Submitted:
26 March 2026
Posted:
27 March 2026
You are already at the latest version
Abstract
Background: Emery–Dreifuss muscular dystrophy (EDMD) is a rare inherited neuromuscular disorder within the spectrum of nuclear envelope diseases, classically characterized by early musculo-tendinous contractures, slowly progressive myopathy and cardiac involvement dominated by conduction disease and arrhythmias, with variable evolution toward cardiomyopathy and heart failure. Methods: We performed a narrative synthesis of the contemporary literature, focusing on clinically relevant and high-impact evidence. Particular attention was given to diagnostic strategies, risk stratification, and therapeutic approaches applicable in real-world clinical settings. Results: Cardiac involvement in EDMD encompasses a broad spectrum, including atrial disease and conduction disturbances, ventricular arrhythmias, dilated cardiomyopathy, thromboembolic complications, and sudden cardiac death. The heterogeneity of phenotypic expression reflects underlying genetic diversity. Early recognition and systematic cardiovascular surveillance are essential to guide timely intervention, including device therapy and heart failure management. Despite growing awareness, significant gaps remain in risk prediction and standardized management pathways. Conclusions: EDMD represents a paradigmatic model of cardiomyopathy with prominent electrical instability and systemic implications. A structured, genotype- and phenotype-aware strategy, centered on early surveillance, targeted rhythm and thromboembolic risk management, and timely device therapy, can improve clinical decision-making in real-world settings. Future perspectives include the integration of precision medicine and the development of gene-targeted therapies, with the potential to shift from symptomatic management toward disease-modifying strategies. This narrative review aims to provide an updated and comprehensive, clinically actionable narrative synthesis of cardiovascular manifestations across EDMD genotypes and phenotypes, integrating rare and under-recognized high-impact presentations, and to outline pragmatic diagnostic and therapeutic pathways for real-world care while highlighting unmet needs and future directions.
Keywords:
Emery-Dreifuss muscular dystrophy
; nuclear envelope diseases
; cardiomyopathy
; cardiac conduction disorders
; atrial fibrillation
; heart failure
; ventricular arrhythmias
; sudden cardiac death
; implantable cardioverter-defibrillator
1. Introduction
In 1966, the neurologists Alan E. Emery and Fritz E. Dreifuss first identified a clinical phenotype distinct from other progressive muscular dystrophies. In their original report, Emery and Dreifuss described a benign X-linked neuromuscular phenotype characterized by a distinctive clinical triad [1]:
- early contractures (particularly involving the elbows, Achilles tendons, and cervical spine), often preceding the onset of muscle weakness;
- slowly progressive myopathy with a scapulohumeral and peroneal distribution;
- cardiac involvement with conduction system disease, responsible for syncope and sudden cardiac death, already recognized as an integral component of the disorder
Since the 1960s, the term “Emery-Dreifuss syndrome” has been used to describe a group of rare inherited syndromes sharing muscular, joint, and cardiac manifestations and caused by different types of genetic mutations. They are considered neuromuscular degenerative disorders, similar to Duchenne and Becker muscular dystrophies (DMD and BMD), but belonging to the spectrum of nuclear envelope diseases and with a more favorable course [2]. From a gene-level perspective, according to the corresponding mutations, different types of Emery-Dreifuss muscular dystrophy (EDMD) can be distinguished [3,4]:
- EDMD1 (X-linked transmission), the most frequently diagnosed form, linked to mutations in the EMD gene, which encodes the protein “emerin”, a component of the inner nuclear membrane;
- EDMD2 (laminopathy, autosomical dominant transmission), caused by mutations in the lamin gene (LMNA);
- EDMD4 and EDMD5, caused by mutations in the SYNE1 or SYNE2 genes, respectively, encoding nesprin-1 or nesprin-2.
