Submitted:
23 January 2023
Posted:
25 January 2023
You are already at the latest version
Abstract
DNA is stored in the nucleus of a cell in a folded state; however, only the necessary genetic information is extracted from the required group of genes. The key to extracting genetic information is chromatin ambivalence. Depending on the chromosomal region, chromatin is characterized into low-density "euchromatin" and high-density "heterochromatin", with various factors being involved in its regulation. Here, we focus on chromatin regulation and gene expression by the yeast FACT complex, which functions in both euchromatin and heterochromatin. FACT is known as a histone H2A/H2B chaperone and initially reported as an elongation factor associated with RNA polymerase II. In budding yeast, FACT activates promoter chromatin by interacting with the transcriptional activators SBF/MBF via the regulation of G1/S cell cycle genes. In fission yeast, FACT plays an important role in the formation of higher-order chromatin structures and transcriptional repression by binding to Swi6, an HP1 family protein, at heterochromatin. This FACT property, which refers to the alternate chromatin-regulation depending on the binding partner, is an interesting phenomenon. Further analysis of nucleosome regulation within heterochromatin is expected in future studies.
Keywords:
FACT
; Spt16
; Pob3
; Nhp6
; SBF
; Heterochromatin
; Swi6
; Clr4
; H3K9me
; Cell cycle
1. Introduction
Gene expression requires the binding of transcriptional activators that recognize and bind specific upstream activating sequences of DNA on the promoter of each gene or enhancer of distal sites [1,2,3]. In most cases, chromatin remodeling factors, histone chaperones, and histone acetyl-transferase complexes are recruited by gene specific transcriptional activators to loosen the core promoter chromatin around the TATA-box and transcriptional start site for the formation of a preinitiation complex with the general transcription factors, TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH, and RNA polymerase II adjacent to the +1 nucleosome [4,5,6,7,8]. The sequential scheme of this transcription initiation indicates the repressive feature of the chromatin structure itself against gene expression, and that the regulatory mechanism of relaxing and closing the chromatin structure is closely related to the regulation of gene expression [9,10]. The landscape of nucleosome occupancy is being analyzed by ChIP-seq or MNase-seq, continuously revealing the genome-wide positioning of nucleosomes and profiling of gene expression [11,12,13,14]. In terms of chromatin structure, there is euchromatin with loose chromatin structure and heterochromatin with complex higher-order chromatin structure [15], with posttranslational modifications of histone proteins playing a key role in maintaining each structure [16,17]. In budding yeast, the SIR complex, Sir2, Sir3, and Sir4, performs chromatin silencing through a molecular mechanism that does not depend on posttranslational modifications by histone H3K9 methyltransferase, whereas in fission yeast, heterochromatin is formed and maintained through high histone H3K9 methylation and low histone H3K4 methylation by histone H3K9 methyltransferase, histone H3K4 de-methylase, and HP1 as in mammal cells [18,19,20,21,22,23,24,25].
2. FACT plays a multifunctional role in transcriptional regulation
The facilitate chromatin transcription (FACT) complex, a heterodimer of Spt16 and SSRP1, was isolated as a transcriptional elongation factor required for chromatin transcription [26,27,28,29]. Unlike other chromatin remodeling factors, FACT has no ATPase domain and performs chromatin remodeling in an ATP-independent manner. The nucleosome-binding activity of human FACT is low in vitro, but through some destabilization in the contact between core histones and nucleosomal DNA, FACT starts interacting with the core region of the histone covered by DNA [30,31]. FACT is a highly conserved histone chaperone between divergent eukaryotic species (Figure 1).
In yeast, it consists of an Spt16/Pob3 heterodimer and the high mobility group box (HMGB) protein Nhp6 [32,33,34,35]. Spt16 was originally isolated as CDC68, a gene responsible for causing G1 arrest [36]. Pob3 and Nhp6 are bipartite analogs of SSRP1 [37,38]. The intracellular roles of FACT in budding and fission yeast appear to be somewhat different. First, the copy number of the budding yeast NHP6 gene and fission yeast nhp6+ gene is different: there are 2 copies of NHP6 in budding yeast, NHP6A and NHP6B [39], whereas there is a single copy of nhp6+ in fission yeast [40]. Second, SPT16 and POB3 are essential genes, whereas NHP6A/B are nonessential genes in budding yeast [34,41]. In fission yeast, spt16+ is an essential gene, whereas pob3+ and nhp6+ are nonessential genes [40], allowing the disruption of pob3+ and nhp6+, which are functional bipartite analogues of SSRP1 in multicellular organisms, exceptionally through eukaryotic species. Given that the function of Spt16 is required even in the absence of Pob3 and Nhp6, it is likely that it exerts a certain chromatin regulatory role, which is specific to Spt16 in fission yeast. As suggesting a role for each FACT component, human SSRP1 has Spt16-dependent and independent roles in transcriptional regulation [42,43]. In budding yeast, Pob3 forms a stable heterodimer with Spt16 via their dimerization domains [33,38]. Biochemical studies have exhibited that Nhp6 plays an essential role in binding the Spt16/Pob3 heterodimer to the nucleosome [35], with the required amount of Nhp6 appearing to be in stoichiometrically excess to that of the Spt16/Pob3 heterodimer [44]. This might suggest that when the HMGB DNA binding domain is fused to the FACT, as in SSRP1, it enhances nucleosome recognition for the efficient H2A/H2B dimer eviction from the nucleosome.
Another FACT isoform, in which Pob3 and Nhp6 are expressed as a fusion SSRP1 protein, has also been analyzed in vivo. In budding yeast, Nhp6 is expressed from NHP6A and NHP6B, and strains in which both NHP6A and NHP6B are simultaneously disrupted show growth defects. Under this condition, the expression of the POB3-NHP6 fusion gene was found to complement the growth defect shown by the nhp6ab∆ strain [37]. Moreover, the Pob3-Nhp6-fused FACT has been reported to be involved in nucleosome regulation, as indicated by biochemical analyses. Single or multiple HMGB modules were fused to Pob3 to mimic SSRP1 for evaluating its nucleosome binding capacity. Human SSRP1 and a yeast Pob3-Nhp6 fusion both required free Nhp6 to support nucleosome reorganization. This result indicated that a single intrinsic DNA-binding HMGB was not sufficient for intact FACT nucleosome reorganizing activity. Whereas, triple HMGB modules at the C-terminus of Pob3 supported FACT activity without free Nhp6; however, this FACT variant was not efficiently released from nucleosomes, in turn exhibiting toxicity in yeast [45]. Recent cryo-EM structure analysis revealed that human Spt16 bound to histones in a subnucleosome and tethered H2A/H2B through its C-terminal acidic tail by acting as a placeholder for DNA, with no electron density being observed at the HMGB domain of SSRP1 [46]. Phosphorylation of the Spt16 C-terminal acidic tail is required for its binding to H2A/H2B in nucleosome [47,48], suggesting the involvement of CKII [49,50]. FACT was also reported to displace H2A/H2B dimers from the nucleosome through the tandem PH domain of Spt16 and histone H3/H4 binding of the Spt16 peptidase-like domain with the help of Nhp6[51,52,53]. Apart from its DNA binding activity, cryo-EM analysis revealed that Nhp6 binds to both C-terminal acidic tails of Spt16 and Pob3 to unfold the FACT complex structure for the activation of efficient nucleosome reorganization [54]. These results suggested the importance of Nhp6 for chromatin remodeling [46].
3. Working models of FACT for nucleosome dynamics
Various molecular models have been proposed to explain the means by which FACT transforms the nucleosome [47]. In the case of the fission yeast FACT, the histone H3/H4 binding activity of the peptidase like domain at the N-terminus of Spt16, the histone H2A/H2B chaperone activity of the tandem PH domain in the central region, the histone H3/H4 binding activity of the tandem PH domain in the C-terminal region of the Pob3 tandem PH domain in the central region, and the DNA binding activity of Nhp6 are thought to play key roles in nucleosome recognition. Accordingly, 2 different models by which fission yeast FACT binds to mononucleosome or dinucleosome are shown in Figure 2.
In the case of binding to mononucleosome, the peptidase like domain of Spt16 and the tandem PH domain of Pob3 bind to the 2 histone H3/H4 dimers present in mononucleosome via their dimerization domains, respectively (Figure 2A). After binding of the Spt16/Pob3 heterodimer, the acidic tail at the C-terminus of the 2 proteins competes with the nucleosomal DNA on the surface of histone H2A/H2B. Following this competition, Nhp6 binds to the fluctuated DNA and promotes the divergence of histone H2A/H2B from DNA in the nucleosome, with the tandem PH domain of Spt16 depositing the histone H2A/H2B dimer from the octasome and transforming it to a hexasome or tetrasome. Studies have already reported the histone chaperone activity of both human and budding yeast FACT for histone H2A/H2B, suggesting the induction of a transient dynamic change in chromatin regulation by a similar process of nucleosome conformational change. Whereas, the histone binding properties of the peptidase like domain of Spt16 and tandem PH domain of Pob3 have suggested their binding to dinucleosome (Figure 2B). In this case, the peptidase like domain of Spt16 and tandem PH domain of Pob3 are expected to act separately on 2 neighboring nucleosomes to bridge them; however, the mechanism by which the tandem PH domain of Spt16 acts on histone H2A/H2B in the nucleosome remains undetermined. As we currently lack any biochemical or structural data on the mechanism of action of FACT on dinucleosome, so we do not know whether this bridging activity affects chromatin regulation. Therefore, the histone H2A/H2B chaperone activity of FACT should be examined in the dinucleosome regulatory case.
