Submitted:
25 June 2026
Posted:
25 June 2026
You are already at the latest version
Abstract
Percutaneous coronary intervention (PCI) treats focal coronary obstruction by compressing plaque, injuring the vessel wall and placing a metallic or bioresorbable scaffold. Most treated segments heal, but a small minority enter a prolonged, excessive or unstable repair state that contributes to in-stent restenosis (ISR), in-stent neoatherosclerosis and stent thrombosis (ST). Human evidence is strongest for delayed healing, uncovered struts, macrophage-rich neoatherosclerosis and hypersensitivity-associated late thrombosis, whereas routine immune biomarker-guided care remains unsupported. The narrowed lumen is only the visible endpoint; beneath it sits vascular repair shaped by de-vice-material exposure, local haemodynamics and host immunity. Endothelial denuda-tion and platelet activation initiate fibrin deposition, complement signalling and dam-age-associated molecular-pattern (DAMP) release. Neutrophils, monocytes and macro-phages dominate early, followed by lymphocytes and vascular smooth muscle cells that remodel the repair compartment. Drug-eluting stents (DES) have markedly reduced early neointimal hyperplasia, yet selected late failures still involve delayed endothelial recovery, chronic peristrut inflammation, hypersensitivity and neoatherosclerotic transformation. Immune biology is useful at the bedside only when interpreted with procedural context, device design and patient phenotype. Imaging-defined endpoints and paired immune phenotyping are therefore needed to guide treatment by mechanism instead of angiography or isolated biomarkers.
Keywords:
percutaneous coronary intervention
; drug-eluting stent
; in-stent restenosis
; stent thrombosis
; neoatherosclerosis
; vascular inflammation
; endothelial healing
; hypersensitivity
; macrophages
; immunothrombosis
1. Introduction
Coronary stents have changed interventional cardiology, but they have not removed the vascular cost of percutaneous coronary intervention (PCI). Contemporary drug-eluting stents (DES) have reduced the restenosis rates seen after balloon angioplasty and bare-metal stents (BMS). Target-lesion failure still matters, however, because it presents as recurrent angina, acute coronary syndrome or sudden thrombotic occlusion [1,2,3,4]. PCI does not simply restore lumen diameter but it delivers a controlled injury to a diseased arterial segment that may contain lipid, calcium and necrotic core, with inflammatory cells and disturbed flow adding further complexity.
In routine practice, stent failure is usually divided into in-stent restenosis (ISR), in-stent neoatherosclerosis and stent thrombosis (ST). The classification is useful at the bedside, but it can hide common repair pathways. ISR is often described as excessive neointimal growth, neoatherosclerosis as accelerated atherosclerosis within the neointima, and ST as thrombus formation within or adjacent to the stented segment. These phenotypes overlap through endothelial disruption, platelet-leukocyte interaction, innate immune activation and smooth muscle-cell phenotypic switching. Extracellular matrix remodelling, delayed re-endothelialisation and persistent hypersensitivity to device components add further shared mechanisms [5,6,7,8].
Immunity does not explain every failure after stenting. Technical factors such as underexpansion, malapposition, edge dissection and fracture are decisive in many cases, but mechanical and immunological mechanisms often interact. Poor deployment amplifies injury and disturbs flow, while persistent inflammation slows endothelial recovery and leaves the segment more prone to neoatherosclerosis or thrombosis. The stented coronary artery is a useful human setting in which sterile injury, foreign-body exposure and failed tissue repair intersect with immunothrombosis.
The discussion traces the path from PCI injury to failed repair, giving greatest weight to human pathology, intravascular imaging and interventional consensus. Reproducible biomarker associations are treated as intermediate evidence, whereas preclinical data, small pathway studies and indirect extrapolation from systemic anti-inflammatory cardiovascular trials are exploratory. To keep the discussion accessible outside interventional cardiology, malapposition denotes incomplete contact between stent struts and the vessel wall, and geographic miss denotes incomplete coverage of the diseased or injured segment. IVUS denotes intravascular ultrasound, and OCT denotes optical coherence tomography, the high-resolution intravascular imaging method used to assess strut coverage, thrombus, tissue pattern and neoatherosclerosis.
The proposed immune-vascular pathway from PCI-induced injury to stent failure is summarised in Figure 1.
2. PCI-Induced Vascular Injury as Sterile Immune Activation
PCI begins by fracturing plaque and stripping endothelium, with stretch injury imposed on the vessel wall. Balloon inflation and stent expansion expose subendothelial matrix, including collagen and tissue factor, creating a thrombogenic surface. Platelets then adhere and release adenosine diphosphate, thromboxane and serotonin alongside chemokines and growth factors. Fibrin deposition and complement activation reinforce leukocyte recruitment as part of vascular repair after iatrogenic injury, not as an aberrant response [5,6].
Complement sits early in the sterile-injury response. Local activation generates C3a and C5a, increases endothelial and leukocyte activation, amplifies monocyte recruitment and supports platelet-leukocyte crosstalk. In most patients the response resolves, but around uncovered struts, polymer residues or necrotic plaque, persistent complement activity amplifies delayed healing and thrombotic susceptibility. Direct stent-specific clinical evidence is still limited, so complement is not a proven standalone cause of stent failure [9,10].
Failed repair develops when the local repair response is excessive, poorly organised or persistent around stent struts. Struts projecting into the lumen alter shear stress and create areas of platelet activation and delayed endothelial coverage. Deep penetration into necrotic core, severe calcification, long treated segments and overlapping stents increase the volume of injured tissue and foreign material. The local immune response is shaped partly by PCI geometry and by the quality of the final result. Stent implantation also directly triggers leukocyte integrin Mac-1 upregulation on circulating neutrophils and monocytes, an effect linked to subsequent neointimal thickening in human studies [11].
Sterile vascular injury also releases DAMPs from damaged endothelial cells, smooth muscle cells and plaque components, thereby activating pattern-recognition pathways in neutrophils and monocyte-macrophage lineages. Neutrophils contribute early through protease release, oxidative burst and, in some contexts, neutrophil extracellular trap formation. Monocytes enter later and differentiate into macrophages that clear debris, release cytokines and influence smooth muscle-cell behaviour. A resolved reaction produces a stable neointima and endothelial layer, whereas persistence of the same biology drives ISR, neoatherosclerosis or thrombosis.
