Submitted:
13 January 2023
Posted:
17 January 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The outbreak of Coronavirus, as well as its emerging potential consequences, has become a global challenge worldwide, demanding effective and controlled therapeutic strategies. Potential drug candidates could achieve that with minimal toxicity. The current investigation selected 2-Deoxy-D-glucose prescribed by Defense Research and Development Organization (DRDO), India. The derivative and modified form was tested through in silico analysis against COVID-19 main protease complex with N3 inhibitor. The derived form of 2-Deoxy-D-glucose was generated by replacing the hydroxy group with a hydrogen atom, and Cypate 2-Deoxy-D-glucose was chosen as a derivative against the COVID-19 main protease complex. A molecular docking approach was adopted to identify the stable and competent form among the modified and derivative forms of 2-Deoxy-D-glucose based on binding energy. By further promoting the stabilized complex, these compounds' toxicity was also scrutinized through ADMET analysis to predict the potential candidate. The current investigation suggested that the modified version of 2-Deoxy-D-glucose was more stable with minimal toxicity against COVID-19 main protease.
Keywords:
Covid-19
; Deoxy-D-glucose
; molecular docking
; ADMET
1. Introduction
COVID-19, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with clinical complexities including inflammation, cytokine storm, and multi-organ dysfunction, established as an outbreak and gained significant attention worldwide [1]. The approximate number of deaths all over the world reached approximately 2.3 million by February 2021 [2]. Nevertheless, controlled and therapeutic strategies are essential requisites to deal with the rising cases of infections. Before this pandemic situation, a group of researchers working in the Institute of Nuclear Medicine and Allied Sciences under the Defense Research and Development Organisation (DRDO) India developed a 2-Deoxy-D-glucose drug with the help of a pharmaceutical group to treat patients with moderate and severe COVID-19 symptoms. The past studies examined critically affected COVID patients by implementing Low Dose Radiation Therapy (LDRT) as a plausible method in pneumonia conditions [7]. The constraints of the therapeutic strategies in COVID-19 were raised due to evolving side effects of antibodies and anti-viral agents like remdesivir, lopinavir, and other drugs, including heparin and atazanavir which have been investigated during drug-protein and protein-protein interactions [3,4]. The possibility of representing 2-Deoxy-D-glucose, previously designed for cancer, arose as a result of a potential antiviral drug against SARS-CoV-2. The influential role of 2-Deoxy-D-glucose was evident as a conjugate named Hyaluronic acid-2-Deoxy-D-Glucose against SARS-CoV-2 viral protein with better biodegradability and biocompatibility and minimal toxicity than 2-Deoxy-D-Glucose [5]. This analogue of glucose plays a crucial role in the glycolytic pathway and leads to the inhibition of glycolysis.
Nonetheless, the adversity of 2-Deoxy-D-Glucose was conquered by combining with natural polymers, metal complexes, and azido analogue, which was able to produce catastrophic oxidative stress in acute conditions of critically affected COVID-19 patients [6,7]. The effectiveness of 2-Deoxy-D-Glucose as a dietary component was previously scrutinized in in vitro and in vivo conditions by exploring its radio and chemo-sensitizing effects [8]. Nonetheless, the role of 2-Deoxy-D-Glucose in glycolysis acts as a competitive antagonist and inhibits viral replication and tumour cell proliferation [9]. Since the original 2-Deoxy-D-Glucose was generated from d-glucose by the elimination of the hydroxyl group at the C-2 position [10]; the current study aimed to explore the potentiality of modified 2-Deoxy-D-Glucose against COVID-19. Previously, the removal of the hydroxyl group was compensated by hydrogen atoms and led to the inhibition of glycolysis followed by the prevention of the glucose-6- phosphate production [11]. As the study's objective was to identify the potential compound similar to 2-Deoxy-D-Glucose with less toxicity, the derivative was also considered for this investigation. Among the derivatives of 2-Deoxy-D-Glucose, Cypate 2-Deoxy-D-Glucose was chosen as a COVID-19 inhibitor due to its optical fluorescence imaging ability. Cypate is a reactive carbocyanine and lipophilic dye where the evaluation of the compound was reported in tumor-bearing mice in vivo condition through NIR optical imaging [12,13].
This study tested the potentiality of 2-Deoxy-D-Glucose molecules against COVID-A19 main protease complex with N3 inhibitor (PDB ID: 6LU7) through in silico model. The target main protease (Mpro) of SARS-CoV-2: Mpro emerged as a critical enzyme of coronavirus and has a crucial role in regulating viral replication and transcription [14]. DockThor is an automated web server based on a global docking method utilized for flexible docking [15]. Furthermore, the potential candidate among the modified and derivative forms of 2-Deoxy-D-Glucose against COVID-19 main protease complex was identified based on minimal toxicity compared to the parent compound 2-Deoxy-D-Glucose.
2. Results and Discussion
2.1. Structural Evaluation and Optimization of COVID-19 Main Protease Complex
The structure of COVID-19 main protease in complex with an inhibitor N3 (PDB 6LU7), formerly known as SARS-CoV-2 Mpro, plays a vital role in regulating viral replication and transcription, which could be used as a drug target to elucidate the binding mechanism. [16,17]. Due to the functional importance of Mpro in the viral life cycle, it can be implemented as a potential target for developing antiviral drugs after the structural evaluation and submission of 3D structure to the RCSB database [14]. Although the enzymatic activity of COVID-19 main protease in Vero cells was measured against potential inhibitors such as N3 and Ebselen [14], in silico model evaluation is still to be explored. The structural validation of the COVID-19 main protease complex is an essential prerequisite before predicting the binding mechanism with 2-Deoxy-Glucose molecules. For this purpose, the ERRAT tool of SAVES v6.0, an online server, was implemented to approve the protein structure based on the overall quality factor, contributing to non-bonded atomic interactions. Within proteins, atoms are distributed non-randomly concerning one another, influenced by complex geometric and energetic considerations [18]. The exceptional was maintained by excluding the residues covalently bonded to each other. The higher score symbolizes the high quality of the structure. The basic acceptance rate should be >50 to establish a high-quality model [19]. However, the overall quality factor of COVID-19 main protease was found to be 99.552, which was within a highly acceptable range (Figure 1). Furthermore, the Ramachandran plot of COVID-19 main protease was evaluated using PROCHECK software (Figure 2). The residues in the most favourable region were found to be 90.6%, while 8.6% were in the allowed region and only 0.4% in the disallowed region, confirming the target protein as a good quality model to perform the docking. PROCHECK also predicts the structure quality based on G-factors, which measure the unusualness [20]. Recently, a group of researchers validated the spike glycoprotein through PROCHECK to obtain immunogenic epitopes [21]. Hence, by correlating the ERRAT value, the model had a 0.12 G-factor value significantly lower than the expected exceptional value (0.5), indicating the model's efficacy. Subsequently, the active site prediction of the COVID-19 main protease complex was performed by CASTp 3.0 server and discovered the amino acid residues present in the active site cavity. Polar uncharged amino acids like Thr dominated the active site of COVID-19's main protease. Followed by Thr, the cavity contained the polar positively charged amino acid His and other amino acids such as Ser, Leu, and Met. Other residues like Phe140, Asn142, Gly143, Cys145, and Glu166 were present in the tunnel of protein (Figure 3). Earlier, Hatada and his group analysed the key residues during complex formation with inhibitors through fragment molecular orbital study and reported that His41, His163, His164, and Glu166 were the critical residues [22]. The active site residues have mouth openings that connect their interior to the outside bulk solution by developing concave surfaces, which aids in presenting the amino acid residues in a suitable configuration for the drug compound binding [23]. Hence, these residues of the COVID-19 main protease complex constructively influence the facilitation and adaptation of binding pockets to regulate binding processes and specific functionalities.
