Submitted:
06 July 2026
Posted:
08 July 2026
You are already at the latest version
Abstract
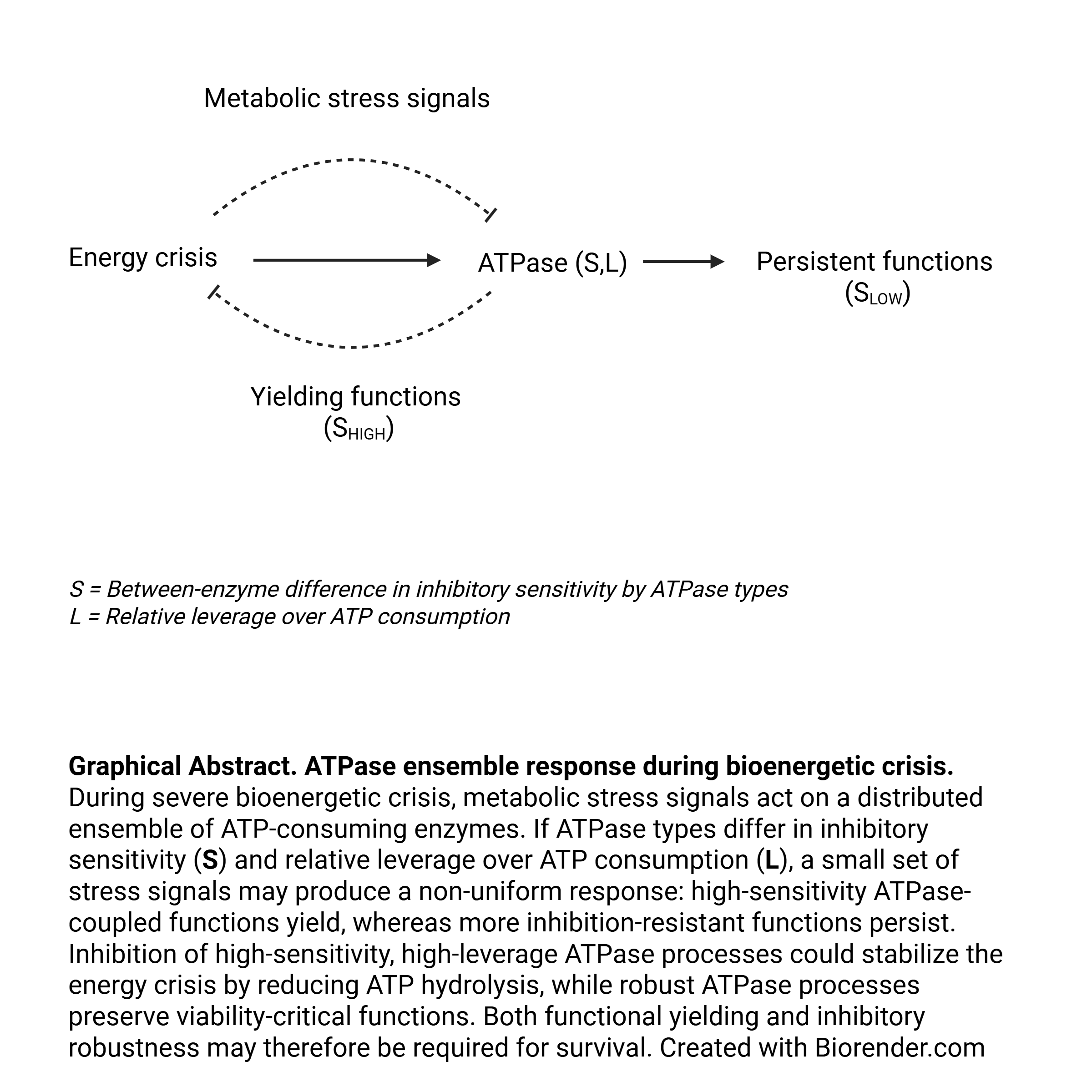
Gibbs free energy (∆G) supports work from an ensemble of ATP-consuming enzymes (ATPases). Data from ischemia and acute organ failure show that functions fail early, whereas ATP and ∆G fail later. Given bioenergetic failure, supply-demand adjustment must occur at the timescale of ATP turnover (10-20 seconds in heart and brain), suggesting rapid energetic adaptation. ATPases are exposed to simple metabolic stress signals that inhibit their function (H+, Pi, ROS, and NO). The altered functional output observed in acute organ failure is stereotypic with limited cell death, suggesting a controlled response. Several ATPases show differential intrinsic inhibitory sensitivity, where these differences in the receiving end of metabolic stress signals could produce a complex response. A distributed, graded inhibitory response of ATP hydrolysis, depending on the ATPase type leverage over ATP consumption, could occur. If ATPases involved in critical functions are robust to inhibition, this allows a routing of bioenergetic failure to fault-tolerant processes, allowing residual ATP hydrolytic capacity for critical functions. Given the capacity for collective specialized organ functions to reduce their functions, unserved workloads may better be absorbed there than in individual cells.
Keywords:
bioenergetics
; enzymes
; ATPases
; critical care
; acute organ failure
; control theory
; ATP hydrolysis regulation
; control inversion
Synopsis
Processes that produce acute organ failure generate supply-side bioenergetic constraint through mitochondrial suppression, hypoxia, hypoperfusion and microcirculatory dysfunction. Although acute organ failure is highly lethal, it is often fully reversible. Cell death is sparse, despite ATP turnover time of only 10-20 seconds in high-flux organs. How cells and organs organize a stable, potentially reversible mode of failure on this timescale is not resolved. In acute ischemia, the most severe bioenergetic crisis, function fails early while ATP and free energy remain relatively preserved for minutes, supported by phosphocreatine. A similar dissociation between function and energetic collapse is observed over hours to days in acute organ failure.
Physiologically, cells operate far from equilibrium with strongly negative Gibbs free energy of ATP hydrolysis (∆G). ATPases can exert considerable metabolic control, but more importantly they control ATP free-energy consumption by coupling ATP hydrolysis to cellular work. In acute organ failure, falling ATP production is accompanied by cellular injury and rising ATP costs, producing a thermodynamic bottleneck. When disturbances such as hypoxia or hypoperfusion develop slowly relative to ATP turnover, the controlled variable (ΔG) may show smaller deviations while adjusted variables such as organ function can exhibit large excursions. Bioenergetic constraints produce rapid local changes in metabolic stress signals. These signals include ATPase kinetic regulators (H+, Pi, NO and ROS), while ATPases may eventually also be constrained by ΔG. Inhibiting consumption can stabilize ∆G under supply-side metabolic constraint. Studies indicate type-specific differences in ATPase inhibitory sensitivity. ATPase types could therefore be distributed along two axes: intrinsic inhibitory sensitivity and leverage over ATP consumption. Inhibiting high-sensitivity/high-leverage ATPases could stabilize ∆G. The aggregate behavior of an ATPase type is proposed to produce a cell-wide inhibitory threshold that reflects structural inhibitory sensitivity more than local embeddedness. Evidence remains limited. High-sensitivity ATPase types may provide controlled points of entry for bioenergetic failure, allowing fault-tolerant functions to yield in order to preserve ∆G. Specialized organ functions have large work-reduction tolerance that can be mobilized as a reserve during bioenergetic constraints, allowing single cells to export unserved workloads, reduce loads on supporting functions and promote viability. According to Ashby, increasing failure variety must be met by a requisite variety of responses. The ATPase system could reduce failure-mode variety by controlling points of entry and provide requisite response variety at the timescale of ATP turnover.
Longer-term inhibition of ATPases and transcriptional responses stabilizes the failure process but can introduce hysteresis and delayed recovery. Hence, when acute bioenergetic failure constrains ATPase-coupled work, acute organ failure may represent a demand-side response that conditionally limits bioenergetic collapse and protects cellular viability.
Background
How a lethal syndrome permits structurally intact recovery after days, weeks or months [1,2,3] remains a paradox of acute organ failure [4]. Autopsy studies show limited necrosis, mitochondrial swelling and reduced, disorganized cristae [5,6]. Acute organ failure of the heart [7], kidney [8] and liver [9] can show complete reversibility after sepsis. Reports describe substantial recovery after prolonged cardiac standstill on extracorporeal membrane oxygenation (ECMO) [10]. In post-cardiac arrest syndrome, patients can recover from cardiogenic shock, severe brain dysfunction and renal failure [11]. Multiorgan dysfunction syndrome (MODS) following trauma shows long-term recovery in a slight majority [3]. Deep behavioral disturbances in delirium often recover [12]. Cell death is limited relative to dysfunction [5]. Reduced O2 extraction has been reported in sepsis [13], post-cardiac arrest syndrome [14] and MODS after trauma [15], with reduced glucose metabolism identified in delirium [16].