3. In-Stent Restenosis: Exaggerated Immune-Repair Responses
At its simplest, ISR reflects excessive repair within a fixed scaffold. After BMS, early restenosis was driven mainly by neointimal hyperplasia: smooth muscle-cell migration and proliferation, extracellular matrix deposition and progressive lumen loss. DES reduced this response through antiproliferative drug delivery, although the injury-repair biology remained [2,3].
Inflammation is not evenly weighted across lesions and devices. Platelet-derived growth factor, transforming growth factor-beta, interleukin signalling, monocyte chemoattractant pathways and matrix metalloproteinase activity influence smooth muscle-cell migration and matrix remodelling [5,6]. Smooth muscle cells do not behave as passive contractile cells that simply proliferate. In the broader vascular smooth muscle plasticity literature, synthetic, osteogenic, macrophage-like and pro-inflammatory states have been described [12,13]. In stented arteries, these transitions shape restenosis and calcific neoatherosclerosis when endothelial recovery is incomplete or inflammation persists around the scaffold. Among the cytokine mediators implicated, elevated plasma monocyte chemoattractant protein-1 has been documented in patients with angiographic restenosis after coronary intervention, including in early post-angioplasty cohorts, and OCT-phenotyped restenotic tissue also links macrophage-rich signal with local inflammatory biomarker elevation [14,15].
Adaptive immunity is less directly characterised than innate-cell responses in stented coronary tissue, but it remains relevant to ISR. Human restenotic tissues after DES implantation show CD45-positive T-lymphocyte accumulation with macrophage-rich inflammation, supporting lymphocyte participation in at least some restenotic phenotypes, not simply background atherosclerosis [16,17]. Th1-skewed responses sustain macrophage activation through interferon-gamma signalling, while Th17-related pathways are linked to neutrophil recruitment, endothelial activation and vascular inflammation [18,19]. Evidence for a protective Treg signal in PCI populations is lacking, and the inference rests on broader vascular-immunology data in which regulatory T cells restrain excessive cytokine signalling and support resolution [18]. B cells and antibody-mediated responses are less consistently studied in ISR, but are relevant to polymer or metal hypersensitivity, immune-complex biology and chronic foreign-body reactions. ISR is, therefore, better framed as a combined innate-adaptive repair disorder than as macrophage and smooth-muscle biology alone [20,21,22].
This heterogeneity matters clinically as underexpansion-related ISR, proliferative neointimal ISR and neoatherosclerotic ISR have different dominant causes. Treating all angiographic ISR as one endpoint weakens clinical trials and translational immunology. Imaging-guided practice is better served by classification according to cause than by percentage diameter stenosis alone. A focal restenotic lesion caused by underexpansion is not a failure of immune regulation. Conversely, recurrent diffuse ISR in a well-expanded stent suggests a stronger host-shaped phenotype, especially in the presence of diabetes, chronic kidney disease, smoking, autoimmune disease, hypersensitivity or persistent systemic inflammation [1,2,3,4].
4. Drug-Eluting Stents, Delayed Healing and Hypersensitivity
DES were designed to suppress neointimal proliferation but this therapeutic success carries a trade-off: the same antiproliferative effect delays endothelial restoration in some settings and alters immune-cell behaviour. Human autopsy studies of first-generation DES described persistent fibrin, incomplete endothelial coverage, local inflammation and late thrombotic risk, particularly in complex lesions and acute myocardial infarction settings [23,24,25,26].
Second-generation DES improved safety through thinner struts, better polymers, refined drug kinetics and greater deliverability, but they did not abolish vascular failure. Late events often reflect residual mechanical problems together with impaired endothelial function, chronic low-grade inflammation, neoatherosclerosis or systemic patient risk. A simple mechanical-versus-immune division is too crude for contemporary DES failure because many cases contain both elements.
Suppressing smooth muscle-cell proliferation reduces early restenosis, while excessive suppression of repair prolongs exposure of thrombogenic struts. A successful device is judged not only by late lumen loss but by durable patency, functional endothelial recovery and low chronic inflammatory activity, and the antiproliferative trade-off does not change that standard.
Hypersensitivity to stent components is still relevant, although it is one of the easiest mechanisms to overstate. First-generation DES raised concern because autopsy and adverse-event analyses reported eosinophilic and giant-cell-rich inflammation, aneurysmal change and late thrombotic events in some patients [27,28]. Metal alloys, durable polymers, antiproliferative drugs and antiplatelet medications have all been implicated, but proving causality in an individual coronary event is difficult.
The best evidence for hypersensitivity comes from selected pathology, thrombus-aspiration and case-based material, not large prospective screening studies. This distinction matters because nickel allergy, for example, is common, whereas proven coronary stent failure caused by metal allergy is uncommon and hard to establish. Patch testing also does not reproduce the coronary environment, where metal, polymer and drug exposure interact with blood flow, endothelial injury and plaque biology.
Hypersensitivity is documented in selected late or recurrent failures but is not a universal explanation for DES failure. The broader issue is foreign-body immune persistence, in which a stent becomes a chronic inflammatory scaffold when endothelial coverage is incomplete, polymer degradation is abnormal, strut apposition is poor or the adjacent plaque stays active [28,29].
5. Neoatherosclerosis: Accelerated In-Stent Atherosclerosis
Neoatherosclerosis represents an atherosclerotic transformation within the neointimal tissue, distinct from simple neointimal thickening, with foamy macrophages, lipid pools, necrotic core formation and calcification. In some lesions, thin-cap fibroatheroma-like morphology develops. Human pathology shows that neoatherosclerosis occurs after both BMS and DES, although it tends to appear earlier after DES [30,31].
The sequence starts with endothelial leak, lipid entry and leukocyte accumulation. A dysfunctional or incomplete endothelial layer permits lipoprotein entry and leukocyte trafficking, after which macrophages take up modified lipids and become foam cells. Oxidative stress, inflammatory cytokines and defective efferocytosis then favour necrotic core formation. Over time, the treated segment resembles accelerated atherosclerosis, although the process develops within a constrained neointimal compartment around metallic struts. Defective efferocytosis sustains this inflammatory environment in native atherosclerosis, where impaired apoptotic cell clearance drives necrotic core expansion and plaque instability, and analogous mechanisms fit the constrained neoatherosclerotic compartment [32,33].
