Submitted:
09 June 2026
Posted:
10 June 2026
You are already at the latest version
Abstract
Rotavirus infection in neonates is occasionally associated with neurological complications, including encephalopathy and white matter injury; however, the underlying mechanisms remain elusive. While direct viral invasion of the central nervous system (CNS) has been proposed, accumulating evidence suggests that indirect mechanisms may also contribute to the pathogenesis. In this review, we propose a barrier-centered, hypothesis-driven framework in which rotavirus nonstructural protein 4 (NSP4), a multifunctional viral protein known for its role in calcium dysregulation and enterotoxicity, may influence neuroimmune function through its effects on endothelial and epithelial barriers. Evidence derived primarily from intestinal and non-neural systems suggests that NSP4 may modulate inflammatory signaling pathways and barrier integrity. We hypothesize that circulating NSP4-related signals could interact with CNS interfaces without requiring widespread direct neuroinvasion. However, most supporting evidence remains indirect, and the relevance of these mechanisms to the neonatal CNS has not been directly established. This framework integrates current evidence while distinguishing between established findings and hypothesis-driven interpretations. Importantly, it also highlights key experimental questions, including whether NSP4 interacts with CNS barrier cells and whether such signaling alters barrier function in vivo. Addressing these questions may improve our understanding of rotavirus-associated neurological disease.
Keywords:
rotavirus
; NSP4
; neonatal encephalopathy
; innate immunity
; blood–brain barrier
; neuroimmune interaction
; cytokines
; barrier dysfunction
; vagal nerve
; hypothesis
1. Introduction
Rotaviruses are non-enveloped, double-stranded RNA viruses belonging to the family Reoviridae. They exhibit a characteristic wheel-like morphology and possess a triple-layered icosahedral capsid enclosing 11 genome segments that encode six structural (VP1–VP4, VP6, and VP7) and six non-structural (NSP1–NSP6) proteins [1]. Rotavirus infection primarily targets intestinal epithelial cells, where robust expression of non-structural glycoprotein 4 (NSP4) occurs. NSP4 functions as a viral enterotoxin and plays a central role in the induction of secretory diarrhea [1]. Despite the availability of effective vaccines, rotavirus infection remains a major global health burden, accounting for more than 200,000 deaths annually among children under five years of age [2].
Currently, four oral live-attenuated rotavirus vaccines are licensed for administration beginning at ≥6 weeks of age [3]. Although these vaccines have substantially reduced the incidence and severity of rotavirus gastroenteritis, neonates remain vulnerable to infection and its associated complications during the early postnatal period. Notably, increasing clinical evidence that rotavirus infection may be accompanied by neurological manifestations.
Neurological complications associated with rotavirus infection are increasingly recognized and appear to occur more frequently in neonates than in older infants and children, with approximately a twofold higher risk reported in the neonatal population [4]. A wide spectrum of central nervous system (CNS) manifestations has been described, including acute cerebellitis [5], encephalopathy and encephalitis [6,7], acute necrotizing encephalitis [8,9], reversible splenial lesion syndrome [10], and neonatal white matter injury or leukoencephalopathy [11,12,13,14]. In one cohort study, CNS complications were identified in 10 of 59 children hospitalized with rotavirus infection [6]. Furthermore, children aged six years with a history of hospitalization for rotavirus gastroenteritis demonstrated lower motor and cognitive performance compared with age-matched controls [15], suggesting the possibility of subclinical or delayed neurodevelopmental sequelae. Despite these observations, the biological mechanisms linking rotavirus infection to CNS dysfunction remain poorly understood. In particular, whether neurological involvement results from direct viral neuroinvasion or from indirect mechanisms such as systemic inflammation, barrier dysfunction, or host immune responses remains unresolved. Addressing this knowledge gap is essential for understanding rotavirus-associated neurological sequelae, especially during the vulnerable neonatal period.
Several hypotheses have been proposed to explain the mechanisms underlying rotavirus-associated neurological manifestations. Direct viral invasion of the CNS has been suggested in rare cases [16,17]. However, consistent evidence supporting neurotropism of rotavirus remains limited. Alternatively, increasing attention has been directed toward indirect mechanisms, including NSP4, serotonin, cytokines, and second messenger molecules, which may secondarily affect the developing brain [18].
Among rotavirus-derived factors, NSP4 has emerged as a multifunctional viral protein with pleiotropic biological effects extending beyond the intestinal epithelium. In addition to its established enterotoxigenic activity, NSP4 induces intracellular calcium dysregulation, endoplasmic reticulum (ER) stress, and inflammatory signaling pathways in infected cells. These properties indicate that NSP4, or its circulating degradation products, may influence vascular endothelial cells and immune responses at extraintestinal sites.
Notably, the neonatal period is characterized by immature blood–brain barrier (BBB) function and heightened sensitivity to inflammatory stimuli. Under such conditions, excessive innate immune activation and endothelial perturbation may render the developing brain particularly vulnerable to systemic viral insults. Based on these considerations, this review focuses on NSP4-centered mechanisms that may contribute to neuroinflammatory vulnerability through innate immune activation and barrier dysfunction, without necessarily requiring direct viral entry into the CNS.
2. Current Hypotheses and the Need for New Perspectives
Historically, the pathogenesis of CNS complications associated with rotavirus infection has primarily been attributed to direct or indirect mechanisms. Proposed routes of direct involvement include viral access through the BBB, blood–cerebrospinal fluid barrier (BCSFB), circumventricular organs (CVOs), enteric nervous system, and lymphatic pathways. Indirect mechanisms have also been suggested, including the effects of circulating proinflammatory cytokines and activated peripheral leukocytes on the developing brain [18,19]. However, consistent evidence supporting direct neuroinvasion of rotavirus remains limited.
In addition to these mechanisms, several hypotheses have focused on the role of the rotavirus NSP4. NSP4 and its peptide derivatives produced in the gastrointestinal tract have been proposed to act as neuroactive mediators within the enteric nervous system, potentially contributing to CNS manifestations [18]. Innate antiviral immune responses and inflammatory signaling, particularly in neonates, may influence CNS vulnerability. These include the potential effects of NSP4 released from rotavirus-infected monocytes on neuronal function, as well as microglial production of monocyte chemoattractant protein-1 (currently designated C–C motif chemokine ligand 2; CCL2) in the context of neuroinflammation [11]. NSP4 and serotonin activate the enteric nervous system and vagal nerves, contributing to CNS symptoms [18]. Despite these diverse hypotheses, most existing models emphasize either direct viral invasion or generalized immune-mediated injury, and no unifying framework has emerged to explain the characteristic features of rotavirus-associated acute encephalopathy and neonatal white matter injury. This gap highlights the need for renewed perspectives that integrate viral protein biology, host innate immune responses, and developmental vulnerability of the neonatal brain. Accordingly, this review proposes potential NSP4-centered mechanisms that extend beyond classical innate immune activation and may contribute to neuroinflammatory susceptibility without requiring direct viral entry into the CNS.
3. Intestinal Origin of NSP4 and Systemic Immune Activation
Rotavirus infection of intestinal epithelial cells induces the production and release of NSP4, which disrupts intracellular calcium homeostasis and activates innate immune signaling, thereby initiating systemic inflammatory responses, including the production of interleukin-6 (IL-6), IL-1β, and tumor necrosis factor (TNF-α) [1]. Rotavirus infection preferentially targets mature villous tip epithelial cells in the small intestine, leading to epithelial injury and malabsorption [20]. During the acute phase of infection, viral antigens and, in some cases, viral RNA can be detected in the systemic circulation, resulting in antigenemia and transient viremia [21]. NSP4, synthesized and secreted by rotavirus-infected enterocytes, is a multifunctional viral protein that modulates host cellular signaling pathways and contributes to viral replication and disease pathogenesis.
NSP4 activates multiple intracellular signaling cascades, including nuclear factor kappa B (NF-κB), c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase pathways, at least in part through toll-like receptor 2 (TLR2)-dependent mechanisms. Activation of these pathways results in the production of proinflammatory cytokines such as interleukin (IL)-6 and tumor necrosis factor-α in macrophage-like cell lines through modulation of TLR2-mediated innate immune responses [22]. Through modulation of TLR2-mediated innate immune responses, NSP4 facilitates rotavirus replication and amplifies local inflammatory signaling during infection [22]. Moreover, NSP1 contributes to evasion of the innate interferon response through signal transducer and activator of transcription 1 inhibition [1]. In addition to secretory diarrhea, which is caused by the effect of NSP4-serotonin axis-mediated enteric nervous system activation, osmotic diarrhea due to malabsorption is important in rotavirus gastroenteric symptoms [1]. Specifically, NSP2 induces the degradation of diacylglycerol O-acyltransferase 1, which disrupts lipid metabolism and facilitates viral replication [23]. These findings emphasize the complex interplay of multiple viral proteins in host responses.
The diarrheal phenotype characteristic of rotavirus gastroenteritis is primarily attributed to the biological activity of NSP4 produced by infected intestinal epithelial cells [24,25]. NSP4 disrupts intracellular calcium homeostasis and activates multiple signaling pathways in intestinal epithelial cells, contributing to diarrhea and innate immune activation.
In intestinal enterocytes, elevated intracellular calcium promotes chloride secretion and water efflux into the intestinal lumen, resulting in secretory diarrhea. Accordingly, NSP4 is widely recognized as a viral enterotoxin [1]. In addition to full-length NSP4 [27,28], cleavage products such as NSP4 (112–175) are released from rotavirus-infected epithelial cells in vitro [29]. Secreted NSP4 assembles into oligomeric soluble lipoprotein complexes [28] capable of binding integrin α1 and α2 I domains [30] on the cell surface. Engagement of integrin α2 by extracellular NSP4 increases cytosolic calcium levels in epithelial cell lines through phospholipase C activation and inositol 1,4,5-trisphosphate production [31]. Moreover, NSP4 exposure induces reactive oxygen species generation and chloride secretion in Caco-2 cells [32]. Collectively, both intracellular and extracellular forms of NSP4 disrupt epithelial function, impair ion transport, and damage villous architecture, thereby contributing to diarrhea [33]. Beyond epithelial injury, NSP4 also modulates innate immune gene expression. Transfection of NSP4 into MA104 cells induces upregulation of NF-κB–dependent and innate immune–related genes, including CCL20, C-X-C motif chemokine ligand 8 (IL-8), IL-6, CCL2, and nucleotide-binding oligomerization domain-containing protein 2 (NOD2) [25]. NOD2 is a key regulator of nucleotide-binding domain leucine-rich repeat-containing receptor family pyrin domain-containing protein 3 (NLRP3) inflammasome activation, which can lead to IL-1β production in brain endothelial cells (BECs) [34].
Taken together, these findings establish NSP4 as a potent mediator of epithelial dysfunction and innate immune activation in the intestine. Given the shared signaling pathways between intestinal epithelial cells, vascular endothelial cells, and innate immune cells, it is biologically plausible that NSP4 and its degradation products may exert similar immunomodulatory effects at extraintestinal sites. In neonates, where BBB and choroid plexus barrier functions are immature, such mechanisms may contribute to neuroinflammatory susceptibility. Elucidating whether and how NSP4 interacts with BECs, choroid plexus epithelial cells, and innate immune cells remains a critical area for future investigation. Collectively, these findings support the intestine as the primary source of NSP4-driven systemic immune activation during rotavirus infection.
