Submitted:
12 February 2023
Posted:
16 February 2023
You are already at the latest version
Abstract
Controlling the selectivity of detonation initiation reaction of explosive is a Holy Grail, and it seems to be an “idiot's daydream”, by strengthening external electric field, to reduce the explosive sensitivity. The effects of external electric fields on the initiation reactions in NH2NO2∙∙∙NH3, a model system of the nitroamine explosive with alkaline additive, were investigated at the MP2/6-311++G(2d,p) and CCSD/6-311++G(2d,p) level. The concerted effect in the intermolecular hydrogen exchange is characterized by an index of the imaginary vibrations. Due to the weakened concerted effects by the electric field along the −x-direction opposite to the “reaction axis”, the dominant reaction changes from the hydrogen exchange to 1,3-intramolecular hydrogen transference with the increase of the field strengths. Furthermore, the stronger the field strengths, the higher the barrier heights become, indicating the lower sensitivities. Therefore, by strengthening the field strength and adjusting the orientation between the field and “reaction axis”, not only can the reaction selectivity be controlled, but also the sensitivity can be reduced, in particular under a super-strong field. Thus, a traditional concept, which the explosive is dangerous under the super-strong external electric field, is broken theoretically. Compared to the neutral medium, the low sensitivity of the explosive with alkaline can be achieved under the stronger field. Employing atoms in molecules, reduced density gradient and surface electrostatic potentials, the origin of the reaction selectivity and sensitivity change is revealed. This work provides a new idea to the technical improvement for adding the external electric field into the explosive system.
Keywords:
External electric field
; reaction selectivity
; explosive sensitivity
; intermolecular hydrogen exchange concerted reaction
; surface electrostatic potential
1. Introduction
Explosive sensitivity is an index to measure the stability of explosive to external stimuli. Today it has been imperative to explore the effective ways to reduce the sensitivity of the explosive under the external electric field. This is mainly initiated and driven by two factors: (1) technical improvement of the micro-electric explosion and (2) avoidance of the accidental explosion from electrostatic spark. The micro-electric explosion is a new explosion technology that the external electric field is added into the ignition and initiation system to improve the performance, such as detonation heat, detonation velocity and detonation pressure, etc., and it has aroused great interest [1,2,3,4]. However, the sensitivities of the explosives loaded in the wire or slapper plate explosive detonators could be increased under the external electric fields, leading to the serious obstacles in the technical improvement to adapt to the modern bad war environment. Moreover, the static electricity can not be avoided, and for explosives the potential catastrophic accidental explosions from electrostatic spark are always companied anywhere and anytime. It is generally believed that, the stronger the external electric field, the more easily the detonation initiation occurs, leading to the higher explosive sensitivity and higher risk of the accidental explosion [5,6,7,8]. Therefore, under the strong external electric field, it seems to be an “idiot's daydream” to achieve a low sensitivity of the high-energetic explosive or even a sensitivity much lower than that without electric field.
Fortunately, the external electric field has been widely used as a “smart reagent” to control the reactivity by adjusting the orientation of the electric field to “reaction axis” for over two decades [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25].“Reaction axis” could be defined as a direction along which chemical bonds are formed or broken. When the direction of the external electric field is the same as that of the “reaction axis”, the field will promote the electronic reorganization beneficial to the reaction from reactants’ to products’ bonding, leading to a decreased barrier height and an acceleration of reaction. On the contrary, when the direction of the external electric field is opposite, the field will inhibits the formation of the bonding mode from the reactant to product, leading to an increased barrier height [26,27]. Therefore, by adjusting the orientation of the external electric field to “reaction axis”, the barrier heights and reaction paths could be changed, and thus the regioselectivity and reaction rate could be controlled [14,15,16,17,18,19,20,21,22,23,24,25,26,27]. A lot of theoretical [19] and experimental investigations [20,28] have been carried out to control the reactivity by the adjustment of the orientation of the external electric fields, extending from the general catalytic reactions [23,29] to the selectivity of enzymatic-like bond activations [12,22], DNA damage [11], proton transfer [9], photochemical CO2 capture [25], etc.. In recent perspective articles [13,26], Shaik et al. demonstrated that the external electric field can control redox or nonredox reactions, and emphasized that the barrier height could be increased and the reaction could be inhibited by enforcing the electron transfers of the reactant from the acceptor to donor along/against the external-electric-field direction. Furthermore, it is well-known that there are often multiple reaction axes, and the changes of reactivities along the different reaction axes are often asynchronous with the changes of the external electric field strengths and directions [30]. Some are sensitive and some are insensitive to the external electric fields. This gives us an inspiration that, by the adjustment of the external field strengths and the orientations of the electric fields to the “reaction axes” of the explosive molecules, such as the reaction axes involving the homolysis of the C−NO2, N−NO2 and O−NO2 trigger-linkages, the hydrogen transference, etc., as the first step in the initiation reaction [31,32], the reaction paths could be changed and the barrier heights could be increased, leading to the decreased explosive sensitivities.
In fact, it has been as a Holy Grail to reduce the explosive sensitivity by adjusting the external electric field to control the selectivity of the detonation initiation reaction in experiment [33] and theory [4−6]. An electromagnetic pulse effect was tested during the bridge wire electric explosion [2]. We have found that, for the simple (pure) explosives, the bond dissociation energies (BDEs) of the “trigger linkages” and barrier heights of the initiation reactions could be increased under a certain external electric fields, and the selectivity of the detonation initiation reaction could be controlled and the explosive sensitivities could be reduced by the adjustment of the strength or orientation of the external electric fields [34,35,36]. For the mixture of nitroamine explosive and H2O, NH2NO2∙∙∙H2O as a model system, the transilience of the activation energies between the intermolecular and 1,3-intramolecular hydrogen transfer reactions was confirmed. Thus we predicted that, by controlling the external electric field strengths or orientations, the explosive sensitivity could be reduced in neutral environment, such as aqueous solution [30].
Alkaline agents and additives with the –NH2 group have more influence on the decomposition rates and sensitivities of explosives than the neutral or even acidic species [37,38]. Using a special Bourdon manometer, Shu et al. investigated the thermal decomposition of a series of mixtures with the concentration of RDX (cyclotrimethylene trinitramine) in the range of 0.1% ~ 2%. They found that the decomposition rate of RDX was hardly affected in the neutral medium, such as benzene, isooctane and naphthalene, etc., while it was increased in the alkaline medium, such as the agent with the ‒NH2 group [37]. We have found that, in alkaline environment, the explosive stability was changed greatly due to either the modified BDEs of the “trigger bonds”, or the changed activation energies or hydrogen-transfer reaction paths induced by the intermolecular interactions between the explosives and additives [39,40]. Therefore, the mechanism of the sensitivity change is more unpredictable for the explosive in the alkaline environment, and it is more imperative to reveal the essence of the explosive sensitivity and reduce it by adjusting the external electric field for the explosive in the alkaline environment than that in the neutral or acidic medium.
The nitramine explosive is one of the most widely used energetic materials. In this work, NH2NO2 and NH3 are selected as the model compounds of the nitroamine explosive and alkaline additive to reveal the essence of the explosive sensitivity under the external electric fields, and explore the ways to reduce sensitivity of explosive in the alkaline environment. For NH2NO2, it has been confirmed the initiation reaction fragments (i.e., O•, NH2NO•, NH2N•, •NH2, •NO2, NO2 and NO) [41] and initial competitive reaction between the N‒NO2 bond cleavage and NH2NO2→NH2ONO rearrangement [42]. For NH2NO2∙∙∙NH3 under the external electric field, the first or rate-determining step of the detonation reaction may be the homolysis of the N−NO2 bond, rearrangement, 1,3-intramolecular hydrogen transfer, or intermolecular hydrogen exchange between NH2NO2 and NH3. Therefore, the effects of the external electric fields on the hydrogen exchange and 1,3-intramolecular hydrogen transfer as well as homolysis of the N−NO2 bond in NH2NO2∙∙∙NH3 will be mainly investigated by the theoretical method, accompanied by a comparison with those of the NH2NO2∙∙∙H2O system. One of our goals is to clarify whether or not, by strengthening external electric field, the sensitivity of the high-energetic nitroamine explosive in the alkaline environment can be reduced to much lower than that without electric field. This work must be useful for the micro-electric explosion technology to design rationally the equipments and add efficiently the external electric fields into the nitramine explosive systems with the alkaline agents or additives, and to avoid the catastrophic explosions of them in the process of preparation, transportation and use under the external electric fields.
2. Computational Details
All the calculations were carried out with Gaussian 09 programs [43]. The molecular geometries of the reactant and transition state (TS) were fully optimized using the MP2/6-311++G(2d,p) method in the absence and presence of the different external electric fields. The energy minima were judged by the criteria of lacking imaginary frequency or only one imaginary frequency in which two atoms vibrate along the direction of forming or breaking chemical bonds. The activation energies (Ea) were calculated at the MP2/6-311++G(2d,p) and CCSD(T)/6-311++G(2d,p)//MP2/6-311++G(2d,p) level, respectively.
For the coordinate systems, the N1 atom of the −NH2 group is at the origin and the N2 atom of the −NO2 moiety is along the +z-axis (see Figure 1); and the x-axis is in the N−NO2 plane and the N atom of NH3 is approximately along the −x-axis; and thus the y-axis is defined as the direction perpendicular to the xy-plane. In three orthogonal directions, the field strengths of ±0.002, ±0.004, ±0.006, ±0.008 and ±0.010 a.u. were considered. In order to find out a possible conversion of the activation energies between the intermolecular hydrogen-exchange and 1,3-intramolecular hydrogen transference reactions, the dynamics was also considered with the field strengths of −0.012, −0.014, −0.016, −0.018, −0.019 and −0.020 a.u. along the −x-direction.
At the MP2/6-311++G(2d,p) level, the rate constants k(T) and Wigner tunneling-corrected rate constants kC(T) [44] were estimated at 298.15 and 688.0 K by the conventional transition state theory [45,46], as is expressed as:
where kB, κ(T), T, h, ΔG≠ and R are the Boltzmann constant, Wigner tunneling correction factor, absolute temperature, Planck,s constant, Gibbs energy change of activation and universal gas constant, respectively. κ(T) was given as follows [47]:
where Im(v≠) is the imaginary frequency corresponding to TS.
3. Results and Discussion
Two conformations of NH2NO2∙∙∙NH3 were optimized. In one conformation, NH3 is not only as a H-bonded donor, but also as a H-bonded acceptor (or lewis base), shown by the intermolecular N−H∙∙∙O2N and N−H∙∙∙NH3 H-bonds (see Figure 1). In another conformation, NH3 is only as a H-bonded donor with one intermolecular N−H∙∙∙O2N H-bond. The former is 32.0 kJ/mol more stable in energy than the latter at the MP2/6-311++G(2d,p) level, so the former was chosen as the reactant.
Two feasible initial reactions were predicted: intermolecular hydrogen exchange with TS1 and 1,3-intramolecular hydrogen transference with TS2. The hydrogen exchange is a concerted reaction, in which one of the H atoms of NH3 is transferred to –NO2 and simultaneously one of the H atoms of –NH2 moiety in NH2N(O)OH• is transferred to •NH2 radical, i.e., NH2NO2∙∙∙NH3↔NH2N(O)OH•∙∙∙•NH2 (biradical) ↔ NHN(O)OH∙∙∙NH3 (see Figure 1). For 1,3-intramolecular hydrogen transference, the H atom of the –NH2 moiety is transferred to −NO2 to form NHN(O)OH∙∙∙NH3. In no field, the barrier of TS1 is far lower than that of TS2 or BDE of the N–NO2 bond (see Table S1), as the “trigger linkage” of nitroamine explosive [52]. The hydrogen exchange occurs preferentially, as is similar to the detonation initiation mechanism of the CH3NO2∙∙∙H2O [53] and NH2NO2∙∙∙H2O systems [30]. The following will give a comparison of the effect of the external electric field on the kinetics of the intermolecular hydrogen exchange with 1,3-intramolecular hydrogen transference and N‒NO2 bond cleavage. Although the NH2NO2→NH2ONO rearrangement was confirmed [42], the barrier is too high (about 380.0 kJ/mol at the MP2/6-311++G(2d,p) level), so it is not considered in this work.