For patients, neoatherosclerosis matters because it links restenosis and thrombosis. A lipid-rich neointima narrows the lumen gradually and causes recurrent ischaemia, while rupture or erosion of neoatherosclerotic tissue may also trigger late or very late ST. OCT has made this process visible in vivo, although OCT is not histology. Macrophage-rich signal, lipid-laden neointima, calcification and thrombus support a lesion-level interpretation that still requires clinical and procedural context [31,34,35,36]. Published OCT reproducibility data are reassuring at cohort level, but individual restenotic specimens still show enough heterogeneity that a single label often feels more precise than the tissue deserves.
Table 1 summarises the practical distinction between neointimal hyperplasia and neoatherosclerosis, while recognising that both processes may coexist within the same stented segment.
Both processes coexist in the same stented segment. Macrophage-rich lesions suggest higher late instability, while OCT serves as a useful but imperfect substitute for pathology. No validated neoatherosclerosis-specific immune therapy exists yet.
6. Stent Thrombosis and Immunothrombosis
ST is usually classified as acute, subacute, late or very late, but timing alone does not explain the underlying cause. Acute and subacute ST often reflect deployment problems, inadequate antiplatelet effect, residual dissection, tissue protrusion, thrombus burden or early interruption of therapy. Late and very late ST more often involve delayed healing, uncovered struts, malapposition, neoatherosclerosis, persistent fibrin and, in selected cases, hypersensitivity [1,4,23,24,25,26].
In this setting, immunothrombosis describes a process well beyond platelet aggregation, as neutrophils, monocytes, tissue factor, complement, endothelial cells and extracellular traps all influence thrombus composition and stability. Neutrophil extracellular traps (NETs) provide a physical and biochemical scaffold made of decondensed chromatin and histones, with neutrophil elastase, myeloperoxidase and other granule proteins adding biochemical activity. This scaffold traps platelets and red cells, concentrates tissue factor, activates factor XII, impairs endogenous fibrinolysis and intensifies endothelial injury. The pathobiological case is reasonable, though causality in individual stent thrombosis events is unproven [37,38,39].
In the stented segment, NET formation is most likely when several triggers coexist, including uncovered or malapposed struts, residual thrombus, high local shear disturbance, necrotic plaque protrusion, infection or systemic inflammatory priming. NETs interact with complement and platelet activation, creating a feed-forward loop in which neutrophil activation promotes thrombin generation, and thrombin further recruits inflammatory cells. NETs warrant study as part of a defined thrombotic process rather than as a universal explanation for ST, with thrombus aspiration, OCT-defined lesion context and circulating markers of NETosis belonging in the same future stent-failure cohorts [37,38]. In practice, the loop becomes tangible when a late thrombotic occlusion occurs in a previously treated segment and OCT shows uncovered or malapposed struts, residual thrombus and inflamed-looking neoatherosclerotic tissue despite apparently adequate systemic antiplatelet therapy.
A stent thrombosis event warrants cause-based investigation rather than management as a single uniform diagnosis. The relevant question is whether the dominant process is mechanical, pharmacological, inflammatory or mixed. Immunothrombosis framing adds value when tied to an imaged lesion, not when it replaces the conventional timing taxonomy. OCT or IVUS identifies underexpansion, malapposition, edge dissection and tissue prolapse, while uncovered struts and neoatherosclerosis identify the healing context. Without imaging, events are easily labelled as antiplatelet failure or non-adherence when the underlying cause is mechanical or inflammatory.
7. Patient-Level Immune Phenotypes and Emerging Biomarkers
Stent healing varies between patients, and diabetes, chronic kidney disease and acute coronary syndrome presentation influence endothelial repair, macrophage polarisation, smooth muscle-cell switching and thrombosis risk. Autoimmune disease, persistent high-sensitivity C-reactive protein and haematological inflammatory traits add further host-level signals. Targeted DES-ISR data also show that diabetes and chronic kidney disease shape restenotic presentation and post-ISR outcomes, supporting attention to host biology in addition to device mechanics [40]. These systemic states do not cause poor implantation, but they make an apparently acceptable PCI result vulnerable to failed healing.
Composite blood inflammatory indices, including the systemic immune-inflammation index and the systemic inflammation response index, have been associated with DES-ISR and related post-stent failure phenotypes in recent observational studies [41,42]. The signal is interesting but not ready for routine decision-making. These indices are non-specific, affected by infection, malignancy and renal disease, further confounded by physiological stress and medication exposure, and reported with thresholds that differ across populations.
Clonal haematopoiesis of indeterminate potential (CHIP) links immunology and cardiovascular disease through clonal myeloid biology. CHIP mutations, particularly involving TET2 and DNMT3A, skew monocyte-macrophage biology toward a more inflammatory phenotype and have been associated with atherosclerotic cardiovascular risk. No robust evidence shows that CHIP should change PCI technique or stent choice. It may help explain exaggerated vascular inflammation in some older patients despite similar angiographic results, and it merits inclusion in future studies linking blood immune phenotypes with OCT-defined restenosis, neoatherosclerosis and thrombosis [43,44,45,46,47].
Single-marker studies alone are unlikely to move the field and, therefore, the next step is to pair OCT/IVUS-defined stent-failure mechanisms with blood immune-cell profiles, proteomics and lipid metrics. Medication exposure and procedural data belong in the same analysis and without that integration, biomarkers may remain statistically significant but weak at the bedside.
8. Imaging and Biomarker Assessment
Angiography identifies lumen compromise, but it explains little about why a stent has failed. That limitation matters in immune-mediated stent-failure research. A 70% restenosis caused by underexpansion is not equivalent to a 70% restenosis caused by macrophage-rich neoatherosclerosis, although both are often coded simply as ISR in registries and trials.