Figure 1. Classical clinical triad and major genetic determinants of Emery-Dreifuss muscular dystrophy. Emery-Dreifuss muscular dystrophy is classically defined by the association of early musculo-tendinous contractures, slowly progressive humeroperoneal muscle weakness, and cardiac involvement characterized by conduction disease, arrhythmias, and variable cardiomyopathy. The figure also highlights the principal genes implicated in EDMD, including EMD, LMNA, and other nuclear-envelope/LINC-complex–related genes, underscoring the genetic heterogeneity of the disorder and the close link between molecular substrate and clinical phenotype. Abbreviations: EDMD, Emery-Dreifuss muscular dystrophy; EMD, emerin gene; LMNA, lamin A/C gene; LINC, linker of nucleoskeleton and cytoskeleton.
A large-scale study conducted on cells from affected patients showed that myogenic, metabolic, and fibrotic signaling pathways are strongly implicated. It was demonstrated that alpha smooth muscle actin positive myofibroblasts are overrepresented and that the profibrotic miRNA-21 is upregulated. Consistent with this, correction of the sequence carrying the mutation was associated with reduced expression of fibrogenic molecules [4]. From a clinical standpoint, disease progression is often characterized by the so-called “triad,” with early development of contractures, followed by fatigue and muscle atrophy, and finally the development of cardiac abnormalities [5]. There is no absolute correlation between muscular manifestations and cardiac manifestations. Cardiac involvement in EDMD may follow an independent course and can even precede neuromuscular symptoms [6]. In some cases, cardiomyopathy may present with syncope or sudden death, which can be the first symptom in rare instances in certain forms [7]. Symptomatic treatments mitigate orthopedic and cardiac complications, but no curative therapy is currently available for these diseases [4]. This lack of association between cardiac and muscular symptoms was emphasized in a study by Ishikawa et al., which introduced the concept of “cardiac emerinopathy” to describe patients carrying the EDM mutation who exhibit an isolated cardiac phenotype, without clinically relevant neuromuscular manifestations [8]. The 2023 ESC Guidelines for the management of cardiomyopathies explicitly recognize laminopathies, including EDMD, as important causes of genetic cardiomyopathy and emphasize the need for an aetiology-driven, multidisciplinary diagnostic approach rather than a purely morphological classification [9].
Figure 1.
summarizes the classical clinical triad of EDMD together with the main genes involved in its pathogenesis.
Figure 1.
summarizes the classical clinical triad of EDMD together with the main genes involved in its pathogenesis.

The pathophysiological link between the underlying genetic defect and the major cardiovascular manifestations of Emery-Dreifuss muscular dystrophy is schematically illustrated in Figure 2.
2. Materials and Methods
A targeted literature search was conducted in PubMed, Embase, and Web of Science to identify original studies, case series, registries, guideline documents, and high-quality reviews addressing cardiovascular involvement in EDMD. Particular attention was given to genotype–phenotype correlations (e.g., EMD, LMNA), arrhythmic risk, conduction disease, cardiomyopathy progression, and device-based and pharmacological management strategies.
The retrieved evidence was selected and synthesized in a narrative manner, prioritizing clinical relevance, methodological rigor, and consistency with contemporary recommendations for inherited cardiomyopathies and neuromuscular disorders. Rare and under-recognized clinical presentations were also included to provide a comprehensive and practice-oriented perspective.
This review does not present new or unpublished data. The manuscript was prepared in accordance with the International Committee of Medical Journal Editors (ICMJE) recommendations for the conduct, reporting, editing, and publication of scholarly work in medical journals. Artificial intelligence tools were used exclusively for the generation of illustrative figures. All content was fully reviewed, verified, and curated by the authors, who take full responsibility for the accuracy and integrity of the work. No AI was used for data generation, analysis, or interpretation.
