Submitted:
31 May 2026
Posted:
02 June 2026
You are already at the latest version
Abstract
Pregnancy is a physiological state of transient, reversible insulin resistance accompanied by major adaptations in glucose and lipid metabolism. Although placental hormones are key drivers of gestational insulin resistance, the mechanisms responsible for the rapid restoration of insulin sensitivity after delivery remain unclear.
This review presents a dual-axis model in which fibroblast growth factor 21 (FGF21) and sex hormone–binding globulin (SHBG) represent complementary hepatic pathways regulating maternal metabolic plasticity. During late pregnancy, FGF21 may function as a metabolic resilience factor by promoting fatty acid oxidation, limiting lipid overload, and supporting mitochondrial flexibility through AMPK–PPARα signaling. Reduced SHBG, in contrast, may reflect hepatic insulin resistance and altered HNF4α-dependent transcription. After delivery, restoration of the HNF4α–SHBG axis may indicate recovery of hepatic insulin responsiveness, while persistent adaptive FGF21 signaling may facilitate metabolic reprogramming.
Postpartum metabolic resetting may therefore represent an actively regulated process rather than a passive consequence of placental hormone withdrawal. Disruption of FGF21- and SHBG-mediated pathways may contribute to persistent insulin resistance and increased cardiometabolic risk after gestational diabetes. Understanding hepatokine-mediated regulation of maternal metabolic flexibility may provide new insights into risk stratification, early biomarkers, and therapeutic targets for the prevention of type 2 diabetes after pregnancy.
Keywords:
pregnancy
; insulin resistance
; hepatokines
; FGF21
; SHBG
; gestational diabetes mellitus
; postpartum metabolic adaptation
; metabolic reprogramming
; type 2 diabetes
1. Introduction
1.1. Pregnancy as a Model of Reversible Insulin Resistance
Pregnancy is a unique physiological model of transient and reversible insulin resistance associated with maternal metabolic remodeling, in which insulin sensitivity decreases by more than 50% during the second and third trimesters. This phenomenon represents an evolutionarily programmed strategy for the redistribution of energy substrates to the fetoplacental unit. Placental hormonal activity, particularly placental growth hormone (PGH) and human placental lactogen (hPL), leads to a systemic reprogramming of maternal metabolism.
Glucose-stimulated insulin secretion and pancreatic β-cell mass are increased by hPL and prolactin [1,2,3,4], thereby supporting compensatory hyperinsulinemia during normal pregnancy.
In late pregnancy, the placenta releases pro-inflammatory cytokines, including tumor necrosis factor alpha (TNF-α), which modulate insulin receptor substrate-1 (IRS-1) phosphorylation, thereby enhancing peripheral insulin resistance [5,6]. This process reduces glucose uptake by maternal tissues and ensures its availability for the fetus.
Importantly, pregnancy-associated insulin resistance rapidly resolves after delivery, highlighting the highly dynamic and reversible nature of maternal metabolic adaptation [7].
1.2. Differences Between Physiological and Pathological Insulin Resistance
The key distinction between normal pregnancy and gestational diabetes mellitus (GDM) lies in the compensatory capacity of pancreatic β-cells [8]. Whereas normal pregnancy is characterized by adaptive hyperinsulinemia that maintains glucose homeostasis, GDM develops when β-cell compensation becomes insufficient in the presence of increasing metabolic demands and insulin resistance [6,9,10].
Activation of c-Jun N-terminal kinase (JNK) and inhibitor of κB kinase β (IKKβ) pathways, induced by TNF-α and free fatty acids, contributes to cellular stress and impaired insulin signaling [11]. In parallel, enhanced lipolysis promotes the accumulation of ceramides and diacylglycerols in skeletal muscle and liver, further exacerbating metabolic dysfunction. This process is accompanied by chronic lipotoxicity and low-grade metabolic inflammation [12,13,14].
1.3. Research Gap
A defining feature of pregnancy-associated insulin resistance is its rapid reversal after delivery, often occurring within days postpartum. Defining the mechanisms underlying this process is crucial, as impaired postpartum metabolic adaptation may contribute to the progression of type 2 diabetes mellitus (T2DM) [15].
However, the molecular mechanisms governing this postpartum “metabolic reset” remain incompletely understood.
1.4. Two Hepatokine Axes
During pregnancy, the liver functions as an endocrine organ integrating metabolic, inflammatory, and hormonal signals [17].
Among hepatokines, fibroblast growth factor 21 (FGF21) and sex hormone-binding globulin (SHBG) are of particular importance, reflecting different aspects of metabolic adaptation and hepatic insulin sensitivity.
We propose that maternal metabolic adaptation during pregnancy and postpartum restoration of insulin sensitivity are coordinated by two complementary axes:
- the FGF21 axis, responsible for adaptation to metabolic stress and lipid overload,
- the hepatocyte nuclear factor 4 alpha (HNF4α)–SHBG axis, potentially reflecting restoration of hepatocellular function after delivery.
The pathogenesis of GDM is associated with disruption of these axes and activation of inflammatory signaling pathways, which may include mediators such as fetuin-A, contributing to inhibition of insulin signaling via toll-like receptor 4 (TLR4) [19,20].
Collectively, we hypothesize that postpartum “metabolic reset” represents an active, hepatokine-driven process involving coordinated signaling through the FGF21 axis and the HNF4α–SHBG pathway.
2. Maternal Metabolic Adaptation and Postpartum Recovery
2.1. Metabolic Remodeling in Pregnancy: Glucose and Lipids
At the beginning of pregnancy, glucose and insulin levels do not differ significantly from pre-pregnancy values. In the second and third trimesters, insulin sensitivity decreases progressively by 50–60%, increasing substrate availability for the fetus [21,22].
Maintenance of glucose homeostasis in late pregnancy requires increased hepatic gluconeogenesis, providing alanine as a key substrate. In GDM, impaired autophagy, accumulation of dysfunctional mitochondria, and endoplasmic reticulum stress are observed, suggesting compromised cellular metabolic flexibility [23,24].
Advanced pregnancy is characterized by hyperlipidemia with elevated triglycerides, very-low-density lipoprotein (VLDL), and low-density lipoprotein (LDL) fractions. This process is regulated by nuclear lipid receptors, including farnesoid X receptor (FXR). Rising estrogen levels increase VLDL synthesis, while lipoprotein lipase (LPL) activity in adipose tissue is reduced [17,25].
Pregnancy is associated with “accelerated starvation,” leading to enhanced ketogenesis during short-term fasting, resulting in increased circulating free fatty acids and ketone bodies after several hours of fasting [4,26,27].
In late pregnancy, enhanced lipolysis driven by placental hormones and increased non-esterified fatty acids (NEFA) shifts metabolism toward lipid oxidation. However, in GDM, this adaptive increase in lipid utilization is impaired, with reduced mitochondrial function and activation of pro-inflammatory fatty acid oxidation pathways in the placenta [12,28,29,30].
Hepatic insulin resistance and impaired gluconeogenesis regulation persist in GDM, accompanied by an abnormal adipokine profile and elevated triglycerides [31,32,33]. Collectively, these changes highlight the central role of the liver in integrating glucose and lipid metabolic adaptations during pregnancy and their dysregulation in GDM.
2.2. Hormonal Regulation of Pregnancy-Induced Insulin Resistance
The main regulators of maternal metabolic adaptation are PGH and hPL. PGH is secreted continuously and impairs insulin signaling through modulation of IRS-1, thereby contributing to reduced maternal glucose uptake and increased substrate availability for the fetus [3]. hPL promotes lipolysis and redistribution of energy substrates toward the fetus, while cortisol further enhances gluconeogenesis and lipolysis [24]. Together, these hormones coordinate a progressive shift in maternal metabolism toward increased reliance on lipid-derived fuels.
Estrogens and progesterone modulate glucose transporter type 4 (GLUT4) expression and insulin signaling in skeletal muscle. Progesterone may additionally induce oxidative stress and β-cell apoptosis in the pancreas [34,35,36].
Taken together, these hormonal signals converge on insulin signaling pathways and systemic substrate partitioning, establishing the endocrine framework underlying pregnancy-associated insulin resistance.
2.3. Reversibility of Insulin Resistance After Delivery
Delivery leads to a rapid decline in diabetogenic hormones, including progesterone and hPL, initiating the rapid restoration of insulin sensitivity [37].
In women with GDM, abnormal IRS phosphorylation may maintain insulin resistance despite the disappearance of placental stimuli, suggesting partially persistent impairments in insulin signaling pathways.
In most patients, glycemia normalizes; however, some individuals exhibit residual β-cell dysfunction and low-grade chronic inflammation, increasing the risk of T2DM. Reduction in inflammatory cytokines improves glucose homeostasis and supports return to normoglycemia [3,38,39].
Postpartum metabolic recovery involves remodeling of hepatic and peripheral immune-metabolic functions [40], with gradual resolution of insulin-resistance-promoting signals and hepatocyte secretory remodeling. This transition reflects a coordinated shift from a pregnancy-adapted to a post-pregnancy metabolic state.
During pregnancy, liver volume increases by up to 20%, while postpartum involution occurs alongside remodeling of blood flow and amino acid metabolism [41]. Disturbances in this process may contribute to persistent metabolic dysfunction and increased risk of metabolic syndrome and non-alcoholic fatty liver disease (NAFLD) [19,20,27,42]. Together, these observations highlight the liver as a central organ in both the establishment and resolution of pregnancy-associated insulin resistance.
3. The Liver as a Central Integrator of Maternal Metabolic Plasticity
3.1. The Liver as an Endocrine Organ and Metabolic Hub
The liver is not only a metabolic organ but also an important endocrine regulator integrating hormonal, immunological, and metabolic signals controlling systemic energy homeostasis. It participates in thyroid hormone metabolism, steroid metabolism, glucagon-like peptide-1 (GLP-1) signaling, hepatokine production, detoxification, and energy substrate redistribution [43,44]. Importantly, these functions position the liver as a key coordinator of systemic metabolic adaptation rather than a passive metabolic reservoir.
It communicates with endocrine organs such as the pancreas, pituitary gland, thyroid, intestine, bone, and adrenal glands, while hormones themselves modulate hepatic synthetic and metabolic functions [17,20,43,45]. This bidirectional crosstalk enables dynamic regulation of whole-body energy balance.
Due to its unique vascularization, the liver is constantly exposed to circulating and gut-derived signals, enabling rapid metabolic adaptation. Despite its relatively small mass (~2.5% of body weight), it receives up to 25% of cardiac output at rest [43,46]. This anatomical arrangement ensures continuous integration of nutrient, hormonal, and inflammatory signals.
3.2. Hepatokines as a Systemic Metabolic Communication Interface
Hepatokines are active mediators of inter-organ communication rather than mere metabolic biomarkers. They regulate insulin signaling, lipid metabolism, and inflammatory responses, maintaining systemic glucose and energy homeostasis. They act as molecular messengers conveying information about hepatic energy status to peripheral tissues [20,47], linking liver-derived signals with whole-body metabolic regulation.
Selected hepatokines, such as selenoprotein P and fetuin-A, promote insulin resistance by modulating signaling in muscle and adipose tissue and influencing endothelial function and cardiometabolic risk. These biomolecules are generally associated with maladaptive metabolic signaling states.
3.3. Liver–Peripheral Tissue Axis as an Energy Substrate Distribution System
Communication between the liver, adipose tissue, skeletal muscle, and pancreas is a fundamental mechanism of metabolic adaptation in pregnancy. This inter-organ network enables coordinated regulation of systemic energy allocation in response to changing metabolic demands.
Hepatokines induced by placental signals, such as selenoprotein P (SeP), inhibit glucose transport in muscle via suppression of protein kinase B (Akt) activity [17,18], thereby contributing to reduced peripheral glucose utilization and enhanced substrate availability for the fetoplacental unit. This mechanism supports hierarchical redistribution of energy substrates during pregnancy.
In insulin-resistant states, compensatory β-cell proliferation occurs, partly regulated by hepatokines such as serpin B1, linking hepatic metabolic status to pancreatic adaptive capacity.