2.2. Molecular docking of COVID-19 main protease complex with a modified and derivative form of 2-Deoxy-Glucose
The optimized and validated COVID-19 main protease complex structure by SAVES v6.0 was exposed to molecular docking to uncover the binding mechanism against DRDO's prescribed anti-covid potential drug 2-Deoxy-Glucose along with its modified and derivative forms. The results obtained from this binding analysis could help in establishing the opening of new research avenues. By performing the docking study of the COVID-19 main protease complex with 2-Deoxy-Glucose ligands using the DockThor, the top pose with the lowest binding energy and affinity was selected (Figure 4). The docking station, DockThor, was used to predict the specific binding of ligands to the receptor using empirical scoring functions [24]. The comparative analysis among different forms of 2-Deoxy-Glucose with target protein was done to identify the competent compound against COVID viral infection. Initially, the parental compound, 2-Deoxy-Glucose retrieved from the PubChem database, was examined through docking, where the binding affinity was found to be -5.68 kcal/mol (Table 1). The Vander Waal and electrostatic energy were estimated by 0.76 and -34.78 kcal/mol, respectively. The binding energy of the complex formed by COVID-19 main protease and 2-Deoxy-Glucose was determined by -2.065 kcal/mol. The binding mechanism explored the critical amino acid residues that were participating in forming a complex with 2-Deoxy-Glucose. Mainly, Gln127, Ala129, Lys137 and Glu290 were found to be involved in forming hydrogen bonds with the oxygen atom of 2-Deoxy-Glucose at O1, O2, and O4 positions (Table 2). However, the specific amino acid residues, such as Lys5 and Cys128, assisted in fabricating the hydrophobic region around the complex. Gln127 and Glu290 amplified the stability of the complex by forming the hydrogen bond with a 2.6 Å distance. In addition to the parental compound 2-Deoxy-Glucose, the modification was made to investigate the compound's efficacy against COVID-19 main protease by removing hydroxyl groups and replacing them with hydrogen atoms.
Interestingly, the binding mechanism was indistinguishable to some extent from the parent compound, 2-Deoxy-Glucose (Figure 5). The residues such as Ala129, Lys137, and Glu290 were also found in common. Nevertheless, the bond length and the compound's orientation were changed by altering the position of the oxygen atom. Apart from these residues, Lys5 and Cys128 were found to be involved in complex formation by establishing the bond with 2.75 and 3.06 Å, respectively (Table 2). Tyr126 and Gln127 generated the hydrophobic region around the complex. And the Vander Waal and electrostatic energy were measured by -2.62 and -19.69 kcal/mol (Table 1).
On the other hand, the binding energy was found to be exceptionally distinctive from the parent compound, i.e., -39.26 kcal/mol. It contributed the maximum to the stability of the complex. The binding energy or free energy evaluation was a key factor that helped identify critical residues against potential inhibitors design [25]. The reported studies on COVID-19 main protease described the binding mechanism with few natural, synthetic compounds such as flavonoids, peptides, terpenes and quinolines. These complex formations were dominated by hydrogen and hydrophobic interaction involving key residues such as Gln89, Asn147, Glu166, and Gln189 [26,27]. Subsequently, the derivative form, Cypate 2-Deoxy-D-glucose, was also taken for comparative analysis against COVID-19 main protease protein (Figure 6). The critical residues of the complex and the hydrophobic region were also generated in Ligplot software (Figure 7). Moreover, the binding mechanism was more or less identical to the compounds mentioned above, with divergent orientations and positions of the compound. Although the affinity was found to be -9.18 kcal/mol, the binding energy with -8.01 kcal/mol indicated a less stable complex formation compared to the modified form. The exceptional residues such as Gln110, Thr111, Asn151 and Cys160 were found to be involved through hydrogen bonding with nitrogen and oxygen atoms.
Potential Mpro inhibitors with high binding energy, such as saquinavir, aclarubicin, TMC-310911, and faldaprevir, contributed to the complex via hydrogen bonding by involving specific residues like Asn142, Ser144, and Gly143 [28]. By critically comparing the above three compounds on in silico basis screening, the modified 2-Deoxy-D-glucose was the best compound against the COVID-19 main protease complex. Recent studies discovered potential inhibitors against COVID-19 main protease, including the phytocompounds from Houttuynia cordata, marine bioactive compounds with antiviral activities, and marine polycyclic guanidine alkaloids [29,30]. Hence modified 2-Deoxy-D-glucose can be further examined as a suitable candidate drug against the critical condition of COVID-19. The results of this analysis may be fruitful in opening helpful research avenues against COVID during this pandemic.
2.3. ADMET prediction of 2-Deoxy-Glucose ligands for toxicity assessment
The growing demand for therapeutic strategy development and the discovery of competent drugs against COVID is essential in this pandemic. Consequently, the toxicity of these compounds was screened by the web server ADMETlab 2.0, and the properties were evaluated based on parameters like Absorption, Distribution, Metabolism, Excretion, and Toxicity. Regarding the physicochemical aspect, a few important parameters were considered vital in predicting efficient drug candidates against COVID-19 main protease. The detailed parameters are described in Table 3. The toxicity profile of 2-Deoxy-D-glucose and its various forms was evaluated based on a few potential parameters: molecular weight, which is critical for controlled and systematic drug delivery. These compounds' molecular weight was considered because they are crucial in the medication and oral bioavailability [31]. The result obtained from the analysis showed that the modified 2-Deoxy-D-glucose with the lowest molecular weight, i.e., 132.08 Da, may indicate rapid absorption and excretion rate compared to the other two compounds. Controlled and systematic delivery of low molecular weight drugs through brain barrier modulation prevented retinal degeneration and suppressed laser-induced choroidal neovascularisation in neurodegenerative conditions [32]. In addition, logP of the modified 2-Deoxy-D-glucose was found to be within an optimal range from 0 to 3 while 2-Deoxy-D-glucose and Cypate 2-Deoxy-D-glucose were in the < 0 and > 3 range. According to the boundaries set by Lipinski’s Rule, no violation was found when the partition coefficient logP < 5 [33]. The reduced bioavailability of the compound beyond these boundaries indicated poor absorption or permeation [34]. Similarly, the solubility of the compound, indicated by the logS value, improved the absorption at high rates. Apart from derivative and parent forms, the modified version evolved as an active inhibitor and increased the adsorption rate. The number of hydrogen bond acceptors and donors within the threshold values was ≤ 10 and ≤ 5, respectively. Another competent aspect, such as HIA, acted as an indicator of oral bioavailability, which was required to determine the efficacy of any drug [35]. Table 3 showed the efficacy of modified 2-Deoxy-D-glucose and concluded that the revised version was preferable to the other two. The HIA category is restricted from 0 to 1, whereas 0 to 0.3 resides in the excellent category, 0.3 to 0.7 belongs to the medium, and 0.7 to 1.0 belongs to the poor category [36]. Similar to HIA, F30% or human oral bioavailability by 30% was acted as a correlated factor and found to be within the moderate region, i.e., 0.513 for modified 2-Deoxy-D-glucose while the other two fell in the poor category (a range within 0-0.3: excellent; 0.3-0.7: medium; 0.7-1.0: poor). The fraction unbound in plasma, Fu, is a critical component for determining drug efficacy in drug pharmacokinetic studies because of its relation with unbound states which alter the cellular response [37]. On the other hand, PPB may not significantly alter drug development strategy but must be considered during the analysis of pharmacokinetic profiles in vivo conditions. The value of PPB ≤ 90% is considered a preferable category, while above 90% belongs to a poor level. The values were 6.58% of PPB in modified 2-Deoxy-D-glucose and 97.58% in Cypate. PPB has an influential impact on oral bioavailability/distribution and clearance of the drug. Earlier studies reported the consequence of high PPB on the antiviral activity of NVR 3-778, a sulfamoylbenzamide capsid assembly modulator [38]. The clearance of a drug for a modified one was found to be maximum. The threshold value of clearance of a drug is set by five while more than it belongs to the favorable region and lesser to the low category [39]. Subsequently, half-life or T1/2 was related by clearance of the drug and was found to be within a moderate range for the molecules except for the derivative forms (0-0.3: excellent; 0.3-0.7: medium; 0.7-1.0: poor). The safety of these compounds was also evaluated, where both the parent and modified molecule had no mutagenic effect. However, the derivative molecule had a toxic effect. Similarly, the modified form's drug-induced liver injury or DILI effect was less than Cypate 2-Deoxy-D-glucose. A high level of DILI has serious clinical consequences that cause hepatotoxicity or acute liver failure and has been cited as one of the major reasons for drug removal from the market [40,41]. Thus, the modified 2-Deoxy-D-glucose had favorable ADMET parameters and could be considered a suitable drug candidate during the treatment of viral infection. Hence, by combining these parameters, the current findings indicated that the potentiality of 2-Deoxy-D-glucose was improved following structural modification. Thus, the modified forms have the potential to be used as a therapeutic strategy against virally infected cells by inhibiting or restricting energy production in viral cells. The in vivo study with the potential drug could be a game changer in the fight against pandemics like COVID.
3. Materials and Methods
3.1. The target PDB Structure Selection and Validation
The target protein considered for this investigation was the crystal structure of COVID-19 main protease in complex with an inhibitor N3 with PDB ID: 6LU7. The structure with N3 inhibitor (2.16 Å resolutions) was downloaded from the RCSB database (https://www.rcsb.org/search). The selection was based on the importance of the Main protease (Mpro) of SARS-CoV-2 in mediating viral replication and transcription having close homologous with humans. After the protein retrieval from the database, the protein's validity was evaluated using SAVESv6.0 - Structure Validation Server. The overall quality factor was determined by ERRAT tool of SAVES v6.0 to recognize the correct regions of protein structures based on atomic interaction. Further, the overall G factor was determined by PROCHECK tool to analyze the quality of the selected target protein model. The degrees of unusualness and normality were predicted by G-factor based on stereochemical properties.