4. Systemic Dissemination of NSP4 and Barrier-Associated Mechanisms
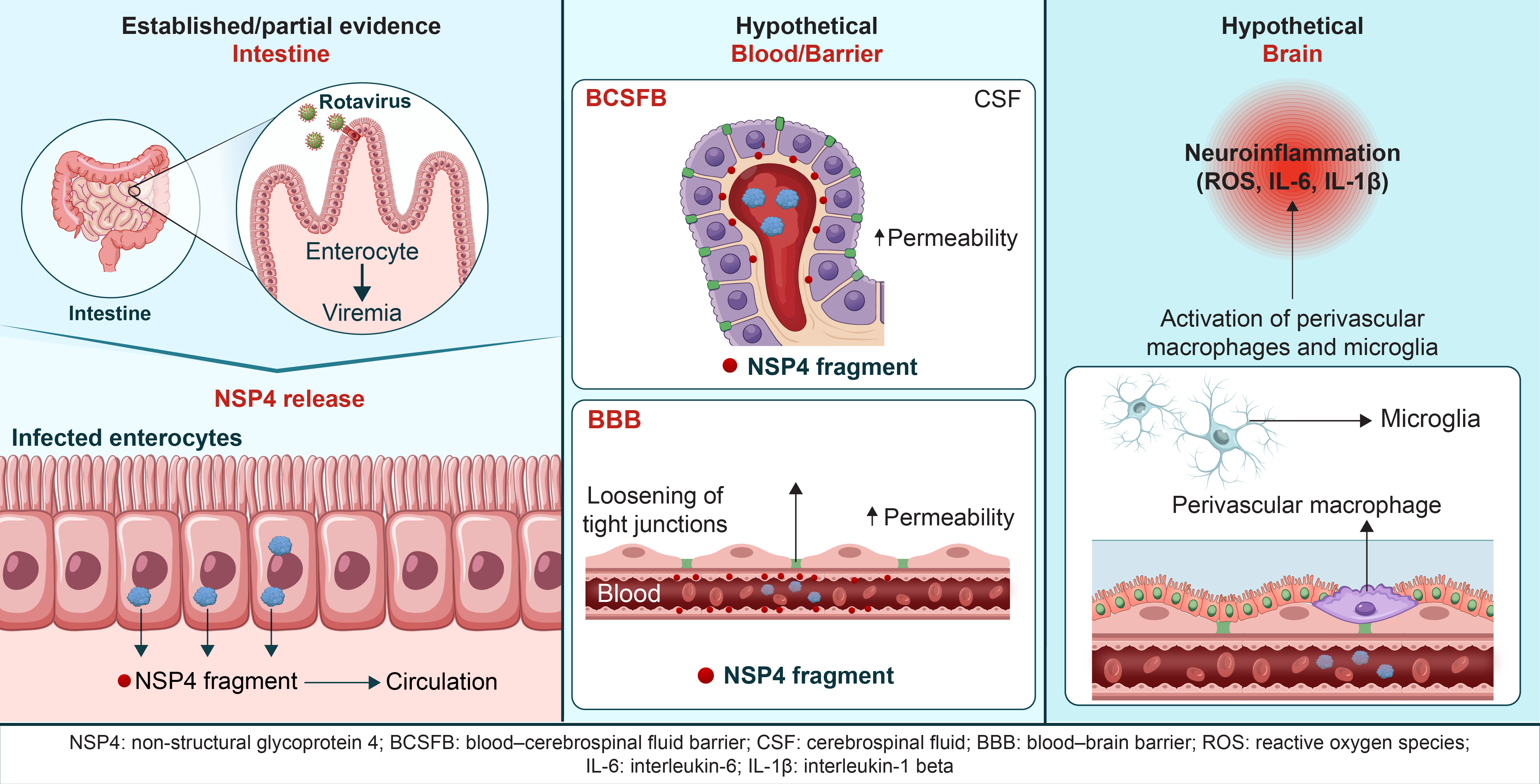
The following sections outline a set of interconnected and hypothesis-driven mechanisms, derived from established findings in intestinal rotavirus infection, innate immune signaling, and developmental neurobiology. Although direct experimental evidence in the CNS is limited for many of these pathways, they are presented here as biologically plausible models intended to stimulate future experimental investigation rather than to assert definitive pathogenic mechanisms. Following intestinal infection, NSP4 and inflammatory mediators disseminate systemically, where the unique immunological features of neonates may predispose to exaggerated or dysregulated innate immune activation. The proposed inflammatory pathways underlying this pathology are illustrated in Figure 1.
NSP4, a rotaviral enterotoxin, induces calcium dysregulation and inflammatory signaling in intestinal epithelial cells. Circulating NSP4-related factors and cytokines may interact with the BBB, potentially leading to neuroimmune activation. These CNS-related mechanisms remain speculative and require experimental validation. Solid lines indicate mechanisms supported by experimental evidence, primarily in intestinal systems. Dashed lines indicate hypothetical pathways that have not been directly demonstrated in the neonatal central nervous system.
The mechanisms illustrated in this figure are further categorized according to their level of supporting evidence in Table 1 and Table 2.
4.1. NSP4 and NSP4 Fragments
Although direct demonstration of the release of NSP4 and the NSP4 fragment into the systemic circulation of patients with rotavirus gastroenteritis is not known, the presence of anti-NSP4 IgG antibodies in the serum of afflicted children [35] suggests that NSP4, which is synthesized within rotavirus-infected enterocytes, may disseminate into the systemic circulation [19,36]. The cleavage product of NSP4, namely NSP4 112–175 [29], exhibits adjuvant activity [37], presumably via TLR2 [22], whereas the full-length NSP4 [28] is discharged from rotavirus-infected cells. Moreover, the NSP4 protein produced by rotavirus-infected intestinal cells can be secreted from the apical side [27] and interacts with basement membrane components, such as fibronectin and laminin-3, in infected neonatal mice [38]. It could enter the circulation from the tissue beneath the basement membrane [38]. Therefore, it is speculated that free and fibronectin/laminin-β3-bound NSP4 and NSP4 fragments are released from infected enterocytes into the bloodstream. These proteins could then potentially interact with BECs, choroid plexus epithelial cells, and innate immune cells, thereby contributing to inflammatory signaling and cytotoxic stress within the CNS. It is crucial to ascertain whether these disseminated factors can interact with BECs, choroid plexus epithelial cells (CPECs), and innate immune cells, inducing an inflammatory response on the CNS.
4.2. Innate Immune Activation of BECs
NSP4 may directly activate BECs through innate immune pathways, leading to cytokine production and increased BBB permeability in the neonatal period. BECs express TLR2 [39], and the expression of TLR2 and the other innate immune signaling proteins is enhanced in the neonatal mouse brain [40]. We previously hypothesized innate immune activation in severe acute respiratory syndrome coronavirus 2 infection in the neonatal brain under enhanced expression of innate immune molecules [41]. Similarly, we propose the mechanism considering the innate immune response to rotavirus infection in the neonatal brain.
Activation of TLR2 by Pam3Cys and the gram-positive bacterial cell component peptidoglycan-associated lipoprotein induces IL-6 production in in vitro cultures of human endothelial cells [42]. Moreover, IL-6 production by BECs plays a significant role in the TNF-α-dependent generation of reactive oxygen species and the downregulation of adherens and tight junction proteins, ultimately disrupting BBB integrity in experimental models [43]. Similarly, the release of the NSP4 fragment into the circulation may directly interact with BECs, potentially increasing permeability via TLR2.
TLR2 activation in macrophages by NSP4 [22] can trigger the same immune response in circulating monocytes, including IL-6 and TNF-α production. Dendritic cells (DCs) are activated via TLR2, producing IL-6 and TNF-α [44]. Thus, perivascular immature DCs, when activated by NSP4 and its fragment, may result in similar responses within the neonatal brain. Circulating monocytes can interact with perivascular macrophages and, inpathological conditions, replace them [45]. Therefore, it is conceivable that NSP4- and NSP4-fragment-activated monocytes activate BECs, brain perivascular macrophages, and perivascular immature DCs, producing pro-inflammatory cytokines and chemokines. This, in turn, may heighten BBB permeability. NSP4-induced innate immune activation in BECs provides a plausible mechanism for BBB dysfunction in neonatal rotavirus infection.
4.3. Interleukin-8
Children with rotavirus gastroenteritis who exhibit antigenemia and extraintestinal manifestations have elevated serum IL-8 levels [81]. Moreover, neonates affected by rotavirus infection-induced white matter injury display elevated CSF levels of IL-8 [11]. Rotavirus boosts IL-8 gene expression by activating inhibitor of NF-κB kinase in HT-29 cells [82], whereas NSP4 induces IL-8 secretion in TLR2-expressing HEK293 cells [22]. IL-8 plays a crucial role in disrupting the integrity of tight junctions in microvascular endothelial cells by activating the C-X-C chemokine receptor (CXCR)1 and CXCR2 [84]. These observations may suggest that rotavirus infection may initiate IL-8 signaling in BECs and CPECs, ultimately resulting in increased barrier permeability. NSP4 is a multifunctional rotaviral protein that has been shown to disrupt intracellular calcium homeostasis [31], induce ER stress [96], and modulate innate immune signaling in infected cells [22]. These effects are well documented in intestinal epithelial systems and are thought to contribute to rotavirus-induced diarrhea. However, most of these findings are derived from in vitro systems or non-neural tissues, and their direct relevance to the neonatal CNS remains uncertain. Based on these observations, we propose a barrier-centered hypothesis in which circulating NSP4 or NSP4-related signaling molecules may influence the function of endothelial and epithelial barriers, including the BBB and the BCSFB. These interactions may, in turn, alter barrier integrity and promote neuroimmune activation.
Although direct evidence in the neonatal central nervous system remains limited, several potential mechanisms may be considered to explain how NSP4 and rotavirus-associated signaling could influence barrier integrity. These include modulation of tight junction proteins [63,64,65,66,67,69,70,111], activation of TLR2 signaling pathways in the macrophages [22] and choroid plexus [53,54,55], TLR signaling mediated inflammasome activation [88], decreased regulatory T cells/Th17 cells ratio [48], disruption of barrier integrity by IL-8 [11,84] and TLR3 [112,113], induction of ER stress [96,114], and oxidative stress responses [32,115]. However, these mechanisms have not been directly demonstrated in BBB and BCSFB and should therefore be considered hypothetical.
Similarly, possible routes by which rotavirus, NSP4 or its fragments could damage and/or access the CNS—such as plasma membrane permeabilization by NSP4 48–91 [62], integrin interactions [56,71,72,73,75] and receptor-mediated transport [116], or caveolae-dependent transcytosis [57,117]—remain speculative and require experimental validation. However, if NSP4 and NSP4 fragments increase and/or are taken up by BECs and CPECs via the endocytic mechanism, it may be important to determine whether an innate immune response can be induced, as occurs in intestinal epithelial cells [25,34].
Notably, these mechanisms have been primarily characterized in intestinal or non-neural systems, where NSP4-related effects on epithelial barrier function are better established. By contrast, whether similar processes occur at central nervous system barriers, such as the BBB or BCSFB, remains unclear. Therefore, the extension of these mechanisms to the neonatal brain should be regarded as hypothetical and requires further experimental validation.
5. Possible Direct Penetration of Rotavirus into the CNS
Although immune-mediated mechanisms are likely primary, direct viral penetration may occur in a limited subset of cases. Rotavirus RNA has been detected in barrier-associated brain cells [74], but direct viral invasion of the CNS appears to play a limited role compared with barrier-centric inflammatory mechanisms. Although direct viral invasion is unlikely to represent the primary mechanism underlying rotavirus-associated neurological complications, pathological and experimental evidence has suggested that it may occur as a secondary process under specific conditions. Rotavirus causes extraintestinal dissemination in neonatal rat models [118], and viremia is relatively common during rotavirus infection in children [21]. Nevertheless, direct CNS invasion is rare [119].
One proposed mechanism involves CCL2-mediated recruitment of rotavirus-infected monocytes into the brain [11]. The BBB and BCSFB disruption mechanisms may create a permissive environment for such viral entry. Supporting this notion, in children with systemic endothelialitis associated with rotavirus infection, viral RNA has been detected predominantly in BECs, as well as in perivascular mononuclear cells, neurons, and microglia [74]. This distribution suggests that, when direct CNS invasion does occur, the BBB may represent the principal route of entry. While it has not been established that rotavirus infects BECs and CPECs, we hypothesize that rotavirus and NSP4, which are encapsuled within exosomes released into blood stream from infected intestinal epithelial cells, may enter these cells, because some viruses can be transmitted by this mechanism [120]. NSP4 is known to be secreted from rotavirus-infected cells as an extracellular oligomeric lipoprotein capable of binding non-infected cells through cell-surface glycosaminoglycans [28]. In addition, rotavirus components have been reported to associate with extracellular vesicles (EVs) containing exosome markers (such as CD63) [121], and EVs can interact with brain endothelial cells and undergo endocytic/transcytotic trafficking at the BBB [122]. Although there is currently no direct evidence that NSP4 and rotavirus themselves are EV-packaged and delivered to BECs, these findings suggest a plausible mechanism by which circulating NSP4 as well as rotavirus, either in soluble form or potentially in association with EVs, could access the BBB and modulate BEC function. Future studies should determine whether NSP4 and rotavirus are incorporated into EVs released from infected intestinal epithelial cells and whether such EVs are taken up by BECs to alter barrier integrity or inflammatory signaling, and/or penetrate into the CNS.
Importantly, intestinal TLR3 responses in neonates are developmentally limited and do not effectively restrict rotavirus replication [100]. On the contrary, TLR3 expression in the neonatal brain is developmentally increased [101]. TLR3 signaling in the immature brain may be biphasic and context-dependent: transient activation before ischemia may induce tolerance [101,123], whereas sustained activation during ongoing neonatal viral exposure may promote neuroinflammation. Therefore, during neonatal rotavirus infection, insufficient intestinal antiviral control may permit prolonged exposure of the immature brain to rotavirus and viral products including NSP4, where sustained and enhanced TLR3-related signaling in BECs, neurons, and pre-oligodendrocyte-rich white matter may contribute to injury rather than tolerance.
The frequent absence of rotavirus RNA in CSF does not preclude CNS involvement, as endothelial or perivascular localization may trigger neuroimmune responses without detectable viral shedding into CSF. Although direct CNS invasion by rotavirus may occur, available pathological evidence suggests it contributes only modestly to overall disease pathogenesis.
