Submitted:
29 January 2019
Posted:
30 January 2019
You are already at the latest version
Abstract
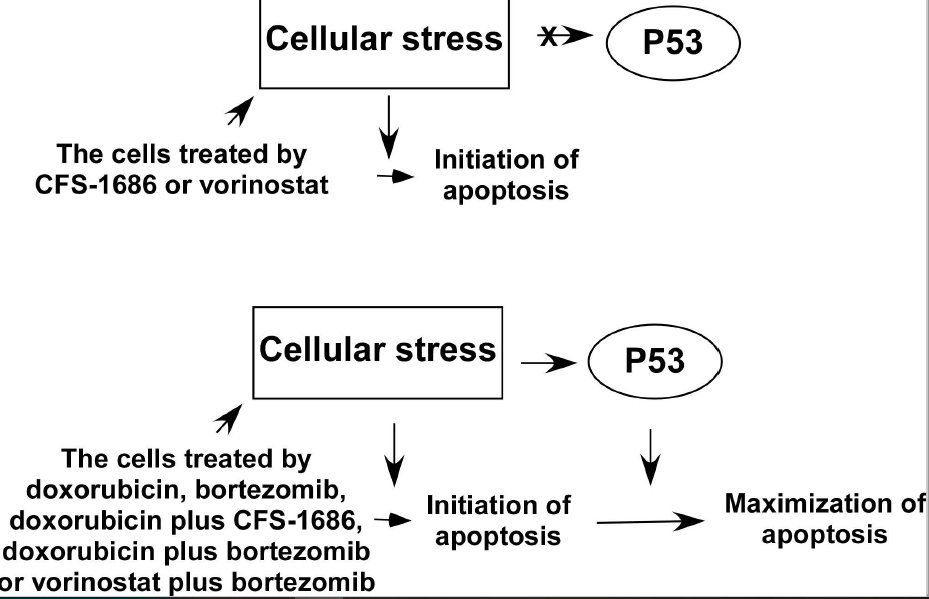
In prostate cancer, p53 maximizes apoptosis in response to severe DNA damage, not DNA replication stress. Here, we examined the apoptotic response of two glioblastoma cells, p53-wild type U87 and a p53-mutated T98G cell, for the same stresses. We ascertained that p53 intensified apoptosis in response to severe DNA damage, not DNA replication stress in glioblastoma. We further asked if p53-mediated apoptosis can be induced by cellular stress other than severe DNA damage. We analyzed two compounds, bortezomib and vorinostat, respective inhibitors of 26S proteasome and histone deacetylase, to evaluate their capacity to activate p53-mediated apoptosis. The cellular stress incited by bortezomib, not vorinostat, activated p53-mediated apoptosis. Next, we asked if the cellular stress generated by combining the two compounds had a synergistic effect on apoptosis. Our results demonstrated that doxorubicin with bortezomib or CFS-1686, or bortezomib with vorinostat have a significant synergistic effect on apoptosis only in p53-wild type cell. Under high stress, p53 translocates from cytosol into the nucleus to cause apoptosis possibly. Together, p53 maximizes apoptosis for cellular stress caused by severe DNA damage, disruption of protein turnover, and for the stress induced by drug combination including doxorubicin with bortezomib or CFS-1686, and bortezomib with vorinostat.
Keywords:
glioblastoma
; p53
; apoptosis
; doxorubicin
; bortezomib
; vorinostat.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