3.1. Cooperativity of H-Bonds in Reactant under External Electric Field
The fields parallel to the z- and x-axis directions affect the structures of NH2NO2∙∙∙NH3 considerably more than those parallel to the y-axis. Since NH2NO2 and NH3 are both the electron donor and acceptor, and both the O8∙∙∙H7 and N6∙∙∙H5 H-bonds are in the xz-plane, the effects of the fields along the z- and x-axes on the electron transfers corresponding to the O8∙∙∙H7 H-bond are opposite to N6∙∙∙H5. Thus, the field effects on their distances are also opposite. For example, the fields along the +z-direction lengthen the O8∙∙∙H7 distance while it is shortened in the −z-direction. Therefore, when one H-bonding interaction is strengthened, the other will be weakened, and vice versa. The phenomenon that multiple intermolecular interactions enhance each others’ strength when they work simultaneously in a system is termed as the cooperativity effect [54]. Due to the effects of the external electric fields, the cooperativity effect of two H-bonding interactions is weakened, leading to an unobvious change of the total intermolecular interaction.
3.2. Concerted Effect of Intermolecular Hydrogen Exchange in External Electric Field
(1) Structures of TS1
The intermolecular hydrogen exchange is mainly accompanied by the changes of the activation O8∙∙∙H7, H7∙∙∙N6, N6∙∙∙H5 and H5∙∙∙N1 distances involving TS1 (see Figure 1). Since the y-direction is approximately perpendicular to the plane of the motion regions of the activation atoms H7 and H5, the activation distances are more affected by the external electric fields parallel to the x- and z-axis directions than by those parallel to the y-axis, and the effects of the fields parallel to the x-axis are the most notable (see Table S2). A good (R2 > 0.9900) linear correlation is found between the change of the N6∙∙∙H5 distances and field strengths along z-axis (see Figure S1).
On the whole, with the increased electric field along the +x-direction, the H5∙∙∙N1 and O8∙∙∙H7 distances are increased while the N6∙∙∙H5 and H7∙∙∙N6 distances are decreased, and the reverse trend occurs along the −x-directions. Some structural changes inconsistent with the overall trend have attracted interest. Along the +x-direction, when the field strength is lager than +0.004 a.u., the O8∙∙∙H7 and H7∙∙∙N6 distances are hardly changed, indicating that the effect of the external electric field is “immune” to the reaction involving the activation H7 atom. Along the −x-direction, several transience values are confirmed in the activation distances. For example, when the field strength is up to –0.010 a.u., the O8∙∙∙H7 and H5∙∙∙N1 distances are increased suddenly and the N6∙∙∙H5 distances are decreased, a local “bump” appearing in the overall trend. Another transience of the O8∙∙∙H7 distance is found (increased suddenly) under the field with the strength of –0.019 a.u.. When the field strength is larger than –0.014 a.u., the change trend of the H5∙∙∙N1 and N6∙∙∙H5 distances is the same as that with the field strength lower than –0.008 a.u.. These results indicate that the change of the activation distances is transient and the trend is zigzag under the external electric fields with the field strengths larger than −0.010 a.u.. This may lead to uncertainty or diversification of the chemical reaction path under the “strong” external electric fields, which, to our knowledge, has not been clearly emphasized in the experimental literature.
The field effects on the activation distances are confirmed by the AIM results (see Figure 2). The changes of the electron densities ρ of the bond critical point (BCP) (3, -1) corresponding to O8∙∙∙H7, H7∙∙∙N6, N6∙∙∙H5 and H5∙∙∙N1 in the fields parallel to the y-axis are not significant in comparison with those in the fields along the x- and z-axes, among which the change corresponding to O8∙∙∙H7 is the most notable under the field along the −x-axis. The changes of the ρ values within the bond paths related to the activation distances are more notable than those corresponding to other bond paths. The transient change of the interatomic activation distances and the zigzag trend are also confirmed by the AIM results. For example, along the −x-direction, an extremely sharp abrupt change of ρ(O8∙∙∙H7) from 0.2792 a.u. with the field strength of −0.018 a.u. to 0.1632 a.u. with the strength of −0.019 a.u., then it is increased to 0.3028 a.u. with the strength of −0.020 a.u.
(2)Barrier, imaginary vibration and rate constant of hydrogen exchange
Both the energies of TS1 and reactant decrease synchronously (become more negative) with the increase of the external electric field strength in z-directions, so the changes of activation energies are not obvious (see Figure 3 and Table S1). Although the energies of TS1 decrease and those of the reactant increase along the +x- and y-direction, the changes of them are slight, so the decreased barrier heights are also not obvious. With the field strength of 0.010 or −0.010 a.u., the change of the barrier compared with that in no field is no more than 25.0 kJ/mol in the field along the z-, +x-, or y-axis direction at two levels of theory. However, since the energies of TS1 increase greatly while those of the reactant decrease, the significant increased barrier heights are found along the −x-direction. With the field strengths from 0.000 to −0.020 a.u., the barrier heights soar up from 68.6 to 291.3 kJ/mol at the MP2/6-311++G(2d,p) level. The change is up to more than 100.0 or 220.0 kJ/mol with the field strength of −0.010 or −0.020 a.u., accompanied by a relative value more than 140.0% or 300.0%. In general, the change of the barrier compared with that in no field is no more than 15.0 kJ/mol with the field strength of −0.010 or +0.010 a.u. [34−36, 52].
The changes of the barriers induced by the external electric fields are also reflected in the imaginary vibrations, Gibbs energies and rate constants. The field parallels to the x-direction has the more notable effect on the magnitude than that along the z- or y-direction (see Figure 3 and Table 1). In most cases, the imaginary vibration and rate constant are hardly changed along the z- or y-direction. For example, the changes of the imaginary vibration are not more than 50 cm-1 with the strongest field strength. However, the imaginary vibrations are decreased or increased significantly in the fields along the +x- or −x-direction, in particular in the fields with strengths more than −0.014 a.u.. In comparison with the value in no field, the change is up to 755.9 and 1270.1 cm−1 with the field strength of −0.014 and −0.020 a.u., respectively. The rate constants at 298.15 K (k298.15 K) are decreased remarkably with the increased field strength, from 2.86×100 s-1 in no field to 4.41×10-20 s-1 with the field strength of −0.010 a.u. and 1.15×10-36 s-1 with −0.020 a.u.. Similar to k298.15 K, for the rate constants at 688.0 K (k688.0 K), an obvious decrease is also found, reduced by 1.87×1021 times from the field strength of 0.000 to −0.020 a.u.. Except for the field strength more than −0.014 a.u., the Wigner corrections are small and the tunneling-corrected effects on the rate constants could be ignored.
(3) Concerted reaction and reaction axis of hydrogen exchange
The concerted reaction refers to the reaction in which there is only one transition state involving all the coexistent multiple reactions enhanced each other, and the breaking of chemical bond and the formation of new bond occur simultaneously. For a concerted reaction, each of the coexistent multiple reactions corresponds to one “reaction axis”. For a concerted reaction under the external electric field, the effect of the field on each of the coexisting reactions is different: for the reaction in which the direction of the “reaction axis” is consistent with that of the external electric field, the external electric field will accelerate it, while the external field will inhibit the reaction in which the direction of “reaction axis” is opposite to that of the external electric field. The comprehensive effect of the external field on all the coexistent reactions will be reflected in the change of the barrier height of the concerted reaction.
The intermolecular hydrogen exchange is in essence a concerted (cooperative) process of two hydrogen transfers (i.e., NH2NO2∙∙∙NH3↔NH2N(O)OH•∙∙∙•NH2 and NH2N(O)OH•∙∙∙•NH2↔NHN(O)OH∙∙∙NH3), and it can be shown clearly from the changes of the imaginary vibrations under the external electric field. In order to evaluate the vibration intensity of the H7 or H5 atom, the average amplitude of H in the imaginary vibration is defined as follows:
where means the average amplitude of H, and ΔX, ΔY and ΔZ are the maximum value of the H atom vibration along the x-, y-and z-axis direction, respectively. Along the –x-direction field, both the and values simultaneously increase with the field strengths from 0.000 to −0.010 a.u., and decrease from −0.012 to −0.016 a.u., and again increase from −0.018 to −0.020 a.u., showing a synchronous zigzag trend (see Table S3). Thus, the concerted effect between the NH2NO2∙∙∙NH3 ↔ NH2N(O)OH•∙∙∙•NH2 and NH2N(O)OH•∙∙∙•NH2↔NHN(O)OH∙∙∙NH3 reactions is confirmed from the synchronous changes of and .
Above two hydrogen transfer processes in the concerted reaction correspond to two “reaction axes” along N6→H7→O8 and N1→H5→N6 (see Figure 1). For the reaction axis along N6→H7→O8, the smaller the electron density of N6, and the higher the electron density of O8, the more likely the activation H7 atom is transferred from N6 to O8. On the contrary, H7 is easy to be transferred toward N6. When the direction of the external electric field is the same as that of the N6→H7→O8 reaction axis, the electron density of N6 will be decreased and that of O8 will be increased. As a result, the electric field will induce the electronic reorganization which is favorable for the transformation from NH2NO2∙∙∙NH3 to NH2N(O)OH•∙∙∙•NH2. Conversely, the external field hinders this transformation. Similar to N6→H7→O8, for the reaction axis along N1→H5→N6, the smaller the electron density of N1, and the higher the electron density of N6, the more likely H5 is transferred from N1 to N6. The external electric field which follows the direction of N1→H5→N6 can induce the transformation from NH2N(O)OH•∙∙∙•NH2 to NHN(O)OH∙∙∙NH3. The electric field in the opposite direction leads to the inhibition of the formation of NHN(O)OH∙∙∙NH3.
Under the external electric field, the change of the atomic charge is often complicated due to the influence of the molecular dipole (internal electric field). From the Mulliken charges collected in Table S3, along the −x-direction with the field strengths no more than −0.008 a.u., although both of the negative charges of N6 and O8 increase with the increase of the field strengths, the change of N6 is far larger than that of O8. When the field strengths are more than −0.014 a.u., the negative charges of N6 increase while those of O8 decrease, leading the more notable negative charge of N6 than that of O8. With the field strengths of −0.008 ~ −0.014 a.u., the charge of N6 is more negative than that of O8. Therefore, the H7 atom with the positive charge would rather bind to N6 than O8. In other words, the −x-direction of the external electric field is opposite to that of the “reaction axis” along N6→H7→O8, and it is not beneficial to the hydrogen transfer from NH2NO2∙∙∙NH3 to NH2N(O)OH•∙∙∙•NH2. In particular, although the positive charges of H7 and H5 increase with the increase of the electric field along the −x-direction (except for −0.010 ~ −0.014 a.u.), the change of H7 is larger than that of H5, shown by the Mulliken charges from 0.495 to 0.634, and from 0.546 to 0.620, respectively. Furthermore, the change of N6 is also far larger than that of N1 (from −0.786 to −1.195 vs. from −0.656 to −0.610). These results indicate that the effect of external electric field on the N6→H7→O8 reaction is more significant than that on N1→H5→N6. Thus, although the −x-direction is the same as that of the N1→H5→N6 reaction axis and it is beneficial to the intermolecular hydrogen transfer of NH2N(O)OH•∙∙∙•NH2 → NHN(O)OH∙∙∙NH3 since the negative charge of N6 increases while that of N1 decrease, this electric field is unfavorable to the total reaction, i.e., the NH2NO2∙∙∙NH3→NHN(O)OH∙∙∙NH3 concerted reaction. In the absence of electric field, there is a concerted reaction between the N6→H7→O8 and N1→H5→N6 reactions, which promotes the intermolecular hydrogen exchange reaction of NH2NO2∙∙∙NH3→NHN(O)OH∙∙∙NH3. However, under the electric field along the −x-direction, the N6→H7→O8 reaction is dominant while it is hindered. Thus, the concerted effect is weakened, leading to the increased barrier heights of the hydrogen exchange reaction. Furthermore, the stronger the external electric field along the −x-direction, the more seriously the concerted effect is weakened and the higher the barrier heights become. When the field strength reaches to be −0.016 a.u., the Mulliken charge of N6 increases to be −1.023 while that of O8 decreases to be −0.452, suggesting that, in this case, H7 will hardly bind to O8 and the concerted effect almost disappears. In sum, the −x-direction of the electric field is opposite to that of the “reaction axis” of the dominant reaction in the intermolecular hydrogen exchange, leading to the inhibition of the reaction from NH2NO2∙∙∙NH3 to NH2N(O)OH•∙∙∙•NH2. Thus, the concerted effect of the hydrogen exchange is weakened, and the barrier height is increased in comparison with that in no field. In one word, it is the weakening or even breaking of the concerted effect in the hydrogen exchange that makes the barrier heights increase dramatically along the −x-direction.