6. BBB and BCSFB as Immunologically Active Interfaces in Neonates
6.1. Barrier-Centered Innate Immune Activation
Rotavirus-associated neuroinflammation is initiated at CNS interfaces (BBB, BCSFB, and CVOs), where perivascular innate immune cells amplify NSP4-driven inflammatory signaling without deep parenchymal viral invasion. Accumulating evidence has suggested that rotavirus-associated neurological complications are not primarily driven by widespread viral invasion of the brain parenchyma but rather by exaggerated innate immune activation at CNS interfaces. In neonates, the BBB, BCSFB, and CVOs constitute immunologically active zones enriched with perivascular macrophages, dendritic cells, and other innate immune sentinels. Rotavirus infection, particularly through the actions of the viral enterotoxin NSP4 and its degradation products, may perturb these barrier-associated cells by engaging pattern recognition receptors and inflammatory signaling pathways. This results in the amplification of cytokine and chemokine cascades, such as IFN-γ, CCL2, high mobility group box 1 (HMGB1), and IL-17, which propagate neuroinflammation without requiring extensive viral replication within neurons or glial cells. Such a barrier-centric model reconciles neuropathological findings showing preferential viral RNA localization in brain endothelial and perivascular cells with clinical observations of encephalopathy occurring in the absence of overt CNS viral burden. In the neonatal brain, where immune regulation and barrier integrity are developmentally immature, this interface-focused inflammatory response may represent a critical mechanism underlying rotavirus-associated neurological vulnerability.
6.2. Barrier-Adjacent Innate Immunity
6.2.1. Perivascular Macrophages
Rotavirus can infect and replicate in pneumocytes and macrophages in the lungs and livers in the neonatal rat [118]. It can target macrophages in the livers of mice with a congenital biliary atresia model and release macrophage inflammatory protein 2 and Cxcl2 which recruits neutrophils [124]. Thus, we hypothesize that rotavirus may infect perivascular macrophages in the neonatal brain and recruit neutrophils. Rotavirus-infected macrophages produce IL-6 and IL-1β through mitochondrial-associated protein signaling [125]. Therefore, if rotavirus crosses the BBB and BCSFB, it may be captured by brain perivascular macrophages, inducing an innate immune-inflammatory response. Moreover, as discussed above, NSP4 and its fragment may induce TLR2-mediated TNF-α production in macrophages. Circulating TLR2-activated monocytes may also activate brain perivascular macrophages.
6.2.2. Perivascular Dendritic Cells
Rotavirus infects neonatal DCs, inducing IL-6 and TNF-α, and sustaining viral replication while also promoting the proliferation of IFN-γ-producing CD4+ T cells [94]. Particularly, immature DCs are more capable of adhering to TNF-α-stimulated BECs and crossing the BBB than mature DCs [126]. Furthermore, CD14+ monocytes migrate across BECs. Activated BECs secrete granulocyte-macrophage colony-stimulating factor and transforming growth factor β, which promote differentiation and maturation of peripheral blood CD14+ monocytes into perivascular DCs [127]. These DCs produce IL-6 and IL-12p70, which promote the development of IFN-γ-producing Th1 cells and IL-17-producing Th17 CD4+ T lymphocytes [127]. Thus, rotavirus infection may activate perivascular DCs in the brain, inducing IFN-γ and IL-17-mediated neuroinflammation, which will be discussed later. However, if rotavirus replicates in brain perivascular macrophages and DCs, then viral genomes may not be detected in the CSF using polymerase chain reaction.
6.2.3. Circumventricular Organs
CVOs have highly permeable, fenestrated capillaries that play an important role in bidirectional communication between peripheral blood and brain parenchyma. The peripheral administration of the TLR2 ligand Pam3CSK4 activates NF-κB to induce cytokine/chemokine production in perivascular macrophages and microglia in the CVOs [128]. Moreover, peripheral TLR2 activation increases c-Fos expression in astrocytes and neurons in the CVOs and adjacent regions [128]. Thus, we propose a mechanism whereby circulating NSP4 and NSP4 fragments reach the CVOs, where they induce neuroinflammation. Furthermore, pretreatment with the TLR4 ligand lipopolysaccharide (LPS) increases TLR2 expression in the CVOs [118], and systemic administration of LPS upregulates Tlr2 mRNA levels in the choroid plexus and activates NF-κB in adjacent microglial cells [129]. In a rotavirus-induced experimental model of biliary atresia, rotavirus disrupts tolerance to physiological levels of LPS, thereby increasing the expression of the TLR4/NF-κB pathway and metalloproteinase-7. Thus, in rotavirus infection, although the NSP4 fragment alone exhibits weak TLR2 activity, TLR2- and TLR4-mediated inflammatory signaling may be activated in the choroid plexus and CVOs, particularly in the presence of LPS.
6.2.4. Junctional Adhesion Molecule-A
Given that astrocytic Junctional adhesion molecule-A (JAM-A) contributes to immune cell trafficking into the CNS by increasing metalloproteinase-2 levels [69], rotavirus binding to JAM-A [63] could activate immune cell entry into the perivascular space. The entry of activated T cells into the brain is also increased by damage to JAM-A [69].
6.3. Amplifiers of Neuroinflammation
6.3.1. Natural Killer Cells
In a low-dose rotavirus-induced congenital biliary atresia model, persistent activation of natural killer (NK) and CD8+ T cells leads to long-term hepatobiliary inflammation, which is associated with enhanced mRNA levels of Th1 cytokines, Ifnγ, Cxcl9, and Cxcl10 [130]. In the rotavirus-associated congenital biliary atresia model, paucity of Tregs in the liver decreases suppressive control on NK cells [131]. Thus, suppressed Treg activity in the brain could activate NK cells, causing enhanced and persistent neuroinflammation.
6.3.2. Interferon-Gamma
Rotavirus increases IFN-γ mRNA expression in peripheral blood mononuclear cells [51], and IFN-γ levels in the CSF of neonates with leukoencephalopathy are high [132]. Rotavirus induces IFN-γ production by infecting neonatal DCs [94]. T cells produce IFN-γ via α2β1 integrin [133], suggesting rotavirus as well as NSP4 may trigger IFN-γ production in BECs-crossing T cells by activating α2β1 integrin signaling. Moreover, NK cells migrate across the BBB and produce IFN-γ by interacting with activated microglia [134]. Furthermore, IFN-γ primes the TLR-mediated inflammatory responses by macrophages [135] and activates TNF-α/NF-κB signaling in microglia [136]. Although IFN-γ and TLR play an important role in enhancing microglial activation [137], IFN-γ plus transforming growth factor β contributes to inflammation and mucosal damage in gastrointestinal inflammation [138]. The same responses may occur in the BBB, BCSFB, and microglia during rotavirus infection, contributing to barrier disruption and enhanced neuroinflammation.
6.3.3. Chemokine Ligand 2
Microglial production in response to rotavirus infection hypothetically increases levels of CCL2 production by microglia, contributing to white matter injury in neonates [11]. The precise mechanism of increased CCL2 levels in the brain and its role remains unclear. TNF-α and IFN-γ increase CCL2 production by BECs [139]. In the intestine, CCL2 recruits inflammatory monocytes via Nod2, which senses pathogens [140]. Therefore, the increased expression of Nod2 in developing mice brains [40] may lead to enhanced peripheral monocyte recruitment in rotavirus infection.
Peripherally stimulated TNF receptor 1-mediated signaling via BECs activates microglia to produce CCL2 [139]. Moreover, IFN-γ activates the interaction of CD40 with CD40 ligand in microglia and CCL2 production [141]. CCR2+monocytes enter the CNS via the choroid plexus, transform into microglia-like cells that promote neuroinflammation in the hypoxic-ischemic injury neonatal mice model [142]. Astrocytes are stimulated by IL-1β and TNF-α to release CCL2 [143], and astrocytes-derived CCL2 also evolves neuroinflammation [144]. Thus, BECs, microglia, and astrocytes may release CCL2, recruiting CCR2+monocytes, which contribute to neuroinflammation.
6.3.4. High Mobility Group Box 1
Elevated levels of high mobility group box 1 (HMGB1) have been reported in the CSF of patients with acute encephalopathy due to viral infection, including rotavirus infection [145]. Rotavirus-infected macrophages in the biliary tract release HMGB1 [146], which is also released through double-stranded RNA-dependent protein kinase-mediated inflammasome activation [147]. Similarly, it seems possible that rotavirus-infected brain perivascular macrophages release HMGB1, contributing to inflammasome activation and neuroinflammation. The elevated IL-1β levels observed in the CSF of patients with acute encephalopathy due to rotavirus infection [148] may support this mechanism.
HMGB1 released from rotavirus-infected macrophages activates NK cells via TLR2/TLR4-mitogen activated protein kinase signaling pathways [146]. IFN-γ-activated macrophages also secrete HMGB1, facilitating transendothelial migration of monocytes [139]. Due to limited NK cell activation in neonatal mice, rotavirus can persistently infect cholangiocytes [146]. The same mechanism may also occur in brain perivascular macrophages, leading to persistent HMGB1 production and NK cell activation. Therefore, rotavirus-infected and IFN-γ-activated macrophages may induce monocyte migration and NK cell activation, contributing to persistent neuroinflammation.
HMGB1 activates astrocytes and induces the production of inflammatory mediators, including cyclooxygenase-2 and chemokines [149]. Thus, we propose the initial activation of brain perivascular macrophages by rotavirus and IFN-γ triggers inflammatory reactions in microglia and astrocytes.
6.3.5. Interleukin-17
Children with rotavirus enteritis have elevated serum levels of IL-17 and circulating Th17 cells [48]. Moreover, IL-17 is higher in the CSF of children with convulsions following gastroenteritis [150]. In the rotavirus-associated congenital biliary atresia model, IL-17-producing γδ T cells infiltrating the periportal area in the liver contribute to inflammation, and anti-IL-17 therapy ameliorated infiltration of inflammatory cells [151]. In the brain, IL-17-producing γδ T cells increase BBB permeability [152]. Therefore, rotavirus may induce IL-17-mediated neuroinflammation in the neonatal brain by inducing γδ T cells around the BBB and BCSFB, as in the neonatal liver. The binding of rotavirus to integrin on BECs may initiate γδ T cells-mediated barrier disruption and neuroinflammation.
Moreover, as discussed above, rotavirus-infected BECs and brain perivascular macrophages may release HMGB1, which upregulates TLR2 on CD14+ monocytes and promotes Th17 cell differentiation [153]. TNF-α and IL-17 facilitate Th17 cells’ adhesion to BECs and transmigration of Th17 cells across the BBB [154]. In addition to decreased Treg/Th17 ratio in the blood in rotavirus infection [48], the transmigration of Tregs across the BBB can be suppressed by IL-6 production by perivascular DCs, macrophages, and BECs. Thus, rotavirus infection may relatively increase Th17 cells’ migration across the BBB than Treg cells, decreasing the Treg/Th17 ratio in the CNS. IL-1β stimulates microglia to produce IL-17 using an autocrine mechanism [155]. IL-17 also causes astrocytes to produce IL-6 and IL-1β [156]. Therefore, microglia and astrocytes may persistently be activated, unless IL-1β and IL-17 are blocked.
6.4. Resident Glial Response
6.4.1. Microglia and Astrocytes
Microglia are activated by TLR2/NF-κB and p38 mitogen activated protein kinase to produce reactive oxygen species (ROS) [157]. In microglia, activating the TLR2/myeloid differentiation primary response protein 88 pathway contributes to neurodegeneration [158]. In astrocytes, TNF-α increases TLR2 expression and nitric oxide release via an NF-κB-dependent pathway, acting in an autocrine and paracrine manner [159]. Thus, TNF-α and ROS-mediated mechanisms in microglia and astrocytes may contribute to neuroinflammation and neuronal damage in rotavirus infection. However, the activity of TNF-α may not be reflected in its concentration in CSF because of its autocrine-paracrine action. Rotavirus-infected epithelial cells release adenosine diphosphate [77]. Extracellular adenosine diphosphate activates the NLRP3 inflammasome in microglia, as well as the NF-κB/IL-6 and IL-1β pathways [160]. Moreover, because viral RNA was detected in microglia in a pathological study [74], TLR3-mediated inflammatory cytokine production may be induced.
7. Neuroimmune Consequences in the Developing Brain
Neonatal rotavirus infection has been associated with neurological complications, including encephalopathy and white matter injury. However, the mechanisms underlying these manifestations remain incompletely understood.
We propose that barrier dysfunction and systemic inflammation may contribute to neuroimmune activation in the developing brain. In this context, NSP4-induced calcium dysregulation [31] and ER stress [115]—well characterized in intestinal cells—may have analogous effects if similar pathways are engaged in barrier-associated or neural cells. However, direct evidence for such effects in the neonatal CNS is currently lacking.
TLR3 recognizes rotavirus double-stranded RNA [112]. TLR3 is developmentally upregulated in the neonatal brain [101], where it regulates neural [161] and oligodendrocyte maturation [113], whereas in the neonatal intestine its expression and antiviral function is impaired [100].
Human neurons induce IL-6, TNF-α, CCL5, and CXCL10 production via TLR3 in response to double-stranded RNA [164]. Moreover, peripheral IL-1β stimulation in mice induces TLR3 upregulation in immature oligodendrocytes [113]. While intestinal TLR3 expressions and function are immature in early life, resulting in insufficient antiviral interferon responses and prolonged viral replication, the developing brain exhibits relatively heightened TLR3 expression and responsiveness. This imbalance allows persistent viral components, including double-stranded RNA and NSP4, to access the systemic circulation and potentially prolong interaction with the immature BBB. Therefore, a key feature of neonatal rotavirus-associated encephalopathy and white matter injury may lie in the developmental mismatch of TLR3 signaling between the intestine and the brain. On the contrary, TLR3 activation has a protective role in hypoxic-ischemic injury in neonates [123]. Thus, the developmentally enhanced expression of TLR2 [40] and TLR3 [101] in the neonatal brain as well as their pleomorphism, may hypothetically modify anti-rotaviral innate immune responses, including interferon, cytokine, and chemokine synthesis, thereby damaging the BBB and neurons in the neonatal brain, by a similar mechanism. Overall, we propose that rotavirus-associated neurological complications may arise from a combination of barrier dysregulation and neuroimmune activation, rather than widespread direct viral invasion. This model remains hypothetical and requires experimental validation.