3. Results
3.1. Epidemiology, Diagnostic Criteria, and Neuromuscular Involvement
EDMD is a rare disorder whose real prevalence is uncertain and estimates vary widely across sources and subtypes, ranging from ~1:400,000 to ~1.3–2 per 100,000 (≈ 1:50,000–1:77,000). In the epidemiological literature, it has been estimated from ~0.39 per 100,000 (≈ 1:256,000) to 1:250.000 births, while X-linked EDMD has been reported at ~0.13 per 100,000 in a regional population study and commonly at ~1:100,000 male births [5,10,11,12,13,14]. Despite its classical description, the epidemiology remains imprecise, largely due to marked genetic heterogeneity, incomplete penetrance, and frequent under-recognition, particularly in patients presenting with an apparently isolated cardiac phenotype [9,15].
3.1.1. Age and Sex Distribution
Age at presentation and sex distribution are strongly dependent on the underlying gene and mode of inheritance. In X-linked EDMD (EDMD1) due to pathogenic variants, the full clinical phenotype is observed almost exclusively in males, whereas heterozygous females typically show a milder and later-onset presentation [13,16]. In a recent large multicentre cohort, males with EDMD1 had a mean age of 33.4 ± 13.3 years at clinical evaluation and exhibited a substantial risk of malignant ventricular arrhythmias and progression to end-stage heart failure. In contrast, female carriers were older (43.3 ± 16.8 years) and, although approximately 40-45% developed cardiac involvement during follow-up, this typically occurred later in life (median age ~58.6 years) and was associated with a markedly lower incidence of life-threatening ventricular arrhythmias [16]. Female carriers are often asymptomatic but may present cardiac symptoms after age 50, especially for the conduction anomalies [12]. These data have direct clinical implications, supporting lifelong cardiac surveillance even in apparently asymptomatic female carriers. In autosomal dominant EDMD (EDMD2) associated with LMNA variants, both sexes are affected, and clinical expression is considerably more heterogeneous [17,18]. In these patients, disease onset frequently occurs in adulthood and may be dominated by a cardiac-first phenotype, characterized by early conduction disease, atrial arrhythmias, or dilated cardiomyopathy, while skeletal muscle involvement may be subtle or clinically unapparent in the early stages [9,17]. This presentation fully aligns with the ESC 2023 concept that early arrhythmias and conduction disturbances may represent sentinel manifestations of genetic or syndromic cardiomyopathies [9].
3.1.2. Neuromuscular Involvement: The Diagnostic Core of EDMD
Despite increasing attention to the cardiac phenotype, neuromuscular involvement remains the most characteristic and diagnostically specific feature of EDMD, particularly in classical forms [5,13]. Early and selective musculo-tendinous contractures are often the first clinical manifestation and may appear in childhood or adolescence [13]. Typical sites include the elbows (loss of extension), Achilles tendons (limited dorsiflexion or equinus deformity), and cervical and paraspinal muscles, leading to progressive spinal rigidity (“rigid spine”) [5,19]. These contractures frequently precede overt muscle weakness and represent one of the most discriminating clinical features of EDMD compared with other muscular dystrophies [19]. The associated myopathy is slowly progressive and displays a highly characteristic humeroperoneal distribution, with involvement of the scapulo-humeral muscles in the upper limbs (often with scapular winging) and distal/peroneal compartments in the lower limbs, resulting in foot drop and steppage gait [5,13]. Progression is typically slow, and muscle strength may remain relatively preserved for years, a feature that may delay diagnosis unless contractures are actively sought [18]. Spinal rigidity constitutes an additional hallmark. Progressive limitation of cervical and thoracic mobility may be disproportionate to the degree of muscle weakness and contributes significantly to functional impairment. Together, the triad of early contractures, humeroperoneal weakness, and rigid spine constitutes a powerful pattern-recognition framework for clinical diagnosis [13,18]. From a laboratory and electrophysiological standpoint, serum creatine kinase levels are often normal or only mildly elevated and therefore lack exclusionary value. Electromyography typically demonstrates non-specific myopathic changes. Muscle biopsy, historically used in the diagnostic work-up, is now reserved for selected cases, as genetic testing has become the diagnostic gold standard [9,13].