The weakened concerted effect could also be seen from the changes of the activation distances induced by the external electric fields. Along the z- and x-axes, the external electric field effect on the O8∙∙∙H7 and H7∙∙∙N6 distances are just opposite each other, as is also found for the N6∙∙∙H5 and H5∙∙∙N1 distances. For example, with the increased electric fields along the +z-direction, the O8∙∙∙H7 and H5∙∙∙N1 distances are increased, while the N6∙∙∙H5 and H7∙∙∙N6 distances are decreased. In particular, although the significant effects on the activation distances are achieved along the −x-direction, such as transient and zigzag features, the transilience of the barrier is not found. This can be explained as follows: the change of the barrier in the hydrogen exchange reaction is a result of the concerted effect of the two reactions. Although the molecular and electronic structures of the species corresponding to one of the reactions change greatly in a certain electric field, the change of them in the other reaction is not obvious, and even their changes have opposite effects on each other's barriers, which leads to the weakening of the concerted effect and acts as a buffer effect for the fluctuation of the barriers in the total hydrogen exchange reaction.
3.3. 1,3-Intramolecular Hydrogen Transfer in External Electric Field
The structures of TS2 in 1,3-intramolecular hydrogen transfer are shown in Figure 1. Similar to the intermolecular hydrogen exchange, the fields parallel to the z- and x-axes affect the activation distances considerably more than those parallel to the y-axis. Along the −z-direction, the changes of the activation N1∙∙∙H4 and H4∙∙∙O3 distances are irregular. With the increased electric field along the +z-direction, the N1∙∙∙H4 distance is increased while the H4∙∙∙O3 distance is decreased. Along the +x-direction, the N1∙∙∙H4 and H4∙∙∙O3 distances are increased, and the opposite trend is found along the −x-direction. Different from the hydrogen exchange, the abrupt changes of the activation N1∙∙∙H4 or H4∙∙∙O3 distance are not found under the strong fields, and they are increased or decreased gently. Moreover, the changes of the activation distances are far less than those of the intermolecular O8∙∙∙H7 and N6∙∙∙H5 distances. For example, the O8∙∙∙H7 distance is up to more than 3.200 Å under the field along the +z-axis, indicating that the O8∙∙∙H7 interaction disappears. The changes of the activation distances are confirmed by the RDG results (see Figure 4). A good (R2 > 0.9850) linear correlation is found between the changes of the activation H4∙∙∙O3 distances and field strengths Ex (see Figure S1). These structural changes are confirmed by the electron densities of the bond critical points (BCP) (3, -1).
The fields parallel to the z- and x-axes affect the energies considerably more than those parallel to the y-axis. Along the +z-axis, although the energies of TS2 and reactants decrease with the increase of the electric field, the changes of the former are more significant than those of the latter, leading to the decreased barrier heights (see Table S1). Along the −z-axis, due to the decreased energies of the reactant while increased energies of TS2 with the increase of the electric field, the barrier heights increase tremendously, up to more than 230.0 kJ/mol with the field strength of more than −0.010 a.u.. Since the energies of TS2 and reactant synchronously increase along the +x-axis direction, the change of the barrier height is not obvious. Along the −x-axis, although the energies of TS2 and reactant decrease, the change of the latter is far more significant than that of the former, leading to the significant increase of the barriers.
There is one “reaction axis” along N1→H4→O3 in the 1,3-intramolecular hydrogen transfer from NH2NO2∙∙∙NH3 to NHN(O)OH∙∙∙NH3. The smaller the electron density of N1, and the higher the electron density of O3, the more likely the activation H4 atom is transferred from N1 to O3, and the smaller the barrier height is. Conversely, the hydrogen transfer to O3 becomes difficult. From the Mulliken and APT charges in Table S4, with the increase of field strength along the −x-axis, although the negative charges of N1 and O3 increase, the negative charges of N1 are always greater than that of O3, which leads to H4 preferring to bond to N1. Thus, the N1→H4→O3 reaction is inhibited, leading to the increased barriers.
The fields parallels to the z- and x-directions have the more notable effect on the magnitudes of the imaginary vibration, Gibbs energies and rate constant of the 1,3-intramolecular hydrogen transfer than those along the y-direction (see Table 2). The imaginary vibrations are increased under the electric fields along the −z- and +x-directions, and the opposite trend is found in the +z- and −x-directions, and the decreased values are notable along −x-direction, up to 48.6 and 111.2 cm−1 with the field strength of −0.010 and −0.020 a.u., respectively. A low imaginary vibration could lead to a small curvature at the region near the TS on the potential energy surface (PES) [55], and thus the fields along the −z- and +x-directions flatten the PES near TS2. Different from TS1, the Wigner corrections are up to 4.829 at 298.15 K or 1.719 at 688.0 K, suggesting that the tunneling-corrected effects on the rate constants are notable at 298.15 K. The rate constant k298.15 K is reduced by 1.19×1013 times from the field strength of 0.000 to −0.020 a.u., and the change is smaller than that of the intermolecular hydrogen exchange.
3.4. Prediction of Explosive Sensitivity under External Electric Field
As mentioned above, the explosive sensitivity in no field is mainly influened by the barrier of the intermolecular hydrogen exchange. Due to the concerted effect between the NH2NO2∙∙∙NH3→NH2N(O)OH•∙∙∙•NH2 and NH2N(O)OH•∙∙∙•NH2→NHN(O)OH∙∙∙NH3 reactions, the barrier is very low, suggesting a very high explosive sensitivity, as was cofirmed by an experimental result from the nitramine explosive in the alkaline environment [37].
Along the y-, +x- and +z-directions of the electric fields, the variation trends of the barrier heights of the intermolecular hydrogen exchange and 1,3-intramolecular hydrogen transferences are synchronous, and the barriers of the former are always far lower than those of the latter. Along the −z-direction, although the trends of them are opposite, the barrier heights of hydrogen exchange are also far lower than those of 1,3-hydrogen transference. Furthermore, with the field strengths of −0.010 ~ +0.010 a.u., the barrier heights of TS1 are also far lower than the BDEs of the N−NO2 bond. Therefore, the hydrogen exchange is always dominant with the field strengths of −0.010 ~ +0.010 a.u. along the y-, +x- and z-directions, and the initiation reaction is the intermolecular hydrogen exchange, which controls the explosive sensitivity. The barriers of the hydrogen exchange are insensitive to the external electric fields along the y-, +x- and z-directions, i.e., the barriers change slowly (see Table S1). Therefore, the explosive sensitivities of NH2NO2∙∙∙H3O are almost unchanged, close to those in no fied. In other words, by adjusting the field strengths and orientations between the “reaction axes” and external electric fields along the y-, +x- or z-directions, the selectivities of the reactions could not be changed or controlled, and the sensitivities could not be reduced obviously.
The conversion of the dominant reactions occur along the −x-direction (see Figure 3, Table 1 and Table 2). The intersection point of the two curves, the barriers of the TS1 and TS2 versus field strengths, is found with the field strengths between −0.008 and −0.010 a.u., and between −0.012 and −0.014 a.u. at the CCSD(T)/6-311++G(2d,p) level. At the MP2/6-311++G(2d,p) level, it is also found between −0.012 and −0.014 a.u.. Although botht the barrier heights of TS1and TS2 increase with the enhanced field strength along the −x-direction, due to the weakened concerted effect in the hydrogen exchange by the strong fields, the change of the barrier heights of TS1 is more notable than that of TS2. When the field strength is lower than −0.008 a.u., the barrier height of the TS1 are lower than those of TS2. The corresponding rate constants of TS1 are more than 3.89×104 times those of TS2 at the MP2/6-311++G(2d,p) level, and the intermolecular hydrogen exchange is dominant. Due to the significant increase of the barriers of the hydrogen exchange, the sensitivities are reduced obviously. When the field strength is more than −0.014 a.u., the barrier heights of TS1 are higher than those of TS2. The rate constants of TS2 are more than 1.21×108 times those of TS1, and the 1,3-intramolecular hydrogen transference is dominant. Due to the increase of the barriers of the 1,3-intramolecular hydrogen transference, the sensitivities are also reduced. Although the dominant reaction can not be identified explicitly within the field strengths of −0.008 a.u. ~ −0.014 a.u., the barriers of the hydrogen exchange and 1,3-intramolecular hydrogen transference are both increased, leading to the reduced sensitivities, too. Therefore, by adjusting the field strengths and orientations between the “reaction axes” and external electric fields along the −x-direction, not only can the selectivities of the reaction involving the explosive system NH2NO2∙∙∙NH3 be controlled, but also the explosive sensitivities can be reduced. Thus, it can be inferred that,by adjusting the external field orientation and strengthening electric field, the sensitivity of the high-energetic nitroamine explosive in the alkaline environment can be reduced greatly to much lower than that without electric field.
In particular, along the −x-direction, the stronger the field strengths, the higher the barrier heights of the dominant reaction, suggesting that the significantly the explosive sensitivities are reduced! For example, compared with the barrier height in no field, the significant increased barrier height is found under the super-strong external electric field, such as −0.020 a.u., suggesting the significantly decreased explosive sensitivities under the super-strong external electric field. This is one of the most coveted expectations in the field of the explosives: significant reduction of the explosive sensitivity induced by super-strong external electric field! It is well known that the introduction of external electric field into energetic material can increase the energy, and thus accelerate the detonation velocity and increase the detonation pressure [33,56,57]. Then, if simultaneously the stability can also be increased, the inherent contradiction between the performance and stability of explosive will be solved, and not only will a traditional concept, in which the explosives are dangerous under the super-strong external electric field, be broken theoretically, but also by strengthening external electric field, the sensitivity of the high-energetic explosive can be reduced or even reduced to much lower than that without electric field. This will be of great significance to the improvement of the technologythat the external electric fields are added safely into the energetic material system to enhance the explosive performance!
Noted that, although theoretically the sensitivity can be reduced by adjusting the external field orientation and strengths, in practice, the technology may be very difficult because the direction of molecular motion is disorderly, and the control the direction of the external electric field to the “reaction axis” is one of the technical bottlenecks. Furthermore, the imposed external field strengths can not exceed the dielectric strengths of the explosive system. Otherwise, the explosive device may be broken down, and the work to control the selectivity of the reaction and reduce the explosive sensitivity by adjusting the external electric fields will not be practical.
3.5. Surface Electrostatic Potentials of TS1 under the Field Along −x-Orientation
The electrostatic potential (ESP) shows the characteristics of electron density distribution by means of the local parameters and global statistical quantities, and it can be used to evaluate the possibility of chemical reaction [58,59]. For example for the electron-rich atom, the more notable local negative surface ESPs (Vs−), the more significant the electron sufficiencies within the localized region that is prone to the electrophilic reaction become. According to the global parameters, the larger the variance or , the greater possibility of the nucleophilic or electrophilic reaction becomes [50,60,61,62]. In order to further reveal the essence of the sharp increase of the barriers of TS1 under the field along −x-orientation, the surface electrostatic potentials involving TS1 are shown in Figure 5 and Table 3.