Intravascular imaging provides the necessary separation. IVUS is useful for assessing expansion, vessel size, plaque burden and calcific constraint. OCT has higher axial resolution and is particularly useful for strut coverage, malapposition, thrombus, neointimal pattern and neoatherosclerotic features. Dedicated OCT consensus standards and stent-failure imaging reviews provide the methodological basis for using OCT to separate underexpansion, uncovered struts, thrombus and neoatherosclerosis instead of treating ISR or ST as purely angiographic labels [1,48,49]. A DES-era intravascular imaging review further supports the role of IVUS and OCT in lesion preparation, stent optimisation and mechanism-based evaluation of failure [50]. European Society of Cardiology guidelines for both chronic and acute coronary syndromes endorse intravascular imaging to guide management of stent failure [51,52].
Blood biomarkers sit at an earlier translational stage. High-sensitivity C-reactive protein, interleukin-6 and neutrophil-to-lymphocyte ratio reflect systemic inflammatory risk. Eosinophil count, platelet reactivity and composite immune indices provide additional but non-specific signals. Since none distinguish underexpansion from neoatherosclerosis or hypersensitivity, their strongest role at present is research stratification rather than routine clinical decision-making.
Table 2 summarises how angiography, intravascular imaging, circulating biomarkers and tissue-based assessment contribute differently to mechanism-based evaluation of stent failure.
9. Therapeutic Implications
Preventable stent failure is reduced most reliably by excellent PCI technique. Adequate lesion preparation, calcium modification where needed, correct sizing and complete expansion reduce avoidable injury and disturbed flow. Avoidance of geographic miss, minimisation of overlapping metal layers and imaging-guided optimisation reinforce the same principle because immune modulation does not compensate for preventable mechanical problems.
Device design remains central. Thinner struts, biocompatible or biodegradable polymers, polymer-free platforms, improved drug-release kinetics and better endothelial compatibility all respond to the foreign-body and delayed-healing problem. Each design choice involves trade-offs between radial strength, deliverability and antiproliferative efficacy, while endothelial recovery and thrombogenicity create further constraints. An optimal device is not judged by the smallest neointimal area at six months alone. Durable patency, a functional endothelial surface and low chronic inflammatory activity are the correct endpoints. Bioresorbable scaffolds test this principle directly, yet longer-term follow-up data show that scaffold resorption does not guarantee full vascular restoration of the treated segment, with residual concerns about endothelial function and late thrombotic risk persisting beyond the resorption phase [53,54].
Drug-coated balloons (DCB) are attractive in ISR because they deliver antiproliferative therapy without adding another permanent metal layer, which is especially relevant in recurrent ISR, small vessels and multilayer stents. Their immunological appeal lies in leaving no permanent scaffold behind, although DCB efficacy still depends on lesion preparation, drug transfer, tissue retention and the biology of the restenotic neointima. DES and DCB are best chosen by the cause of failure, not by habit, and pooled individual patient data show comparable outcomes between DCB and repeat DES across both BMS-ISR and DES-ISR, with lesion preparation and restenotic biology among the key determinants of response [55,56].
Systemic anti-inflammatory therapy is harder to place. Canakinumab and established colchicine trials support the broader inflammatory hypothesis in atherosclerotic cardiovascular disease [57,58,59]. The recent neutral acute myocardial infarction colchicine data are still relevant because they caution against assuming that systemic anti-inflammatory benefit in atherosclerosis will automatically translate into better PCI-healing outcomes [60]. A smaller trial in diabetic patients with BMS suggested that colchicine could reduce restenosis [61]. These data do not prove that routine anti-inflammatory treatment prevents modern DES failure in all PCI patients. Translation is likely to require high-inflammatory-risk phenotypes, careful timing and duration, and imaging-defined endpoints instead of angiography alone.
10. Evidence Strength, Limitations and Future Directions
Overall, failure after stenting is better understood as a problem of repair and interpretation than as a purely angiographic event. PCI causes vascular injury and inflammation, and delayed endothelial healing and uncovered struts are important causes of late thrombotic risk. Neoatherosclerosis is established within stents and contributes to both late ISR and ST in selected lesions. Intravascular imaging improves interpretation compared with angiography alone [34,35,48,49,50].
The next generation of studies should analyse mechanical and inflammatory failure together. A useful classification would distinguish underexpansion-dominant, hyperplasia-dominant and neoatherosclerosis-dominant phenotypes while still accommodating hypersensitivity-dominant, thrombosis-dominant and mixed patterns. Prospective cohorts are strongest when they pair OCT/IVUS-defined mechanisms with blood immune-cell profiles, including T-cell subsets, B-cell or antibody signatures, complement activation fragments and NETosis markers. CHIP status, lipid metrics, platelet function, medication adherence and procedural data belong in the same dataset. A prospective immune-mechanical registry is the next necessary step because it forces biomarkers to be interpreted alongside the implanted scaffold. Such a registry would address immune-vascular mechanisms more rigorously than studies reporting angiographic ISR against a single blood count-derived index.
Therapeutic translation is the least settled part of the field. Anti-inflammatory drugs or immune-compatible coatings are attractive only if they improve patient outcomes without increasing infection, bleeding, delayed healing or other off-target effects. Future trials warrant stratification by OCT/IVUS findings, systemic inflammatory phenotype and device type, with comparable attention to time from implantation and antithrombotic context. Otherwise, a precise immune-targeted therapy may look ineffective simply because it was tested in a mixed population. When angiographic ISR is used as the primary endpoint across several stent generations and lesion complexities, the published result loses the distinction between a poorly expanded scaffold, a drug-resistant proliferative lesion and late neoatherosclerotic failure.
Device studies need healing-quality endpoints, not only late lumen loss. Functional endothelialisation, strut coverage, macrophage-rich neointima, thrombus signal and neoatherosclerosis belong in reporting at the level of healing biology. Anti-inflammatory interventions are best tested in enriched groups such as recurrent diffuse ISR, imaging-confirmed neoatherosclerosis, high residual inflammatory risk or suspected hypersensitivity, not as blanket therapy after uncomplicated PCI.
11. Conclusions
The most actionable implication is that recurrent ISR, late neoatherosclerosis and ST require cause-based assessment instead of angiographic labelling alone. This is especially true when intravascular imaging shows delayed healing, macrophage-rich neoatherosclerosis, uncovered struts or thrombotic material in a host with persistent inflammatory risk. Prevention rests on precise PCI technique, device selection, aggressive secondary prevention and selective immune phenotyping.