8. Implications for Prevention and Therapeutic Targeting
Understanding NSP4-mediated barrier dysfunction and innate immune activation suggests a conceptual framework for preventive strategies and therapeutic interventions aimed at reducing neurological sequelae.
9. Research Gaps
Based on this review, we propose a hypothetical model in which NSP4 and NSP4 fragments trigger multiple innate immune responses in the brain. Elucidating the mechanisms of encephalopathy, particularly in neonates, requires understanding the bioactivity and neurotoxicity of NSP4 and its fragments, as well as their effects on neural cells and immune molecule activation in the developing brain. The development of treatments for preventing sequelae is expected to continue in the future.
10. Limitations
This review is inherently limited by its hypothesis-generating nature and by assumptions derived from known biological activities of rotavirus and NSP4. At present, direct experimental evidence establishing a causal relationship between NSP4 and neonatal CNS injury has not been published, and this manuscript does not demonstrate direct neurotoxicity or definitive CNS invasion by NSP4 or its peptide fragments. Furthermore, it remains unclear whether NSP4 or NSP4-derived peptides can directly induce TLR2-mediated inflammatory signaling in BECs, microglia, or astrocytes under physiological conditions. The frequent absence of detectable rotavirus RNA in CSF in clinical cases further complicates the interpretation of CNS involvement and highlights the difficulty of linking peripheral infection to central pathology.
Experimental validation is therefore required to determine whether peripheral administration of NSP4 or its fragments can elicit innate immune activation within the neonatal brain in vivo, and whether such responses are sufficient to disrupt BBB integrity or white matter development. In addition, in silico molecular dynamics analyses may suggest insights into potential interactions between NSP4, BECs, innate immune cells, neurons, astrocytes, and oligodendrocytes; however, such approaches remain predictive and require biological confirmation.
Despite these limitations, this review suggests a conceptual framework that integrates existing clinical, pathological, and molecular observations and aims to generate testable hypotheses for future experimental and translational studies, particularly in the neonatal period.
11. Discussion
The proposed mechanisms and their levels of supporting evidence are summarized in Table 1. This framework integrates findings from intestinal systems, immune signaling studies, and limited CNS-related observations, and distinguishes between mechanisms that are experimentally supported and those that remain indirect or hypothetical in the context of the neonatal brain.
Accumulating evidence from intestinal and other non-neural systems suggests that NSP4 may disrupt epithelial integrity and modulate host responses through multiple pathways, including calcium signaling, cytokine induction, and barrier dysfunction. These findings provide a basis for considering whether NSP4-related factors may enter the circulation and interact with distal tissues. However, direct evidence supporting such systemic effects, particularly in the context of the neonatal CNS, remains limited.
Although several mechanisms may be considered to explain how NSP4 and rotavirus-associated signaling could influence barrier integrity—such as modulation of tight junction proteins, activation of TLR-mediated pathways, induction of ER stress, and oxidative stress responses—these processes have been primarily characterized in intestinal or non-neural systems. Whether similar processes occur at CNS barriers, such as the BBB or BCSFB, remains unclear. Therefore, their relevance to the neonatal brain should be regarded as hypothetical and requires further experimental validation.
Importantly, the neonatal period is characterized by developmental immaturity of barrier systems and heightened sensitivity to inflammatory and metabolic perturbations. In this context, even indirect or transient systemic signals may have disproportionate effects on barrier function and neuroimmune regulation. This raises the possibility that NSP4-associated signaling, even in the absence of direct CNS invasion, could influence brain development through secondary mechanisms involving circulating mediators and barrier-associated immune activation.
Notably, some pathways, such as TLR3 signaling, may exert context-dependent effects, including potentially protective roles in the immature brain, and therefore should not be interpreted as uniformly pathogenic. This highlights the importance of carefully distinguishing between protective and detrimental immune responses in the developing CNS.
The strength and limitations of the proposed mechanisms are further categorized in Table 2. While several pathways, including NSP4-induced calcium signaling and cytokine responses, are supported by experimental data in intestinal or immune systems, their direct relevance to the neonatal CNS remains uncertain. In particular, mechanisms involving BBB permeability changes, infection of brain endothelial cells, and immune cell activation within the CNS are supported only by limited or indirect evidence and should therefore be considered highly speculative. This distinction underscores the importance of separating established biological observations from hypothesis-driven extrapolations.
Importantly, several key experimental questions arise from this framework and should be prioritized in future studies. First, it remains to be determined whether NSP4 or its circulating fragments can directly interact with brain endothelial cells or choroid plexus epithelial cells under physiological conditions. Second, it is essential to clarify whether NSP4-related signaling is sufficient to induce measurable changes in BBB permeability in neonatal models in vivo. Third, the relative contributions of systemic cytokine signaling and neural pathways, including vagal activation, to neuroimmune responses in the developing brain should be quantitatively evaluated. Fourth, the extent to which innate immune activation at CNS interfaces can occur in the absence of direct viral invasion remains unclear and requires experimental validation.
Several limitations should be acknowledged. Most of the mechanistic insights discussed here are derived from non-neural or in vitro systems, and their applicability to the neonatal CNS remains uncertain. In addition, direct experimental evidence linking NSP4 to CNS barrier dysfunction is currently lacking. These limitations emphasize that the present model should be regarded as hypothesis-generating rather than definitive.
In conclusion, this perspective outlines a barrier-centered, hypothesis-driven model linking rotavirus-associated signaling to potential neuroimmune effects in neonates. This framework may suggest a conceptual basis for future mechanistic and translational studies in neonatal viral neurobiology.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Acknowledgments
The author would like to thank Editage (www.editage.jp) for English language editing and figure preparation. The author used ChatGPT-5.2 (OpenAI, San Francisco, CA, USA) to improve the language and logical structure of the manuscript. The author reviewed and edited the content as needed and takes full responsibility for the content of the publication. This is a revised version of the manuscript, which has been updated to incorporate valuable feedback and comments received during a peer-review process.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| BBB | blood-brain barrier |
| BCSFB | blood–cerebrospinal fluid barrier |
| BECs | brain endothelial cells |
| CCR2 | C-C chemokine receptor type 2 |
| CCL2 | C-C motif chemokine ligand 2 |
| Ca2+ | calcium ion |
| CD | cluster of differentiation |
| CNS | central nervous system |
| CPECs | choroid plexus epithelial cells |
| CVOs | circumventricular organs |
| DCs | dendritic cells |
| ER | endoplasmic reticulum |
| HMGB | high-mobility group box |
| IFN | interferon |
| IL | interleukin |
| JAM-A | junctional adhesion molecule-A |
| LPS | lipopolysaccharide |
| Macrophage | MΦ |
| Mo | monocyte |
| NF-κB | nuclear factor kappa B |
| NK cells | natural killer cells |
| NLRP | NLR family pyrin domain-containing protein |
| NOD | nucleotide-binding oligomerization domain protein |
| NSP4-F | NSP4 fragment |
| ROS | reactive oxygen species |
| Th1 | T helper 1 |
| Th17 | T helper 17 |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| Tregs | regulatory T cells |
References
- Crawford, S.E.; Ramani, S.; Tate, J.E.; Parashar, U.D.; Svensson, L.; Hagbom, M.; Franco, M.A.; Greenberg, H.B.; O’Ryan, M.; Kang, G.; et al. Rotavirus infection. Nat. Rev. Dis. Prim. 2017, 3, 17083. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Kennedy, M.A.; Wang, D.; Li, F. The role and implication of rotavirus VP8 * in viral infection and vaccine development. Virology 2025, 609, 110563. [Google Scholar] [CrossRef]
- World Health Organization. Immunization, Vaccines and Biologicals: Rotavirus. Available online: https://www.who.int/teams/immunization-vaccines-and-biologicals/diseases/rotavirus (accessed on 17 April 2026).
- Xu, X.; Luo, Y.; He, C.; Dian, Z.; Mi, H.; Yang, J.; Feng, Y.; Miao, Z.; Xia, X. Increased risk of neurological disease following pediatric rotavirus infection: A two-center case-control study. J. Infect. Dis. 2023, 227, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Gowdie, P.J.; Kirkwood, C.D.; Doherty, R.R.; Fahey, M. Rotavirus cerebellitis: New aspects to an old foe? Pediatr. Neurol. 2012, 46, 48–50. [Google Scholar] [CrossRef]
- Slotboom, D.E.F.; Peeters, D.; Groeneweg, S.; van Rijn-Klink, A.; Jacobs, E.; Schoenaker, M.H.D.; van Veen, M. Neurologic complications of rotavirus infections in children. Pediatr. Infect. Dis. J. 2023, 42, 533–536. [Google Scholar] [CrossRef]
- Meyer, A.; Mazzara, C.; Lava, S.A.G.; Treglia, G.; Bianchetti, M.G.; Goeggel Simonetti, B.; Simonetti, G.D. Neurological complications of rotavirus infection in children: A systematic review and meta-analysis. Acta Paediatr. 2023, 112, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Kirton, A.; Busche, K.; Ross, C.; Wirrell, E. Acute necrotizing encephalopathy in caucasian children: Two cases and review of the literature. J. Child Neurol. 2005, 20, 527–532. [Google Scholar] [CrossRef]
- Poojari, V.S.; Shah, I.; Shetty, N.S. Acute necrotising encephalopathy of childhood secondary to rotaviral diarrhoea. Indian Pediatr. 2021, 58, 491. [Google Scholar] [CrossRef]
- Jiang, L.; Mao, S.; Xu, J.; Gao, F. Reversible splenial lesion syndrome in children with benign convulsions associated with mild gastroenteritis: A retrospective study of five cases. Brain Dev. 2019, 41, 271–275. [Google Scholar] [CrossRef]
- Yeom, J.S.; Jo, J.Y.; Park, J.S.; Kim, Y.S.; Chung, J.Y.; Han, T.H.; Seo, J.H.; Park, E.S.; Lim, J.Y.; Woo, H.O.; et al. Monocyte chemoattractant protein (MCP)-1 in rotavirus-associated white matter injury in newborns. Neuropediatrics 2019, 50, 228–234. [Google Scholar] [CrossRef]
- Yeom, J.S.; Kim, Y.S.; Seo, J.H.; Park, J.S.; Park, E.S.; Lim, J.Y.; Woo, H.O.; Youn, H.S.; Choi, D.S.; Chung, J.Y.; et al. Distinctive pattern of white matter injury in neonates with rotavirus infection. Neurology 2015, 84, 21–27. [Google Scholar] [CrossRef]