Because the negative ESP of N6 in the NH3 moiety is shielded tightly by four positive ESPs of the H atoms, the changes of the chemical bonding modes of the activation H7 to N6 and O8 are explored through the changes of the negative ESP of O8 () under the electric field. With the increase of the electric field strength along −x-orientation, the value of decreases obviously, indicating that the possibility of the formation of the chemical bond between H7 and O8 is reduced greatly. Thus, the concerted effect is weakened in the hydrogen exchange and the barrier is increased dramatically along the −x-direction, as is consistent with the barrier result and charge analysis.
From the overall trend, with the increase of electric field strength, the absolute values of , , and are decreased. However, within the field strengths between –0.008 and –0.012 a.u., except for , the other values are all increased suddenly and then decreased, as is also found in the change of the polar surface area (|ESP| > 10 kcal/mol).
The changes of the global statistical quantities of ESPs, such as , , and polar surface area, are more consistent with the those of the activation O8∙∙∙H7, H7∙∙∙N6, N6∙∙∙H5 and H5∙∙∙N1 distances induced by the external electric fields, while the changes of the local parameter and global statistical quantity are more consistent with the trend of the barriers of the concerted reaction. This suggests that the essence of the barrier changes in the concerted reaction is not only originated from the changes of the local electronic properties induced by the external electric fields, but also related to the changes of the global electronic structures. As mentioned above, a concerted reaction means the reaction in which there is only one transition state involving all the coexistent multiple reactions, and the barrier changes of each of the coexistent reactions must be closely related to the changes of the local electronic structures under the external electric fields. In one word, the coupling effect of the changes of the local and global ESPs, induced by the external electric fields, together controls the possibility of the concerted reaction, and thus the explosive sensitivity.
For NH2NO2∙∙∙H2O, with the increase of the field strength along the −x-orientation, the negative surface ESPs of the O6 atom of the neutral H2O were decreased slightly, and increased suddenly with the strength of –0.012 a.u., and then decreased gradually. Simultaneously, the positive surface ESPs of the H7 atom are increased slightly, and increased dramatically with the field strength of –0.013 a.u., and then increased slowly. The electrophilicity of O6 and the nucleophilicity of H7 suddenly are increased dramatically with the strength of –0.012 a.u. or –0.013 a.u., leading a suddenly and extremely increased barrier of the reaction in which the intermolecular hydrogen transfer from the O6 atom of H2O to the O8 atom of the –NO2 group. However, for the alkalescent NH3 in NH2NO2∙∙∙NH3, as mentioned above, the negative surface ESP of the N6 atom is shielded by the positive surface ESPs of four H atoms. The change of the electrophilicity of N6 in NH3 is less notable than that of O6 in H2O induced by the external electric field. Therefore, the barrier of NH2NO2∙∙∙NH3→NHN(O)OH∙∙∙NH3 is increased more gently than that of NH2NO2∙∙∙NH2→NHN(O)OH∙∙∙NH2, and the sensitivity of the nitramine explosive in the alkaline environment is decreased more gently than that in the neutral medium. In other words, for the nitramine explosive with the neutral medium, the low sensitivity can be achieved with the weak external electric field, for the explosive in the alkaline environment, the low sensitivity can only be achieved under the super-strong field.
4. Conclusions
In order to clarify whether or not, by strengthening external electric field, the sensitivity of the nitroamine explosive in alkaline environment can be reduced, the effects of the external electric fields on the initiation reactions in NH2NO2∙∙∙NH3 were investigated by the theoretical method.
The cooperativity effect of the intermolecular H-bonding interactions of reactant is weakened under the external electric fields.
The activation distances and barriers of TS1 and TS2 are more affected by the external electric fields parallel to the x- and z-axes than by those parallel to the y-axis, and the effects along the x-axis are the most notable. The electric field along the −x-direction is unfavorable to the NH2NO2∙∙∙NH3→NHN(O)OH∙∙∙NH3 reaction.
The intermolecular hydrogen exchange is in essence a concerted process and it can be shown from the change of the imaginary vibrations under the external electric field. The −x-direction of the electric field is opposite to that of the “reaction axis” of the dominant reaction in the hydrogen exchange, leading to an inhibition of the reaction from NH2NO2∙∙∙NH3 to NH2N(O)OH•∙∙∙•NH2. Thus, the concerted effect of the hydrogen exchange is weakened obviously and the barrier height is increased sharply. It is the weakening or even breaking of the concerted effect in the hydrogen exchange that makes the barrier heights increase dramatically along the −x-direction.
The intermolecular hydrogen exchange is always dominant with the field strengths of −0.010 ~ +0.010 a.u. along the y-, +x- and z-directions, and it controls the explosive sensitivity. Due to the unobvious barrier changes, the explosive sensitivities are almost unchanged and remains at a high state, close to those in no fied.
However, the conversion of the dominant reaction occurs along the −x-direction. The hydrogen exchange and 1,3-intramolecular hydrogen transference are dominant with the field strength lower than −0.008 a.u. and more than −0.014 a.u., respectively. The barriers of both reactions are increased significantly with the increase of the field strengths along the −x-direction. Furthermore, the stronger the field strengths, the higher the barrier heights become, suggesting the more significantly the explosive sensitivities are reduced. Therefore, by strengthening the field strengths and adjusting orientations between the “reaction axes” and external electric fields, not only can the selectivities of the reaction be controlled, but also the explosive sensitivities can be reduced significantly, in particular under the super-strong external electric field. Thus, a traditional concept, in which the explosive is dangerous under the super-strong external electric field, is broken theoretically.
For the nitramine explosive with the neutral medium, the low sensitivity can be achieved with the weak external electric field, and for the explosive in the alkaline environment, the low sensitivity can only be achieved under the super-strong field.
This work will be of great significance to the improvement of the technology that the external electric fields are added safely into the energetic material system to enhance the explosive performance.
Ethical Statement
We allow the journal to review all the data, and we confirm the validity of results. There is none of the financial relationships. This work was not published previously and it is not submitted to more than one journal. It is also not split up into several parts to submit. No data have been fabricated or manipulated.
Availability of data and material
We confirm the availability of all the data and materials in this manuscript.
Supplementary Materials
The barriers, bond dissociation energies, geometrical parameters, Mulliken charges and APT charges, index of imaginary vibration, changes of the activation distances versus field strengths in the different field strengths and orientations are in Supporting Information.
Author Contributions
Fu-de Ren: Conceptualization, Project management, Investigation, Data curation, Writing-original draft; Ying-zhe Liu: Data curation; Xiao-lei Wang: Investigation; Li-li Qiu: Data curation; Zi-hui Meng: Writing—review and editing; Xiang Cheng: Software; Yong-xiang Li: Calculation.
Funding
This research received no external funding.
Data Availability Statement
The data related to this research can be accessed upon a reasonable request at email: fdren888@126.com.
Acknowledgments
The authors are grateful for the financial support from the Shanxi Province Natural Science Foundation of China (No. 201801D121067).
Consent for publication
All the authors agree to publish the manuscript.
Conflicts of interest
The authors declare no competing financial interest.
References
- Abdulazeem, M.S.; Alhasan, A.M.; Abdulrahmann, S. Initiation of solid explosives by laser. Int. J. Therm. Sci., 2011, 50, 2117–2121.
- Zhao, L.; Yi, T.; Zhu, H.; Fu, Q.; Sun, X.; Yang, S.; Zheng, W.; Jiang, S. Electromagnetic pulse effect during the bridge wire electric explosion. Chin. J. Energ. Mater., 2019, 27, 481–486.
- Borisenok, V.A.; Mikhailov, A.S.; Bragunets, V.A. Investigation of the polarization of explosives during impact and the influence of an external electric field on the impact sensitivity of superfine PETN. Russ. J. Phys. Chem. B+, 2011, 5, 628–639.
- Rodzevich, A.P.; Gazenaur, E.G.; Kuzmina, L.V.; Krasheninin, V.I.; Gazenaur, N.V. The effect of electric field in the explosive sensitivity of silver azide. J. Phys. Conf. Ser., 2017, 830, 012131.
- Wang, W.J.; Sun, X.J.; Zhang, L.; Lei, F.; Guo, F.; Yang, S.; Fu, Q.B. Sub-microsecond interferometry diagnostic and 3D dynamic simulation of the bridgewire electrical explosion. Chin. J. Energ. Mater., 2019, 27, 473–480.
- Politzer, P.; Murray, J.S.; Concha, M.C.; Lane, P. Effects of electric fields upon energetic molecules: nitromethane and dimethylnitramine. Cent. Eur. J. Energ. Mat., 2007, 4, 3–21.
- Politzer, P.; Murray, J.S.; Lane, P. Computational determination of effects of electric fields upon “trigger linkages” of prototypical energetic molecules. Int. J. Quant. Chem., 2009, 109, 534–539.
- Politzer, P.; Murray, J.S. Computed effects of electric fields upon the C–NO2 and N–NO2 bonds of nitromethane and dimethylnitramine. Int. J. Quant. Chem., 2009, 109, 3–7.
- Zhou, Z.J.; Li, X.P.; Liu, Z.B.; Li, Z.R.; Huang, X.R.; Sun, C.C. Electric field-driven acid-base chemistry: proton transfer from acid (HCl) to Base (NH3/H2O). J. Phys. Chem. A, 2011, 115, 1418–1422.
- Venelin, E.; Valentin, M.; Nadezhda, M.; Marin, R.; Silvia, A.; Milena, S. A model system with intramolecular hydrogen bonding: Effect of external electric field on the tautomeric conversion and electronic structures. Comput. Theor. Chem., 2013, 1006, 113–122.
- Cerón-Carrasco, J.P.; Jacquemin, D. Electric field induced DNA damage: an open door for selective mutations. Chem. Commun., 2013, 49, 7578–7580.
- Shaik, S., de Visser, S.P.; Kumar, D. External electric field will control the selectivity of enzymatic-like bond activations. J. Am. Chem. Soc., 2004, 126, 11746–11749.
- Shaik, S.; Mandal, D.; Ramanan, R. Oriented electric fields as future smart reagents in chemistry. Nat. Chem., 2016, 8, 1091−1098.
- Shaik, S.; Danovich, D.; Joy, J.; Wang, Z.; Stuyver, T. Electric-field mediated chemistry: Uncovering and exploiting the potential of (oriented) electric fields to exert chemical catalysis and reaction control. J. Am. Chem. Soc., 2020, 142, 12551−12562.
- Welborn, V.V.; Head-Gordon, T. Computational design of synthetic enzymes. Chem. Rev., 2019, 119, 6613−6630.
- Wang, C.; Danovich, D.; Chen, H.; Shaik, S. Oriented external electric fields: Tweezers and catalysts for reactivity in halogen-bond complexes. J. Am. Chem. Soc., 2019, 141, 7122−7136.
- Laconsay, C.J.; Tsui, K.Y.; Tantillo, D.J. Tipping the balance: Theoretical interrogation of divergent extended heterolytic fragmentations. Chem. Sci., 2020, 11, 2231−2242.
- Zang, Y.; Zou, Q.; Fu, T.; Ng, F.; Fowler, B.; Yang, J.; Li, H.; Steigerwald, M.L.; Nuckolls, C.; Venkataraman, L. Directing isomerization reactions of cumulenes with electric fields. Nat. Commun., 2019, 10, 4482.
- Dubey, K.D.; Stuyver, T.; Kalita, S.; Shaik, S. Solvent-organization and rate-regulation of a menshutkin reaction by oriented-external electric fields are revealed by combined MD and QM/MM calculations. J. Am. Chem. Soc., 2020, 142, 9955−9965.