3.2. Active Site Prediction
The active site prediction of COVID-19 main protease was examined by CASTp 3.0 server using a Computational Geometry algorithm with basic ingredients such as Delaunay triangulation, alpha shape, and discrete flow (http://sts.bioe.uic.edu/castp/index.html). The area and the volume of the active site occupied by the residues were determined.
3.3. Ligand Files Preparation and Optimization
The 3D structure of 2-Deoxy-Glucose (C6H12O5) and its derivative Cypate-2-Deoxy-D-glucose (C53H63N4O12 ) treated as candidate ligands against the target protein COVID-19 mthe ain protease. The structures were retrieved from the PubChem database in SDF format and converted to PDB format using BIOVIA Discovery studio (https://pubchem.ncbi.nlm.nih.gov/). The modified 2-Deoxy-Glucose was prepared in Argus lab (4.0.1) by replacing two hydroxyl groups with polar hydrogen molecules. The structure was optimised by performing energy minimisation using the UFF force field. The energy-minimized ligands were further analysed in molecular docking experiments.
3.4. Molecular Docking
DockThor, a web tool for ligand-protein docking, was implemented to perform the molecular docking between the modified and derivative of 2-Deoxy-Glucose against COVID-19 main protease; both the receptor and ligands were optimised by applying the same force field known as MMFF94 Force Field throughout the docking experiments. DockThor program implemented the phenotypic crowding-based multiple solution steady-state genetic algorithm as the search method during molecular docking. As the blind docking was performed, the grid box parameter was set automatically by setting the centres X, Y, and Z as 22 Å. The complex's total energy (Etotal) was calculated based on intermolecular and intramolecular interaction energy by summing up the Van der Waals and electrostatic potentials between the protein-ligand atom pairs. All docking poses were generated during the docking step by utilizing the standard algorithm and then clustered with the help of the DTStatistics tool. The number of docking runs was set by 24; for each docking run, maximum 20 numbers of clusters were generated. The top energy poses of each cluster were screened as best reprehensive of 2-Deoxy-Glucose ligands against COVID-19 main protease.
3.5. Prediction of ADMET Properties
An approaching compound's toxicity and clinical safety concerns are significant problems in drug discovery. Hence, the pharmacokinetics and toxicity of candidate ligands against COVID-19 main protease were screened by ADMETlab 2.0: an integrated online platform that helps evaluate accurate and comprehensive ADMET properties. The SMILES chemical structures of the parent, modified, and derivative of 2-Deoxy-Glucose was submitted to the server. The complete ADMET profiles of ligands were created after the generation of the model framework. The ligands described here were subjected to ADMET prediction to investigate the physiochemical properties of these compounds during the interaction with COVID-19 main protease. For the binding purpose, physicochemical descriptors with vital roles were selected, including molecular weight, logP, logS, hydrogen acceptor and donor, polar surface area, F30% human oral bioavailability, and Human intestinal absorption (HIA). These are the critical and essential prerequisites to determine its apparent efficacy and related to oral bioavailability. Plasma protein binding (PPB) acts as a significant mechanism of drug uptake and distribution by binding to plasma proteins indicating a powerful influence on pharmacodynamic behaviour. Other properties such as drug clearance (CL) and half-life (T1/2) are important pharmacokinetic parameters to study the influence on drugs. Drug-induced liver injury (DILI) and mutagenicity are vital descriptors to evaluate the safety of medicines. These descriptors helped correlate the binding pose and binding energy formed during the formation of the COVID-19 main protease complex with 2-Deoxy-Glucose molecules. ADMET analysis provided a better mechanistic understanding of binding relationships with COVID-19 main protease and determined the suitable ligand candidate for implementation as the essential medication and strategy to treat a viral infection like COVID in this pandemic.
4. Conclusions
The current study investigated the potentiality of 2-Deoxy-D-glucose and its modified and derivative forms against the COVID-19 main protease complex. Compared to the derivative and parent compound, the report concluded that the modified form of 2-Deoxy-D-glucose by replacing the hydroxyl group with hydrogen atoms evolved as a promising candidate. The docking result demonstrated that the minimal energy of modified 2-Deoxy-D-glucose during the complex formation with COVID-19 main protease indicated the stability of the complex during the interaction. By supporting the docking results, ADMET analysis favored the potentiality of modified 2-Deoxy-D-glucose with minimal toxicity, while the derivative form of 2-Deoxy-D-glucose was found to exhibit higher toxicity. Hence, the replacement of the hydroxyl group of 2-Deoxy-D-glucose can be used as a further effective strategy to treat the COVID-19 outbreak.
Author Contributions
Conceptualisation, R.R.S., and A.K.D.; resources A.B.J, R.R.S., and S.S. writing—original draft preparation, A.B.J, R.R.S, and C.S.M.; writing—review and editing, R.R.S., A.K.D; visualization, A.B.J, R.R.S., and C.S.M.; supervision, A.K.D.; All authors have read and agreed to the published version of the manuscript.
Funding
No funding was available.
Ethics approval (include appropriate permissions or waivers)
Not applicable.
Consent to participate (include relevant statements)
Not applicable.
Consent for publication
Not applicable.
Availability of data and material (data transparency)
Available on request.
Code availability (software application or custom code)
Not applicable.
Conflicts of interest/Competing interests
All authors express no conflicts of interest.
References
- Cimolai, N. The Complexity of Co-Infections in the Era of COVID-19. SN Compr. Clin. Med. 2021, 3, 1502–1514. [Google Scholar] [CrossRef] [PubMed]
- Bandala, E.R.; Kruger, B.R.; Cesarino, I.; Leao, A.L.; Wijesiri, B.; Goonetilleke, A. Impacts of COVID-19 Pandemic on the Wastewater Pathway into Surface Water: A Review. Sci. Total Environ. 2021, 774, 145586. [Google Scholar] [CrossRef] [PubMed]
- Aygün, İ.; Kaya, M.; Alhajj, R. Identifying Side Effects of Commonly Used Drugs in the Treatment of Covid 19. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Barlow, A.; Landolf, K.M.; Barlow, B.; Yeung, S.Y.A.; Heavner, J.J.; Claassen, C.W.; Heavner, M.S. Review of Emerging Pharmacotherapy for the Treatment of Coronavirus Disease 2019. Pharmacotherapy 2020, 40, 416–437. [Google Scholar] [CrossRef] [PubMed]
- Rathinavel, T.; Aroulmoji, V. Hyaluronic Acid - 2-Deoxy-D-Glucose Conjugate Act as a Promising Targeted Drug Delivery Option for the Treatment of COVID-19. Int. J. Adv. Sci. Eng. 2021, 7, 2–13. [Google Scholar] [CrossRef]
- Mi, Q.; Ma, Y.; Gao, X.; Liu, R.; Liu, P.; Mi, Y.; Fu, X.; Gao, Q. 2-Deoxyglucose Conjugated Platinum (II) Complexes for Targeted Therapy: Design, Synthesis, and Antitumor Activity. J. Biomol. Struct. Dyn. 2016, 34, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Adhikary, A.; Woloschak, G.; Dwarakanath, B.S.; Papineni, R.V.L. A Combinatorial Approach of a Polypharmacological Adjuvant 2-Deoxy-D-Glucose with Low Dose Radiation Therapy to Quell the Cytokine Storm in COVID-19 Management. Int. J. Radiat. Biol. 2020, 96, 1323–1328. [Google Scholar] [CrossRef]
- Dwarakanath, B.S. Cytotoxicity, Radiosensitization, and Chemosensitization of Tumor Cells by 2-Deoxy-D-Glucose in Vitro. J. Cancer Res. Ther. 2009, 5 Suppl 1, 27–31. [Google Scholar] [CrossRef]
- Passalacqua, K.D.; Lu, J.; Goodfellow, I.; Kolawole, A.O.; Arche, J.R.; Maddox, R.J.; Carnahan, K.E.; O’riordan, M.X.D.; Wobus, C.E. Glycolysis Is an Intrinsic Factor for Optimal Replication of a Norovirus. MBio 2019, 10. [Google Scholar] [CrossRef]
- Fokt, I.; Ziemniak, M.; Ja, A. 2-Deoxy- d -Glucose and Its Analogs : From Diagnostic to Therapeutic Agents. 2020.
