Submitted:
09 January 2019
Posted:
10 January 2019
You are already at the latest version
Abstract
2-Hydroxypropyl-β-cyclodextrin (HPβCD) has unique properties to enhance the stability and the solubility of low water-soluble compounds by inclusion complexation. Understanding of the structural properties of HPβCD and its derivatives based on the number of 2-hydroxypropyl (HP) substituents at the a-D-glucopyranose subunits is rather important. In this work, replica exchange molecular dynamics simulations were performed to investigate the conformational changes of single- and double-sided HP-substitution called as 6-HPβCDs and 2,6-HPβCDs, respectively. The results show that glucose subunits in both 6-HPβCDs and 2,6-HPβCDs have lower chance to flip than in βCD. Also, HP groups are occasionally blocking the hydrophobic cavity of HPβCDs, thus hindering the drug inclusion. We found that HPβCDs with high number of HP-substitutions are more likely to be blocked, while HPβCDs with double-sided HP-substitution are even more probable to be blocked. Overall, 6-HPβCDs with three and four HP-substitutions are highlighted as the most suitable structures for guest encapsulation based on our conformational analyses such as structural distortion, radius of gyration, circularity and cavity self-closure of the HPβCDs.
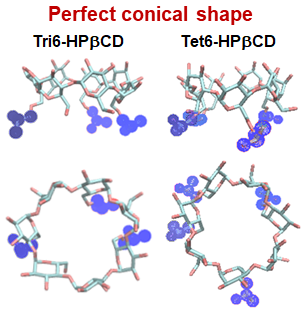
Keywords:
2-Hydroxypropyl-β-cyclodextrin (HPβCD)
; Replica exchange molecular dynamics (REMD)
; Conformational change
; Cavity self-closure
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