- Joy, J.; Stuyver, T.; Shaik, S. Oriented external electric fields and ionic additives elicit catalysis and mechanistic crossover in oxidative addition reactions. J. Am. Chem. Soc., 2020, 142, 3836−3850.
- Starr, R.L.; Fu, T.; Doud, E.A.; Stone, I.; Roy, X.; Venkataraman, L. Gold-carbon contacts from addition of aryl Iodides. J. Am. Chem. Soc., 2020, 142, 7128−7133.
- Stuyver, T.; Huang, J.; Mallick, D.; Danovich, D.; Shaik, S. TITAN: A code for modeling and generating electric fields features and applications to enzymatic reactivity. J. Comput. Chem., 2020, 41, 74−82.
- Stuyver, T.; Danovich, D.; De Proft, F.; Shaik, S. Electrophilic aromatic substitution reactions: Mechanistic landscape, electrostatic and electric-field control of reaction rates, and mechanistic crossovers. J. Am. Chem. Soc., 2019, 141, 9719−9730.
- Yu, L.-J.; Coote, M.L. Electrostatic switching between SN1 and SN2 pathways. J. Phys. Chem. A, 2019, 123, 582−589.
- Blyth, M.T.; Noble, B.B.; Russell, I.C.; Coote, M.L. Oriented internal electrostatic fields cooperatively promote ground- and excited-state reactivity: A case study in photochemical CO2 capture. J. Am. Chem. Soc., 2020, 142, 606–613.
- Shaik, S.; Ramanan, R.; Danovich, D.; Mandal, D. Structure and reactivity/selectivity control by oriented-external electric fields. Chem. Soc. Rev., 2018, 47, 5125−5145.
- Stuyver, T.; Danovich, D.; Joy, J.; Shaik, S. External electric field effects on chemical structure and reactivity. WIRes. Comput. Mol. Sci., 2020, 10, e1438.
- Alemani, M.; Peters, M.V.; Hecht, S.; Rieder, K.H.; Moresco, F.; Grill, L. Electric field-induced isomerization of azobenzene by STM. J. Am. Chem. Soc., 2006, 128, 14446–14447.
- Meir, R.; Chen, H.; Lai, W.; Shaik, S. Oriented electric fields accelerate diels−alder reactions and control the endo/exo selectivity. Chem. Phys. Chem., 2010, 11, 301−310.
- Ren, F.-d.; Shi, W.-j.; Cao, D.-l.; Li, Y.-x.; Zhang, D.-h.; Wang, X.-f.; Shi, Z.-y. External electric field reduces the explosive sensitivity: a theoretical investigation into the hydrogen transference kinetics of the NH2NO2∙∙∙H2O complex. J. Mol. Model., 2020, 26, 351.
- Cabalo, J.; Sausa, R. Theoretical and experimental study of the C–H stretching overtones of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12 hexaazaisowurtzitane (CL-20). J. Phys. Chem. A, 2013, 117, 9039–9046.
- Macharla, A.K.; Parimi, A.; Anuj, A.V. Decomposition mechanism of hexanitrohexaazaisowurtzitane (CL-20) by coupled computational and experimental study. J. Phys. Chem. A, 2019, 123, 4014−4020.
- Demske, D. The experimental aspects of coupling electrical energy into a dense detonation wave: Part 1. NSWC TR, 1982, pp79–143.
- Wang, Y.; Ren, F.; Cao, D. A dynamic and electrostatic potential prediction of the prototropic tautomerism between imidazole 3-oxide and 1-hydroxyimidazole in external electric field. J. Mol. Model., 2019, 25, 330.
- Ren, F.; Cao, D.; Shi, W.; You, M. A dynamic prediction of stability for nitromethane in external electric field. RSC Adv., 2017, 74, 47063–47072.
- Ren, F.; Cao, D.; Shi, W. A dynamics prediction of nitromethane → methyl nitrite isomerization in external electric field. J. Mol. Model., 2016, 22, 96.
- Shu, Y.J.; Dubikhin, V.V.; Nazin, G.M.; Manelis, G.B. Effect of solvents on thermal decomposition of RDX. Chin. J. Energ. Mater., 2000, 8, 108–110.
- Wang, H.B.; Shi, W.J.; Ren, F.D.; Yang, L.; Wang, J.L. A B3LYP and MP2(full) theoretical investigation into explosive sensitivity upon the formation of the intermolecular hydrogen-bonding interaction between the nitro group of RNO2(R = –CH3, –NH2, –OCH3) and HF, HCl or HBr. Comput. Theor. Chem., 2012, 994, 73–80.
- Li, B.H.; Shi, W.J.; Ren, F.D.; Wang, Y. A B3LYP and MP2(full) theoretical investigation into the strength of the C–NO2 bond upon the formation of the intermolecular hydrogen-bonding interaction between HF and the nitro group of nitrotriazole or its methyl derivatives. J. Mol. Model., 2013, 19, 511–519.
- Qiu, W.; Ren, F.D.; Shi, W.J.; Wang, Y.H. A theoretical study on the strength of the C–NO2 bond and ring strain upon the formation of the intermolecular H-bonding interaction between HF and nitro group in nitrocyclopropane, nitrocyclobutane, nitrocyclopentane or nitrocyclohexane. J. Mol. Model., 2015, 21, 114–122.
- Zhou, S.Q.; Zhao, F.Q.; Ju, X.H.; Cheng, X.C.; Yi, J.H. A density functional theory study of adsorption and decomposition of nitroamine molecules on the Al(111) surface. J Phys Chem C 114:9390–9397. J. Phys. Chem. C, 2010, 114, 9390–9397.
- Ju, G.Z.; Ju, Q. The theoretically thermodynamic and kinetic studies on the scission and rearrangement of NH2NO2. Chem. J. Chin. Univ., 1991, 12, 1669–1671.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery Jr, J.A.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell A.;, Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J.M.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, O.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian 09, Revision B.01, Gaussian, Inc.; USA: Wallingford CT, 2010.
- Wigner, E.P. Über das überschreiten von potential schwellen bei chemischen reaktionen. Z. Phys. Chem. B, 1932, 19, 203–216.
- Cramer, C.J. Essentials of computational chemistry: Theories and models, John Wiley & Sons, Ltd., 2002, New York.
- Arabi, A.A.; Matta, C.F. Effects of external electric fields on double proton transfer kinetics in the formic acid dimer. Phys. Chem. Chem. Phys., 2011, 13, 13738–13748.
- Eyring, H.; Eyring, E.M. Modern chemical kinetics, Reinhold Publishing Corporation, 1963, New York.
- Biegler-König, F.W.; Bader, R.F.W.; Tang, T.H. Calculation of the average properties of atoms in molecules. II., J. Comput. Chem., 1982, 3, 317–328.
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc., 2010, 132, 6498–6506.
- Politzer, P.; Murray, J.S. Some molecular/crystalline factors that affect the sensitivities of energetic materials: molecular surface electrostatic potentials, lattice free space and maximum heat of detonation per unit volume. J. Mol. Model., 2015, 21, 25−35.
- Lu, T. Multiwfn: a multifunctional wavefunction analyzer, Version 3.3.5. 2014, Beijing.
- Ren, F.; Cao, D.; Shi, W.; You, M.; Li, M. A theoretical prediction of the possible trigger linkage of CH3NO2 and NH2NO2 in an external electric field. J. Mol. Model., 2015, 21, 145.
- Zeman, S.; Atalar, T.; Friedl, Z.; Ju, X.H. Accounts of the new aspects of nitromethane initiation reactivity. Cent. Eur. J. Energ. Mat., 2009, 6, 119–133.
- Mahadevi, A.S.; Sastry, G.N. Cooperativity in noncovalent interactions. Chem. Rev., 2016, 116, 2775−2825.
- Glasstone, S.; Laidler, K.J.; Eyring, H. The theory of rate processes, McGraw-Hill Book Company, Inc., 1941, New York, 1st edn.
- Tasker, D. The properties of condensed explosives for electromagnetic energy coupling. NSWC TR, 1985, pp85–360.
- Piehler, T.; Hummer, C.; Benjamin, R.; Summers, E.; Mc Nesby, K.; Boyle, V. Preliminary study of coupling electrical energy to detonation reaction zone of primasheet-1000 explosive, 27th international symposium on ballistics, freiburg, Germany, 2013, pp 22–26.
- Politzer, P.; Murray, J.S. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc., 2002, 108, 134–142.
- Rice, B.M.; Hare, J.J. A quantum mechanical investigation of the relation between impact sensitivity and the charge distribution in energetic molecules. J. Phys. Chem. A, 2002, 106, 1770–1783.
- Politzer, P.; Murray, J.S. Impact sensitivity and crystal lattice compressibility/free space. J. Mol. Model., 2014, 20, 2223–2230.
- Murray, J.S.; Concha, M.C.; Politzer, P. Links between surface electrostatic potentials of energetic molecules, impact sensitivities and C–NO2/N–NO2 bond dissociation energies. Mol. Phys., 2009, 107, 89–97.
- Politzer, P.; Murray, J.S. in: Brinck T (ed) Green Energetic Materials, Wiley, Chichester, UK, ch. 2014, 3:45–62.
Figure 1.
Selected structures of the reactant (R), transition states (TS) in the different external electric field strengths and orientations (including those in the absence of field) at the MP2/6-311++G(2d,p) level (geometric parameters are in Å). The “reaction axes” are N6→H7→O8, N1→H5→N6 and N1→H4→O3. .
Figure 1.
Selected structures of the reactant (R), transition states (TS) in the different external electric field strengths and orientations (including those in the absence of field) at the MP2/6-311++G(2d,p) level (geometric parameters are in Å). The “reaction axes” are N6→H7→O8, N1→H5→N6 and N1→H4→O3. .
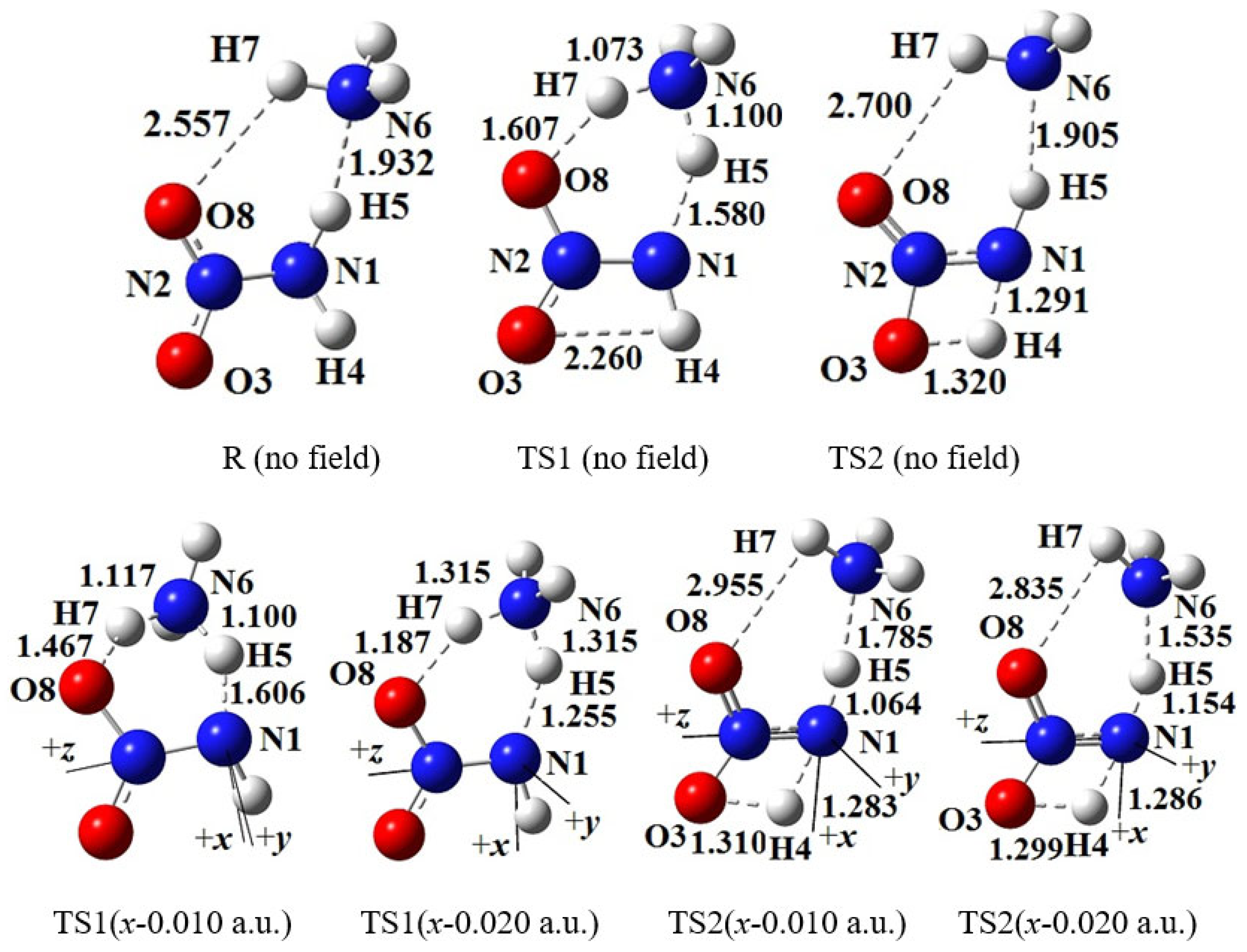
Figure 2.
