
Submitted:
07 February 2023
Posted:
09 February 2023
You are already at the latest version
Abstract
The mechanisms of treatment-resistant epilepsy remain unclear. We have previously shown that frontline administration of therapeutic doses of lamotrigine (LTG), which preferentially inhibits the fast-inactivation state of sodium channels, during corneal kindling of mice promotes cross-resistance to several other antiseizure medicines (ASMs). However, whether this phenomenon extends to monotherapy with ASMs that stabilize the slow inactivation state of sodium channels is unknown. Therefore, this study assessed whether lacosamide (LCM) monotherapy during corneal kindling would promote future development of drug-resistant focal seizures in mice. Male CF-1 mice (n=40/group; 18-25 g) were administered an anticonvulsant dose of LCM (4.5 mg/kg, ip), LTG (8.5 mg/kg, ip), or vehicle (0.5% methylcellulose) twice daily for two weeks during kindling. A subset of mice (n=10/group) were euthanized 1 day after kindling for immunohistochemical assessment of astrogliosis, neurogenesis, and neuropathology. The dose-related antiseizure efficacy of distinct ASMs, LTG, LCM, carbamazepine, levetiracetam, gabapentin, perampanel, valproic acid, phenobarbital, and topiramate was then assessed in remaining kindled mice. Neither LCM nor LTG administration prevented kindling: 29/39 vehicle-exposed mice were kindled; 33/40 LTG-exposed mice were kindled; and 31/40 LCM-exposed mice were kindled. Mice administered LCM or LTG during kindling became resistant to escalating doses of LCM, LTG, and carbamazepine. Perampanel, valproic acid, and phenobarbital were less potent in LTG- and LCM-kindled mice, whereas levetiracetam and gabapentin retained equivalent potency across groups. Notable differences in reactive gliosis and neurogenesis were also appreciated. This study indicates that early, repeated administration of sodium channel blocking ASMs, regardless of inactivation state preference, promotes pharmacoresistant chronic seizures. Inappropriate ASM monotherapy in newly diagnosed epilepsy may thus be one driver of future drug-resistance, with resistance being highly ASM class-specific.
Keywords:
carbamazepine
; levetiracetam
; gabapentin
; perampanel
; valproic acid
; phenobarbital
; topiramate
; neurogenesis
; Ki-67
1. Introduction
The percentage of patients with pharmacoresistant epilepsy has remained unchanged despite the availability of over 30 antiseizure medicines (ASMs), yet the precise mechanisms underlying development and prevalence of treatment-resistant epilepsy (TRE) is relatively unclear [1]. Understanding how frontline ASM monotherapy promotes the onset of future pharmacoresistant seizures would be of immense value to inform the appropriate selection an ASM in newly diagnosed epilepsy. Further, this information provides critical differentiation between currently available ASMs for frontline prescribing practice upon epilepsy diagnosis. Use of an inappropriate ASM monotherapy may adversely affect disease trajectory, but the randomized clinical trials to prospectively address this hypothesis have so far failed to be completed, in part because of the limited preclinical evidence needed to support such a complex study, as well as the ethical limitations associated with potentially imparting drug-resistance on a person with epilepsy. Preclinical studies of the impact of first line ASM monotherapy on subsequent pharmacoresistance can thus guide rational ASM selection after clinical epilepsy diagnosis.
The traditional 60 Hz corneal kindled mouse (CKM) represents a well-characterized preclinical model of temporal lobe epilepsy (TLE) that has contributed to the identification and development of numerous ASMs [2,3], including levetiracetam [2]. The National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP) has prioritized early drug screening in this model to incorporate more etiologically relevant epilepsy models earlier in the drug discovery process [4,5]. The 60 Hz CKM exhibits behavioral deficits [6,7,8] and pathophysiology consistent with human TLE (e.g. reactive gliosis in hippocampal structures [9]), thus making it useful to evaluate behavioral and pathophysiological changes associated with chronic seizures. Moreover, because of the ease in which many mice with uniform seizure and drug exposure history can be generated and precisely experimentally manipulated, this model is incredibly valuable to explore pharmacotherapeutic drivers of pharmacoresistance in early epileptogenesis and define whether specific frontline ASMs may induce future drug-resistant epilepsy.
Clinical guidance recommends frontline use of lamotrigine (LTG) and lacosamide (LCM) in newly diagnosed epilepsy indications. We have previously extensively characterized the pharmacological and behavioral profile of the LTG-resistant 60 Hz CKM based on the larger, more resource-intensive LTG-resistant amygdala-kindled rat [10,11]. Chronic administration of anticonvulsant doses of LTG during kindling acquisition, essentially the epileptogenesis period [12,13], does not delay acquisition of the fully kindled state but instead leads to a kindled mouse with marked resistance to retigabine, carbamazepine, and valproic acid, reflective of a suitable drug-resistant epilepsy model [8]. Inappropriate ASM monotherapy early in the disease course may thus drastically influence the onset of future pharmacosensitivity; a finding that reported in monozygotic twins with epilepsy [14] and also in resected brain tissues from people with drug-resistant TLE [15]. The LTG-resistant CKM is thus an appropriate preclinical model to interrogate the factors that drive the development of pharmacoresistant epilepsy, as well as the sequelae associated with pharmacoresistant TLE. Further, CKM facilitates complex drug efficacy tests with a reduced demand on animal welfare, addressing a major objective of the 3Rs in animal research [16].
It is currently unclear whether the development of drug-resistance in the available preclinical models [8,10,11] is due to sodium channel blocking ASM administration, or a function of LTG monotherapy alone. LTG is mechanistically similar to other sodium channel-blocking ASMs, i.e. carbamazepine and phenytoin, in that it preferentially inhibits the fast inactivation state, whereas LCM is an ASM broadly residing in the sodium channel-blocking class, but dissimilar in that it preferentially stabilizes the slow inactivation state and conformational change of sodium channels [17]. We thus hypothesized that frontline monotherapy with inactivation state-specific sodium channel blocking ASMs would not impact the subsequent development of pharmacoresistance of the corneal kindled seizure, reflecting a class-specific effect of chronic sodium channel modulation rather than an effect isolated to LTG administration alone. This study therefore provides the preclinical evidence that inappropriate selection of sodium channel blocking ASMs in early epileptogenesis can alone elicit the development of future drug-resistant epilepsy.
2. Results
2.1. Administration of Neither LTG nor LCM at Anticonvulsant Doses Delays Kindling Acquistion
We have previously demonstrated that administration of an anticonvulsant LTG dose does not delay kindling acquisition in male CF-1 mice [8]. LCM has been found to delay amygdala-kindling acquisition in male rats in a dose-related manner, but the high dose that delayed kindling also exerted signficant motor impairment [18]. Therefore, our first objective was to define the median anticonvulsant dose of LCM in male CF-1 mice in the maximal electroshock test (Table 1) to define the anticonvulsant dose needed to match our earlier studies with LTG [8]. We quantified the i.p. median effective dose (ED50) of LCM in male CF-1 mice derived from Envigo as 4.05 mg/kg [3.65-4.80; Table 1], consistent with the previously reported ED50 for this agent in CF-1 mice from Charles River [19]. Therefore, a 4.5 mg/kg (i.p.) dose was subsequently acutely administered prior to each twice daily corneal stimulation session to determine whether an anticonvulsant dose of LCM would similarly delay corneal kindling (Figure 1).
2.2. Administration of either LTG or LCM at Anticonvulsant doses During Kindling Reduces Subsequent Kindled Seizure Cross-Sensitivity
We have earlier shown that administration of an anticonvulsant dose of LTG twice/day during corneal kindling to male CF-1 mice does not delay kindling and instead leads to the subsequent development of a highly drug-resistant kindled seizure [8]. We thus tested the hypothesis that cross-tolerance to other anstiseizure medicines would arise in mice similarly exposed to LCM. We quantified the seizure suppressive effects of a diversity of ASMs in mice that received either LTG or LCM during the kindling process to determine whether this observation could similarly extend to mice exposed to LCM (Figure 2).
Following a 5-7 days seizure-free period, fully kindled mice within each kindling treatment group were first challenged to determine their sensitivity to a doubling of the initially-exposed dose of each sodium channel-blocking ASM (i.e., 17 mg/kg for LTG and 9 mg/kg for LCM) matching our prior methods in corneal kindled mice exposed to LTG [20], as well as approaching the previously determined median behaviorally impairing dose for each agent (50 mg/kg for LTG [20] and 26.5 mg/kg for LCM [19,21]). This strategy reflects a dose-escalation approach in a clinical setting for treatment-resistant epilepsy.
Acute administration of LTG to fully kindled mice conferred a drug dose x kindling group interaction on mean seizure score (F(4, 42) = 2.635; p=0.0473; Figure 2A). Within kindling groups, LTG exerted a dose-related anticonvulsant protective effect in VEH-kindled mice (Table 2) and high dose LTG signficantly reduced mean seizure score in these mice (p<0.0001). However, LTG administration to LTG-kindled mice lead to a significant reduction in the total number of mice that were protected from seizure at the highest dose administered (50 mg/kg; Table 2), albeit there was a small, but significant, reduction in mean seizure score (Figure 2A; p=0.048). Similarly, administration of escalating doses of LCM to LTG-kindled mice lead to a significant reduction in the total number of mice that were protected from seizure at the highest dose administered (Table 2) but there was no significant reduction in mean seizure score (Figure 2). This demonstrates that cross tolerance to LCM occurs subsequent to LTG exposure during early epileptogenesis.
Analogously, acute administration of LCM to fully kindled mice conferred a significant main effect of drug administration on mean seizure score (F(2, 41) = 16.58; p<0.0001; Figure 2B). LCM exerted a dose-related anticonvulsant effect in VEH-kindled mice (Table 2) and LCM signficantly reduced mean seizure score in these mice in a dose-related manner (p<0.01 at both 9 and 26.5 mg/kg). However, LCM administration to LTG-kindled mice did not significantly reduce the total number of mice that were protected from seizure at either dose administered (Table 2), albeit there was a small, but significant, reduction in mean seizure score at the highest dose of LTG administered to LCM-kindled mice (Figure 2B; p=0.018). Similarly, administration of two doses of LCM to LCM-kindled mice did not significantly reduce the total number of mice that were protected from seizure at either dose administered (Table 2) and there was no significant reduction in mean seizure score (Figure 2B). These data illustrate that early exposure to therapeutic doses of the ASMs LTG or LCM ultimately leads to reduced sensitivity to dose escalation of the same ASM, as well as significant cross-tolerance to the alternative agent (i.e. LTG switched to LCM is ineffective).