- Oh, K.W.; Moon, C.H.; Lee, K.Y. Association of rotavirus with seizures accompanied by cerebral white matter injury in neonates. J. Child Neurol. 2015, 30, 1433–1439. [Google Scholar] [CrossRef]
- Lee, K.Y.; Oh, K.W.; Weon, Y.C.; Choi, S.H. Neonatal seizures accompanied by diffuse cerebral white matter lesions on diffusion-weighted imaging are associated with rotavirus infection. Eur. J. Paediatr. Neurol. 2014, 18, 624–631. [Google Scholar] [CrossRef]
- Ha, E.K.; Kim, J.H.; Han, B.; Shin, J.; Lee, E.; Rhie, S.; Lee, W.S.; Lee, S.; Han, M.Y. Rotavirus hospitalization in early childhood: Fine motor skills and cognition at 6 years old in a population-based cohort study. J. Infect. Dis. 2024, 230, 1167–1176. [Google Scholar] [CrossRef]
- Medici, M.C.; Abelli, L.A.; Guerra, P.; Dodi, I.; Dettori, G.; Chezzi, C. Case report: Detection of rotavirus RNA in the cerebrospinal fluid of a child with rotavirus gastroenteritis and meningism. J. Med. Virol. 2011, 83, 1637–1640. [Google Scholar] [CrossRef]
- Liu, B.; Fujita, Y.; Arakawa, C.; Kohira, R.; Fuchigami, T.; Mugishima, H.; Kuzuya, M. Detection of rotavirus RNA and antigens in serum and cerebrospinal fluid samples from diarrheic children with seizures. Jpn. J. Infect. Dis. 2009, 62, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Hellysaz, A.; Hagbom, M. Understanding the central nervous system symptoms of rotavirus: A qualitative review. Viruses 2021, 13, 658. [Google Scholar] [CrossRef]
- Lee, K.Y. Rotavirus infection-associated central nervous system complications: Clinicoradiological features and potential mechanisms. Clin. Exp. Pediatr. 2022, 65, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Bass, D.M. Rotaviruses, caliciviruseses, and astroviruses. In Nelson textbook of pediatrics, 21st ed.; Kliegman, R.M., St. Geme, J.W., III, Blum, N.J., Tasker, R.C., Wilson, K.M., Eds.; Elsevier: Amsterdam, 2020; Volume 1, pp. 1745–1747. [Google Scholar]
- Blutt, S.E.; Kirkwood, C.D.; Parreno, V.; Warfield, K.L.; Ciarlet, M.; Estes, M.K.; Bok, K.; Bishop, R.F.; Conner, M.E. Rotavirus antigenaemia and viraemia: A common event? Lancet. 2003, 362, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Mansell, A.; Ussher, J.E.; Brooks, A.E.; Manning, K.; Wang, C.J.; Taylor, J.A. Rotavirus NSP4 triggers secretion of proinflammatory cytokines from macrophages via toll-Like receptor 2. J. Virol. 2013, 87, 11160–11167. [Google Scholar] [CrossRef]
- Liu, Z.; Smith, H.; Criglar, J.M.; Valentin, A.J.; Karandikar, U.; Zeng, X.L.; Estes, M.K.; Crawford, S.E. Rotavirus-mediated DGAT1 degradation: A pathophysiological mechanism of viral-induced malabsorptive diarrhea. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2302161120. [Google Scholar] [CrossRef]
- Hyser, J.M.; Collinson-Pautz, M.R.; Utama, B.; Estes, M.K. Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. mBio. 2010, 1, e00265-00210. [Google Scholar] [CrossRef] [PubMed]
- Gebert, J.T.; Scribano, F.J.; Engevik, K.A.; Huleatt, E.M.; Eledge, M.R.; Dorn, L.E.; Philip, A.A.; Kawagishi, T.; Greenberg, H.B.; Patton, J.T.; et al. Viroporin activity is necessary for intercellular calcium signals that contribute to viral pathogenesis. Sci. Adv. 2025, 11, eadq8115. [Google Scholar] [CrossRef] [PubMed]
- Chang-Graham, A.L.; Perry, J.L.; Strtak, A.C.; Ramachandran, N.K.; Criglar, J.M.; Philip, A.A.; Patton, J.T.; Estes, M.K.; Hyser, J.M. Rotavirus calcium dysregulation manifests as dynamic calcium signaling in the cytoplasm and endoplasmic reticulum. Sci. Rep. 2019, 9, 10822. [Google Scholar] [CrossRef]
- Bugarcic, A.; Taylor, J.A. Rotavirus nonstructural glycoprotein NSP4 is secreted from the apical surfaces of polarized epithelial cells. J. Virol. 2006, 80, 12343–12349. [Google Scholar] [CrossRef]
- Didsbury, A.; Wang, C.; Verdon, D.; Sewell, M.A.; McIntosh, J.D.; Taylor, J.A. Rotavirus NSP4 is secreted from infected cells as an oligomeric lipoprotein and binds to glycosaminoglycans on the surface of non-infected cells. Virol. J. 2011, 8, 551. [Google Scholar] [CrossRef]
- Zhang, M.; Zeng, C.Q.; Morris, A.P.; Estes, M.K. A functional NSP4 enterotoxin peptide secreted from rotavirus-infected cells. J. Virol. 2000, 74, 11663–11670. [Google Scholar] [CrossRef]
- Seo, N.S.; Zeng, C.Q.; Hyser, J.M.; Utama, B.; Crawford, S.E.; Kim, K.J.; Hook, M.; Estes, M.K. Integrins α1β1 and α2β1 are receptors for the rotavirus enterotoxin. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8811–8818. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zeng, C.Q.; Ball, J.M.; Estes, M.K.; Morris, A.P. The rotavirus enterotoxin NSP4 mobilizes intracellular calcium in human intestinal cells by stimulating phospholipase C-mediated inositol 1,4,5-trisphosphate production. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 3960–3965. [Google Scholar] [CrossRef]
- Buccigrossi, V.; Laudiero, G.; Russo, C.; Miele, E.; Sofia, M.; Monini, M.; Ruggeri, F.M.; Guarino, A. Chloride secretion induced by rotavirus is oxidative stress-dependent and inhibited by Saccharomyces boulardii in human enterocytes. PLoS ONE 2014, 9, e99830. [Google Scholar] [CrossRef]
- Lorrot, M.; Vasseur, M. How do the rotavirus NSP4 and bacterial enterotoxins lead differently to diarrhea? Virol. J. 2007, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Nagyőszi, P.; Nyúl-Tóth, Á.; Fazakas, C.; Wilhelm, I.; Kozma, M.; Molnár, J.; Hasko, J.; Krizbai, I.A. Regulation of NOD-like receptors and inflammasome activation in cerebral endothelial cells. J. Neurochem. 2015, 135, 551–564. [Google Scholar] [CrossRef]
- Johansen, K.; Hinkula, J.; Espinoza, F.; Levi, M.; Zeng, C.; Ruden, U.; Vesikari, T.; Estes, M.; Svensson, L. Humoral and cell-mediated immune responses in humans to the NSP4 enterotoxin of rotavirus. J. Med. Virol. 1999, 59, 369–377. [Google Scholar] [CrossRef]
- Yeom, J.S.; Kim, Y.S.; Jun, J.S.; Do, H.J.; Park, J.S.; Seo, J.H.; Park, E.S.; Lim, J.Y.; Woo, H.O.; Park, C.H.; et al. NSP4 antibody levels in rotavirus gastroenteritis patients with seizures. Eur. J. Paediatr. Neurol. 2017, 21, 367–373. [Google Scholar] [CrossRef]
- Kavanagh, O.V.; Ajami, N.J.; Cheng, E.; Ciarlet, M.; Guerrero, R.A.; Zeng, C.Q.; Crawford, S.E.; Estes, M.K. Rotavirus enterotoxin NSP4 has mucosal adjuvant properties. Vaccine. 2010, 28, 3106–3111. [Google Scholar] [CrossRef]
- Boshuizen, J.A.; Rossen, J.W.; Sitaram, C.K.; Kimenai, F.F.; Simons-Oosterhuis, Y.; Laffeber, C.; Buller, H.A.; Einerhand, A.W. Rotavirus enterotoxin NSP4 binds to the extracellular matrix proteins laminin-β3 and fibronectin. J. Virol. 2004, 78, 10045–10053. [Google Scholar] [CrossRef] [PubMed]
- Nagyoszi, P.; Wilhelm, I.; Farkas, A.E.; Fazakas, C.; Dung, N.T.; Hasko, J.; Krizbai, I.A. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem. Int. 2010, 57, 556–564. [Google Scholar] [CrossRef]
- Arentsen, T.; Qian, Y.; Gkotzis, S.; Femenia, T.; Wang, T.; Udekwu, K.; Forssberg, H.; Diaz Heijtz, R. The bacterial peptidoglycan-sensing molecule Pglyrp2 modulates brain development and behavior. Mol. Psychiatry. 2017, 22, 257–266. [Google Scholar] [CrossRef]
- Nobutoki, T. Long-term neurological implications of severe acute respiratory syndrome coronavirus 2 infections in neonates: Innate immune memory and chronic neuroinflammation. In Med. Hypotheses.; 2023. [Google Scholar]
- Shin, H.S.; Xu, F.; Bagchi, A.; Herrup, E.; Prakash, A.; Valentine, C.; Kulkarni, H.; Wilhelmsen, K.; Warren, S.; Hellman, J. Bacterial lipoprotein TLR2 agonists broadly modulate endothelial function and coagulation pathways in vitro and in vivo. J. Immunol. 2011, 186, 1119–1130. [Google Scholar] [CrossRef]
- Rochfort, K.D.; Collins, L.E.; McLoughlin, A.; Cummins, P.M. Tumour necrosis factor-alpha-mediated disruption of cerebrovascular endothelial barrier integrity in vitro involves the production of proinflammatory interleukin-6. J. Neurochem. 2016, 136, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Re, F.; Strominger, J.L. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J. Biol. Chem. 2001, 276, 37692–37699. [Google Scholar] [CrossRef]
- Wen, W.; Cheng, J.; Tang, Y. Brain perivascular macrophages: Current understanding and future prospects. Brain 2024, 147, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, R.; Sakaguchi, S. T(reg) cells augment self-tolerance during infection. Nat. Immunol. 2025, 26, 650–652. [Google Scholar] [CrossRef]
- Piao, W.; Lee, Z.L.; Zapas, G.; Wu, L.; Jewell, C.M.; Abdi, R.; Bromberg, J.S. Regulatory T cell and endothelial cell crosstalk. Nat. Rev. Immunol. 2025, 25, 588–607. [Google Scholar] [CrossRef]
- Dong, H.; Qu, S.; Chen, X.; Zhu, H.; Tai, X.; Pan, J. Changes in the cytokine expression of peripheral Treg and Th17 cells in children with rotavirus enteritis. Exp. Ther. Med. 2015, 10, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef]
- Liston, A.; Pasciuto, E.; Fitzgerald, D.C.; Yshii, L. Brain regulatory T cells. Nat. Rev. Immunol. 2024, 24, 326–337. [Google Scholar] [CrossRef]
- Xu, J.; Yang, Y.; Sun, J.; Ding, Y.; Su, L.; Shao, C.; Jiang, B. Expression of Toll-like receptors and their association with cytokine responses in peripheral blood mononuclear cells of children with acute rotavirus diarrhoea. Clin. Exp. Immunol. 2006, 144, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shipley, F.B.; Shannon, M.L.; Alturkistani, O.; Dani, N.; Webb, M.D.; Sugden, A.U.; Andermann, M.L.; Lehtinen, M.K. Inflammation of the embryonic choroid plexus barrier following maternal immune activation. Dev. Cell. 2020, 55, 617–628 e616. [Google Scholar] [CrossRef]
- Mottahedin, A.; Joakim Ek, C.; Truve, K.; Hagberg, H.; Mallard, C. Choroid plexus transcriptome and ultrastructure analysis reveals a TLR2-specific chemotaxis signature and cytoskeleton remodeling in leukocyte trafficking. Brain. Behav. Immun. 2019, 79, 216–227. [Google Scholar] [CrossRef]
- Mottahedin, A.; Smith, P.L.; Hagberg, H.; Ek, C.J.; Mallard, C. TLR2-mediated leukocyte trafficking to the developing brain. J. Leukoc. Biol. 2017, 101, 297–305. [Google Scholar] [CrossRef]
- Rayasam, A.; Faustino, J.; Lecuyer, M.; Vexler, Z.S. Neonatal stroke and TLR1/2 ligand recruit myeloid cells through the choroid plexus in a CX3CR1-CCR2- and context-specific manner. J. Neurosci. 2020, 40, 3849–3861. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Druet, V.A.; Sibilia, J.; Klein, J.P.; Quesniaux, V.; Wachsmann, D. Cross talk between MyD88 and focal adhesion kinase pathways. J. Immunol. 2005, 174, 7393–7397. [Google Scholar] [CrossRef]
- Parr, R.D.; Storey, S.M.; Mitchell, D.M.; McIntosh, A.L.; Zhou, M.; Mir, K.D.; Ball, J.M. The rotavirus enterotoxin NSP4 directly interacts with the caveolar structural protein caveolin-1. J. Virol. 2006, 80, 2842–2854. [Google Scholar] [CrossRef]
- D’Alessio, A. Unraveling the cave: A seventy-year journey into the caveolar network, cellular signaling, and human disease. Cells 2023, 12. [Google Scholar] [CrossRef]
- Ikezu, T.; Ueda, H.; Trapp, B.D.; Nishiyama, K.; Sha, J.F.; Volonte, D.; Galbiati, F.; Byrd, A.L.; Bassell, G.; Serizawa, H.; et al. Affinity-purification and characterization of caveolins from the brain: Differential expression of caveolin-1, -2, and -3 in brain endothelial and astroglial cell types. Brain Res. 1998, 804, 177–192. [Google Scholar] [CrossRef]