Cytokines and nitric oxide (NO) correlate with disease severity [17], mediate vasodilation and inhibit the electron transport chain (ETC) [18]. Most trials of cytokine inhibition have failed in sepsis and acute organ failure [19] while non-selective NO synthase inhibitors are associated with increased mortality despite less hypotension [20]. In sepsis, ETC inhibition is considered an important mechanism of mitochondrial suppression, decreasing O2 consumption and ATP production [4]. Hans Selye cautioned against attributing causality to stress mediators [21]. Currently, observational studies and trials do not align on whether cytokines and NO are causes of acute organ failure. Current models of acute organ failure emphasize hypoxia, hypoperfusion [22], cellular injury [23] or bioenergetic failure [24] which impose a reduced cellular capacity, with less focus on cellular adaptations. The reversibility paradox led Singer to reframe acute organ failure as pro-survival cellular hypometabolism [4,25,26], a known survival strategy in hypoxia-tolerant and heterotherm species [25,27,28]. Although the evidence of a bioenergetic crisis following mitochondrial suppression in critical illness is substantial [4,29], it remains less clear how cells tolerate this crisis and later recover.
The ATP Flux Constraint
ATP turnover times are about 10-20 seconds in cardiomyocytes and neurons [30,31]. Hence, the free energy available for ATP hydrolysis (∆G) collapses rapidly following uncompensated supply failure [32]. Cells can respond by signaling for increased perfusion or by reducing ATP hydrolysis (cellular work). ATP flux constrains the maximal tolerable delay in supply-demand matching over time.
In ischemia, the EEG becomes nearly isoelectric after 20-40 seconds, whereas cell death occurs after 5-10 minutes [33]. In ischemic myocardium, contractility falls dramatically before ATP declines; phosphocreatine (PCr) depletion and falling pH precede ATP depletion [34]. During the initial phase of hypoperfusion, cardiac energetics in rat models change minimally (PCr, ATP and ADP) [35]. Less negative ∆G has been associated with reduced cardiac function during anoxia [36]. Synapses fail during hypoxia [37]. During perturbations, glycolysis lacks sufficient ATP-generating capacity to sustain unrestricted high-flux physiology in most mammalian tissues [38]. However, reduced function of lactate dehydrogenase A (LDHA) results in NAD+ collapse, ATP depletion, oxidative stress and necrosis during hypoxia [39].
Dissipative byproducts, some of which are also metabolic stress signals, are generated in proportion to ATP turnover, such as protons (H+), intracellular phosphate (Pi), and adenosine diphosphate (ADP) [38]. Calcium (Ca2+) cycling coordinates cellular work [40,41]. Reactive oxygen species (ROS) production is related to flux depending on the mitochondrial membrane potential (∆ψm) and redox status [42].
With low Michaelis-Menten constants (Km), ATPases are mostly not substrate-limited under physiological conditions [43] where Na⁺/K⁺ ATPase has a turnover rate of ~50 ATP per second [44]. Although ATP synthase can work more rapidly, it may run considerably below capacity at physiological ATP/ADP concentrations [45,46,47].
The PCr–creatine kinase (CK) buffer helps maintain ATP and ADP concentrations steady despite rapid turnover [43]. ATP is therefore not a resource to draw down but a flow to stabilize. Organ functions depend on continuous ATPase-coupled cellular work, constrained by the timescale of ATP turnover. In supply-constrained critical illness, how long could organs continue their pre-illness work rates?
Thermodynamic and Biochemical Considerations
At steady state, ATP hydrolysis and ATP resynthesis must be mass-balanced across finite adenine nucleotide and PCr pools; sustained mismatch necessarily changes ATP, ADP, Pi, phosphocreatine and therefore ΔG [38]. In acute organ failure, ATP supply becomes constraining while ATP costs increase, producing a thermodynamic bottleneck (Figure 1) [4]. Bioenergetic failure does not fully explain how ∆G for ATPases is adequately stabilized in the viable range over hours to days (indicated by survival), as membrane leaks, injury and repair raise the free-energy cost of maintaining biological order [48].
From the second law, a reaction proceeds spontaneously only when ∆G is negative [38]; the magnitude of ∆G for ATP hydrolysis therefore determines which ATPase-driven processes remain thermodynamically permissible (not considering load). This could make ∆G, determined by ATP, ADP, Pi, H+, an ordering variable for ATPases if their intrinsic ∆G tolerances differ. Kinetic regulation could also provide an ordering of ATPase activity. H+ and Pi are both enzyme-kinetic regulators and contributors to the effective ∆G of ATP hydrolysis [38].
Time Scale and Controllers
Theories of acute organ failure imply loss of control as central to pathology [23]. Cells have only limited control over energy supply but near-immediate control over metabolic flux. ATP turnover operates on a much more rapid timescale than critical perturbation develops (minutes, hours for hypoxia and hypoperfusion).
According to control theory, a controller should operate at a bandwidth that matches or exceeds the bandwidth of the process it controls [49,50]. High controller bandwidth relative to the disturbance can provide robust control, but introduces fragilities elsewhere [49,51]. Cytokine responses act over minutes and hours, as does transcriptional iNOS [18], and thus cannot provide direct feedback control over ATP turnover.
Processes coupled with ATP turnover that operate on the same time scale are implicated in causing reduced organ function. In control-theoretic terms, Pi, ADP and ΔG provide negative feedback to ATPases, as may H⁺, although H⁺ generated during the glycolytic shift may also act as a feedforward-like constraint on ATPase-coupled work during metabolic stress [36,38,52,53,54]. ROS can inhibit ATP production and provide feedforward redox inhibition of ATPases, whereas Ca2+ cycling suppression and NO provide feedforward-like constraints on production and consumption [38,41,42]. Buffering (PCr-CK) [43] makes ATP and ADP likely later regulators with AMP amplified early [38]. This control-theoretic classification is not intended to describe the full complexity of the ATP/ADP moiety cycle. This simplified overview is presented in Supplementary Table 1.
Cells contain multiple pathways producing a vast failure mode variety upon perturbation. A regulator must have the requisite variety to absorb the failure modes to be effective [55]. Here, failure modes refer broadly to the number of accessible failure configurations and trajectories in essential variables relevant to viability. These include but are not limited to pH, membrane integrity, ion pumps, intact organelles, membrane potentials, including mitochondrial (∆ψm), genome integrity, redox, proteostasis and ∆G. The requisite response variety does not have to be equal in number to failure modes but must cover the relevant variance and is linked to biological robustness [56].
Viability theory asks which states still leave a system able to remain within life-compatible limits [57]. A common clinical assumption in acute organ failure is that preserving organ function preserves viability.
Selected paradoxes of acute organ failure are presented inTable 1.
Cellular Work as the Survival Variable
Cytokines inhibit several specialized organ functions [58,59,60,61]. Activation of AMP-activated protein kinase (AMPK) and the integrated stress response (ISR) lowers energy demand [25,62]. Adenosine inhibits synaptic transmission [63], and reduces glomerular filtration [64]. Triiodothyronine (T3) stimulates oxidative phosphorylation (OXPHOS), increases Na+/K+ ATPase activity and protein turnover [65]. Cytokines inhibit the conversion of thyroxine (T4) to T3 in critical illness [66]. Mild acidosis conserves energy in cardiomyocytes [67]. The adrenergic system provides rapid, high-temporal control of vascular tone, centralizing perfusion to vital organs and counter-regulating the vasodilation produced by inflammatory activation [68]. Multiple stress mediators inhibit cellular work.
Cellular work emerges from 107-108 ATP hydrolysis events per second in mammalian cells [69]. Lower ATP, reduced Ca2+ cycling, increased ADP, Pi, H+ and ROS, or altered Ca2+ availability can provide a less permissive environment for ATPases through kinetic regulation or ΔG constraints [38,43,70]. Glycolysis-derived H+ [52,71] and stress-induced Pi inhibit ATPases kinetically [53,72] with ROS inhibiting ion pumps and NO having context-dependent actions [40,54,73]. ROS generation following OXPHOS inhibition is complex. Inhibition only reduces ROS if the ∆ψm is also lowered. A slow ETC flux with high ∆ψm promotes ROS formation [74]. Mitochondrial suppression can limit cellular metabolic needs during hypoxia and hypoperfusion, but in modern critical care, patients often have cytopathic hypoxia alongside hyperglycemia [75]. There is thus debate on whether this is more adaptive than alternatives, over-adaptation or simply harmful [4]. Either way, the consequences for ATP demand must be tolerated.
Energy First
Control Inversion of Cellular Work
Prigogine showed that cells maintain rapid metabolic flux far from equilibrium [76]. Under physiological conditions, ADP is a major determinant of OXPHOS and glycolytic rates [38]. Low ADP is maintained by the respiratory chain operating mostly near equilibrium, with small ∆G driving its net flux. Here, the ATPase work rate partly determines ATP synthesis, with ATP and PCr pools buffered against demand [38], supported by metabolic autoregulation [77]. A highly negative ∆G and supportive enzyme kinetics form a permissive environment for ATPases [78].