The liver–pancreas axis integrates β-cell secretory function with hepatocyte metabolic status [45]. Its disruption promotes insulin resistance and hepatic metabolic dysfunction, including steatosis with a pro-inflammatory hepatokine profile [20,48,49]. This underscores the bidirectional regulatory nature of liver–pancreas crosstalk in maintaining systemic metabolic homeostasis.
3.4. Pregnancy as a Model of Hepatokine Plasticity and the FGF21–SHBG Axis
Pregnancy represents a unique model of physiological metabolic overload in which the liver integrates fetal energy demands with maternal metabolic capacity [32]. This creates a dynamic state of coordinated metabolic stress adaptation and subsequent recovery.
This process requires tightly regulated endocrine control involving hepatokine secretion and coordinated remodeling of metabolic pathways [17].
Hepatokines coordinate energy substrate distribution between maternal tissues and the fetoplacental unit, enabling transient but reversible insulin resistance. This controlled insulin-resistant state is essential for maintaining fetal nutrient supply while preserving maternal metabolic flexibility.
Unlike obesity and T2DM, pregnancy allows observation of a complete and time-limited metabolic adaptation–de-adaptation cycle, providing a natural model of reversible systemic insulin resistance.
In this framework, the FGF21 axis mediates adaptation to metabolic stress and lipid overload during late pregnancy, whereas the hepatocyte nuclear factor 4α (HNF4α)–SHBG axis reflects restoration and reactivation of hepatocyte insulin-sensitive transcriptional programs after delivery [17].
4. FGF21 in Pregnancy: Adaptive Metabolic Stress Signaling
4.1. The PPARα–FGF21 Axis as a Lipid Stress Sensor
The hepatic peroxisome proliferator-activated receptor alpha (PPARα) acts as a master lipid sensor and transcriptional regulator of genes involved in energy homeostasis and inflammatory responses [50,51]. It is activated in highly oxidative tissues, primarily the liver, where it integrates metabolic signals related to substrate availability. During pregnancy, this pathway contributes to maternal metabolic resilience by coordinating adaptive responses to progressive lipid mobilization and fluctuating energy demand.
In this framework, PPARα functions primarily as a hepatic metabolic stress sensor that enables coordinated adaptation to increased lipid flux rather than as a generic lipid regulator.
PPARα activity depends on heterodimerization with retinoid X receptor (RXR), enabling regulation of genes involved in lipid and glucose metabolism, repair processes, and cell differentiation [50]. Its ligands include free fatty acids and endogenous molecules such as phospholipids and bilirubin. This ligand-dependent signaling network enables PPARα to function as an interface between nutrient sensing, endocrine adaptation, and hepatic metabolic programming during gestation.
Collectively, this positions PPARα as an upstream regulatory node integrating nutrient availability with hepatic transcriptional adaptation.
In this context, FGF21 is a key component of the adaptive response to metabolic stress, and its expression is tightly linked to PPARα activity [50,52]. FGF21 represents a downstream endocrine effector of PPARα signaling that coordinates systemic metabolic adaptation to hepatic lipid stress. Activation of the PPARα–FGF21 axis is particularly important during fasting-like states and late gestation, when maternal tissues increasingly rely on fatty acid oxidation and ketogenesis to preserve glucose availability for the fetus. Disruption of this axis may impair FGF21 induction, promoting hepatic steatosis and metabolic dysfunction.
Importantly, this pathway should be viewed as one of several adaptive mechanisms supporting metabolic flexibility during pregnancy rather than a singular determinant of lipid homeostasis.
4.2. FGF21 as a Metabolic Stress Hormone
FGF21 is a hepatokine secreted in response to energy deficit, fasting, and changes in substrate availability. It acts as an adaptive hormone integrating metabolic signals and regulating appetite and energy fuel utilization [13,30,51]. In pregnancy, FGF21 may function as a systemic stress-adaptation signal that coordinates maternal energy flexibility under conditions of physiological metabolic overload.
In this context, FGF21 is best understood as an endocrine stress-response effector linking hepatic nutrient sensing to systemic metabolic regulation.
Its expression increases during PPARα activation, particularly during fasting, ketogenic diet, and elevated NEFA, while it is suppressed after feeding [12,14,30,53]. These fluctuations reflect its role as a nutrient-sensitive regulator of substrate utilization.
FGF21 promotes utilization of alternative energy sources such as ketone bodies and fatty acids, sparing glucose for fetal needs [4,25,27]. It also improves insulin sensitivity and reduces lipotoxicity, exerting protective effects under metabolic overload.
Through these mechanisms, FGF21 supports metabolic flexibility by optimizing substrate utilization and limiting lipid-induced cellular stress.
In energy excess states, its circulating levels are also elevated in obesity and T2DM, where they correlate with insulin resistance and lipid disturbances [54,55]. This paradoxical elevation may reflect a compensatory response to chronic metabolic stress and progressive FGF21 resistance.
Thus, elevated FGF21 in chronic metabolic disease likely reflects adaptive compensation that is insufficient to restore metabolic homeostasis, consistent with the concept of FGF21 resistance.
4.3. FGF21 as a Response to Pregnancy-Induced Lipid Overload
In the third trimester, physiological hyperlipidemia and intense mobilization of fatty acids from adipose tissue occur, a process termed “lipid overflow” [30]. This state represents a physiological form of metabolic stress in which maternal tissues must adapt to sustained lipid exposure while maintaining metabolic homeostasis.
In this context, the liver–adipose axis must continuously balance lipid disposal with prevention of ectopic lipid accumulation.
FGF21 acts as a protective mechanism by limiting lipotoxicity through inhibition of de novo lipogenesis (including suppression of sterol regulatory element-binding protein 1c, SREBP-1c), thereby reducing hepatic metabolic burden [27]. This positions FGF21 as a hepatic stress-response factor that restrains lipid overaccumulation and preserves metabolic integrity under conditions of sustained lipid flux.
It also promotes browning of adipose tissue and increases thermogenesis, facilitating dissipation of excess energy as heat [12,13,14]. This adaptive thermogenic response enables conversion of excess metabolic substrates into energy expenditure rather than ectopic lipid storage.
Accordingly, FGF21 coordinates inter-organ energy redistribution by coupling hepatic lipid sensing to adipose tissue thermogenic capacity.
This mechanism creates a feedback loop between liver and adipose tissue, stabilizing circulating lipid levels and preventing transition from physiological hyperlipidemia to pathological states [13,30]. Accordingly, the FGF21 axis may represent a central component of interorgan communication linking hepatic lipid sensing with adipose tissue metabolic adaptation during pregnancy.
In conditions of excessive or prolonged metabolic stress, these adaptive mechanisms may become insufficient, potentially contributing to progression toward maladaptive metabolic states [12]. Failure of these adaptive mechanisms may therefore contribute to progression from physiological insulin resistance toward pathological metabolic dysfunction.
4.4. FGFR1/β-Klotho Signaling and FGF21 Resistance
Full FGF21 activity requires cooperation with the co-receptor β-Klotho and fibroblast growth factor receptors (FGFRs), which activate signaling cascades in target tissues [13,27,56]. These signaling pathways are particularly important in adipose tissue, which becomes a major metabolic target of FGF21 during late pregnancy.
In this context, the FGFR1/β-Klotho complex functions as the essential signaling interface translating circulating FGF21 into intracellular metabolic responses.
FGFR1/β-Klotho activation induces autophosphorylation and kinase signaling [13,14,56], supporting transcriptional programs involved in lipid utilization and thermogenic regulation. Sustained activation of this pathway may be required to maintain metabolic flexibility under conditions of gestational lipid overload and progressive insulin resistance.
Although FGF21 levels increase in late pregnancy, its signaling may be impaired in GDM. One early marker of resistance is reduced β-Klotho expression in adipose tissue, limiting thermogenic response [51]. Reduced receptor availability may therefore compromise adaptive energy dissipation and contribute to metabolic instability before overt hyperglycemia becomes clinically apparent.
Accordingly, impaired FGF21 action in GDM is increasingly interpreted as a state of signaling resistance characterized by reduced receptor–cofactor availability rather than decreased ligand production.
FGF21 resistance may therefore arise not from ligand deficiency but from impaired receptor–cofactor availability, resulting in uncoupling of endocrine signal from downstream metabolic response.
4.5. The FGF21–AMPK Axis as a Regulator of Energy Flexibility
FGF21 activates adenosine monophosphate-activated protein kinase (AMPK), which increases GLUT4 translocation in skeletal muscle, enhancing insulin-independent glucose uptake [27]. This mechanism may help maintain maternal glucose homeostasis despite the physiological decline in insulin sensitivity observed during pregnancy. In this context, AMPK functions as a downstream energy sensor translating FGF21 signaling into acute adjustments in cellular fuel uptake.
AMPK simultaneously inhibits acetyl-CoA carboxylase (ACC) and activates carnitine palmitoyltransferase 1 (CPT1), promoting mitochondrial β-oxidation of fatty acids [27,57]. As a result, maternal tissues become more capable of utilizing lipids as alternative energy substrates during periods of increased metabolic demand. These coordinated effects shift cellular metabolism toward fatty acid utilization while reducing lipid synthesis, thereby supporting substrate switching under gestational metabolic stress.
The FGF21–AMPK interaction forms a regulatory network controlling autophagy and mitophagy, reducing cellular stress and supporting metabolic adaptation under increased energy demand. Through these mechanisms, the axis supports cellular homeostasis by maintaining mitochondrial integrity under conditions of persistent metabolic pressure.
Adiponectin enhances this signaling axis, integrating communication between liver and adipose tissue [14,17,57,58]. Together, the FGF21–AMPK–adiponectin network may represent a central regulator of maternal metabolic flexibility and adaptive energy redistribution during gestation.
Collectively, this network integrates endocrine and cellular energy-sensing pathways to coordinate systemic substrate allocation between liver, muscle, and adipose tissue.
4.6. Mitochondria as Effectors of FGF21 Action
FGF21 promotes mitochondrial biogenesis via activation of the sirtuin 1 (SIRT1)–peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) axis, increasing mitochondrial number and ATP production efficiency [58]. These adaptations may support the increased energetic demands of maternal tissues during late pregnancy and lactation.
In this context, the SIRT1–PGC-1α pathway represents a primary downstream effector mechanism linking FGF21 signaling to enhanced mitochondrial capacity.
It also reduces reactive oxygen species (ROS) production and improves electron transport chain efficiency, maintaining redox balance under metabolic stress. These effects reflect improved mitochondrial functional efficiency, resulting in reduced oxidative stress during sustained lipid utilization.
FGF21 induces uncoupling proteins (UCP1–UCP3), enabling controlled energy dissipation as heat and reducing lipotoxicity [12,30]. This mechanism may prevent mitochondrial overload during physiological “lipid overflow” and facilitate safe disposal of excess energy substrates.
Accordingly, uncoupling activity provides an additional adaptive layer that limits excessive mitochondrial substrate pressure by converting surplus energy into heat.
4.7. FGF21 as a Lipotoxicity Buffer and Regulator of Postpartum Adaptation
In late pregnancy, FGF21 exerts cytoprotective effects, protecting the liver, heart, and peripheral tissues from excess free fatty acids via activation of the SIRT1–AMPK axis [12,30,58]. In this context, FGF21 functions as a stress-buffering endocrine signal that limits lipotoxic injury during sustained gestational lipid overload.
It has anti-inflammatory and antioxidant properties, including induction of superoxide dismutase 2 (SOD2) and reduction of oxidative stress in cardiomyocytes [59]. These protective effects may be particularly important in tissues vulnerable to oxidative injury during late gestation, including the maternal cardiovascular system. Collectively, these actions reflect a coordinated reduction of lipid-induced oxidative and inflammatory stress across multiple maternal tissues.
After delivery, elevated FGF21 supports lipid mobilization and metabolic adaptation to lactation, although its long-term role in improving insulin sensitivity remains unclear [30,60]. The postpartum increase in FGF21 may therefore represent continuation of the adaptive metabolic program initiated during late pregnancy. In this phase, FGF21 likely reflects ongoing metabolic reprogramming associated with the transition to the lactation state rather than a purely protective signal.