4. SBF recruits FACT for promoter chromatin activation in budding yeast
Previous studies have revealed that FACT alters chromatin structure dynamically, transiently evicting nucleosomes for the passage of RNA polymerase II in vivo [53]. Following nucleosome eviction and the passage of RNA polymerase II, FACT deposits nucleosomes to close the transiently loosened chromatin structure [55,56]. In addition to functioning as a transcriptional elongation factor, previous studies have demonstrated that FACT binds to the G1/S START transcription factors SBF and MBF (Figure 3A), which are analogs of E2F in budding yeast. SBF enters the nucleus in late M/early G1 phase [57,58,59], and binds to the G1 gene promoter. In turn, the FACT transiently evicts nucleosomes from the promoter of G1/S regulatory genes before initiation of transcription by RNA polymerase II [60,61,62]. In budding yeast, SBF acts at the START checkpoint in the G1/S phase regulating the expression of the CLN1 and CLN2 genes [63,64,65] (Figure 3A). Cell-cycle-related gene transcription is regulated by the competition between positive and negative regulators of chromatin. In early G1 phase, the activity of SBF is repressed by Whi5 after binding to the "CACGAAAA" promoter element in the UAS until it is activated to initiate transcription at the proper timing. Whi5 recruits the histone deacetyl-transferase complex Rpd3(L) to keep the promoter chromatin at a silent state [60]. Cyclin kinase Cdk1/Cln1,2 phosphorylates Whi5 during the progression of the G1 phase to remove it from SBF [66], which is then converted to its activated state [67,68]. After the removal of Whi5 and Rpd3(L) from the promoter, the SBF-recruited FACT functions to change the promoter chromatin structure [60]; however, the detailed molecular mechanism by which FACT recognizes SBF/MBF remains undetermined. The expression timing of G1 genes during G1 phase is also different, with variations observed during the transition from early G1 to late G1/S. Even though they are regulated by the same transcription factor SBF/MBF, it is assumed that the reason for this is the differences in the chromatin structure of the promoters of each gene. In addition to the G1 cyclin gene, the chromatin structure of the homothallic switching (HO) gene promoter is regulated by SFB and FACT (Figure 3B). The HO gene on chromosome IV encodes the Ho endonuclease.
Budding yeast strains commonly used in laboratories contain a mutation in HO gene that results in defective nuclease activity in vitro and in vivo [69]. Wild type Ho endonuclease induces a double-strand break that targets a DNA element in the MAT locus, causing mating-type switching via gene conversion of the MAT decision cassette [70,71,72,73]. This phenomenon occurs asymmetrically during cell division, with expression of the HO gene in the mother cell and transcriptional repression of the HO gene in the daughter cell. This asymmetry is generated by Ash1, a component of Rpd3(L) HDAC [74,75] that is mainly expressed in daughter cells, and binds to the promoter of the HO gene, thereby strongly repressing transcription [62,76,77,78,79,80]. In addition to this asymmetric expression, the expression of HO needs to be strictly regulated in the mother cell. The promoter structure of the HO gene is relatively long and complex compared with that of common yeast genes, and consists of two sequential regions, URS1 and URS2 (Figure 3B). The combination of URS1 and URS2, approximately 1.0 kbp each, regulates cell cycle-dependent transcriptional initiation [61,81,82,83,84]. URS1 contains two binding sites for Swi5, which is expressed at the M/G1 phase boundary. Swi5 is phosphorylated by the Cdk1 kinase, and is transported into the nucleus from the end of M to the early G1 phase [85,86,87]. A SBF binding site has also been identified at the 3′ side of URS1; however, mutation of this SBF binding sequence does not affect the expression of the HO gene, suggesting that it is not a functionally essential element [62]. The SAGA complex, Swi/Snf complex, and SRB mediator complex are then recruited onto URS1 by Swi5 to loosen the chromatin structure of URS1. In turn, FACT and SBF loosen the chromatin structure from the 3′ side of URS1 to the 5′ side of URS2 and recruit additional SBF activators for the recruitment of the SAGA complex, Swi/Snf complex, and SRB mediator complex downstream of UAR2 and TATA-box [60,61,81,82]. FACT is assumed to be the factor that causes the nucleosome eviction from the 3′ side of URS1 to the 5′ side of URS2 in the sequence of chromatin conformational changes involved in this transcriptional activation.
5. Wave of nucleosome eviction, and the site where FACT functions in HO promoter in budding yeast
A schematic representation of the FACT working region in the HO promoter and the dynamic changes in chromatin along cell cycle progression is shown in Figure 4. In wild type budding yeast strains, nucleosome eviction at URS1 is triggered by the Swi5 activator, and the recruited SAGA complex, Swi/Snf complex, and SRB mediators. FACT-induced nucleosome eviction is then triggered from downstream URS1 to upstream URS2, with the wave of nucleosome eviction being propagated downstream to URS2 and the core promoter, where the chromatin around the TATA-box is finally opened to promote the transcription of the HO gene by RNA polymerase II (Figure 4A). The nucleosomes of URS1 are quickly repositioned, presumably due to polyubiquitination and proteolysis of Swi5 [86]. However, in the FACT mutant yeast strain, nucleosomes are evicted from URS1 by the Swi5 activator and the recruited SAGA complex, Swi/Snf complex, and SRB mediators as in wild type, but this eviction is not propagated downstream from the URS2 to the TATA-box during cell cycle progression (Figure 4B). ChIP analysis of FACT exhibited a biased binding pattern to the upstream of URS2 of the HO gene promoter, suggesting that FACT does not bind to the promoter solely through the SBF recruitment [61]. The reason for this biased promoter binding activity of FACT remains unclear; there might be a characteristic chromatin structure upstream of URS2 that FACT prefers.
6. FACT dependent heterochromatic silencing in fission yeast
Like many other eukaryotes, heterochromatin in fission yeast is formed in a histone H3K9 methlation-dependent manner. The mechanism of formation of constitutive heterochromatin at pericentromeres, subtelomeres, and the MAT locus is very complex [19,88,89,90], with the molecular mechanism of heterochromatin formation being distinct in these 3 regions [91]. Histone H3K9 methylation-dependent higher-order chromatin structures cannot be stably maintained unless the various effector factors function at the correct timing. Methylation of histone H3K9 is the most important key for heterochromatin formation. Although multicellular eukaryotes possess multiple histone H3K9 methyltransferases, in fission yeast, Clr4 is the sole source of methylase of histone H3K9 via its SET domain [92] A recent study revealed that automethylation of Clr4 stimulates its enzymatic activity and maintains its epigenetic stability [93]. At least 2 recruitment mechanisms are known for Clr4 in the establishment of pericentromeric heterochromatin. One is the direct association of Clr4 with HP1/Swi6 [94], and the other is an RNAi-dependent recruitment onto heterochromatin [95,96] (Figure 5). Although heterochromatin formation and transcription of noncoding RNAs (ncRNAs) sound contradictory, HP1/Swi6 is strongly bound by Epe1, a JmjC protein [97,98]. Epe1 carries the acidic activation domain at the N-terminus and stimulates the transcription of heterochromatic ncRNAs by RNA polymerase II [98]. This transcription in the heterochromatin is assumed to be slow and suspendable, and creates a scaffold retaining the nascent ncRNA on heterochromatin for RNAi-related effectors on heterochromatin [99,100,101,102].
In addition to the SET domain, Clr4 itself also has a chromodomain (CD) at its N-terminus that recognizes histone H3K9me, and following recognition exerts its self-propagation ability to methylate H3K9 on the adjacent nucleosome [23,103]. Swi6 and Chp2 are known as fission yeast HP1, which bind to H3K9me-containing nucleosomes, forming homodimers via their chromo-shadow domain (CSD) [104,105]. Heterochromatin is stably maintained by 2 homodimers, Swi6 and Chp2 [106,107], which attract different silencing effectors [108,109,110], with Swi6 being a more versatile HP1, potentially important for the formation and maintenance of stable heterochromatin. In addition to the different roles of the 2 HP1 family proteins in fission yeast, posttranslational modifications of HP1 also affect HP1 heterochromatin formation. For instance, Swi6 has been reported to phosphorylated, and mutations at the phosphorylation site were reported to disrupt heterochromatin formation [111,112,113].
Genetic analysis and ChIP-qPCR showed that fission yeast strains lacking pob3+ (pob3∆) had comparable levels of histone H3K9 methylation and Swi6 localization in the heterochromatic region to those of the wild type strain, but with high levels of heterochromatic expression of ncRNAs. Phenotypic analysis of the pob3∆ strain indicated that heterochromatic silencing was defective in heterochromatin without significant loss of levels of histone H3K9 methylation and HP1/Swi6 binding [114,115]. ChIP analysis of Spt16 exhibited that the binding level of Spt16 to the heterochromatic region in the pob3∆ strain was decreased to half that of the wild type, suggesting the existence of a Pob3-independent recruitment mechanism of Spt16 onto heterochromatin. Genetic analysis also revealed that pob3∆swi6∆ double disruption exhibited an additive silencing defect compared with that shown in each of the pob3∆ and swi6∆ single mutant strains [115]. Therefore, we assumed that the recruitment of Spt16 onto heterochromatin is partially dependent on Swi6. To test this hypothesis, we performed biochemical analysis using recombinant Spt16 and the fission yeast HP1 family, Swi6 and Chp2. We found that the peptidase like domain of Spt16 directly binds to the dimerized chromo-shadow domain (CSD) of Swi6, but not to Chp2-CSD [106,115,116]. Although the "PxVxL/I" hydrophobic amino acid sequence of the CSD binding motif is necessary for stable CSD binding [117,118,119], this motif is not conserved in the peptidase like domain of Spt16. Further Spt16-Swi6 binding experiments revealed that the binding of Spt16 and Swi6 was easily compromised by increasing salt concentration in the binding buffer in vitro. This property is not seen in the hydrophobic interaction via the PxVxL motif. These results are suggesting the existence of a novel binding mode between Spt16 and Swi6.
To elucidate this binding mode between Spt16 and Swi6, we carefully compared the primary sequences of Swi6 and Chp2 in the CSD, and found a difference in the β1-β2 connecting loop, which forms the protrusion in the CSD homodimer. We identified a charge-biased "RKDD" cluster in the β1-β2 connecting loop of Swi6-CSD, but no such charge-biased cluster in the β1-β2 connecting loop of Chp2-CSD. Other HP1 family proteins were also examined for the presence of charge-biased clusters in the β1-β2 connecting loop, but no charge-biased clusters were found. Physical interaction between HP1c and SSRP1 for transcriptional activation on euchromatin has been reported in Drosophila melanogaster, but physical interactions between HP1 and Spt16 have not been reported in other species so far. The charge-biased cluster in the β1-β2 connecting loop of the Swi6-CSD might be a specific property of the formation of heterochromatin in fission yeast.