- Song, L.; Ge, S.; Pachter, J.S. Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood. 2007, 109, 1515–1523. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Bjorn, C.; Ekblom, J. Antimicrobial peptides as therapeutic agents: Opportunities and challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P.; Bellamy, A.R.; Taylor, J.A. Membrane-destabilizing activity of rotavirus NSP4 is mediated by a membrane-proximal amphipathic domain. J. Gen. Virol. 2000, 81, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Torres-Flores, J.M.; Silva-Ayala, D.; Espinoza, M.A.; López, S.; Arias, C.F. The tight junction protein JAM-A functions as coreceptor for rotavirus entry into MA104 cells. Virology 2015, 475, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.M.; Anderson, J.M.; Vanltallie, C.M.; Doctrow, S.R. The tight-junction-specific protein ZO-1 is a component of the human and rat blood-brain barriers. Neurosci. Lett. 1991, 129, 6–10. [Google Scholar] [CrossRef]
- Hirase, T.; Staddon, J.M.; Saitou, M.; Ando-Akatsuka, Y.; Itoh, M.; Furuse, M.; Fujimoto, K.; Tsukita, S.; Rubin, L.L. Occludin as a possible determinant of tight junction permeability in endothelial cells. J. Cell Sci. 1997, 110 Pt 14, 1603–1613. [Google Scholar] [CrossRef]
- Dickman, K.G.; Hempson, S.J.; Anderson, J.; Lippe, S.; Zhao, L.; Burakoff, R.; Shaw, R.D. Rotavirus alters paracellular permeability and energy metabolism in Caco-2 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G757–766. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, I.; Vasiljevic, A.; Rey, C.; Fevre-Montange, M.; Saunders, N.; Strazielle, N.; Ghersi-Egea, J.F. Complexity and developmental changes in the expression pattern of claudins at the blood-CSF barrier. Histochem. Cell Biol. 2012, 138, 861–879. [Google Scholar] [CrossRef]
- Nava, P.; Lopez, S.; Arias, C.F.; Islas, S.; Gonzalez-Mariscal, L. The rotavirus surface protein VP8 modulates the gate and fence function of tight junctions in epithelial cells. J. Cell Sci. 2004, 117, 5509–5519. [Google Scholar] [CrossRef]
- Amatruda, M.; Chapouly, C.; Woo, V.; Safavi, F.; Zhang, J.; Dai, D.; Therattil, A.; Moon, C.; Villavicencio, J.; Gordon, A.; et al. Astrocytic junctional adhesion molecule-A regulates T-cell entry past the glia limitans to promote central nervous system autoimmune attack. Brain Commun. 2022, 4, fcac044. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Graham, K.L.; Halasz, P.; Tan, Y.; Hewish, M.J.; Takada, Y.; Mackow, E.R.; Robinson, M.K.; Coulson, B.S. Integrin-using rotaviruses bind α2β1 integrin α2 I domain via VP4 DGE sequence and recognize αXβ2 and αVβ3 by using VP7 during cell entry. J. Virol. 2003, 77, 9969–9978. [Google Scholar] [CrossRef]
- Halasz, P.; Holloway, G.; Turner, S.J.; Coulson, B.S. Rotavirus replication in intestinal cells differentially regulates integrin expression by a phosphatidylinositol 3-kinase-dependent pathway, resulting in increased cell adhesion and virus yield. J. Virol. 2008, 82, 148–160. [Google Scholar] [CrossRef]
- Jafri, M.; Donnelly, B.; Allen, S.; Bondoc, A.; McNeal, M.; Rennert, P.D.; Weinreb, P.H.; Ward, R.; Tiao, G. Cholangiocyte expression of α2β1-integrin confers susceptibility to rotavirus-induced experimental biliary atresia. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G16–G26. [Google Scholar] [CrossRef]
- Morrison, C.; Gilson, T.; Nuovo, G.J. Histologic distribution of fatal rotaviral infection: An immunohistochemical and reverse transcriptase in situ polymerase chain reaction analysis. Hum. Pathol. 2001, 32, 216–221. [Google Scholar] [CrossRef]
- Banerjee, A.; Kim, B.J.; Carmona, E.M.; Cutting, A.S.; Gurney, M.A.; Carlos, C.; Feuer, R.; Prasadarao, N.V.; Doran, K.S. Bacterial Pili exploit integrin machinery to promote immune activation and efficient blood-brain barrier penetration. Nat. Commun. 2011, 2, 462. [Google Scholar] [CrossRef]
- Brown, R.C.; Davis, T.P. Calcium modulation of adherens and tight junction function: A potential mechanism for blood-brain barrier disruption after stroke. Stroke. 2002, 33, 1706–1711. [Google Scholar] [CrossRef]
- Chang-Graham, A.L.; Perry, J.L.; Engevik, M.A.; Engevik, K.A.; Scribano, F.J.; Gebert, J.T.; Danhof, H.A.; Nelson, J.C.; Kellen, J.S.; Strtak, A.C.; et al. Rotavirus induces intercellular calcium waves through ADP signaling. Science 2020, 370. [Google Scholar] [CrossRef]
- Frelin, C.; Breittmayer, J.P.; Vigne, P. ADP induces inositol phosphate-independent intracellular Ca2+ mobilization in brain capillary endothelial cells. J. Biol. Chem. 1993, 268, 8787–8792. [Google Scholar] [CrossRef]
- Hess, C.N.; Kou, R.; Johnson, R.P.; Li, G.K.; Michel, T. ADP signaling in vascular endothelial cells: ADP-dependent activation of the endothelial isoform of nitric-oxide synthase requires the expression but not the kinase activity of AMP-activated protein kinase. J. Biol. Chem. 2009, 284, 32209–32224. [Google Scholar] [CrossRef]
- Beauchesne, E.; Desjardins, P.; Hazell, A.S.; Butterworth, R.F. eNOS gene deletion restores blood-brain barrier integrity and attenuates neurodegeneration in the thiamine-deficient mouse brain. J. Neurochem. 2009, 111, 452–459. [Google Scholar] [CrossRef]
- Sugata, K.; Taniguchi, K.; Yui, A.; Miyake, F.; Suga, S.; Asano, Y.; Ohashi, M.; Suzuki, K.; Nishimura, N.; Ozaki, T.; et al. Analysis of rotavirus antigenemia and extraintestinal manifestations in children with rotavirus gastroenteritis. Pediatrics 2008, 122, 392–397. [Google Scholar] [CrossRef]
- Casola, A.; Garofalo, R.P.; Crawford, S.E.; Estes, M.K.; Mercurio, F.; Crowe, S.E.; Brasier, A.R. Interleukin-8 gene regulation in intestinal epithelial cells infected with rotavirus: Role of viral-induced IκB kinase activation. Virology 2002, 298, 8–19. [Google Scholar] [CrossRef]
- Lund, B.T.; Ashikian, N.; Ta, H.Q.; Chakryan, Y.; Manoukian, K.; Groshen, S.; Gilmore, W.; Cheema, G.S.; Stohl, W.; Burnett, M.E.; et al. Increased CXCL8 (IL-8) expression in multiple sclerosis. J. Neuroimmunol. 2004, 155, 161–171. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Chung, J.; Burger, M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1094–1103. [Google Scholar] [CrossRef]
- Franchi, L.; Munoz-Planillo, R.; Nunez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef]
- Mullins, B.; Chen, J. NLRP9 in innate immunity and inflammation. Immunology 2021, 162, 262–267. [Google Scholar] [CrossRef]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.F.; Yu, T.; Chu, X.M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef]
- Acioglu, C.; Elkabes, S. Innate immune sensors and regulators at the blood brain barrier: Focus on toll-like receptors and inflammasomes as mediators of neuro-immune crosstalk and inflammation. J. Neuroinflammation 2025, 22, 39. [Google Scholar] [CrossRef]
- Parthun, M.; Long, M.E.; Hemann, E.A. Established and emerging roles of DEAD/H-box helicases in regulating infection and immunity. Immunol. Rev. 2025, 329, e13426. [Google Scholar] [CrossRef]
- Zhu, S.; Ding, S.; Wang, P.; Wei, Z.; Pan, W.; Palm, N.W.; Yang, Y.; Yu, H.; Li, H.B.; Wang, G.; et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017, 546, 667–670. [Google Scholar] [CrossRef]
- Mosallanejad, K.; Sekine, Y.; Ishikura-Kinoshita, S.; Kumagai, K.; Nagano, T.; Matsuzawa, A.; Takeda, K.; Naguro, I.; Ichijo, H. The DEAH-box RNA helicase DHX15 activates NF-kappaB and MAPK signaling downstream of MAVS during antiviral responses. Sci. Signal. 2014, 7, ra40. [Google Scholar] [CrossRef]
- Lu, H.; Lu, N.; Weng, L.; Yuan, B.; Liu, Y.J.; Zhang, Z. DHX15 senses double-stranded RNA in myeloid dendritic cells. J. Immunol. 2014, 193, 1364–1372. [Google Scholar] [CrossRef]
- Zhang, Z.; Zou, J.; Shi, Z.; Zhang, B.; Etienne-Mesmin, L.; Wang, Y.; Shi, X.; Shao, F.; Chassaing, B.; Gewirtz, A.T. IL-22-induced cell extrusion and IL-18-induced cell death prevent and cure rotavirus infection. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Rosales-Martinez, D.; Gutierrez-Xicotencatl, L.; Badillo-Godinez, O.; Lopez-Guerrero, D.; Santana-Calderon, A.; Cortez-Gomez, R.; Ramirez-Pliego, O.; Esquivel-Guadarrama, F. Rotavirus activates dendritic cells derived from umbilical cord blood monocytes. Microb. Pathog. 2016, 99, 162–172. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic reticulum Ca2+ handling in excitable cells in health and disease. Pharmacol. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef]
- Goodarzi, Z.; Soleimanjahi, H.; Arefian, E.; Saberfar, E. The effect of bovine rotavirus and its nonstructural protein 4 on ER stress-mediated apoptosis in HeLa and HT-29 cells. Tumour Biol. 2016, 37, 3155–3161. [Google Scholar] [CrossRef]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nuñez, G.; He, Y.; Yin, X.M.; O’Riordan, M.X. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef]
- Banerjee, S.; Sarkar, R.; Mukherjee, A.; Mitra, S.; Gope, A.; Chawla-Sarkar, M. Rotavirus-induced lncRNA SLC7A11-AS1 promotes ferroptosis by targeting cystine/glutamate antiporter xCT (SLC7A11) to facilitate virus infection. Virus Res. 2024, 339, 199261. [Google Scholar] [CrossRef]
- Song, J.; Kang, S.M.; Lee, W.T.; Park, K.A.; Lee, K.M.; Lee, J.E. Glutathione protects brain endothelial cells from hydrogen peroxide-induced oxidative stress by increasing nrf2 expression. Exp. Neurobiol. 2014, 23, 93–103. [Google Scholar] [CrossRef]
- Pott, J.; Stockinger, S.; Torow, N.; Smoczek, A.; Lindner, C.; McInerney, G.; Backhed, F.; Baumann, U.; Pabst, O.; Bleich, A.; et al. Age-dependent TLR3 expression of the intestinal epithelium contributes to rotavirus susceptibility. PLoS Pathog. 2012, 8, e1002670. [Google Scholar] [CrossRef]
- Shi, H.; Gabarin, N.; Hickey, E.; Askalan, R. TLR-3 receptor activation protects the very immature brain from ischemic injury. J. Neuroinflammation 2013, 10, 104. [Google Scholar] [CrossRef]
- Zimmer, S.; Steinmetz, M.; Asdonk, T.; Motz, I.; Coch, C.; Hartmann, E.; Barchet, W.; Wassmann, S.; Hartmann, G.; Nickenig, G. Activation of endothelial toll-like receptor 3 impairs endothelial function. Circ. Res. 2011, 108, 1358–1366. [Google Scholar] [CrossRef]
- Vargas-Caraveo, A.; Sayd, A.; Maus, S.R.; Caso, J.R.; Madrigal, J.L.M.; Garcia-Bueno, B.; Leza, J.C. Lipopolysaccharide enters the rat brain by a lipoprotein-mediated transport mechanism in physiological conditions. Sci. Rep. 2017, 7, 13113. [Google Scholar] [CrossRef]
- Moreno-Layseca, P.; Icha, J.; Hamidi, H.; Ivaska, J. Integrin trafficking in cells and tissues. Nat. Cell Biol. 2019, 21, 122–132. [Google Scholar] [CrossRef]
- Preston, J.E.; Joan Abbott, N.; Begley, D.J. Transcytosis of macromolecules at the blood-brain barrier. Adv. Pharmacol. 2014, 71, 147–163. [Google Scholar] [CrossRef]
- Unkmeir, A.; Latsch, K.; Dietrich, G.; Wintermeyer, E.; Schinke, B.; Schwender, S.; Kim, K.S.; Eigenthaler, M.; Frosch, M. Fibronectin mediates Opc-dependent internalization of Neisseria meningitidis in human brain microvascular endothelial cells. Mol. Microbiol. 2002, 46, 933–946. [Google Scholar] [CrossRef]
- Ribeiro, M.M.; Domingues, M.M.; Freire, J.M.; Santos, N.C.; Castanho, M.A. Translocating the blood-brain barrier using electrostatics. Front. Cell. Neurosci. 2012, 6, 44. [Google Scholar] [CrossRef]
- Stalmans, S.; Bracke, N.; Wynendaele, E.; Gevaert, B.; Peremans, K.; Burvenich, C.; Polis, I.; De Spiegeleer, B. Cell-penetrating peptides selectively cross the blood-brain barrier in vivo. PLoS ONE 2015, 10, e0139652. [Google Scholar] [CrossRef]
- Huang, H.; Schroeder, F.; Estes, M.K.; McPherson, T.; Ball, J.M. Interaction(s) of rotavirus non-structural protein 4 (NSP4) C-terminal peptides with model membranes. Biochem. J. 2004, 380, 723–733. [Google Scholar] [CrossRef]