- Laussel, C.; Léon, S. Cellular Toxicity of the Metabolic Inhibitor 2-Deoxyglucose and Associated Resistance Mechanisms. Biochem. Pharmacol. 2020, 182. [Google Scholar] [CrossRef]
- Guo, J.; Du, C.; Shan, L.; Zhu, H.; Xue, B.; Qian, Z.; Achilefu, S.; Gu, Y. Comparison of Near-Infrared Fluorescent Deoxyglucose Probes with Different Dyes for Tumor Diagnosis in Vivo. Contrast Media Mol. Imaging 2012, 7, 289–301. [Google Scholar] [CrossRef]
- Ye, Y.; Bloch, S.; Kao, J.; Achilefu, S. Multivalent Carbocyanine Molecular Probes: Synthesis and Applications. Bioconjug. Chem. 2005, 16, 51–61. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Santos, K.B.; Guedes, I.A.; Karl, A.L.M.; Dardenne, L.E. Highly Flexible Ligand Docking: Benchmarking of the DockThor Program on the LEADS-PEP Protein-Peptide Data Set. J. Chem. Inf. Model. 2020, 60, 667–683. [Google Scholar] [CrossRef]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The Crystal Structures of Severe Acute Respiratory Syndrome Virus Main Protease and Its Complex with an Inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 13190–13195. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of Coronavirus Main Proteinase Reveals Combination of a Chymotrypsin Fold with an Extra α-Helical Domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Messaoudi, A.; Belguith, H.; Ben Hamida, J. Homology Modeling and Virtual Screening Approaches to Identify Potent Inhibitors of VEB-1 β-Lactamase. Theor. Biol. Med. Model. 2013, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Sisakht, M.; Mahmoodzadeh, A.; Darabian, M. Plant-Derived Chemicals as Potential Inhibitors of SARS-CoV-2 Main Protease (6LU7), a Virtual Screening Study. Phyther. Res. 2021, 1–13. [Google Scholar] [CrossRef]
- Hatada, R.; Okuwaki, K.; Mochizuki, Y.; Handa, Y.; Fukuzawa, K.; Komeiji, Y.; Okiyama, Y.; Tanaka, S. Fragment Molecular Orbital Based Interaction Analyses on COVID-19 Main Protease - Inhibitor N3 Complex (PDB ID: 6LU7). J. Chem. Inf. Model. 2020, 60, 3593–3602. [Google Scholar] [CrossRef] [PubMed]
- Stank, A.; Kokh, D.B.; Fuller, J.C.; Wade, R.C. Protein Binding Pocket Dynamics. Acc. Chem. Res. 2016, 49, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Guedes, I.A.; Barreto, A.M.S.; Marinho, D.; Krempser, E.; Kuenemann, M.A.; Sperandio, O.; Dardenne, L.E.; Miteva, M.A. New Machine Learning and Physics-Based Scoring Functions for Drug Discovery. Sci. Rep. 2021, 11, 1–19. [Google Scholar] [CrossRef]
- Su, J.G.; Jin Xu, X.; Hua Li, C.; Chen, W.Z.; Wang, C.X. Identification of Key Residues for Protein Conformational Transition Using Elastic Network Model. J. Chem. Phys. 2011, 135. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.I.; Shaikh, M. ; Atia-Tul-Wahab; Atta-Ur-Rahman In Silico Identification of Potential Inhibitors of Key SARS-CoV-2 3CL Hydrolase (Mpro) via Molecular Docking, MMGBSA Predictive Binding Energy Calculations, and Molecular Dynamics Simulation. PLoS One 2020, 15, 1–15. [Google Scholar] [CrossRef]
- Singh, E.; Khan, R.J.; Jha, R.K.; Amera, G.M.; Jain, M.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. A Comprehensive Review on Promising Anti-Viral Therapeutic Candidates Identified against Main Protease from SARS-CoV-2 through Various Computational Methods. J. Genet. Eng. Biotechnol. 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational Approach toward COVID-19 Main Protease Inhibitors via Molecular Docking, Molecular Dynamics Simulation and Free Energy Calculation. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Mahanta, S.; Tanti, B.; Tag, H.; Hui, P.K. Identification of Phytocompounds from Houttuynia Cordata Thunb. as Potential Inhibitors for SARS-CoV-2 Replication Proteins through GC–MS/LC–MS Characterization, Molecular Docking and Molecular Dynamics Simulation. Mol. Divers. 2021,. [CrossRef]
- El-Demerdash, A.; Metwaly, A.M.; Hassan, A.; El-Aziz, T.M.A.; Elkaeed, E.B.; Eissa, I.H.; Arafa, R.K.; Stockand, J.D. Comprehensive Virtual Screening of the Antiviral Potentialities of Marine Polycyclic Guanidine Alkaloids against Sars-Cov-2 (Covid-19). Biomolecules 2021, 11, 1–25. [Google Scholar] [CrossRef]
- Ya’u Ibrahim, Z.; Uzairu, A.; Shallangwa, G.; Abechi, S. Molecular Docking Studies, Drug-Likeness and in-Silico ADMET Prediction of Some Novel β-Amino Alcohol Grafted 1,4,5-Trisubstituted 1,2,3-Triazoles Derivatives as Elevators of P53 Protein Levels. Sci. African 2020, 10, e00570. [Google Scholar] [CrossRef]
- Campbell, M.; Humphries, M.M.; Kiang, A.S.; Nguyen, A.T.H.; Gobbo, O.L.; Tam, L.C.S.; Suzuki, M.; Hanrahan, F.; Ozaki, E.; Farrar, G.J.; et al. Systemic Low-Molecular Weight Drug Delivery to Pre-Selected Neuronal Regions. EMBO Mol. Med. 2011, 3, 235–245. [Google Scholar] [CrossRef]
- Schneider, G. Prediction of Drug-Like Properties The “ Drug-Likeness ” Concept. 2000, 1–34.
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.; Wang, Z.; Cai, Z. Prediction of Human Intestinal Absorption by GA Feature Selection and Support Vector Machine Regression. Int. J. Mol. Sci. 2008, 9, 1961–1976. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wang, N.N.; Yao, Z.J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.P.; Cao, D.S. Admetlab: A Platform for Systematic ADMET Evaluation Based on a Comprehensively Collected ADMET Database. J. Cheminform. 2018, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Esaki, T.; Kawashima, H.; Natsume-Kitatani, Y.; Nagao, C.; Ohashi, R.; Mizuguchi, K. Predicting Fraction Unbound in Human Plasma from Chemical Structure: Improved Accuracy in the Low Value Ranges. Mol. Pharm. 2018, 15, 5302–5311. [Google Scholar] [CrossRef] [PubMed]
- Helsen, N.; Vervoort, T.; Vandenbossche, J.; Lenz, O.; Monshouwer, M.; Pauwels, F.; Snoeys, J. Effect of Plasma Protein Binding on the Anti-Hepatitis B Virus Activity and Pharmacokinetic Properties of NVR 3-778. Antimicrob. Agents Chemother. 2018, 62, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 1–10. [Google Scholar] [CrossRef]
- Walker, P.A.; Ryder, S.; Lavado, A.; Dilworth, C.; Riley, R.J. The Evolution of Strategies to Minimise the Risk of Human Drug-Induced Liver Injury (DILI) in Drug Discovery and Development. Arch. Toxicol. 2020, 94, 2559–2585. [Google Scholar] [CrossRef]
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-Induced Liver Injury: Interactions between Drug Properties and Host Factors. J. Hepatol. 2015, 63, 503–514. [Google Scholar] [CrossRef]
Figure 1.
ERRAT plot for the COVID-19 main protease model. Regions that can be rejected at the
99% level are shown in red. Gray bars indicates the error region between 95% and 99%, and regions
of the structure that can be rejected at the 95% confidence level are in yellow.
Figure 1.
ERRAT plot for the COVID-19 main protease model. Regions that can be rejected at the
99% level are shown in red. Gray bars indicates the error region between 95% and 99%, and regions
of the structure that can be rejected at the 95% confidence level are in yellow.