Control of respiratory flux is distributed between ATP producing and consuming processes [79]. Under local OXPHOS inhibition, ATP synthesis falls below the rate of ATP hydrolysis, allowing inhibitory signals to rise and constrain ATPases [38]. If ATP/ADP deterioration continues, the ΔG of ATP hydrolysis can become less negative, first affecting ATPases that require a highly negative ΔG [38]. The respiratory chain now operates with a larger driving force, accompanied by a rise in ADP [80].
From a control-theory perspective, a regulatory process that is much faster than the disturbance is expected to produce only minor deviations in the regulated variables [49,50]. In the initial phase of ischemia, the disturbance, reduced oxygen delivery and perfusion, often develops on a timescale slower than ATP turnover, PCr-CK buffering, and local metabolic signaling. The relative preservation of ATP, supported by PCr buffering, and ultimately ΔG therefore suggests that these energetic variables are regulated under constraint [50,51]. Metabolic stress signals that inhibit ATPases to reduce ATP demand would be expected to show larger deviations than the regulated energetic variables [50,81]. Relative preservation of ATP and ΔG despite early and marked reductions in functional output is consistent with regulated suppression of ATP hydrolysis.
In contracting muscle, dominant control inverts from demand to supply-side as workload increases [38,82]. Considering supply and demand blocks of metabolism, it has been shown that when flux is controlled by one block, the other block stabilizes the linking metabolites [83], such as ATP, ADP and Pi. Conceptually, with supply-side constraint, energy is no longer in service of functions; functions are in service of energy, an “energy-first” regime. Hence, the lower ATP concentrations found in muscle in severe sepsis [24] could reflect a regulated constrained state rather than a pure depletion marker.
Cellular Injuries and Energy Costs
Hours after immune activation in sepsis, immune cells transmigrate to tissues, where NADPH oxidases (NOX) produce ROS for pathogen clearance [84]. ROS and NO damage cell membranes; the resulting leaks increase ATP costs [85] on top of immune activation, antioxidant defense, autophagy, and macromolecule repair [86,87].
ATPase Failure Hierarchy
An ATP consumption hierarchy has been proposed in thymocytes. Macromolecular biosynthesis was most sensitive to supply limitation, followed by Na+ cycling, Ca2+ cycling and mitochondrial proton leak [88,89]. By contrast, gradient-maintaining ATPases, including Na+/K+ ATPase, sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA), plasma membrane Ca2+ ATPase (PMCA)) can operate over a wide range of ∆G and pH [52,90]. Synaptic transmission fails before membranes in ischemia [91], and contractile function is also highly sensitive [92]. In neurons, vacuolar type H+ ATPase (V-ATPase) has a high ATP consumption and contributes to nerve terminal intolerance to low-fuel conditions [93]. Myosin is sensitive to H+ and Pi [54].
From Energy Constraints to Ordered Failure
ATPases as Sensor-Effectors
Under physiological conditions, ATPase-coupled processes are regulated by multiple signaling pathways and transcriptional programs, while bioenergetic stress introduces direct inhibitory and thermodynamic constraints. The total ATPase flux represents the sum of ATP hydrolysis across ATPase-coupled processes. Uniform ATPase sensitivity to feedback inhibition would create a tipping point at which failure mode variety increases sharply as response capacity becomes constrained. A statistical null model following a random distribution of ATPase inhibitory sensitivity would tend to affect essential and non-essential functions indiscriminately. Affecting one of several critical cell functions early will sharply increase failure variety. Because ∆G supports all ATPase-coupled processes concurrently, its maintenance has high priority [25,82,94]. It is proposed that survival from bioenergetic crisis results from a combined product of robust ATPases whose functions persist and ATPases whose inhibition stabilizes ∆G.
Conceptually, each ATPase samples the local inhibitory conditions (kinetic regulators and ∆G) and loads as it channels free energy into work [95]. If local conditions are favorable, ATP is irreversibly hydrolyzed. ATP-consuming enzymes produce an integral output reflecting load, intrinsic properties and local milieu of regulators (kinetic and ∆G). Local ATP-hydrolysis products, including H⁺, Pi and ADP, decrease with ATPase inhibition. ATP-utilizing enzymes are both the coupling gates through which ATP-derived free energy enters work, and the generators of ADP and Pi that drive ATP resynthesis. Their cycling therefore gives ATP-utilizing enzymes substantial influence over ATP turnover.
Inhibitory Sensitivity of ATP Consuming Enzyme Types
ATPase-mediated work is distributed across many subcellular locations and processes, but each ATPase has only one observable work state in its local milieu. Under bioenergetic constraint, multiple inhibitory inputs converge on this state. The inhibitory sensitivity of all ATPases is unknown and although differences have been reviewed above, it is treated here as a theoretical, type-specific property. As many ATPase types occur in large copy numbers local variability may self-average according to their intrinsic inhibitory sensitivity [96]. This could make the mean functional inhibition of each type reflect shared structural sensitivity more than local noise. However, synchronization of microdomains during critical stress could reduce self-averaging [97]. Differential type-specific ATPase-inhibition to metabolic stress signals could therefore partition ATP moiety cycles and potentially route ATP hydrolysis away from selected work processes based on their self-averaged mean inhibitory sensitivity.
Type-specific ATPase Sensitivity and Leverage
ATPase types will also differ in leverage, defined here as the fall in total ATP consumption produced by inhibiting a given ATPase type. This is conceptually different from flux control coefficients [98] as the regulated variable is moiety cycles [99]. High-sensitivity/high-leverage ATPase types are well-suited for rapidly stabilizing a falling ΔG, whereas low-sensitivity ATPase types support functional persistence. Broad inhibitory range may be important, allowing inhibition to occur from mild to severe bioenergetic stress, such as for Na+/K+-ATPase [52,90]. If robust ATPases still respond to load, they can increase flux.
PCr buffers ATP and ADP while generating Pi, potentially inhibiting myosin and SERCA, which could slow Ca2+ cycling [38] and indirectly the maintenance of membrane potential, an example of potential process-coupled leverage. Shared nodes among pathways may form a hierarchy among cellular processes [100]. Because Na⁺/K⁺ ATPase and SERCA activity integrate multiple processes, the leverage from inhibition is an emergent property of the process network, a likely occurrence with many ATPase types [101,102].
Highly correlated sensitivity and leverage between ATPase types, positive or negative, would either decrease ATP conservation or functional persistence capacities, thus a partly uncoupled relationship under physiological conditions is proposed, as conceptually illustrated in Figure 2. The actual ATPase type univariate or bivariate distributions are undetermined. However, increasing inhibition will skew the distribution towards less sensitive ATPases.
The ATPase Ensemble as a Response Layer
Broadcast signals can produce ordered population responses when receivers have distributed response thresholds. This is observed in motor-neuron recruitment (Henneman’s size principle) [103], morphogenesis [104] and differential sensitivity of proteins to pH [105]. ATPases are already spatially distributed in cells, processes and networks, coupled to different functions. This makes differential inhibitory sensitivity to metabolic stress signals a plausible mechanism for organizing a complex response to bioenergetic constraint. This would provide a simple implementation of rapid ATP hydrolysis regulation, because the response could emerge from intrinsic between-enzyme differences. The ordering mechanism in vivo may differ; the key point is that ordering itself diversifies the response by enzyme and temporal sequence. The inhibitory control proposed here is not primarily directed at metabolite flux through pathways. Rather, it concerns the fraction of ATP hydrolysis permitted through different ATPase types, allowing a few metabolic stress signals to partition ATPase-coupled work. Coupling of processes through a shared ATP/ADP pool is supported by metabolic control theory of moiety-conserved cycles [106].
Yield Points and Unserved Workloads
The existence of energy reallocation coupled to ordered functional failure is consistent with early functional loss despite relative preservation of ΔG and PCr during hypoxia, hypoperfusion and ischemia [34,35,37]. High-sensitivity ATPases whose inhibited functions are tolerated represent dynamic functional yield points (DFYP): new yield points emerge in the ensemble as a dynamic process of inhibition.
Inhibiting ATPases leaves unserved workloads, which must be tolerated without critical or cascading errors. Cellular viability therefore requires fault tolerance to reduced activity in several ATPase-coupled processes. Well-suited inhibitory targets are processes whose inhibition also reduces downstream loads on other ATPases and cellular processes, producing cascading load relief. Contractile activity, synaptic activity, hepatic protein synthesis and proximal tubule reabsorption are examples where inhibition can reduce ion traffic and thereby lower ion-pump workload [38]. DFYP are of benefit if the accompanying loss of function poses a lower threat to viability than the bioenergetic crisis it resolves. With progressing failure, residual ATP hydrolysis may be routed through fewer and less fault-tolerant functions.
Costs of Higher Inhibitory Thresholds
After significant energy reallocation, the remaining ATPase distribution will be skewed toward less sensitive types, reducing the capacity for further reallocation. Accumulating ATPase-inhibitory signals can become dangerous, promoting Ca2+ triggered mitochondrial membrane permeability transition [41], acidosis [38] and oxidative damage [107]. Ordered inhibition therefore has an intrinsic cost. Further suppression must act on less sensitive ATPase layers, requiring stronger inhibitory signals.