A prospective immune-mechanical registry linking OCT/IVUS phenotype with immune-cell profiling, CHIP status, platelet function and patient outcomes is the study design most likely to move the field beyond generic inflammation. The practical goal is to identify the patient whose stent fails not because the lumen was treated, but because vascular repair never became stable.
Author Contributions
Conceptualization, S.K. and G.P.G.; methodology, S.K., G.Z., E.O.B. and G.P.G.; investigation, S.K., A.K., M.N., P.G., P.A., F.T., G.Z. and E.O.B.; resources, P.G., P.A., F.T., G.Z., E.O.B. and G.P.G.; data curation, S.K., A.K. and M.N.; formal analysis, S.K., G.Z., E.O.B. and G.P.G.; validation, A.K., M.N., P.G., P.A., F.T., G.Z., E.O.B. and G.P.G.; writing—original draft preparation, S.K.; writing—review and editing, S.K., A.K., M.N., P.G., P.A., F.T., G.Z., E.O.B. and G.P.G.; visualization, S.K., G.Z. and E.O.B.; supervision, F.T. and G.P.G.; project administration, S.K. and G.P.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analysed in this study. Data sharing is not applicable to this article.
Acknowledgments
During the preparation of this manuscript, the authors used OpenAI ChatGPT only to assist with the preparation and refinement of Figure 1 and the graphical abstract. The manuscript text, scientific interpretation, reference selection and final editorial decisions were prepared and verified by the authors. The authors reviewed and edited all AI-assisted outputs and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACS | Acute coronary syndrome |
| BMS | Bare-metal stent |
| CHIP | Clonal haematopoiesis of indeterminate potential |
| DAMPs | Damage-associated molecular patterns |
| DCB | Drug-coated balloon |
| DES | Drug-eluting stent |
| ECM | Extracellular matrix |
| ISR | In-stent restenosis |
| IVUS | Intravascular ultrasound |
| NETs | Neutrophil extracellular traps |
| OCT | Optical coherence tomography |
| PCI | Percutaneous coronary intervention |
| SMC | Smooth muscle cell |
| ST | Stent thrombosis |
| TCFA | Thin-cap fibroatheroma-like change |
| VSMC | Vascular smooth muscle cell |
References
- Klein, L.W.; Nathan, S.; Maehara, A.; et al. SCAI Expert Consensus Statement on Management of In-Stent Restenosis and Stent Thrombosis. J. Soc. Cardiovasc. Angiogr. Interv. 2023, 2, 100971. [Google Scholar] [CrossRef] [PubMed]
- Giustino, G.; Colombo, A.; Camaj, A.; Yasumura, K.; Mehran, R.; Stone, G.W.; Kini, A.; Sharma, S.K. Coronary In-Stent Restenosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 80, 348–372. [Google Scholar] [CrossRef] [PubMed]
- Pelliccia, F.; Zimarino, M.; Niccoli, G.; et al. In-Stent Restenosis after Percutaneous Coronary Intervention: Emerging Knowledge on Biological Pathways. Eur. Heart J. Open 2023, 3. [Google Scholar] [CrossRef] [PubMed]
- Torrado, J.; Buckley, L.; Duran, A.; et al. Restenosis, Stent Thrombosis, and Bleeding Complications: Navigating Between Scylla and Charybdis. J. Am. Coll. Cardiol. 2018, 71, 1676–1695. [Google Scholar] [CrossRef] [PubMed]
- Welt, F.G.P.; Rogers, C. Inflammation and Restenosis in the Stent Era. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Croce, K.; Morooka, T.; Sakuma, M.; Node, K.; Simon, D.I. Vascular Inflammation and Repair: Implications for Re-Endothelialization, Restenosis, and Stent Thrombosis. JACC Cardiovasc. Interv. 2011, 4, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Loscalzo, J.; Ridker, P.M.; et al. Inflammation, Immunity, and Infection in Atherothrombosis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2018, 72, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Byrne, R.A.; Joner, M.; Kastrati, A. Stent Thrombosis and Restenosis: What Have We Learned and Where Are We Going? Eur. Heart J. 2015, 36, 3320–3331. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A Key System for Immune Surveillance and Homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Kizhakkedathu, J.N.; Conway, E.M. Biomaterial and Cellular Implants: Foreign Surfaces Where Immunity and Coagulation Meet. Blood 2022, 139, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Uchida, T.; Yaguchi, I.; et al. Stent-Induced Expression and Activation of the Leukocyte Integrin Mac-1 Is Associated with Neointimal Thickening and Restenosis. Circulation 2003, 107, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Chatterjee, P.; Dave, J.M.; Ostriker, A.C.; Greif, D.M.; Rzucidlo, E.M.; Martin, K.A. Targeting Smooth Muscle Cell Phenotypic Switching in Vascular Disease. J. Clin. Invest. 2021, 131, e142213. [Google Scholar] [CrossRef]
- Cao, G.; Xuan, X.; Hu, J.; Zhang, R.; Jin, H.; Dong, H. How Vascular Smooth Muscle Cell Phenotype Switching Contributes to Vascular Disease. Cell Commun. Signal. 2022, 20, 180. [Google Scholar] [CrossRef] [PubMed]