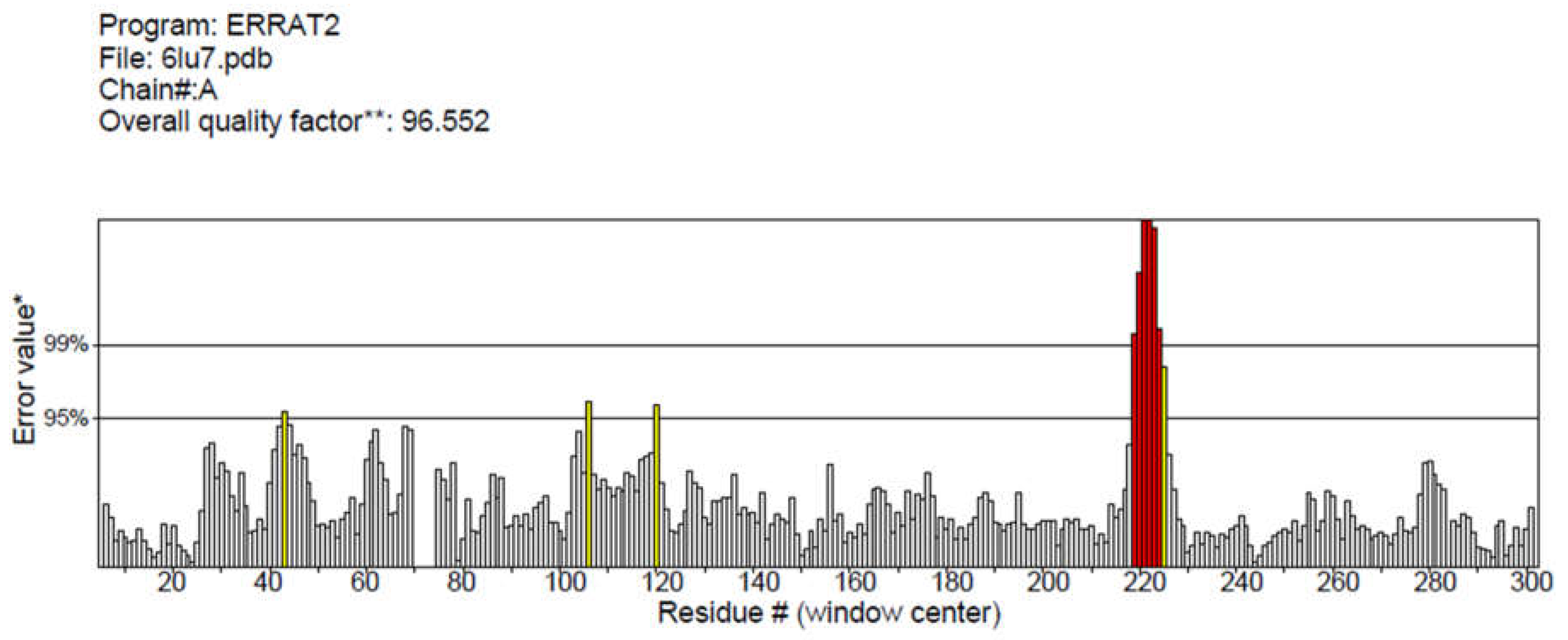
Figure 2.
Ramachandran plot of COVID-19 main protease generated by PROCHECK validation server showing the stereochemical quality with statistical value.
Figure 2.
Ramachandran plot of COVID-19 main protease generated by PROCHECK validation server showing the stereochemical quality with statistical value.
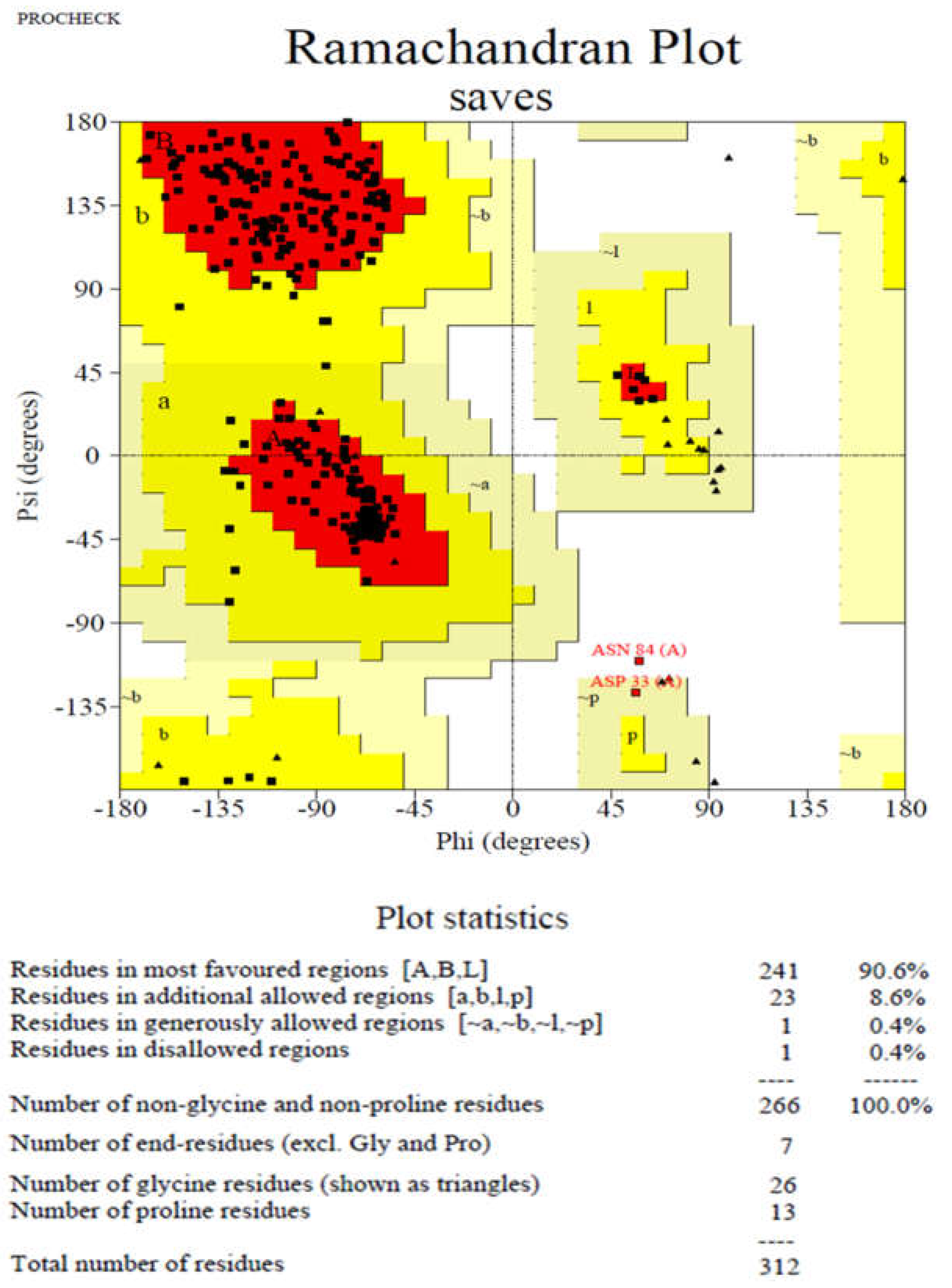
Figure 3.
Active site of COVID-19 main protease complex with N3 inhibitor predicted by CASTp
3.0 server. The area in red color represents the active site of protein and residues were represented
in ball and stick manner. The sequence of COVID-19 main protease was presented by CASTp where
the highlighted residues in both chain A and C represents the active amino acid residues.
Figure 3.
Active site of COVID-19 main protease complex with N3 inhibitor predicted by CASTp
3.0 server. The area in red color represents the active site of protein and residues were represented
in ball and stick manner. The sequence of COVID-19 main protease was presented by CASTp where
the highlighted residues in both chain A and C represents the active amino acid residues.