3.1.3. Peripheral Neuropathy: Accessory Feature and Overlap Phenotypes
In contrast to the prominent myopathic features, peripheral neuropathy is not considered a cardinal feature of classical EDMD [13]. Nevertheless, a limited number of reports, mainly involving LMNA-related disease, have described patients with combined myopathic and neurogenic features on nerve conduction studies and electromyography, suggesting a myopathy-neuropathy overlap phenotype [20,21]. Experimental and pathological data indicate that nuclear envelope dysfunction may, in rare cases, affect peripheral nerves or Schwann cells [21]. Clinically, the presence of mild peripheral neuropathy does not exclude EDMD when typical contractures and humeroperoneal myopathy are present. Conversely, a phenotype dominated by sensory-motor neuropathy in the absence of early contractures should prompt consideration of alternative diagnoses [13].
3.1.4. Integrated Diagnostic Criteria
Although no universally codified major/minor criteria exist, contemporary diagnosis of EDMD relies on an integrated clinical-instrumental-genetic approach, consistent with the ESC 2023 cardiomyopathy framework [9,22]:
- Typical neuromuscular phenotype: early selective contractures plus humeroperoneal myopathy and/or rigid spine.
- Compatible cardiac phenotype: conduction disease, early atrial arrhythmias, ventricular arrhythmias and/or heart failure.
- Genetic confirmation: identification of a pathogenic or likely pathogenic variant in EMD, LMNA, or related nuclear envelope genes, followed by genetic counselling and cascade family screening.
This approach enables accurate diagnosis, prognostic stratification, and early identification of at-risk relatives. An integrated clinical–genetic diagnostic framework for EDMD is summarized in Table 1, highlighting how the combination of early contractures, humeroperoneal myopathy, cardiac involvement, and molecular confirmation supports timely diagnosis and family-based risk stratification.
Genotype-phenotype correlations in EDMD are summarized in Table 2, highlighting the clinically relevant distinction between the typically atrial-dominant course of EMD-related disease and the more malignant ventricular-arrhythmic profile usually observed in LMNA-related phenotypes.
3.2. Cardiac Involvement
3.2.1. Overview of Cardiac Complications in Emery-Dreifuss
Cardiac involvement in EDMD typically becomes clinically apparent from late adolescence to early adulthood, although subclinical abnormalities may precede overt manifestations [5,13,16]. Initial symptoms may include palpitations, presyncope, syncope, or reduced exercise tolerance, reflecting early electrical instability or chronotropic incompetence [2,5]. In some patients, sudden cardiac death may be the first manifestation, highlighting the malignant arrhythmic potential of the disease even in the absence of advanced structural remodeling [16,23].
Conduction system disease is a hallmark of EDMD and may affect multiple levels of the cardiac conduction axis [6,13]. Reported abnormalities include sinus bradycardia, sinoatrial block, atrial standstill, atrioventricular (AV) conduction delay, bundle branch block, and progression to complete AVB, frequently requiring permanent pacemaker implantation [6,8,24].
However, in selected patients with additional markers of arrhythmic risk, an implantable cardioverter-defibrillator (ICD) may be more appropriate than pacing alone [16,25]. Both atrial and ventricular arrhythmias are common during the disease course [26]. Atrial fibrillation and atrial flutter may occur at a young age and contribute substantially to thromboembolic risk, particularly in the setting of atrial mechanical dysfunction [8,27].
Ventricular ectopy and ventricular tachyarrhythmias also occur and are major determinants of prognosis [16,26]. With disease progression, structural cardiomyopathy and heart failure may emerge, most commonly with a dilated phenotype, while hypertrophic forms have been reported less frequently [16,28]. Importantly, sudden death may occur despite prior pacemaker implantation, indicating that pacing does not abolish arrhythmic risk [16,23,24]. In advanced cases, progression to end-stage heart failure may ultimately require heart transplantation as a definitive therapeutic option [16,29].
The progressive cardiac trajectory of EDMD is illustrated in Figure 3, highlighting the transition from early atrial myopathy and conduction disease to atrial standstill, ventricular arrhythmias, cardiomyopathy, and major clinical outcomes.