High Work Microdomains
Mitochondria are clustered in high-work regions of neurons, renal proximal tubular cells, cardiomyocytes, and hepatocytes [108] where ATPase loads are high. Microdomains show substantial local variation in Ca2+, H+, and ROS [70,109,110,111,112,113] which could increase the probability of reaching inhibitory thresholds in local ATPases. The PCr-CK kinase system [38] allows local stabilization of ATP and ADP. However, Pi remains locally available, potentially inhibiting ATPases and making ∆G less favorable [38,87]. During stress, NO can limit the ceiling of ATP production while promoting ATPase inhibitory signals [114]. Local variance can promote threshold crossing, whereas self-averaging promotes low cell-wide variance in ∆G and kinetic regulators [115].
Transcriptional Programs and the Cost of Adaptation
Cellular Stress Response Pathways
AMPK and ISR produce lasting kinetic and transcriptional responses that stabilize ATP supply and demand after perturbation [117,118]. Both are high-gain pathways with significant leverage over ATP demand, inhibiting protein synthesis [87], where the unserved workload is compensated temporarily by the existing proteome [119].
AMPK is activated within seconds by increases in AMP relative to ATP, reducing cellular work by inhibiting ATP-consuming processes by rapid kinetic regulation [120]. AMPK also activates slow-working transcriptional programs that increase substrate supply, mitochondrial function and NAD⁺ salvage over time [120]. Tissue-specific or pharmacological inhibition worsens inflammation, cell death, organ function and outcomes in sepsis models [121], whereas AMPK activation promotes survival [122,123]. Together with hypoxia-inducible factor alpha (HIF-1α), AMPK reduces cellular dependence on O2 by inducing glycolysis [120,124].
The ISR is activated by Ca²⁺, ROS, endoplasmic reticulum (ER) stress and nutrient deficiency, rapidly suppressing protein translation and initiating autophagy [125]. Several knockout (KO) models are lethal. KO of related factors and tissue-specific deletions of eukaryotic initiation factor 2α (eIF2α) show increased tissue injury, ER stress, ROS, mitochondrial injury and dysregulated cytokine responses in sepsis. Cell injury and dysfunction occur across organs with higher mortality [125,126].
The transcriptional adaptive programs of AMPK, ISR and HIF-1α activation seem not to have the regulatory bandwidth to regulate flux directly [49,50] but mitigate upcoming risk.
Allostasis: a Language for Describing Acute Organ Failure
Allostasis describes stability through changing physiological setpoints during stress [127]. Rather than preserving fixed internal conditions, allostatic responses prioritize survival by reallocating resources. The cost of sustaining such adaptive states is referred to as allostatic load (or overload when pathological), originally developed for chronic disease [127,128]. Inhibition of protein synthesis together with activation of autophagy contracts the cellular proteome over time [119], delaying recovery due to hysteresis. Several allostatic costs occur more rapidly in acute organ failure, most significantly reductions in specialized organ functions. The same processes that reduce ATP demand progressively reduce the system’s capacity to sustain that reduction.
A New Perspective on Acute Organ Failure
Acute organ failure may be understood as the visible consequence of adjustment to a deeper bioenergetic crisis unfolding on a rapid timescale. When ΔG becomes sufficiently constrained, local microdomains may become coupled through a shared energetic limitation, increasing failure-mode variety sharply, as suggested by interdependent network models [129]. To defend ΔG, between-enzyme differences in ATPase inhibitory sensitivity are proposed to route ATP hydrolysis away from non-critical work and toward viability-critical processes. This could provide the kinetic priority and control bandwidth needed to meet bioenergetic failure on the timescale of ATP turnover. Further, it could reduce failure-mode variety at two levels: globally, by stabilizing ΔG across ATPase-coupled processes, and functionally, by routing failure toward more fault-tolerant outputs. In this way, the ATPase ensemble could provide the requisite variety to defend vital cellular functions. It is thus proposed that there may be a parallel control layer that regulates ATP hydrolysis under metabolic stress rather than the metabolism of compounds. Yielding may therefore regulate where failure enters the system, lowering failure-mode variety. A rapid control system for ∆G by ordered ATPase-mediated inhibition of ATP hydrolysis can potentially adapt to more slow-evolving perturbations producing bioenergetic constraints by adjusting functional output [49,50].
If present, ordered ATPase inhibition has favorable kinetic and spatial properties: it is broadly distributed, immediately available and activated at the point of consumption. Redox sensitivity of Na+/K+ ATPase varies by isoform [130] and conserved motifs for S-nitrosylation have been identified in proteins [131]. ATPases involved in specialized organ functions can be redox sensitive [132,133], although no broad mapping has been performed. It is hypothesized that selection may have acted on bioenergetic tolerance, with between-enzyme differences in ATPase inhibitory sensitivity serving as a molecular substrate for routing suppression of ATP hydrolysis toward fault-tolerant processes. ATPases are distributed across cells and organs [87]. Because metabolic stress signals are decoded within individual cells, a coherent multicellular pattern of acute organ failure would require repeated sensor-effector rules with sufficiently conserved ATPase responses across cells [134,135]. Notably, ion pumps have an additional role beyond maintaining critical gradients. Because their load is continuously imposed, they also maintain ADP production [38].
Such a system would also have costs. As inhibition progresses into less sensitive ATPase layers, stronger inhibitory signals are required, increasing the risk of Ca2+ toxicity, acidosis and oxidative injury. Organ functions could fall below viable thresholds. NO and ROS can induce nitrosylation and oxidative changes that may be brief, last minutes to hours during redox stress, or last longer with post-translational modifications [107,136]. Transcriptional responses can last much longer [87]. Hence, the current output from the ATPase ensemble could also reflect prior metabolic stress [87,137]. This could contribute to hysteresis with delayed functional recovery in acute organ failure [7], together with mitochondrial suppression [138]. This raises the possibility that cytopathic hypoxia could reflect not only mitochondrial suppression [139], but reduced ADP availability from ATPase suppression. If present, this allostatic overload would resemble hypoxia-tolerance, a response in other species, linked to a regulated reduction in ATPase work [25].
Work-Reduction Tolerance
A robust system must maintain robustness capital: for organs, this may mean controllability below baseline function, not merely unused functional reserve [51]. When an individual cell reallocates energy, some unserved work can have immediate consequences. However, the contribution of a single cell to overall organ function is usually negligible. Collective organ work may tolerate certain unserved workloads better than individual cells, depending on redundancy and functional reserve. Reducing input to specialized organ functions can cascade load relief to ion cycling, secretion and transport [87]. Hence, there is an asymmetric benefit for cells to export unserved work to redundant specialized organ functions when those functions have residual capacity and fault-tolerance. This also allows cell-level temporal buffering through organ functions, as cells can re-enter specialized work once internal conditions improve, for example after activating transcriptional programs.
Each organ is proposed to have work-reduction tolerance: a residual below-baseline capacity for unserved workloads during energy reallocation. This reserve may be mobilized to stabilize a supply-demand bioenergetic crisis with limited cell death, at the cost of collective function, even under shared constraints. Outside ischemia, the brain often survives critical illness despite limited metabolic reserves [31]. However, it may have a large reserve in work-reduction tolerance with respect to vital functions that could be recruited during delirium and metabolic coma [16]. Reversible dysfunction rather than cell death is observed after sublethal ischemia in myocardial stunning and hibernating myocardium [140].
The Reversibility Paradox and Irreversibility Thresholds
Reduced function stabilizes a bioenergetic crisis but at the same time the consumption of functional margins can be as lethal as the underlying illness. The thresholds at which reversibility is lost remain to be defined empirically. Relevant candidates are mostly in a causal chain. These include critically low ∆G, loss of ∆ψm, NAD+ depletion [139], inability to export dissipation products, ROS bursts [42], failing ion gradients or organ function [85].
Other Frameworks of Acute Organ Failure
Singer (2004, 2014) proposed an adaptive hibernation-like state involving NO and the sick-euthyroid response [4,26]. Contemporary models increasingly recognize adaptive metabolic suppression and disease tolerance [141]. This framework proposes how tolerance to bioenergetic failure could be organized during constraint. Uncompensated supply-demand mismatch is likely followed by cell death, with histopathological consequences. Necrosis takes hours to become histologically visible [85] and is a frequent cause of cell-death following uncontrolled energy collapse [142]. Cellular injuries can concurrently cause reversible dysfunction, but such cells must retain sufficient reserve to bear the energetic costs of injury and ongoing repair.
Table 2lists predictions and falsifiers from this framework.