These data have suggested that the "RKDD" sequence in the β1-β2 connecting loop of Swi6-CSD might function as a binding surface of the peptidase like domain of Spt16. To this end, a recombinant Swi6-4A mutant ("RKDD" to "AAAA") was used to test the binding activity of Spt16 in vitro. As expected, Swi6-4A lost its ability to bind to the peptidase like domain of Spt16. Interestingly, heterochromatin was significantly disordered in Swi6-4A mutant in fission yeast. Although FACT targets the β1-β2 connecting loop of Swi6, other effectors might bind to the "RKDD" sequence in the β1-β2 connecting loop of Swi6-CSD in fission yeast. This scenario requires further examination in future heterochromatin studies. To evaluate the effect of eliminating FACT in heterochromatin, a peptidase-like domain truncated Spt16 was expressed in a pob3∆ strain. Different from the pob3∆ strain, a major decrease in the levels of histone H3K9 methylation and Swi6 binding was observed. This indicated that FACT plays a critical role in the establishment and maintenance of heterochromatin.
7. Mechanism of action of FACT on nucleosomes within heterochromatin for formation and maintenance of heterochromatin in fission yeast
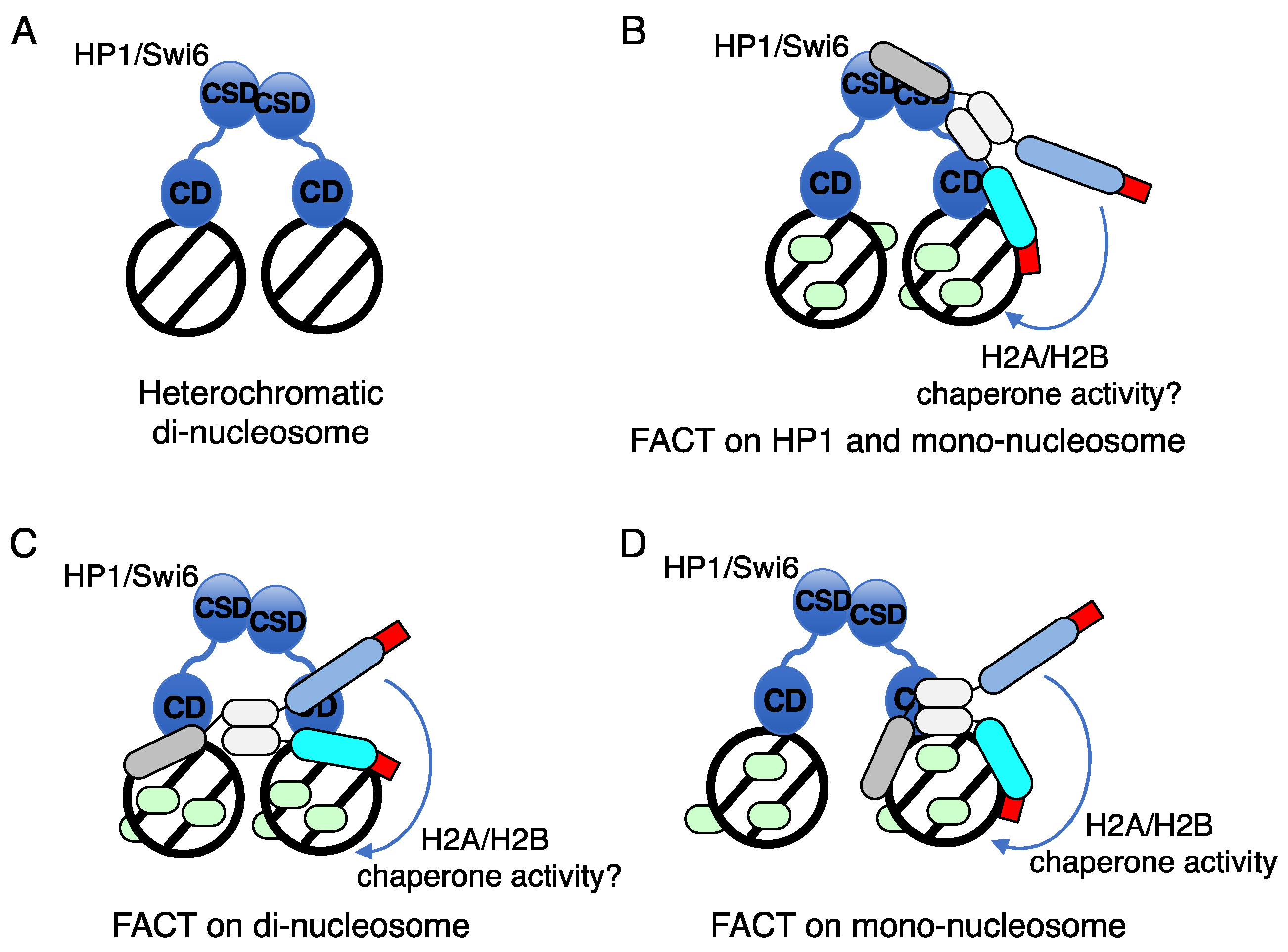
Our analysis revealed the molecular mechanism by which FACT is recruited onto heterochromatin for the dynamic regulation of the H2A/H2B dimer and optimal management of nucleosomes in heterochromatin [115]. In pob3∆ cells, impaired histone H2A/H2B dimer reposition is promoted in the heterochromatin. As the dynamic properties of histone H2A/H2B within heterochromatin, we also reported that the histone H2A.Z/H2B dimer contributes to this heterochromatic silencing to a certain extend [120]. Other groups have reported that FACT strongly suppresses histone turnover [121,122,123], and the mechanism by which FACT regulates nucleosomes in heterochromatin is an important aspect to be considered. To predict the means by which FACT regulates heterochromatic nucleosomes in fission yeast, we proposed a hypothetical model shown in Figure 6.
Within the heterochromatin, 2 nucleosomes are bridged by the HP1/Swi6 heterodimer via histone H3K9me (Figure 6A). Structural analysis revealed that it is not sufficient to enumerate the dinucleosome with a HP1 homodimer alone, and the linker DNA is exposed to allow the access of silencing effectors [124]. In fission yeast, the Swi6-CSD homodimer physically binds to the peptidase like domain of Spt16 to recruit FACT onto the heterochromatin. Concomitantly, the tandem PH domain of Pob3 binds to the H3/H4 dimer in a heterochromatic nucleosome (Figure 6B). After the recruitment of FACT by Swi6, the peptidase-like domain of Spt16, which recognized Swi6-CSD, shifts its scaffold to histone H3/H4 in the nucleosome [115]. This scaffold shift can be divided into 2 modes: binding to dinucleosome as a bridge (Figure 6C) and binding to mononucleosome (Figure 6D), shown as euchromatic nucleosome regulation in Figure 2. In the heterochromatic dinucleosome bridging model, FACT helps Swi6 enhance nucleosome condensation and formation of higher order chromatin structure (Figure 6C). In the heterochromatic mononucleosome binding model, the peptidase like domain of Spt16 and the tandem PH domain of Pob3 clip the histone H3/H4 tetramer in the heterochromatic nucleosome to stably tether FACT on a single nucleosome (Figure 6D). Under the stable FACT-nucleosome binding, deposition and reposition of the histone H2A/H2B dimer occurs via the chaperon activity of the Spt16 tandem PH domain. Histone H2A/H2B ChIP analysis in the pob3∆ strain indicated that reposition of the H2A/H2B dimer was dependent on the recognition of the stable H3/H4 dimer in nucleosomes by the tandem PH domain of Pob3 [115].
8. Summary and Perspective
FACT is an important transcription stimulating factor that transiently relaxes chromatin structure through its histone H2A/H2B chaperone activity. The transcription-activating properties of FACT are especially demonstrated in START, the checkpoint of the G1/S phase, through binding to SBF/MBF in budding yeast. However, the mechanism by which FACT contributes to START activation by the mammalian E2F family remains unclear. In addition to transcriptional activation in budding yeast, studies using fission yeast have shown that FACT contributes to the formation and maintenance of histone H3K9-mediated heterochromatin. In particular, the molecular mechanism by which FACT is recruited onto heterochromatin was previously analyzed in detail by the authors, and the means by which FACT regulates nucleosomes in heterochromatin will be a subject of future research.
Funding
This work was supported by KAKENHI (17K07496) from JSPS; MEXT KAKENHI (221S0002; a Grant-in-Aid for Scientific Research on Innovative Areas “Transcription Cycle”) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan to S.T.; and a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (19H05742) and a Grant-in-Aid for Scientific Research (A) from JSPS (19H00973) to Y.M.
Acknowledgments
We thank our laboratory members for many helpful discussions.
Conflicts of Interest
The authors declare no competing of interests.
References
- Zabidi, M.A.; Stark, A. Regulatory Enhancer-Core-Promoter Communication via Transcription Factors and Cofactors. Trends Genet 2016, 32, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Roeder, R.G. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet 2010, 11, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.L.; Sen, R.; Roeder, R.G. Enhancer-promoter communication and transcriptional regulation of Igh. Trends Immunol 2011, 32, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Schilbach, S.; Ninov, M.; Urlaub, H.; Cramer, P. Structures of transcription preinitiation complex engaged with the +1 nucleosome. Nat Struct Mol Biol 2022. [Google Scholar] [CrossRef] [PubMed]
- Schier, A.C.; Taatjes, D.J. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev 2020, 34, 465–488. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S. Structure and mechanism of the RNA polymerase II transcription machinery. Nat Struct Mol Biol 2004, 11, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Svaren, J.; Horz, W. Transcription factors vs nucleosomes: regulation of the PHO5 promoter in yeast. Trends Biochem Sci 1997, 22, 93–97. [Google Scholar] [CrossRef]
- Roeder, R.G. Role of general and gene-specific cofactors in the regulation of eukaryotic transcription. Cold Spring Harb Symp Quant Biol 1998, 63, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Shibata, Y.; Rao, B.; Strahl, B.D.; Lieb, J.D. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet 2004, 36, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wong, Y.C.; Elgin, S.C. The chromatin structure of specific genes: II. Disruption of chromatin structure during gene activity. Cell 1979, 16, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Pugh, B.F. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 2009, 10, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Chereji, R.V.; Ocampo, J.; Clark, D.J. MNase-Sensitive Complexes in Yeast: Nucleosomes and Non-histone Barriers. Mol Cell 2017, 65, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Givens, R.M.; Lai, W.K.; Rizzo, J.M.; Bard, J.E.; Mieczkowski, P.A.; Leatherwood, J.; Huberman, J.A.; Buck, M.J. Chromatin architectures at fission yeast transcriptional promoters and replication origins. Nucleic Acids Res 2012, 40, 7176–7189. [Google Scholar] [CrossRef] [PubMed]
- Lantermann, A.B.; Straub, T.; Stralfors, A.; Yuan, G.C.; Ekwall, K.; Korber, P. Schizosaccharomyces pombe genome-wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae. Nat Struct Mol Biol 2010, 17, 251–257. [Google Scholar] [CrossRef]
- Morrison, O.; Thakur, J. Molecular Complexes at Euchromatin, Heterochromatin and Centromeric Chromatin. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Shahid, Z.; Simpson, B.; Miao, K.H.; Singh, G. Genetics, Histone Code. In StatPearls; Treasure Island (FL), 2022.