In women with prior GDM, it may play a compensatory role; however, there is no conclusive evidence for its protective effect against the development of T2DM [60,61]. Whether persistent elevation of FGF21 after pregnancy reflects successful metabolic recovery or compensatory stress signaling remains unresolved.
Accordingly, sustained postpartum FGF21 elevation should be interpreted as a context-dependent marker that may indicate either adaptive remodeling or unresolved metabolic stress.
5. SHBG Beyond Steroid Transport: Indicator and Mediator of Hepatic Insulin Sensitivity
5.1. Hepatic Regulation of SHBG
Sex steroid hormones, including androgens and estrogens, are important not only for reproductive function but also for energy metabolism [62]. They affect insulin secretion, β-cell function, insulin sensitivity, and glucose uptake [63]. According to the free hormone hypothesis, only unbound sex hormones are biologically active [64]. Since hormone activity depends on their bioavailability, SHBG plays a key role in regulating their effects [65].
SHBG is a glycoprotein produced mainly in the liver. Its expression is regulated by the transcription factor hepatocyte nuclear factor 4α (HNF4α) [66]. HNF4α is increasingly recognized not only as a regulator of SHBG transcription but also as an important determinant of hepatocyte metabolic identity, integrating glucose and lipid metabolism with endocrine signaling. Thus, HNF4α represents a central node linking hepatocyte transcriptional identity with endocrine output, including SHBG synthesis.
Several metabolic pathways, including AMP-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor γ (PPARγ), influence HNF4α activity [67,68]. HNF4α levels are also affected by adiponectin, the inflammatory cytokines TNF-α and IL-1β, and the sugars glucose and fructose [69,70,71]. These factors are linked to hepatic lipid accumulation and lipogenesis, suggesting that excessive sugar intake may lower SHBG production [72]. Collectively, these signals converge on HNF4α as an integrative sensor of hepatic metabolic and inflammatory status.
Insulin resistance reduces SHBG production through interconnected metabolic and inflammatory pathways, mainly by decreasing hepatic HNF-4α expression. High intake of fructose and sucrose stimulates sterol regulatory element binding protein 1c (SREBP-1c), promotes de novo lipogenesis, and contributes to hepatic fat accumulation, which suppresses HNF-4α activity and decreases SHBG production [73,74]. In insulin-resistant states, this regulatory network shifts toward enhanced lipogenesis and reduced HNF4α-driven transcriptional activity, resulting in decreased SHBG synthesis.
Obesity and insulin resistance also increase adipose tissue lipolysis, resulting in elevated delivery of free fatty acids to the liver. This further promotes hepatic lipogenesis and gluconeogenesis, contributing to lower HNF-4α expression and reduced SHBG synthesis [75]. At the molecular level, HNF-4α stimulates SHBG gene transcription by binding to regions within the SHBG promoter. Increased hepatic lipogenesis also enhances PPAR-γ expression, which competes with HNF-4α and further inhibits SHBG transcription [68,75]. This establishes a feed-forward metabolic loop in which lipid flux, inflammation, and transcriptional reprogramming jointly suppress SHBG production.
Together, these mechanisms explain how insulin resistance, obesity, inflammation, and hepatic fat accumulation contribute to decreased SHBG expression and secretion, supporting the concept that low SHBG reflects impaired hepatic metabolic regulation rather than an isolated hormonal abnormality.
Accordingly, SHBG should be interpreted as a functional surrogate marker of hepatic metabolic and transcriptional state rather than a passive transport protein.
5.2. SHBG Dynamics During Pregnancy and Postpartum
Pregnancy causes profound hormonal and metabolic changes that strongly affect SHBG levels. One of the main reasons for the increase in SHBG during pregnancy is the rising concentration of estrogens, which stimulate hepatic SHBG production [76]. SHBG levels increase early in pregnancy and continue to rise throughout gestation, reaching peak values in the third trimester. Higher SHBG concentrations help regulate the amount of free sex hormones in circulation and support hormonal balance during pregnancy [77].
The pregnancy-associated rise in SHBG represents a physiological paradox in which estrogen-driven stimulation of SHBG synthesis partially counterbalances the suppressive effects of insulin resistance on hepatic SHBG production. These endocrine adaptations contribute to the maintenance of pregnancy [78].
Abnormal sex steroid concentrations during the peripartum period may be associated with maternal complications, including preeclampsia, gestational diabetes, altered autoimmune disease activity, impaired cognition, structural brain changes, and increased breast cancer risk later in life [79].
Pregnancy is also characterized by progressive insulin resistance, particularly in the second and third trimesters. Although insulin resistance generally suppresses SHBG production, the strong stimulatory effect of estrogens predominates during pregnancy, resulting in persistently elevated SHBG levels [77].
Nevertheless, women with obesity, GDM, or excessive gestational weight gain often have lower SHBG levels than healthy pregnant women [80]. This suggests that metabolic disturbances may attenuate the physiological pregnancy-related rise in SHBG. In pregnancies complicated by GDM or pregestational obesity, insulin-mediated suppression of hepatic SHBG synthesis may become more apparent despite sustained estrogenic stimulation.
After delivery, placental separation leads to a rapid decline in hormone concentrations and substantial endocrine changes in the postpartum period. Estrogen and progesterone levels decrease, and SHBG concentrations gradually return toward pre-pregnancy values over subsequent weeks and months [77]. This decline reflects both reduced estrogen stimulation and postpartum metabolic adaptations [79].
5.3. SHBG as a Surrogate of Hepatic Insulin Responsiveness
SHBG is increasingly recognized as a marker of metabolic health and hepatic insulin sensitivity. Because SHBG is produced mainly in hepatocytes, circulating SHBG levels reflect changes in liver metabolism and insulin signaling. Clinical and experimental studies have shown that low SHBG concentrations are associated with insulin resistance, obesity, T2DM, metabolic dysfunction-associated steatotic liver disease (MASLD), and polycystic ovary syndrome (PCOS) [69,75,83,84,85]. These findings suggest that SHBG may represent a marker of hepatic metabolic dysfunction rather than merely a transport protein for sex hormones.
Accumulating evidence indicates that the relationship between insulin resistance and SHBG is strongly linked to hepatic lipid metabolism. In vitro and animal studies demonstrated that monosaccharides such as glucose and fructose are converted into palmitate, which suppresses HNF4α and subsequently decreases SHBG synthesis. This suggests that low SHBG concentrations may reflect hepatic lipid overload and impaired hepatic insulin responsiveness rather than the isolated effect of hyperinsulinemia [84].
Current evidence also supports the concept of selective hepatic insulin resistance, in which insulin loses its ability to suppress hepatic gluconeogenesis while lipogenic pathways remain active or become overstimulated [86,87]. As a result, hepatic triglyceride synthesis and liver fat accumulation continue despite systemic insulin resistance [88]. Persistent activation of lipogenic pathways may therefore contribute to reduced SHBG production.
Lower SHBG levels are associated not only with hepatic fat accumulation but also with markers of liver injury and impaired metabolic homeostasis [85]. Inflammation and adipose tissue dysfunction further contribute to SHBG regulation. Obesity and insulin resistance are associated with chronic low-grade inflammation, elevated TNF-α and IL-1β concentrations, and reduced adiponectin levels [89,90]. These mediators influence HNF4α expression and suppress SHBG synthesis. Adiponectin appears to support HNF4α activity through AMPK, whereas inflammatory cytokines inhibit HNF4α through NF-κB and MAPK signaling pathways [67]. Consequently, reduced SHBG concentrations may integrate several components of metabolic dysfunction, including hepatic insulin resistance, inflammation, adipose tissue dysfunction, and lipid accumulation.
Genome-wide association studies (GWAS) identified several loci associated with circulating SHBG levels in genes involved in hepatic glucose and lipid metabolism, including GCKR, GCK, MLXIPL/ChREBP, HNF4A, and PNPLA3 [91]. One of the strongest associations was identified for the GCKR variant rs1260326, which encodes glucokinase regulatory protein (GKRP), a regulator of hepatic glucokinase activity. Studies showed that this variant reduces the inhibitory effect of GKRP on glucokinase, leading to increased hepatic glucose uptake, enhanced glycolysis, and increased de novo lipogenesis with subsequent liver fat accumulation [92,93].
Lower SHBG levels were also associated with variants in GCK, encoding glucokinase, and MLXIPL, encoding carbohydrate response element-binding protein (ChREBP), a key transcription factor regulating lipogenic enzymes [94].
From a systems biology perspective, SHBG may be viewed as an integrative marker of hepatic insulin signaling and metabolic status, reflecting the balance between lipogenesis, inflammation, and HNF4α-dependent transcriptional regulation. Accordingly, circulating SHBG concentrations may capture a hepatic phenotype characterized by selective insulin resistance, in which gluconeogenic suppression is impaired while lipogenic pathways remain active. In this context, SHBG is best interpreted as a dynamic biomarker of hepatic metabolic state rather than a direct mediator of systemic insulin sensitivity.
Taken together, these findings suggest that circulating SHBG levels are linked to pathways regulating hepatic glucose metabolism, lipogenesis, and intrahepatic lipid accumulation. They further support the concept that SHBG may serve as a surrogate marker of hepatic insulin responsiveness. Low SHBG concentrations appear to reflect impaired hepatic insulin sensitivity, increased de novo lipogenesis, liver fat accumulation, chronic inflammation, and adipose tissue dysfunction. Although the underlying mechanisms continue to be investigated, SHBG is increasingly viewed as a clinically useful marker of liver-related metabolic dysfunction.
5.4. Restoration of the HNF4α–SHBG Axis After Delivery
After delivery, declining placental hormone levels initiate a gradual reversal of pregnancy-related metabolic adaptations. During the postpartum period, hepatic metabolism progressively shifts toward the pre-pregnancy state, including improved insulin sensitivity, reduced hepatic lipogenesis, and normalization of glucose and lipid metabolism [95,96].
Although these changes are expected after childbirth, the extent to which the HNF4α–SHBG axis normalizes postpartum has not been well characterized in humans. Current evidence is indirect and derives mainly from studies on GDM, postpartum glucose intolerance, and the relationship between SHBG and insulin resistance [67,83,97,98], rather than from dedicated longitudinal investigations of postpartum hepatic transcriptional recovery.
Restoration of HNF4α activity appears to contribute to recovery of SHBG synthesis. Accordingly, SHBG should be considered not only as a reproductive hormone-binding protein but also as a hepatic marker linked to metabolic regulation [99].
Several studies demonstrated associations between SHBG levels, hyperinsulinemia, and GDM [100,101]. Women with previous GDM also remain at increased risk of postpartum glucose intolerance and later T2DM [102], suggesting that metabolic recovery after pregnancy may be incomplete in some patients.
Persistently low postpartum SHBG concentrations may therefore reflect incomplete restoration of HNF4α-dependent transcriptional regulation or ongoing hepatic insulin resistance. Postpartum normalization of SHBG levels may indicate not only hormonal withdrawal but also progressive recovery of hepatocyte metabolic function through reactivation of HNF4α-dependent pathways.
In this context, the HNF4α–SHBG axis may serve as a systemic marker of hepatic metabolic reprogramming during the transition from the gestational lipogenic state to the postnatal metabolic phenotype. However, whether restoration of SHBG actively contributes to metabolic recovery or merely reflects upstream hepatic normalization remains to be determined and requires further longitudinal mechanistic studies.
Postpartum changes in the HNF4α–SHBG axis may therefore reflect recovery of coordinated hepatic glucose metabolism, lipid handling, and insulin signaling after pregnancy. Nevertheless, this interpretation remains largely inferential because direct postpartum studies simultaneously assessing HNF4α activity and SHBG dynamics remain limited and are currently lacking in large longitudinal cohorts.