Future Perspectives
This demand-side perspective does not distinguish whether mitochondrial suppression in internal organs is adaptive or maladaptive [143]. Pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) are detected by pattern recognition receptors (PRRs), molecular sensors of infection and tissue injury that are active in sepsis, trauma and post-cardiac arrest syndrome [144]. These sensors may provide early-warning systems. High efficiency, high-performance systems are typically hypersensitive to unexpected perturbations [145]. Cytokines act slowly relative to ATP turnover, but can induce NO, ROS and transcriptional programs that alter the ATPase milieu before critical bioenergetic constraint occurs from supply deficiency [119,125,146,147,148]. At the same time, cytokines can trigger hypoperfusion. Slow-acting cytokines can induce features of the acute organ failure phenotype in experimental models [149,150]. Not all failure pathways will be viable [57]. Failure trajectories dominated by OXPHOS inhibition may be more reversible than trajectories dominated by structural damage during thermodynamic bottlenecks. Distinguishing supply-side from demand-side behavior during acute organ failure may help separate triggers and constraints from adaptive responses, allostatic loads and functional output.
Current treatments that alleviate supply-side constraint may reduce the need for ATPase-mediated workload suppression and thereby preserve organ function. Such interventions are likely to be most effective early, before sustained ATPase inhibition, transcriptional adaptation and tissue-level hysteresis have developed. Procedures such as cardiac unloading may be cytoprotective in severe acute heart failure when they reduce ATPase-coupled cellular work. Demand-side biology may therefore define a future therapeutic field, although intervention will require a clearer understanding of which workload reductions are protective and which are harmful.
Strengths and Limitations
This perspective focuses on demand-side constraints during acute organ failure. It should first be evaluated by whether kinetics, thermodynamics, control theory and requisite variety identify constraints that cells and organs must satisfy for acute organ failure to remain reversible. The clinical course strongly supports preservation of cell viability, with high rates of reversibility [4]. The ordered classification of inhibitory sensitivity used here is theoretical and must be assessed empirically. It is currently supported by a limited number of studies. Combinatorial effects will add much complexity to individual ATPase inhibition. Overall, biological plausibility could be incomplete. The framework produces testable hypotheses. The proposals here cannot be used for treatment decisions. Respiration, like the immune system, increases work during critical illness and has not been addressed. Post-cardiac arrest syndrome, sepsis and MODS are the primary clinical considerations but, as the mechanisms are broad, other types of acute organ failure may be relevant. The framework does not apply when massive cell death causes dysfunction.
Conclusions
This Perspective separates supply-side bioenergetic constraint from demand-side workload adjustment in acute organ failure. It identifies constraints that limit the possible demand-side responses to bioenergetic failure. Among cellular systems, ATPase-coupled work is uniquely positioned to respond on the timescale of ATP turnover, limit supply-demand mismatch, and provide sufficient response variety to organize failure while defending bioenergetic stability during perturbations. Further, during perturbations, the ATPase system cannot remain free to set demand; it must be constrained to avoid supply-demand mismatch. The specific biological implementation cannot be inferred from these constraints beyond directionality and convergence on a differentiated response architecture for ATPase-coupled work. It is proposed that type-specific differences in ATPase sensitivity to simple metabolic stress signals could provide the requisite variety needed to regulate ATP hydrolysis acutely. Unserved workloads may be dangerous when they affect cell-autonomous survival functions, but tolerable or even protective when they reduce downstream loads on basic processes such as ion pumping. Specialized organ functions, in contrast, are distributed across many cells and often operate above the minimum required for survival. This asymmetry could make it advantageous for cells to export unserved workloads to collective organ functions rather than absorb them within viability-critical cellular processes. Where lower organ function remains tolerable, preserving cell viability may be the most effective way to preserve organ function over time. Taken together, this proposed response could explain the reversibility paradox: marked organ dysfunction, limited cell death and the potential for full recovery observed in acute and multiorgan failure. In this framework, acute organ dysfunction may be understood as the allostatic cost of bioenergetic constraint: organ function is adjusted to defend cellular viability. An imperfect face of survival.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
The author thanks Jan Erik Nordrehaug for patient mentoring in working with ideas, concepts, and scientific writing. The research environment at Haraldsplass Deaconess Hospital led by Petter Thornam has been immensely important, as has reflections formed during clinical duties at the highly professional Medical Intensive Care Unit at Haukeland University Hospital. Thank you to Håvard Melvær for detailed discussions on control-theory.
References
- Forni, L.G.; Darmon, M.; Ostermann, M.; Straaten, H.M.O.-V.; Pettilä, V.; Prowle, J.R.; Schetz, M.; Joannidis, M. Renal recovery after acute kidney injury. Intensiv. Care Med. 2017, 43, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Guirgis, F.W.; Khadpe, J.D.; Kuntz, G.M.; Wears, R.L.; Kalynych, C.J.; Jones, A.E. Persistent organ dysfunction after severe sepsis: A systematic review. J. Crit. Care 2014, 29, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Ulvik, A.; Kvåle, R.; Wentzel-Larsen, T.; Flaatten, H. Multiple organ failure after trauma affects even long-term survival and functional status. Crit. Care 2007, 11, R95–R95. [Google Scholar] [CrossRef] [PubMed]
- Singer, M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 2013, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, R.E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of Cardiac and Renal Dysfunction in Patients Dying of Sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, A.M.; Lorente-Ros, M.; Goncalvez, G.; Carriedo, D.; Ballén-Barragán, A.; Villar-Fernández, A.; Peñuelas, Ó.; Herrero, R.; Granados-Carreño, R.; Lorente, J.A. Histopathological changes of organ dysfunction in sepsis. Intensiv. Care Med. Exp. 2019, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.M.; Shelhamer, J.H.; Bacharach, S.L.; Green, M.V.; Natanson, C.; Frederick, T.M.; Damske, B.A.; Parrillo, J.E. Profound but Reversible Myocardial Depression in Patients with Septic Shock. Ann. Intern. Med. 1984, 100, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute Renal Failure in Critically Ill PatientsA Multinational, Multicenter Study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; Bassegoda, O.; Toapanta, D.; Bernal, W. Acute liver failure: A practical update. JHEP Rep. 2024, 6, 101131. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Cavarocchi, N.; Hirose, H. Management of cardiac standstill on veno-arterial extracorporeal membrane oxygenation using a high flow strategy. AME Med. J. 2018, 3, 110–110. [Google Scholar] [CrossRef]
- Neumar, R.W.; Nolan, J.P.; Adrie, C.; Aibiki, M.; Berg, R.A.; Böttiger, B.W.; Callaway, C.; Clark, R.S.B.; Geocadin, R.G.; Jauch, E.C.; et al. Post-cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation 2008, 118, 2452–2483. [Google Scholar] [CrossRef] [PubMed]