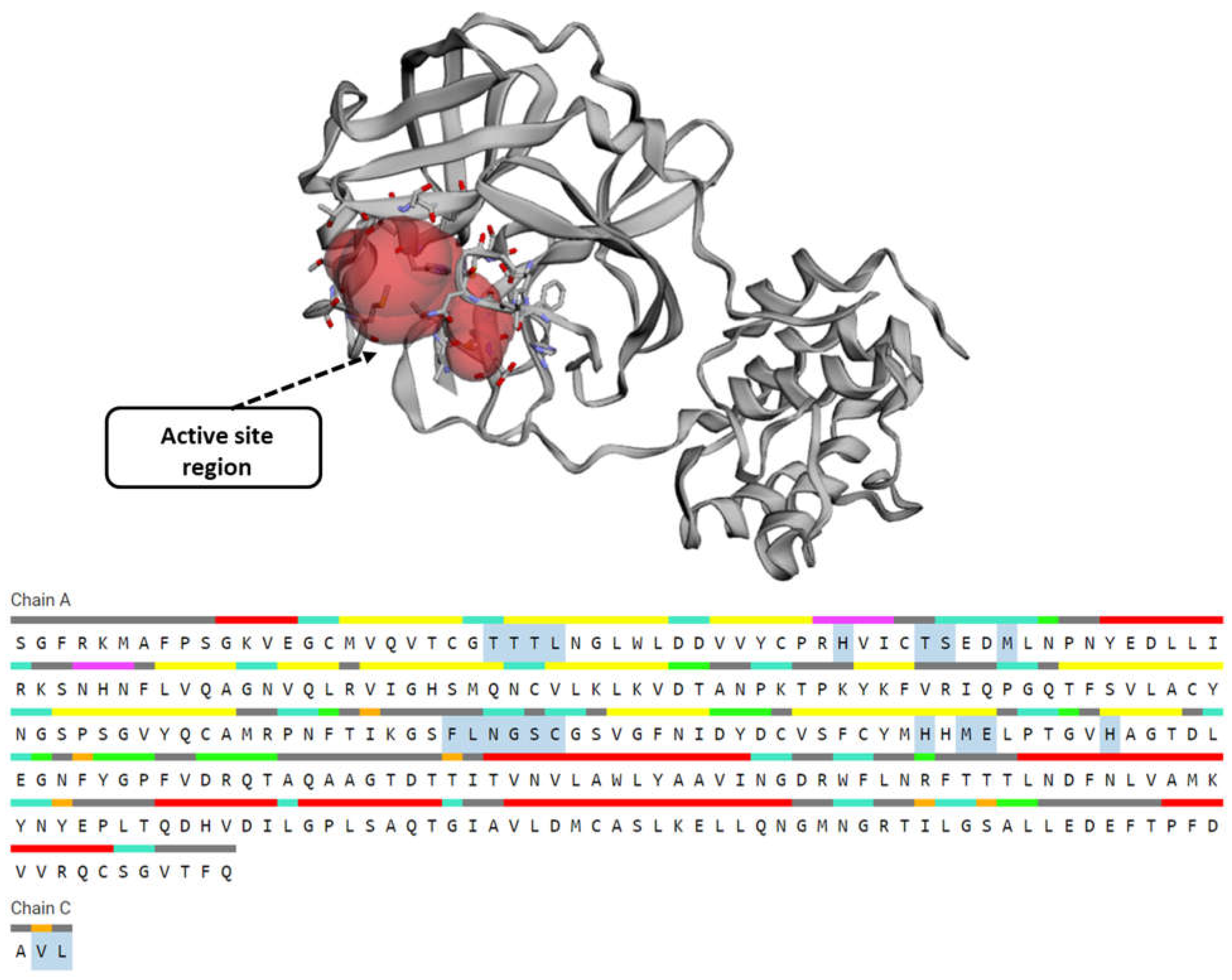
Figure 4.
Molecular docking of the crystal structure of COVID-19 main protease complex (PDB
ID:6LU7) with 2-Desoxy-D-glucose. The COVID-19 main protease complex was shown in cyan color
in ribbon form and 2-Desoxy-D-glucose was represented in ball and stick model in orange color
surface. The enlarged version showing the binding mode of top ranked docked poses and interacting
important residues of COVID-19 main protease with 2-Desoxy-D-glucose by forming hydrogen
bonds. The docking was performed in AutoDock 4.2 and visualization of the complex was done by
UCSF Chimera software.
Figure 4.
Molecular docking of the crystal structure of COVID-19 main protease complex (PDB
ID:6LU7) with 2-Desoxy-D-glucose. The COVID-19 main protease complex was shown in cyan color
in ribbon form and 2-Desoxy-D-glucose was represented in ball and stick model in orange color
surface. The enlarged version showing the binding mode of top ranked docked poses and interacting
important residues of COVID-19 main protease with 2-Desoxy-D-glucose by forming hydrogen
bonds. The docking was performed in AutoDock 4.2 and visualization of the complex was done by
UCSF Chimera software.
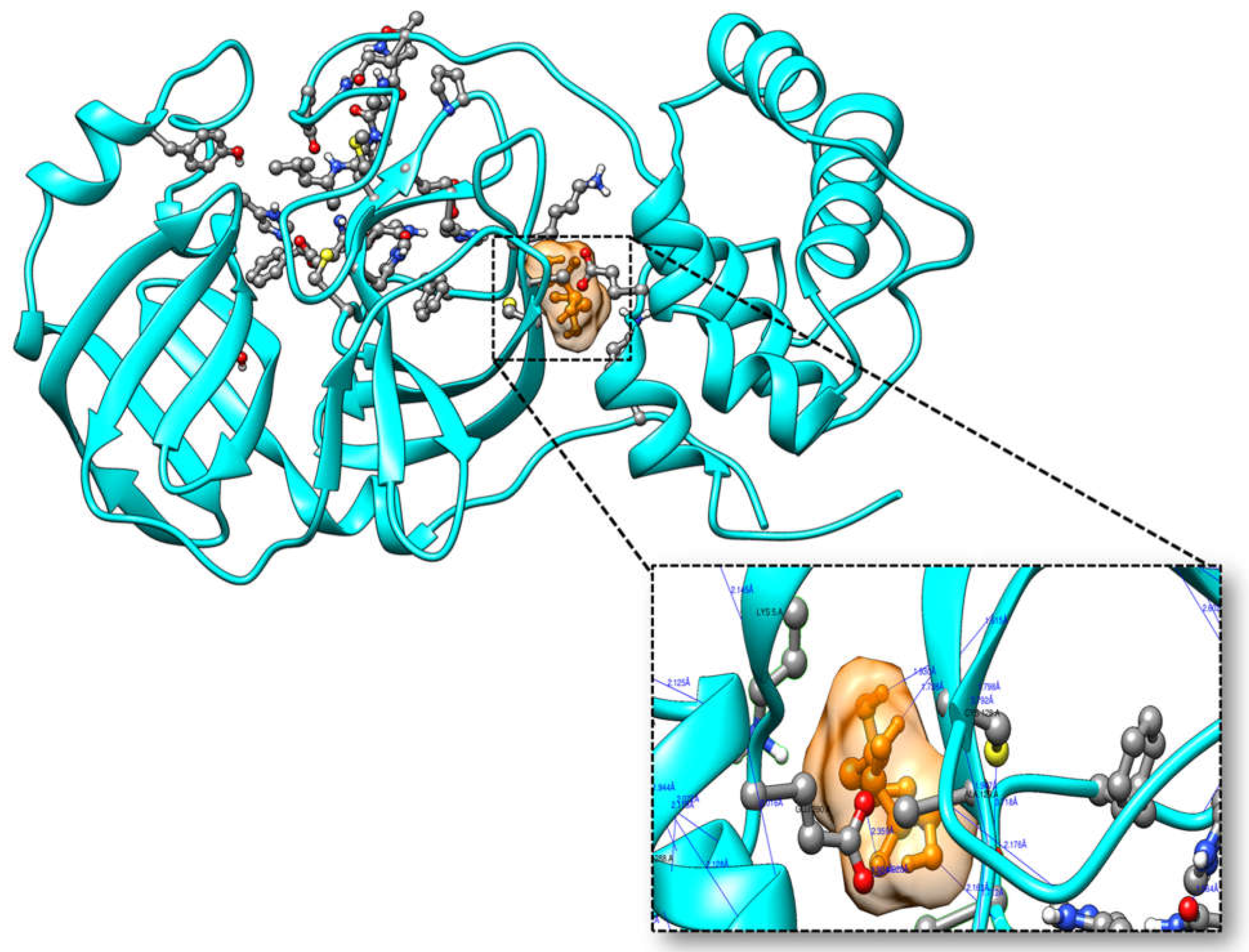
Figure 5.
Molecular docking of the crystal structure of COVID-19 main protease complex (PDB
ID:6LU7) with modified 2-Desoxy-D-glucose by removing two hydroxyl group. The COVID-19
main protease complex was shown in cyan color in ribbon form and modified 2-Desoxy-D-glucose
was represented in ball and stick model in green color surface. The enlarged version showing the
binding mode of top ranked docked poses and interacting important residues of COVID-19 main
protease with modified 2-Desoxy-D-glucose by forming hydrogen bonds. The docking was performed
in AutoDock 4.2 and visualization of the complex was done by UCSF Chimera software.
Figure 5.
Molecular docking of the crystal structure of COVID-19 main protease complex (PDB
ID:6LU7) with modified 2-Desoxy-D-glucose by removing two hydroxyl group. The COVID-19
main protease complex was shown in cyan color in ribbon form and modified 2-Desoxy-D-glucose
was represented in ball and stick model in green color surface. The enlarged version showing the
binding mode of top ranked docked poses and interacting important residues of COVID-19 main
protease with modified 2-Desoxy-D-glucose by forming hydrogen bonds. The docking was performed
in AutoDock 4.2 and visualization of the complex was done by UCSF Chimera software.