6. Integrated Molecular Model: Coordinated Hepatokine Control of Postpartum Insulin Sensitivity
6.1. Converging Molecular Pathways Regulating Metabolic Plasticity
Pregnancy constitutes a dynamic metabolic state characterized by progressive insulin resistance, enhanced lipolysis, and increased hepatic glucose output. Although these adaptations are physiologically required to ensure adequate nutrient supply to the fetus, they impose significant metabolic stress on maternal tissues [32]. Restoration of insulin sensitivity after delivery is not solely driven by the withdrawal of placental hormones, but rather reflects hepatic metabolic reprogramming involving hepatokine signaling [103,104].
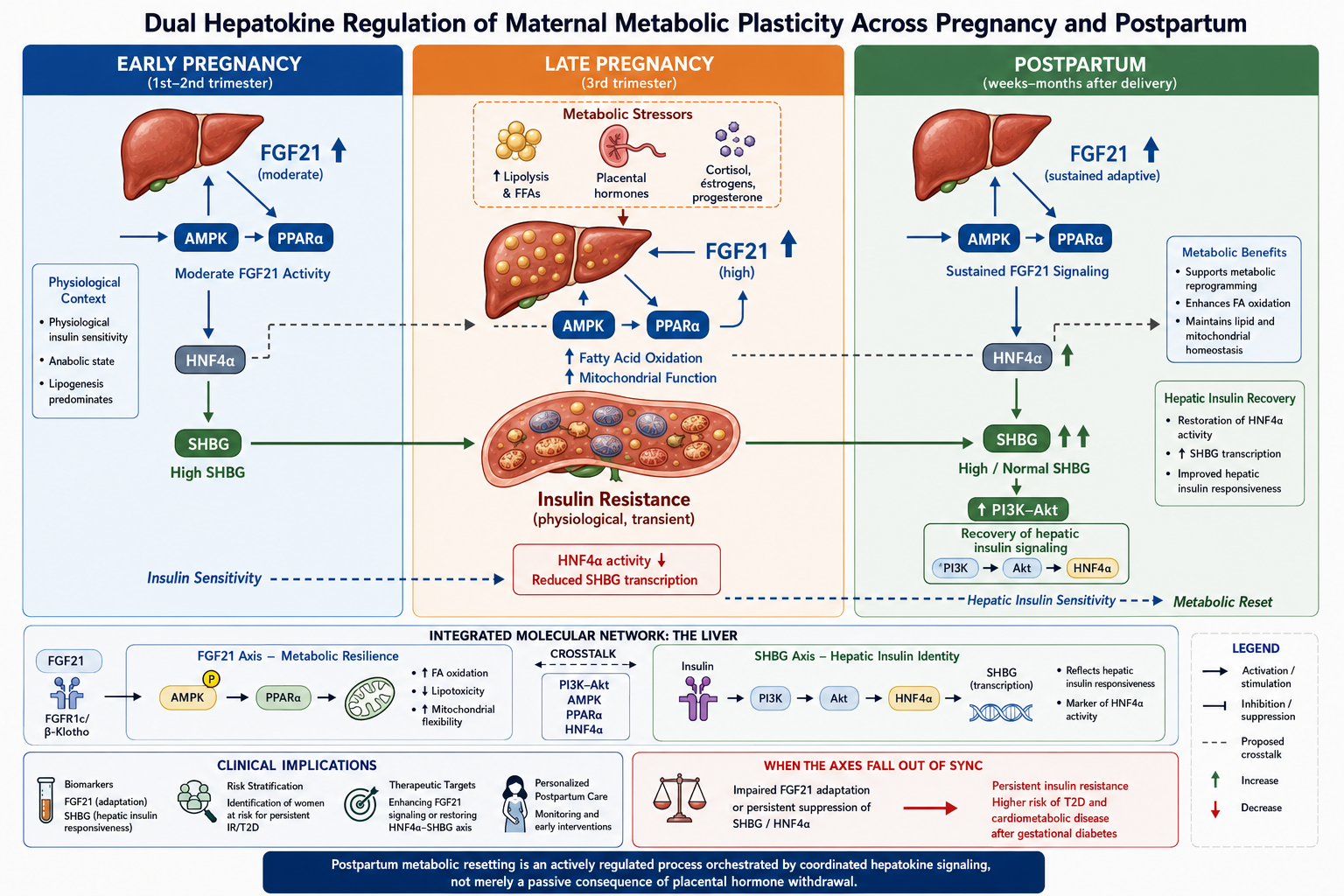
Within this framework, the FGF21-associated axis is interpreted as reflecting metabolic stress response capacity, whereas SHBG-associated variation is considered a proxy of hepatic insulin sensitivity and transcriptional state, as reflected in circulating SHBG levels [105,106]. Importantly, these axes operate at different temporal scales: FGF21 represents a rapid stress-responsive adaptive endocrine response system, while SHBG reflects slower transcriptional reprogramming of hepatic identity. A summary of the integrated signaling nodes is provided in Table 1.
Several intracellular nodes integrate these axes:
- AMPK, acting as a cellular energy sensor, is activated under metabolic stress and promotes fatty acid oxidation, mitochondrial biogenesis, and improved insulin signaling. FGF21 has been shown to enhance AMPK activity in peripheral tissues, thereby supporting adaptive fuel utilization during periods of increased lipid flux [107];
- PPARα, a key transcriptional regulator of hepatic β-oxidation, drives FGF21 expression in response to lipid overflow and fasting-like signals characteristic of late pregnancy. Activation of the PPARα–FGF21 pathway may therefore buffer lipotoxic stress and limit ectopic lipid accumulation [108];
These pathways primarily mediate acute energetic adaptation and lipid handling.
- PI3K–Akt signaling, central to insulin action, is progressively attenuated during gestational insulin resistance. Restoration of this pathway postpartum likely reflects recovery of hepatic insulin responsiveness. SHBG production, which is suppressed by hyperinsulinemia, may serve as a functional indicator of hepatic insulin sensitivity and HNF4α-dependent transcriptional activity, thereby reflecting hepatic insulin action [109];
- HNF4α, a master regulator of hepatocyte metabolic identity, controls SHBG transcription and coordinates genes involved in glucose and lipid metabolism. Impaired HNF4α activity under conditions of insulin resistance and hepatic steatosis may contribute to reduced SHBG levels, whereas postpartum normalization of hepatic nutrient handling and endocrine signaling may restore HNF4α-driven transcriptional programs [73].
In this context, HNF4α can be interpreted not only as a transcription factor, but as a central determinant of hepatic “metabolic identity stability” across pregnancy and postpartum transition.
Together, these interconnected pathways suggest that FGF21 and SHBG operate within a shared regulatory network governing hepatic adaptive plasticity, with complementary roles in acute metabolic buffering and long-term transcriptional reprogramming.
However, direct causal evidence linking coordinated FGF21–SHBG dynamics to postpartum insulin sensitivity recovery remains limited. Accordingly, the model should be interpreted as a systems-level hypothesis rather than a proven coordinated signaling axis.
6.2. A Dynamic Three-Phase Model of Hepatokine-Mediated Adaptation
We propose a dynamic, phase-dependent model of hepatokine regulation across pregnancy and the postpartum period.
Early Pregnancy: Relative Insulin Sensitivity and Metabolic Priming
In early gestation, maternal insulin sensitivity is relatively preserved or even transiently enhanced. Hepatic energetic and transcriptional programs remain largely balanced, and SHBG levels may reflect intact HNF4α activity and insulin signaling [73]. FGF21 activity is moderate, consistent with limited metabolic stress.
This phase represents a relative baseline state preceding progressive placental-driven insulin resistance.
Late Pregnancy: Adaptive Insulin Resistance and Metabolic Resilience
As gestation progresses, placental hormones promote systemic insulin resistance, increased adipose tissue lipolysis, and elevated circulating free fatty acids. This lipid overflow challenges hepatic metabolic capacity [110].
In this context, activation of the PPARα–FGF21 axis may function as a compensatory mechanism that:
- enhances fatty acid oxidation,
- supports ketogenesis when required,
- limits hepatic lipotoxicity,
- maintains mitochondrial function.
Thus, FGF21 may act as a resilience factor that permits physiological insulin resistance without irreversible metabolic damage [111,112].
Concurrently, hyperinsulinemia and altered hepatic lipid flux may be associated with reduced SHBG production, potentially reflecting reduced hepatic insulin sensitivity via impaired HNF4α-dependent transcription.
This divergence highlights a key feature of the model: stress-adaptive metabolic signaling may remain active while markers of hepatic transcriptional identity progressively decline.
Postpartum Period: Active Metabolic Resetting
Following delivery, the rapid decline in placental hormones reduces diabetogenic pressure. However, we propose that full restoration of insulin sensitivity requires coordinated hepatic reprogramming rather than passive hormonal normalization alone [49].
Postpartum recovery may involve:
- Attenuation of lipid overflow and inflammatory signaling.
- Persistence or recalibration of FGF21-mediated metabolic adaptations, facilitating transition from a lipolytic to a more balanced metabolic state.
In this model, SHBG serves as an accessible systemic indicator of hepatic metabolic recovery, while FGF21 reflects adaptive metabolic stress signaling capacity.
Postpartum recovery is therefore conceptualized as a two-layer process: metabolic recalibration followed by transcriptional identity restoration (Figure 1).
6.3. Pathophysiological Implications: When Synchronization Fails
A critical feature of this integrated model is the temporal synchronization between FGF21-driven metabolic stress adaptation and restoration of hepatic insulin signaling reflected by SHBG [115].
We hypothesize that disruption of this coordination may underlie persistent insulin resistance after GDM. For example:
- Insufficient or dysregulated FGF21 signaling may be associated with impaired buffering of lipid-induced stress during late pregnancy, potentially contributing to hepatic steatosis and persistent metabolic dysfunction.
- Delayed normalization of the HNF4α–SHBG axis may reflect persistent hepatic insulin resistance, as suggested by sustained reductions in circulating SHBG despite resolution of placental endocrine signals.
Such dysregulation could contribute to the well-established increased risk of T2DM and cardiometabolic disease in women with prior GDM [117].
Within this framework, postpartum metabolic disease may be viewed as a state of impaired coordination between acute metabolic stress adaptation and longer-term hepatic transcriptional reprogramming.
6.4. Conceptual Implications
This integrated hepatokine model reframes pregnancy as a physiological experiment in reversible insulin resistance, in which the liver plays an active, endocrine role in determining whether metabolic stress resolves or progresses toward chronic disease [95].
Rather than viewing postpartum insulin sensitivity as a passive rebound phenomenon, our framework proposes that coordinated hepatokine signaling - through the metabolic resilience axis (FGF21) and the hepatic insulin identity axis (HNF4α–SHBG) - may contribute to successful metabolic recovery.
Accordingly, pregnancy and postpartum transition should be understood as a continuum of regulated hepatic endocrine plasticity, rather than two discrete metabolic states.
From a translational perspective, this framework may help guide future development of postpartum risk stratification strategies integrating hepatokine profiling, hepatic insulin sensitivity markers, and longitudinal metabolic surveillance following GDM. In particular, combined assessment of FGF21 and SHBG trajectories may potentially assist in identifying women at increased risk for persistent insulin resistance and future T2DM progression.
Future longitudinal and mechanistic studies are required to determine whether these pathways merely reflect or actively influence postpartum metabolic trajectories.
7. Clinical Implications of the Proposed Two-Axis Hepatokine Framework
7.1. Integrative Framing
Building on the previously described roles of FGF21 in metabolic stress adaptation and SHBG as a marker of hepatic insulin sensitivity, this section integrates these hepatokines into a unified conceptual framework of dynamic hepatic adaptation during pregnancy.
Within this model, FGF21-associated signaling is interpreted as reflecting metabolic stress response capacity, whereas SHBG-associated variation is considered a proxy of hepatic insulin sensitivity and transcriptional function, as reflected in circulating SHBG levels [118,119]. Importantly, this framework is intended as a systems-level synthesis of established biological pathways rather than evidence of direct coordinated signaling between these axes.
Accordingly, the following sections explore the potential clinical implications of this dual-axis construct in risk prediction, longitudinal disease trajectory, and therapeutic hypothesis generation.