2.3. Administration of either LTG or LCM at Anticonvulsant doses During Kindling Leads to Significant Drug-Resistance Across a Range of ASMs with Diverse Mechanims
Patients with epilepsy who fail their first ASM are transitioned to an alternative agent but there is often little consideration for the mechanism of action of the following or add-on treatment. Therefore, we wanted to determine whether the efficacy of any ASM class was particularly subsequently impacted in LTG- or LCM-kindled mice following confirmation of the lost sensitivity to these ASMs (Figure 3). The resulting profile of the selected ASMs tested in LTG- and LCM-kindled mice can be classified into three principle profiles: no change in potency; loss of potency; loss of efficacy (Figure 3 and Table 2).

Table 2.
Repeated early exposure to anticonvulsant doses of sodium channel-blocking ASMs, LTG (8.5 mg/kg) and LCM (4.5 mg/kg), during kindling leads to cross-resistance to both ASMs and dose-escalation of the same ASM. Early LTG and LCM monotherapy promotes resistance to several other ASMs, except levetiracetam (LEV) and gabapentin (GBP). Gradient shading indicates degree of protection greater in than 50% of animals tested, with Racine stage 2 or lower seizure score considered “protected” 1.
Table 2.
Repeated early exposure to anticonvulsant doses of sodium channel-blocking ASMs, LTG (8.5 mg/kg) and LCM (4.5 mg/kg), during kindling leads to cross-resistance to both ASMs and dose-escalation of the same ASM. Early LTG and LCM monotherapy promotes resistance to several other ASMs, except levetiracetam (LEV) and gabapentin (GBP). Gradient shading indicates degree of protection greater in than 50% of animals tested, with Racine stage 2 or lower seizure score considered “protected” 1.
| ASM | Acute Low and High Doses Tested (mg/kg, i.p.) | VEH-Kindled Mice (N protected/F tested) | LTG-Kindled Mice (N protected/F tested) | LCM-Kindled Mice (N protected/F tested) | |||
|---|---|---|---|---|---|---|---|
| Low Dose | High Dose | Low Dose | High Dose | Low Dose | High Dose | ||
| LTG | 17; 50 | 2/8 | 7/8 | 2/8 | 2/8 | 1/8 | 1/8 |
| LCM | 9; 26.5 | 3/8 | 4/8 | 0/8 | 3/8 | 2/8 | 3/8 |
| LEV | 60; 180 | 7/8 | 7/8 | 6/8 | 7/8 | 6/8 | 5/8 |
| GBP | 75; 300 | 2/8 | 6/8 | 3/8 | 5/8 | 3/8 | 5/8 |
| PER | 1; 4 | 6/8 | 8/8 | 3/8 | 7/8 | 1/8 | 6/8 |
| VPA | 75; 300 | 4/8 | 7/8 | 3/8 | 5/8 | 2/8 | 5/8 |
| PB | 15; 45 | 7/8 | 7/8 | 3/8 | 4/8 | 2/8 | 4/8 |
| CBZ | 10; 40 | 5/8 | 7/8 | 1/8 | 3/8 | 1/8 | 2/8 |
| TPM | 8; 20 | 1/8 | 0/8 | 1/8 | 0/8 | 1/7 | 2/8 |
1 Gradient shading indicates increasing proportion of mice protected from Racine stage 3 or greater behavioral seizure following administration of each ASM in the previously exposed kindling treatment groups. ASMs were delivered by the i.p. route and tested at the previously defined time of effect for each agent.
Two of the ASMs tested demonstrated no change in potency across the three kindling groups. Levetiracetam (LEV) administration at escalating doses was associated with a significant main dose effect on mean seizure score (F(2, 64) = 83.46, p<0.0001), and post-hoc analysis indicated that regardless of kindling group, the low and high doses of LEV resulted in significant reductions in mean seizure score (p<0.0001 for groups; Figure 3A). Further, protection scores did not significantly differ between treatment groups (Table 2). Similarly acute administration of, gabapentin (GBP) was associated with a significant main effect of ASM dose (F(2, 66) = 24.68, p<0.0001). While low-dose GBP did not significantly reduce mean seizure score (Figure 3B) or protect mice from seizures (Table 2), there was a significant reduction in mean seizure score and total number of protected mice with high dose GBP administration, regardless of kindling group (Figure 3B, p<0.01 for all). Further, high dose GBP led to a significant increase in the number of mice protected from seizures in all kindling groups (Table 2; p<0.01 for all). Thus, animals that were kindled in the presence of anticonvulsant doses of LTG or LCM remained sensitive to escalating doses of LEV and GBP.
Several other ASMs tested demonstrated reduced potency to block the fully kindled seizure in the LTG- and LCM-kindled mice. Specifically, administration of low-dose perampanel (PER) significantly reduced mean seizure score (Figure 3C; F(2, 66) = 79.50, p<0.0001) and post-hoc tests indicated that VEH-treated mice had significant score reductions at both the low and high doses tested (p<0.0001). PER administration also significantly increased the number of mice protected from seizure in VEH-kindled mice (Table 2). However, only high-dose PER significantly reduced mean seizure score in LTG- and LCM-kindled mice (Figure 3C; p<0.001 for all) and significantly increased protection from seizures (Table 2). Similarly, valproic acid (VPA) confered a main dose effect (F(2, 42) = 27.57, p<0.0001) and post-hoc analysis demonstrated that VEH-kindled mice had dose-related reductions in mean seizure score following VEH administration (Figure 3D). There was also a significant dose-related increase in the number of protected mice (Table 2). VPA, however, lost potency in LTG- and LCM-kindled mice; only the 300 mg/kg dose reduced mean seizure score (Figure 3) and increased the proportion of protected mice (Table 2). Finally, phenobarbital (PB) also significantly reduced mean seizure score (Figure 3E; dose effect (F(2, 42) = 40.22, p<0.0001), with post-hoc analysis revealing dose-related reductions in mean seizure score (Figure 3E) in VEH-kindled mice. PB also significantly increased the number of protected mice at both doses tested (Table 2). Yet there was a loss of potency in LTG- and LCM-kindled mice; only the 45 mg/kg PB dose reduced mean seizure score (Figure 3; p<0.001) and increased the proportion of protected mice (Table 2). Therefore, these three mechanistically-distinct ASMs lost potency in mice kindled in the presence of therapeutic doses of LTG or LCM.
Exposure to LCM during the kindling process led to cross-resistance to escalating doses of carbamazepine (CBZ; Figure 3F and Table 2). Like LTG and LCM, CBZ is another sodium channel blocking ASM and we have previously demonstrated that neither CBZ, nor phenytoin, retain potency in LTG-exposed CKM [8]; an effect that is similar to LTG-resistant amygdala kindled rats [10,11]. These There was a main dose effect in all treatment groups (F(2, 42) = 24.07; p<0.0001; Figure 3F), with VEH-kindled mice demonstrating significant reductions in seizure score at both doses tested (p<0.01). However post-hoc tests demonstrated that mean seizure score in LTG-kindled mice was not significantly reduced at either dose tested. Conversely, mice kindled in the presence of LCM only demonstrated a modest, albeit statistically significant, reduction in mean seizure score at the highest CBZ dose tested (p<0.05). This did not, however, significantly impact the total number of protected mice in either LTG- or LCM-kindled mice. Further, topiramate (TPM), which has a mixed mechanism of action [17] and has not been previously reported to be effective in the corneal kindled mouse [22], was not effective in any of the kindling groups at any dose tested (Figure 3G and Table 2).
2.4. LTG Administration during Corneal Kindling Blunts Chronic Seizure-Induced Increases in Reactive Gliosis in Area CA1 of Dorsal Hippocampus
Our earlier studies demonstrated that chronic kindled seizures promote increased astrogliosis as early as one day after achieving the fully kindled state and that this increase is not due to astroglial proliferation [9,23]. Thus we sought to assess whether LTG- or LCM-administration during the kindling process would impact glial fibrillary acid protein (GFAP), a marker of astroglial reactivity in response to seizure activity [9,23], immunoreactivity at this same time point (Figure 4). Consistent with our prior reports, there was significant kindling-induced inreases in GFAP immunoreactivity in area CA1 (Figure 4A; F=4.13, p=0.016), but no other region exhibited reactive gliosis (Figure 4B and 4C). Further, post-hoc analysis demonstrated significant increases in GFAP immunoreactivity in VEH-kindled mice (p=0.022) and LCM-kindled mice (p=0.012) in area CA1. However, there was no signficant difference from sham in LTG-kindled mice in this brain region. Thus, treatment with LTG, but not LCM, may blunt chronic seizure-induced increases in reactive gliosis in area CA1 of dorsal hippocampus.
2.5. LCM Administration during Corneal Kindling Blunts Chronic Seizure-Induced Increases in Neuronal Density in Dorsal Hippocampus
We also wanted to determine whether anticonvulsant administration of LTG or LCM during corneal kindling could influence neuronal density, as assessed by NeuN immunoreactivity in dorsal hippocampal structures (Figure 5), which would potentially suggest possible changes in neuropathology or neurodegeneration. We observed significant seizure-induced increases in NeuN immunoreactivity in area CA1 (F= 3.108, p=0.042; Figure 5A) and dentate gyrus (F= 5.694, p=0.004; Figure 5C). There was no signficant effect of kindling on NeuN levels in CA3 (Figure 5B). Post-hoc analysis in CA1 revealed that only VEH- and LTG-kindled mice had significant increases in total field area with NeuN immunoreactivity (p<0.05); an effect that was also evident in dentate gyrus (p<0.01). Thus, NeuN immunoreactivity was significantly increased in area CA1 and dentate gyrus in VEH- and LTG-kindled mice only.
2.6. LCM Administration during Corneal Kindling is Associated with Increased Neurogenesis in Dentate Gyrus
Considering the findings of increased NeuN immunoreactivity in VEH- and LTG-kindled mice, we sought to determine whether this increase was potentially the result of neurogenesis as measured by the molecular marker of cellular proliferation, Ki67 (Figure 6) [24]. There was a significant main effect of kindling on the number of colocalized Ki67+ and NeuN+ cells (F= 5.848, p=0.003) within the dentate gyrus only; no other region of dorsal hippocampus demonstrated significant Ki67+ labeling (not shown). However, post-hoc analysis revealed a significant increase in the number of Ki67+/NeuN+ colocalized cells only in mice kindled the presence of LCM (Figure 6A). Thus, only LCM administration during the corneal kindling process significantly influenced dentate gyrus neurogenesis.
