
Submitted:
14 December 2020
Posted:
15 December 2020
You are already at the latest version
Abstract
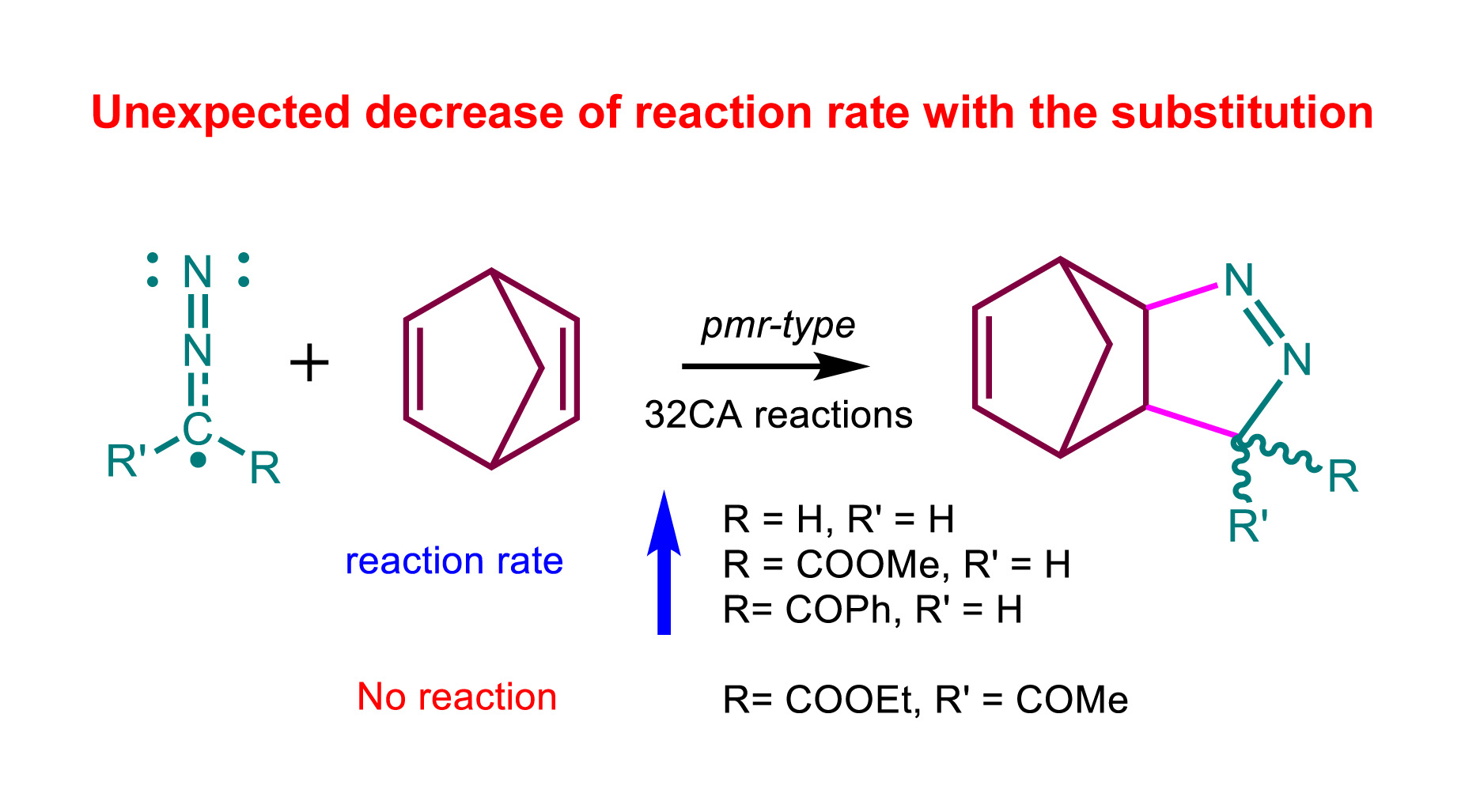
The [3+2] cycloaddition (32CA) reactions of strongly nucleophilic norbornadiene (NBD) with simplest diazoalkane (DAA) and three DAAs of increased electrophilicity have been studied within the Molecular Electron Density Theory (MEDT) at the MPWB1K/6-311G(d,p) computational level. These pmr-type 32CA reactions follow an asynchronous one-step mechanism with activation enthalpies ranging from 17.7 to 27.9 kcal·mol-1 in acetonitrile. The high exergonic character of these reactions makes them irreversible. The presence of electron-withdrawing (EW) substituents in the DAA increases the activation enthalpies, in complete agreement with the experimental slowing-down of the reactions, but contrary to the Conceptual DFT prediction. Despite the nucleophilic and electrophilic character of the reagents, the global electron density transfer at the TSs indicates rather non-polar 32CA reactions. The present MEDT study allows establishing that the depopulation of the NNC core in this series of DAAs with the increase of the EW character of the substituents present at the carbon center is responsible for the experimentally found deceleration.
Keywords:
Molecular Electron Density Theory
; Norbornadiene
; Diazoalkanes
; Conceptual DFT
; Electron Localization Function.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



