
Submitted:
02 November 2018
Posted:
05 November 2018
You are already at the latest version
Abstract
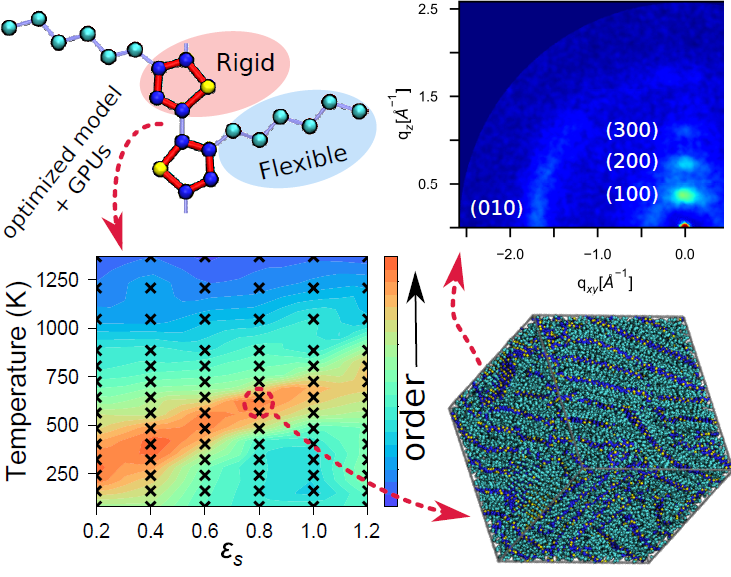
We develop an optimized force-field for poly(3-hexylthiophene) (P3HT) and demonstrate its utility for predicting thermodynamic self-assembly. In particular, we consider short oligomer chains, model electrostatics and solvent implicitly, and coarsely model solvent evaporation. We quantify the performance of our model to determine what the optimal system sizes are for exploring self-assembly at combinations of state variables. We perform molecular dynamics simulations to predict the self-assembly of P3HT at ~350 combinations of temperature and solvent quality. Our structural calculations predict that the highest degrees of order are obtained with good solvents just below the melting temperature. We find our model produces the most accurate structural predictions to date, as measured by agreement with grazing incident X-ray scattering experiments.
Keywords:
organic photovoltaics
; self-assembly
; thermodynamics
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