- Khakshur, A.A.; Khodaverdi, E.; Kamali, H.; Nokhodchi, A. An insight into cell-penetrating peptides: Perspectives on design, optimization, and targeting in drug delivery systems. Pharm. Dev. Technol. 2025, 30, 521–547. [Google Scholar] [CrossRef]
- Tafazoli, F.; Zeng, C.Q.; Estes, M.K.; Magnusson, K.E.; Svensson, L. NSP4 enterotoxin of rotavirus induces paracellular leakage in polarized epithelial cells. J. Virol. 2001, 75, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Wei, H.; Sun, R.; Tian, Z. Recognition of double-stranded RNA by TLR3 induces severe small intestinal injury in mice. J. Immunol. 2007, 178, 4548–4556. [Google Scholar] [CrossRef] [PubMed]
- Boccazzi, M.; Van Steenwinckel, J.; Schang, A.L.; Faivre, V.; Le Charpentier, T.; Bokobza, C.; Csaba, Z.; Verderio, C.; Fumagalli, M.; Mani, S.; et al. The immune-inflammatory response of oligodendrocytes in a murine model of preterm white matter injury: The role of TLR3 activation. Cell Death Dis. 2021, 12, 166. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, J.L.; Ettayebi, K.; Maaty, W.S.; Faunce, N.R.; Bothner, B.; Hardy, M.E. Rotavirus infection activates the UPR but modulates its activity. Virol. J. 2011, 8, 359. [Google Scholar] [CrossRef]
- Guerrero, C.A.; Acosta, O. Inflammatory and oxidative stress in rotavirus infection. World J. Virol. 2016, 5, 38–62. [Google Scholar] [CrossRef]
- Huang, C.W.; Chuang, C.P.; Chen, Y.J.; Wang, H.Y.; Lin, J.J.; Huang, C.Y.; Wei, K.C.; Huang, F.T. Integrin alpha(2)beta(1)-targeting ferritin nanocarrier traverses the blood-brain barrier for effective glioma chemotherapy. J. Nanobiotechnol. 2021, 19, 180. [Google Scholar] [CrossRef] [PubMed]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron. 2017, 94, 581–594 e585. [Google Scholar] [CrossRef]
- Crawford, S.E.; Patel, D.G.; Cheng, E.; Berkova, Z.; Hyser, J.M.; Ciarlet, M.; Finegold, M.J.; Conner, M.E.; Estes, M.K. Rotavirus viremia and extraintestinal viral infection in the neonatal rat model. J. Virol. 2006, 80, 4820–4832. [Google Scholar] [CrossRef]
- Takanashi, J.; Miyamoto, T.; Ando, N.; Kubota, T.; Oka, M.; Kato, Z.; Hamano, S.; Hirabayashi, S.; Kikuchi, M.; Barkovich, A.J. Clinical and radiological features of rotavirus cerebellitis. AJNR Am. J. Neuroradiol. 2010, 31, 1591–1595. [Google Scholar] [CrossRef]
- Mardi, N.; Haiaty, S.; Rahbarghazi, R.; Mobarak, H.; Milani, M.; Zarebkohan, A.; Nouri, M. Exosomal transmission of viruses, a two-edged biological sword. Cell Commun. Signal 2023, 21, 19. [Google Scholar] [CrossRef] [PubMed]
- Isa, P.; Perez-Delgado, A.; Quevedo, I.R.; Lopez, S.; Arias, C.F. Rotaviruses associate with distinct types of extracellular vesicles. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Osaid, Z.; Haider, M.; Hamoudi, R.; Harati, R. Exosomes interactions with the blood-brain barrier: Implications for cerebral disorders and therapeutics. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Stridh, L.; Mottahedin, A.; Johansson, M.E.; Valdez, R.C.; Northington, F.; Wang, X.; Mallard, C. Toll-like receptor-3 activation increases the vulnerability of the neonatal brain to hypoxia-ischemia. J. Neurosci. 2013, 33, 12041–12051. [Google Scholar] [CrossRef]
- Mohanty, S.K.; Ivantes, C.A.; Mourya, R.; Pacheco, C.; Bezerra, J.A. Macrophages are targeted by rotavirus in experimental biliary atresia and induce neutrophil chemotaxis by Mip2/Cxcl2. Pediatr. Res. 2010, 67, 345–351. [Google Scholar] [CrossRef]
- Di Fiore, I.J.; Holloway, G.; Coulson, B.S. Innate immune responses to rotavirus infection in macrophages depend on MAVS but involve neither the NLRP3 inflammasome nor JNK and p38 signaling pathways. Virus Res. 2015, 208, 89–97. [Google Scholar] [CrossRef]
- Arjmandi, A.; Liu, K.; Dorovini-Zis, K. Dendritic cell adhesion to cerebral endothelium: Role of endothelial cell adhesion molecules and their ligands. J. Neuropathol. Exp. Neurol. 2009, 68, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Ifergan, I.; Kebir, H.; Bernard, M.; Wosik, K.; Dodelet-Devillers, A.; Cayrol, R.; Arbour, N.; Prat, A. The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain 2008, 131, 785–799. [Google Scholar] [CrossRef]
- Murayama, S.; Kurganov, E.; Miyata, S. Activation of microglia and macrophages in the circumventricular organs of the mouse brain during TLR2-induced fever and sickness responses. J. Neuroimmunol. 2019, 334, 576973. [Google Scholar] [CrossRef]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Shivakumar, P.; Mourya, R.; Bessho, K.; Walters, S.; Bezerra, J.A. Natural killer cells promote long-term hepatobiliary inflammation in a low-dose rotavirus model of experimental biliary atresia. PLoS ONE 2015, 10, e0127191. [Google Scholar] [CrossRef]
- Miethke, A.G.; Saxena, V.; Shivakumar, P.; Sabla, G.E.; Simmons, J.; Chougnet, C.A. Post-natal paucity of regulatory T cells and control of NK cell activation in experimental biliary atresia. J. Hepatol. 2010, 52, 718–726. [Google Scholar] [CrossRef]
- Lee, K.Y.; Moon, C.H.; Choi, S.H. Type I interferon and proinflammatory cytokine levels in cerebrospinal fluid of newborns with rotavirus-associated leukoencephalopathy. Brain Dev. 2018, 40, 211–217. [Google Scholar] [CrossRef]
- Boisvert, M.; Gendron, S.; Chetoui, N.; Aoudjit, F. Alpha2 beta1 integrin signaling augments T cell receptor-dependent production of interferon-gamma in human T cells. Mol. Immunol. 2007, 44, 3732–3740. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, I.E.; Lewen, A.; Galow, L.V.; Cesetti, T.; Scheffel, J.; Regen, T.; Hanisch, U.K.; Kann, O. TLR4-activated microglia require IFN-γ to induce severe neuronal dysfunction and death in situ. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Sweet, M.J.; Hume, D.A. Signal integration between IFNγ and TLR signalling pathways in macrophages. Immunobiology 2006, 211, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Benveniste, E.N. Critical role of tumor necrosis factor-alpha and NF-kappa B in interferon-gamma -induced CD40 expression in microglia/macrophages. J. Biol. Chem. 2002, 277, 13796–13803. [Google Scholar] [CrossRef]
- Kann, O.; Almouhanna, F.; Chausse, B. Interferon gamma: A master cytokine in microglia-mediated neural network dysfunction and neurodegeneration. Trends Neurosci. 2022, 45, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Kelsall, B.; Fuss, I.; Marth, T.; Ludviksson, B.; Ehrhardt, R.; Neurath, M. Reciprocal IFN-gamma and TGF-beta responses regulate the occurrence of mucosal inflammation. Immunol. Today 1997, 18, 61–64. [Google Scholar] [CrossRef]
- Chui, R.; Dorovini-Zis, K. Regulation of CCL2 and CCL3 expression in human brain endothelial cells by cytokines and lipopolysaccharide. J. Neuroinflammation 2010, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Kamada, N.; Shaw, M.H.; Warner, N.; Chen, G.Y.; Franchi, L.; Nunez, G. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 2011, 34, 769–780. [Google Scholar] [CrossRef]
- D’Aversa, T.G.; Weidenheim, K.M.; Berman, J.W. CD40-CD40L interactions induce chemokine expression by human microglia: Implications for human immunodeficiency virus encephalitis and multiple sclerosis. Am. J. Pathol. 2002, 160, 559–567. [Google Scholar] [CrossRef]
- Chen, H.R.; Chen, C.W.; Kuo, Y.M.; Chen, B.; Kuan, I.S.; Huang, H.; Lee, J.; Anthony, N.; Kuan, C.Y.; Sun, Y.Y. Monocytes promote acute neuroinflammation and become pathological microglia in neonatal hypoxic-ischemic brain injury. Theranostics 2022, 12, 512–529. [Google Scholar] [CrossRef]
- Thompson, W.L.; Van Eldik, L.J. Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-3 through NFkB and MAPK dependent pathways in rat astrocytes [corrected]. Brain Res. 2009, 1287, 47–57. [Google Scholar] [CrossRef]
- Shrestha, B.; Ge, S.; Pachter, J.S. Resolution of central nervous system astrocytic and endothelial sources of CCL2 gene expression during evolving neuroinflammation. Fluids Barriers CNS 2014, 11, 6. [Google Scholar] [CrossRef]
- Morichi, S.; Yamanaka, G.; Watanabe, Y.; Takamatsu, T.; Kasuga, A.; Takeshita, M.; Go, S.; Ishida, Y.; Oana, S.; Kashiwagi, Y.; et al. High mobility group box 1 and angiogenetic growth factor levels in children with central nerve system infections. J. Infect. Chemother. 2021, 27, 840–844. [Google Scholar] [CrossRef]
- Qiu, Y.; Yang, J.; Wang, W.; Zhao, W.; Peng, F.; Xiang, Y.; Chen, G.; Chen, T.; Chai, C.; Zheng, S.; et al. HMGB1-promoted and TLR2/4-dependent NK cell maturation and activation take part in rotavirus-induced murine biliary atresia. PLoS Pathog. 2014, 10, e1004011. [Google Scholar] [CrossRef]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef]
- Suzuki, S.; Kashiwagi, Y.; Kawashima, H. PO-0250 cerebrospinal fluid cytokine in central nervous system involvement associated with rotavirus infection. Arch. Dis. Child. 2014, 99, A326.321–A326. [Google Scholar] [CrossRef]
- Pedrazzi, M.; Patrone, M.; Passalacqua, M.; Ranzato, E.; Colamassaro, D.; Sparatore, B.; Pontremoli, S.; Melloni, E. Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. J. Immunol. 2007, 179, 8525–8532. [Google Scholar] [CrossRef] [PubMed]
- Morichi, S.; Urabe, T.; Morishita, N.; Takeshita, M.; Ishida, Y.; Oana, S.; Yamanaka, G.; Kashiwagi, Y.; Kawashima, H. Pathological analysis of children with childhood central nervous system infection based on changes in chemokines and interleukin-17 family cytokines in cerebrospinal fluid. J. Clin. Lab. Anal. 2018, 32. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.; Schröder, A.; Dreier, A.; Möhn, N.; Dippel, S.; Winterberg, T.; Wilde, A.; Yu, Y.; Thorenz, A.; Gueler, F.; et al. Interleukin 17, produced by γδ T cells, contributes to hepatic inflammation in a mouse model of biliary atresia and is increased in livers of patients. Gastroenterology 2016, 150, 229–241. e225. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, X.; Li, C.; Chen, J.; Xia, S.; Bao, X.; Ge, J.; Cao, X.; Xu, Y. gammadelta T cells aggravate blood-brain-barrier injury via IL-17A in experimental ischemic stroke. Neurosci. Lett. 2022, 776, 136563. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Shotorbani, S.S.; Jiao, Z.; Su, Z.; Tong, J.; Liu, Y.; Shen, P.; Ma, J.; Gao, J.; Wang, T.; et al. HMGB1 promotes the differentiation of Th17 via up-regulating TLR2 and IL-23 of CD14+ monocytes from patients with rheumatoid arthritis. Scand. J. Immunol. 2012, 76, 483–490. [Google Scholar] [CrossRef]
- Wojkowska, D.W.; Szpakowski, P.; Glabinski, A. Interleukin 17A promotes lymphocytes adhesion and induces CCL2 and CXCL1 release from brain endothelial cells. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Kawanokuchi, J.; Shimizu, K.; Nitta, A.; Yamada, K.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Production and functions of IL-17 in microglia. J. Neuroimmunol. 2008, 194, 54–61. [Google Scholar] [CrossRef]
- Trajkovic, V.; Stosic-Grujicic, S.; Samardzic, T.; Markovic, M.; Miljkovic, D.; Ramic, Z.; Mostarica Stojkovic, M. Interleukin-17 stimulates inducible nitric oxide synthase activation in rodent astrocytes. J. Neuroimmunol. 2001, 119, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Kim, D.; Lee, S.J. Toll-like receptor 2 mediates peripheral nerve injury-induced NADPH oxidase 2 expression in spinal cord microglia. J. Biol. Chem. 2013, 288, 7572–7579. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S.; Henneke, P.; Lien, E.; Kasper, D.L.; Volpe, J.J.; Bechmann, I.; Nitsch, R.; Weber, J.R.; Golenbock, D.T.; Vartanian, T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J. Immunol. 2006, 177, 583–592. [Google Scholar] [CrossRef]
