Submitted:
01 July 2026
Posted:
01 July 2026
You are already at the latest version
Abstract
In contemporary cardiac surgery, preservation of cognitive function has become the principal criterion of success; however, postoperative cognitive dysfunction (POCD), documented in 30–80% of patients, negates the benefits of the operation. Traditional risk factors explain only part of the genesis of this deficit. A key link is clonal haematopoiesis of indeterminate potential (CHIP): somatic mutations in haematopoietic stem cells lead to expansion of mutant immunocytes with hyperproduction of IL-1β and IL-6. Under cardiopulmonary bypass conditions, CHIP potentiates the systemic cytokine response, initiating neuroinflammation — from blood–brain barrier disruption to microglial activation and neuronal death. The prevalence of CHIP reaches 10–20% in the general population over 70 years of age and 44% among cardiac surgery patients; an allelic burden ≥2% is associated with a 2- to 3-fold increased risk of stroke and persistent POCD. This review proposes a new paradigm, viewing CHIP-associated inflammation as a systemic driver of neurological risk. Molecular mechanisms, clinical epidemiology and prospects for targeted therapy (IL-1β, IL-6 and JAK2 inhibitors) and cellular correction of the bone marrow niche are discussed. The strategic conclusion is that controlling CHIP opens a new front in the battle to preserve patients' intellect after cardiac surgical interventions.
Keywords:
clonal haematopoiesis of indeterminate potential (CHIP)
; neuroinflammation
; cardiac surgery
; blood–brain barrier (BBB)
; microglia
; targeted therapy
; cell therapy
; inflammaging
Introduction
Modern cardiac surgery has reached impressive heights: open-heart operations, which half a century ago carried fatal risk, are now performed even in elderly patients. However, progress has given rise to a paradox: survival is no longer the sole criterion of success. Quality of life [1] has come to the fore, the core component of which is preservation of cognitive function. For the cardiac surgical patient, loss of intellect can be more devastating [2,3] than the cardiac defect itself — it nullifies the clinical success of a technically flawless operation, turning it into a personal and social catastrophe.
Postoperative cognitive dysfunction (POCD) is documented in 30–80% of patients at hospital discharge [4], and persists in 42% at 5 years. Traditional perioperative risk factors — duration of cardiopulmonary bypass (CPB), systemic hypoperfusion, microembolism — explain only part of the genesis of cognitive deficit [5,6]. Patients of the same chronological age demonstrate radically different neurological outcomes [7], pointing to the key role of biological ageing, which is phenotypically not identical to chronological age [8]. The pathogenetic core of this process is chronic subclinical inflammation — the phenomenon of "inflammaging", a potential trigger of which is clonal haematopoiesis of indeterminate potential (CHIP) [9].
CHIP is characterised by the accumulation of somatic mutations in driver genes of haematopoietic stem cells (HSCs) in the bone marrow, leading to expansion of mutant immunocytes prone to hyperproduction of pro-inflammatory cytokines [10,11]. The prevalence of CHIP correlates with age, reaching 10–20% in the population over 70 years, and no less than 44% among cardiac surgery patients [12,13]. The phenomenon may also manifest in relatively young patients with aggressive cardiovascular disease [14]. Different mutations determine unique cytokine profiles: clones with DNMT3A mutation are sustained by IL-6 hyperproduction [15], TET2-mutant cells depend on IL-1β [16], expansion of ASXL1-mutant macrophages is mediated by IL-10 [17], and the JAK2-mutant clone is stimulated by TNFα [18]. This polymorphism of the cytokine response creates an individual trajectory of inflammation for each patient.
Cardiac surgery under CPB inevitably provokes a massive systemic cytokine response. In the presence of CHIP, this response is potentiated many times over, creating a "cytokine storm" effect [19,20,21,22], which destabilises the blood–brain barrier, activates microglia and triggers a cascade of inflammatory reactions leading to hippocampal neuronal apoptosis and persistent cognitive deficit (Figure 1). Mutant myeloid cells penetrate the brain parenchyma [23,24], replacing resident microglia and sustaining chronic neuroinflammation even after resolution of the systemic response.
Traditionally, CHIP was considered in the context of cardiovascular catastrophes, whereas its role in brain injury remained underestimated. This review proposes a new paradigm: CHIP-associated inflammation is a systemic driver of neurological risk in cardiac surgery. We analyse the molecular mechanisms of clonal expansion, their link to neuroinflammation, clinical epidemiology of neurological outcomes across different types of interventions, and discuss prospects for targeted pharmacotherapy and innovative cellular strategies for bone marrow niche correction. The strategic conclusion of this work is that controlling CHIP-associated inflammation opens a new front in the battle to preserve patients' intellect after cardiac surgery [25].
Section 1. Neurological Complications in Cardiac Surgery: Frequency and Pathogenetic Aspects
Up to one-third of cardiac operations [26] are accompanied by neurological complications. The American Heart Association (AHA) subdivides them into two main categories: focal and diffuse brain injuries [27]. Focal injuries include stroke, transient ischaemic attack (TIA), covert (clinically silent) stroke verifiable only by magnetic resonance imaging (MRI), as well as fatal brain injury. Diffuse injuries include postoperative delirium, postoperative cognitive dysfunction (POCD), coma and postoperative encephalopathy with gross intellectual impairment. This division, however, is rather arbitrary: clinical manifestations often do not correspond to the true extent of injury, and the dynamic nature of symptoms complicates timely diagnosis.
Stroke [28] remains one of the most unfavourable complications in cardiac surgery. According to Mario Gaudino and colleagues, it increases the risk of in-hospital death by 5–10 fold and the likelihood of severe cognitive impairment one year after surgery: up to 81% of patients would decline surgical treatment [29] to avoid stroke. However, data on the scale of stroke in cardiac surgery are highly contradictory. In the North American population, the incidence of stroke in coronary artery bypass grafting (CABG) reaches 6%, and in valve surgery — 10%. In the European population [30], isolated CABG is associated with stroke in 1.7% of patients; addition of valve surgery increases the risk to 3.3%; multiple valve procedures raise this figure to 6.7%. A particularly high-risk group comprises patients requiring reoperations: the incidence of stroke in repeat CABG ranges from 1.5% to 5.1%.
Ischaemic stroke predominates; haemorrhagic stroke is less common. In clinical practice, intraoperative stroke, identified on awakening from anaesthesia, is distinguished from postoperative stroke, which manifests later. Causes of intraoperative stroke include thromboembolism (up to 80% of cases) and hypoperfusion (30% of cases). Hypoperfusion strokes occur predominantly in border-zone (watershed) areas of the brain, due to impaired cerebrovascular autoregulation when mean arterial pressure is maintained around 60 mmHg — a critically low value for patients with chronic arterial hypertension [31]. Among the aetiological factors of postoperative stroke, atrial fibrillation and hypercoagulability play key roles [32].
Diffuse brain injuries represent an equally serious problem. Postoperative delirium develops in 7–52% of cardiac surgery patients. Postoperative cognitive dysfunction (POCD) affects up to 80% of patients in the early postoperative period and persists at 5 years in 42% of patients [4]. The particular insidiousness of diffuse injuries lies in their dynamic nature and the frequent mismatch between clinical manifestations and the true extent of injury. Thus, near-complete regression of focal symptoms within 24 hours may be erroneously interpreted as TIA, whereas large-focal ischaemic stroke with subsequent fatal consequences may develop in clinically silent areas [33]. Conversely, akinetic mutism or apallic syndrome on emergence from anaesthesia directly indicates severe injury to the higher centres of the central nervous system.
It is evident that the existing literature [29] underestimates the true scale of neurological impairment in cardiac surgical practice, primarily due to the absence of mandatory neuroimaging in this patient cohort. Diffusion-weighted MRI after CABG reveals signs of acute perioperative cerebral ischaemia in 18–26% of low-risk patients and in 45–62% of high-risk patients. New postoperative MRI lesions are found in 43% of patients after isolated valve replacement, with a substantial proportion demonstrating cognitive impairment of varying severity throughout the entire postoperative follow-up period.
The risk of brain injury [34] is determined both by iatrogenic factors (rapidity of rewarming on emergence from hypothermia, blood gas–temperature relationships, duration of hypoglycaemia and cardiopulmonary bypass time) and by individual patient characteristics. However, patients of the same chronological age often show radically different neurological outcomes. This observation indicates that chronological age is a relative marker; biological ageing, phenotypically not identical to chronological age, is of true significance.
Ageing is a dynamic process of continuous accumulation of changes in the organism, one of whose key [9] manifestations is chronic subclinical inflammation — the "inflammaging" phenomenon. Among the leading mechanisms [35] of this phenomenon is clonal haematopoiesis of indeterminate potential (CHIP). In CHIP carriers, the risk of coronary heart disease increases 2-fold, early myocardial infarction 4-fold, and stroke 2.5–3.5-fold [11]. The V617F mutation in the JAK2 gene is associated with a 12-fold increase in cardiovascular catastrophes [36]. These data allow CHIP to be regarded not merely as a genetic accident, but as an objective marker of ageing and a powerful perioperative risk factor.
Thus, traditional risk factors explain only part of the pathogenesis of neurological complications in cardiac surgery. Individual variability in outcomes is determined by biological age and the presence of CHIP.
Section 2. CHIP: Structure of Genetic Abnormalities and Mechanisms of Neural Tissue Injury
In the young healthy organism [37], haematopoiesis is sustained by a pool of haematopoietic stem cells (HSCs), the total number of which in the red bone marrow reaches 200,000. Over time, random somatic mutations accumulate in these cells, which, without conferring selective advantages, generate genetic heterogeneity. Under physiological conditions, all blood cells, including immune cells, derive from multiple such HSC clones — a state defined as polyclonal haematopoiesis.
However, somatic mutations in one of the HSC driver genes associated with myeloid neoplasms confer upon the clone a survival advantage and promote its expansion, inhibiting other clones [11]. The mutant cells retain the capacity to differentiate and populate the peripheral blood. In the absence of cytopenia or dysplasia, this state is termed [10] clonal haematopoiesis of indeterminate potential (CHIP). The prevalence of CHIP correlates with age: in the population under 50 years, the figure does not exceed 0.5%, whereas by 70 years it rises to 10–20% [12].
In the majority of individuals with CHIP [38], mutations are found in a limited set of genes involved in epigenetic regulation of HSC differentiation: DNMT3A, TET2 and ASXL1. Five other genes — JAK2, TP53, PPM1D, SF3B1 and SRSF2 — account for a further 10–20% of CHIP-associated mutations [39]. Each mutation creates a unique cytokine profile that determines the character of systemic inflammation and the spectrum of clinical risks. Below we examine in detail the major driver mutations and their link to neural tissue injury.
DNMT3A mutation. The most common mutation is in the DNMT3A gene [40]. Its expression product — DNA methyltransferase 3A — catalyses DNA methylation and coordinates epigenetic programmes of stem cell differentiation. The key cytokine sustaining the clonal advantage of DNMT3A-mutant immunocytes is IL-6 [15]. In neuroinflammation, IL-6 plays a dual role, acting simultaneously as a provoker of brain injury and a neuroprotective factor; owing to this polarity it is often termed a "Janus-faced" cytokine [41]. At high concentration, IL-6 binds to soluble receptors (sIL-6R) directly in the intercellular space, triggering chronic neuroinflammation. In carriers of DNMT3A-CHIP, the risk of coronary heart disease is increased 2-fold, and early myocardial infarction 4-fold [11].