3. Discussion
The percentage of patients with drug-resistant epilepsy has remained unchanged for over 20 years despite the availability of over 30 ASMs [25,26]; there is thus a high unmet need for agents that are effective in individuals with pharmacoresistant epilepsy. Further, understanding how frontline ASM selection in early epileptogenesis impacts upon future onset of pharmacoresistance would be of immense value to appropriately selecting an initial ASM in the clinical setting, as well as differentiating currently available ASMs for use in newly diagnosed epilepsy. This present study confirms that exposure to sodium channel blocking ASMs, regardless of inactivation state preference, during corneal kindling leads to a highly drug-resistant chronic seizure model. This drug-resistant seizure model is both resistant to subsequent dose escalation with the same agent, as well as resistant to cross-over of mechanistically related ASMs (i.e., LTG, LCM, and CBZ are all sodium channel blocking ASMs). Furthermore, we presently reveal stark differences in neuroinflammation and neurogenesis between mice exposed to anticonvulsant doses of LTG versus LCM during kindling, suggesting that ASM monotherapy during kindling can dramatically alter the resulting neuropathology in a chronic seizure model. These findings extend our earlier work to indicate that exposure of mice to LTG during corneal kindling induces a state of subsequent pharmacoresistance [8], which closely aligns with findings following chronic administration of LTG to either an amygdala-kindled rat [10,11] or pentylenetetrazol kindled mouse [27]. Importantly, we now demonstrate that there is a significant loss of potency with the ASMs VPA, PB, and PER, or an altogether loss of anticonvulsant efficacy with mechanistically related agents (in the case of CBZ). This work clearly indicates that inappropriate ASM monotherapy can dramatically alter later ASM sensitivity and instead likely lays the foundation for future drug-resistant epilepsy. Our work carries substantial translational impact for the management of treatment-resistant epilepsy. Further, this study suggests a possible point of clinical caution in the frontline selection of ASM regimen for a newly diagnosed patient with epilepsy. Our present findings suggest that inappropriate ASM selection in early epileptogenesis could be one contributing mechanism to influence the subsequent onset of treatment-resistant epilepsy. While many contributing factors have been proposed as driving the development of pharmacoresistant epilepsy [1], the idea that ASM monotherapy alone may in any way contribute to the ultimate acquisition of drug-resistant epilepsy has been less extensively clinically investigated.
Treatment-resistant epilepsy likely arises due to several inciting mechanisms [1]. Drug-refractory epilepsy is likely the result of a complex combination of mechanisms, including modifications of therapeutic targets [28], changes in multidrug transporters at the blood brain barrier (BBB)[29], intrinsic differences in seizure severity [30], and even failures in medication adherence [31]. Although we did not exclusively evaluate sodium channel subunit composition, function, or density in this study, it is possible that the subsequent treatment resistance observed in both LTG- and LCM-kindled mice may arise because of modifications in sodium channels themselves. LTG and CBZ share the same binding site [32], although LCM preferentially stabilizes the slow inactivation state conformation of sodium channels [33,34,35], suggesting that a global modification of the sodium channel availability or function underlies the presently observed treatment resistant chronic seizure model. Consistent with our earlier findings in the LTG-resistant CKM [8], mice exposed to LCM in this study also are resistant to PER, PB, and VPA, and altogether lose sensitivity to CBZ (Figure 3 and Table 2). CBZ, LTG, and VPA all exert some anticonvulsant effects through sodium channels [17,36]. PHT shares the same binding site on neuronal sodium channels as LTG and CBZ [32], however we did not test the activity of this agent in LCM-kindled mice because we have already documented lack of activity with this agent in LTG-resistant CKM [8]. Clinically, there is a strong indication that mutations in the Nav1.1 subunit of sodium channels are heavily involved in treatment-resistant seizures (i.e., Dravet syndrome [37]). Additionally, sodium channels and KCNQ potassium channels can co-localize and are positioned in proximity within the neuronal membrane [38,39], with KCNQ2 encephalopathy representing another highly treatment-resistant epilepsy syndrome [40]. Whether potassium channel activators would be ineffective in the LCM-resistant CKM, as we have previously reported with ezogabine administration in LTG-resistant CKM [8], requires future studies but would further bolster the hypothesis that modifications in the availability or intrinsic properties of sodium channels specifically evokes onset of drug-resistant epilepsy. Alternative contributors to treatment resistance may underly the presently reported shift in ASM efficacy in mice with a history of LTG or LCM exposure during kindling. Efflux transporter changes at the BBB, e.g. P-glycoprotein (PgP), may also promote pharmacoresistance [28]. The PgP inhibitor, tariquidar, has successfully rescued drug sensitivity in PB-resistant rats with epilepsy [29], directly supporting the transporter hypothesis. PB is a PgP substrate [29,41], as are LTG and LEV [41], but only PB and LTG efficacy were significantly diminished following LCM or LTG administration during kindling. LEV retained efficacy in this present study, as well as in our previously published work with the LTG-resistant CKM [8]. CBZ is also not a substrate for PgP [41]. Thus, our present and prior studies clearly rule out changes in drug transporters as a major contributor to drug-resistance in this model and instead strongly points to changes in sodium channels themselves as a key factor promoting ASM resistance.
One core goal of this study was to define how LTG versus LCM monotherapy changes hippocampal neuropathology and neurogenesis at a cellular level. We found that LTG or LCM as a frontline monotherapy led to substantial changes in cellular and behavioral outcomes. One novel finding of our present study is the observation for differential impacts on astroglia immunoreactivity in area CA1 of dorsal hippocampus following LTG versus LCM administration during kindling (Figure 4A). LTG-administration did not lead to marked changes in astrocyte immunoreactivity relative to sham-treated mice, whereas both VEH- and LCM-treated mice exhibited significant astrogliosis in this brain region. Moreover, the density of NeuN immunoreactivity, a marker of mature, post-mitotic neurons [42], was only significantly increased versus sham- in VEH- and LTG-treated mice in areas CA1 and DG of dorsal hippocampus (Figure 5A and 5C). LCM-kindled mice did not demonstrate significant increases in NeuN immunoreactivity in any brain region relative to sham-kindled mice (Figure 5). We then assessed whether these changes in NeuN immunoreactivity were due to changes in neurogenesis within dentate gyrus using the protein marker, Ki-67. This protein is expressed in all phases of the cell cycle except the resting phase and has been documented to be a suitable marker of mitosis on par with BrdU [24]. Ki-67 positive/NeuN positive cells were only robustly upregulated with LCM administration (Figure 6). Repeated kindled seizures induce premature stem cell differentiation and neurogenesis [43], which may itself further drive mossy fiber sprouting and formation of an epileptic network. Considering that our study was not powered to determine median effective doses of ASMs in LCM- versus LTG-treated CKM, we cannot presently conclude whether either ASM monotherapy led to any differential ASM potency within a drug type. Instead, the present study demonstrates that ASM monotherapy alone can strikingly influence the resulting neuropathology that warrants further detailed study.
From a clinical perspective, our present findings need additional detailed analysis in a real-world setting to determine whether initial ASM monotherapy influences subsequent treatment continuity or treatment switching, reflecting drug-resistance. Inappropriate ASM monotherapy early in the disease course may drastically alter the trajectory of future pharmacosensitivity, as has been observed in a clinical case report from monozygotic twins [14]. However, few studies have been completed to determine real-world ASM prescribing practices and the subsequent impact of ASM monotherapy on patient outcomes. Faught and colleagues previously reported that within a population of 12,975 US patients between 2010-2013, LEV was the frontline ASM of choice in 44.4% of all individuals, regardless of seizure type [44], but sodium channel blocking ASMs, including PHT, LTG, and oxcarbazepine (OXC, a CBZ analogue) represented the next most common choice (18.5% of patients). When patients were stratified by epilepsy diagnosis (focal versus generalized) and insurance type (commercial and Medicare claims versus Medicaid), the ASM use distribution in focal epilepsy patients shifted to 49.7% (commercial/Medicare) and 42.6% (Medicaid) of patients receiving frontline LEV versus 23.8% (commercial/Medicare) and 23.5% (Medicaid) of patients receiving PHT, LTG, and OXC [44]. Conversely, the distribution of generalized epilepsy distribution was 46.0% (commercial/Medicare) and 39.1% (Medicaid) of patients receiving frontline LEV, versus 17.4% (commercial/Medicare) and 14.5% (Medicaid) of patients receiving PHT, LTG, and OXC. Interestingly, that earlier study demonstrated that patients who initially received PHT were the least likely to stay on this monotherapy regimen and the most likely to switch to another ASM treatment [44]. The retrospective clinical study suggests that while sodium channel-blocking ASMs are not the most commonly prescribed frontline agent in newly diagnosed epilepsy, the frontline prescribing of sodium channel-blocking ASMs, regardless of epilepsy diagnosis, is likely to be poorly tolerated and lead to ASMs switching [44], reflective of poorly controlled or drug-resistant epilepsy. Our present study offers valuable insight to future clinical efforts to optimize ASM selection. Further, we justified selecting LTG and LCM, two newer sodium channel-blocking ASMs, based on studies from the UK to indicate that LTG and LEV prescribing has steadily increased in the last several decades, whereas PHT has declined and CBZ has remained steady [45]. This retrospective study of over 63,586 UK patients indicates that sodium channel-blocking ASMs accounted for over 72% of all prescriptions between the period of 1993-2008 [45]. These clinical studies in consort with our present preclinical work indicates that sodium channel-blocking ASMs are commonly prescribed in clinical practice, yet if seizure control is not successfully attained with these agents, the patient may be set up for future onset of treatment-resistant epilepsy.
4. Materials and Methods
Animals: All animal experimentation was approved by the University of Washington Institutional Animal Care and Use Committee under approval number 4387-01 (MBH; approval date 5/5/2019), University of Washington Public Health Service (PHS) Assurance issued by the Office of Laboratory Animal Welfare (OLAW) assurance number D16-00292, and University of Washington AAALAC accreditation number #000523. Male CF-1 mice (4-8 weeks; Envigo Laboratories, Indianapolis, IN, USA) were housed 5 mice/cage in a temperature-controlled vivarium on a 14:10 light/dark cycle. Mice had free access to irradiated chow (Picolab 5053) and water, except during periods of behavioral seizure testing, as previously detailed [46]. Mice were allowed a minimum of 4 days habituation to the housing facility, given a minimum of 1 hour to acclimate to the procedure room prior to all experimentation, and euthanized by decapitation at the completion of all in-life studies. Upon euthanasia at the designated time point after corneal kindling (or sham kindling), the brain was rapidly removed and hippocampus isolated and flash-frozen on dry ice. Brain samples were stored at -80°C until analytical processing.