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Bi, X. Heterochromatin structure: lessons from the budding yeast. IUBMB Life 2014, 66, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.I.; Jia, S. Heterochromatin revisited. Nat Rev Genet 2007, 8, 35–46. [Google Scholar] [CrossRef]
- Noma, K.; Allis, C.D.; Grewal, S.I. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 2001, 293, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef]
- Stewart, M.D.; Li, J.; Wong, J. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol Cell Biol 2005, 25, 2525–2538. [Google Scholar] [CrossRef] [PubMed]
- Cutter DiPiazza, A.R.; Taneja, N.; Dhakshnamoorthy, J.; Wheeler, D.; Holla, S.; Grewal, S.I.S. Spreading and epigenetic inheritance of heterochromatin require a critical density of histone H3 lysine 9 tri-methylation. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.H.; Mermoud, J.E.; O'Carroll, D.; Pagani, M.; Schweizer, D.; Brockdorff, N.; Jenuwein, T. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Genet 2002, 30, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huarte, M.; Zaratiegui, M.; Vaughn, M.W.; Shi, Y.; Martienssen, R.; Cande, W.Z. Lid2 is required for coordinating H3K4 and H3K9 methylation of heterochromatin and euchromatin. Cell 2008, 135, 272–283. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, G.; Orphanides, G.; Lane, W.S.; Reinberg, D. Requirement of RSF and FACT for transcription of chromatin templates in vitro. Science 1998, 282, 1900–1904. [Google Scholar] [CrossRef] [PubMed]
- Orphanides, G.; LeRoy, G.; Chang, C.H.; Luse, D.S.; Reinberg, D. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 1998, 92, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Orphanides, G.; Wu, W.H.; Lane, W.S.; Hampsey, M.; Reinberg, D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 1999, 400, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskaya, R.; Reinberg, D. Facts about FACT and transcript elongation through chromatin. Curr Opin Genet Dev 2004, 14, 139–146. [Google Scholar] [CrossRef]
- Valieva, M.E.; Gerasimova, N.S.; Kudryashova, K.S.; Kozlova, A.L.; Kirpichnikov, M.P.; Hu, Q.; Botuyan, M.V.; Mer, G.; Feofanov, A.V.; Studitsky, V.M. Stabilization of Nucleosomes by Histone Tails and by FACT Revealed by spFRET Microscopy. Cancers (Basel) 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Tsunaka, Y.; Fujiwara, Y.; Oyama, T.; Hirose, S.; Morikawa, K. Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev 2016, 30, 673–686. [Google Scholar] [CrossRef]
- Wittmeyer, J.; Formosa, T. The Saccharomyces cerevisiae DNA polymerase alpha catalytic subunit interacts with Cdc68/Spt16 and with Pob3, a protein similar to an HMG1-like protein. Mol Cell Biol 1997, 17, 4178–4190. [Google Scholar] [CrossRef] [PubMed]
- Brewster, N.K.; Johnston, G.C.; Singer, R.A. Characterization of the CP complex, an abundant dimer of Cdc68 and Pob3 proteins that regulates yeast transcriptional activation and chromatin repression. J Biol Chem 1998, 273, 21972–21979. [Google Scholar] [CrossRef] [PubMed]
- Wittmeyer, J.; Joss, L.; Formosa, T. Spt16 and Pob3 of Saccharomyces cerevisiae form an essential, abundant heterodimer that is nuclear, chromatin-associated, and copurifies with DNA polymerase alpha. Biochemistry 1999, 38, 8961–8971. [Google Scholar] [CrossRef] [PubMed]
- Formosa, T.; Eriksson, P.; Wittmeyer, J.; Ginn, J.; Yu, Y.; Stillman, D.J. Spt16-Pob3 and the HMG protein Nhp6 combine to form the nucleosome-binding factor SPN. EMBO J 2001, 20, 3506–3517. [Google Scholar] [CrossRef] [PubMed]
- Rowley, A.; Singer, R.A.; Johnston, G.C. CDC68, a yeast gene that affects regulation of cell proliferation and transcription, encodes a protein with a highly acidic carboxyl terminus. Mol Cell Biol 1991, 11, 5718–5726. [Google Scholar] [CrossRef] [PubMed]
- Brewster, N.K.; Johnston, G.C.; Singer, R.A. A bipartite yeast SSRP1 analog comprised of Pob3 and Nhp6 proteins modulates transcription. Mol Cell Biol 2001, 21, 3491–3502. [Google Scholar] [CrossRef] [PubMed]
- Formosa, T. FACT and the reorganized nucleosome. Mol Biosyst 2008, 4, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Costigan, C.; Kolodrubetz, D.; Snyder, M. NHP6A and NHP6B, which encode HMG1-like proteins, are candidates for downstream components of the yeast SLT2 mitogen-activated protein kinase pathway. Mol Cell Biol 1994, 14, 2391–2403. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.U.; Hayles, J.; Kim, D.; Wood, V.; Park, H.O.; Won, M.; Yoo, H.S.; Duhig, T.; Nam, M.; Palmer, G.; et al. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol 2010, 28, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Kolodrubetz, D.; Kruppa, M.; Burgum, A. Gene dosage affects the expression of the duplicated NHP6 genes of Saccharomyces cerevisiae. Gene 2001, 272, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zeng, S.X.; Landais, I.; Lu, H. Human SSRP1 has Spt16-dependent and -independent roles in gene transcription. J Biol Chem 2007, 282, 6936–6945. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zeng, F.; Liu, Y.; Shao, C.; Li, S.; Lv, H.; Shi, Y.; Niu, L.; Teng, M.; Li, X. Crystal Structure of Human SSRP1 Middle Domain Reveals a Role in DNA Binding. Sci Rep 2015, 5, 18688. [Google Scholar] [CrossRef]
- Ruone, S.; Rhoades, A.R.; Formosa, T. Multiple Nhp6 molecules are required to recruit Spt16-Pob3 to form yFACT complexes and to reorganize nucleosomes. J Biol Chem 2003, 278, 45288–45295. [Google Scholar] [CrossRef] [PubMed]
- McCullough, L.L.; Connell, Z.; Xin, H.; Studitsky, V.M.; Feofanov, A.V.; Valieva, M.E.; Formosa, T. Functional roles of the DNA-binding HMGB domain in the histone chaperone FACT in nucleosome reorganization. J Biol Chem 2018, 293, 6121–6133. [Google Scholar] [CrossRef] [PubMed]
- Stillman, D.J. Nhp6: a small but powerful effector of chromatin structure in Saccharomyces cerevisiae. Biochim Biophys Acta 2010, 1799, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Mayanagi, K.; Saikusa, K.; Miyazaki, N.; Akashi, S.; Iwasaki, K.; Nishimura, Y.; Morikawa, K.; Tsunaka, Y. Structural visualization of key steps in nucleosome reorganization by human FACT. Sci Rep 2019, 9, 10183. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Crickard, J.B.; Srikanth, A.; Reese, J.C. A highly conserved region within H2B is important for FACT to act on nucleosomes. Mol Cell Biol 2014, 34, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Michl-Holzinger, P.; Obermeyer, S.; Markusch, H.; Pfab, A.; Ettner, A.; Bruckmann, A.; Babl, S.; Langst, G.; Schwartz, U.; Tvardovskiy, A.; et al. Phosphorylation of the FACT histone chaperone subunit SPT16 affects chromatin at RNA polymerase II transcriptional start sites in Arabidopsis. Nucleic Acids Res 2022, 50, 5014–5028. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Keller, D.M.; Scott, J.D.; Lu, H. CK2 phosphorylates SSRP1 and inhibits its DNA-binding activity. J Biol Chem 2005, 280, 11869–11875. [Google Scholar] [CrossRef] [PubMed]
- Hondele, M.; Stuwe, T.; Hassler, M.; Halbach, F.; Bowman, A.; Zhang, E.T.; Nijmeijer, B.; Kotthoff, C.; Rybin, V.; Amlacher, S.; et al. Structural basis of histone H2A-H2B recognition by the essential chaperone FACT. Nature 2013, 499, 111–114. [Google Scholar] [CrossRef]
- Stuwe, T.; Hothorn, M.; Lejeune, E.; Rybin, V.; Bortfeld, M.; Scheffzek, K.; Ladurner, A.G. The FACT Spt16 "peptidase" domain is a histone H3-H4 binding module. Proc Natl Acad Sci U S A 2008, 105, 8884–8889. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Takahata, S.; Blanksma, M.; McCullough, L.; Stillman, D.J.; Formosa, T. yFACT induces global accessibility of nucleosomal DNA without H2A-H2B displacement. Mol Cell 2009, 35, 365–376. [Google Scholar] [CrossRef]