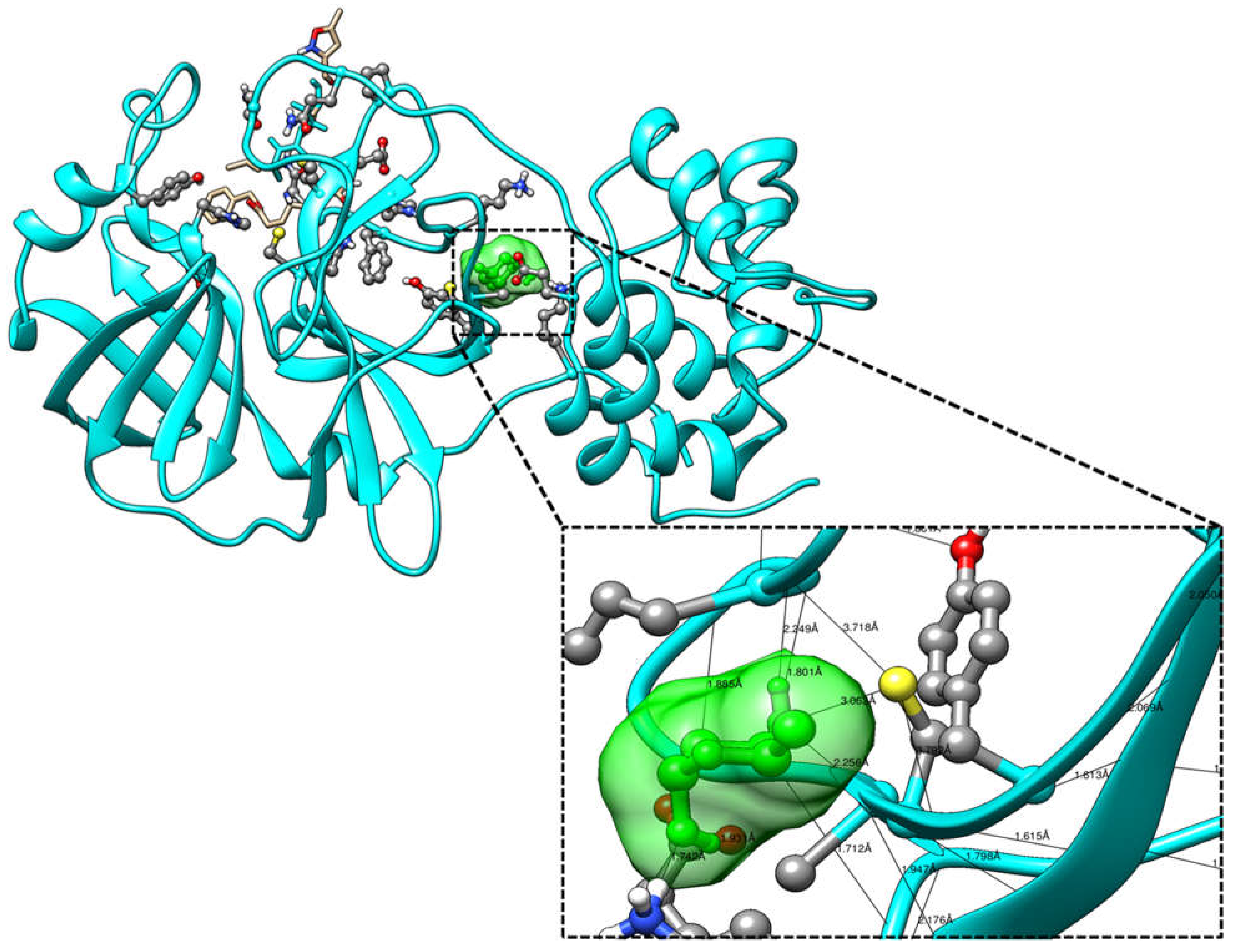
Figure 6.
Molecular docking of the crystal structure of COVID-19 main protease complex (PDB
ID:6LU7) with Cypate-2-Deoxy-D-glucose. The COVID-19 main protease complex was shown in
cyan color in ribbon form and modified 2-Desoxy-D-glucose was represented in ball and stick model
in pink color surface. The enlarged version showing the binding mode of top ranked docked poses
and interacting important residues of COVID-19 main protease with Cypate-2-Deoxy-D-glucose by
forming hydrogen bonds. The docking was performed in AutoDock 4.2 and visualization of the
complex was done by UCSF Chimera software.
Figure 6.
Molecular docking of the crystal structure of COVID-19 main protease complex (PDB
ID:6LU7) with Cypate-2-Deoxy-D-glucose. The COVID-19 main protease complex was shown in
cyan color in ribbon form and modified 2-Desoxy-D-glucose was represented in ball and stick model
in pink color surface. The enlarged version showing the binding mode of top ranked docked poses
and interacting important residues of COVID-19 main protease with Cypate-2-Deoxy-D-glucose by
forming hydrogen bonds. The docking was performed in AutoDock 4.2 and visualization of the
complex was done by UCSF Chimera software.
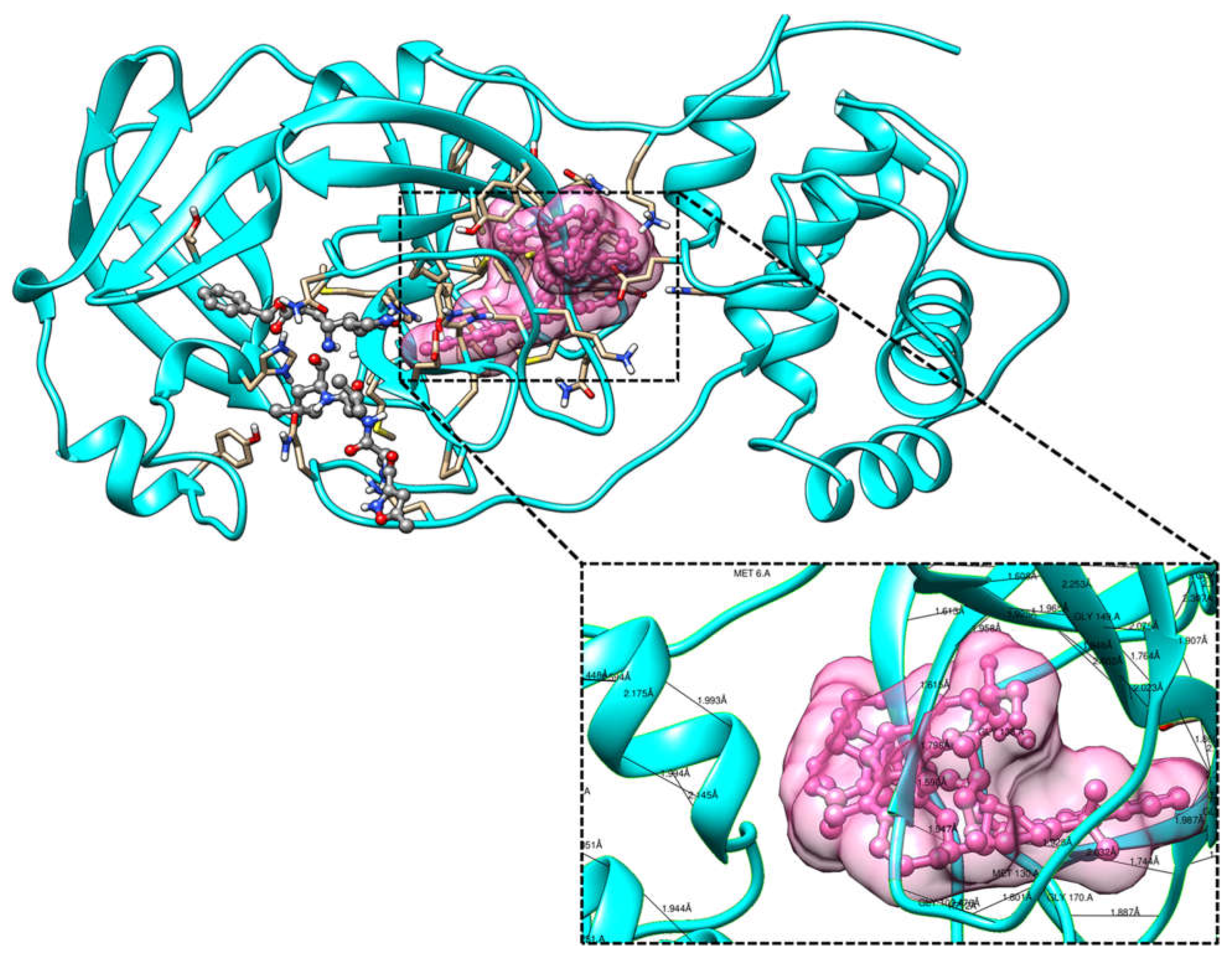
Figure 7.