7.2. Prediction of GDM and Early Metabolic Risk Stratification
The proposed two-axis framework (FGF21-associated metabolic resilience axis and SHBG-associated hepatic insulin sensitivity axis) provides a conceptual extension of traditional static glycemic markers for early risk stratification in pregnancy.
In this context, circulating FGF21 may reflect early compensatory metabolic stress in response to lipid overload and mitochondrial strain, whereas SHBG levels may serve as a surrogate marker of hepatic insulin sensitivity and HNF4α-associated transcriptional activity [75,120,121].
Combined assessment of these hepatokine-associated pathways may hypothetically improve early identification of women transitioning from physiological insulin resistance to pathological gestational glucose dysregulation [98]. However, it remains to be determined whether combined FGF21 and SHBG profiling improves predictive performance beyond established clinical risk models.
Rather than representing a unified signaling pathway, these biomarkers should be interpreted as complementary indicators of distinct but potentially interacting aspects of hepatic metabolic adaptation, with currently limited evidence for coordinated predictive utility.
7.3. Prediction of Long-Term Risk of Type 2 Diabetes After Pregnancy
Pregnancy complicated by GDM represents a period of transient metabolic stress during which underlying hepatic insulin resistance may become clinically apparent.
Within the proposed framework, persistent dysregulation of FGF21 signaling after delivery may indicate incomplete resolution of metabolic stress adaptation [122], whereas sustained reductions in SHBG may reflect persistent impairment of hepatic insulin sensitivity and hepatic transcriptional function [123].
Longitudinal trajectories of these markers may therefore be explored as potential contributors to risk stratification for women at increased risk of progression to T2DM and cardiometabolic disease [124]. Importantly, this interpretation is hypothesis-generating and requires validation in prospective postpartum cohorts with long-term follow-up.
At present, these markers should be considered exploratory rather than clinically validated predictors of long-term metabolic outcomes.
7.4. Therapeutic Hypotheses Targeting the FGF21-Associated Axis
Interventions targeting metabolic stress pathways may modulate the FGF21-associated axis. These include pharmacologic FGF21 analogs [125], as well as lifestyle-based interventions such as dietary modification and structured physical activity [126]. From a mechanistic perspective, such interventions may enhance FGF21–PPARα–AMPK signaling, thereby improving lipid oxidation, mitochondrial function, and resistance to lipotoxic stress. Importantly, these effects are likely to reflect downstream metabolic recalibration rather than direct amplification of FGF21 signaling alone.
At present, the relevance of these interventions in pregnancy and postpartum populations remains exploratory, and their clinical application should be considered within the context of ongoing or future clinical trials. In particular, safety and timing considerations in gestation and early postpartum remain critical limiting factors for translational application.
7.5. Modulation of Hepatic Insulin Sensitivity and the SHBG-Associated Axis
Improvement of hepatic insulin sensitivity represents a potential mechanism for normalization of SHBG levels during and after pregnancy.
Possible pathways include reduction of hepatic lipogenesis, attenuation of inflammatory signaling, and restoration of insulin-regulated hepatic transcriptional programs, potentially involving HNF4α-associated networks [127]. Collectively, these processes converge on improved hepatocyte metabolic function and reduced lipogenic drive, which may permit reactivation of SHBG transcriptional output.
Given its clinical accessibility, SHBG may serve as a pragmatic biomarker for monitoring hepatic insulin sensitivity dynamics during postpartum metabolic recovery. However, its role as a direct surrogate of hepatic transcriptional integrity remains indirect and not yet mechanistically validated in longitudinal human studies.
In this context, SHBG should be interpreted primarily as a circulating biomarker reflecting hepatic insulin sensitivity and HNF4α-dependent transcriptional activity rather than an active mediator of insulin signaling. Accordingly, changes in SHBG levels are best understood as downstream consequences of hepatic metabolic reprogramming rather than drivers of insulin action.
7.6. Combined Preventive and Therapeutic Strategy
Integrated modulation of both proposed axes may offer a more comprehensive approach to metabolic risk reduction than targeting either pathway alone.
Within this conceptual framework, simultaneous support of FGF21-associated metabolic resilience and SHBG-associated hepatic insulin sensitivity is more appropriately interpreted as reflecting coordinated changes in acute metabolic stress responses and longer-term hepatic metabolic reprogramming.
This model reframes pregnancy as a dynamic period of hepatic endocrine adaptation with potential long-term cardiometabolic consequences extending beyond glycemic regulation alone.
However, the clinical utility of integrated FGF21-SHBG profiling remains to be established. At present, this framework should be interpreted as a systems-level hypothesis requiring validation in longitudinal clinical and mechanistic studies. In particular, its immediate translational value is currently limited to risk stratification and hypothesis generation rather than interventional guidance.
8. Conclusions
Pregnancy represents a unique physiological state of coordinated metabolic stress and adaptation, characterized by a reversible shift toward systemic insulin resistance that ensures adequate nutrient allocation to the developing fetus. Rather than being a passive consequence of placental hormone action, this state emerges from an integrated, dynamic network of hepatic signaling pathways that actively regulate maternal metabolic flexibility.
In this framework, the liver functions as a central endocrine organ orchestrating systemic energy distribution through hepatokine-mediated communication. Two complementary regulatory axes can be distinguished: a rapid metabolic resilience axis centered on FGF21, and a slower hepatic identity axis governed by HNF4α and reflected systemically by SHBG. Together, these pathways integrate lipid flux, glucose homeostasis, inflammatory tone, and transcriptional control of hepatic function.
FGF21 acts as an adaptive stress-response hormone that buffers lipid overload, enhances fatty acid oxidation, and supports mitochondrial function through AMPK- and PPARα-dependent signaling. In contrast, SHBG serves as a downstream indicator of hepatic insulin sensitivity and transcriptional integrity, reflecting the functional state of HNF4α-dependent metabolic programming. While FGF21 captures dynamic metabolic stress adaptation, SHBG provides a more stable circulating readout of hepatic insulin responsiveness and HNF4α-dependent hepatic metabolic state.
The coordinated interaction between these two axes offers a unified model of gestational insulin resistance and postpartum metabolic recovery. During pregnancy, physiological insulin resistance is maintained within adaptive limits through FGF21-mediated metabolic buffering, while SHBG dynamics reflect progressive modulation of hepatic insulin signaling. After delivery, restoration of metabolic homeostasis requires not only withdrawal of placental hormones but also progressive restoration of hepatic metabolic regulation, involving recalibration of FGF21 signaling and reactivation of the HNF4α–SHBG axis.
Disruption of this coordinated system may contribute to pathological outcomes, including GDM and increased long-term risk of T2DM. In such cases, insufficient metabolic flexibility, persistent hepatic insulin resistance, or incomplete restoration of hepatic transcriptional identity may prevent full postpartum metabolic recovery.
Overall, the proposed two-axis hepatokine model reframes pregnancy and the postpartum period as a continuum of regulated hepatic endocrine adaptation. This perspective shifts the interpretation of insulin resistance from a purely pathological feature to a context-dependent physiological program with clearly defined failure modes. Future longitudinal and mechanistic studies are required to determine whether FGF21 and SHBG are merely biomarkers of these processes or direct mediators or downstream indicators of maternal metabolic trajectories.
Author Contributions
Conceptualization, K.P.-B. and Ż.K-T.; methodology, K.P.-B. and Ż.K.-T.; writing – original draft preparation, K.P.-B., K.T., A.M., A.B.; visualization, K.T.; supervision, Ż.K.-T. and B.L.-G..; project administration, K.P.-B. and Ż.K.-T.; funding acquisition, Ż.K.-T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef]
- Lain, K.Y.; Catalano, P.M. Metabolic Changes in Pregnancy. Clin. Obstet. Gynecol. 2007, 50, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Stern, C.; Schwarz, S.; Moser, G.; Cvitic, S.; Jantscher-Krenn, E.; Gauster, M.; Hiden, U. Placental Endocrine Activity: Adaptation and Disruption of Maternal Glucose Metabolism in Pregnancy and the Influence of Fetal Sex. Int. J. Mol. Sci. 2021, 22, 12722. [Google Scholar] [CrossRef] [PubMed]
- Karcz, K.; Królak-Olejnik, B. Impact of Gestational Diabetes Mellitus on Fetal Growth and Nutritional Status in Newborns. Nutrients 2024, 16, 4093. [Google Scholar] [CrossRef]
- Friedman, J.E.; Ishizuka, T.; Shao, J.; Huston, L.; Highman, T.; Catalano, P. Impaired Glucose Transport and Insulin Receptor Tyrosine Phosphorylation in Skeletal Muscle from Obese Women with Gestational Diabetes. Diabetes 1999, 48, 1807–1814. [Google Scholar] [CrossRef]
- Colomiere, M.; Permezel, M.; Lappas, M. Diabetes and Obesity during Pregnancy Alter Insulin Signalling and Glucose Transporter Expression in Maternal Skeletal Muscle and Subcutaneous Adipose Tissue. J. Mol. Endocrinol. 2010, 44, 213–223. [Google Scholar] [CrossRef]
- Kampmann, U.; Knorr, S.; Fuglsang, J.; Ovesen, P. Determinants of Maternal Insulin Resistance during Pregnancy: An Updated Overview. J. Diabetes Res. 2019, 2019, 5320156. [Google Scholar] [CrossRef]
- Poon, L.C.; McIntyre, H.D.; Hyett, J.A.; da Fonseca, E.B.; Hod, M. FIGO Pregnancy and NCD Committee The First-Trimester of Pregnancy - A Window of Opportunity for Prediction and Prevention of Pregnancy Complications and Future Life. Diabetes Res. Clin. Pract. 2018, 145, 20–30. [Google Scholar] [CrossRef]
- Du, R.; Wang, F.; Li, L.; Wang, Q. The Double-Edged Sword of Gestational Insulin Resistance: Navigating Maternal Adaptation and Its Risks for Pregnancy and Offspring Health. Obes. Rev. 2026, 27, e70048. [Google Scholar] [CrossRef]
- Leoni, M.; Padilla, N.; Fabbri, A.; Della-Morte, D.; Ricordi, C.; Infante, M. Mechanisms of Insulin Resistance during Pregnancy. In Evolving Concepts in Insulin Resistance; IntechOpen, 2022; ISBN 978-1-80355-502-7. [Google Scholar]