- Kiely, D.K.; Marcantonio, E.R.; Inouye, S.K.; Shaffer, M.L.; Bergmann, M.A.; Yang, F.M.; Fearing, M.A.; Jones, R.N. Persistent Delirium Predicts Greater Mortality. J. Am. Geriatr. Soc. 2008, 57, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Textoris, J.; Fouché, L.; Wiramus, S.; Antonini, F.; Tho, S.; Martin, C.; Leone, M. High central venous oxygen saturation in the latter stages of septic shock is associated with increased mortality. Crit. Care 2011, 15, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rivers, E.P.; Rady, M.Y.; Martin, G.B.; Fenn, N.M.; Smithline, H.A.; Alexander, M.E.; Nowak, R.M. Venous Hyperoxia after Cardiac Arrest. Chest 1992, 102, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Cairns, C.B.; Moore, F.A.; Haenel, J.B.R.; Gallea, B.L.; Ortner, J.P.B.; Rose, S.J.B.; Moore, E.E. Evidence for Early Supply Independent Mitochondrial Dysfunction in Patients Developing Multiple Organ Failure after Trauma. J. Trauma Inj. Infect. Crit. Care 1997, 42, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Nitchingham, A.; Pereira, J.V.; Wegner, E.A.; Oxenham, V.; Close, J.; Caplan, G.A. Regional cerebral hypometabolism on 18F-FDG PET/CT scan in delirium is independent of acute illness and dementia. Alzheimer's Dement. 2022, 19, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Vella, R.; Panci, D.; Carini, F.; Malta, G.; Vieni, S.; David, S.; Albano, G.D.; Puntarello, M.; Zerbo, S.; Argo, A. Cytokines in sepsis: a critical review of the literature on systemic inflammation and multiple organ dysfunction. Front. Immunol. 2025, 16, 1682306. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D.; Moncada, S. Nitric Oxide and Mitochondrial Signaling: From physiology to pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2524–2531. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 2013, 13, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Ramirez, J.; Koutrouvelis, A. The nitric oxide pathway antagonists in septic shock: Meta-analysis of controlled clinical trials. J. Crit. Care 2019, 51, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Selye, H. Stress and Disease. Science 1955, 122, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Grootendorst, A.F. Hemodynamic aspects of multiple organ failure. Intensiv. Care Med. 1990, 16, S165–S167. [Google Scholar] [CrossRef] [PubMed]
- Jarczak, D.; Kluge, S.; Nierhaus, A. Sepsis—Pathophysiology and Therapeutic Concepts. Front. Med. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Hochachka, P.W.; Buck, L.T.; Doll, C.J.; Land, S.C. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc. Natl. Acad. Sci. 1996, 93, 9493–9498. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; De Santis, V.; Vitale, D.; Jeffcoate, W. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet 2004, 364, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Jun, J.C. Does Hypoxia Decrease the Metabolic Rate? Front. Endocrinol. 2018, 9, 668. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, Z.A.; Dulac, C. Sickness and the brain. Curr. Biol. 2025, 35, R973–R979. [Google Scholar] [CrossRef] [PubMed]
- Omar, Y.G.; Massey, M.; Andersen, L.W.; Giberson, T.A.; Berg, K.; Cocchi, M.N.; Shapiro, N.I.; Donnino, M.W. Sublingual microcirculation is impaired in post-cardiac arrest patients. Resuscitation 2013, 84, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, M.R.; Tian, R.; Arany, Z. Human cardiac metabolism. Cell Metab. 2024, 36, 1456–1481. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-H.; Qiao, H.; Du, F.; Xiong, Q.; Liu, X.; Zhang, X.; Ugurbil, K.; Chen, W. Quantitative imaging of energy expenditure in human brain. NeuroImage 2012, 60, 2107–2117. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Heiden, M.G.V. Understanding Metabolic Regulation and Its Influence on Cell Physiology. Mol. Cell 2013, 49, 388–398. [Google Scholar] [CrossRef] [PubMed]
- van Putten, M.J.A.M.; Hofmeijer, J. EEG Monitoring in Cerebral Ischemia. J. Clin. Neurophysiol. 2016, 33, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.C.; Smith, G.L.; A Eisner, D.; Allen, D.G. Metabolic changes during ischaemia and their role in contractile failure in isolated ferret hearts. J. Physiol. 1992, 454, 467–490. [Google Scholar] [CrossRef] [PubMed]
- Saupe, K.W.; Eberli, F.R.; Ingwall, J.S.; Apstein, C.S.; Stecyk, J.A.W.; Bock, C.; Overgaard, J.; Wang, T.; Farrell, A.P.; Pörtner, H.-O.; et al. Hypoperfusion-induced contractile failure does not require changes in cardiac energetics. Am. J. Physiol. Circ. Physiol. 1999, 276, H1715–H1723. [Google Scholar] [CrossRef] [PubMed]
- Kammermeier, H.; Schmidt, P.; Jüngling, E. Free energy change of ATP-hydrolysis: a causal factor of early hypoxic failure of the myocardium? J. Mol. Cell. Cardiol. 1982, 14, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Fedorovich, S.; Hofmeijer, J.; van Putten, M.J.A.M.; le Feber, J. Reduced Synaptic Vesicle Recycling during Hypoxia in Cultured Cortical Neurons. Front. Cell. Neurosci. 2017, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Ferguson, S.J. Bioenergetics; Academic press, 2013. [Google Scholar]
- Lin, Y.; Wang, Y.; Li, P.-F. Mutual regulation of lactate dehydrogenase and redox robustness. Front. Physiol. 2022, 13, 1038421. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Rokita, A.G.; Anderson, M.E.; Maier, L.S. Redox Regulation of Sodium and Calcium Handling. Antioxid. Redox Signal. 2013, 18, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Balaban, R.S. Role of Mitochondrial Ca2+ in the Regulation of Cellular Energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, Y.; Juhaszova, M.; Nuss, H.B.; Wang, S.; Zorov, D.B.; Lakatta, E.G.; Sollott, S.J. Matching ATP supply and demand in mammalian heart. Ann. N. Y. Acad. Sci. 2010, 1188, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Lüpfert, C.; Grell, E.; Pintschovius, V.; Apell, H.-J.; Cornelius, F.; Clarke, R.J. Rate Limitation of the Na+,K+-ATPase Pump Cycle. Biophys. J. 2001, 81, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.J.; Carey, M.F.; Kemper, W.F.; Conley, K.E. Control of glycolysis in contracting skeletal muscle. I. Turning it on. Am. J. Physiol. Metab. 2002, 282, E67–E73. [Google Scholar] [CrossRef] [PubMed]
- Walsh, B.; Howlett, R.A.; Stary, C.M.; Kindig, C.A.; Hogan, M.C. Determinants of Oxidative Phosphorylation Onset Kinetics in Isolated Myocytes. Med. Sci. Sports Exerc. 2005, 37, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Meyrat, A.; von Ballmoos, C. ATP synthesis at physiological nucleotide concentrations. Sci. Rep. 2019, 9, 3070. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Kay, J. Life as a manifestation of the second law of thermodynamics. Math. Comput. Model. 1994, 19, 25–48. [Google Scholar] [CrossRef]
- ström, K.J.; Murray, R. Feedback systems: an introduction for scientists and engineers; Princeton university press, 2008. [Google Scholar]
- Skogestad, S.; Postlethwaite, I. Multivariable feedback control: analysis and design; john Wiley & sons, 2005. [Google Scholar]
- El-Samad, H. Biological feedback control—Respect the loops. Cell Syst. 2021, 12, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, F.; Tsunekawa, N.; Toyoshima, C. Distinct pH dependencies of Na+/K+ selectivity at the two faces of Na,K-ATPase. J. Biol. Chem. 2018, 293, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Stehle, R. Phosphate rebinding induces force reversal via slow backward cycling of cross-bridges. Front. Physiol. 2025, 15, 1476876. [Google Scholar] [CrossRef] [PubMed]
- Woodward, M.; Debold, E.P. Acidosis and Phosphate Directly Reduce Myosin’s Force-Generating Capacity Through Distinct Molecular Mechanisms. Front. Physiol. 2018, 9, 862. [Google Scholar] [CrossRef] [PubMed]
- Ashby. [CrossRef] [PubMed]
- Kitano, H. Biological robustness. Nat. Rev. Genet. 2004, 5, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Aubin, J.-P.; Bayen, A.M.; Saint-Pierre, P. Viability theory: new directions; Springer Science & Business Media, 2011. [Google Scholar]
- Zipp, F.; Bittner, S.; Schafer, D.P. Cytokines as emerging regulators of central nervous system synapses. Immunity 2023, 56, 914–925. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D. Cytokine-Induced Modulation of Cardiac Function. Circ. Res. 2004, 95, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Morrell, E.D.; Kellum, J.A.; Hallows, K.R.; Pastor-Soler, N.M. Epithelial transport during septic acute kidney injury. Nephrol. Dial. Transplant. 2013, 29, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Moshage, H. Cytokines and the hepatic acute phase response. J. Pathol. 1997, 181, 257–266. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Physiol. 2011, 300, C385–C393. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-G.; Saggau, P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron 1994, 12, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Granger, J.P. Adenosine alters glomerular filtration control by angiotensin II. Am. J. Physiol. Physiol. 1986, 250, F917–F923. [Google Scholar] [CrossRef] [PubMed]