TET2 mutation. The second most prevalent gene associated with CHIP is TET2 [42]. Its expression product — tetramethylcytosine dioxygenase 2 — initiates active DNA demethylation and regulates chromatin architecture. The specific cytokine sustaining expansion of the TET2-mutant clone is IL-1β [16]. In neuroinflammation, IL-1β acts as the principal molecular trigger, whose effects are predominantly destructive [43]. It binds to IL-1R receptors on microglia, causing avalanche-like production of IL-1β, IL-6 and TNF-α. Under the influence of this cytokine, blood–brain barrier (BBB) permeability increases sharply, glutamate production is enhanced, leading to excitotoxicity and neuronal apoptosis, and long-term memory mechanisms in the hippocampus are blocked [44].
ASXL1 mutation. The third most frequent CHIP driver gene is ASXL1 [45]. Its expression product — a protein stabilising Polycomb complexes (PRC1 and PRC2) — comprises epigenetic regulatory systems [46] that mediate long-term transcriptional repression without altering the DNA sequence. ASXL1 mutations actively promote clonal expansion, increasing the risk of cardiovascular disease 2- to 3-fold or more [17]. The phlogogenic effect of ASXL1 mutation is mediated by NF-κB activation, increased production of IL-1β, IL-6 and TNFα in macrophages, accompanied by enhanced IL-10 production, which plays a major role in expanding the mutant clone by protecting it from apoptosis [47]. Elevated IL-10 is particularly notable in gliomas, where it enables neoplastic cells to escape immune responses.
JAK2 mutation. The key cytokine associated with expansion of the JAK2-mutant clone is TNFα [18]. The V617F mutation in the JAK2 gene promotes the formation of neutrophil extracellular traps (NETosis) that damage the endothelium and activate the coagulation cascade [48,49]. In JAK2-CHIP, the risk of both arterial and venous thromboses is increased many times over.
Other genes. Mutations in TP53 (the "guardian of the genome") and PPM1D are associated with the DNA damage response; they often arise after chemotherapy and significantly increase the risk of secondary myeloid neoplasms [50,51]. TP53 mutation represents the most dangerous CHIP variant, since even a small clone with a tumour suppressor gene mutation can drive therapy-related myelodysplasia [52]. The genes SF3B1 and SRSF2 are spliceosomal and disrupt RNA processing [39].

Table 1.
Major CHIP driver mutations, associated cytokines and clinical risks.
| Mutation |
Frequency in CHIP |
Associated cytokine |
Principal mechanism of injury | Clinical risk | Mutation |
| DNMT3A | Most common (~50–60%) | IL-6 [5] | Disrupted DNA methylation, NF-κB activation |
CHD (↑ 2-fold), early MI (↑ 4-fold) [11] | DNMT3A |
| TET2 | Second most common (~20%) | IL-1β [16] | Disrupted DNA demethylation, microglial activation |
Atherosclerosis, neuroinflammation, memory deficit [43] |
TET2 |
| ASXL1 | Third most common (~10%) | IL-10, IL-1β, IL-6, TNFα [7,47] | NF-κB activation, Polycomb stabilisation |
CVD (↑ 2–3-fold), fibrosis, valve calcification [53] | ASXL1 |
| JAK2 V617F | ~5–10% | TNFα [18] | NETosis, coagulation activation, endothelial injury | CVD (↑ 12-fold), thrombosis, stroke [36,48] | JAK2 V617F |
| TP53 | ~3–5% | — (stress response) | Impaired DNA repair, genomic instability | Risk of therapy-related myeloid neoplasms [50,52] | TP53 |
| PPM1D | ~2–4% | — (stress response) | Impaired DNA damage response |
Risk after chemotherapy [51] |
PPM1D |
| SF3B1/SRSF2 | ~3–5% | — (spliceosomal) | Disrupted RNA processing |
Haematological abnormalities, CVD [39] |
SF3B1/SRSF2 |
In addition to the above genes, other rarer mutations associated with CHIP have been described. Each accounts for less than 1% of cases, but they are important for understanding the full picture of clonal heterogeneity (Table 2).
Of particular note is the link between TERT-associated CHIP and telomere length. This mutation is not included in most diagnostic panels and often falls into researchers' "blind spot". Yet [57] it is associated with increased telomere length and rejuvenation of the actively proliferating HSC population. However, here a significant paradox becomes apparent: increased telomere length raises the risk of clonal expansion [58], excessive expansion of which leads to telomere exhaustion and shortening.
Furthermore, mosaic structural variations — loss of segments or entire sex chromosomes — can also lead to clonal haematopoiesis. Mosaic loss of chromosome Y (mLOY) is found in 40–50% of men aged 70, and loss of chromosome X (mLOX) in 10–20% of women of the same age [59]. These chromosomal abnormalities can drive clonal expansion, with risk increasing with age [60].
Not only the mutations themselves are important, but also context-dependent effects. According to Joshua Weinstock and colleagues, the TCL1A mutation is not associated with CHIP, as it is characterised by slow expansion of the mutant clone. However, the clinical effect of this mutation depends on the driver gene: in TCL1A carriers, a marked reduction in the growth rate of clones with mutations in TET2, ASXL1, SF3B1 and SRSF2 is observed, but this effect was not seen in clones with DNMT3A mutations [61]. Conversely, Michael Kessler and colleagues [62] demonstrate opposing effects with respect to JAK2V617F-CHIP.
Environmental factors also play a critical role in CHIP clone expansion. Smoking increases the overall probability of CHIP by 20%, with the risk of ASXL1- and PPM1D-CHIP doubling [63,64]. Following cytostatic therapy, clones with mutations in TP53, PPM1D and CHEK2 expand [50,51,52]. Risk is highest for patients receiving high cumulative drug doses [65]. Unfavourable factors also include sleep hygiene disturbances, radon exposure, nutrient deficiencies and pharmacologically uncontrolled arterial hypertension [63,64,65].
Gender factors modulate the architecture of CHIP. In women with natural menopause [66] before age 40, the risk of CHIP is 1.4–1.7-fold higher, with the strongest association observed for DNMT3A mutations. The competitive activity of mutant progeny cells depends on oestrogen receptor alpha (ERα): knockout of the Esr1 gene encoding this receptor abolishes the competitive advantage of mutant cells [67]. The sharp drop in oestrogen levels at menopause alters the systemic inflammatory milieu, accelerating mutant clone growth. However, CHIP is more common in men than in women [68]. The U2AF1 mutation is considered a male-specific mutation; male sex correlates with increased risk of expansion of ASXL1-associated clones. In turn, andropause is associated with more frequent detection of JAK2 mutations and increased systemic inflammation.
Thus, CHIP represents a fundamental mechanism of biological ageing and a qualitatively new cardiovascular risk factor [69,70]. However, its significance extends far beyond the coronary bed. Mutant myeloid cells [71], circulating in the blood, secrete pro-inflammatory cytokines (IL-1β, IL-6, TNFα), which systemically alter the organism's inflammatory background. During cardiac surgery, this background triggers a "double-hit" mechanism, which on contact with cardiopulmonary bypass transforms into a cytokine storm.
Section 3. Adaptive Immunity and Myeloid CHIP: A Feedback Loop
Traditionally, CHIP is viewed as a phenomenon confined to the myeloid lineage, giving rise to components of the innate immune system. This selectivity is due to heterogeneity of the HSC pool: with age, the proportion of myeloid-biased HSCs increases, and somatic mutations are preferentially fixed in this pool [72]. Lymphoid-biased HSCs remain relatively intact, providing mutation-free lymphopoiesis [73]. However, the myeloid paradigm of CHIP overlooks feedback mechanisms from cells of the adaptive immune system, which act as architects of the bone marrow niche [74].
Lymphocytes, in addition to their standard adaptive immune functions, survey genomic integrity and regulate stem cell activity within bone marrow niches. Regulatory T cells (Treg) create zones of immune privilege, protecting HSCs from oxidative stress and cytokine attack through secretion of IL-10 and TGFβ [75]. These cytokines maintain local homeostasis, inducing apoptosis of cells with critically damaged genomes and modulating HSC receptor interactions with the extracellular matrix. With ageing, Treg function declines, "opening the gates" to the inflammatory advantage of CHIP clones [76].
Crucially, HSCs themselves are capable of presenting antigens to T lymphocytes, which then eliminate aberrant cells with damaged genomes. Such presentation of immunogenic antigens initiates bidirectional interaction between HSCs and antigen-specific CD4+ T cells, causing stem cell proliferation, differentiation and specific exhaustion of aberrant HSCs [77]. Immunosenescence reduces the ability of CD4+ T cells to recognise neo-antigens arising from somatic mutagenesis in HSCs. Whereas in youth a CHIP clone with high metabolic or mutational load is promptly eliminated, in the ageing organism the "escape" mechanism becomes systemic [78]. In effect, the mutant myeloid lineage begins to dominate because the adaptive arm can no longer verify and constrain cellular atypia at the bone marrow level.
In addition to the waning of Treg suppressor function, age-related rearrangement of the cytotoxic T-lymphocyte pool becomes a critical factor. Terminally differentiated effector T lymphocytes of the TEM and TEMRA phenotypes, having lost the CD28 antigen, infiltrate the bone marrow and alter its humoral landscape, secreting interferon-gamma (IFN-γ) and TNFα [79]. Local cytokine stress proves lethal to healthy HSCs, whereas mutant clones possess selective molecular resistance to inflammatory pro-apoptotic signals. Thus, TEM and TEMRA T-effectors, though not carrying primary mutations, create a microenvironment that suppresses polyclonal haematopoiesis and induces expansion of mutant myeloid clones.
Reduced functional activity of lymphoid-biased HSCs leads to progressive deficiency of early B-cell precursors in the niche [80]. However, B lymphocytes at early developmental stages perform an important regulatory role: they locally produce factors that sustain endothelial and mesenchymal stem cells. The decline in B-cell precursor numbers leads to stromal remodelling and reduced expression of retention factors such as CXCL12 and SCF, destabilising cell–matrix interactions [81].
Thus, CHIP emerges as a consequence of deformation of the interaction algorithm between adaptive memory and haematopoietic progenitors. Mutant myeloid cells established in the niche (Figure 2) begin to oversecrete IL-1β and IL-6, which systemically stimulates terminal differentiation of new T-effectors in the periphery and sustains their continuous influx into the bone marrow. This mechanism closes the vicious circle of autoinflammation, in which the CHIP clone is simultaneously both consequence and driving force of immune imbalance.
Section 4. Factors Promoting the Emergence and Expansion of CHIP
CHIP is a complex process occurring under synergistic influence of environmental and biological factors. Age remains the major risk factor: in the population under 50 years, CHIP prevalence does not exceed 0.5%, whereas by 70 years it rises to 20% [82]. However, biological ageing can be substantially accelerated by external triggers.
Smoking is a major catalyst of clonal expansion. It increases the overall probability of CHIP by 20%, with the risk of ASXL1- and PPM1D-CHIP doubling [63,64]. Chemotherapy also dramatically affects clonal dynamics: following cytostatic therapy, clones with mutations in TP53, PPM1D and CHEK2 expand [50,51,52]. The presence of CHIP before treatment significantly increases the risk of developing secondary therapy-related myeloid neoplasms.