Corneal Kindled Mouse (CKM): For the 60 Hz corneal kindling protocol, mice (n=40) were stimulated with an initially benign electrical current (60 Hz, sinusoidal pulse; 3.0 mA) delivered for 3 sec via corneal electrodes or sham-kindled (n=10). Seizures were scored upon a 5-point rating scale consistent with the Racine scale in amygdala-kindled rats and routinely used by our group for corneal kindled mice [47], wherein; 1 = jaw chomping and vibrissae twitching, 2 = head bobbing and Straub tail, 3 = unilateral forelimb clonus, 4 = bilateral forelimb clonus and hind-limb rearing, 5 = bilateral forelimb clonus and rearing followed by loss of righting reflex. Twice daily stimulations continued for each mouse until it achieved the criterion of 5 consecutive Racine stage 5 seizures, whereby the mouse was considered “fully kindled”. Any mouse not achieving the fully kindled state was not included for further study.
In Vivo Experimental Reagents: The following antiseizure medicines were commercially purchased: lamotrigine – AK Scientific, K499; lacosamide – Cayman Chemical Co 10012592; carbamazepine – Sigma Aldrich, C4024; gabapentin – Tokyo Chemical Industry, G0318; levetiracetam – Tokyo Chemical Industry, L0234; perampanel – Cayman Chemical Co. 23003; topiramate – Tokyo Chemical Industry, T2755; valproic acid - Sigma Aldrich, P4543. Tetracaine HCl (Sigma Aldrich, T3937) was formulated in 0.9% saline. All ASMs were formulated in 0.5% methylcellulose (Sigma Aldrich, M0430). Brains were rapidly collected following in vivo experimentation and flash frozen in 2-methylbutane overlying dry ice [48].
Antiseizure Medication Efficacy Studies in Fully Kindled Mice: Antiseizure medication (ASM) efficacy studies commenced at least 5-7 days after achieving kindling criterion. Seizure scores of 2 or lower were considered “protected”. Mice were administered escalating doses of each ASM surrounding known median effective doses (ED50) in male WT kindled mice at previously determined time of peak pharmacodynamic effect [18,20]. Mice were allowed a minimum of 3 days’ washout between a dose of each ASM. ASMs were administered in a cross-over drug administration protocol to account for drug and seizure history [21].
Cryosectioning for Immunohistochemistry: Mice for histology were euthanized 24 hours after meeting kindling criterion. Brains were flash frozen and maintained at –80°C until sectioning on a cryostat (Leica DM1860). Four consecutive 20 mm-thick sections/mouse from the dorsal hippocampus (AP from Bregma: -1.58 to -2.18 mm) were slide mounted on SuperFrost slides (Fisher). Slides were stored at –80°C until immunohistochemical processing.
Immunohistochemistry: Brains of all mice were processed for quantitative assessment of molecular markers of reactive gliosis (GFAP; Sigma Aldrich catalogue), neuronal integrity (NeuN; Millipore catalogue), and neurogenesis (Ki67; Abcam) at the conclusion of corneal kindling (n = 9-10 mice/group). The GFAP/NeuN labeling followed previously reported protocols [9,49].
Neurogenesis in hippocampal structures was assessed by Ki-67 labeling (Abcam ab16667) with a goat anti-rabbit secondary antibody conjugated to a 555 nm Cy3 fluorophore (Abcam ab150078). Briefly, slides labeled for Ki67 were removed from –80°C directly into fixative (4% paraformaldehyde; FD Neurotechnologies) for 10 min. Slides were then washed 3x10 min in 1xPBS at RT before being incubation with a blocking reagent (10% goat serum with 0.1% Triton-X in 1x PBS) for 2 hrs at RT in a humid chamber. Slides were then incubated with the Ki67 antibody (1:300) in 1% BSA in 1x PBS/0.1% Triton-X over night at 4°C in a humid chamber. The following day, slides were again washed 3x10 min in 1x PBS before incubation with the goat-anti rabbit secondary antibody (1:500) and NeuN (1:300) in 1x PBS/0.1% Triton-X for 1 hour at RT in a humid chamber. Slides were then washed (3x10 min in 1x PBS) and mounted with ProlongGold with DAPI nuclear counterstain (Invitrogen P36935). Slides were allowed to cure for 24 hours at RT before imaging.
Photomicrographs were captured with a fluorescent microscope (Leica DM-4) with a 20x objective (80x final magnification). Acquisition settings were held constant throughout. NeuN, GFAP, and Ki67 expression levels, given as average area percentage, were automatically measured using Leica Thunder software. Ki67-positive cells were also hand-counted from all hippocampal structures imaged (CA1, CA3, and dentate gyrus) by two investigators blinded to treatment, as well as by the automated cell counting feature of the Leica Thunder imaging software. Cell counts were averaged from all three reviewers and concordance determined by a coefficient of variance.
Statistics: Percent of fully kindled mice was compared between experimental cohorts by a X2 test. The total numbers of stimulations needed to attain the fully kindled state between the treatment groups were compared by one-way ANOVA. Statistical differences in ASM response in fully kindled mice and immunohistochemical detection of protein expression were assessed by one-way ANOVA, with Dunnett’s post-hoc tests. For all statistical measures, p < 0.05 was considered significant and all analysis was performed with Prism version 8.0 or later (GraphPad, San Diego, CA, USA).
Author Contributions
Conceptualization, D.Z., S.M. and M.B.H.; methodology, D.Z., S.M. and M.B.H..; formal analysis, D.Z., S.M. and M.B.H..; investigation D.Z., S.M. and M.B.H.; writing—original draft preparation, D.Z. and M.B.H.; writing—review and editing, M.B.H.; visualization, D.Z. and M.B.H.; supervision, M.B.H.; project administration, M.B.H.; funding acquisition, M.B.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the University of Washington Department of Pharmacy and ITHS KL2 (NCATS KL2 TR002317 to M.B.H.). The APC was funded by a journal voucher from MDPI.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of University of Washington (protocol 4387-01 and 05/05/2019).
Data Availability Statement
In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Please refer to suggested Data Availability Statements in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics. If the study did not report any data, you might add “Not applicable” here.
Acknowledgments
The authors are grateful for the technical assistance of Zach Koneval, Kevin Knox, and Rami Koutobi. The authors are grateful for editorial input of Aaron del Pozo.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front Neurol 2017, 8, 301. [CrossRef]
- Matagne, A.; Klitgaard, H. Validation of corneally kindled mice: a sensitive screening model for partial epilepsy in man. Epilepsy research 1998, 31, 59-71. [CrossRef]
- Klitgaard, H.; Matagne, A.; Gobert, J.; Wulfert, E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur J Pharmacol 1998, 353, 191-206.
- Barker-Haliski, M.L.; Johnson, K.; Billingsley, P.; Huff, J.; Handy, L.J.; Khaleel, R.; Lu, Z.; Mau, M.J.; Pruess, T.H.; Rueda, C.; et al. Validation of a Preclinical Drug Screening Platform for Pharmacoresistant Epilepsy. Neurochemical research 2017, 42, 1904-1918. [CrossRef]
- Kehne, J.H.; Klein, B.D.; Raeissi, S.; Sharma, S. The National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP). Neurochemical research 2017. [CrossRef]
- Barker-Haliski, M.L.; Vanegas, F.; Mau, M.J.; Underwood, T.K.; White, H.S. Acute cognitive impact of antiseizure drugs in naive rodents and corneal-kindled mice. Epilepsia 2016, 57, 1386-1397. [CrossRef]
- Remigio, G.J.; Loewen, J.L.; Heuston, S.; Helgeson, C.; White, H.S.; Wilcox, K.S.; West, P.J. Corneal kindled C57BL/6 mice exhibit saturated dentate gyrus long-term potentiation and associated memory deficits in the absence of overt neuron loss. Neurobiology of disease 2017, 105, 221-234. [CrossRef]
- Koneval, Z.; Knox, K.M.; White, H.S.; Barker-Haliski, M. Lamotrigine-resistant corneal-kindled mice: A model of pharmacoresistant partial epilepsy for moderate-throughput drug discovery. Epilepsia 2018, 59, 1245-1256. [CrossRef]
- Loewen, J.L.; Barker-Haliski, M.L.; Dahle, E.J.; White, H.S.; Wilcox, K.S. Neuronal Injury, Gliosis, and Glial Proliferation in Two Models of Temporal Lobe Epilepsy. Journal of neuropathology and experimental neurology 2016, 75, 366-378. [CrossRef]
- Srivastava, A.K.; White, H.S. Carbamazepine, but not valproate, displays pharmacoresistance in lamotrigine-resistant amygdala kindled rats. Epilepsy research 2013, 104, 26-34. [CrossRef]
- Metcalf, C.S.; Huff, J.; Thomson, K.E.; Johnson, K.; Edwards, S.F.; Wilcox, K.S. Evaluation of antiseizure drug efficacy and tolerability in the rat lamotrigine-resistant amygdala kindling model. Epilepsia Open 2019, 4, 452-463. [CrossRef]
- Sutula, T.P.; Kotloski, R.J. Kindling: A Model and Phenomenon of Epilepsy. In Models of Seizure and Epilepsy, 2nd ed.; Pitkanen, A., Buckmaster, P.S., Galanopoulou, A.S., Moshe, S.L., Eds.; Academic Press: 2017; pp. 813-825.
- Sutula, T.P. Secondary epileptogenesis, kindling, and intractable epilepsy: a reappraisal from the perspective of neural plasticity. Int Rev Neurobiol 2001, 45, 355-386.