- Ismail-Beigi, F. Thyroid hormone regulation of Na,K-ATPase expression. Trends Endocrinol. Metab. 1993, 4, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Savvidis, C.; Ragia, D.; Kallistrou, E.; Kouroglou, E.; Tsiama, V.; Proikaki, S.; Belis, K.; Ilias, I. Critical illness-implications of non-thyroidal illness syndrome and thyroxine therapy. World J. Crit. Care Med. 2025, 14, 102577. [Google Scholar] [CrossRef] [PubMed]
- Koop, A. Protection of energy status of hypoxic cardiomyocytes by mild acidosis. J. Mol. Cell. Cardiol. 1992, 24, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Landry, D.W.; Oliver, J.A. The Pathogenesis of Vasodilatory Shock. New Engl. J. Med. 2001, 345, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Flamholz, A.; Phillips, R.; Milo, R. The quantified cell. Mol. Biol. Cell 2014, 25, 3497–3500. [Google Scholar] [CrossRef] [PubMed]
- Llinás, R.; Sugimori, M.; Silver, R. The concept of calcium concentration microdomains in synaptic transmission. Neuropharmacology 1995, 34, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Pluteanu, F.; Musset, B.; Rinne, A. Ca2+ Signaling in Striated Muscle Cells During Intracellular Acidosis. Biomolecules 2025, 15, 1244. [Google Scholar] [CrossRef] [PubMed]
- Guzman, N.J.; Fang, M.Z.; Tang, S.S.; Ingelfinger, J.R.; Garg, L.C. Autocrine inhibition of Na+/K(+)-ATPase by nitric oxide in mouse proximal tubule epithelial cells. J. Clin. Investig. 1995, 95, 2083–2088. [Google Scholar] [CrossRef] [PubMed]
- Figtree, G.A.; Karimi, G.K.; Liu, C.-C.; Rasmussen, H.H. Oxidative regulation of the Na+–K+ pump in the cardiovascular system. Free. Radic. Biol. Med. 2012, 53, 2263–2268. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.P. Cytopathic Hypoxia. Crit. Care Clin. 2001, 17, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Prigogine, I. Time, Structure, and Fluctuations. Science 1978, 201, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Segal, S.S. Regulation of Blood Flow in the Microcirculation. Microcirculation 2005, 12, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Williams, G. RESPIRATORY ENZYMES IN OXIDATIVE PHOSPHORYLATION. J. Biol. Chem. 1955, 217, 409–427. [Google Scholar] [CrossRef]
- Balaban, R.S.; Kantor, H.L.; Katz, L.A.; Briggs, R.W. Relation Between Work and Phosphate Metabolite in the in Vivo Paced Mammalian Heart. Science 1986, 232, 1121–1123. [Google Scholar] [CrossRef] [PubMed]
- Wikström, M.; Springett, R. Thermodynamic efficiency, reversibility, and degree of coupling in energy conservation by the mitochondrial respiratory chain. Commun. Biol. 2020, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Åström, K. Limitations on Control System Performance. Eur. J. Control. 2000, 6, 2–20. [Google Scholar] [CrossRef]
- Jeneson, J.A.L.; Westerhoff, H.V.; Kushmerick, M.J. A metabolic control analysis of kinetic controls in ATP free energy metabolism in contracting skeletal muscle. Am. J. Physiol. Physiol. 2000, 279, C813–C832. [Google Scholar] [CrossRef] [PubMed]
- Hofmeyr, J.-H.S.; Cornish-Bowden, A. Regulating the cellular economy of supply and demand. FEBS Lett. 2000, 476, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.N.; Nessel, I.; Puthucheary, Z.; Henson, S.M. Sepsis and the immunometabolic inflammatory response. npj Metab. Heal. Dis. 2026, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- A Hoda, S.; Hoda, R.S. Robbins and Cotran Pathologic Basis of Disease. Adv. Anat. Pathol. 2005, 12, 103. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Heald, R.; Johnson, A.; et al. Molecular Biology of the Cell (Seventh Edition); W. W. Norton, Incorporated, 2022. [Google Scholar]
- Wieser, W.; Krumschnabel, G. Hierarchies of ATP-consuming processes: direct compared with indirect measurements, and comparative aspects. Biochem. J. 2001, 355, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Brand, M.D. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995, 312 Pt 1, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Salonikidis, P.S.; Kirichenko, S.N.; Tatjanenko, L.V.; Schwarz, W.; Vasilets, L.A. Extracellular pH modulates kinetics of the Na +,K + -ATPase. Biochim. Et. Biophys. Acta (BBA) -Biomembr. 2000, 1509, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Hofmeijer, J.; van Putten, M.J. Ischemic Cerebral Damage. Stroke 2012, 43, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.W.; Chen, H.; Boyle, J.A.; Bamburg, J.R. Formation of actin-ADF/cofilin rods transiently retards decline of mitochondrial potential and ATP in stressed neurons. Am. J. Physiol. Physiol. 2006, 291, C828–C839. [Google Scholar] [CrossRef] [PubMed]
- Pulido, C.; Ryan, T.A. Synaptic vesicle pools are a major hidden resting metabolic burden of nerve terminals. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, D.E. Energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 1968, 7, 4030–4034. [Google Scholar] [CrossRef] [PubMed]
- Krupka, R. Channelling free energy into work in biological processes. Exp. Physiol. 1998, 83, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Van Kampen, N.G. Stochastic Processes in Physics and Chemistry, 3rd ed.; North Holland: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Aharony, A.; Harris, A.B. Absence of Self-Averaging and Universal Fluctuations in Random Systems near Critical Points. Phys. Rev. Lett. 1996, 77, 3700–3703. [Google Scholar] [CrossRef] [PubMed]
- Kacser, H.; Burns, J.A. The control of flux. Symp. Soc. Exp. Biol. 1973, 27, 65–104. [Google Scholar] [PubMed]
- Kholodenko, B.N.; Lyubarev, A.E.; Kurganov, B.I. Control of the metabolic flux in a system with high enzyme concentrations and moiety-conserved cycles. Eur. J. Biochem. 1992, 210, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.; Landau, E.M.; Iyengar, R. Signaling Networks. Cell 2000, 103, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, B.M.; Fontes, C.F.L.; Meyer-Fernandes, J.R. Molecular Basis of Na, K–ATPase Regulation of Diseases: Hormone and FXYD2 Interactions. Int. J. Mol. Sci. 2024, 25, 13398. [Google Scholar] [CrossRef] [PubMed]
- Gorski, P.A.; Ceholski, D.K.; Young, H.S. Structure-function relationship of the serca pump and its regulation by phosphol-amban and sarcolipin. In Membrane Dynamics and Calcium Signaling; Krebs, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 77–119. [Google Scholar] [CrossRef] [PubMed]
- Henneman, E. Relation between Size of Neurons and Their Susceptibility to Discharge. Science 1957, 126, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Wolpert, L. Positional information and the spatial pattern of cellular differentiation. J. Theor. Biol. 1969, 25, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Schönichen, A.; Webb, B.A.; Jacobson, M.P.; Barber, D.L. Considering Protonation as a Posttranslational Modification Regulating Protein Structure and Function. Annu. Rev. Biophys. 2013, 42, 289–314. [Google Scholar] [CrossRef] [PubMed]
- Hofmeyr, J.S.; Kacser, H.; van der Merwe, K.J. Metabolic control analysis of moiety-conserved cycles. Eur. J. Biochem. 1986, 155, 631–640. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Zuo, L. Redox Roles of Reactive Oxygen Species in Cardiovascular Diseases. Int. J. Mol. Sci. 2015, 16, 27770–27780. [Google Scholar] [CrossRef] [PubMed]
- L Mescher, A. Junqueira’s basic histology: text and atlas; 2013. [Google Scholar]
- López-Doménech, G.; Kittler, J.T. Mitochondrial regulation of local supply of energy in neurons. Curr. Opin. Neurobiol. 2023, 81, 102747. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Veksler, V.; Novotova, M.; Ventura-Clapier, R. Energetic Interactions Between Subcellular Organelles in Striated Muscles. Front. Cell Dev. Biol. 2020, 8, 581045. [Google Scholar] [CrossRef] [PubMed]
- Koide, T.; Giles, W.R.; Kondo, R.; Imaizumi, Y.; Yamamura, H.; Suzuki, Y. Ca2+ microdomain-based excitation-transcription coupling in cardiac myocytes and vascular smooth muscle cells. Inflamm. Regen. 2025, 45, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Deshwal, S.; Di Lisa, F. Reactive oxygen species and redox compartmentalization. Front. Physiol. 2014, 5, 285. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2009, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Borutaite, V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc. Res. 2007, 75, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, T.G. The Relationship between Stochastic and Deterministic Models for Chemical Reactions; CONFERENCE NAME, LOCATION OF CONFERENCE, COUNTRYDATE OF CONFERENCE; pp. 2976–2978.