Gender factors modulate the architecture of CHIP. In women with early natural menopause (before age 40), the risk of CHIP is 1.4–1.7-fold higher, particularly for DNMT3A mutations [66]. Experimentally, the competitive activity of mutant progeny cells has been shown to depend on oestrogen receptor alpha (ERα); Esr1 knockout abolishes the competitive advantage of mutant cells [67]. However, CHIP is more common in men than in women [68]. U2AF1 is considered a male-specific mutation, and male sex correlates with increased risk of ASXL1 clone expansion. Andropause is associated with more frequent detection of JAK2 mutations and increased systemic inflammation. Racial differences have also been noted: individuals of Asian descent are less predisposed to CHIP compared with Caucasians.
Section 5. CHIP Clone Size and Clinical Consequences of Its Expansion
CHIP is an independent risk factor for the development of atherosclerosis and coronary heart disease [83]. The risk of disease is higher in individuals with larger clone size (Variant Allele Frequency, VAF) ≥ 0.1%, while in CHIP carriers [84] with accelerated epigenetic age, the CHD risk is increased 3.24-fold. A clone with VAF 2% (corresponding to a clone comprising no less than 4% of blood cells) is associated with early atherosclerosis [85], which constitutes one of the most dangerous signs of epigenetic ageing observed in 40% of CHIP carriers.
The mechanism of CHIP-induced atherosclerosis has mutation-specific features. Tet2-mutant macrophages increase atherosclerotic plaque instability [86], enlarging its necrotic core and sharply elevating the risk of thrombosis. DNMT3A-mutant monocytes and macrophages with impaired efferocytosis accumulate in the vascular intima [87], creating a self-sustaining inflammatory microenvironment through persistent NF-κB activation. This explains the multi-fold, nearly order-of-magnitude, increase in CHD and thrombosis risk in JAK2-CHIP carriers. ASXL1 and TP53 mutations lead to accumulation of low-density lipoproteins in the vessel wall, induce secretion of profibrotic factors, predisposing to valve calcification and myocardial fibrosis [53].
Of particular importance for cardiac surgery is the link between clone size and neurological outcomes. According to Zhiwei Zeng and colleagues, the presence of CHIP is associated with a 20% increase in stroke risk [88]. Sandro Ninni and colleagues established that CHIP is present in 44% of cardiac surgery patients and is associated with a doubling of stroke risk. Moreover, increasing clone size to ≥2% is accompanied by an additional 2.4-fold increase in risk, whereas CHIP with small clones (VAF 0.1–2%) showed no significant association with long-term outcomes [13]. These data allow the focus to shift from mere detection of CHIP to assessment of clone size and specific mutation, which has direct clinical relevance for risk stratification before cardiac surgery.
Section 6. CHIP and the Pathogenesis of Neurological Disorders in Cardiac Surgery
Systemic inflammatory expansion induced by CHIP triggers a cascade of destructive changes extending far beyond the vascular bed. One of the main targets of this process is the cerebral endothelium. Under the influence of high concentrations of IL-6 and IL-1β, degradation of tight junction proteins that maintain blood–brain barrier (BBB) integrity occurs. A "window of vulnerability" is formed: the BBB becomes permeable to pro-inflammatory molecules and activated immune cells, radically altering the neuronal microenvironment [19,89].
A key link in pathogenesis is activation of resident microglia. Cytokines penetrating the BBB shift microgliocytes into a reactive state, in which they themselves become sources of phlogogenic cytokines [20]. This gives rise to the phenomenon of "sterile" neuroinflammation — an autonomous, self-sustaining process that continues to destroy neural tissue even after resolution of the systemic inflammatory response [90]. Mutant monocytes, actively penetrating the BBB under conditions of age-related inflammatory injury, differentiate into M1-phenotype macrophages, which intensify the inflammatory milieu [23].
Hind Bouzid and colleagues identified in microglial cells of the brain in seven of eight CHIP carriers the same mutations [24] as in peripheral blood immunocytes. This confirms that mutant blood cells can replace a substantial portion of the microglial pool. These "new" cells possess an altered epigenetic profile and release increased amounts of reactive oxygen species and proteases, directly damaging neurons and sustaining the vicious circle of neuroinflammation.
A special role in this cascade is assigned to IL-18 — a marker cytokine of CHIP-associated inflammation. It initiates hippocampal neuronal apoptosis, blocks long-term potentiation mechanisms and undermines synaptic plasticity, disrupting neurogenesis in the subventricular zone [21]. It is this pathochemical link that explains the high correlation between IL-18 levels and the development of persistent POCD. Elevated postoperative IL-18 levels are associated with atrophy of the nucleus basalis of Meynert three months after surgery, directly linking this cytokine to persistent structural changes in the brain [22].
Cardiac surgery under cardiopulmonary bypass imposes an additional insult on an already compromised brain. Ischaemia–reperfusion during cardioplegia, microembolism and the systemic cytokine response create a "double-hit" situation: CHIP-induced inflammation sharply increases BBB permeability, opening the door to infiltration of mutant myeloid cells directly into the brain parenchyma. Once there, CHIP-mutant cells sustain persistent local neuroinflammation that continues long after completion of surgery, accounting for the high frequency and persistence of POCD.
It is important to note that the role of CHIP in neuroinflammation is not unequivocally destructive. According to Katie Matatall and colleagues, TET2-mutant cells enhance phagocytosis of pathological proteins and restrain progression of Alzheimer's disease [91]. However, in cardiac surgery patients, where operative stress is superimposed on a pre-existing pro-inflammatory background, this protective mechanism appears to be overridden, with destructive neuroinflammation coming to the fore.
Section 7. Statistics of Neurological Complications in Cardiac Surgery and CHIP
Coronary Artery Bypass Grafting (CABG)
Patients undergoing CABG remain the most studied group. The incidence of clinically overt stroke in the postoperative period ranges from 0.8 to 5.2%, which may be related to racial, sex and age differences [92,93]. In the European population, isolated CABG is associated with stroke in 1.7% of patients; addition of valve surgery increases the risk to 3.3%; multiple valve procedures — to 6.7%. A particularly high-risk group comprises patients undergoing reoperations: the incidence of stroke in repeat CABG reaches 5.1% [30].
However, clinically manifest symptoms represent only the tip of the iceberg. Diffusion-weighted MRI after CABG reveals signs of acute perioperative cerebral ischaemia in 18–26% of low-risk patients and in 45–62% of high-risk patients. Jürgen Puchinger and colleagues identify intraoperative elevation of IL-6 as a major risk factor for neurological impairment [94]. The presence of CHIP increases the likelihood of adverse cardiovascular events, including stroke, by 1.5- to 3-fold compared with mutation-free patients [88]. Spencer Flynn and colleagues regard CHIP as a major risk factor for acute heart failure and death after CABG; moderate cognitive impairment is observed in 40–80% of patients, and CHIP is associated with a tendency toward severe encephalopathy and prolonged cognitive deficit [95].
Aortic Surgery
Aortic operations are among the most complex and are inextricably linked to neurological complications. Postoperative stroke in aortic arch interventions is recorded at a frequency of 5–10% or more. Christopher Pereira and colleagues link this to elevated levels of 6 out of 10 measured cytokines in cerebrospinal fluid, with a 63-fold increase in IL-6 potentially serving as an independent CHIP marker [96].
For prevention of neurological complications, selective antegrade cerebral perfusion is employed; however, in the presence of an inflammatory cytokine background, there is a risk of exacerbating cerebral oedema. Eridan Rocha-Ferreira and colleagues showed in experimental models that high baseline levels of inflammatory cytokines in cerebrospinal fluid are associated with adverse neurological outcomes, with hypothermia and other neuroprotective factors proving largely ineffective [97]. This allows the patient's cytokine status to be considered an independent criterion of neurological risk in aortic surgery.
Isolated Valve Surgery
The proportion of severe cerebral complications in patients after valve replacement is approximately 14%, substantially higher than in CABG. This is related to greater technical complexity, longer cardiopulmonary bypass duration and higher embolic risk. The incidence of clinical stroke after isolated valve reconstruction is 2–5%, whereas other types of neurological injury — encephalopathy, seizure syndrome — are observed in 8–20% of patients.
MRI reveals foci of clinically silent infarcts in 41% of patients after isolated aortic or mitral valve replacement, with neurocognitive impairment persisting at 4–6 weeks after surgery in 35% of these. New postoperative MRI lesions are found in 43% of patients, a substantial proportion demonstrating cognitive impairment of varying severity throughout the entire follow-up period. Valve surgery provokes a more powerful release of inflammatory cytokines due to extensive tissue trauma and prolonged blood contact with CPB circuit surfaces. According to Rodríguez-Montolio and colleagues, IL-6 and IL-8 levels in such patients are independent predictors of fatal outcome and neurological catastrophes [98,99].
Thus, the severity of neurological impairment is directly dependent on the patient's cytokine status, with blood inflammatory cytokine levels rising with increasing operative volume and peaking in CHIP. These data dictate the need for development of new therapeutic approaches to correction of this condition.
Section 8. Pharmacotherapeutic Approaches to the Treatment of Neurological Complications
Despite the enormous clinical significance of the problem, no unified approaches to its solution have been developed. Many authors advocate symptomatic prevention of neurological complications, relying on standard agents: acetylsalicylic acid, statins, barbiturates, anaesthetics, lidocaine, NMDA receptor antagonists, magnesium, nimodipine, corticosteroids and aprotinin [100]. However, the evidence base for such claims is extremely limited or absent.
Statins (atorvastatin, rosuvastatin) have been regarded since their advent as the most promising group, yet they have been shown to produce only a marginal difference in stroke incidence. Preoperative statin administration does not reduce the risk of stroke or other neurological complications [101].
α2-adrenoceptor agonists (dexmedetomidine) reduce the risk of postoperative delirium by 39% and the incidence of POCD by up to 60% [102]; however, their use is associated with increased risk of bradycardia and requires close monitoring. In a study by Bjørn Erik Neerland and colleagues, the potential of dexmedetomidine for prevention of delirium and cognitive impairment after open-heart surgery was demonstrated [103]. Combination with melatonin showed a 69% reduction in delirium risk, but the evidence is of low certainty due to the small number of direct comparisons [104].
Ketamine reduces the severity of POCD symptoms from 81% to 27% one week after surgery [105]. Gastrodin reduced delirium risk from 35.9% to 19.5% in patients after CABG [106]. Memantine reduces serum S100-B levels, which may indicate reduced brain injury [107]. However, all these data require confirmation in large randomised trials.
Glucocorticosteroids, theoretically capable of reducing the systemic inflammatory response, do not reduce the risk of death, stroke or postoperative delirium [108,109]. Their use is associated with hyperglycaemia and infectious complications. In paediatric cardiac surgery, steroids are used more frequently, but consensus has not been reached.
Citicoline, edaravone, magnesium preparations, cerebrolysin — all these agents show conflicting results. Citicoline significantly reduced infarct volume compared with placebo, but controlled trials have shown only modest potential [110]. Edaravone blocks the damaging effects of free radicals, but large clinical trials are lacking [111].
The most effective "neuroprotection" today is not pharmacological but systemic in nature. It includes maintenance of adequate cerebral perfusion pressure, strict glycaemic control, prevention of hyperthermia and minimisation of microembolism. Pharmacological support is considered merely an adjunctive component.
The key problem with traditional strategies is that they do not address the root cause — the mutant clone and its cytokine footprint. In the presence of CHIP, standard anti-inflammatory therapy does not eliminate the source of cytokine hyperproduction, but merely temporarily masks symptoms. This makes targeted therapy (IL-1β, IL-6 and JAK2 inhibitors) not simply an alternative, but the only pathogenetically justified approach. At the same time, prolonged cytokine blockade renders the patient vulnerable to opportunistic infections, allowing targeted therapy to be considered a temporary measure for the acute phase; long-term correction of CHIP will likely require the development of cellular technologies.