- Pawluski, J.L.; Kuchenbuch, M.; Hadjadj, S.; Dieuset, G.; Costet, N.; Vercueil, L.; Biraben, A.; Martin, B. Long-term negative impact of an inappropriate first antiepileptic medication on the efficacy of a second antiepileptic medication in mice. Epilepsia 2018, 59, e109-e113. [CrossRef]
- Jandova, K.; Pasler, D.; Antonio, L.L.; Raue, C.; Ji, S.; Njunting, M.; Kann, O.; Kovacs, R.; Meencke, H.J.; Cavalheiro, E.A.; et al. Carbamazepine-resistance in the epileptic dentate gyrus of human hippocampal slices. Brain : a journal of neurology 2006, 129, 3290-3306. [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G.; Group, N.C.R.R.G.W. Animal research: reporting in vivo experiments: the ARRIVE guidelines. J Gene Med 2010, 12, 561-563. [CrossRef]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [CrossRef]
- Brandt, C.; Heile, A.; Potschka, H.; Stoehr, T.; Loscher, W. Effects of the novel antiepileptic drug lacosamide on the development of amygdala kindling in rats. Epilepsia 2006, 47, 1803-1809. [CrossRef]
- Stohr, T.; Kupferberg, H.J.; Stables, J.P.; Choi, D.; Harris, R.H.; Kohn, H.; Walton, N.; White, H.S. Lacosamide, a novel anti-convulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy research 2007, 74, 147-154. [CrossRef]
- Koneval, Z.; Knox, K.; Memon, A.; Zierath, D.K.; White, H.S.; Barker-Haliski, M. Antiseizure drug efficacy and tolerability in established and novel drug discovery seizure models in outbred versus inbred mice. Epilepsia 2020, In press.
- Florek-Luszczki, M.; Zagaja, M.; Luszczki, J.J. Influence of WIN 55,212-2 on the anticonvulsant and acute neurotoxic potential of clobazam and lacosamide in the maximal electroshock-induced seizure model and chimney test in mice. Epilepsy research 2014, 108, 1728-1733. [CrossRef]
- Rowley, N.M.; White, H.S. Comparative anticonvulsant efficacy in the corneal kindled mouse model of partial epilepsy: Correlation with other seizure and epilepsy models. Epilepsy research 2010, 92, 163-169.
- Knox, K.M.; Beckman, M.; Smith, C.L.; Jayadev, S.; Barker-Haliski, M. Chronic seizures induce sex-specific cognitive deficits with loss of presenilin 2 function. Experimental neurology 2023, 114321. [CrossRef]
- Kee, N.; Sivalingam, S.; Boonstra, R.; Wojtowicz, J.M. The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods 2002, 115, 97-105. [CrossRef]
- Chen, Z.; Brodie, M.J.; Liew, D.; Kwan, P. Treatment Outcomes in Patients With Newly Diagnosed Epilepsy Treated With Established and New Antiepileptic Drugs: A 30-Year Longitudinal Cohort Study. JAMA Neurol 2018, 75, 279-286. [CrossRef]
- Kwan, P.; Brodie, M.J. Epilepsy after the first drug fails: substitution or add-on? Seizure 2000, 9, 464-468. [CrossRef]
- Kumar, S.; Goel, R.K. Pharmacokinetic, pharmacodynamic, and neurochemical investigations of lamotrigine-pentylenetetrazole kindled mice to ascertain it as a reliable model for clinical drug-resistant epilepsy. Animal Model Exp Med 2020, 3, 245-255. [CrossRef]
- Remy, S.; Beck, H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain : a journal of neurology 2006, 129, 18-35. [CrossRef]
- Brandt, C.; Bethmann, K.; Gastens, A.M.; Loscher, W. The multidrug transporter hypothesis of drug resistance in epilepsy: Proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiology of disease 2006, 24, 202-211. [CrossRef]
- Rogawski, M.A.; Johnson, M.R. Intrinsic severity as a determinant of antiepileptic drug refractoriness. Epilepsy currents / American Epilepsy Society 2008, 8, 127-130. [CrossRef]
- Thomson, K.E.; Modi, A.; Glauser, T.A.; White, H.S. The Impact of Nonadherence to Antiseizure Drugs on Seizure Outcomes in an Animal Model of Epilepsy. Epilepsia 2017, In Press.
- Kuo, C.C. A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal Na+ channels. Molecular pharmacology 1998, 54, 712-721.
- Hebeisen, S.; Pires, N.; Loureiro, A.I.; Bonifacio, M.J.; Palma, N.; Whyment, A.; Spanswick, D.; Soares-da-Silva, P. Eslicarbazepine and the enhancement of slow inactivation of voltage-gated sodium channels: a comparison with carbamazepine, oxcarbazepine and lacosamide. Neuropharmacology 2015, 89, 122-135. [CrossRef]
- Curia, G.; Biagini, G.; Perucca, E.; Avoli, M. Lacosamide: a new approach to target voltage-gated sodium currents in epileptic disorders. CNS drugs 2009, 23, 555-568. [CrossRef]
- Beyreuther, B.K.; Freitag, J.; Heers, C.; Krebsfanger, N.; Scharfenecker, U.; Stohr, T. Lacosamide: a review of preclinical properties. CNS Drug Rev 2007, 13, 21-42. [CrossRef]
- Remy, S.; Urban, B.W.; Elger, C.E.; Beck, H. Anticonvulsant pharmacology of voltage-gated Na+ channels in hippocampal neurons of control and chronically epileptic rats. Eur J Neurosci 2003, 17, 2648-2658.
- Catterall, W.A.; Kalume, F.; Oakley, J.C. NaV1.1 channels and epilepsy. J Physiol 2010, 588, 1849-1859. [CrossRef]
- Pan, Z.; Kao, T.; Horvath, Z.; Lemos, J.; Sul, J.Y.; Cranstoun, S.D.; Bennett, V.; Scherer, S.S.; Cooper, E.C. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. The Journal of neuroscience : the official journal of the Society for Neuroscience 2006, 26, 2599-2613. [CrossRef]
- Nguyen, H.M.; Miyazaki, H.; Hoshi, N.; Smith, B.J.; Nukina, N.; Goldin, A.L.; Chandy, K.G. Modulation of voltage-gated K+ channels by the sodium channel beta1 subunit. Proc Natl Acad Sci U S A 2012, 109, 18577-18582. [CrossRef]
- Pisano, T.; Numis, A.L.; Heavin, S.B.; Weckhuysen, S.; Angriman, M.; Suls, A.; Podesta, B.; Thibert, R.L.; Shapiro, K.A.; Guerrini, R.; et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 2015, 56, 685-691. [CrossRef]
- Luna-Tortos, C.; Fedrowitz, M.; Loscher, W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology 2008, 55, 1364-1375. [CrossRef]
- Duan, W.; Zhang, Y.P.; Hou, Z.; Huang, C.; Zhu, H.; Zhang, C.Q.; Yin, Q. Novel Insights into NeuN: from Neuronal Marker to Splicing Regulator. Mol Neurobiol 2016, 53, 1637-1647. [CrossRef]
- Parent, J.M.; Lowenstein, D.H. Seizure-induced neurogenesis: are more new neurons good for an adult brain? Progress in brain research 2002, 135, 121-131. [CrossRef]
- Faught, E.; Helmers, S.; Thurman, D.; Kim, H.; Kalilani, L. Patient characteristics and treatment patterns in patients with newly diagnosed epilepsy: A US database analysis. Epilepsy & behavior : E&B 2018, 85, 37-44. [CrossRef]
- Nicholas, J.M.; Ridsdale, L.; Richardson, M.P.; Ashworth, M.; Gulliford, M.C. Trends in antiepileptic drug utilisation in UK primary care 1993-2008: cohort study using the General Practice Research Database. Seizure 2012, 21, 466-470. [CrossRef]
- Meeker, S.; Beckman, M.; Knox, K.M.; Treuting, P.M.; Barker-Haliski, M. Repeated Intraperitoneal Administration of Low-Concentration Methylcellulose Leads to Systemic Histologic Lesions Without Loss of Preclinical Phenotype. The Journal of pharmacology and experimental therapeutics 2019. [CrossRef]
- Cho, C.; Zeigler, M.; Mizuno, S.; Morrison, R.S.; Totah, R.A.; Barker-Haliski, M. Reductions in Hydrogen Sulfide and Changes in Mitochondrial Quality Control Proteins Are Evident in the Early Phases of the Corneally Kindled Mouse Model of Epilepsy. Int J Mol Sci 2022, 23. [CrossRef]
- Barker-Haliski, M.L.; Oldenburger, K.; Keefe, K.A. Disruption of subcellular Arc/Arg 3.1 mRNA expression in striatal efferent neurons following partial monoamine loss induced by methamphetamine. Journal of neurochemistry 2012, 123, 845-855. [CrossRef]
- Knox, K.M.; Zierath, D.K.; White, H.S.; Barker-Haliski, M. Continuous seizure emergency evoked in mice with pharmacological, electrographic, and pathological features distinct from status epilepticus. Epilepsia 2021. [CrossRef]
Figure 1.
Repeated administration of neither lamotrigine (LTG - 8.5 mg/kg, i.p.) nor lacosamide (LCM – 4.5 mg/kg, i.p.) to male CF-1 mice at the respective time of peak effect prior to each twice daily transcorneal 60 Hz stimulation over 2 weeks prevents the development of the fully corneal kindled state. (A,B) Electrical stimulation of the corneas first does not evoke a behavioral seizure, but over the course of several days, the seizure severity becomes progressively more severe and the percent of mice with a Stage 5 seizure increases, (B) as does the percentage of mice that meet kindling criterion of five consecutive Racine stage 5 seizures. There is no difference in proportion of fully kindled mice in either monotherapy group vs VEH-kindled mice. (C) The mean seizure score is plotted for all treatment groups over time. (D) The change in body weight from baseline (kindling session 1) is generally blunted in kindled mice, but there is no significant treatment effect across kindled groups (* indicates p<0.05).
Figure 1.
Repeated administration of neither lamotrigine (LTG - 8.5 mg/kg, i.p.) nor lacosamide (LCM – 4.5 mg/kg, i.p.) to male CF-1 mice at the respective time of peak effect prior to each twice daily transcorneal 60 Hz stimulation over 2 weeks prevents the development of the fully corneal kindled state. (A,B) Electrical stimulation of the corneas first does not evoke a behavioral seizure, but over the course of several days, the seizure severity becomes progressively more severe and the percent of mice with a Stage 5 seizure increases, (B) as does the percentage of mice that meet kindling criterion of five consecutive Racine stage 5 seizures. There is no difference in proportion of fully kindled mice in either monotherapy group vs VEH-kindled mice. (C) The mean seizure score is plotted for all treatment groups over time. (D) The change in body weight from baseline (kindling session 1) is generally blunted in kindled mice, but there is no significant treatment effect across kindled groups (* indicates p<0.05).