- Phulwani, N.K.; Esen, N.; Syed, M.M.; Kielian, T. TLR2 expression in astrocytes is induced by TNF-α- and NF-κB-dependent pathways. J. Immunol. 2008, 181, 3841–3849. [Google Scholar] [CrossRef]
- Suzuki, T.; Kohyama, K.; Moriyama, K.; Ozaki, M.; Hasegawa, S.; Ueno, T.; Saitoe, M.; Morio, T.; Hayashi, M.; Sakuma, H. Extracellular ADP augments microglial inflammasome and NF-κB activation via the P2Y12 receptor. Eur. J. Immunol. 2020, 50, 205–219. [Google Scholar] [CrossRef]
- Lathia, J.D.; Okun, E.; Tang, S.C.; Griffioen, K.; Cheng, A.; Mughal, M.R.; Laryea, G.; Selvaraj, P.K.; ffrench-Constant, C.; Magnus, T.; et al. Toll-like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. J. Neurosci. 2008, 28, 13978–13984. [Google Scholar] [CrossRef]
- Weclewicz, K.; Svensson, L.; Kristensson, K. Targeting of endoplasmic reticulum-associated proteins to axons and dendrites in rotavirus-infected neurons. Brain Res. Bull. 1998, 46, 353–360. [Google Scholar] [CrossRef]
- Chandra, P.; Patra, U.; Mukhopadhyay, U.; Mukherjee, A.; Halder, P.; Koley, H.; Chawla-Sarkar, M. Rotavirus non-structural protein 4 usurps host cellular RIPK1-RIPK3 complex to induce MLKL-dependent necroptotic cell death. Biochim Biophys. Acta Mol. Cell Res. 2024, 1871, 119745. [Google Scholar] [CrossRef]
- Lafon, M.; Megret, F.; Lafage, M.; Prehaud, C. The innate immune facet of brain: Human neurons express TLR-3 and sense viral dsRNA. J. Mol. Neurosci. 2006, 29, 185–194. [Google Scholar] [CrossRef]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.; Khatiwada, S.; Costa-Mattioli, M.; Hetz, C. ER proteostasis control of neuronal physiology and synaptic function. Trends Neurosci. 2018, 41, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Liu, H.; Niu, F.; Kang, B.; Luo, X.; Yang, H.; Tian, Z.; Yang, J. Endoplasmic reticulum stress promotes neuronal damage in neonatal hypoxic-ischemic brain damage by inducing ferroptosis. Mol. Biotechnol. 2025, 67, 805–815. [Google Scholar] [CrossRef]
- Lin, W.; Harding, H.P.; Ron, D.; Popko, B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J. Cell Biol. 2005, 169, 603–612. [Google Scholar] [CrossRef]
- Liu, D.; Ke, Z.; Luo, J. Thiamine deficiency and neurodegeneration: The interplay among oxidative stress, endoplasmic reticulum stress, and autophagy. Mol. Neurobiol. 2017, 54, 5440–5448. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H.; Inage, Y.; Ogihara, M.; Kashiwagi, Y.; Takekuma, K.; Hoshika, A.; Mori, T.; Watanabe, Y. Serum and cerebrospinal fluid nitrite/nitrate levels in patients with rotavirus gastroenteritis induced convulsion. Life Sci. 2004, 74, 1397–1405. [Google Scholar] [CrossRef]
- Angelis, D.; Savani, R.; Chalak, L. Nitric oxide and the brain. Part 1: Mechanisms of regulation, transport and effects on the developing brain. Pediatr. Res. 2021, 89, 738–745. [Google Scholar] [CrossRef]
- Blais, V.; Rivest, S. Effects of TNF-alpha and IFN-gamma on nitric oxide-induced neurotoxicity in the mouse brain. J. Immunol. 2004, 172, 7043–7052. [Google Scholar] [CrossRef]
- Kaufer, C.; Chhatbar, C.; Broer, S.; Waltl, I.; Ghita, L.; Gerhauser, I.; Kalinke, U.; Loscher, W. Chemokine receptors CCR2 and CX3CR1 regulate viral encephalitis-induced hippocampal damage but not seizures. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E8929–E8938. [Google Scholar] [CrossRef]
- Errede, M.; Annese, T.; Petrosino, V.; Longo, G.; Girolamo, F.; de Trizio, I.; d’Amati, A.; Uccelli, A.; Kerlero de Rosbo, N.; Virgintino, D. Microglia-derived CCL2 has a prime role in neocortex neuroinflammation. Fluids Barriers CNS 2022, 19, 68. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, J.; Zhao, H.; Yang, X.; Gao, F. White matter anomaly associated cognitive impairment during systemic inflammation is related to CX3CR1 mediated microglia-node interactions that impacts the conductive function of axons. J. Inflamm. Res. 2025, 18, 8477–8492. [Google Scholar] [CrossRef] [PubMed]
- Ronzano, R.; Roux, T.; Thetiot, M.; Aigrot, M.S.; Richard, L.; Lejeune, F.X.; Mazuir, E.; Vallat, J.M.; Lubetzki, C.; Desmazieres, A. Microglia-neuron interaction at nodes of Ranvier depends on neuronal activity through potassium release and contributes to remyelination. Nat. Commun. 2021, 12, 5219. [Google Scholar] [CrossRef]
- Albertsson, A.M.; Zhang, X.; Vontell, R.; Bi, D.; Bronson, R.T.; Supramaniam, V.; Baburamani, A.A.; Hua, S.; Nazmi, A.; Cardell, S.; et al. γδ T cells contribute to injury in the developing brain. Am. J. Pathol. 2018, 188, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Derkow, K.; Kruger, C.; Dembny, P.; Lehnardt, S. Microglia induce neurotoxic IL-17+ γδ T cells dependent on TLR2, TLR4, and TLR9 activation. PLoS ONE 2015, 10, e0135898. [Google Scholar] [CrossRef]
- Liu, Q.; Sanai, N.; Jin, W.N.; La Cava, A.; Van Kaer, L.; Shi, F.D. Neural stem cells sustain natural killer cells that dictate recovery from brain inflammation. Nat. Neurosci. 2016, 19, 243–252. [Google Scholar] [CrossRef]
- Benarroch, E. What are the functions of caveolins and their role in neurologic disorders? Neurology 2025, 104, e213341. [Google Scholar] [CrossRef]
- Yang, W.; Wu, W.; Zhao, Y.; Li, Y.; Zhang, C.; Zhang, J.; Chen, C.; Cui, S. Caveolin-1 suppresses hippocampal neuron apoptosis via the regulation of HIF1alpha in hypoxia in naked mole-rats. Cell Biol. Int. 2022, 46, 2060–2074. [Google Scholar] [CrossRef]
- Trevino, T.N.; Almousawi, A.A.; Robinson, K.F.; Fogel, A.B.; Class, J.; Minshall, R.D.; Tai, L.M.; Richner, J.M.; Lutz, S.E. Caveolin-1 mediates blood-brain barrier permeability, neuroinflammation, and cognitive impairment in SARS-CoV-2 infection. J. Neuroimmunol. 2024, 388, 578309. [Google Scholar] [CrossRef]
- Cao, D.; Li, B.; Cao, C.; Zhang, J.; Li, X.; Li, H.; Yu, Z.; Shen, H.; Ye, M. Caveolin-1 aggravates neurological deficits by activating neuroinflammation following experimental intracerebral hemorrhage in rats. Exp. Neurol. 2023, 368, 114508. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.H.; Tsai, C.N.; Lai, M.W.; Chen, C.C.; Chao, H.C.; Lin, C.W.; Chiu, C.H.; Chen, S.Y. Antigenemia and cytokine expression in rotavirus gastroenteritis in children. J. Microbiol. Immunol. Infect. 2012, 45, 265–270. [Google Scholar] [CrossRef]
- Steelman, A.J.; Li, J. Poly(I:C) promotes TNFα/TNFR1-dependent oligodendrocyte death in mixed glial cultures. J. Neuroinflammation. 2011, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef]
- Krall, R.F.; Tzounopoulos, T.; Aizenman, E. The function and regulation of zinc in the brain. Neuroscience 2021, 457, 235–258. [Google Scholar] [CrossRef]
- Zhang, Y.; Aizenman, E.; DeFranco, D.B.; Rosenberg, P.A. Intracellular zinc release, 12-lipoxygenase activation and MAPK dependent neuronal and oligodendroglial death. In Mol. Med.; Zhang, Ed.; 2007; Volume 13, pp. 350–355. [Google Scholar] [CrossRef]
- do Rosario, M.C.; Bey, G.R.; Nmezi, B.; Liu, F.; Oranburg, T.; Cohen, A.S.A.; Coffman, K.A.; Brown, M.R.; Kiselyov, K.; Waisfisz, Q.; et al. Variants in the zinc transporter TMEM163 cause a hypomyelinating leukodystrophy. Brain. 2022, 145, 4202–4209. [Google Scholar] [CrossRef]
- Stork, C.J.; Li, Y.V. Zinc release from thapsigargin/IP3-sensitive stores in cultured cortical neurons. J. Mol. Signal. 2010, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.E.; Richards, L.L.; Votyakova, T.V.; Reynolds, I.J. Zinc causes loss of membrane potential and elevates reactive oxygen species in rat brain mitochondria. Mitochondrion. 2005, 5, 55–65. [Google Scholar] [CrossRef] [PubMed]
- McCord, M.C.; Aizenman, E. The role of intracellular zinc release in aging, oxidative stress, and Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 77. [Google Scholar] [CrossRef]
Figure 1.
Proposed barrier-centered model of rotavirus-associated neuroimmune dysregulation.


Table 1.
Summary of proposed mechanisms and levels of supporting evidence in relation to neonatal CNS involvement.
Table 1.
Summary of proposed mechanisms and levels of supporting evidence in relation to neonatal CNS involvement.
| Mechanism | Evidence Type | Experimental System | Relevance to neonatal CNS | Interpretation | |
| NSP4-induced Ca2+ dysregulation | Direct | Intestinal epithelial cells | Indirect | Established in gut, unknown in CNS | |
| ER stress induction | Direct | In vitro (non-neural) | Indirect | Plausible but unverified in BBB | |
| TLR2 activation | Indirect | Immune cells | Indirect | Possible systemic role | |
| TLR3 signaling | Mixed | CNS & immune | Unclear | Context-dependent (protective vs harmful) | |
| Tight junction modulation | Indirect | Epithelial/endothelial | Indirect | Hypothetical in BBB | |
| Oxidative stress | Indirect | Various | Indirect | Non-specific mechanism | |
| BBB permeability changes | Limited | Animal models | Partial | Not NSP4-specific | |
| NSP4 circulation | Limited | Clinical/experimental | Unclear | Requires validation | |
| Direct CNS invasion | Limited | Case reports | Weak | Not primary mechanism | |
| Interpretation: Direct = experimentally demonstrated Indirect = inferred from related systems Limited = sparse or indirect evidence Note: BBB, blood-brain barrier; Ca²⁺, calcium ion; ER, endoplasmic reticulum; CNS, central nervous system; DC, dendritic cells; IL-6, interleukin-6; NK, natural killer cells; NSP4, nonstructural protein 4; TLR, toll-like receptor |
|||||
Note: BBB, blood-brain barrier; BECs, brain endothelial cells; CCL2, C-C motif chemokine ligand 2; CD, cluster of differentiation; DCs, dendritic cells; HMGB, high-mobility group box; IFN, interferon; IL, interleukin; Macrophage, MΦ; Mo, monocyte; NK cells, natural killer cells; NSP4, nonstructural protein 4; NSP4-F, NSP4 fragment; Th1, T helper 1; Th17, T helper 17; TLR, Toll-like receptor; TNF, tumor necrosis factor; Tregs, regulatory T cells;.
Table 2.
Classification of evidence supporting proposed rotavirus- and NSP4-associated mechanisms in neonatal neuroimmune dysregulation.
Table 2.
Classification of evidence supporting proposed rotavirus- and NSP4-associated mechanisms in neonatal neuroimmune dysregulation.
| Mechanism | Rotavirus (in vivo/clinical) | NSP4 (experimental) | NSP4 fragments | Evidence type | Relevance to neonatal CNS |
| Enterocyte damage / diarrhea | ✔ Established | ✔ | ✖ | Human+ animal | Indirect (systemic effects) |
| Ca²⁺ dysregulation | ✔ (indirect) | ✔ Strong | ✔ | Invitro / animal | Unknown in CNS |
| TLR2 activation | ✖ | ✔ | ✔ | In vitro | Speculative |
| TLR3 activation | ✔ (context-dependent) | ✖ | ✖ | Animal | Unclear (protective vs harmful) |
| ER stress | ✖ | ✔ | ✔ | In vitro | Speculative in BECs |
| Tight junction disruption | ✔ (gut) | ✔ | ✔ | In vitro | Not demonstrated in BBB |
| BBB permeability changes | Limited | ✖ | ✖ | Indirect | Highly speculative |
| Infection of BECs | Controversial | ✖ | ✖ | Limited reports | Not established |
| Cytokine induction (IL-6, IL-8) | ✔ | ✔ | ✔ | Mixed | Possible indirect effects |
| Immune cell activation (Treg, DC, NK) | Limited | ✖ | ✖ | Indirect | Hypothetical |
| White matter injury | ✔ (clinical association) | ✖ | ✖ | Clinical | Mechanism unknown |
| Note: BBB, blood-brain barrier; BECs, brain endothelial cells; Ca²⁺, calcium ion; ER, endoplasmic reticulum; CNS, central nervous system; DC, dendritic cells; IL-6, interleukin-6; NK, natural killer cells; NSP4, nonstructural protein 4; regulatory T cell; Treg, TLR, toll-like receptor | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