- Kirwan, J.P.; Hauguel-De Mouzon, S.; Lepercq, J.; Challier, J.-C.; Huston-Presley, L.; Friedman, J.E.; Kalhan, S.C.; Catalano, P.M. TNF-Alpha Is a Predictor of Insulin Resistance in Human Pregnancy. Diabetes 2002, 51, 2207–2213. [Google Scholar] [CrossRef] [PubMed]
- Jarvie, E.; Hauguel-de-Mouzon, S.; Nelson, S.M.; Sattar, N.; Catalano, P.M.; Freeman, D.J. Lipotoxicity in Obese Pregnancy and Its Potential Role in Adverse Pregnancy Outcome and Obesity in the Offspring. Clin. Sci. (Lond) 2010, 119, 123–129. [Google Scholar] [CrossRef]
- Hua, L.; Yang, Y.; Zhang, H.; Jiang, X.; Jin, C.; Feng, B.; Che, L.; Xu, S.; Lin, Y.; Wu, D.; et al. Adipocyte FGF21 Signaling Defect Aggravated Adipose Tissue Inflammation in Gestational Diabetes Mellitus. Nutrients 2024, 16, 3826. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Morgan, D.A.; Rahmouni, K.; Sonoda, J.; Fu, X.; Burgess, S.C.; Holland, W.L.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19, FGF21 and an FGFR1/β-Klotho-Activating Antibody Act on the Nervous System to Regulate Body Weight and Glycemia. Cell Metab. 2017, 26, 709–718.e3. [Google Scholar] [CrossRef] [PubMed]
- Waters, T.P.; Kim, S.Y.; Sharma, A.J.; Schnellinger, P.; Bobo, J.K.; Woodruff, R.T.; Cubbins, L.A.; Haghiac, M.; Minium, J.; Presley, L.; et al. Longitudinal Changes in Glucose Metabolism in Women with Gestational Diabetes, from Late Pregnancy to the Postpartum Period. Diabetologia 2020, 63, 385–394. [Google Scholar] [CrossRef]
- Abbassi-Ghanavati, M.; Greer, L.G.; Cunningham, F.G. Pregnancy and Laboratory Studies: A Reference Table for Clinicians. Obstet. Gynecol. 2009, 114, 1326–1331. [Google Scholar] [CrossRef]
- Chen, C.; Xie, L.; Zhang, M.; Shama, null; Cheng, K.K.Y.; Jia, W. The Interplay between the Muscle and Liver in the Regulation of Glucolipid Metabolism. J. Mol. Cell Biol. 2024, 15, mjad073. [Google Scholar] [CrossRef]
- Misu, H. Identification of Hepatokines Involved in Pathology of Type 2 Diabetes and Obesity. Endocr. J. 2019, 66, 659–662. [Google Scholar] [CrossRef]
- Miao, X.; Alidadipour, A.; Saed, V.; Sayyadi, F.; Jadidi, Y.; Davoudi, M.; Amraee, F.; Jadidi, N.; Afrisham, R. Hepatokines: Unveiling the Molecular and Cellular Mechanisms Connecting Hepatic Tissue to Insulin Resistance and Inflammation. Acta Diabetol. 2024, 61, 1339–1361. [Google Scholar] [CrossRef] [PubMed]
- Jensen-Cody, S.O.; Potthoff, M.J. Hepatokines and Metabolism: Deciphering Communication from the Liver. Mol. Metab. 2021, 44, 101138. [Google Scholar] [CrossRef]
- Hadden, D.R.; McLaughlin, C. Normal and Abnormal Maternal Metabolism during Pregnancy. Semin Fetal Neonatal Med. 2009, 14, 66–71. [Google Scholar] [CrossRef]
- Koukkou, E.; Watts, G.F.; Lowy, C. Serum Lipid, Lipoprotein and Apolipoprotein Changes in Gestational Diabetes Mellitus: A Cross-Sectional and Prospective Study. J. Clin. Pathol. 1996, 49, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Lim, Y.-M.; Lee, M.-S. Role of Autophagy in Diabetes and Endoplasmic Reticulum Stress of Pancreatic β-Cells. Exp. Mol. Med. 2012, 44, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Torres-Torres, J.; Monroy-Muñoz, I.E.; Perez-Duran, J.; Solis-Paredes, J.M.; Camacho-Martinez, Z.A.; Baca, D.; Espino-Y-Sosa, S.; Martinez-Portilla, R.; Rojas-Zepeda, L.; Borboa-Olivares, H.; et al. Cellular and Molecular Pathophysiology of Gestational Diabetes. Int. J. Mol. Sci. 2024, 25, 11641. [Google Scholar] [CrossRef] [PubMed]
- Duttaroy, A.K.; Basak, S. Maternal Fatty Acid Metabolism in Pregnancy and Its Consequences in the Feto-Placental Development. Front Physiol. 2021, 12, 787848. [Google Scholar] [CrossRef]
- Kim, T.H.; Hong, D.-G.; Yang, Y.M. Hepatokines and Non-Alcoholic Fatty Liver Disease: Linking Liver Pathophysiology to Metabolism. Biomedicines 2021, 9, 1903. [Google Scholar] [CrossRef]
- Yano, K.; Yamaguchi, K.; Seko, Y.; Okishio, S.; Ishiba, H.; Tochiki, N.; Takahashi, A.; Kataoka, S.; Okuda, K.; Liu, Y.; et al. Hepatocyte-Specific Fibroblast Growth Factor 21 Overexpression Ameliorates High-Fat Diet-Induced Obesity and Liver Steatosis in Mice. Lab Invest 2022, 102, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-P.; Lin, S.; Xie, B.-Y.; Zhao, H.-F. Recent Progress in Metabolic Reprogramming in Gestational Diabetes Mellitus: A Review. Front Endocrinol. 2023, 14, 1284160. [Google Scholar] [CrossRef]
- Visiedo, F.; Vázquez-Fonseca, L.; Ábalos-Martínez, J.; Broullón-Molanes, J.R.; Quintero-Prado, R.; Mateos, R.M.; Bugatto, F. Maternal Elevated Inflammation Impairs Placental Fatty Acids β-Oxidation in Women with Gestational Diabetes Mellitus. Front Endocrinol. 2023, 14, 1146574. [Google Scholar] [CrossRef]
- Buell-Acosta, J.D.; Garces, M.F.; Parada-Baños, A.J.; Angel-Muller, E.; Paez, M.C.; Eslava-Schmalbach, J.; Escobar-Cordoba, F.; Caminos-Cepeda, S.A.; Lacunza, E.; Castaño, J.P.; et al. Maternal Fibroblast Growth Factor 21 Levels Decrease during Early Pregnancy in Normotensive Pregnant Women but Are Higher in Preeclamptic Women—A Longitudinal Study. Cells 2022, 11, 2251. [Google Scholar] [CrossRef]
- Butte, N.F. Carbohydrate and Lipid Metabolism in Pregnancy: Normal Compared with Gestational Diabetes Mellitus. Am. J. Clin. Nutr. 2000, 71, 1256S–61S. [Google Scholar] [CrossRef]
- Parrettini, S.; Caroli, A.; Torlone, E. Nutrition and Metabolic Adaptations in Physiological and Complicated Pregnancy: Focus on Obesity and Gestational Diabetes. Front Endocrinol. 2020, 11, 611929. [Google Scholar] [CrossRef]
- Moyce Gruber, B.L.; Dolinsky, V.W. The Role of Adiponectin during Pregnancy and Gestational Diabetes. Life 2023, 13, 301. [Google Scholar] [CrossRef] [PubMed]
- Barros, R.P.D.A.; Morani, A.; Moriscot, A.; Machado, U.F. Insulin Resistance of Pregnancy Involves Estrogen-Induced Repression of Muscle GLUT4. Mol. Cell Endocrinol. 2008, 295, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, T.; Takata, Y.; Aki, N.; Kunimi, K.; Satoh, M.; Nii, M.; Izumi, Y.; Otoda, T.; Hashida, S.; Osawa, H.; et al. Insulin Receptor Cleavage Induced by Estrogen Impairs Insulin Signaling. BMJ Open Diabetes Res. Care 2021, 9, e002467. [Google Scholar] [CrossRef]
- Khara, M.; Korda, I.; Kulyanda, O.; Podilska, T. PATHOPHYSIOLOGICAL ASPECTS OF INSULIN RESISTANCE IN GESTATIONAL DIABETES MELLITUS: A LITERATURE REVIEW. Neonatol. Surg. Perinat. Med. 2025, 15, 189–193. [Google Scholar] [CrossRef]
- Bar, A.; Moran, R.; Mendelsohn-Cohen, N.; Korem Kohanim, Y.; Mayo, A.; Toledano, Y.; Alon, U. Pregnancy and Postpartum Dynamics Revealed by Millions of Lab Tests. Sci. Adv. 2025, 11, eadr7922. [Google Scholar] [CrossRef]
- Usman, T.O.; Chhetri, G.; Yeh, H.; Dong, H.H. Beta-Cell Compensation and Gestational Diabetes. J. Biol. Chem. 2023, 299, 105405. [Google Scholar] [CrossRef]
- Mittal, R.; Prasad, K.; Lemos, J.R.N.; Arevalo, G.; Hirani, K. Unveiling Gestational Diabetes: An Overview of Pathophysiology and Management. Int. J. Mol. Sci. 2025, 26, 2320. [Google Scholar] [CrossRef] [PubMed]
- Ander, S.E.; Diamond, M.S.; Coyne, C.B. Immune Responses at the Maternal-Fetal Interface. Sci. Immunol. 2019, 4, eaat6114. [Google Scholar] [CrossRef]
- Yang, L.; Meng, Y.; Shi, Y.; Fang, H.; Zhang, L. Maternal Hepatic Immunology during Pregnancy. Front Immunol. 2023, 14, 1220323. [Google Scholar] [CrossRef]
- Yan, Y.-S.; Feng, C.; Yu, D.-Q.; Tian, S.; Zhou, Y.; Huang, Y.-T.; Cai, Y.-T.; Chen, J.; Zhu, M.-M.; Jin, M. Long-Term Outcomes and Potential Mechanisms of Offspring Exposed to Intrauterine Hyperglycemia. Front Nutr. 2023, 10, 1067282. [Google Scholar] [CrossRef]
- Rhyu, J.; Yu, R. Newly Discovered Endocrine Functions of the Liver. World J. Hepatol. 2021, 13, 1611–1628. [Google Scholar] [CrossRef]
- Bikle, D.D. The Free Hormone Hypothesis: When, Why, and How to Measure the Free Hormone Levels to Assess Vitamin D, Thyroid, Sex Hormone, and Cortisol Status. JBMR Plus 2021, 5, e10418. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef] [PubMed]
- Eipel, C.; Abshagen, K.; Vollmar, B. Regulation of Hepatic Blood Flow: The Hepatic Arterial Buffer Response Revisited. World J. Gastroenterol. 2010, 16, 6046–6057. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Häring, H.-U. The Role of Hepatokines in Metabolism. Nat. Rev. Endocrinol. 2013, 9, 144–152. [Google Scholar] [CrossRef]
- Wu, P.; Wang, Y.; Ye, Y.; Yang, X.; Huang, Y.; Ye, Y.; Lai, Y.; Ouyang, J.; Wu, L.; Xu, J.; et al. Liver Biomarkers, Lipid Metabolites, and Risk of Gestational Diabetes Mellitus in a Prospective Study among Chinese Pregnant Women. BMC Med. 2023, 21, 150. [Google Scholar] [CrossRef]
- Abdalla, M.M.I.; Ismail-Khan, M. Liver as a Metabolic Sensor in Gestational Diabetes: Implications for Offspring’s Liver and Diabetes Risk. World J. Hepatol. 2025, 17, 110185. [Google Scholar] [CrossRef]
- Fougerat, A.; Bruse, J.; Polizzi, A.; Montagner, A.; Guillou, H.; Wahli, W. Lipid Sensing by PPARα: Role in Controlling Hepatocyte Gene Regulatory Networks and the Metabolic Response to Fasting. Prog. Lipid Res. 2024, 96, 101303. [Google Scholar] [CrossRef]
- Zdrojewicz, Z.; Pawlus, K.; Horochowska, M.; Jagiełło, J. Fibroblast growth factor 21 - current point of view on its role in physiology of the organism and the pathogenesis of diabetes mellitus and obesity. Pediatr. Endocrinol. Diabetes Metab. 2015, 21, 184–187. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty Acids and Eicosanoids Regulate Gene Expression through Direct Interactions with Peroxisome Proliferator-Activated Receptors Alpha and Gamma. Proc. Natl. Acad. Sci. U S A 1997, 94, 4318–4323. [Google Scholar] [CrossRef]
- Zhang, X.; Yeung, D.C.Y.; Karpisek, M.; Stejskal, D.; Zhou, Z.-G.; Liu, F.; Wong, R.L.C.; Chow, W.-S.; Tso, A.W.K.; Lam, K.S.L.; et al. Serum FGF21 Levels Are Increased in Obesity and Are Independently Associated with the Metabolic Syndrome in Humans. Diabetes 2008, 57, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.G.; Guo, Y.Z.; Kong, Y.Z.; Dai, S.; Zhao, B.Y. High Non-Esterified Fatty Acid Concentrations Promote Expression and Secretion of Fibroblast Growth Factor 21 in Calf Hepatocytes Cultured in Vitro. J. Anim. Physiol. Anim. Nutr. (Berl) 2018, 102, e476–e481. [Google Scholar] [CrossRef]
- Sutton, E.F.; Morrison, C.D.; Stephens, J.M.; Redman, L.M. Fibroblast Growth Factor 21, Adiposity, and Macronutrient Balance in a Healthy, Pregnant Population with Overweight and Obesity. Endocr. Res. 2018, 43, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Corrales, P.; Vidal-Puig, A.; Medina-Gómez, G. Obesity and Pregnancy, the Perfect Metabolic Storm. Eur. J. Clin. Nutr. 2021, 75, 1723–1734. [Google Scholar] [CrossRef]
- Yang, W.; Wang, L.; Wang, F.; Yuan, S. Roles of AMP-Activated Protein Kinase (AMPK) in Mammalian Reproduction. Front Cell Dev. Biol. 2020, 8, 593005. [Google Scholar] [CrossRef]
- Chau, M.D.L.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast Growth Factor 21 Regulates Energy Metabolism by Activating the AMPK-SIRT1-PGC-1alpha Pathway. Proc. Natl. Acad. Sci. U S A 2010, 107, 12553–12558. [Google Scholar] [CrossRef]
- Wpływ Czynników Wzrostu Fibroblastów Na Układ Sercowo-Naczyniowy | Heleniak | Choroby Serca i Naczyń. Available online: https://journals.viamedica.pl/choroby_serca_i_naczyn/article/view/60040 (accessed on 19 May 2026).