The broad spectrum of cardiac involvement in EDMD is outlined in Table 3, underscoring that atrial disease, conduction abnormalities, ventricular arrhythmias, and progressive cardiomyopathy represent interconnected components of a single evolving cardio-neuromuscular phenotype.
3.2.2. Cardiac Imaging: Echocardiography and Cardiac Magnetic Resonance
Cardiac imaging, particularly echocardiography and cardiac magnetic resonance (CMR), plays a central role within a structured management strategy [2]. Transthoracic echocardiography commonly demonstrates a phenotype in which atrial remodelling may predominate over ventricular dilatation, with variable degrees of LV systolic impairment and occasional diastolic dysfunction. In a comparative echocardiographic study including both major genetic subgroups (EDMD1/EMD and EDMD2/LMNA), chamber enlargement (particularly atrial) was frequent, and a substantial subset had LVEF below the normal range, supporting the concept that structural and functional abnormalities may be present even in clinically “mild” skeletal phenotypes. Earlier echocardiographic series likewise documented that LV function can be subtly reduced compared with controls and that a minority may fulfil criteria for dilated cardiomyopathy, reinforcing the need for systematic baseline and longitudinal echocardiographic surveillance [28,30]. A case-control echocardiographic study evaluated left ventricular (LV) morphology and function in 27 men with confirmed EDMD (age 16–40 years; mean ~26 years), including 23 X-linked and 4 autosomal-dominant cases, compared with 16 age-matched healthy male controls (mean ~25 years). LV end-diastolic diameter was similar between groups, although three X-linked patients showed LV dilatation. Despite comparable wall thickness, EDMD patients had evidence of LV remodeling, with higher relative wall thickness and a higher LV mass index. LV systolic function was significantly reduced in EDMD, with a lower mean ejection fraction and 6/27 (22.2%) showing LVEF <50%; the most severe impairment (EF 19%, 32%, 34%) occurred in X-linked disease, and overt heart failure was documented in one patient. Doppler assessment (in those in sinus rhythm) indicated impaired relaxation, with prolonged isovolumetric relaxation time, higher A-wave velocity, and reduced E/A ratio; applying age-specific criteria, slow early filling was present in four patients and a restrictive pattern in two with advanced systolic dysfunction. Overall, subclinical LV dysfunction was frequent even in young adults. A substantial proportion, often minimally symptomatic, shows LV dysfunction, supporting the need for ongoing cardiology and echocardiographic surveillance [30].
Beyond standard parameters, deformation-based assessment suggests that myocardial dysfunction may be detectable at an early, subclinical stage. In autosomal dominant EDMD (LMNA-related), tissue Doppler and CMR-based strain identified abnormal LV functional indices despite preserved conventional chamber dimensions and the absence of late gadolinium enhancement, implying that functional impairment can precede overt scar formation [31]. In broader LMNA cardiomyopathy cohorts (highly relevant to EDMD2 and overlapping laminopathy phenotypes), speckle-tracking–derived mechanical dispersion has been proposed as a marker associated with ventricular arrhythmic risk beyond LVEF, supporting the rationale for incorporating strain mechanics into integrated phenotyping and follow-up when available [32]. CMR data are very limited but it is increasingly used to define an early myocardial phenotype beyond conventional echocardiography. In the single-centre cohort described by Ditaranto et al., clinical trajectories differed according to neuromuscular onset, supporting the concept that LMNA-related disease spans heterogeneous cardiac phenotypes and progression patterns, with imaging playing a role in longitudinal characterization [33]. CMR is increasingly emphasized as the reference technique in muscular dystrophies to detect subclinical myocardial involvement and to support risk stratification, particularly through tissue characterization (e.g., LGE and mapping) and functional assessment [34]. Extending EDMD relevance to EMD variants, Bulmer et al. reported a large family with X-linked isolated DCM due to an EMD missense variant, where CMR demonstrated late gadolinium enhancement with mixed ischaemic and non-ischaemic patterns, alongside conduction disease and ventricular arrhythmias, highlighting that fibrosis/structural substrate may be present even when skeletal muscle involvement is minimal [35]. In EDMD2, feature-tracking/strain analyses have reported reduced inferior wall contractility versus controls, suggesting that regional dysfunction may precede clinical cardiac manifestations; the absence of LGE argues against scar-driven impairment, unlike Duchenne muscular dystrophy where LGE is common [31,36]. In LMNA mutation carriers, CMR more consistently identifies fibrosis: in a small cohort of asymptomatic or mildly symptomatic individuals, 88% had non-coronary, predominantly mid-myocardial basal septal LGE, often linear and involving <50% of the affected segment [37]. Even with preserved LV systolic function, LMNA carriers may show longer phantom-normalized T2, higher ECV, more LGE, and impaired radial strain, consistent with subclinical cardiomyopathy; greater LGE burden and worse myocardial mechanics independently predicted higher MACE risk [36,38].