- Sivkina, A.L.; Karlova, M.G.; Valieva, M.E.; McCullough, L.L.; Formosa, T.; Shaytan, A.K.; Feofanov, A.V.; Kirpichnikov, M.P.; Sokolova, O.S.; Studitsky, V.M. Electron microscopy analysis of ATP-independent nucleosome unfolding by FACT. Commun Biol 2022, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Jamai, A.; Puglisi, A.; Strubin, M. Histone chaperone spt16 promotes redeposition of the original h3-h4 histones evicted by elongating RNA polymerase. Mol Cell 2009, 35, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Voth, W.P.; Takahata, S.; Nishikawa, J.L.; Metcalfe, B.M.; Naar, A.M.; Stillman, D.J. A role for FACT in repopulation of nucleosomes at inducible genes. PLoS One 2014, 9, e84092. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, J.M.; Mikesell, G.E.; Breeden, L.L. Cell cycle-regulated phosphorylation of Swi6 controls its nuclear localization. Mol Biol Cell 1995, 6, 1641–1658. [Google Scholar] [CrossRef] [PubMed]
- Moll, T.; Dirick, L.; Auer, H.; Bonkovsky, J.; Nasmyth, K. SWI6 is a regulatory subunit of two different cell cycle START-dependent transcription factors in Saccharomyces cerevisiae. J Cell Sci Suppl 1992, 16, 87–96. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Takahata, S.; Yu, Y.; Stillman, D.J. The E2F functional analogue SBF recruits the Rpd3(L) HDAC, via Whi5 and Stb1, and the FACT chromatin reorganizer, to yeast G1 cyclin promoters. EMBO J 2009, 28, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Takahata, S.; Yu, Y.; Stillman, D.J. FACT and Asf1 regulate nucleosome dynamics and coactivator binding at the HO promoter. Mol Cell 2009, 34, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Takahata, S.; Yu, Y.; Stillman, D.J. Repressive chromatin affects factor binding at yeast HO (homothallic switching) promoter. J Biol Chem 2011, 286, 34809–34819. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Nishikawa, J.L.; Tang, X.; Millman, J.S.; Schub, O.; Breitkreuz, K.; Dewar, D.; Rupes, I.; Andrews, B.; Tyers, M. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 2004, 117, 899–913. [Google Scholar] [CrossRef] [PubMed]
- Wittenberg, C.; Reed, S.I. Cell cycle-dependent transcription in yeast: promoters, transcription factors, and transcriptomes. Oncogene 2005, 24, 2746–2755. [Google Scholar] [CrossRef] [PubMed]
- Dirick, L.; Moll, T.; Auer, H.; Nasmyth, K. A central role for SWI6 in modulating cell cycle Start-specific transcription in yeast. Nature 1992, 357, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Brocca, S.; Sacco, E.; Spinelli, M.; Papaleo, E.; Lambrughi, M.; Alberghina, L.; Vanoni, M. A comparative study of Whi5 and retinoblastoma proteins: from sequence and structure analysis to intracellular networks. Front Physiol 2013, 4, 315. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.V.; Smolka, M.B.; de Bruin, R.A.; Zhou, H.; Wittenberg, C.; Dowdy, S.F. Whi5 regulation by site specific CDK-phosphorylation in Saccharomyces cerevisiae. PLoS One 2009, 4, e4300. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.A.; McDonald, W.H.; Kalashnikova, T.I.; Yates, J., 3rd; Wittenberg, C. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 2004, 117, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A.; Chen, E.Y.; Heffron, F. A 24-base-pair DNA sequence from the MAT locus stimulates intergenic recombination in yeast. Proc Natl Acad Sci U S A 1986, 83, 7831–7835. [Google Scholar] [CrossRef]
- Kostriken, R.; Strathern, J.N.; Klar, A.J.; Hicks, J.B.; Heffron, F. A site-specific endonuclease essential for mating-type switching in Saccharomyces cerevisiae. Cell 1983, 35, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Nasmyth, K. Molecular analysis of a cell lineage. Nature 1983, 302, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Klar, A.J.; Hicks, J.B.; Strathern, J.N. Directionality of yeast mating-type interconversion. Cell 1982, 28, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Strathern, J.N.; Klar, A.J.; Hicks, J.B.; Abraham, J.A.; Ivy, J.M.; Nasmyth, K.A.; McGill, C. Homothallic switching of yeast mating type cassettes is initiated by a double-stranded cut in the MAT locus. Cell 1982, 31, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, M.J.; Florens, L.; Swanson, S.K.; Shia, W.J.; Anderson, S.; Yates, J.; Washburn, M.P.; Workman, J.L. Stable incorporation of sequence specific repressors Ash1 and Ume6 into the Rpd3L complex. Biochim Biophys Acta 2005, 1731, 77–87; discussion 75–76. [Google Scholar] [CrossRef] [PubMed]
- Sardiu, M.E.; Gilmore, J.M.; Carrozza, M.J.; Li, B.; Workman, J.L.; Florens, L.; Washburn, M.P. Determining protein complex connectivity using a probabilistic deletion network derived from quantitative proteomics. PLoS One 2009, 4, e7310. [Google Scholar] [CrossRef] [PubMed]
- Bohl, F.; Kruse, C.; Frank, A.; Ferring, D.; Jansen, R.P. She2p, a novel RNA-binding protein tethers ASH1 mRNA to the Myo4p myosin motor via She3p. EMBO J 2000, 19, 5514–5524. [Google Scholar] [CrossRef] [PubMed]
- Darzacq, X.; Powrie, E.; Gu, W.; Singer, R.H.; Zenklusen, D. RNA asymmetric distribution and daughter/mother differentiation in yeast. Curr Opin Microbiol 2003, 6, 614–620. [Google Scholar] [CrossRef]
- Yu, Y.; Yarrington, R.M.; Stillman, D.J. FACT and Ash1 promote long-range and bidirectional nucleosome eviction at the HO promoter. Nucleic Acids Res 2020, 48, 10877–10889. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.J.; Sil, A.; Measday, V.; Yu, Y.; Moffat, J.; Maxon, M.E.; Herskowitz, I.; Andrews, B.; Stillman, D.J. The protein kinase Pho85 is required for asymmetric accumulation of the Ash1 protein in Saccharomyces cerevisiae. Mol Microbiol 2001, 42, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Cosma, M.P. Daughter-specific repression of Saccharomyces cerevisiae HO: Ash1 is the commander. EMBO Rep 2004, 5, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Cosma, M.P.; Tanaka, T.; Nasmyth, K. Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell 1999, 97, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Cosma, M.P.; Panizza, S.; Nasmyth, K. Cdk1 triggers association of RNA polymerase to cell cycle promoters only after recruitment of the mediator by SBF. Mol Cell 2001, 7, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Stillman, D.J.; Bankier, A.T.; Seddon, A.; Groenhout, E.G.; Nasmyth, K.A. Characterization of a transcription factor involved in mother cell specific transcription of the yeast HO gene. EMBO J 1988, 7, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Stillman, D.J. Dancing the cell cycle two-step: regulation of yeast G1-cell-cycle genes by chromatin structure. Trends Biochem Sci 2013, 38, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Moll, T.; Tebb, G.; Surana, U.; Robitsch, H.; Nasmyth, K. The role of phosphorylation and the CDC28 protein kinase in cell cycle-regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell 1991, 66, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Ikeda, A.; Koyama, N.; Fukada, J.; Nagao, R. A refined two-hybrid system reveals that SCF(Cdc4)-dependent degradation of Swi5 contributes to the regulatory mechanism of S-phase entry. Proc Natl Acad Sci U S A 2008, 105, 14497–14502. [Google Scholar] [CrossRef] [PubMed]
- Dohrmann, P.R.; Butler, G.; Tamai, K.; Dorland, S.; Greene, J.R.; Thiele, D.J.; Stillman, D.J. Parallel pathways of gene regulation: homologous regulators SWI5 and ACE2 differentially control transcription of HO and chitinase. Genes Dev 1992, 6, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Allshire, R.C.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 2018, 19, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Martienssen, R.; Moazed, D. RNAi and heterochromatin assembly. Cold Spring Harb Perspect Biol 2015, 7, a019323. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, T.; Barrowman, J.; Grewal, S.I. Chromosome domain architecture and dynamic organization of the fission yeast genome. FEBS Lett 2015, 589, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Yeom, S.; Park, J.; Lee, J.S. The regional sequestration of heterochromatin structural proteins is critical to form and maintain silent chromatin. Epigenetics Chromatin 2022, 15, 5. [Google Scholar] [CrossRef]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat Rev Mol Cell Biol 2022, 23, 623–640. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, N.; Currie, M.A.; Jih, G.; Paulo, J.A.; Siuti, N.; Kalocsay, M.; Gygi, S.P.; Moazed, D. Automethylation-induced conformational switch in Clr4 (Suv39h) maintains epigenetic stability. Nature 2018, 560, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Saini, A.; Nanda, J.S.; Saini, S.; Singh, J. Role of Swi6/HP1 self-association-mediated recruitment of Clr4/Suv39 in establishment and maintenance of heterochromatin in fission yeast. J Biol Chem 2011, 286, 9308–9320. [Google Scholar] [CrossRef] [PubMed]