- Wang, N.; Sun, B.; Guo, H.; Jing, Y.; Ruan, Q.; Wang, M.; Mi, Y.; Chen, H.; Song, L.; Cui, W. Association of Elevated Plasma FGF21 and Activated FGF21 Signaling in Visceral White Adipose Tissue and Improved Insulin Sensitivity in Gestational Diabetes Mellitus Subtype: A Case-Control Study. Front Endocrinol. 2021, 12, 795520. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, W.; Zhou, L.; Ma, L.; An, Y.; Xu, W.; Li, T.; Yu, Y.; Li, D.; Liu, Y. Fibroblast Growth Factor 21 Expressions in White Blood Cells and Sera of Patients with Gestational Diabetes Mellitus during Gestation and Postpartum. Endocrine 2015, 48, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Tramunt, B.; Smati, S.; Grandgeorge, N.; Lenfant, F.; Arnal, J.-F.; Montagner, A.; Gourdy, P. Sex Differences in Metabolic Regulation and Diabetes Susceptibility. Diabetologia 2020, 63, 453–461. [Google Scholar] [CrossRef]
- Kautzky-Willer, A.; Leutner, M.; Harreiter, J. Sex Differences in Type 2 Diabetes. Diabetologia 2023, 66, 986–1002. [Google Scholar] [CrossRef]
- Mendel, C.M. The Free Hormone Hypothesis: A Physiologically Based Mathematical Model. Endocr. Rev. 1989, 10, 232–274. [Google Scholar] [CrossRef]
- Goldman, A.L.; Bhasin, S.; Wu, F.C.W.; Krishna, M.; Matsumoto, A.M.; Jasuja, R. A Reappraisal of Testosterone’s Binding in Circulation: Physiological and Clinical Implications. Endocr. Rev. 2017, 38, 302–324. [Google Scholar] [CrossRef]
- Jänne, M.; Hammond, G.L. Hepatocyte Nuclear Factor-4 Controls Transcription from a TATA-Less Human Sex Hormone-Binding Globulin Gene Promoter. J. Biol. Chem. 1998, 273, 34105–34114. [Google Scholar] [CrossRef]
- Lu, H. Crosstalk of HNF4α with Extracellular and Intracellular Signaling Pathways in the Regulation of Hepatic Metabolism of Drugs and Lipids. Acta Pharm. Sin. B 2016, 6, 393–408. [Google Scholar] [CrossRef]
- Selva, D.M.; Hammond, G.L. Peroxisome-Proliferator Receptor Gamma Represses Hepatic Sex Hormone-Binding Globulin Expression. Endocrinology 2009, 150, 2183–2189. [Google Scholar] [CrossRef]
- Selva, D.M.; Hogeveen, K.N.; Innis, S.M.; Hammond, G.L. Monosaccharide-Induced Lipogenesis Regulates the Human Hepatic Sex Hormone-Binding Globulin Gene. J. Clin. Invest 2007, 117, 3979–3987. [Google Scholar] [CrossRef]
- Simó, R.; Saez-Lopez, C.; Lecube, A.; Hernandez, C.; Fort, J.M.; Selva, D.M. Adiponectin Upregulates SHBG Production: Molecular Mechanisms and Potential Implications. Endocrinology 2014, 155, 2820–2830. [Google Scholar] [CrossRef]
- Simó, R.; Barbosa-Desongles, A.; Sáez-Lopez, C.; Lecube, A.; Hernandez, C.; Selva, D.M. Molecular Mechanism of TNFα-Induced Down-Regulation of SHBG Expression. Mol. Endocrinol. 2012, 26, 438–446. [Google Scholar] [CrossRef]
- Brouwers, M.C.G.J. Fructose 1-Phosphate, an Evolutionary Signaling Molecule of Abundancy. Trends Endocrinol. Metab. 2022, 33, 680–689. [Google Scholar] [CrossRef]
- Winters, S.J.; Scoggins, C.R.; Appiah, D.; Ghooray, D.T. The Hepatic Lipidome and HNF4α and SHBG Expression in Human Liver. Endocr. Connect 2020, 9, 1009–1018. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Qu, X.; Donnelly, R. Sex Hormone-Binding Globulin (SHBG) as an Early Biomarker and Therapeutic Target in Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2020, 21, 8191. [Google Scholar] [CrossRef]
- Hammond, G.L. Diverse Roles for Sex Hormone-Binding Globulin in Reproduction. Biol. Reprod. 2011, 85, 431–441. [Google Scholar] [CrossRef]
- Kuijper, E.A.M.; Ket, J.C.F.; Caanen, M.R.; Lambalk, C.B. Reproductive Hormone Concentrations in Pregnancy and Neonates: A Systematic Review. Reprod. Biomed. Online 2013, 27, 33–63. [Google Scholar] [CrossRef]
- Soma-Pillay, P.; Catherine, N.-P.; Tolppanen, H.; Mebazaa, A.; Tolppanen, H.; Mebazaa, A. Physiological Changes in Pregnancy. Cardiovasc J. Afr. 2016, 27, 89–94. [Google Scholar] [CrossRef]
- Dukic, J.; Ehlert, U. Longitudinal Course of Sex Steroids From Pregnancy to Postpartum. Endocrinology 2023, 164, bqad108. [Google Scholar] [CrossRef] [PubMed]
- Xargay-Torrent, S.; Carreras-Badosa, G.; Borrat-Padrosa, S.; Prats-Puig, A.; Soriano, P.; Álvarez-Castaño, E.; Ferri, M.J.; De Zegher, F.; Ibáñez, L.; López-Bermejo, A.; et al. Circulating Sex Hormone Binding Globulin: An Integrating Biomarker for an Adverse Cardio-Metabolic Profile in Obese Pregnant Women. PLoS ONE 2018, 13, e0205592. [Google Scholar] [CrossRef]
- Gunderson, E.P.; Hurston, S.R.; Ning, X.; Lo, J.C.; Crites, Y.; Walton, D.; Dewey, K.G.; Azevedo, R.A.; Young, S.; Fox, G.; et al. Lactation and Progression to Type 2 Diabetes Mellitus After Gestational Diabetes Mellitus. Ann. Intern Med. 2015, 163, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Stuebe, A.M.; Rich-Edwards, J.W.; Willett, W.C.; Manson, J.E.; Michels, K.B. Duration of Lactation and Incidence of Type 2 Diabetes. JAMA 2005, 294, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Kajaia, N.; Binder, H.; Dittrich, R.; Oppelt, P.G.; Flor, B.; Cupisti, S.; Beckmann, M.W.; Mueller, A. Low Sex Hormone-Binding Globulin as a Predictive Marker for Insulin Resistance in Women with Hyperandrogenic Syndrome. Eur. J. Endocrinol. 2007, 157, 499–507. [Google Scholar] [CrossRef]
- Simons, P.I.H.G.; Valkenburg, O.; Stehouwer, C.D.A.; Brouwers, M.C.G.J. Sex Hormone–Binding Globulin: Biomarker and Hepatokine? Trends Endocrinol. Metab. 2021, 32, 544–553. [Google Scholar] [CrossRef]
- Scoditti, E.; Sabatini, S.; Carli, F.; Gastaldelli, A. Hepatic Glucose Metabolism in the Steatotic Liver. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 319–334. [Google Scholar] [CrossRef]
- Bo, T.; Gao, L.; Yao, Z.; Shao, S.; Wang, X.; Proud, C.G.; Zhao, J. Hepatic Selective Insulin Resistance at the Intersection of Insulin Signaling and Metabolic Dysfunction-Associated Steatotic Liver Disease. Cell Metab. 2024, 36, 947–968. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Miao, J.; Chanda, D.; Wang, Y.; Zhao, E.; Haas, M.E.; Hirschey, M.; Vaitheesvaran, B.; Farese, R.V.; Kurland, I.J.; et al. Hepatic Insulin Signaling Is Required for Obesity-Dependent Expression of SREBP-1c mRNA but Not for Feeding-Dependent Expression. Cell Metab. 2012, 15, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Matsuda, M.; Hammer, R.E.; Bashmakov, Y.; Brown, M.S.; Goldstein, J.L. Decreased IRS-2 and Increased SREBP-1c Lead to Mixed Insulin Resistance and Sensitivity in Livers of Lipodystrophic and Ob/Ob Mice. Mol. Cell 2000, 6, 77–86. [Google Scholar] [CrossRef] [PubMed]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of Obesity-Induced Inflammation and Insulin Resistance: Insights into the Emerging Role of Nutritional Strategies. Front Endocrinol. 2013, 4, 52. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef]
- Coviello, A.D.; Haring, R.; Wellons, M.; Vaidya, D.; Lehtimäki, T.; Keildson, S.; Lunetta, K.L.; He, C.; Fornage, M.; Lagou, V.; et al. A Genome-Wide Association Meta-Analysis of Circulating Sex Hormone–Binding Globulin Reveals Multiple Loci Implicated in Sex Steroid Hormone Regulation. PLoS Genet. 2012, 8, e1002805. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, M.C.G.J.; Jacobs, C.; Bast, A.; Stehouwer, C.D.A.; Schaper, N.C. Modulation of Glucokinase Regulatory Protein: A Double-Edged Sword? Trends Mol. Med. 2015, 21, 583–594. [Google Scholar] [CrossRef]
- Santoro, N.; Caprio, S.; Pierpont, B.; Van Name, M.; Savoye, M.; Parks, E.J. Hepatic de Novo Lipogenesis in Obese Youth Is Modulated by a Common Variant in the GCKR Gene. J. Clin. Endocrinol. Metab. 2015, 100, E1125–E1132. [Google Scholar] [CrossRef]
- Ruth, K.S.; Day, F.R.; Tyrrell, J.; Thompson, D.J.; Wood, A.R.; Mahajan, A.; Beaumont, R.N.; Wittemans, L.; Martin, S.; Busch, A.S.; et al. Using Human Genetics to Understand the Disease Impacts of Testosterone in Men and Women. Nat. Med. 2020, 26, 252–258. [Google Scholar] [CrossRef]
- Fang, H.; Li, Q.; Wang, H.; Ren, Y.; Zhang, L.; Yang, L. Maternal Nutrient Metabolism in the Liver during Pregnancy. Front. Endocrinol. 2024, 15. [Google Scholar] [CrossRef]
- Skajaa, G.O.; Fuglsang, J.; Knorr, S.; Møller, N.; Ovesen, P.; Kampmann, U. Changes in Insulin Sensitivity and Insulin Secretion during Pregnancy and Post Partum in Women with Gestational Diabetes. BMJ Open Diabetes Res. Care 2020, 8, e001728. [Google Scholar] [CrossRef]
- Wallace, I.R.; McKinley, M.C.; Bell, P.M.; Hunter, S.J. Sex Hormone Binding Globulin and Insulin Resistance. Clin. Endocrinol. (Oxf) 2013, 78, 321–329. [Google Scholar] [CrossRef]
- Kim, C.; Sen, A.; Osborne, E.; Lee, J.M.; Richardson, C.R. Associations between Glucose Tolerance and Sex Hormone Binding Globulin among Women with Recent Gestational Diabetes Mellitus. Diabetes Res. Clin. Pract. 2011, 93, e110–e112. [Google Scholar] [CrossRef] [PubMed]
- Kotulkar, M.; Robarts, D.R.; Apte, U. HNF4α in Hepatocyte Health and Disease. Semin Liver Dis. 2023, 43, 234–244. [Google Scholar] [CrossRef]
- van Bree, B.E.; van der Heijden, D.M.B.; van Golde, R.J.T.; Brouwers, M.C.G.J.; Spaanderman, M.E.A.; Valkenburg, O. Sex Hormone-Binding Globulin as a Biomarker for Metabolic Risk in European Women with Polycystic Ovary Syndrome. Gynecol. Endocrinol. 2025, 41, 2500462. [Google Scholar] [CrossRef] [PubMed]