- Cipollone, F.; Marini, M.; Fazia, M.; Pini, B.; Iezzi, A.; Reale, M.; Paloscia, L.; Materazzo, G.; D'Annunzio, E.; Conti, P.; Chiarelli, F.; Cuccurullo, F.; Mezzetti, A. Elevated circulating levels of monocyte chemoattractant protein-1 in patients with restenosis after coronary angioplasty. Arterioscler. Thromb. Vasc. Biol. 2001, 21(3), 327–34. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, G.; Dato, I.; Imaeva, A.E.; et al. Association between Inflammatory Biomarkers and In-Stent Restenosis Tissue Features: An Optical Coherence Tomography Study. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, S.; Abe, S.; Kanaya, T.; Oda, K.; Nishino, S.; Kageyama, M.; Taguchi, I.; Masawa, N.; Inoue, T. Late-Phase Inflammatory Response as a Feature of In-Stent Restenosis after Drug-Eluting Stent Implantation. Coron. Artery Dis. 2013, 24, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Lozano, I.; Bangueses, R.; Rodríguez, I.; et al. In-Stent Restenosis Is Associated with Proliferative Skin Healing and Specific Immune and Endothelial Cell Profiles: Results from the RACHEL Trial. Front. Immunol. 2023, 14, 1138247. [Google Scholar] [CrossRef] [PubMed]
- Foks, A.C.; Lichtman, A.H.; Kuiper, J. Treating Atherosclerosis with Regulatory T Cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.E.; Rao, D.A.; Zhou, J.; et al. Interleukin-17 and Interferon-Gamma Are Produced Concomitantly by Human Coronary Artery-Infiltrating T Cells and Act Synergistically on Vascular Smooth Muscle Cells. Circulation 2009, 119, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The Immune System in Atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in Atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Joner, M.; Finn, A.V.; Farb, A.; et al. Pathology of Drug-Eluting Stents in Humans: Delayed Healing and Late Thrombotic Risk. J. Am. Coll. Cardiol. 2006, 48, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Finn, A.V.; Joner, M.; Nakazawa, G.; et al. Pathological Correlates of Late Drug-Eluting Stent Thrombosis: Strut Coverage as a Marker of Endothelialization. Circulation 2007, 115, 2435–2441. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, G.; Finn, A.V.; Joner, M.; Ladich, E.; Kutys, R.; Mont, E.K.; Gold, H.K.; Burke, A.P.; Kolodgie, F.D.; Virmani, R. Delayed Arterial Healing and Increased Late Stent Thrombosis at Culprit Sites after Drug-Eluting Stent Placement for Acute Myocardial Infarction Patients: An Autopsy Study. Circulation 2008, 118, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, G.; Finn, A.V.; Vorpahl, M.; Ladich, E.R.; Kolodgie, F.D.; Virmani, R. Coronary Responses and Differential Mechanisms of Late Stent Thrombosis Attributed to First-Generation Sirolimus- and Paclitaxel-Eluting Stents. J. Am. Coll. Cardiol. 2011, 57, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Nebeker, J.R.; Virmani, R.; Bennett, C.L.; et al. Hypersensitivity Cases Associated with Drug-Eluting Coronary Stents: A Review of Available Cases from the Research on Adverse Drug Events and Reports Project. J. Am. Coll. Cardiol. 2006, 47, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Guagliumi, G.; Farb, A.; et al. Localized Hypersensitivity and Late Coronary Thrombosis Secondary to a Sirolimus-Eluting Stent: Should We Be Cautious? Circulation 2004, 109, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, K.; Kubo, S.; Inoue, K.; et al. Association of Localized Hypersensitivity and In-Stent Neoatherosclerosis with Very Late Drug-Eluting Stent Thrombosis. PLoS ONE 2014, 9, e113870. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, G.; Otsuka, F.; Nakano, M.; Vorpahl, M.; Yazdani, S.K.; Ladich, E.; Kolodgie, F.D.; Finn, A.V.; Virmani, R. The Pathology of Neoatherosclerosis in Human Coronary Implants: Bare-Metal and Drug-Eluting Stents. J. Am. Coll. Cardiol. 2011, 57, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kang, S.J.; Virmani, R.; Nakano, M.; Ueda, Y. In-Stent Neoatherosclerosis: A Final Common Pathway of Late Stent Failure. J. Am. Coll. Cardiol. 2012, 59, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Yurdagul, A., Jr.; Doran, A.C.; Cai, B.; Fredman, G.; Tabas, I.A. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front. Cardiovasc. Med. 2017, 4, 86. [Google Scholar] [CrossRef] [PubMed]
- Adkar, S.S.; Leeper, N.J. Efferocytosis in Atherosclerosis. Nat. Rev. Cardiol. 2024, 21, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.E.; Yahagi, K.; Kolodgie, F.D.; Virmani, R. Neoatherosclerosis from a Pathologist's Point of View. Arterioscler. Thromb. Vasc. Biol. 2015, 35, e43–e49. [Google Scholar] [CrossRef] [PubMed]
- Nusca, A.; Viscusi, M.M.; Piccirillo, F.; et al. In Stent Neo-Atherosclerosis: Pathophysiology, Clinical Implications, Prevention, and Therapeutic Approaches. Life 2022, 12, 393. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, F.; Byrne, R.A.; Yahagi, K.; et al. Neoatherosclerosis: Overview of Histopathologic Findings and Implications for Intravascular Imaging Assessment. Eur. Heart J. 2015, 36, 2147–2159. [Google Scholar] [CrossRef] [PubMed]
