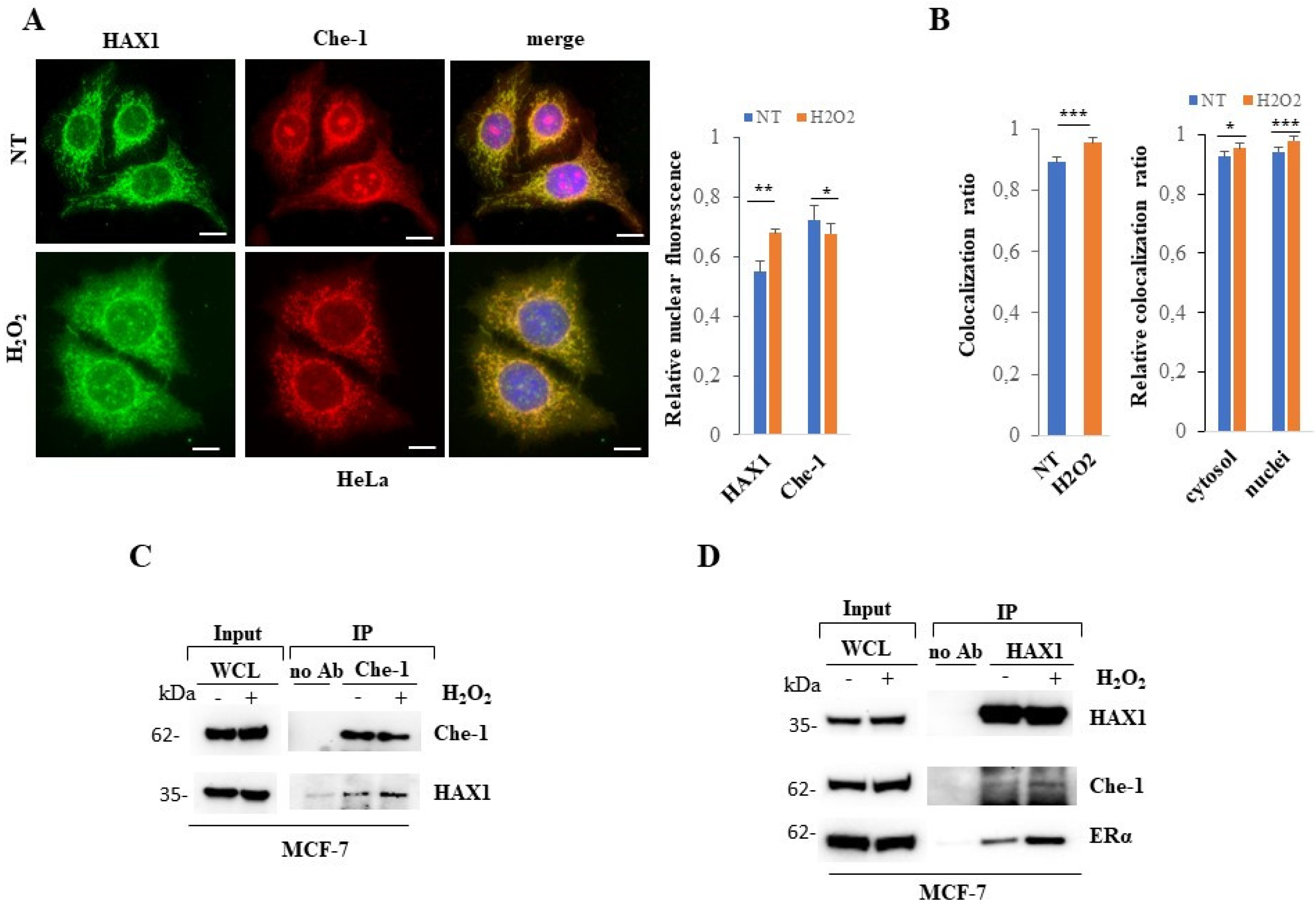
Figure 4.
Knockdown of Che-1 induces downregulation of HAX1. (A): Quantitative real time RT-PCR (qPCR) analysis of the HAX1 mRNA in MCF-7 cells upon Che-1-silencing (siControl, siChe-1), untreated (NT) or treated with H2O2 (1 mM, 3 h). The gene expression ratio of HAX1 mRNA, normalized as indicated, is shown as the mean ± SD from at least three independent experiments performed in triplicate (*p≤0.05; **p ≤0.01; ***p≤0.001). (B): Visualization of HAX1 mRNA (red dots) upon Che-1-silencing in MCF-7 cells (siControl, siChe-1) by padlock probe-based exponential rolling circle amplification. Nuclei were stained with DAPI (blue). Scale bar, 50 μm. Histogram of HAX1 mRNA amplicons per cell detected (left). Data represents the mean ± S.D. of at least three independent experiments (*p≤0.05; **p≤0.01; ***p≤0.001). (C): Representative western blot of MCF-7 cells upon Che-1-silencing (siControl, siChe-1), untreated or treated with H2O2 (1 mM, 3 h). The antibodies used are indicated. Densitometric analysis represents the mean ± S.D. of at least four independent experiments (right panel). (D): Representative fluorescence images of MCF-7 cells upon Che-1-silencing (siControl, siChe-1), untreated or treated with H2O2 (1 mM, 3 h). Indirect immunofluorescence was performed with the anti-HAX1 rabbit polyclonal antibody (green). Nuclei were stained with DAPI (blue). Scale bar, 50 μm.
Figure 4.
Knockdown of Che-1 induces downregulation of HAX1. (A): Quantitative real time RT-PCR (qPCR) analysis of the HAX1 mRNA in MCF-7 cells upon Che-1-silencing (siControl, siChe-1), untreated (NT) or treated with H2O2 (1 mM, 3 h). The gene expression ratio of HAX1 mRNA, normalized as indicated, is shown as the mean ± SD from at least three independent experiments performed in triplicate (*p≤0.05; **p ≤0.01; ***p≤0.001). (B): Visualization of HAX1 mRNA (red dots) upon Che-1-silencing in MCF-7 cells (siControl, siChe-1) by padlock probe-based exponential rolling circle amplification. Nuclei were stained with DAPI (blue). Scale bar, 50 μm. Histogram of HAX1 mRNA amplicons per cell detected (left). Data represents the mean ± S.D. of at least three independent experiments (*p≤0.05; **p≤0.01; ***p≤0.001). (C): Representative western blot of MCF-7 cells upon Che-1-silencing (siControl, siChe-1), untreated or treated with H2O2 (1 mM, 3 h). The antibodies used are indicated. Densitometric analysis represents the mean ± S.D. of at least four independent experiments (right panel). (D): Representative fluorescence images of MCF-7 cells upon Che-1-silencing (siControl, siChe-1), untreated or treated with H2O2 (1 mM, 3 h). Indirect immunofluorescence was performed with the anti-HAX1 rabbit polyclonal antibody (green). Nuclei were stained with DAPI (blue). Scale bar, 50 μm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