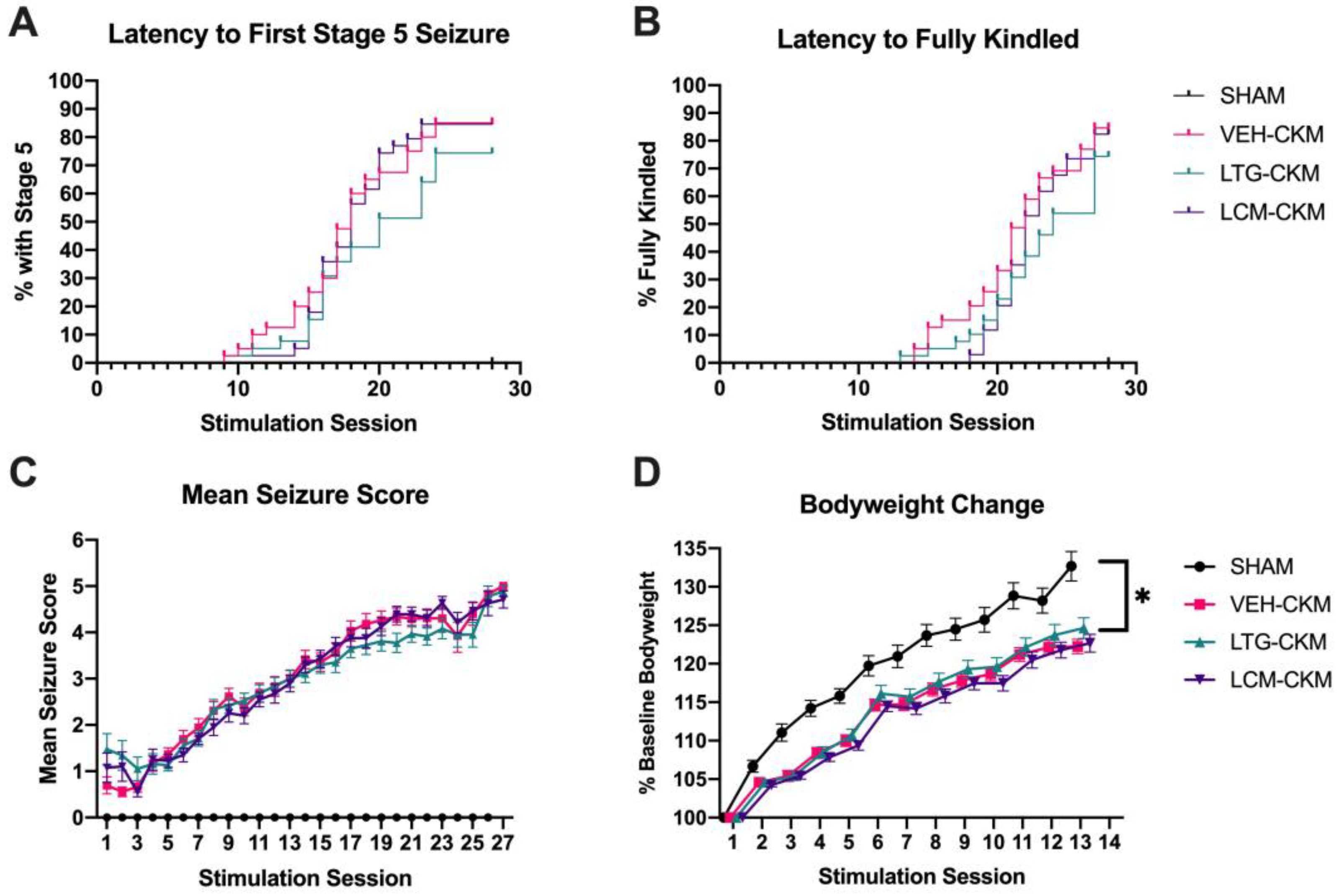
Figure 2.
Repeated administration of anticonvulsant doses of either lamotrigine (LTG; 8.5 mg/kg) or lacosamide (LCM; 4.5 mg/kg) to male CF-1 mice twice daily for 2 weeks during 60 Hz corneal kindling leads to cross-resistance to each sodium channel-blocking ASM at a subsequent doubling or behaviorally impairing dose of each agent (50 mg/kg, LTG; 26.5 mg/kg, LCM). Following a 2 week kindling procedure, mice were allowed a 5-7 day stimulation free period before baseline seizure score was assessed (Baseline) to confirm stable presentation of the Racine stage 5 seizure. The following day, the fully kindled VEH-, LTG-, or LCM-CKM were challenged with one of two doses of either (A) LTG or (B) LCM and mean seizure score assessed at the time of peak effect of each ASM. * indicates significantly different from baseline seizure score, p<0.05.
Figure 2.
Repeated administration of anticonvulsant doses of either lamotrigine (LTG; 8.5 mg/kg) or lacosamide (LCM; 4.5 mg/kg) to male CF-1 mice twice daily for 2 weeks during 60 Hz corneal kindling leads to cross-resistance to each sodium channel-blocking ASM at a subsequent doubling or behaviorally impairing dose of each agent (50 mg/kg, LTG; 26.5 mg/kg, LCM). Following a 2 week kindling procedure, mice were allowed a 5-7 day stimulation free period before baseline seizure score was assessed (Baseline) to confirm stable presentation of the Racine stage 5 seizure. The following day, the fully kindled VEH-, LTG-, or LCM-CKM were challenged with one of two doses of either (A) LTG or (B) LCM and mean seizure score assessed at the time of peak effect of each ASM. * indicates significantly different from baseline seizure score, p<0.05.
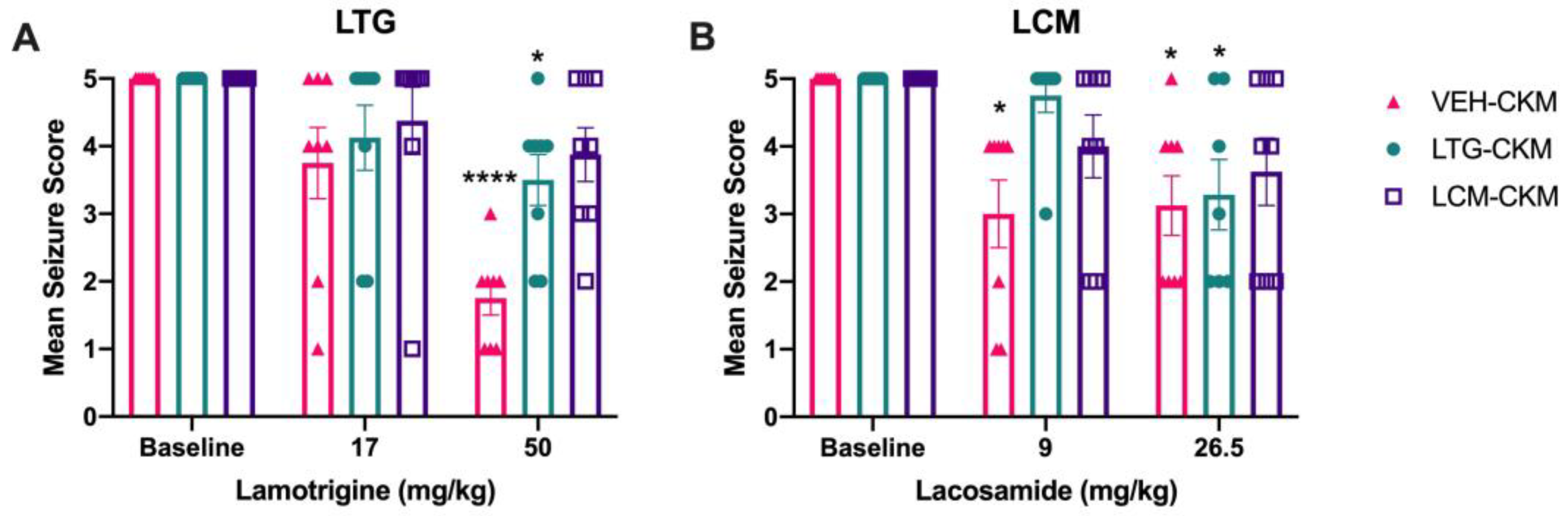
Figure 3.
Repeated administration of either lamotrigine (LTG) or lacosamide (LCM) to male CF-1 mice during 60 Hz corneal kindling leads to subsequent development of a drug-resistant acute secondarily generalized focal seizure model. Only escalating doses of the ASMs A) levetiracetam (LEV) and B) gabapentin (GBP) retained the same degree of anticonvulsant efficacy across VEH-, LTG- and LCM-kindled mice. Several ASMs lost potency in LTG- and LCM-kindled mice relative to that observed in VEH-kindled animals, including C) perampanel (PER), valproic acid (VPA), and phenobarbital (PB). These agents were ineffective in LTG- and LCM-kindled mice at the low dose, whereas VEH-kindled mice demonstrated significant suppression of mean seizure score at this low dose. However, when the higher, nearly maximum tolerated dose was acutely administered to LTG- and LCM-kindled mice, there was a significant reduction of mean seizure score, consistent with the effect of that same dose in VEH-kindled mice. The sodium channel blocking ASM, F) carbamazepine (CBZ) completely lost potency in LTG- and LCM-kindled mice at both low and high doses tested. Finally, G) topiramate (TPM) was not effective at either dose tested in any kindling cohort, consistent with prior reports in corneal kindled mice [22]. * indicates significantly different from baseline seizure score within kindling treatment group (* p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001).
Figure 3.
Repeated administration of either lamotrigine (LTG) or lacosamide (LCM) to male CF-1 mice during 60 Hz corneal kindling leads to subsequent development of a drug-resistant acute secondarily generalized focal seizure model. Only escalating doses of the ASMs A) levetiracetam (LEV) and B) gabapentin (GBP) retained the same degree of anticonvulsant efficacy across VEH-, LTG- and LCM-kindled mice. Several ASMs lost potency in LTG- and LCM-kindled mice relative to that observed in VEH-kindled animals, including C) perampanel (PER), valproic acid (VPA), and phenobarbital (PB). These agents were ineffective in LTG- and LCM-kindled mice at the low dose, whereas VEH-kindled mice demonstrated significant suppression of mean seizure score at this low dose. However, when the higher, nearly maximum tolerated dose was acutely administered to LTG- and LCM-kindled mice, there was a significant reduction of mean seizure score, consistent with the effect of that same dose in VEH-kindled mice. The sodium channel blocking ASM, F) carbamazepine (CBZ) completely lost potency in LTG- and LCM-kindled mice at both low and high doses tested. Finally, G) topiramate (TPM) was not effective at either dose tested in any kindling cohort, consistent with prior reports in corneal kindled mice [22]. * indicates significantly different from baseline seizure score within kindling treatment group (* p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001).