Section 9. CHIP and Gene-Engineered Targeted Pharmacotherapy
For CHIP correction, the timing of intervention is critically important. The extent of the planned surgical procedure and clone size must be assessed. At VAF > 2%, targeted therapy may be one means of preventing CHIP-associated disorders.
IL-6 Inhibitors (Tocilizumab)
IL-6 is a key cytokine associated with CHIP and postoperative delirium. In Mendelian randomisation studies, the rs2228145 polymorphism, which reduces IL-6 signalling, is associated with a 55% reduction in cardiovascular risk in carriers of large DNMT3A and TET2 clones [15]. Tocilizumab, an IL-6 receptor inhibitor, may be effective in DNMT3A-like phenotypes and possibly in ASXL1-like phenotypes.
IL-1β Inhibitors (Canakinumab)
The IL-1β inhibitor canakinumab reduced cardiovascular event rates in experimental models by 62%, particularly in patients with prior myocardial infarction [112]. The effect was maximal in TET2-CHIP carriers: a 62% risk reduction versus 7% in those without CHIP [113]. Experimentally, blockade of IL-1β or NLRP3 abolished the atherogenic effect of Tet2 and Jak2 mutations. Given the key role of IL-1β in BBB disruption and microglial activation, canakinumab may be considered a potential agent for prevention of delirium and POCD in patients with TET2-like phenotype.
JAK1/2 Inhibitors (Ruxolitinib)
Ruxolitinib in experimental models prevented the development of abdominal aortic aneurysm, inferior vena cava thrombosis and pulmonary hypertension in animals with JAK2 mutation [114]. In stimulated neutrophils from patients with JAK2 V617F mutation, ruxolitinib reduced NETosis formation [115]. In JAK2-like phenotypes with extremely high TNFα and high thrombotic risk, ruxolitinib may be the most justified choice, capable of preventing aneurysm, thrombosis and pulmonary hypertension [116].
NLRP3 Inhibitors (MCC950)
In mouse models, infusion of the NLRP3 inhibitor MCC950 reduced aortic atherosclerotic plaque size by approximately 50% in mice with Tet2-deficient bone marrow. In heart failure models, NLRP3 inhibitor protected against heart failure development in Tet2- and Ppm1d-deficient mice [117].
It is important to emphasise that all these strategies currently have evidence only for cardiovascular outcomes. Their application for prevention of neurological complications in cardiac surgery is a theoretical extrapolation requiring clinical studies. Nevertheless, the pathophysiological plausibility (BBB disruption, neuroinflammation, neuroapoptosis as direct consequences of the same cytokines) makes this extrapolation highly promising. However, all these approaches have substantial drawbacks: immunosuppression does not kill mutant cells, but merely "sedates" them; there is a high risk of relapse of expansion after therapy withdrawal. Prolonged cytokine blockade renders the patient vulnerable to opportunistic infections, allowing this therapy to be considered a temporary measure for the acute phase. Further treatment of CHIP will require the development of biomedical cellular technologies.
Section 10. Strategies for CHIP Suppression: From Hypotheses to Cell Therapy
Despite the absence of approved protocols for eradication of mutant HSC clones, contemporary biomedical strategies for CHIP correction are actively developing in the context of biomedical gene and cellular technologies. Theoretically, as Friederike Christen and colleagues assert, CRISPR/Cas9 could correct the mutation in HSCs [118] and solve the problem. However, practical application of this technology for correcting mutations in CHIP faces several serious obstacles. First [119], for correcting a mutation rather than simply "knocking out" a gene, CRISPR/Cas9 utilises the homology-directed repair (HDR) mechanism, which is most active predominantly in dividing cells. The majority of long-lived HSCs are in a quiescent state (G0 cell cycle), making HDR extremely inefficient. At the same time, the actively operating alternative repair pathway, non-homologous end joining (NHEJ), frequently leads to uncontrolled insertions or deletions at the cleavage site, disrupting gene function and promoting mutagenesis.
Second [120], genome editing is not an ideal process. In the general population of such "edited" HSCs, only a minority of alleles receive the desired correction. The remaining cells may carry a variety of undesirable changes: from small indels to large chromosomal rearrangements. This creates significant genetic heterogeneity, which may not only reduce the therapeutic effect but also carry potential risks, such as malignant transformation, which is fundamentally unacceptable. In addition to off-target mutations, the process of creating double-strand DNA breaks can itself induce cellular senescence and inflammation, reducing their long-term repopulation potential and compromising the safety and efficacy of therapy. But this is only part of the problem.
At the current stage of biomedical development, application of CRISPR/Cas9 is fraught with difficulties in delivering system components to the bone marrow. Electroporation — a fairly popular ex vivo method — may be effective but often causes significant cytotoxicity and is limited to use outside the organism. Development of safe and effective in vivo delivery methods using lipid nanoparticles is in its early stages and cannot be recommended clinically. Furthermore, genome editing is an extremely expensive and technologically complex process in which risks and costs must be carefully weighed against potential benefit, particularly in elderly patients with cardiovascular disease.
At the same time, the idea of genetic editing entails ex vivo modification of HSCs, which must be returned to the patient as a high-technology biomedical product. Transplantation of edited cells includes chemotherapeutic conditioning to free space in the bone marrow and eliminate the mutant clone. However, an attempt simply to "kill" the mutant clone and transplant healthy HSCs is considered unjustifiably risky even in oncological clinical practice. Chemotherapy with autologous HSC transplantation often creates a "bottleneck": most healthy cells die [121], while stress-resistant CHIP mutant clones gain a competitive advantage. In turn, in patients with lymphoma who had CHIP at the time of autologous HSC transplantation, 10-year overall survival was significantly worse, amounting to only 30.4% compared with 60.9% in patients without CHIP [122], not accounting for the risk of death from infectious transplant complications.
A current approach to CHIP correction is an attempt at depletion of the mutant haematopoietic stem cell clone. Mutant cells in CHIP are characterised by increased expression of receptors for inflammatory signals, which underpins their clonal expansion. Theoretically, hyperstimulation with such signals could lead to forced division of mutant HSCs, clone exhaustion and terminal differentiation. Such stimulation could be performed using classical drugs or by manipulating bone marrow cells ex vivo.
A research group led by Benjamin Ebert [123] attempted to deplete the dominant CHIP clone through hyperstimulation with TLR ligands (exogenous DNA, RNA, bacterial lipopolysaccharide, etc.). However, mutant HSCs, particularly those with TET2 and ASXL1 mutations, possess mechanisms of resistance to TLR-mediated exhaustion, so such stimulation may instead provide them with additional competitive advantage. In turn, Paul S. Frenette and colleagues [124] propose an alternative strategy — immobilisation of HSCs in the bloodstream through G-CSF stimulation followed by glucocorticoid therapy. However, G-CSF itself induces systemic inflammation, and mutant HSCs acquire even greater resistance to apoptosis in this context.
Thus, the idea of depleting the dominant clone currently does not withstand criticism. However, the strategy of CHIP correction using biomedical cell products appears fundamentally sound. The aim of this strategy is to render the bone marrow environment less favourable for mutant clones by modifying the HSC niche.
Approaches to this task currently vary considerably: Sevanthy Suresh and colleagues proposed transplantation of mesenchymal stem cells (MSCs) and autologous M2-activated macrophages [125] with high immunosuppressive activity. Such a procedure, hypothetically, could be performed with or without prior cytostatic preconditioning. Carsten Riether [126] and Virginia Camacho and colleagues [127] propose regulation of the HSC niche through transplantation of Treg and myeloid suppressor cells. Karin Prummel and colleagues propose transplantation of MSCs induced toward adipogenic stromal cells [128] expressing CXCL12⁺ to suppress inflammatory niche remodelling. Ideas have been put forward for managing the HSC niche [129,130,131] through revitalisation of autologous MSCs with simultaneous use of synergistic effects of statins [132] and other pharmacological agents.
Critically, virtually all the strategies described above for correcting the mutant HSC niche attempt to recapitulate mechanisms most studied in models of cancer stem cell interaction with the local microenvironment. The essence of such models is maximal imitation of the immunosuppressive influences exerted by niche cells on HSCs, which may restrain the rate of mutant clone expansion.
In this light, the idea of immunosuppressive modulation of HSCs through TGF-β treatment, partially outlined in the work of Grant Challen and colleagues [133], appears rather promising. TGF-β can indeed deepen functional differences between different HSC subpopulations. At concentrations of 10 ng/ml, TGF-β inhibits proliferation of both myeloid-oriented and lymphoid-oriented HSCs, suggesting that the cytokine may produce a balancing effect on different HSC subclones. At low concentrations, approximately 10 pg/ml, TGF-β stimulates proliferation of myeloid-oriented HSCs but suppresses all other clones. In this context, it is evident that age-related decline in TGF-β levels promotes preferential accumulation of myeloid-biased HSCs.
In light of the above, theoretically, by manipulating TGF-β concentration one could suppress hyperproliferative myeloid clones, thereby restoring haematopoietic balance. It is important to consider that in the real CHIP situation, the action of TGF-β may be more complex. Mutations in CHIP-associated genes (e.g., DNMT3A, TET2, ASXL1) may alter cellular sensitivity to TGF-β or even disrupt the signalling pathway itself. Therefore, the "balancing" effect may vary depending on the specific mutant clone. In this regard, a two-stage approach appears most promising: first, blockade of inflammation using targeted agents (pro-inflammatory cytokine inhibitors deprive mutant cells of their main proliferative stimulus), then stimulation of polyclonality using TGF-β at a concentration of 10 ng/ml, with niche modulator support, to activate dormant healthy stem cells [134].
Such therapy could, of course, only be performed ex vivo, since excess TGF-β could provoke bone marrow fibrosis, accelerate atherothrombosis or trigger CHIP progression to myelodysplasia.
Section 11. Tactical and Methodological Approaches to the Creation of a Biomedical Cell Product for CHIP Correction
Addressing the treatment of CHIP-associated haematopoietic disorders and prevention of related neurological complications in cardiac surgery marks a turning point toward personalised medicine. Such an approach cannot be formulaic; it goes beyond symptomatic surgical care — undoubtedly strategically important in the treatment of cardiovascular disease — and requires development of fundamentally new diagnostic tools and therapeutic products of a new type, which allow us to step further than traditional cardiac surgery.