Selected bond critical point (BCP) of AIM in TS1 and TS2. .
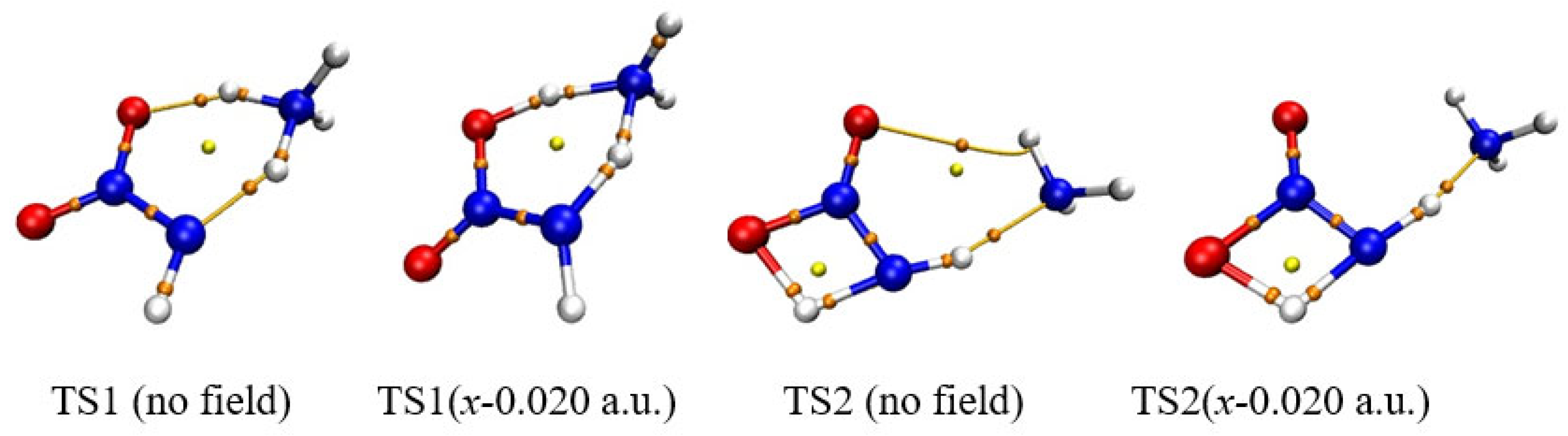
Figure 3.
Gibbs energies (∆G), barriers (Ea) or N−NO2 BDEs versus field strengths along the different field orientations (Ex, Ey and Ez).
Figure 3.
Gibbs energies (∆G), barriers (Ea) or N−NO2 BDEs versus field strengths along the different field orientations (Ex, Ey and Ez).
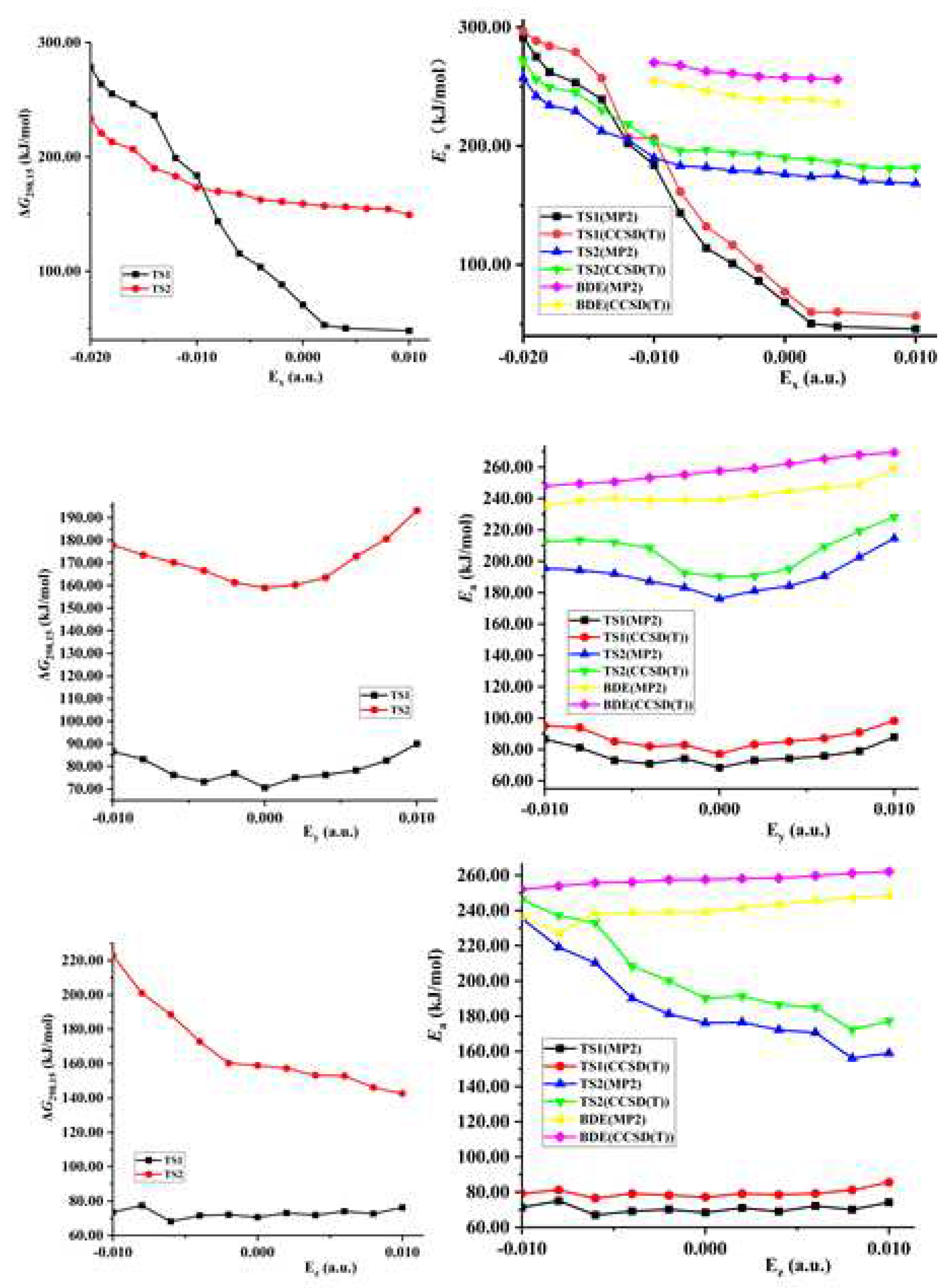
Figure 4.
Plots of the RDG versus the electron density multiplied by the sign of the second Hessian eigenvalue (λ2) for TS1 and TS2. Green indicates strong attractive interaction, blue-brown indicates weak attractive interaction or vdW interaction, and red indicates steric effect.
Figure 4.
Plots of the RDG versus the electron density multiplied by the sign of the second Hessian eigenvalue (λ2) for TS1 and TS2. Green indicates strong attractive interaction, blue-brown indicates weak attractive interaction or vdW interaction, and red indicates steric effect.
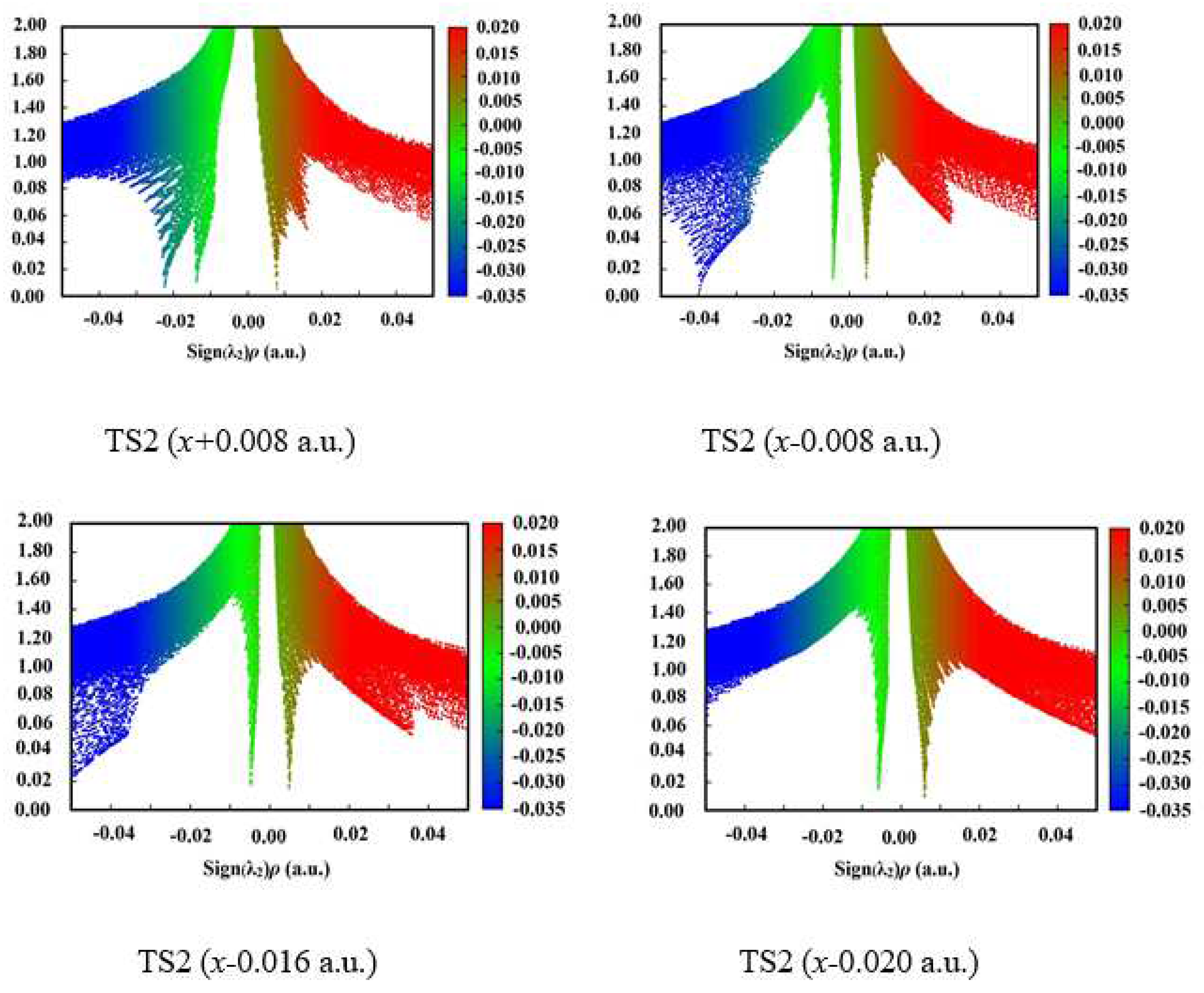
Figure 5.