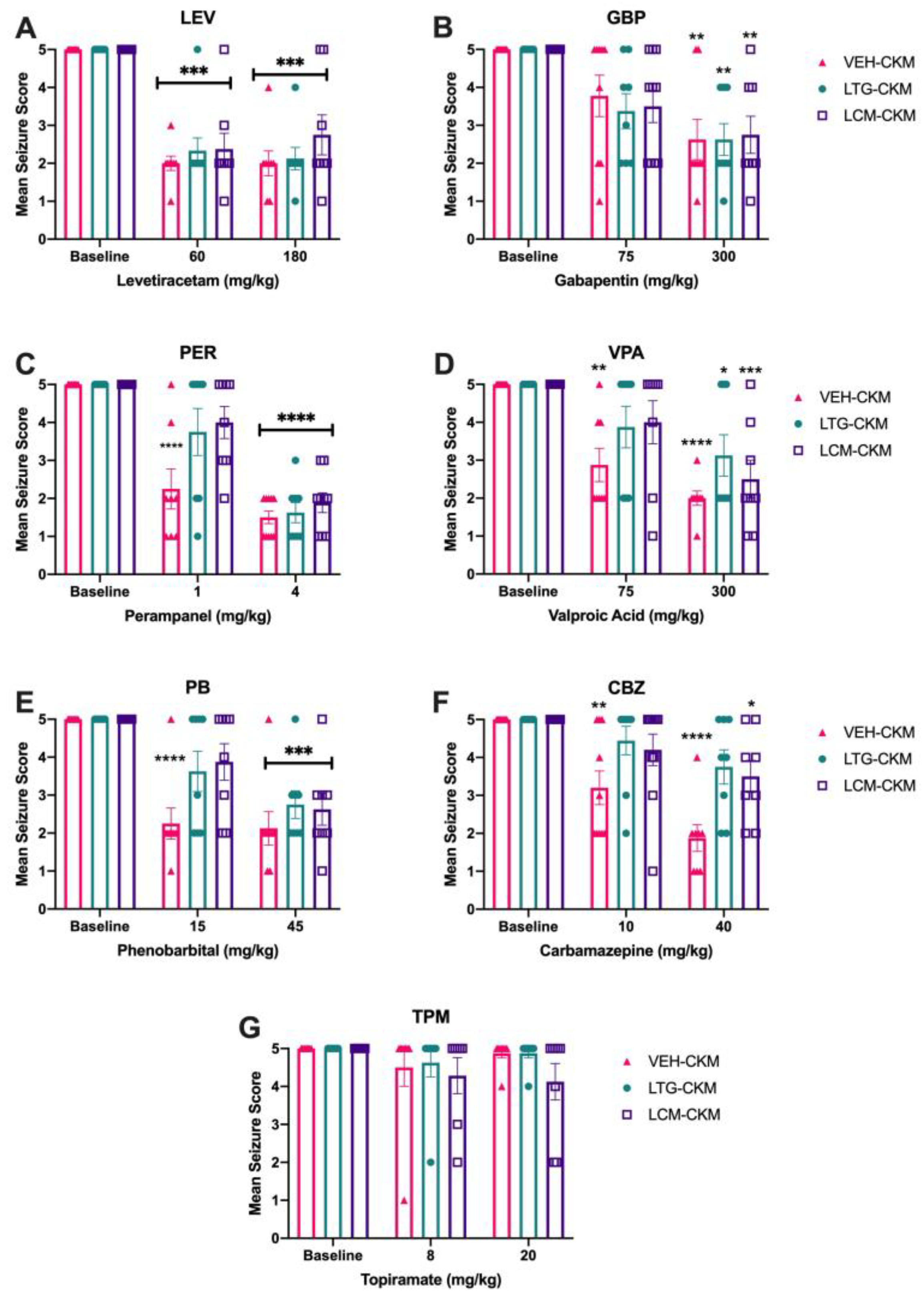
Figure 4.
Administration of LTG (8.5 mg/kg) and LCM (4.5 mg/kg) during corneal kindling differentially impacts subsequent development of reactive astrogliosis in dorsal hippocampus. (A) There is increased GFAP immunoreactivity in area CA1 of dorsal hippocampus of VEH- and LCM-kindled mice versus SHAM-kindled mice, consistent with earlier findings in other CKM [9]. There is no significant change in reactive gliosis in VEH- or LCM-kindled mice in either (B) area CA3 or (C) dentate gyrus. (A–C) LTG-kindled mice do not demonstrate significant reactive gliosis in any region of dorsal hippocampus. (D) Representative photomicrographs of GFAP immunoreactivity in hippocampal CA1 demonstrate increased reactive gliosis in VEH- and LCM-kindled mice in this brain region. Red is GFAP; green is NeuN. * p<0.05 relative to sham-kindled mice (p<0.05).
Figure 4.
Administration of LTG (8.5 mg/kg) and LCM (4.5 mg/kg) during corneal kindling differentially impacts subsequent development of reactive astrogliosis in dorsal hippocampus. (A) There is increased GFAP immunoreactivity in area CA1 of dorsal hippocampus of VEH- and LCM-kindled mice versus SHAM-kindled mice, consistent with earlier findings in other CKM [9]. There is no significant change in reactive gliosis in VEH- or LCM-kindled mice in either (B) area CA3 or (C) dentate gyrus. (A–C) LTG-kindled mice do not demonstrate significant reactive gliosis in any region of dorsal hippocampus. (D) Representative photomicrographs of GFAP immunoreactivity in hippocampal CA1 demonstrate increased reactive gliosis in VEH- and LCM-kindled mice in this brain region. Red is GFAP; green is NeuN. * p<0.05 relative to sham-kindled mice (p<0.05).
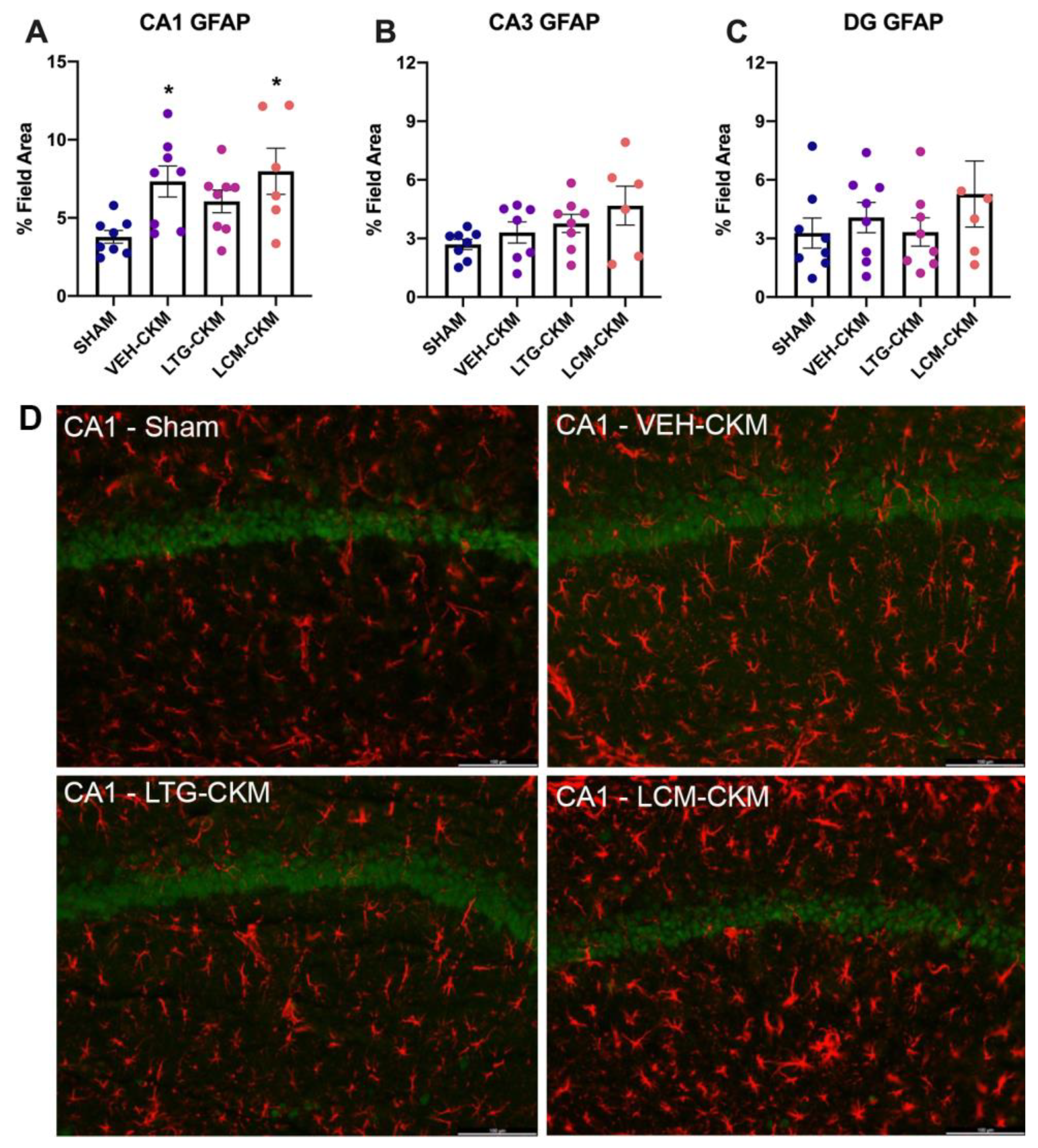
Figure 5.
Neuronal density in vehicle (VEH)- and lamotrigine (LTG) versus lacosamide (LCM) kindled mice was assessed in dorsal hippocampal structures one day after acquisition of the fully kindled state. Administration of LCM during the corneal kindling process reduces chronic seizure-induced increases in NeuN immunoreactivity in hippocampal structures. (A) In area CA1, VEH-kindled mice and mice kindled in the presence of LTG exhibit significant increases in NeuN immunoreactivity (p<0.05); an effect that is not observed in mice kindled in the presence of LCM. (B) There was no significant change in NeuN immunoreactivity in area CA3 in any group versus sham-kindled mice. (C) Chronic kindled seizures led to increased NeuN immunoreactivity in dentate gyrus versus sham-kindled mice (p<0.01). Mice kindled in the presence of LTG demonstrate similar chronic seizure-induced increases in NeuN immunoreactivity (p<0.01); an effect that was not evident in LCM-kindled mice. * indicates significantly different from sham-kindled mice, p<0.05.
Figure 5.