As summarized in Table 4, multimodality imaging in EDMD extends beyond conventional assessment of chamber size and ejection fraction, enabling earlier detection of subclinical myocardial dysfunction and more refined phenotypic characterization.
3.2.3. Heart Failure and Heart Transplantation
Unlike young patients with X-linked muscular dystrophies such as Duchenne muscular dystrophy, only a smaller proportion of individuals with EDMD develop dilated cardiomyopathy and heart failure [39]. In a longitudinal study of 18 patients with EDMD, progression to severe heart failure occurred in only one individual with an autosomal dominant form and severe limb-girdle muscular dystrophy, with markedly reduced LVEF (30%), atrial flutter, and AVB requiring pacemaker implantation. Owing to refractory heart failure, the patient was ultimately referred for heart transplantation [27].
A non-negligible proportion of patients already exhibited advanced heart failure symptoms (NYHA functional class ≥III–IV) at the time of diagnosis, and they tended to progress further during follow-up. In some patients, heart failure severity may progress to the point of requiring heart transplantation. Most deaths in individuals with EDMD are attributable to sudden cardiac death or advanced heart failure [26].
Reversible forms of right-sided heart failure have been reported in patients with EDMD, particularly in the setting of venous congestion due to AVB with a very low heart rate, with improvement after pacemaker pacing [27].
3.2.4. ECG Abnormalities, Conduction Alterations and Brady-Arrhythmias
Atrial fibrillation and AV conduction disorders represent the most frequently observed arrhythmias, whereas life-threatening ventricular arrhythmias and sudden cardiac death occur less often but remain major complications [36].
Clinically relevant conduction abnormalities are commonly present already after the first cardiological assessment and tend to progress over time toward more advanced degrees of conduction block during longitudinal follow-up [40]. The surface ECG may show baseline abnormalities reflecting progressive involvement of the cardiac conduction system and/or an associated cardiomyopathic substrate, even in the absence of symptomatic rhythm events. Typical findings include low amplitude P wave, AV conduction delay (PR prolongation up to higher-degree AV block) and intraventricular conduction disease with QRS widening (bundle-branch block patterns or non-specific intraventricular conduction delay). Low QRS voltages and non-specific repolarization abnormalities (ST-T changes) may also be observed and should be interpreted in the broader clinical and imaging context, supporting the need for serial ECG surveillance as part of cardiac follow-up in EDMD/laminopathy phenotypes [16,25,41,42,43].
Often, sinus node disease and conduction abnormalities occur before the development of ventricular dysfunction [44]. Moreover, different degrees of AVB may occur [3,41]. However, in a systematic review by Valenti et al., sinus node disease or sinoatrial block was reported at baseline in only a small proportion of the included studies evaluating patients with cardiomyopathy [26].