Surface electrostatic potentials of TS1 and TS2 in the different field strengths (a.u.) along −x-orientation (Small gold and blue spheres represent the most positive and most negative surface electrostatic potentials, respectively).
Figure 5.
Surface electrostatic potentials of TS1 and TS2 in the different field strengths (a.u.) along −x-orientation (Small gold and blue spheres represent the most positive and most negative surface electrostatic potentials, respectively).
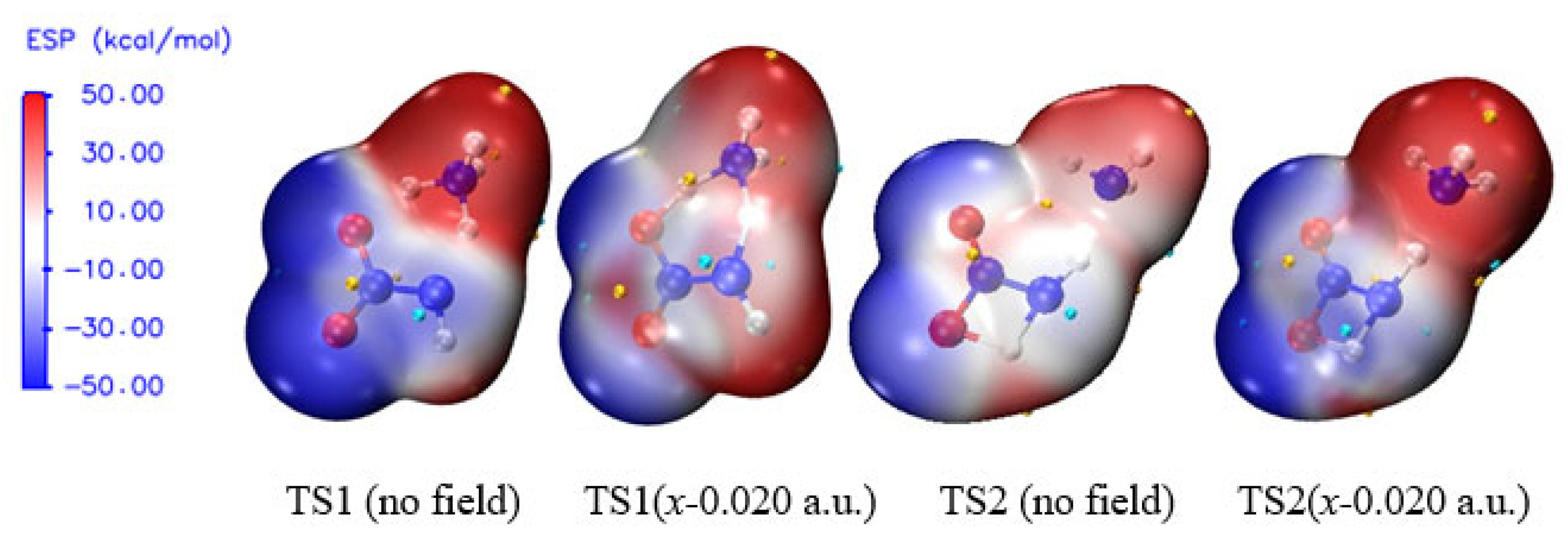

Table 1.
Transition states imaginary frequencies (Imν, cm-1), Gibbs energies (∆G, kJ/mol), reaction rate constants (k, s-1) and corrected reaction rate constants (k298.15 K,C and k688 K,C, s-1), Wigner tunneling corrections (κ) in the absence and presence of fields of varying strengths and directions for the intermolecular hydrogen exchange path (TS1) at the MP2/6-311++G(2d,p) level.
Table 1.
Transition states imaginary frequencies (Imν, cm-1), Gibbs energies (∆G, kJ/mol), reaction rate constants (k, s-1) and corrected reaction rate constants (k298.15 K,C and k688 K,C, s-1), Wigner tunneling corrections (κ) in the absence and presence of fields of varying strengths and directions for the intermolecular hydrogen exchange path (TS1) at the MP2/6-311++G(2d,p) level.
| Field | Imv | ΔG298.15 | k298.15 K | κ298.15 K | k298.15 K,C | ∆G688 K | k688 K | κ688 K | k688 K,C |
|---|---|---|---|---|---|---|---|---|---|
| No field | 368.4 | 70.58 | 2.86×100 | 1.132 | 3.24×100 | 88.62 | 2.68×106 | 1.025 | 2.74×106 |
| z-0.010 | 915.8 | 73.52 | 8.73×10-1 | 1.815 | 1.59×100 | 92.55 | 1.35×106 | 1.153 | 1.55×106 |
| z-0.008 | 872.7 | 77.39 | 1.83×10-1 | 1.740 | 3.19×10-1 | 93.10 | 1.22×106 | 1.139 | 1.39×106 |
| z-0.006 | 492.0 | 68.13 | 7.68×100 | 1.235 | 9.49×100 | 85.62 | 4.53×106 | 1.044 | 4.73×106 |
| z-0.004 | 379.3 | 71.55 | 1.93×100 | 1.140 | 2.20×100 | 84.18 | 5.82×106 | 1.026 | 5.97×106 |
| z-0.002 | 378.1 | 72.08 | 1.56×100 | 1.139 | 1.78×100 | 86.03 | 4.21×106 | 1.026 | 4.32×106 |
| z+0.002 | 355.6 | 73.02 | 1.07×100 | 1.123 | 1.20×100 | 88.92 | 2.54×106 | 1.023 | 2.60×106 |
| z+0.004 | 320.8 | 71.83 | 1.73×100 | 1.100 | 1.90×100 | 90.41 | 1.96×106 | 1.019 | 2.00×106 |
| z+0.006 | 337.0 | 74.03 | 7.11×10-1 | 1.110 | 7.90×10-1 | 92.50 | 1.36×106 | 1.021 | 1.39×106 |
| z+0.008 | 293.5 | 72.59 | 1.27×100 | 1.084 | 1.38×100 | 87.37 | 3.33×106 | 1.016 | 3.39×106 |
| z+0.010 | 271.9 | 76.27 | 2.88×10-1 | 1.072 | 3.09×10-1 | 95.52 | 8.02×105 | 1.013 | 8.13×105 |
| y-0.010 | 350.6 | 86.55 | 4.56×10-3 | 1.119 | 5.10×10-3 | 110.38 | 5.97×104 | 1.022 | 6.10×104 |
| y-0.008 | 327.9 | 83.18 | 1.77×10-2 | 1.104 | 1.96×10-2 | 106.26 | 1.23×105 | 1.020 | 1.25×105 |
| y-0.006 | 342.8 | 76.18 | 2.99×10-1 | 1.114 | 3.33×10-1 | 95.01 | 8.76×105 | 1.021 | 8.95×105 |
| y-0.004 | 360.7 | 73.18 | 1.00×100 | 1.126 | 1.13×100 | 89.17 | 2.43×106 | 1.024 | 2.49×106 |
| y-0.002 | 367.6 | 76.92 | 2.22×10-1 | 1.131 | 2.51×10-1 | 92.35 | 1.40×106 | 1.025 | 1.43×106 |
| y+0.002 | 335.1 | 75.00 | 4.81×10-1 | 1.109 | 5.33×10-1 | 93.25 | 1.19×106 | 1.020 | 1.22×106 |
| y+0.004 | 320.7 | 76.24 | 2.92×10-1 | 1.100 | 3.21×10-1 | 95.18 | 8.51×105 | 1.019 | 8.67×105 |
| y+0.006 | 303.5 | 78.24 | 1.30×10-1 | 1.090 | 1.42×10-1 | 91.00 | 1.77×106 | 1.017 | 1.80×106 |
| y+0.008 | 349.2 | 82.66 | 2.19×10-2 | 1.118 | 2.45×10-2 | 106.36 | 1.20×105 | 1.022 | 1.23×105 |
| y+0.010 | 341.8 | 90.03 | 1.12×10-3 | 1.114 | 1.25×10-3 | 108.27 | 8.63×104 | 1.021 | 8.81×104 |
| x+0.010 | 262.0 | 48.04 | 2.54×104 | 1.067 | 2.71×104 | 62.18 | 2.72×108 | 1.013 | 2.76×108 |
| x+0.004 | 260.0 | 50.17 | 1.08×104 | 1.066 | 1.15×104 | 60.54 | 3.63×108 | 1.012 | 3.67×108 |
| x+0.002 | 310.4 | 52.99 | 3.45×103 | 1.094 | 3.77×103 | 62.15 | 2.74×108 | 1.018 | 2.79×108 |
| x-0.002 | 445.0 | 88.37 | 2.19×10-3 | 1.192 | 2.61×10-3 | 112.26 | 4.30×104 | 1.036 | 4.45×104 |
| x-0.004 | 547.3 | 103.71 | 4.49×10-6 | 1.291 | 5.80×10-6 | 128.71 | 2.42×103 | 1.055 | 2.55×103 |
| x-0.006 | 689.6 | 115.53 | 3.82×10-8 | 1.462 | 5.58×10-8 | 138.53 | 4.35×102 | 1.087 | 4.73×102 |
| x-0.008 | 868.6 | 143.57 | 4.67×10-13 | 1.733 | 8.09×10-13 | 180.02 | 3.08×10-1 | 1.138 | 3.50×10-1 |
| x-0.010 | 649.2 | 183.67 | 4.41×10-20 | 1.410 | 6.21×10-20 | 227.18 | 8.08×10-5 | 1.077 | 8.71×10-5 |
| x-0.012 | 420.2 | 199.01 | 9.05×10-23 | 1.172 | 1.06×10-22 | 248.26 | 2.03×10-6 | 1.032 | 2.09×10-6 |
| x-0.014 | 1124.3 | 236.28 | 2.68×10-29 | 2.228 | 5.96×10-29 | 288.33 | 1.84×10-9 | 1.231 | 2.26×10-9 |
| x-0.016 | 1157.9 | 246.19 | 2.91×10-31 | 2.303 | 1.13×10-30 | 336.05 | 4.38×10-13 | 1.245 | 5.45×10-13 |
| x-0.018 | 1528.7 | 255.11 | 1.34×10-32 | 3.271 | 4.40×10-32 | 340.29 | 2.09×10-13 | 1.426 | 2.98×10-13 |
| x-0.019 | 1633.0 | 263.62 | 4.34×10-34 | 3.591 | 1.56×10-33 | 365.03 | 2.76×10-15 | 1.487 | 4.11×10-15 |
| x-0.020 | 1638.5 | 278.33 | 1.15×10-36 | 3.609 | 4.15×10-36 | 368.81 | 1.43×10-15 | 1.490 | 2.13×10-15 |
Table 2.
Imaginary frequencies (Imν, cm-1), Gibbs energies (∆G, kJ/mol), reaction rate constants (k, s-1) and corrected reaction rate constants (k298.15 K,C and k688 K,C, s-1), Wigner tunneling corrections (κ) in the absence and presence of fields of varying strengths and directions for the 1,3-intramolecular hydrogen transference path (TS2) at the MP2/6-311++G(2d,p) level.
Table 2.