Neuronal density in vehicle (VEH)- and lamotrigine (LTG) versus lacosamide (LCM) kindled mice was assessed in dorsal hippocampal structures one day after acquisition of the fully kindled state. Administration of LCM during the corneal kindling process reduces chronic seizure-induced increases in NeuN immunoreactivity in hippocampal structures. (A) In area CA1, VEH-kindled mice and mice kindled in the presence of LTG exhibit significant increases in NeuN immunoreactivity (p<0.05); an effect that is not observed in mice kindled in the presence of LCM. (B) There was no significant change in NeuN immunoreactivity in area CA3 in any group versus sham-kindled mice. (C) Chronic kindled seizures led to increased NeuN immunoreactivity in dentate gyrus versus sham-kindled mice (p<0.01). Mice kindled in the presence of LTG demonstrate similar chronic seizure-induced increases in NeuN immunoreactivity (p<0.01); an effect that was not evident in LCM-kindled mice. * indicates significantly different from sham-kindled mice, p<0.05.
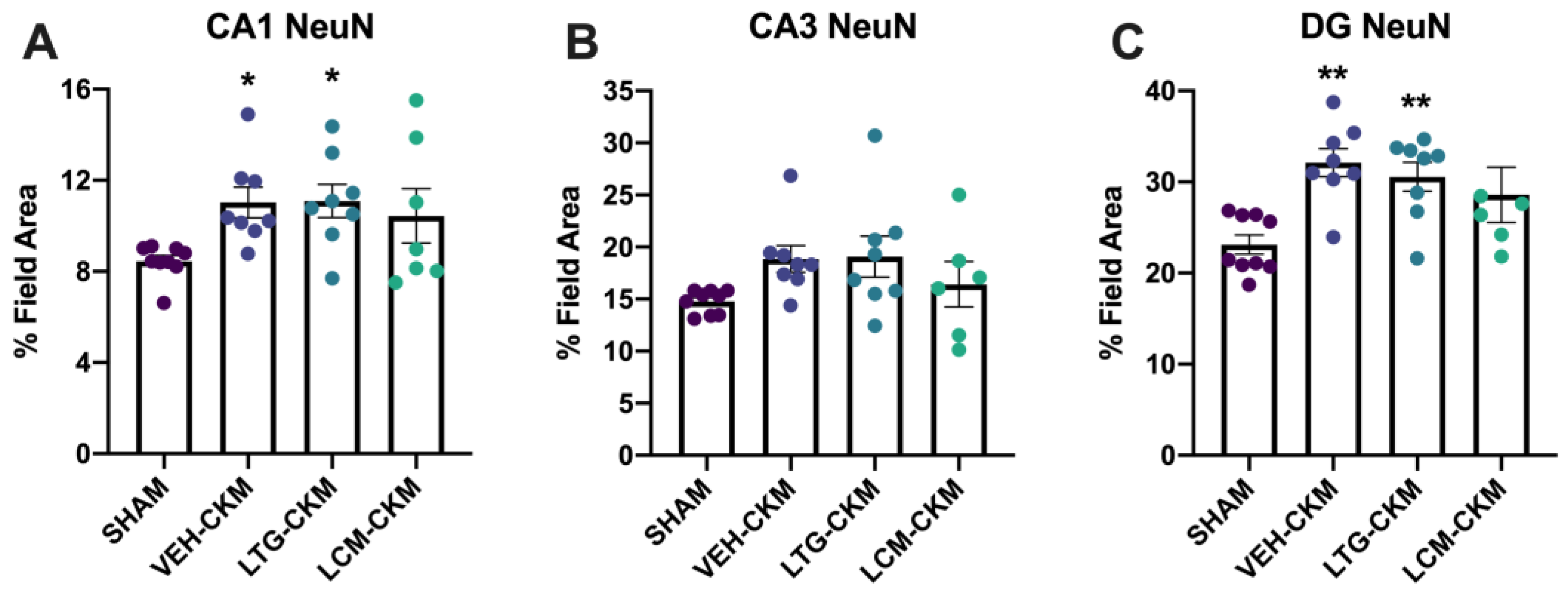
Figure 6.
There is enhanced Ki67 immunoreactivity in NeuN+ cells, presumed mature neurons, in the dentate gyrus of mice kindled in the presence of LCM relative to sham-kindled mice. (A) Neither VEH-kindled nor LTG-kindled mice demonstrate similar increases in Ki67+/NeuN+ cell counts in this brain region. (B) Representative photomicrographs of Ki67+ cells and merged Ki67+NeuN+ immunoreactivity in dentate gyrus (DG). Red indicates Ki67-positive cells, green is NeuN, and blue is DAPI nuclear counterstain. * indicates significantly different from sham-kindled mice (p<0.01).
Figure 6.
There is enhanced Ki67 immunoreactivity in NeuN+ cells, presumed mature neurons, in the dentate gyrus of mice kindled in the presence of LCM relative to sham-kindled mice. (A) Neither VEH-kindled nor LTG-kindled mice demonstrate similar increases in Ki67+/NeuN+ cell counts in this brain region. (B) Representative photomicrographs of Ki67+ cells and merged Ki67+NeuN+ immunoreactivity in dentate gyrus (DG). Red indicates Ki67-positive cells, green is NeuN, and blue is DAPI nuclear counterstain. * indicates significantly different from sham-kindled mice (p<0.01).
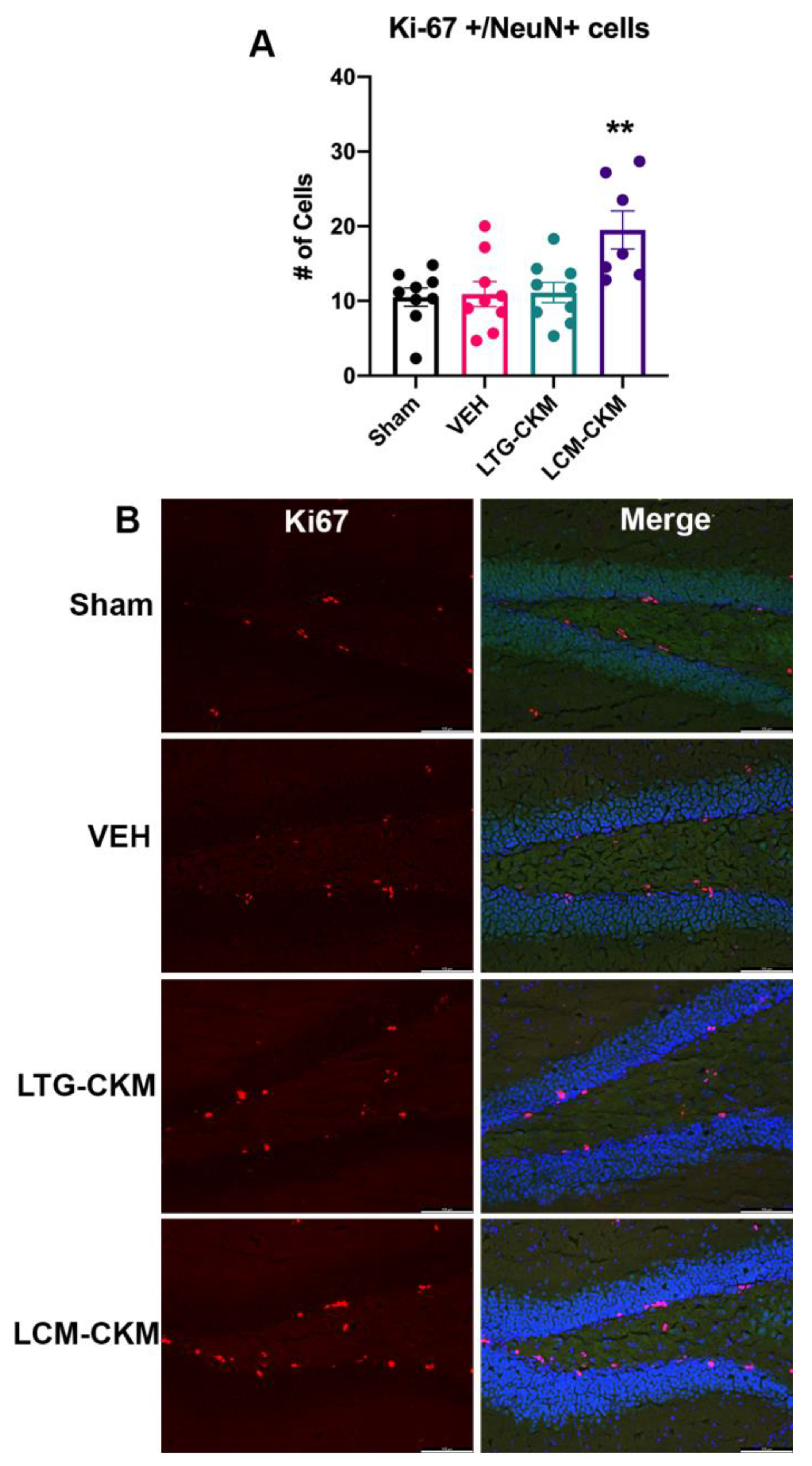
Table 1.
LCM demonstrates dose-related anticonvulsant efficacy in male CF-1 in the maximal electroshock test when administered 1 hour prior to testing. Mice were challenged on the fixed-speed rotarod immediately prior to MES stimulation to assess effects of LCM on minimal motor impairment. The degree of protection was assessed as the abolition of the hindlimb tonic-extension component of the maximal electroshock-induced seizure. These data were used to quantify an i.p. median effective dose (ED50) of LCM (4.05 mg/kg [3.65-4.80]). Data are expressed as N = number protected in the MES test / F = number tested or T = number impaired on a rotarod / F = number tested.
Table 1.
LCM demonstrates dose-related anticonvulsant efficacy in male CF-1 in the maximal electroshock test when administered 1 hour prior to testing. Mice were challenged on the fixed-speed rotarod immediately prior to MES stimulation to assess effects of LCM on minimal motor impairment. The degree of protection was assessed as the abolition of the hindlimb tonic-extension component of the maximal electroshock-induced seizure. These data were used to quantify an i.p. median effective dose (ED50) of LCM (4.05 mg/kg [3.65-4.80]). Data are expressed as N = number protected in the MES test / F = number tested or T = number impaired on a rotarod / F = number tested.
| Dose (mg/kg, i.p.) | Protected (N/F) | Motor Impairment (T/F) |
| 2.5 | 1/8 | 0/8 |
| 3 | 1/16 | 0/16 |
| 4 | 5/15 | 0/15 |
| 5 | 8/8 | 0/8 |
| 10 | 8/8 | 0/8 |
| 15 | 8/8 | 0/8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