Interacting amino acid residues of COVID-19 main protease complex (PDB ID:6LU7) with
(a) 2-Deoxy-D-glucose (b) modified 2-Deoxy-D-glucose and (c) Cypate-2-Deoxy-D-glucose was analyzed
in LigPlot+ suite. The green lines indicate the hydrogen bonds and red dotted lines indicate
the hydrophobic interactions. The bond formed between the atoms in purple color represents the
ligand bonds and the bond in brown color represents the non-ligand bond.
Figure 7.
Interacting amino acid residues of COVID-19 main protease complex (PDB ID:6LU7) with
(a) 2-Deoxy-D-glucose (b) modified 2-Deoxy-D-glucose and (c) Cypate-2-Deoxy-D-glucose was analyzed
in LigPlot+ suite. The green lines indicate the hydrogen bonds and red dotted lines indicate
the hydrophobic interactions. The bond formed between the atoms in purple color represents the
ligand bonds and the bond in brown color represents the non-ligand bond.
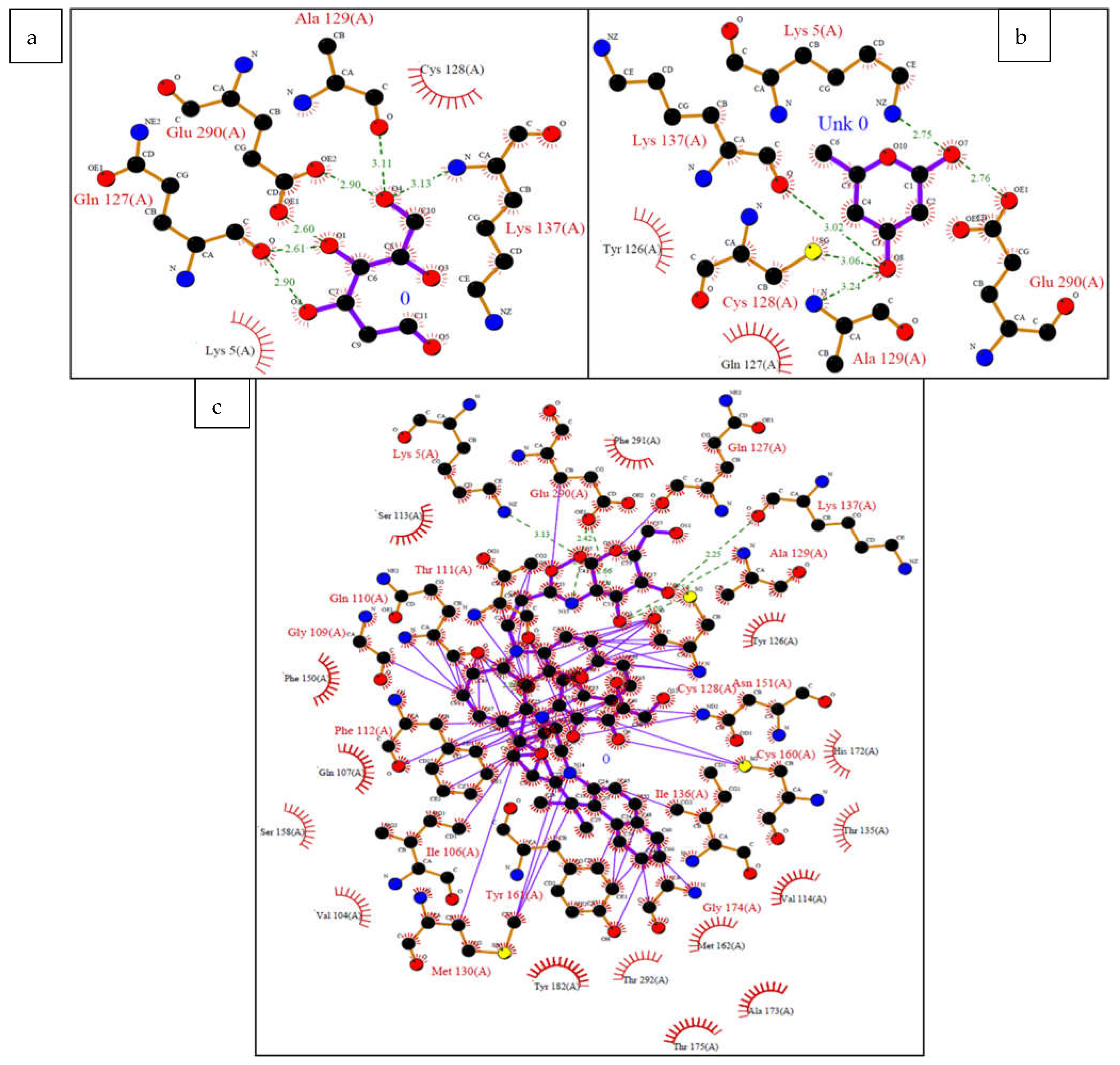

Table 1.
Binding parameters of the complex formed between COVID-19 main protease and ligands in DockThor.
Table 1.
Binding parameters of the complex formed between COVID-19 main protease and ligands in DockThor.
| Ligands | Affinity(kcal/mol) | VDW Energy(kcal/mol) | Electrostatic Energy(kcal/mol) | Binding Energy(kcal/mol) |
|---|---|---|---|---|
| 2-Deoxy-D-glucose | -5.683 | 0.760 | -34.78 | -2.065 |
| Modified 2-Deoxy-D-glucose | -5.821 | -2.622 | -19.69 | -39.263 |
| Cypate 2-Deoxy-D-glucose | -9.18 | -29.34 | -25.43 | -8.01 |
Table 2.
Hydrogen bonds formed between COVID-19 main protease complex and atoms of 2-Deoxy-D-glucose, modified 2-Deoxy-D-glucose and cypate 2-Deoxy-D-glucose.
Table 2.
Hydrogen bonds formed between COVID-19 main protease complex and atoms of 2-Deoxy-D-glucose, modified 2-Deoxy-D-glucose and cypate 2-Deoxy-D-glucose.
| Ligands | Atom Name of Ligands | Residue Name of Receptor | Distance in Å |
|---|---|---|---|
| 2-Deoxy-D-glucose | O2 | Gln127 | 2.9 |
| O1 | Gln127 | 2.6 | |
| O4 | Ala129 | 3.11 | |
| O4 | Lys137 | 3.13 | |
| O1 | Glu290 | 2.6 | |
| O4 | Glu290 | 2.9 | |
| Modified 2-Deoxy-D-glucose | O7 | Lys5 | 2.75 |
| O8 | Cys128 | 3.06 | |
| O8 | Ala129 | 3.24 | |
| O8 | Lys137 | 3.02 | |
| O7 | Glu290 | 2.76 | |
| Cypate 2-Deoxy-D-glucose | O7 | Lys5 | 3.13 |
| N16 | Gln110 | 2.86 | |
| O4 | Thr111 | 1.43 | |
| O3 | Cys128 | 3.20 | |
| O3 | Ala129 | 2.54 | |
| O5 | Lys137 | 2.25 | |
| O4 | Asn151 | 1.89 | |
| O6 | Cys160 | 1.48 | |
| N15 | Glu290 | 2.03 | |
| O3 | Glu290 | 2.66 | |
| O7 | Glu290 | 2.42 |
Table 3.
Prediction of ADMET properties of 2-Deoxy-D-glucose, modified 2-Deoxy-D-glucose and cypate 2-Deoxy-D-gluco.
Table 3.
Prediction of ADMET properties of 2-Deoxy-D-glucose, modified 2-Deoxy-D-glucose and cypate 2-Deoxy-D-gluco.
| Parameters | 2-Deoxy-D-glucose | Modified 2-Deoxy-D-glucose | Cypate 2-Deoxy-D-glucose |
|---|---|---|---|
| Molecular weight | 164.07 | 132.08 | 948.45 |
| logP | -2.035 | 0.029 | 3.383 |
| logS | -0.071 | 0.44 | -3.999 |
| H-Acceptors | 5 | 3 | 16 |
| H-Donors | 4 | 2 | 10 |
| Polar Surface Area | 97.99 | 49.69 | 244.98 |
| HIA | 0.392 | 0.005 | 0.948 |
| F30% | 0.911 | 0.513 | 0.981 |
| Fu | 82.05% | 85.23% | 0.680% |
| PPB | 22.54% | 6.58% | 97.58% |
| CL | 1.954 | 8.07 | 1.13 |
| T1/2 | 0.703 | 0.756 | 0.268 |
| DILI | 0.02 | 0.047 | 0.552 |
| Mutagenic | none | none | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