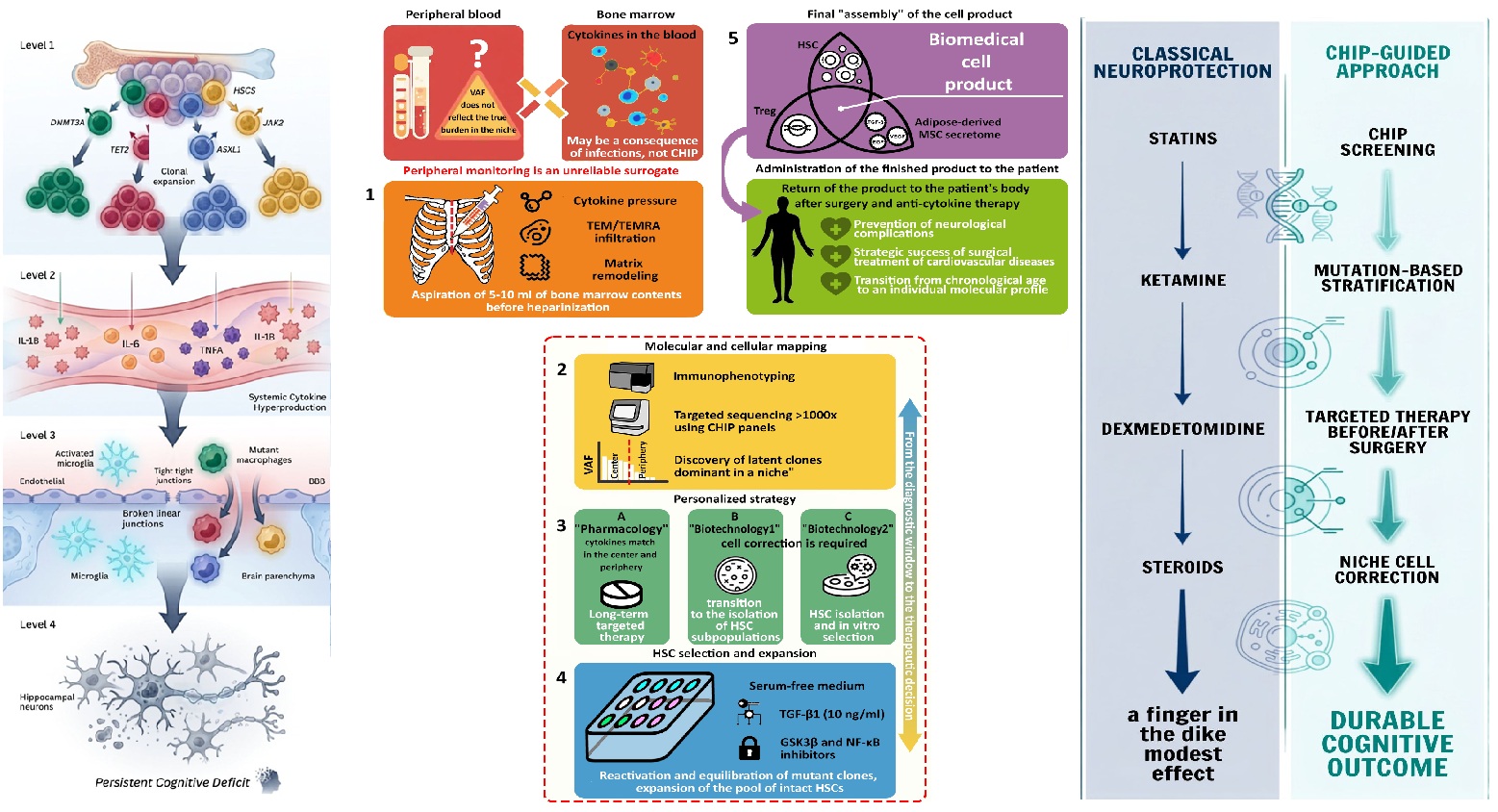
The main limitation in assessing the scale of CHIP in clinical practice is the use of peripheral blood as a surrogate marker, creating a risk of underestimation of the problem. First, peripheral VAF may not reflect the true allelic burden in the central bone marrow niche, where the mutant clone may already be dominant without yet manifesting in the periphery. Second, elevated levels of phlogogenic cytokines in the blood may not be directly related to CHIP but may arise as a consequence of previous infections (e.g., COVID-19) or other diseases. Therefore, the first step toward solving the problem should be determination of cytokine concentration directly in bone marrow tissue.
Direct intraoperative access to the cancellous bone of the sternum during sternotomy (Figure 3) provides a unique clinical model, allowing transition from systemic monitoring of consequences to direct study of the bone marrow niche. Collection of 5–10 ml of bone marrow contents should be performed before heparinisation, at the stage of sternal skeletonisation. Given the high prevalence of CHIP among patients with cardiovascular disease (up to 44% [13]), direct aspiration of bone marrow contents enables in each specific case: validation of the hypothesis of selective "cytokine pressure" on HSCs, verification of the density of bone marrow infiltration by terminally differentiated TEM and TEMRA cells, and assessment of the depth of inflammatory remodelling of the tissue matrix.
A second, equally important step is immunophenotyping of the bone marrow biopsy and targeted NGS with read depth >1000× using standard commercial CHIP panels, available from most manufacturers. This will allow comparative analysis of allelic burden in the compartments "centre (bone marrow) — periphery (blood)", enabling: determination of clonal expansion kinetics, identification of the leading HSC clones forming CHIP, and detection of latent CHIP pools that already dominate in the central niche but have not yet manifested high VAF in the periphery.
Critically, such bone marrow mapping could become a crucial step toward development of a personalised CHIP correction strategy. In one case, where the leading cytokine coincides in the centre and periphery, long-term targeted pharmacotherapy is justified; in another, transition to the third, most biotechnological stage — bone marrow restitution — is appropriate. Theoretically, this stage depends substantially on the leading mutation and may include either direct eradication of the dominant HSC clone or balancing of HSC subpopulations through incubation in medium containing 10 ng/ml TGF-β, which may be considered optimal [135] for realising the regulatory properties of HSCs, capable of suppressing hyperproliferative myeloid clones, thereby restoring haematopoietic balance.
A large clone size may require isolation of suppressed HSC clones with subsequent expansion by culturing in serum-free medium supplemented with a cocktail of growth factors, enhanced with GSK3β inhibitors or wnt ligands. Hypothetically, such an HSC population could be further balanced by co-culture in the presence of autologous Treg and MSCs or components of their secretome.
The cellular material thus generated could be returned to the patient as a biomedical cell product after surgical treatment and systemic anti-cytokine therapy. Such an approach could not only prevent neurological complications in cardiac surgery but also become a decisive factor in the strategic success of surgical treatment of cardiovascular disease.
Conclusion
Changes in surgical techniques, including the use of off-pump operations, have not led to a reduction in the incidence of brain injury. This compels us to acknowledge that the root of the problem lies not only in the technical aspects of the operation, but in the fundamental biological processes that determine the individual patient's response to surgical trauma.
CHIP is not merely a genetic accident or a benign marker of ageing. It is a genetically fixed form of inflammatory ageing that transforms peripheral inflammation into a systemic threat reaching the brain. Mutant haematopoietic cell clones, circulating in the blood, secrete pro-inflammatory cytokines, creating a background inflammation that, during cardiac surgery, explodes into a cytokine storm, destabilises the blood–brain barrier, activates microglia and triggers a cascade of neuroinflammation leading to neuronal death and persistent cognitive deficit.
Cardiac surgery, where operative stress is superimposed on a pre-existing pro-inflammatory background, becomes an ideal "model system" for studying this link. Assessment of cytokine profile and genetic mapping of CHIP mutations is the first step toward making hidden inflammation measurable and manageable.
The next step is clinical trials of targeted therapy (IL-1β, IL-6 and JAK2 inhibitors) and the application of biomedical cellular and genetic technologies. The unique opportunity for intraoperative access to the bone marrow via sternotomy opens new horizons for personalised CHIP correction.
The strategic conclusion of our work is unequivocal: controlling CHIP-associated inflammation is not merely an academic interest. It is a new front in the battle to preserve patients' intellect after cardiac surgery. The answer to the key question — can suppression of CHIP-associated inflammation preserve the patient's intellect — demands an immediate transition from theoretical constructs to clinical trials.
References
- Zhang, Z.R.; Li, Y.Z.; Wu, X.Q.; et al. Postoperative cognitive dysfunction in elderly postcardiac surgery patients: progress in rehabilitation application research. Front Rehabil. Sci. 2024, 5, 1525813. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Liu, Z.; Meng, S.; et al. The Effect of Opioid-Free Anesthesia with Esketamine on Postoperative Cognitive Dysfunction in Elderly Patients Undergoing Thoracoscopic Surgery: A Prospective, Randomized, Controlled Trial. Drug Des. Devel Ther. 2025, 19, 11227–11244. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Lei, Y.; Sun, X.; et al. Postoperative cognitive dysfunction in heart transplantation recipients. Clin. Transplant. 2024, 38(5), e15337. [Google Scholar] [CrossRef] [PubMed]
- Majewski, P.; Zegan-Barańska, M.; Karolak, I.; et al. Current Evidence Regarding Biomarkers Used to Aid Postoperative Delirium Diagnosis in the Field of Cardiac Surgery-Review. Medicina 2020, 56(10), 493. [Google Scholar] [CrossRef] [PubMed]
- Staicu, R.E.; Vernic, C.; Ciurescu, S.; et al. Postoperative Delirium and Cognitive Dysfunction After Cardiac Surgery: The Role of Inflammation and Clinical Risk Factors. Diagnostics 2025, 15(7), 844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.; Peng, Y.; Lin, L.; et al. The influencing factors of cognitive dysfunction in patients after cardiac surgery and the construction of a nomogram prediction model. Eur. J. Med. Res. 2025, 30(1), 925. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, F.; Kuwabara, M.; Michihata, N.; et al. Chronological and Biological Age in Predicting Outcomes of Older Patients in Neurosurgery. Neurol. Med. Chir. 2025, 65(12), 541–550. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sato, H.; Sorimachi, T.; et al. Lack of association between chronological age and fisher group and poor outcomes in older patients with severe-grade aneurysmal subarachnoid hemorrhage: a nationwide registry study in Japan. Neurosurg. Rev. 2025, 48(1), 466. [Google Scholar] [CrossRef] [PubMed]
- Nachun, D.; Lu, A.T.; Bick, A.G.; et al. Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell 2021, 20(6), e13366. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Singh, A. Clonal Hematopoiesis of Indeterminate Potential: Current Understanding and Future Directions. Curr. Oncol. Rep. 2023, 25(6), 539–547. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl. J. Med. 2017, 377(2), 111–121. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Li, N.; Ashrafi, E.; et al. Clonal hematopoiesis of indeterminate potential as a prognostic factor: a systematic review and meta-analysis. Blood Adv. 2024, 8(14), 3771–3784. [Google Scholar] [CrossRef] [PubMed]
- Ninni, S.; Vicario, R.; Coisne, A.; et al. Clonal Hematopoiesis Is Associated With Long-Term Adverse Outcomes Following Cardiac Surgery. J. Am. Heart Assoc. 2024, 13(17), e034255. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kopecky, S.L.; Yang, E.H.; Oren, O. Clonal Hematopoiesis of Indeterminate Potential and Cardiovascular Disease. Curr. Oncol. Rep. 2020, 22(9), 87. [Google Scholar] [CrossRef] [PubMed]
- Bick, A.G.; Pirruccello, J.P.; Griffin, G.K.; et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 2020, 141(2), 124–131. [Google Scholar] [CrossRef] [PubMed]
- Caiado, F.; Kovtonyuk, L.V.; Gonullu, N.G.; et al. Aging drives Tet2+/- clonal hematopoiesis via IL-1 signaling. Blood 2023, 141(8), 886–903. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Goyama, S.; Chang, Y.H.; et al. Clonal hematopoiesis-related mutant ASXL1 promotes atherosclerosis in mice via dysregulated innate immunity. Nat. Cardiovasc Res. 2024, 3(12), 1568–1583. [Google Scholar] [CrossRef] [PubMed]
- Rubio, T.; Viana, R.; Moreno-Estellés, M.; et al. TNF and IL6/Jak2 signaling pathways are the main contributors of the glia-derived neuroinflammation present in Lafora disease, a fatal form of progressive myoclonus epilepsy. Neurobiol. Dis. 2023, 176, 105964. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Gu, T.; Yu, P.; et al. The role of microglia in Neuroinflammation associated with cardiopulmonary bypass. Front Hum. Neurosci. 2025, 19, 1695336. [Google Scholar] [CrossRef]
- Yazdanpanah Moghadam, E.; Sonenberg, N.; Packirisamy, M. Alzheimer model chip with microglia BV2 cells. Microsyst. Nanoeng. 2025, 11(1), 135. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, K.; Hata, M.; Gamachi, N.; et al. Molecular Mechanisms of IL18 in Disease. Int. J. Mol. Sci. 2023, 24(24), 17170. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.; Spies, C.; Borchers, F.; et al. Perioperative Levels of IL8 and IL18, but not IL6, are Associated with Nucleus Basalis Magnocellularis Atrophy Three Months after Surgery. J. Neuroimmune Pharmacol. 2024, 19(1), 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Trzebanski, S.; Shin, S.H.; et al. Clonal hematopoiesis-associated motoric deficits caused by monocyte-derived microglia accumulating in aging mice. Cell Rep. 2025, 44(5), 115609. [Google Scholar] [CrossRef] [PubMed]