- Pasqualini, F.S.; Nesmith, A.P.; Horton, R.E.; Sheehy, S.P.; Parker, K.K. Mechanotransduction and Metabolism in Cardiomyocyte Microdomains. BioMed Res. Int. 2016, 2016, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Hawley, S.A.; Scott, J.W. AMP-activated protein kinase – development of the energy sensor concept. J. Physiol. 2006, 574, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Anthony, T.G.; Staschke, K.A. Surviving and Adapting to Stress: Translational Control and the Integrated Stress Response. Antioxid. Redox Signal. 2023, 39, 351–373. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Shaw, R.J. AMPK: restoring metabolic homeostasis over space and time. Mol. Cell 2021, 81, 3677–3690. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Athea, Y.; Mounier, R.; Guigas, B.; Zarrinpashneh, E.; Horman, S.; Lantier, L.; Hebrard, S.; Devin-Leclerc, J.; Beauloye, C.; et al. AMPK: Lessons from transgenic and knockout animals. Front. Biosci. 2009, ume, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, K.; Zhu, S.; Xie, M.; Kang, R.; Cao, L.; Tang, D. AMPK regulates immunometabolism in sepsis. Brain Behav. Immun. 2018, 72, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Ma, Y.; Manrique-Caballero, C.L.; Li, H.; Emlet, D.R.; Li, S.; Baty, C.J.; Wen, X.; Kim-Campbell, N.; Frank, A.; et al. Activation of AMP-activated protein kinase during sepsis/inflammation improves survival by preserving cellular metabolic fitness. FASEB J. 2020, 34, 7036–7057. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, 384–+. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, X.; Rothermel, B.A.; Lavandero, S.; Wang, Z.V. The integrated stress response in ischemic diseases. Cell Death Differ. 2021, 29, 750–757. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Wingfield, J.C. The concept of allostasis in biology and biomedicine. Horm. Behav. 2003, 43, 2–15. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Stellar, E. Stress and the individual. Mechanisms leading to disease. Arch. Intern. Med. 1993, 153, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Buldyrev, S.V.; Parshani, R.; Paul, G.; Stanley, H.E.; Havlin, S. Catastrophic cascade of failures in interdependent networks. Nature 2010, 464, 1025–1028. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, A.; Petrushanko, I.Y.; Hernansanz-Agustín, P.; Martínez-Ruiz, A. “Oxygen Sensing” by Na,K-ATPase: These Miraculous Thiols. Front. Physiol. 2016, 7, 314. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Arif, A.; Terenzi, F.; Willard, B.; Plow, E.F.; Hazen, S.L.; Fox, P.L. Target-Selective Protein S-Nitrosylation by Sequence Motif Recognition. Cell 2014, 159, 623–634. [Google Scholar] [CrossRef]
- Steinberg, S.F. Oxidative Stress and Sarcomeric Proteins. Circ. Res. 2013, 112, 393–405. [Google Scholar] [CrossRef]
- Seidel, T.; Scholl, S.; Krebs, M.; Rienmüller, F.; Marten, I.; Hedrich, R.; Hanitzsch, M.; Janetzki, P.; Dietz, K.-J.; Schumacher, K. Regulation of the V-type ATPase by redox modulation. Biochem. J. 2012, 448, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Galic, M. Self-organization across scales: from molecules to organisms. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170113. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, B.N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 2006, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M.; Forrester, M.T.; Stamler, J.S. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.P.; Xun, L.; Xie, X.S. Single-Molecule Enzymatic Dynamics. 1998, 282, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- Carré, J.E.; Orban, J.-C.; Re, L.; Felsmann, K.; Iffert, W.; Bauer, M.; Suliman, H.B.; Piantadosi, C.A.; Mayhew, T.M.; Breen, P.; et al. Survival in Critical Illness Is Associated with Early Activation of Mitochondrial Biogenesis. Am. J. Respir. Crit. Care Med. 2010, 182, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.P. Bench-to-bedside review: Cytopathic hypoxia. 2002, 6, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Myocardial stunning and hibernation revisited. Nat. Rev. Cardiol. 2021, 18, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Willmann, K.; Moita, L.F. Physiologic disruption and metabolic reprogramming in infection and sepsis. Cell Metab. 2024, 36, 927–946. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Single, B.; Castoldi, A.F.; Kühnle, S.; Nicotera, P. Intracellular Adenosine Triphosphate (ATP) Concentration: A Switch in the Decision Between Apoptosis and Necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Carré, J.E.; Singer, M. Cellular energetic metabolism in sepsis: The need for a systems approach. Biochim. Et. Biophys. Acta (BBA) -Bioenerg. 2008, 1777, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.C.; Schenck, E.J.; Pabon, M.A.; Choi, A.M.K. The Role of Danger Signals in the Pathogenesis and Perpetuation of Critical Illness. Am. J. Respir. Crit. Care Med. 2018, 197, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.M.; Doyle, J. Highly Optimized Tolerance: Robustness and Design in Complex Systems. Phys. Rev. Lett. 2000, 84, 2529–2532. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Et. Biophys. Acta (BBA) -Bioenerg. 2001, 1504, 46–57. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Beutler, B.; Lowry, S.F.; Merryweather, J.; Wolpe, S.; Milsark, I.W.; Hariri, R.J.; Fahey, T.J.; Zentella, A.; Albert, J.D.; et al. Shock and tissue injury induced by recombinant human cachectin. Science 1986, 234, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Okusawa, S.; A Gelfand, J.; Ikejima, T.; Connolly, R.J.; A Dinarello, C. Interleukin 1 induces a shock-like state in rabbits. Synergism with tumor necrosis factor and the effect of cyclooxygenase inhibition. J. Clin. Investig. 1988, 81, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The thermodynamic bottleneck and viability corridor. Critical illness can impose a thermodynamic bottleneck in which ATP production falls while ATP costs rise. In high-flux organs, rapid ATP turnover requires near-continuous supply-demand matching. Yet organs may remain viable, with potential for full reversibility, despite days or weeks of profound dysfunction. How cells and organs remain within a viability corridor under these constraints remains unresolved. The timescale shown illustrates that 3 days of illness corresponds to approximately 17,000 ATP turnover intervals if turnover time is 15 s. Created in biorender.com.
Figure 1.
The thermodynamic bottleneck and viability corridor. Critical illness can impose a thermodynamic bottleneck in which ATP production falls while ATP costs rise. In high-flux organs, rapid ATP turnover requires near-continuous supply-demand matching. Yet organs may remain viable, with potential for full reversibility, despite days or weeks of profound dysfunction. How cells and organs remain within a viability corridor under these constraints remains unresolved. The timescale shown illustrates that 3 days of illness corresponds to approximately 17,000 ATP turnover intervals if turnover time is 15 s. Created in biorender.com.

Figure 2.
Conceptual organization of the ATPase ensemble under bioenergetic constraint. Each bar represents an ATPase type, positioned by inhibitory sensitivity and leverage over total ATP consumption. Under constraint, differential inhibition could produce ordered energy reallocation and ordered functional failure, while inhibition of specialized functions may reduce load on supporting processes such as ion pumps. As inhibition progresses, the remaining active ensemble becomes enriched for less sensitive ATPase types, increasing the cost of further control. Created in biorender.com.
Figure 2.
Conceptual organization of the ATPase ensemble under bioenergetic constraint. Each bar represents an ATPase type, positioned by inhibitory sensitivity and leverage over total ATP consumption. Under constraint, differential inhibition could produce ordered energy reallocation and ordered functional failure, while inhibition of specialized functions may reduce load on supporting processes such as ion pumps. As inhibition progresses, the remaining active ensemble becomes enriched for less sensitive ATPase types, increasing the cost of further control. Created in biorender.com.


Table 1.
Selected Paradoxes in Acute Organ Failure.
| Reversibility paradox | How can acute organ failure be lethal yet leave limited structural damage in many survivors? |
| ATP flux paradox | How can cells experience impaired ATP production yet remain viable for days, given ATP turnover ratesa? |
| Control paradox | How can acute organ failure be stereotyped and reversible if cellular control is primarily dysregulated? |
| Variety paradox | How can failure-mode variety be kept under control if regulatory capacity is reduced? |
| Cytopathic hypoxia paradox | Why do cells show impaired oxygen utilization during critical illness despite recovery of oxygenation and perfusion? |
a 10-20 seconds in brain and heart.
Table 2.
Predictions and Falsifiers.
|
Robustification of high gain ATPases (leverage x inhibitory sensitivity) decreases stress tolerance. Making high-leverage/high-sensitivity ATPases resistant to inhibition reduces stress tolerance and/or increases cell death. Falsifier: No effect on stress tolerance or cell death |
|
Effect modification: Making high-leverage ATPases that are sensitive to inhibition less sensitive while making low-sensitivity ATPases sensitive reduces stress tolerance and/or increases cell death non-linearly compared to either process alone. Falsifier: Additive, absent, or no interaction effects |
|
High-work microdomains: High-work microdomains show earlier and larger relative suppression of ATP hydrolysis under bioenergetic stress. Falsifier: No microdomain gradient |
|
Protective function: Blocking ATP-demand reduction or forcing ATPase-coupled work during bioenergetic stress decreases stress tolerance and/or increases cell death Falsifier: No worsening of stress tolerance or outcomes |
|
Distribution: ATPase types show a non-random, multimodal distribution across inhibitory sensitivity and ATP-consumption leverage, including high-sensitivity/high-leverage yield points. Falsifier: No such distribution |
|
Viability tracks energetics: Cell-death biomarkers correlate more with ATP/PCr/ΔG than dysfunction. Falsifier: Biomarkers primarily track dysfunction. |
|
Metabolism–function mismatch: High metabolism despite low organ function identifies subgroups with higher cell-death biomarkers. Falsifier: No biomarker association with metabolism–function mismatch. |
|
Non-productive ATP costs. Reducing non-productive ATP consumption (e.g., membrane leak) under constrained ATP production increases capacity for specialized cellular work. Falsifier: No increase in specialized work despite confirmed ATP-demand reduction. |
|
Energy-to-function reserve ratio: During transition to energy-first, ATP/PCr/ΔG falls less than EEG, cardiac or renal function, so E/F rises as function declines. Falsifier: Energy and function decline proportionally. |
|
Viability–inhibitory robustness: Viability-critical ATPases have lower inhibitory sensitivity/higher inhibitory thresholds than fault-tolerant ATPase processes, especially after accounting for inhibitory range. Falsifier: No association, or higher sensitivity in viability-critical ATPases. |
|
Inhibitory priority: Early ATPase-coupled yield points are predicted by high inhibitory sensitivity and ATP-consumption leverage, but low viability importance. Falsifier: Early inhibition is unrelated to this variable pattern. |
|
Enriched stress-sensitive sequence divergence: ATPase types show enriched sequence variation/divergence in residues or regions modulating sensitivity to H⁺, ROS or NO-related stress signals, compared with matched non-ATPase enzymes. Falsifier: No ATPase enrichment in stress-sensitive sequence variation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