- Bayne, E.H.; White, S.A.; Kagansky, A.; Bijos, D.A.; Sanchez-Pulido, L.; Hoe, K.L.; Kim, D.U.; Park, H.O.; Ponting, C.P.; Rappsilber, J.; et al. Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell 2010, 140, 666–677. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Pillai, S.S.; Taglini, F.; Li, F.; Ruan, K.; Zhang, J.; Wu, J.; Shi, Y.; Bayne, E.H. Structural analysis of Stc1 provides insights into the coupling of RNAi and chromatin modification. Proc Natl Acad Sci USA 2013, 110, E1879–E1888. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.; Noma, K.; Isaac, S.; Kahan, T.; Grewal, S.I.; Cohen, A. A novel jmjC domain protein modulates heterochromatization in fission yeast. Mol Cell Biol 2003, 23, 4356–4370. [Google Scholar] [CrossRef] [PubMed]
- Zofall, M.; Grewal, S.I. Swi6/HP1 recruits a JmjC domain protein to facilitate transcription of heterochromatic repeats. Mol Cell 2006, 22, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Verdel, A.; Moazed, D. RNAi-directed assembly of heterochromatin in fission yeast. FEBS Lett 2005, 579, 5872–5878. [Google Scholar] [CrossRef] [PubMed]
- Zofall, M.; Grewal, S.I. RNAi-mediated heterochromatin assembly in fission yeast. Cold Spring Harb Symp Quant Biol 2006, 71, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Goto, D.B.; Martienssen, R.A.; Urano, T.; Furukawa, K.; Murakami, Y. RNA polymerase II is required for RNAi-dependent heterochromatin assembly. Science 2005, 309, 467–469. [Google Scholar] [CrossRef]
- Sorida, M.; Hirauchi, T.; Ishizaki, H.; Kaito, W.; Shimada, A.; Mori, C.; Chikashige, Y.; Hiraoka, Y.; Suzuki, Y.; Ohkawa, Y.; et al. Regulation of ectopic heterochromatin-mediated epigenetic diversification by the JmjC family protein Epe1. PLoS Genet 2019, 15, e1008129. [Google Scholar] [CrossRef] [PubMed]
- Zofall, M.; Sandhu, R.; Holla, S.; Wheeler, D.; Grewal, S.I.S. Histone deacetylation primes self-propagation of heterochromatin domains to promote epigenetic inheritance. Nat Struct Mol Biol 2022, 29, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Eissenberg, J.C.; Elgin, S.C. HP1a: a structural chromosomal protein regulating transcription. Trends Genet 2014, 30, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Canzio, D.; Liao, M.; Naber, N.; Pate, E.; Larson, A.; Wu, S.; Marina, D.B.; Garcia, J.F.; Madhani, H.D.; Cooke, R.; et al. A conformational switch in HP1 releases auto-inhibition to drive heterochromatin assembly. Nature 2013, 496, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Cowieson, N.P.; Partridge, J.F.; Allshire, R.C.; McLaughlin, P.J. Dimerisation of a chromo shadow domain and distinctions from the chromodomain as revealed by structural analysis. Curr Biol 2000, 10, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Leopold, K.; Stirpe, A.; Schalch, T. Transcriptional gene silencing requires dedicated interaction between HP1 protein Chp2 and chromatin remodeler Mit1. Genes Dev 2019, 33, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Motamedi, M.R.; Hong, E.J.; Li, X.; Gerber, S.; Denison, C.; Gygi, S.; Moazed, D. HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol Cell 2008, 32, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Cui, B.; Dhakshnamoorthy, J.; Zhou, M.; Rubin, C.; Zofall, M.; Veenstra, T.D.; Grewal, S.I. Diverse roles of HP1 proteins in heterochromatin assembly and functions in fission yeast. Proc Natl Acad Sci U S A 2009, 106, 8998–9003. [Google Scholar] [CrossRef] [PubMed]
- Sadaie, M.; Kawaguchi, R.; Ohtani, Y.; Arisaka, F.; Tanaka, K.; Shirahige, K.; Nakayama, J. Balance between distinct HP1 family proteins controls heterochromatin assembly in fission yeast. Mol Cell Biol 2008, 28, 6973–6988. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Dohke, K.; Sadaie, M.; Shinmyozu, K.; Nakayama, J.; Urano, T.; Murakami, Y. Phosphorylation of Swi6/HP1 regulates transcriptional gene silencing at heterochromatin. Genes Dev 2009, 23, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Eissenberg, J.C.; Ge, Y.W.; Hartnett, T. Increased phosphorylation of HP1, a heterochromatin-associated protein of Drosophila, is correlated with heterochromatin assembly. J Biol Chem 1994, 269, 21315–21321. [Google Scholar] [CrossRef] [PubMed]
- Minc, E.; Allory, Y.; Worman, H.J.; Courvalin, J.C.; Buendia, B. Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma 1999, 108, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, E.; Bortfeld, M.; White, S.A.; Pidoux, A.L.; Ekwall, K.; Allshire, R.C.; Ladurner, A.G. The chromatin-remodeling factor FACT contributes to centromeric heterochromatin independently of RNAi. Curr Biol 2007, 17, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Takahata, S.; Chida, S.; Ohnuma, A.; Ando, M.; Asanuma, T.; Murakami, Y. Two secured FACT recruitment mechanisms are essential for heterochromatin maintenance. Cell Rep 2021, 36, 109540. [Google Scholar] [CrossRef] [PubMed]
- Brasher, S.V.; Smith, B.O.; Fogh, R.H.; Nietlispach, D.; Thiru, A.; Nielsen, P.R.; Broadhurst, R.W.; Ball, L.J.; Murzina, N.V.; Laue, E.D. The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J 2000, 19, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Thiru, A.; Nietlispach, D.; Mott, H.R.; Okuwaki, M.; Lyon, D.; Nielsen, P.R.; Hirshberg, M.; Verreault, A.; Murzina, N.V.; Laue, E.D. Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J 2004, 23, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qin, S.; Lei, M.; Tempel, W.; Zhang, Y.; Loppnau, P.; Li, Y.; Min, J. Peptide recognition by heterochromatin protein 1 (HP1) chromoshadow domains revisited: Plasticity in the pseudosymmetric histone binding site of human HP1. J Biol Chem 2017, 292, 5655–5664. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Myers, M.P.; Xu, R.M. Crystal structure of the HP1-EMSY complex reveals an unusual mode of HP1 binding. Structure 2006, 14, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Tsukii, K.; Takahata, S.; Murakami, Y. Histone variant H2A.Z plays multiple roles in the maintenance of heterochromatin integrity. Genes Cells 2022, 27, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Murawska, M.; Greenstein, R.A.; Schauer, T.; Olsen, K.C.F.; Ng, H.; Ladurner, A.G.; Al-Sady, B.; Braun, S. The histone chaperone FACT facilitates heterochromatin spreading by regulating histone turnover and H3K9 methylation states. Cell Rep 2021, 37, 109944. [Google Scholar] [CrossRef] [PubMed]
- Murawska, M.; Braun, S. Chaperoning heterochromatin: new roles of FACT in chromatin silencing. Trends Genet 2022, 38, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Holla, S.; Dhakshnamoorthy, J.; Folco, H.D.; Balachandran, V.; Xiao, H.; Sun, L.L.; Wheeler, D.; Zofall, M.; Grewal, S.I.S. Positioning Heterochromatin at the Nuclear Periphery Suppresses Histone Turnover to Promote Epigenetic Inheritance. Cell 2020, 180, 150–164 e115. [Google Scholar] [CrossRef] [PubMed]
- Machida, S.; Takizawa, Y.; Ishimaru, M.; Sugita, Y.; Sekine, S.; Nakayama, J.I.; Wolf, M.; Kurumizaka, H. Structural Basis of Heterochromatin Formation by Human HP1. Mol Cell 2018, 69, 385–397 e388. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic structures of FACT complex in yeast and higher eukaryotes. N1: Subdomain of peptidase like domain 1, N2: Subdomain of peptidase like domain 2, DD: dimerization domain, Tandem PH: Tandem Pleckstrin homology domain, PH1: Pleckstrin homology domain 1, PH2: Pleckstrin homology domain 2, Acidic: Acidic amino acid cluster, HMGB: High mobility group box.
Figure 1.
Schematic structures of FACT complex in yeast and higher eukaryotes. N1: Subdomain of peptidase like domain 1, N2: Subdomain of peptidase like domain 2, DD: dimerization domain, Tandem PH: Tandem Pleckstrin homology domain, PH1: Pleckstrin homology domain 1, PH2: Pleckstrin homology domain 2, Acidic: Acidic amino acid cluster, HMGB: High mobility group box.
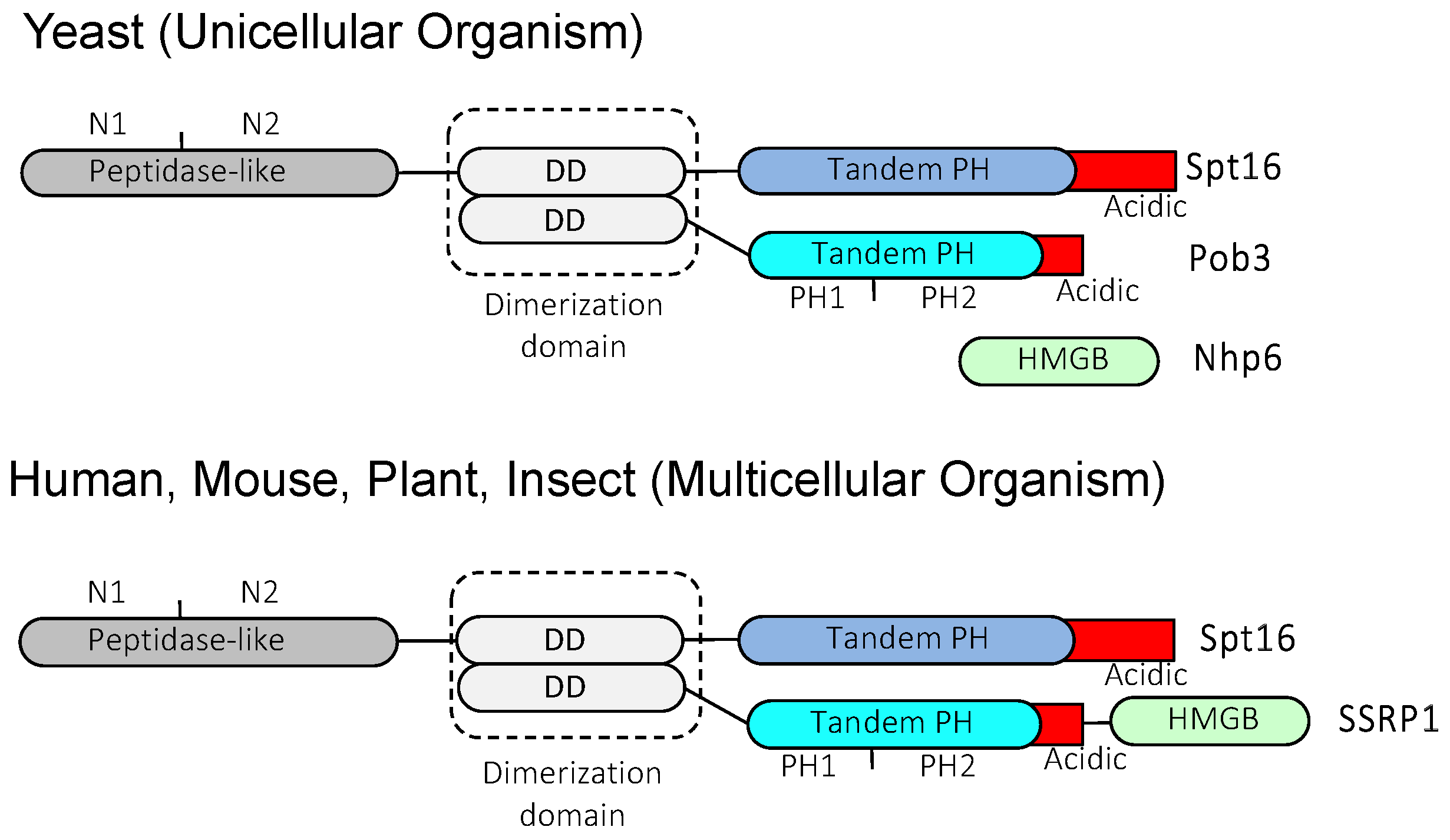
Figure 2.