It has been observed that patients carrying LMNA mutations often undergo pacemaker implantation for conduction disturbances, yet their risk of sudden cardiac death remains unchanged [45]. In EDMD, a true “conduction system disease” is present, manifesting with sinus node dysfunction (including atrial standstill) and AV conduction blocks, such as Mobitz type II AV block and complete AV block, which may ultimately require permanent pacemaker implantation [16]. It is uncommon to observe both bradyarrhythmias and supraventricular tachyarrhythmias concomitantly, particularly in younger patients; nonetheless, individuals with EDMD carry a substantial lifetime risk of developing both. Moreover, given the low escape-rhythm rates that may occur in this patients, bradyarrhythmias have been reported as a potential mechanism of sudden cardiac death [27].
Regarding symptom burden, manifestations associated with complete AVB or sinoatrial block, such as syncope and/or fatigue, appear to be better tolerated in patients with EDMD. This may reflect adaptation to chronically lower heart rates and/or lower habitual physical activity compared with otherwise healthy young individuals [24].
3.2.5. Atrial Arrhythmias
Atrial involvement is a common component of the cardiac phenotype, and supraventricular tachyarrhythmias (SVT) may precede the onset of overt cardiomyopathy, along a continuum ranging from atrial premature beats and atrial tachycardias to atrial fibrillation (AF) or flutter (AFL) and, particularly in more “atrial-dominant” phenotypes, progressing towards “atrial standstill” (atrial paralysis related to fibrosis). Atrial standstill represents an extreme expression of atrial injury, characterized by absence of P waves on ECG, atrial electrical silence with a junctional or ventricular escape rhythm, and lack of mechanical atrial contraction. Atrial ectopy should be interpreted as an early expression of a primary atrial myopathy related to nuclear envelope pathology, rather than merely an isolated electrical trigger [46]. From a pathophysiological perspective, the atrium is the target of progressive structural and electrical remodelling (substrate), upon which atrial premature beats act as markers and/or triggers of tachyarrhythmias. Atrial premature beats (APBs) and other supraventricular arrhythmias in EDMD arise from a distinctive substrate of primary atrial cardiomyopathy driven by mutations in EMD (emerin) or LMNA (lamin A/C). Nuclear lamina proteins play a critical role in nuclear architecture, mechanotransduction, and transcriptional regulation in cardiac cells; their dysfunction promotes interstitial fibrosis, fibro-fatty infiltration, and disruption of the atrial extracellular matrix, thereby generating abnormal conduction substrates with areas of slow conduction and re-entrant circuits that predispose to APBs, atrial tachycardias, and atrial fibrillation/flutter. This atrial remodelling phenotype reduces local refractoriness, facilitates re-entry dependence and abnormal automaticity, and may account for the high prevalence of supraventricular arrhythmias even in young patients, irrespective of the severity of ventricular dysfunction [27]
A systematic review reports that atrial tachyarrhythmias (AF, AFL, atrial tachicardia and atrial standstill) are among the most frequently reported and quantifiable outcomes, consistent with the concept of progressive and clinically relevant atrial damage. [26] In a cohort of 45 patients with muscular dystrophies due to EMD/LMNA mutations and long median follow-up, atrial arrhythmias (including atrial premature beats) were common and often emerged at a young age (in the second or third decade of life), particularly in emerinopathy, suggesting that atrial electrical instability can precede overt heart failure and ventricular remodelling. Conversely, in laminopathy, ventricular arrhythmias appear more prevalent and tend to manifest earlier, underscoring distinct natural histories with an “early atrial-dominant” trajectory in EMD versus an “early ventricular-dominant” trajectory in LMNA. This suggest that atrial electrical instability may precede overt ventricular remodelling by years [46].
The overall reported prevalence of arrhythmias is 89%, with atrial standstill in 31% and AF/AFL in 29%. [46] The incidence rate for AF/AFL/AT ranges from 6.1 to 13.9 events per 100 patient-years, whereas the incidence of atrial standstill ranges from 0 to 2 events per 100 patient-years [26]. Half of the EMD patients develops atrial standstill, whereas no atrial standstill are observed in the LMNA subgroup, suggesting another difference in natural history between emerinopathy and laminopathy [46].