- Bouzid, H.; Belk, J.A.; Jan, M.; Qi, Y.; et al. Clonal hematopoiesis is associated with protection from Alzheimer's disease. Nat. Med. 2023, 29(7), 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Lyu, T.J.; Qiu, X.; Wang, Y.; et al. DNMT3A dysfunction promotes neuroinflammation and exacerbates acute ischemic stroke. MedComm (2020) 2024, 5(7), e652. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, R.F.; McKhann, G.M.; Hogue, C.W. Neurological complications of cardiac surgery. Semin Neurol. 2008, 28(5), 703–15. [Google Scholar] [CrossRef] [PubMed]
- Eagle, K.A.; Guyton, R.A.; Davidoff, R.; et al. ACC/AHA guidelines for coronary artery bypass graft surgery: executive summary and recommendations: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to revise the 1991 guidelines for coronary artery bypass graft surgery). Circulation 1999, 100(13), 1464–80. [Google Scholar] [CrossRef] [PubMed]
- Sultan, I.; Bianco, V.; Kilic, A.; et al. Predictors and Outcomes of Ischemic Stroke After Cardiac Surgery. Ann. Thorac. Surg. 2020, 110(2), 448–456. [Google Scholar] [CrossRef] [PubMed]
- Gaudino, M.; Benesch, C.; Bakaeen, F.; et al. Considerations for Reduction of Risk of Perioperative Stroke in Adult Patients Undergoing Cardiac and Thoracic Aortic Operations: A Scientific Statement From the American Heart Association. Circulation 2020, 142(14), e193–e209. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.D.; Stafford-Smith, M.; White, W.D.; et al. The effect of aprotinin on outcome after coronary-artery bypass grafting. N Engl. J. Med. 2008, 358(8), 784–93. [Google Scholar] [CrossRef] [PubMed]
- Al-Amoodi, A.; Debicki, D.; Sefein, O.; Bainbridge, D. Ischemic Stroke in the Cardiac Surgery Intensive Care Unit: A Quality Improvement Study. J. Cardiothorac. Vasc. Anesth. 2024, 38(7), 1524–1530. [Google Scholar] [CrossRef] [PubMed]
- Caldonazo, T.; Kirov, H.; Rahouma, M.; et al. Atrial fibrillation after cardiac surgery: A systematic review and meta-analysis. J. Thorac. Cardiovasc Surg. 2023, 165(1), 94–103.e24. [Google Scholar] [CrossRef] [PubMed]
- Condello, I.; Dell'Aquila, M.; Condello, S.; et al. Silent Stroke in Adult Cardiac Surgery: Mechanisms, Clinical Impact, and Preventive Strategies. Medicina 2026, 62(4), 675. [Google Scholar] [CrossRef] [PubMed]
- Lackner, I.; Weber, B.; Pressmar, J.; et al. Cardiac alterations following experimental hip fracture - inflammaging as independent risk factor. Front Immunol. 2022, 13, 895888. [Google Scholar] [CrossRef] [PubMed]
- Marnell, C.S.; Bick, A.; Natarajan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cell Cardiol. 2021, 161, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kafeiti, N.; Masarik, K.; et al. Decoding Endothelial MPL and JAK2V617F Mutation: Insight Into Cardiovascular Dysfunction in Myeloproliferative Neoplasms. Arterioscler. Thromb. Vasc. Biol. 2024, 44(9), 1960–1974. [Google Scholar] [CrossRef] [PubMed]
- Schuermans, A.; Honigberg, M.C. Clonal haematopoiesis in cardiovascular disease: prognostic role and novel therapeutic target. Nat. Rev. Cardiol. 2025, 22(11), 845–856. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.A.; Sano, S.; Walsh, K. Cardiovascular Disease, Aging, and Clonal Hematopoiesis. Annu Rev. Pathol. 2020, 15, 419–438. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Smedby, K.E.; Xue, H.; et al. Clonal hematopoiesis of indeterminate potential and the risk of pulmonary embolism: an observational study. EClinicalMedicine 2024, 74, 102753. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Divaris, K.; Pan, B.; et al. Clonal hematopoiesis driven by mutated DNMT3A promotes inflammatory bone loss. Cell 2024, 187(14), 3690–3711.e19. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.D.; Yong, V.W. Neuroinflammation across neurological diseases. Science 2025, 388(6753), eadx0043. [Google Scholar] [CrossRef] [PubMed]
- Saadatagah, S.; Naderian, M.; Uddin, M.; et al. Atrial Fibrillation and Clonal Hematopoiesis in TET2 and ASXL1. JAMA Cardiol. 2024, 9(6), 497–506. [Google Scholar] [CrossRef] [PubMed]
- McClatchy, J.; Strogantsev, R.; Wolfe, E.; et al. Clonal hematopoiesis related TET2 loss-of-function impedes IL1beta-mediated epigenetic reprogramming in hematopoietic stem and progenitor cells. Nat. Commun. 2023, 14(1), 8102. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Huang, Y.; Zhou, R. NLRP3 inflammasome in neuroinflammation and central nervous system diseases. Cell Mol. Immunol. 2025, 22(4), 341–355. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Goyama, S.; Kitamura, T. ASXL1 mutation-related clonal hematopoiesis and age-related diseases: clinical evidence and molecular insights. Int. J. Hematol. 2025, 122(3), 327–340. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.F.; Valencia-Sánchez, M.I.; Tamburri, S.; et al. Structural basis of histone H2A lysine 119 deubiquitination by Polycomb repressive deubiquitinase BAP1/ASXL1. Sci. Adv. 2023, 9(32), eadg9832. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.M.; Neidert, N.; Will, P.; et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat. Commun. 2022, 13(1), 925. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, R.; Sellar, R.S.; Vromman, A.; et al. The clonal hematopoiesis mutation Jak2V617F aggravates endothelial injury and thrombosis in arteries with erosion-like intimas. Int. J. Cardiol. 2024, 409, 132184. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Zhang, J.; Shang, Y.; Liu, S. Multitarget natural compounds taming NETosis: A translational strategy for cancer and inflammatory diseases. Cancer Lett. 2026, 636, 218103. [Google Scholar] [CrossRef] [PubMed]
- Fullin, J.; Topçu, E.; Zielińska, K.A.; et al. The pathogenesis of therapy-related myeloid neoplasms from TP53-mutant clonal hematopoiesis. Leukemia 2026, 40(2), 279–292. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132(11), 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Fandrei, D.; Pegliasco, J.; Pasquier, F.; et al. Clonal Evolution of PPM1D Mutations in the Spectrum of Myeloid Disorders. Clin. Cancer Res. 2025, 31(11), 2241–2253. [Google Scholar] [CrossRef]
- Inkum, F.; Geng, X.; Terkeltaub, R.; Cobo, I. Connecting the dots: Gouty arthritis, clonal haematopoiesis and myeloid activation, in a unified inflammation model for atherosclerosis progression. Jt. Bone Spine 2026, 93(3), 105993. [Google Scholar] [CrossRef] [PubMed]
- Bill, M.; Jentzsch, M.; Bischof, L.; et al. Impact of IDH1 and IDH2 mutation detection at diagnosis and in remission in patients with AML receiving allogeneic transplantation. Blood Adv. 2023, 7(3), 436–444. [Google Scholar] [CrossRef] [PubMed]
- Vobugari, N.; Heuston, C.; Lai, C. Clonal cytopenias of undetermined significance: potential predictor of myeloid malignancies? Clin. Adv. Hematol. Oncol. 2022, 20(6), 375–383. [Google Scholar] [PubMed]
- Loscocco, G.G.; Rotunno, G.; Mannelli, F.; et al. The prognostic contribution of CBL, NRAS, KRAS, RUNX1, and TP53 mutations to mutation-enhanced international prognostic score systems (MIPSS70/plus/plus v2.0) for primary myelofibrosis. Am. J. Hematol. 2024, 99(1), 68–78. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.E.; Chen, Y.Y.; Lu, C.H.; et al. Prevalence and clinical impact of JAK2-CHIP: Association with Parkinsonism and hematologic changes in a population cohort. J. Formos. Med. Assoc. 2025. S0929-6646(25)00523-6. [Google Scholar] [CrossRef]
- Ferrer, A.; Mangaonkar, A.A.; Patnaik, M.M. Clonal Hematopoiesis and Myeloid Neoplasms in the Context of Telomere Biology Disorders. Curr. Hematol. Malig. Rep. 2022, 17(3), 61–68. [Google Scholar] [CrossRef] [PubMed]
- Jakubek, Y.A.; Ma, X.; Stilp, A.M.; et al. Genomic and phenotypic correlates of mosaic loss of chromosome Y in blood. Am. J. Hum. Genet. 2025, 112(2), 276–290. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, A.K.; Brown, D.W.; Machiela, M.J. Clonal hematopoiesis due to mosaic chromosomal alterations: Impact on disease risk and mortality. Leuk. Res. 2023, 126, 107022. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, J.S.; Gopakumar, J.; Burugula, B.B.; et al. Aberrant activation of TCL1A promotes stem cell expansion in clonal haematopoiesis. Nature 2023, 616(7958), 755–763. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.D.; Damask, A.; O'Keeffe, S.; et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 2022, 612(7939), 301–309. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Li, N.; Ashrafi, E.; Thao, L.T.P.; et al. Factors Associated with Risk of Clonal Haematopoiesis of Indeterminate Potential: A Systematic Review and Meta-Analysis. EJHaem 2025, 6(6), e70173. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; An, H.; Koh, Y.; Lee, C.H. Clonal Hematopoiesis of Indeterminate Potential Is Associated with Current Smoking Status and History of Exacerbation in Patients with Chronic Obstructive Pulmonary Disease. Tuberc. Respir. Dis. 2024, 87(3), 309–318. [Google Scholar] [CrossRef] [PubMed]
- Shyr, D.; Pershad, Y.; Zhao, K.; et al. Clonal Hematopoiesis and Cardiovascular Disease Risk After Cancer Therapy in Patients With Solid Tumors. JAMA Oncol. 2026, 12(3), 251–256. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, M.C.; Zekavat, S.M.; Niroula, A.; et al. Premature Menopause, Clonal Hematopoiesis, and Coronary Artery Disease in Postmenopausal Women. Circulation 2021, 143(5), 410–423. [Google Scholar] [CrossRef] [PubMed]
- Stomper, J.; Niroula, A.; Belizaire, R.; et al. Sex differences in DNMT3A-mutant clonal hematopoiesis and the effects of estrogen. Cell Rep. 2025, 44(4), 115494. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, P.; van Zeventer, I.A.; de Graaf, A.O.; et al. Sex Differences in the Spectrum of Clonal Hematopoiesis. Hemasphere 2023, 7(2), e832. [Google Scholar] [CrossRef] [PubMed]
- Páramo Fernández, J.A. Atherosclerosis and clonal hematopoyesis: A new risk factor. Clin. Investig. Arterioscler. 2018, 30(3), 133–136. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.L. Clonal hematopoiesis, inflammaging, and vascular disease: mechanisms, risk stratification, and therapeutic frontiers in older adults. Acta Pharmacol. Sin. 2026. [Google Scholar] [CrossRef] [PubMed]
- Kallai, A.; Ungvari, A.; Csaban, D.; et al. Clonal hematopoiesis of indeterminate potential (CHIP) in cerebromicrovascular aging: implications for vascular contributions to cognitive impairment and dementia (VCID). Geroscience 2025, 47(3), 2739–2775. [Google Scholar] [CrossRef] [PubMed]