FACT working model for mononucleosome and dinucleosome. (A) Fission yeast FACT on mononucleosome. FACT functions as a histone chaperone in this scenario. (B) Fission yeast FACT on dinucleosome. Whether FACT functions as a histone chaperone in this scenario remains unknown.
Figure 2.
FACT working model for mononucleosome and dinucleosome. (A) Fission yeast FACT on mononucleosome. FACT functions as a histone chaperone in this scenario. (B) Fission yeast FACT on dinucleosome. Whether FACT functions as a histone chaperone in this scenario remains unknown.
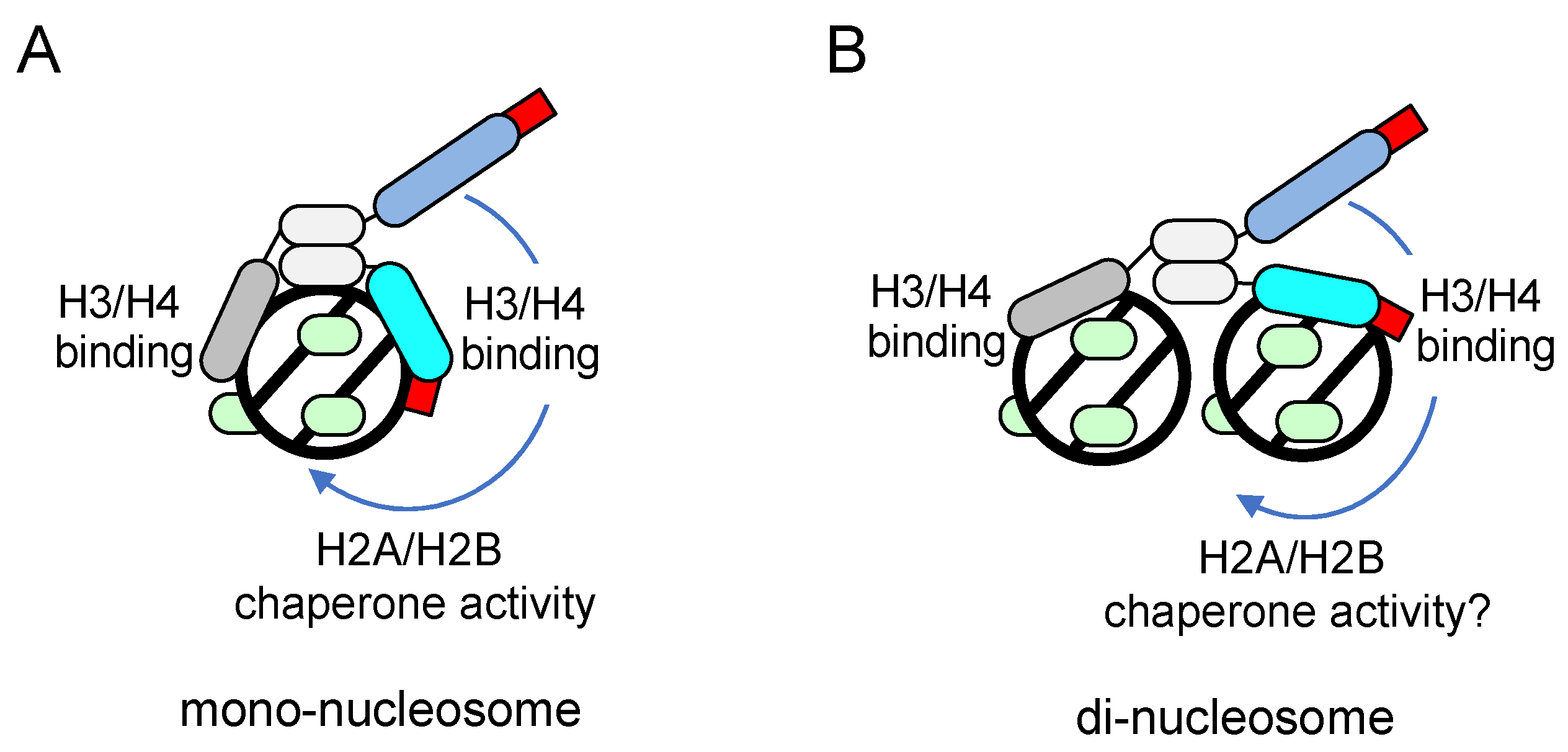
Figure 3.
Schematic diagrams of SBF regulated gene promoter. UAS: Upstream activating sequence, URS1: Upstream regulatory sequence 1, URS2: Upstream regulatory sequence 2. (A) Promoter structure of CLN1 and CLN2 genes. Three SBF binding sites are annotated as arrows. Positive and negative regulators of gene expression are shown below and above the schematic of the gene structure, respectively. FACT is assigned an underline. (B) Promoter structure of the HO gene. Two Swi5 binding sites, one SBF binding site, and one Ash1 binding site are annotated in URS1. Eight SBF binding sites are annotated in URS2. Positive and negative regulators of gene expression are shown below and above the schematic of the gene structure, respectively. FACT is assigned an underline.
Figure 3.
Schematic diagrams of SBF regulated gene promoter. UAS: Upstream activating sequence, URS1: Upstream regulatory sequence 1, URS2: Upstream regulatory sequence 2. (A) Promoter structure of CLN1 and CLN2 genes. Three SBF binding sites are annotated as arrows. Positive and negative regulators of gene expression are shown below and above the schematic of the gene structure, respectively. FACT is assigned an underline. (B) Promoter structure of the HO gene. Two Swi5 binding sites, one SBF binding site, and one Ash1 binding site are annotated in URS1. Eight SBF binding sites are annotated in URS2. Positive and negative regulators of gene expression are shown below and above the schematic of the gene structure, respectively. FACT is assigned an underline.
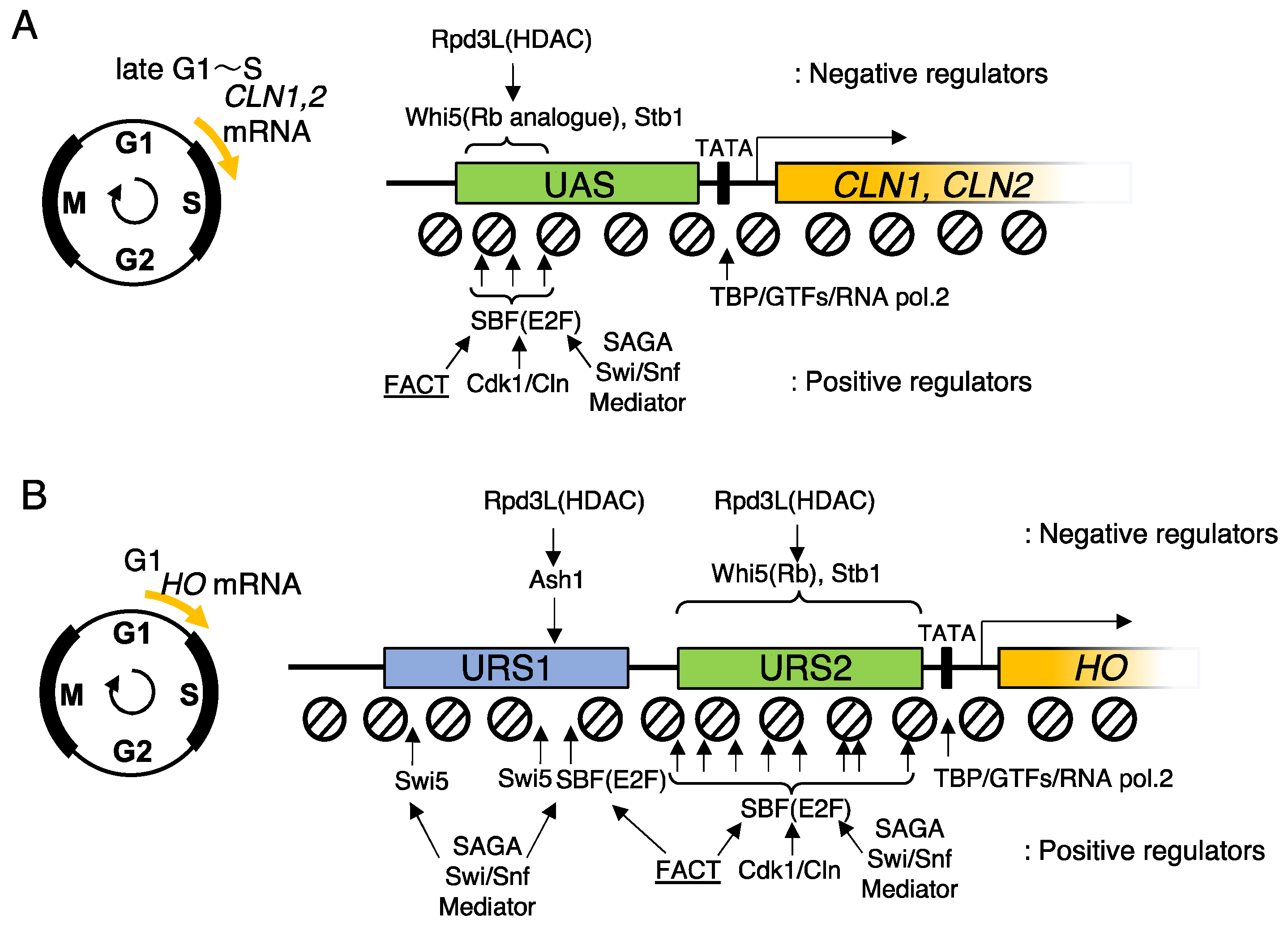
Figure 4.
