
Submitted:
30 August 2021
Posted:
30 August 2021
Read the latest preprint version here
Abstract
The accurate prediction of molecular properties such as lipophilicity and aqueous solubility is of great importance in several stages of the drug discovery pipeline. Machine learning methods like graph-based neural networks have shown exceptionally good performance in predicting these properties. In this work we introduce a novel graph neural network architecture composed of two distinct sub-architectures that achieves an improvement in accuracy over its individual parts employing various learning-, and featurization strategies. We argue that combining models with different key aspects might help make graph neural networks deeper while simultaneously increasing their predictive power. Additionally, we want to highlight the need to move beyond comparing single performance metrics to show machine learning model superiority.
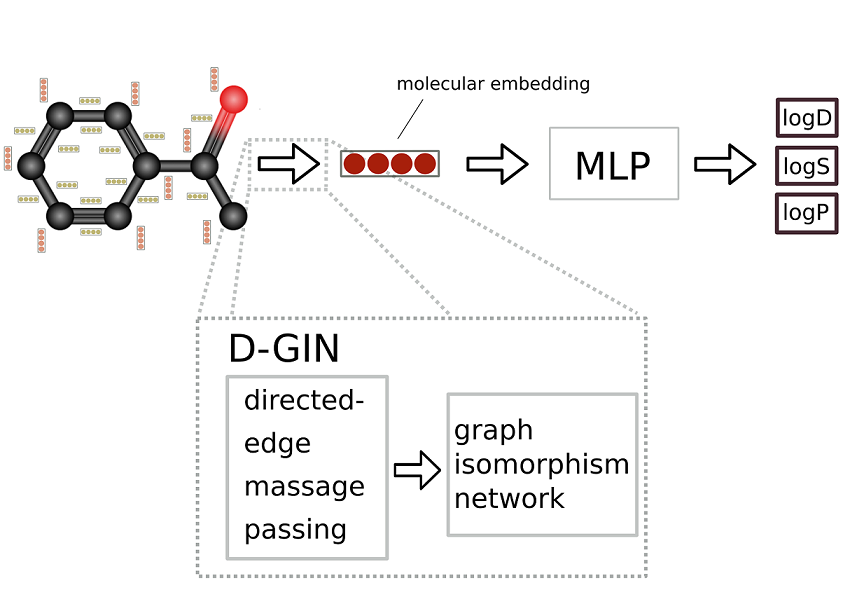
Keywords:
AI
; deep-learning
; neural-networks
; graph neural-networks
; cheminformatics
; molecular property
; machine-learning
; computational chemistry
; lipophilicity
; solubility
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