- Nishi, K.; Sakamaki, T.; Nagasaka, A.; et al. Alteration of long- and short-term hematopoietic stem cell ratio causes myeloid-biased hematopoiesis. Elife 2025, 13, RP95880. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, L.A. Restoring balanced HSC youth. Nat. Immunol. 2025, 26(8), 1213. [Google Scholar] [CrossRef] [PubMed]
- Prummel, K.D.; Woods, K.; Kholmatov, M.; et al. Inflammatory stromal and T cells mediate human bone marrow niche remodeling in clonal hematopoiesis and myelodysplasia. Nat. Commun. 2025, 16(1), 10042. [Google Scholar] [CrossRef] [PubMed]
- Camacho, V.; Matkins, V.R.; Patel, S.B.; et al. Bone marrow Tregs mediate stromal cell function and support hematopoiesis via IL-10. JCI Insight 2020, 5(22), e135681. [Google Scholar] [CrossRef] [PubMed]
- Riether, C. Regulation of hematopoietic and leukemia stem cells by regulatory T cells. Front Immunol. 2022, 13, 1049301. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Malmierca, P.; Vonficht, D.; Schnell, A.; et al. Antigen presentation safeguards the integrity of the hematopoietic stem cell pool. Cell Stem Cell 2022, 29(5), 760–775.e10. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M. Antigen-specific T cells as a potential regulator of hematopoietic stem cell clones. Rinsho Ketsueki 2022, 63(8), 918–927. [Google Scholar] [CrossRef] [PubMed]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Freitas-Cortez, M.A.; Zhang, J.; et al. Periarteriolar niches become inflamed in aging bone marrow, remodeling the stromal microenvironment and depleting lymphoid progenitors. Proc. Natl. Acad. Sci. U S A 2025, 122(11), e2412317122. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Cancelas, J.A. From Marrow to Bone and Fat: Exploring the Multifaceted Roles of Leptin Receptor Positive Bone Marrow Mesenchymal Stromal Cells. Cells 2024, 13(11), 910. [Google Scholar] [CrossRef] [PubMed]
- Haring, B.; Wissel, S.; Manson, J.E. Somatic Mutations and Clonal Hematopoiesis as Drivers of Age-Related Cardiovascular Risk. Curr. Cardiol. Rep. 2022, 24(8), 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Díez-Díez, M.; Ramos-Neble, B.L.; de la Barrera, J.; et al. Unidirectional association of clonal hematopoiesis with atherosclerosis development. Nat. Med. 2024, 30(10), 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Nachun, D.; Lu, A.T.; Bick, A.G.; et al. Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell 2021, 20(6), e13366. [Google Scholar] [CrossRef] [PubMed]
- Senguttuvan, N.B.; Subramanian, V.; Tr, M.; Sankaranarayanan, K.; et al. Clonal hematopoiesis of indeterminate potential and cardiovascular diseases: A review. Indian Heart J. 2025, 77(1), 51–57. [Google Scholar] [CrossRef] [PubMed]
- Maan, M.; Pati, U. CHIP promotes autophagy-mediated degradation of aggregating mutant p53 in hypoxic conditions. FEBS J. 2018, 285(17), 3197–3214. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Ji, J.; Zhan, X.; et al. Tet1-mediated 5hmC regulates hippocampal neuroinflammation via wnt signaling as a novel mechanism in obstructive sleep apnoea leads to cognitive deficit. J. Neuroinflammation 2024, 21(1), 208. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, F.; Gai, S.; et al. The effect of clonal hematopoiesis on long-term outcomes in patients undergoing coronary artery bypass grafting. BMC Med. 2025, 23(1), 322. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, Y.; Gu, W.; Xu, Q. Response of vascular mesenchymal stem/progenitor cells to hyperlipidemia. Cell Mol. Life Sci. 2018, 75(22), 4079–4091. [Google Scholar] [CrossRef] [PubMed]
- Traunmüller, F. Atherosclerosis is a vascular stem cell disease caused by insulin. Med. Hypotheses 2018, 116, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Matatall, K.A.; Wathan, T.K.; Nguyen, M.; et al. TET2-mutant myeloid cells mitigate Alzheimer's disease progression via CNS infiltration and enhanced phagocytosis in mice. Cell Stem Cell 2025, 32(8), 1285–1298.e8. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Grega, M.A.; Borowicz, L.M., Jr.; et al. Stroke and encephalopathy after cardiac surgery: an update. Stroke 2006, 37(2), 562–71. [Google Scholar] [CrossRef] [PubMed]
- Dumitriu LaGrange, D.; Tessitore, E.; Reymond, P.; et al. A systematic review and meta-analysis of differences between men and women in short-term outcomes following coronary artery bypass graft surgery. Sci. Rep. 2024, 14(1), 20682. [Google Scholar] [CrossRef] [PubMed]
- Puchinger, J.; Ryz, S.; Nixdorf, L.; et al. Characteristics of Interleukin-6 Signaling in Elective Cardiac Surgery-A Prospective Cohort Study. J. Clin. Med. 2022, 11(3), 590. [Google Scholar] [CrossRef] [PubMed]
- Flynn, S.; Schuermans, A.; Uddin, M.M.; et al. Clonal Hematopoiesis and Incident Heart Failure. JAMA Cardiol. 2026, 11(2), 126–135. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Perera, A.H.; Rudarakanchana, N.; et al. Cytokine changes in cerebrospinal fluid following vascular surgery on the thoracic aorta. Sci. Rep. 2022, 12(1), 12839. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Ferreira, E.; Kelen, D.; Faulkner, S.; et al. Systemic pro-inflammatory cytokine status following therapeutic hypothermia in a piglet hypoxia-ischemia model. J. Neuroinflammation 2017, 14(1), 44. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Montolio, J.; Meseguer-Gonzalez, D.; Almeida-Zurita, M.; et al. Prevalence of neurological complications in infective endocarditis. Neurologia (Engl Ed) 2024, 39(6), 443–448. [Google Scholar] [CrossRef] [PubMed]
- Das, A.S.; McKeown, M.; Jordan, S.A.; et al. Neurological Complications and Clinical Outcomes of Infective Endocarditis. J. Stroke Cerebrovasc. Dis. 2022, 31(8), 106626. [Google Scholar] [CrossRef]
- Fatima, M.; Bazarbaev, A.; Rana, A.; et al. Neuroprotective Strategies in Coronary Artery Disease Interventions. J. Cardiovasc Dev. Dis. 2025, 12(4), 143. [Google Scholar] [CrossRef] [PubMed]
- Musson, E.N.; Hoade, Y.; Dace, P.; et al. Analysis of the effects of statin therapy on clonal dynamics in clonal haematopoiesis of indeterminate potential: insights from the English Longitudinal Study of Ageing. Leukemia 2026, 40(2), 429–434. [Google Scholar] [CrossRef] [PubMed]
- From the American Association of Neurological Surgeons (AANS); American Society of Neuroradiology (ASNR); Cardiovascular and Interventional Radiology Society of Europe (CIRSE); et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13(6), 612–632. [Google Scholar] [CrossRef] [PubMed]
- Neerland, B.E.; Busund, R.; Haaverstad, R.; et al. Alpha-2-adrenergic receptor agonists for the prevention of delirium and cognitive decline after open heart surgery (ALPHA2PREVENT): protocol for a multicentre randomised controlled trial. BMJ Open 2022, 12(6), e057460. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, I.; Barbosa, L.M.; Gallo Ruelas, M.; et al. Effect of peri-operative pharmacological interventions on postoperative delirium in patients having cardiac surgery: a systematic review and Bayesian network meta-analysis. Anaesthesia 2026, 81(2), 274–287. [Google Scholar] [CrossRef] [PubMed]
- Hudetz, J.A.; Iqbal, Z.; Gandhi, S.D.; et al. Ketamine attenuates post-operative cognitive dysfunction after cardiac surgery. Acta Anaesthesiol. Scand. 2009, 53(7), 864–72. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.X.; Wu, H.L.; Xie, W.L.; et al. Efficacy and safety of gastrodin in preventing postoperative delirium following cardiac surgery: a randomized placebo controlled clinical trial. Crit. Care 2025, 29(1), 108. [Google Scholar] [CrossRef] [PubMed]
- Ziabakhsh Tabary, S.; Ziabakhsh Tabary, P.; Sanei Motlagh, A. Neuroprotective effect of memantine on serum S100-B levels after on-pump coronary artery bypass graft surgery: A randomized clinical trial. Casp. J. Intern Med. 2022, 13(2), 412–417. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, Y.; Qiu, Z.; et al. Efficacy and safety of corticosteroids prophylaxis in cardiac surgery: A protocol for systematic review and meta-analysis. Medicine 2020, 99(50), e23240. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, Y.; Wang, J.; et al. Effects of Glucocorticoids on Postoperative Delirium in Adult Patients Undergoing Cardiac Surgery: A Systematic Review and Meta-analysis. Clin. Ther. 2021, 43(9), 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Vishnu, V.Y.; Sharma, J.; et al. Citicoline in acute ischemic stroke: A randomized controlled trial. PLoS ONE 2022, 17(5), e0269224. [Google Scholar] [CrossRef] [PubMed]
- Dakroub, F.; Awada, B.; Abdelhady, S.; et al. Edaravone: Advances on cytoprotective effects, pharmacological properties, and mechanisms of action. Pharmacol. Rev. 2026, 78(1), 100101. [Google Scholar] [CrossRef] [PubMed]
- Shin, T.H.; Zhou, Y.; Chen, S.; et al. A macaque clonal hematopoiesis model demonstrates expansion of TET2-disrupted clones and utility for testing interventions. Blood 2022, 140(16), 1774–1789. [Google Scholar] [CrossRef] [PubMed]
- Bick, A.G.; Pirruccello, J.P.; Griffin, G.K.; et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 2020, 141(2), 124–131. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, T.; Misaka, T.; Kimishima, Y.; et al. Crucial role of hematopoietic JAK2 V617F in the development of aortic aneurysms. Haematologica 2021, 106(7), 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- Wolach, O.; Sellar, R.S.; Martinod, K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10(436), eaan8292. [Google Scholar] [CrossRef] [PubMed]
- Al-Rifai, R.; Vandestienne, M.; Lavillegrand, J.R.; et al. JAK2V617F mutation drives vascular resident macrophages toward a pathogenic phenotype and promotes dissecting aortic aneurysm. Nat. Commun. 2022, 13(1), 6592. [Google Scholar] [CrossRef] [PubMed]
- Visan, I. Atherosclerosis-prone HSCs. Nat. Immunol. 2017, 18(4), 373. [Google Scholar] [CrossRef] [PubMed]
- Christen, F.; Hablesreiter, R.; Hoyer, K.; et al. Modeling clonal hematopoiesis in umbilical cord blood cells by CRISPR/Cas9. Leukemia 2022, 36(4), 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Nourmohammadi, H.; Babashahi, M.; Panji, M.; Radmehr, S. Gene-edited hematopoietic stem cells for leukemia and lymphoma treatment: a systematic review of preclinical and translational evidence. Discov. Oncol. 2025, 16(1), 1804. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.J.; Yamazaki, S. Understanding genetic heterogeneity in gene-edited hematopoietic stem cell products. Exp. Hematol. 2024, 129, 104133. [Google Scholar] [CrossRef] [PubMed]