Wave of nucleosome deposition and reposition at the HO promoter during cell-cycle progression. Based on GALp::CDC20 arrest/release experiment, nucleosome dynamics were analyzed by time-course histone H3 ChIP-qPCR during M to S phase transition. URS1: Upstream regulatory sequence 1, URS2: Upstream regulatory sequence 2. (A) Schematic representation of nucleosome dynamics along the HO promoter in wild type cells. (B) Schematic representation of nucleosome dynamics along the HO promoter in FACT mutant cells.
Figure 4.
Wave of nucleosome deposition and reposition at the HO promoter during cell-cycle progression. Based on GALp::CDC20 arrest/release experiment, nucleosome dynamics were analyzed by time-course histone H3 ChIP-qPCR during M to S phase transition. URS1: Upstream regulatory sequence 1, URS2: Upstream regulatory sequence 2. (A) Schematic representation of nucleosome dynamics along the HO promoter in wild type cells. (B) Schematic representation of nucleosome dynamics along the HO promoter in FACT mutant cells.
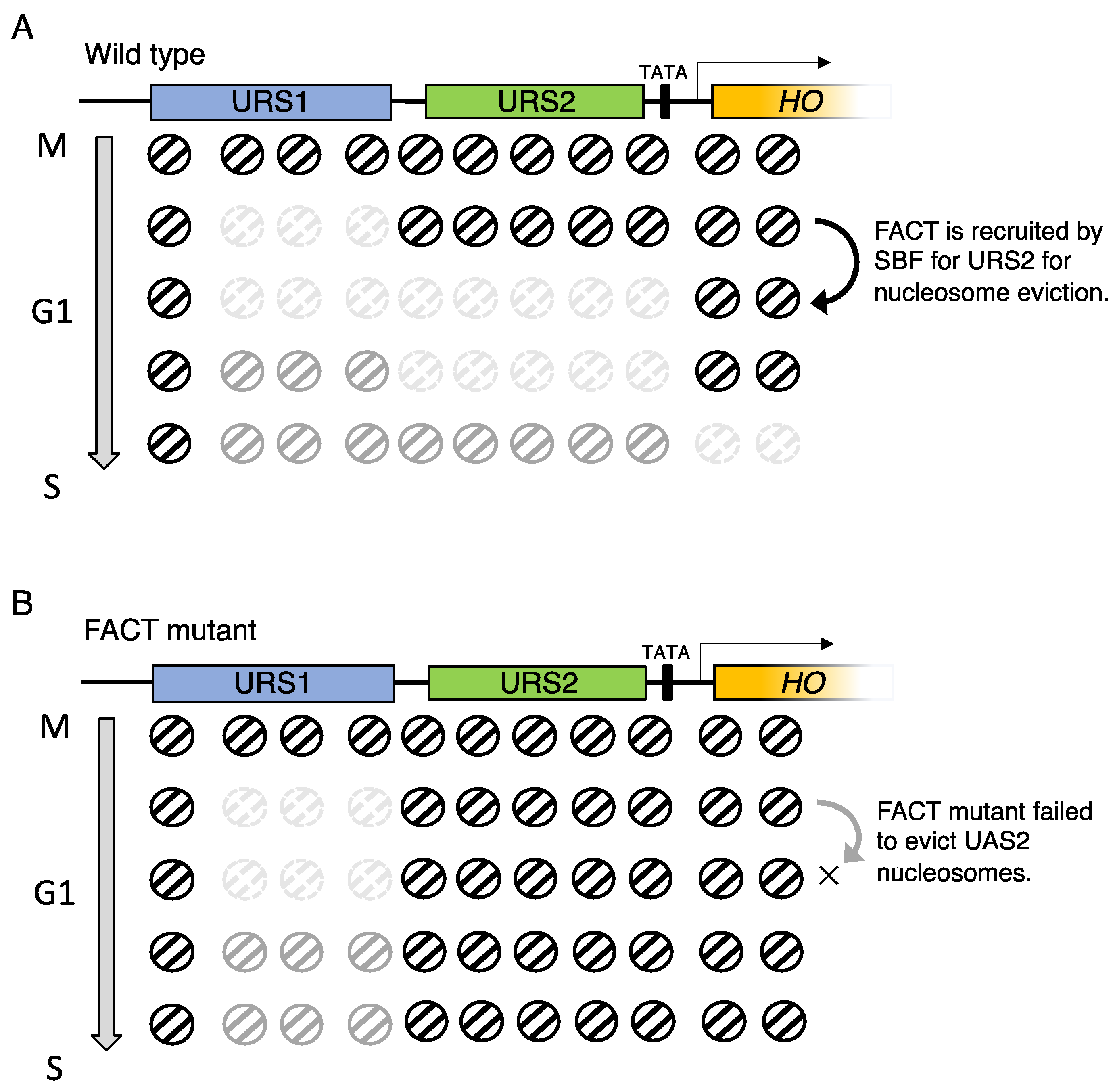
Figure 5.
Schematic diagrams of the FACT recruitment model at pericentromeric heterochromatin.
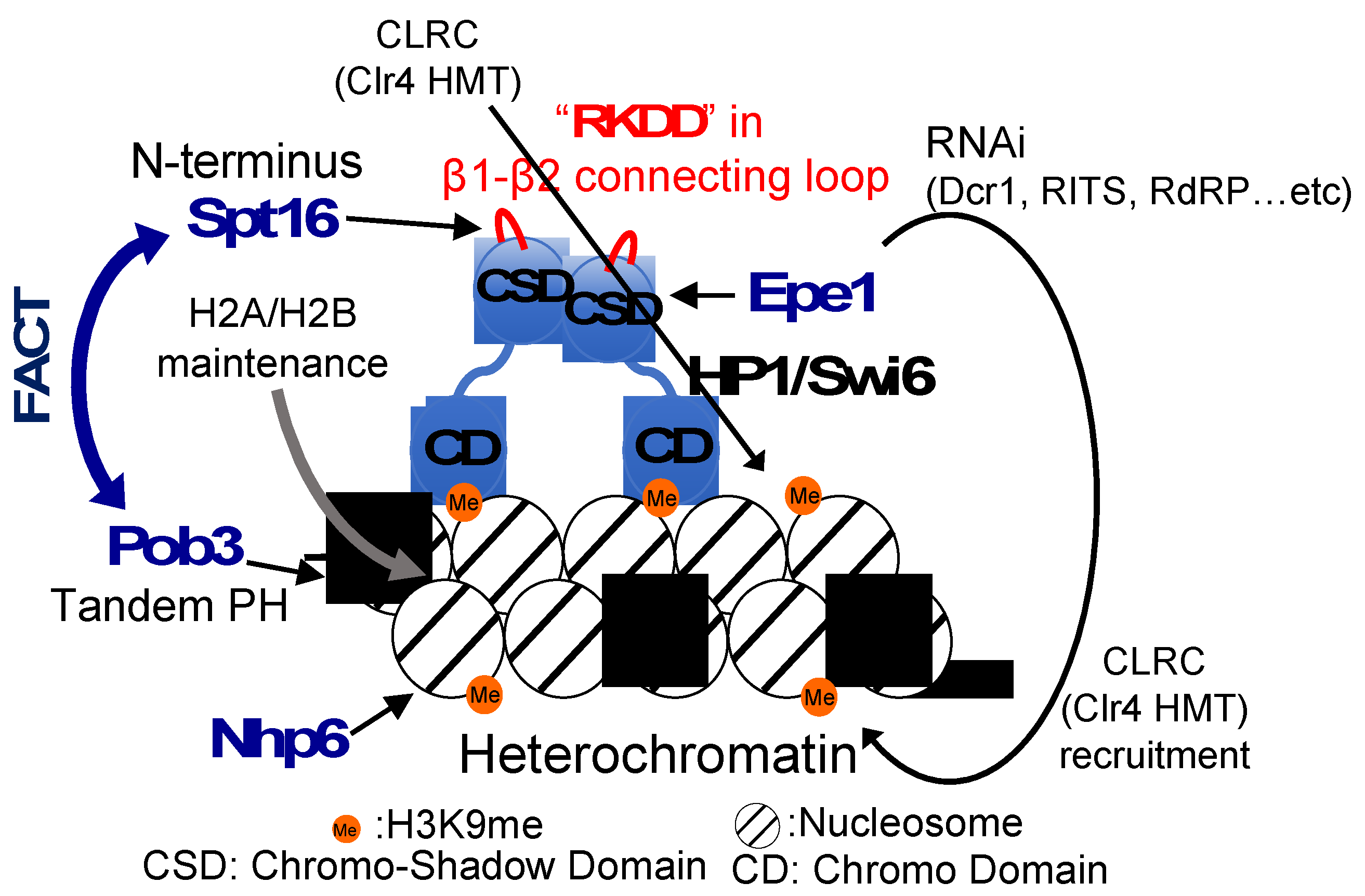
Figure 6.
Hypothetical FACT working model on heterochromatic nucleosomes. (A) HP1/Swi6 bridge dinucleosome. (B) FACT is recruited by a Swi6-CSD homodimer via the peptidase like domain of Spt16. (C) HP1/Swi6 and FACT cooperatively bridge dinucleosome. (D) FACT binds to heterochromatic mononucleosome.
Figure 6.
Hypothetical FACT working model on heterochromatic nucleosomes. (A) HP1/Swi6 bridge dinucleosome. (B) FACT is recruited by a Swi6-CSD homodimer via the peptidase like domain of Spt16. (C) HP1/Swi6 and FACT cooperatively bridge dinucleosome. (D) FACT binds to heterochromatic mononucleosome.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