- Doring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.M.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary Neutrophil Extracellular Trap Burden and Deoxyribonuclease Activity in ST-Elevation Acute Coronary Syndrome Are Predictors of ST-Segment Resolution and Infarct Size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Massberg, S. Interplay between Inflammation and Thrombosis in Cardiovascular Pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Paramasivam, G.; Devasia, T.; Jayaram, A.A.; Abdul Razak, U.K.; Rao, M.S.; Vijayvergiya, R.; Nayak, K. In-Stent Restenosis of Drug-Eluting Stents in Patients with Diabetes Mellitus: Clinical Presentation, Angiographic Features, and Outcomes. Anatol. J. Cardiol. 2020, 23, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Yu, Z.; Xiong, Y.; Wu, Z.; Wu, Y. Systemic Immune-Inflammation Index and In-Stent Restenosis in Patients with Acute Coronary Syndrome: A Single-Center Retrospective Study. Eur. J. Med. Res. 2024, 29, 145. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Cao, Y.; Ren, R.; Zhang, S.; Zhang, C.; Hao, P.; Zhang, M. Usefulness of the Systemic Inflammation Response Index and the Systemic Immune Inflammation Index in Predicting Restenosis after Stent Implantation. J. Inflamm. Res. 2024, 17, 4941–4955. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal Hematopoiesis Associated with TET2 Deficiency Accelerates Atherosclerosis Development in Mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.C.; Madar, A.; Campbell, C.D.; He, Y.; Sultan, M.; Healey, M.L.; Xu, H.; D’Aco, K.; Fernandez, A.; Wache-Mainier, C.; Libby, P.; Ridker, P.M.; Beste, M.T.; Basson, C.T. TET2-Driven Clonal Hematopoiesis and Response to Canakinumab: An Exploratory Analysis of the CANTOS Randomized Clinical Trial. JAMA Cardiol. 2022, 7(5), 521–528. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, P.; Jaiswal, S.; Kathiresan, S. Clonal Hematopoiesis: Somatic Mutations in Blood Cells and Atherosclerosis. Circ. Genom. Precis. Med. 2018, 11, e001926. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; Walsh, K. Somatic Mutations and Clonal Hematopoiesis: Unexpected Potential New Drivers of Age-Related Cardiovascular Disease. Circ. Res. 2018, 122, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Tearney, G.J.; Regar, E.; Akasaka, T.; Adriaenssens, T.; Barlis, P.; Bezerra, H.G.; Bouma, B.; Bruining, N.; Cho, J.M.; Chowdhary, S.; et al. Consensus Standards for Acquisition, Measurement, and Reporting of Intravascular Optical Coherence Tomography Studies: A Report from the International Working Group for Intravascular Optical Coherence Tomography Standardization and Validation. J. Am. Coll. Cardiol. 2012, 59, 1058–1072. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, E.H.; Bajaj, R.; Lansky, A.J.; Mathur, A.; Baumbach, A.; Bourantas, C.V. Intravascular Imaging for Guiding In-Stent Restenosis and Stent Thrombosis Therapy. J. Am. Heart Assoc. 2022, 11, e026492. [Google Scholar] [CrossRef] [PubMed]
- Zeren, G.; Bakır, E.O.; Tufaro, V.; Özkaya, A.N.; Zhou, T.; Kyriakou, S.; Lee, J.-G.; Onuma, Y.; Serruys, P.W.; Bourantas, C.V. Intravascular Imaging for Facilitated Coronary Interventions in DES Era. J. Cardiovasc. Dev. Dis. 2026, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Vrints, C.; Andreotti, F.; Koskinas, K.C.; et al. 2024 ESC Guidelines for the Management of Chronic Coronary Syndromes. Eur. Heart J. 2024, 45, 3415–3537. [Google Scholar] [CrossRef] [PubMed]
- Byrne, R.A.; Rossello, X.; Coughlan, J.J.; et al. 2023 ESC Guidelines for the Management of Acute Coronary Syndromes. Eur. Heart J. 2023, 44, 3720–3826. [Google Scholar] [CrossRef] [PubMed]
- Serruys, P.W.; Chevalier, B.; Sotomi, Y.; et al. Comparison of an Everolimus-Eluting Bioresorbable Scaffold with an Everolimus-Eluting Metallic Stent for the Treatment of Coronary Artery Stenosis (ABSORB II): A 3-Year Randomised Controlled Trial. Lancet 2016, 388, 2479–2491. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.J.; Escarcega, R.O.; Baker, N.C.; et al. Scaffold Thrombosis after Percutaneous Coronary Intervention with ABSORB Bioresorbable Vascular Scaffold: A Systematic Review and Meta-Analysis. JACC Cardiovasc. Interv. 2016, 9, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, D.; Alfonso, F.; Xu, B.; et al. Drug-Coated Balloon Angioplasty versus Drug-Eluting Stent Implantation in Patients with Coronary Stent Restenosis. J. Am. Coll. Cardiol. 2020, 75, 2664–2678. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Münzel, T.; Schmitt, F.C.; et al. Drug-Coated Balloons for Coronary Artery Disease: Third Report of the International DCB Consensus Group. Clin. Res. Cardiol. 2022, 111, 795–808. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Jolly, S.S.; d'Entremont, M.-A.; Lee, S.F.; Cairns, J.A.; Bangdiwala, S.I.; Bignucolo, A.; Igbalode, T.; Mange, P.; Roettger, C.; Roy, A.; et al. Colchicine in Acute Myocardial Infarction. N. Engl. J. Med. 2025, 392, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.; Giannopoulos, G.; Raisakis, K.; et al. Colchicine Treatment for the Prevention of Bare-Metal Stent Restenosis in Diabetic Patients. J. Am. Coll. Cardiol. 2013, 61, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
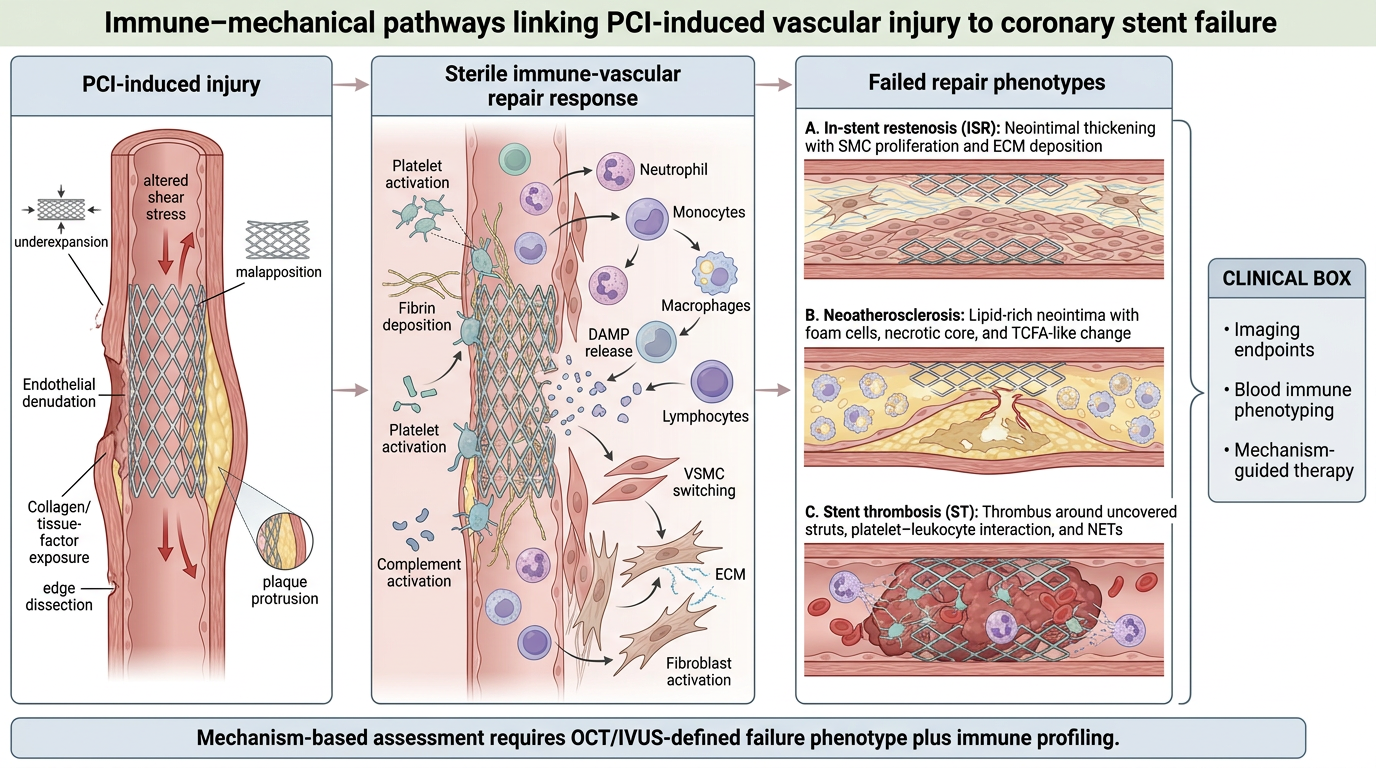
Figure 1.