Imaginary frequencies (Imν, cm-1), Gibbs energies (∆G, kJ/mol), reaction rate constants (k, s-1) and corrected reaction rate constants (k298.15 K,C and k688 K,C, s-1), Wigner tunneling corrections (κ) in the absence and presence of fields of varying strengths and directions for the 1,3-intramolecular hydrogen transference path (TS2) at the MP2/6-311++G(2d,p) level.
| Field | Imv | ΔG298.15 | k298.15 K | κ298.15 K | k298.15 K,C | ∆G688 K | k688 K | κ688 K | k688 K,C |
|---|---|---|---|---|---|---|---|---|---|
| No field | 1955.0 | 158.93 | 9.51×10-16 | 4.714 | 4.48×10-15 | 179.59 | 3.32×10-1 | 1.697 | 5.63×10-1 |
| z-0.010 | 1984.7 | 222.84 | 6.05×10-27 | 4.828 | 2.92×10-26 | 252.80 | 9.17×10-7 | 1.719 | 1.58×10-6 |
| z-0.008 | 1977.6 | 201.03 | 4.01×10-23 | 4.800 | 1.92×10-22 | 221.13 | 2.33×10-4 | 1.714 | 3.99×10-4 |
| z-0.006 | 1970.0 | 188.51 | 6.26×10-21 | 4.771 | 2.98×10-20 | 210.02 | 1.63×10-3 | 1.708 | 2.78×10-3 |
| z-0.004 | 1960.6 | 172.80 | 3.54×10-18 | 4.735 | 1.67×10-17 | 198.26 | 1.27×10-2 | 1.701 | 2.16×10-2 |
| z-0.002 | 1958.1 | 160.25 | 5.59×10-16 | 4.726 | 2.64×10-15 | 182.08 | 2.15×10-1 | 1.700 | 3.65×10-1 |
| z+0.002 | 1949.4 | 157.28 | 1.85×10-15 | 4.693 | 8.69×10-15 | 176.73 | 5.47×10-1 | 1.693 | 9.27×10-1 |
| z+0.004 | 1947.6 | 153.19 | 9.64×10-15 | 4.686 | 4.52×10-14 | 174.10 | 8.66×10-1 | 1.692 | 1.47×100 |
| z+0.006 | 1939.8 | 152.83 | 1.11×10-14 | 4.656 | 5.19×10-14 | 170.70 | 1.57×100 | 1.687 | 2.65×100 |
| z+0.008 | 1934.2 | 146.05 | 1.72×10-13 | 4.635 | 7.96×10-13 | 169.04 | 2.10×100 | 1.683 | 3.53×100 |
| z+0.010 | 1927.1 | 142.64 | 6.79×10-13 | 4.609 | 3.13×10-12 | 163.18 | 5.84×100 | 1.678 | 9.80×100 |
| y-0.010 | 1972.0 | 177.92 | 4.48×10-19 | 4.779 | 2.14×10-18 | 202.05 | 6.54×10-3 | 1.710 | 1.12×10-2 |
| y-0.008 | 1970.3 | 173.51 | 2.66×10-18 | 4.772 | 1.27×10-17 | 195.07 | 2.22×10-2 | 1.708 | 3.79×10-2 |
| y-0.006 | 1962.9 | 170.22 | 1.00×10-17 | 4.744 | 4.75×10-17 | 194.35 | 2.51×10-2 | 1.703 | 4.28×10-2 |
| y-0.004 | 1958.3 | 166.57 | 4.36×10-17 | 4.726 | 2.06×10-16 | 186.22 | 1.04×10-1 | 1.700 | 1.77×10-1 |
| y-0.002 | 1957.2 | 161.39 | 3.53×10-16 | 4.722 | 1.67×10-15 | 183.37 | 1.71×10-1 | 1.699 | 2.91×10-1 |
| y+0.002 | 1958.9 | 160.26 | 5.56×10-16 | 4.729 | 2.63×10-15 | 180.09 | 3.04×10-1 | 1.700 | 5.17×10-1 |
| y+0.004 | 1959.8 | 163.55 | 1.48×10-16 | 4.732 | 6.98×10-16 | 186.81 | 9.39×10-2 | 1.701 | 1.60×10-1 |
| y+0.006 | 1960.2 | 172.89 | 3.41×10-18 | 4.734 | 1.61×10-17 | 196.37 | 1.77×10-2 | 1.701 | 3.01×10-2 |
| y+0.008 | 1969.4 | 180.62 | 1.51×10-19 | 4.769 | 7.19×10-19 | 205.10 | 3.84×10-3 | 1.708 | 6.55×10-3 |
| y+0.010 | 1974.0 | 193.18 | 9.51×10-22 | 4.786 | 4.55×10-21 | 223.29 | 1.59×10-4 | 1.711 | 2.73×10-4 |
| x+0.010 | 1985.2 | 149.23 | 4.76×10-14 | 4.829 | 2.3×10-13 | 165.63 | 3.81×100 | 1.719 | 6.55×100 |
| x+0.008 | 1979.4 | 154.44 | 5.82×10-15 | 4.807 | 2.8×10-14 | 173.52 | 9.59×10-1 | 1.715 | 1.65×100 |
| x+0.006 | 1973.6 | 154.92 | 4.8×10-15 | 4.785 | 2.29×10-14 | 176.06 | 6.15×10-1 | 1.711 | 1.06×100 |
| x+0.004 | 1968.1 | 156.39 | 2.65×10-15 | 4.764 | 1.26×10-14 | 177.72 | 4.60×10-1 | 1.707 | 7.85×10-1 |
| x+0.002 | 1961.5 | 157.08 | 2.01×10-15 | 4.739 | 9.51×10-15 | 178.50 | 4.01×10-1 | 1.702 | 6.83×10-1 |
| x-0.002 | 1947.4 | 160.77 | 4.53×10-16 | 4.685 | 2.12×10-15 | 180.67 | 2.75×10-1 | 1.692 | 4.65×10-1 |
| x-0.004 | 1939.5 | 162.53 | 2.23×10-16 | 4.655 | 1.04×10-15 | 184.66 | 1.37×10-1 | 1.686 | 2.31×10-1 |
| x-0.006 | 1927.1 | 167.62 | 2.86×10-17 | 4.609 | 1.32×10-16 | 188.41 | 7.10×10-2 | 1.678 | 1.19×10-1 |
| x-0.008 | 1917.0 | 169.78 | 1.20×10-17 | 4.571 | 5.46×10-17 | 193.85 | 2.74×10-2 | 1.671 | 4.58×10-2 |
| x-0.010 | 1906.4 | 173.2 | 3.01×10-18 | 4.531 | 1.36×10-17 | 196.72 | 1.66×10-2 | 1.663 | 2.76×10-2 |
| x-0.012 | 1895.5 | 183.20 | 5.33×10-20 | 4.491 | 2.39×10-19 | 210.02 | 1.63×10-3 | 1.656 | 2.69×10-3 |
| x-0.014 | 1884.1 | 190.13 | 3.25×10-21 | 4.449 | 1.45×10-20 | 216.85 | 4.92×10-4 | 1.648 | 8.11×10-4 |
| x-0.016 | 1871.8 | 206.82 | 3.88×10-24 | 4.404 | 1.71×10-23 | 239.71 | 9.05×10-6 | 1.639 | 1.48×10-5 |
| x-0.018 | 1858.7 | 212.95 | 3.27×10-25 | 4.357 | 1.42×10-24 | 241.63 | 6.46×10-6 | 1.630 | 1.05×10-5 |
| x-0.019 | 1851.0 | 220.76 | 2.48×10-28 | 4.329 | 1.07×10-27 | 246.46 | 2.78×10-6 | 1.625 | 4.52×10-6 |
| x-0.020 | 1843.8 | 233.57 | 7.98×10-29 | 4.303 | 3.44×10-28 | 248.93 | 1.80×10-6 | 1.620 | 2.92×10-6 |
Table 3.
The surface electrostatic potentials of the oxygen atoms (VS-,O, kcal/mol), average positive and negative values of the surface potentials (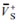 ,
, 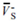 , kcal/mol) and their variances ( and (kcal/mol)2) as well as polar surface area (PSA, %) for TS1 and transition state NH2NO2∙∙∙H2O→NHN(O)OH∙∙∙H2O (TS) in the different field strengths (a.u.) along −x-orientation at the MP2/6-311++G(2d,p) level.
, kcal/mol) and their variances ( and (kcal/mol)2) as well as polar surface area (PSA, %) for TS1 and transition state NH2NO2∙∙∙H2O→NHN(O)OH∙∙∙H2O (TS) in the different field strengths (a.u.) along −x-orientation at the MP2/6-311++G(2d,p) level.
, , kcal/mol) and their variances ( and (kcal/mol)2) as well as polar surface area (PSA, %) for TS1 and transition state NH2NO2∙∙∙H2O→NHN(O)OH∙∙∙H2O (TS) in the different field strengths (a.u.) along −x-orientation at the MP2/6-311++G(2d,p) level.
Table 3.
The surface electrostatic potentials of the oxygen atoms (VS-,O, kcal/mol), average positive and negative values of the surface potentials (, , kcal/mol) and their variances ( and (kcal/mol)2) as well as polar surface area (PSA, %) for TS1 and transition state NH2NO2∙∙∙H2O→NHN(O)OH∙∙∙H2O (TS) in the different field strengths (a.u.) along −x-orientation at the MP2/6-311++G(2d,p) level.
, , kcal/mol) and their variances ( and (kcal/mol)2) as well as polar surface area (PSA, %) for TS1 and transition state NH2NO2∙∙∙H2O→NHN(O)OH∙∙∙H2O (TS) in the different field strengths (a.u.) along −x-orientation at the MP2/6-311++G(2d,p) level.| Field | VS-,O8(TS1) |
(TS1) |
(TS1) |
(TS1) | (TS1) | PSA | VS-,O6(TS)a |
(TS) |
(TS)a
|
(TS)a | (TS)a | PSA(TS) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No Field | -55.8 | 34.8 | -28.2 | 381.9 | 264.3 | 82.7 | -23.2 | 22.0 | -16.1 | 235.9 | 93.5 | 73.2 |
| x-0.002 | -54.4 | 33.1 | -27.5 | 346.8 | 254.8 | 81.7 | -22.4 | 21.5 | -17.3 | 231.8 | 89.5 | 72.1 |
| x-0.004 | -53.0 | 31.5 | -26.6 | 310.2 | 246.0 | 80.6 | -20.3 | 20.8 | -16.5 | 220.3 | 78.3 | 70.8 |
| x-0.006 | -51.7 | 29.8 | -25.8 | 278.7 | 223.5 | 78.3 | -21.6 | 19.6 | -16.6 | 218.1 | 65.7 | 70.3 |
| x-0.008 | -50.7 | 32.5 | -27.2 | 251.6 | 190.7 | 83.9 | -20.3 | 21.3 | -15.1 | 206.2 | 58.3 | 69.5 |
| x-0.010 | -49.6 | 34.4 | -28.2 | 279.9 | 146.2 | 90.6 | -19.7 | 19.7 | -12.2 | 223.4 | 51.4 | 68.8 |
| x-0.012 | -47.8 | 35.2 | -26.7 | 288.3 | 122.8 | 90.8 | -30.2 | 22.5 | -13.6 | 218.7 | 72.1 | 68.1 |
| x-0.013 | -29.8 | 19.3 | -12.8 | 201.3 | 63.6 | 67.3 | ||||||
| x-0.014 | -44.9 | 32.6 | -24.3 | 250.2 | 109.7 | 88.4 | -29.5 | 20.2 | -15.1 | 212.9 | 55.0 | 67.2 |
| x-0.015 | -28.2 | 21.3 | -12.7 | 197.9 | 49.8 | 65.2 | ||||||
| x-0.016 | -41.2 | 28.7 | -21.6 | 211.8 | 111.3 | 84.1 | ||||||
| x-0.018 | -37.6 | 22.0 | -19.9 | 167.6 | 102.5 | 76.5 | ||||||
| x-0.019 | -35.0 | 19.3 | -18.2 | 139.2 | 91.3 | 70.8 | ||||||
| x-0.020 | -33.1 | 16.7 | -15.6 | 116.9 | 90.1 | 68.0% |
a From Ref. 30.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