- Hedderson, M.M.; Xu, F.; Darbinian, J.A.; Quesenberry, C.P.; Sridhar, S.; Kim, C.; Gunderson, E.P.; Ferrara, A. Prepregnancy SHBG Concentrations and Risk for Subsequently Developing Gestational Diabetes Mellitus. Diabetes Care 2014, 37, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Lesniara-Stachon, A.; Cosson, E.; Lacroix, A.; Schenk, S.; Quansah, D.Y.; Puder, J.J. Postpartum Glucose Intolerance after Gestational Diabetes Mellitus: Tailored Prediction According to Data-Driven Clusters and BMI-Categories. Front Endocrinol. 2024, 15, 1381058. [Google Scholar] [CrossRef]
- Son, H.; Moon, J.H.; Choi, S.H.; Cho, N.H.; Kwak, S.H.; Jang, H.C. Amelioration of Insulin Resistance after Delivery Is Associated with Reduced Risk of Postpartum Diabetes in Women with Gestational Diabetes Mellitus. Endocrinol. Metab. 2024, 39, 701–710. [Google Scholar] [CrossRef]
- Iglesias, P. The Endocrine Role of Hepatokines: Implications for Human Health and Disease. Front Endocrinol. 2025, 16, 1663353. [Google Scholar] [CrossRef]
- Yang, X.; Chen, H.; Shen, W.; Chen, Y.; Lin, Z.; Zhuo, J.; Wang, S.; Yang, M.; Li, H.; He, C.; et al. FGF21 Modulates Immunometabolic Homeostasis via the ALOX15/15-HETE Axis in Early Liver Graft Injury. Nat. Commun. 2024, 15, 8578. [Google Scholar] [CrossRef] [PubMed]
- Fodor Duric, L.; Belčić, V.; Oberiter Korbar, A.; Ćurković, S.; Vujicic, B.; Gulin, T.; Muslim, J.; Gulin, M.; Grgurević, M.; Catic Cuti, E. The Role of SHBG as a Marker in Male Patients with Metabolic-Associated Fatty Liver Disease: Insights into Metabolic and Hormonal Status. J. Clin. Med. 2024, 13, 7717. [Google Scholar] [CrossRef]
- Kakoti, B.B.; Alom, S.; Deka, K.; Halder, R.K. AMPK Pathway: An Emerging Target to Control Diabetes Mellitus and Its Related Complications. J. Diabetes Metab. Disord. 2024, 23, 441–459. [Google Scholar] [CrossRef]
- Lundåsen, T.; Hunt, M.C.; Nilsson, L.-M.; Sanyal, S.; Angelin, B.; Alexson, S.E.H.; Rudling, M. PPARalpha Is a Key Regulator of Hepatic FGF21. Biochem Biophys. Res. Commun. 2007, 360, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Jin, Z.; Chi, X.; Zhang, B.; Wang, X.; Sun, L.; Fan, J.; Sun, Q.; Zhang, X. SHBG Expression Is Correlated with PI3K/AKT Pathway Activity in a Cellular Model of Human Insulin Resistance. Gynecol. Endocrinol. 2018, 34, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Siemers, K.M.; Baack, M.L. The Importance of Placental Lipid Metabolism across Gestation in Obese and Non-Obese Pregnancies. Clin. Sci. (Lond) 2023, 137, 31–34. [Google Scholar] [CrossRef]
- Vernia, S.; Cavanagh-Kyros, J.; Garcia-Haro, L.; Sabio, G.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Xu, J.; Shulha, H.P.; Garber, M.; et al. The PPARα-FGF21 Hormone Axis Contributes to Metabolic Regulation by the Hepatic JNK Signaling Pathway. Cell Metab. 2014, 20, 512–525. [Google Scholar] [CrossRef]
- BonDurant, L.D.; Potthoff, M.J. Fibroblast Growth Factor 21: A Versatile Regulator of Metabolic Homeostasis. Annu Rev. Nutr. 2018, 38, 173–196. [Google Scholar] [CrossRef]
- Hribar, K.; Eichhorn, D.; Bongiovanni, L.; Koster, M.H.; Kloosterhuis, N.J.; de Bruin, A.; Oosterveer, M.H.; Kruit, J.K.; van der Beek, E.M. Postpartum Development of Metabolic Dysfunction-Associated Steatotic Liver Disease in a Lean Mouse Model of Gestational Diabetes Mellitus. Sci. Rep. 2024, 14, 14621. [Google Scholar] [CrossRef]
- Régnier, M.; Polizzi, A.; Fougeray, T.; Fougerat, A.; Perrier, P.; Anderson, K.; Lippi, Y.; Smati, S.; Lukowicz, C.; Lasserre, F.; et al. Liver Gene Expression and Its Rewiring in Hepatic Steatosis Are Controlled by PI3Kα-Dependent Hepatocyte Signaling. PLoS Biol. 2025, 23, e3003112. [Google Scholar] [CrossRef]
- Fisher, F.M.; Estall, J.L.; Adams, A.C.; Antonellis, P.J.; Bina, H.A.; Flier, J.S.; Kharitonenkov, A.; Spiegelman, B.M.; Maratos-Flier, E. Integrated Regulation of Hepatic Metabolism by Fibroblast Growth Factor 21 (FGF21) in Vivo. Endocrinology 2011, 152, 2996–3004. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wang, C.; Lai, F.; Xu, C.; Wang, X.; Chen, N.; Li, Y.; Xiao, H.; Cao, X. Delayed Insulin Secretion in the Early Postpartum Period Is Associated with Future Abnormal Glucose Metabolism among Women with a History of Gestational Diabetes Mellitus: A Prospective Cohort Study. BMC Pregnancy Childbirth 2026, 26, 181. [Google Scholar] [CrossRef] [PubMed]
- Shehzad, S.; Shabbir, N.A.; Mumtaz, M.; Um-A-Aiman, null; Shaheen, M.A.; Umer, M.R.; Sahar, A. Gestational Diabetes and Cardiovascular Risk in Women: A Comprehensive Review of Thrombosis, Hemostasis, and Emerging Insights Into Coagulation and Fibrinolysis. Cureus 2025, 17, e89238. [Google Scholar] [CrossRef]
- Szczepańska, E.; Gietka-Czernel, M. FGF21: A Novel Regulator of Glucose and Lipid Metabolism and Whole-Body Energy Balance. Horm. Metab. Res. 2022, 54, 203–211. [Google Scholar] [CrossRef]
- Januszewski, A.S.; Rankin, W.A.; O’Neal, D.N.; Wittert, G.A.; Jenkins, A.J. Sex Hormone Binging Globulin as an Indicator of Insulin Resistance in Type 1 Diabetes. Diabetes Res. Clin. Pract. 2025, 229, 112935. [Google Scholar] [CrossRef] [PubMed]
- Jena, J.; García-Peña, L.M.; Pereira, R.O. The Roles of FGF21 and GDF15 in Mediating the Mitochondrial Integrated Stress Response. Front Endocrinol. 2023, 14, 1264530. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, Y.; Yu, X.; Jin, Y.; Zhao, K.; Hu, Y.; He, Z. Hepatic Mitochondrial Signaling as a Systemic Hub: Inter-Organ Communication Networks in Aging and Aging-Related Diseases. Front Cell Dev. Biol. 14 1745201. [CrossRef]
- Flippo, K.H.; Potthoff, M.J. Metabolic Messengers: FGF21. Nat. Metab. 2021, 3, 309–317. [Google Scholar] [CrossRef]
- Peter, A.; Kantartzis, K.; Machann, J.; Schick, F.; Staiger, H.; Machicao, F.; Schleicher, E.; Fritsche, A.; Häring, H.-U.; Stefan, N. Relationships of Circulating Sex Hormone–Binding Globulin With Metabolic Traits in Humans. Diabetes 2010, 59, 3167–3173. [Google Scholar] [CrossRef]
- Gawlik, K.; Milewicz, T.; Pawlica-Gosiewska, D.; Trznadel-Morawska, I.; Solnica, B. Fibroblast Growth Factor 21 in Gestational Diabetes Mellitus and Type 2 Diabetes Mellitus. J. Diabetes Res. 2023, 2023, 4024877. [Google Scholar] [CrossRef]
- Geng, L.; Lam, K.S.L.; Xu, A. The Therapeutic Potential of FGF21 in Metabolic Diseases: From Bench to Clinic. Nat. Rev. Endocrinol. 2020, 16, 654–667. [Google Scholar] [CrossRef]
- Jin, T. Fibroblast Growth Factor 21 and Dietary Interventions: What We Know and What We Need to Know Next. Med. Rev. 2021, 2, 524–530. [Google Scholar] [CrossRef]
- Szybiak-Skora, W.; Cyna, W.; Lacka, K. New Insights in the Diagnostic Potential of Sex Hormone-Binding Globulin (SHBG)—Clinical Approach. Biomedicines 2025, 13, 1207. [Google Scholar] [CrossRef]
Figure 1.
Integrated model of coordinated hepatokine signaling regulating postpartum insulin sensitivity across pregnancy and postpartum transition. Schematic representation of the FGF21–SHBG regulatory network across early pregnancy, late pregnancy, and postpartum recovery. The model illustrates phase-dependent interactions between AMPK, PPARα, PI3K–Akt, and HNF4α signaling pathways coordinating hepatic adaptation, lipid stress responses, and restoration of insulin sensitivity. During late pregnancy, activation of the PPARα–FGF21 axis supports fatty acid oxidation and mitochondrial adaptation, whereas postpartum recovery is associated with restoration of PI3K–Akt and HNF4α signaling and increased SHBG production.
Figure 1.
Integrated model of coordinated hepatokine signaling regulating postpartum insulin sensitivity across pregnancy and postpartum transition. Schematic representation of the FGF21–SHBG regulatory network across early pregnancy, late pregnancy, and postpartum recovery. The model illustrates phase-dependent interactions between AMPK, PPARα, PI3K–Akt, and HNF4α signaling pathways coordinating hepatic adaptation, lipid stress responses, and restoration of insulin sensitivity. During late pregnancy, activation of the PPARα–FGF21 axis supports fatty acid oxidation and mitochondrial adaptation, whereas postpartum recovery is associated with restoration of PI3K–Akt and HNF4α signaling and increased SHBG production.


Table 1.
Dynamic interpretation of FGF21 and SHBG across pregnancy and postpartum.
| Stage | FGF21 (metabolic axis) | SHBG (hepatic identity axis) | Interpretation |
| Early pregnancy | Low–moderate | Normal | Baseline hepatic metabolic balance |
| Late pregnancy (physiological IR) | High (adaptive stress response) | Moderately reduced or stable (estrogen offset) | Compensated insulin resistance |
| Late pregnancy (GDM risk state) | High but dysregulated / resistant state | Low (reflecting reduced hepatic insulin sensitivity) | Impaired hepatic metabolic adaptation + lipotoxic stress |
| Postpartum recovery | Gradual normalization or transient elevation | Rising toward baseline | Hepatic reprogramming and insulin sensitivity restoration |
| Post-GDM state | Persistently elevated or blunted | Persistently low (reflecting sustained hepatic insulin resistance) | Impaired axis resynchronization → increased T2DM risk |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