- Arabzadeh, M.; Tang, Y.H.; Colin-Leitzinger, C.; et al. Evolution of clonal hematopoiesis during cancer treatment and its impact on outcomes. J. Clin. Invest. 2026, e204429. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.J.; Lindsley, R.C.; Tchekmedyian, V.; et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J. Clin. Oncol. 2017, 35(14), 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Belizaire, R.; Wong, W.J.; Robinette, M.L.; Ebert, B.L. Clonal haematopoiesis and dysregulation of the immune system. Nat. Rev. Immunol. 2023, 23(9), 595–610. [Google Scholar] [CrossRef] [PubMed]
- Pierce, H.; Zhang, D.; Magnon, C.; et al. Cholinergic Signals from the CNS Regulate G-CSF-Mediated HSC Mobilization from Bone Marrow via a Glucocorticoid Signaling Relay. Cell Stem Cell 2017, 20(5), 648–658.e4. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Venkatesan, V.; Thangavel, S.; Marepally, S. Exploring MSC and HSPC interactions: new frontiers in hematopoiesis and transplant medicine. Stem Cell Res. Ther. 2025, 16(1), 640. [Google Scholar] [CrossRef] [PubMed]
- Riether, C. Regulation of hematopoietic and leukemia stem cells by regulatory T cells. Front Immunol. 2022, 13, 1049301. [Google Scholar] [CrossRef] [PubMed]
- Camacho, V.; Matkins, V.R.; Patel, S.B.; et al. Bone marrow Tregs mediate stromal cell function and support hematopoiesis via IL-10. JCI Insight 2020, 5(22), e135681. [Google Scholar] [CrossRef] [PubMed]
- Prummel, K.D.; Woods, K.; Kholmatov, M.; et al. Inflammatory stromal and T cells mediate human bone marrow niche remodeling in clonal hematopoiesis and myelodysplasia. Nat. Commun. 2025, 16(1), 10042. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, F.; Borger, D.K.; Wei, Q.; et al. Engineering a haematopoietic stem cell niche by revitalizing mesenchymal stromal cells. Nat. Cell Biol. 2019, 21(5), 560–567. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, Y.; Gu, W.; Xu, Q. Response of vascular mesenchymal stem/progenitor cells to hyperlipidemia. Cell Mol. Life Sci. 2018, 75(22), 4079–4091. [Google Scholar] [CrossRef] [PubMed]
- Traunmüller, F. Atherosclerosis is a vascular stem cell disease caused by insulin. Med. Hypotheses 2018, 116, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xiong, Y.Y.; Li, Q.; et al. Optimization of Timing and Times for Administration of Atorvastatin-Pretreated Mesenchymal Stem Cells in a Preclinical Model of Acute Myocardial Infarction. Stem Cells Transl. Med. 2019, 8(10), 1068–1083. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Boles, N.C.; Chambers, S.M.; Goodell, M.A. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell 2010, 6(3), 265–78. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, W.E.; Hoag, B.; De Dominici, M.; et al. CHIP: a clonal odyssey of the bone marrow niche. J. Clin. Invest. 2024, 134(15), e180068. [Google Scholar] [CrossRef] [PubMed]
- Milkina, E.; Ponomarenko, A.; Korneyko, M.; et al. Interaction of hematopoietic CD34+ CD45+ stem cells and cancer cells stimulated by TGF-β1 in a model of glioblastoma in vitro. Oncol. Rep. 2018, 40(5), 2595–2607. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Multi-level model of CHIP-associated neuroinflammation in cardiac surgery. The scheme illustrates the cascade from HSC mutation to POCD. Level 1 (bone marrow): somatic mutations (DNMT3A, TET2, ASXL1, JAK2) → clonal expansion (CHIP); variant allele frequency (VAF) ≥ 2% is associated with neurological risk. Level 2 (systemic circulation): hyperproduction of IL-1β, IL-6 and TNFα by mutant immunocytes, establishing a background pro-inflammatory state. Level 3 (BBB and microglia): degradation of endothelial tight junctions, increased blood–brain barrier (BBB) permeability, infiltration by mutant monocytes, microglial activation. Level 4 (neuron): IL-18-mediated hippocampal neuronal apoptosis, impaired synaptic plasticity → persistent cognitive deficit (documented in 42% of patients at 5 years after surgery).
Figure 1.
Multi-level model of CHIP-associated neuroinflammation in cardiac surgery. The scheme illustrates the cascade from HSC mutation to POCD. Level 1 (bone marrow): somatic mutations (DNMT3A, TET2, ASXL1, JAK2) → clonal expansion (CHIP); variant allele frequency (VAF) ≥ 2% is associated with neurological risk. Level 2 (systemic circulation): hyperproduction of IL-1β, IL-6 and TNFα by mutant immunocytes, establishing a background pro-inflammatory state. Level 3 (BBB and microglia): degradation of endothelial tight junctions, increased blood–brain barrier (BBB) permeability, infiltration by mutant monocytes, microglial activation. Level 4 (neuron): IL-18-mediated hippocampal neuronal apoptosis, impaired synaptic plasticity → persistent cognitive deficit (documented in 42% of patients at 5 years after surgery).

Figure 2.
Feedback loop between CHIP and adaptive immunity in the bone marrow niche. Centre (red): HSC with mutation → mutant myeloid cells hyperproducing IL-1β and IL-6. Outer ring: regulatory T cells (Treg, secreting IL-10 and TGFβ) — their suppressor function declines with age; terminally differentiated effector T lymphocytes TEM and TEMRA (secreting IFNγ and TNFα) — create an inflammatory microenvironment harmful to healthy but not mutant HSCs; B-cell precursors — their deficiency remodels the stromal niche. Red feedback arrows: IL-1β and IL-6 stimulate terminal differentiation of new T-effectors in the periphery, sustaining their influx into the bone marrow. The vicious circle closes: CHIP is simultaneously both consequence and driving force of immune imbalance.
Figure 2.
Feedback loop between CHIP and adaptive immunity in the bone marrow niche. Centre (red): HSC with mutation → mutant myeloid cells hyperproducing IL-1β and IL-6. Outer ring: regulatory T cells (Treg, secreting IL-10 and TGFβ) — their suppressor function declines with age; terminally differentiated effector T lymphocytes TEM and TEMRA (secreting IFNγ and TNFα) — create an inflammatory microenvironment harmful to healthy but not mutant HSCs; B-cell precursors — their deficiency remodels the stromal niche. Red feedback arrows: IL-1β and IL-6 stimulate terminal differentiation of new T-effectors in the periphery, sustaining their influx into the bone marrow. The vicious circle closes: CHIP is simultaneously both consequence and driving force of immune imbalance.

Figure 3.
Algorithm for personalised diagnosis and cellular correction of CHIP. The scheme presents a three-stage strategy for transition from non-specific peripheral monitoring to targeted therapy and biomedical cell product. Stage 1 demonstrates the limitations of peripheral blood as a surrogate marker: VAF does not reflect the true allelic burden in the bone marrow niche, and cytokine levels may be a consequence of infections rather than CHIP. Stage 2 — intraoperative collection of 5–10 ml of bone marrow aspirate during sternotomy (before heparinisation) enables assessment of cytokine pressure, infiltration by TEM/TEMRA cells and stromal matrix remodelling. Immunophenotyping and targeted sequencing (NGS, depth >1000×) reveal latent clones already dominant in the niche but not yet manifesting in the periphery. Stage 3 — personalised strategy: when cytokine profiles coincide in the centre and periphery — targeted pharmacotherapy (IL-1β, IL-6 or JAK2 inhibitors); when cellular correction is required — selection and expansion of HSCs in serum-free medium with TGF-β1 (10 ng/ml) and inhibitors of GSK3β and NF-κB. The final "assembly" of the biomedical cell product includes co-culture of HSCs with autologous Treg, mesenchymal stem cells (MSCs) or their secretome (enriched with TGF-β, VEGF, EGF). Reinfusion of the product to the patient after surgical treatment and anti-cytokine therapy ensures prevention of neurological complications and strategic success in the treatment of cardiovascular disease, marking the transition from chronological age to the individual molecular profile.
Figure 3.
Algorithm for personalised diagnosis and cellular correction of CHIP. The scheme presents a three-stage strategy for transition from non-specific peripheral monitoring to targeted therapy and biomedical cell product. Stage 1 demonstrates the limitations of peripheral blood as a surrogate marker: VAF does not reflect the true allelic burden in the bone marrow niche, and cytokine levels may be a consequence of infections rather than CHIP. Stage 2 — intraoperative collection of 5–10 ml of bone marrow aspirate during sternotomy (before heparinisation) enables assessment of cytokine pressure, infiltration by TEM/TEMRA cells and stromal matrix remodelling. Immunophenotyping and targeted sequencing (NGS, depth >1000×) reveal latent clones already dominant in the niche but not yet manifesting in the periphery. Stage 3 — personalised strategy: when cytokine profiles coincide in the centre and periphery — targeted pharmacotherapy (IL-1β, IL-6 or JAK2 inhibitors); when cellular correction is required — selection and expansion of HSCs in serum-free medium with TGF-β1 (10 ng/ml) and inhibitors of GSK3β and NF-κB. The final "assembly" of the biomedical cell product includes co-culture of HSCs with autologous Treg, mesenchymal stem cells (MSCs) or their secretome (enriched with TGF-β, VEGF, EGF). Reinfusion of the product to the patient after surgical treatment and anti-cytokine therapy ensures prevention of neurological complications and strategic success in the treatment of cardiovascular disease, marking the transition from chronological age to the individual molecular profile.

Table 2.
Rare mutations associated with CHIP.
| Gene | Frequency in CHIP | Mechanism/Function | Reference |
| IDH1/2 | <1% | NADP⁺-dependent isocitrate dehydrogenases; mutations lead to production of oncometabolite 2-HG, causing DNA hypermethylation |
[54] |
| KRAS/NRAS | <1% | Components of MAPK signalling pathway; constitutive activation of proliferative processes |
[55] |
| CBL | <1% | Ubiquitin ligase, negative regulator of tyrosine kinase signalling pathways | [56] |
| RUNX1 | <1% | Transcription factor essential for normal haematopoiesis |
[56] |
| U2AF1 | <1% | Spliceosomal component; along with SF3B1 and SRSF2 belongs to spliceosomal genes | [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