Immune-vascular pathways linking PCI-induced injury to coronary stent failure. PCI produces endothelial denudation, plaque fracture, strut implantation, altered shear stress and foreign-material exposure. Mechanical factors, including underexpansion, malapposition, edge dissection, plaque protrusion, necrotic core and calcific constraint, amplify local injury and delay healing. The resulting sterile response involves platelet activation, fibrin deposition, complement activation, DAMP release, leukocyte recruitment, VSMC activation and fibroblast activation. Failed repair may progress to ISR, neoatherosclerosis or ST. Abbreviations: DAMPs, damage-associated molecular patterns; ECM, extracellular matrix; ISR, in-stent restenosis; NETs, neutrophil extracellular traps; PCI, percutaneous coronary intervention; SMC, smooth muscle cell; ST, stent thrombosis; TCFA, thin-cap fibroatheroma; VSMC, vascular smooth muscle cell.
Figure 1.
Immune-vascular pathways linking PCI-induced injury to coronary stent failure. PCI produces endothelial denudation, plaque fracture, strut implantation, altered shear stress and foreign-material exposure. Mechanical factors, including underexpansion, malapposition, edge dissection, plaque protrusion, necrotic core and calcific constraint, amplify local injury and delay healing. The resulting sterile response involves platelet activation, fibrin deposition, complement activation, DAMP release, leukocyte recruitment, VSMC activation and fibroblast activation. Failed repair may progress to ISR, neoatherosclerosis or ST. Abbreviations: DAMPs, damage-associated molecular patterns; ECM, extracellular matrix; ISR, in-stent restenosis; NETs, neutrophil extracellular traps; PCI, percutaneous coronary intervention; SMC, smooth muscle cell; ST, stent thrombosis; TCFA, thin-cap fibroatheroma; VSMC, vascular smooth muscle cell.


Table 1.
Distinguishing neointimal hyperplasia from neoatherosclerosis after coronary stenting. The table contrasts the dominant biology, key cellular features, imaging appearance, clinical consequence and therapeutic implication of two major restenotic phenotypes.
Table 1.
Distinguishing neointimal hyperplasia from neoatherosclerosis after coronary stenting. The table contrasts the dominant biology, key cellular features, imaging appearance, clinical consequence and therapeutic implication of two major restenotic phenotypes.
| Feature | Neointimal hyperplasia | Neoatherosclerosis |
|---|---|---|
| Dominant biology | Repair/proliferation after injury. | Atherosclerotic transformation within neointima. |
| Key cells | Smooth muscle cells and extracellular matrix. | Macrophage foam cells, smooth muscle cells and inflammatory cells. |
| Typical imaging clue | Homogeneous or layered neointima. | Lipidic neointima, calcification, thin-cap-like features or thrombus on OCT. |
| Consequence | Recurrent angina or target-lesion revascularisation. | Late ISR, acute coronary syndrome or very late ST. |
| Therapeutic implication | Optimise expansion and consider DES or drug-coated balloon strategies according to driver. | Aggressive secondary prevention, lipid lowering, imaging-guided assessment and careful antithrombotic decisions. |
Table 2.
Diagnostic tools for immune-mediated coronary stent failure. The table outlines the main role, limitation and research value of commonly used diagnostic approaches, from angiography and intravascular imaging to blood biomarkers and tissue-level analysis.
Table 2.
Diagnostic tools for immune-mediated coronary stent failure. The table outlines the main role, limitation and research value of commonly used diagnostic approaches, from angiography and intravascular imaging to blood biomarkers and tissue-level analysis.
| Tool | Best use | Limitation & Research role |
|---|---|---|
| Angiography | Detects restenosis, occlusion and gross thrombus. |
Limitation: Does not define tissue biology or immune driver. Research role: Endpoint screening and procedural overview. |
| IVUS | Expansion, apposition, vessel size, calcific constraint and plaque burden. |
Limitation: Limited for fine strut coverage or macrophage-rich neointima. Research role: Mechanical-tissue separation in ISR studies. |
| OCT | Strut coverage, thrombus, malapposition, tissue pattern and neoatherosclerosis features. |
Limitation: Does not replace histology, and artefacts and thrombus obscure interpretation. Research role: Phenotyping ISR and ST by tissue context. |
| Blood inflammatory markers | Systemic inflammatory tone and exploratory risk stratification. |
Limitation: Does not localise inflammation to the stented segment. Research role: Enrichment of high-risk patients for prospective studies. |
| Histopathology/thrombus analysis | Direct cellular and material-level evidence. |
Limitation: Available only in selected fatal or aspiration cases. Research role: Validation of imaging and biomarker signatures. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