From a prognostic standpoint, the onset of atrial tachyarrhythmias (and, plausibly, an increasing burden of atrial ectopy) should be regarded as a marker of progression of atrial disease toward atrial paralysis and thromboembolic complications, and therefore as a signal to intensify surveillance and refine the antithrombotic strategy [6,27].
Atrial standstill in EDMD is clinically pivotal because it often represents the end stage of a continuum of atrial tachyarrhythmias and bradyarrhythmias, with direct implications for pacing strategy, anticoagulation decisions, and ischaemic risk. Notably, reports that atrial standstill may emerge after phases of atrial fibrillation or flutter, followed by a progressively electrically “silent” atrium, support an evolution toward extensive atrial fibrosis, low-voltage substrate, and loss of atrial capture, rather than a mere disorder of atrial automaticity [25,27,46].
Operational diagnosis of atrial ectopy and atrial tachycardias should be proactive and longitudinal, combining a baseline 12-lead ECG, 24-h Holter monitoring (preferably extended over multiple days), and, when available, pacemaker/ICD diagnostics, because continuous surveillance can capture nocturnal or intermittent atrial arrhythmias that may be missed during routine outpatient assessment [6,46,47].
Thromboembolic event rates in EDMD have been reported as high as 8.9 events per 100 patient-years, consistent with the substantial ischaemic burden observed in cohort studies and supporting a proactive (non-watchful-waiting) thromboembolic prevention strategy [26]. Anticoagulation should be recommended when AF/AFL occurs in EDMD and should also be considered in atrial standstill, given the consistently reported high thromboembolic risk and its disproportionate prognostic impact relative to patients’ age, despite the absence of EDMD-specific trials or dedicated guidelines, with management therefore extrapolated from standard AF anticoagulation principles in structural cardiomyopathies and individualized to the disease context. The 2022 HRS consensus statement on neuromuscular diseases (including EDMD) emphasizes systematic arrhythmia surveillance and structured thromboembolic risk assessment, explicitly noting that the combination of atrial arrhythmias/atrial standstill and stroke warrants a rigorous, guideline-consistent approach to anticoagulation when indicated [6,25,27].
In a long-term longitudinal study including X-linked and autosomal-dominant EDMD, AF/AFL developed in 11/18 patients (61%), followed by atrial standstill in 5/11 (45%), while embolic stroke occurred in 4/11 (36%) of AF/AFL patients, often with disabling sequelae. This suggests a clinically relevant temporal sequence of “AF/AFL → atrial standstill → stroke” in a non-negligible proportion of cases. Accordingly, the most robust clinical rationale for anticoagulation in EDMD derives from these longitudinal data, where the authors conclude that anti-thromboembolic prophylaxis should be recommended in the presence of AF/AFL or atrial standstill [6,9,25].
In “cardiac emerinopathy” (the X-linked cardiac phenotype related to EMD, even in the absence of a full syndromic presentation), a trajectory of progressive atrial arrhythmias culminating in atrial standstill has been described, frequently in association with left ventricular non-compaction and a familial thromboembolic burden, reinforcing the concept of a structured cardiomyopathy/ atriomyopathy with an intrinsic embolic risk [8].
As shown in a multicentre cohort of EMD variant carriers, even when the earliest clinical manifestations are predominantly atrial, early and structured surveillance remains essential, given the substantial risk of subsequent malignant ventricular arrhythmias and progression to end-stage heart failure [16].
There are no dedicated randomised trials assessing the efficacy of antiarrhythmic drugs for atrial arrhythmias; therefore, pharmacological management is extrapolated from general principles for atrial tachyarrhythmias, with particular attention to the underlying conduction substrate and the frequent coexistence of bradyarrhythmias that characterise these dystrophies. Medical therapy is based on rate and/or rhythm control using antiarrhythmic agents selected according to ventricular function, conduction disturbances, and pro-arrhythmic risk, bearing in mind that management is often device-integrated in the context of a brady-tachy syndrome [6,9,25].
3.2.6. Ventricular Arrhythmias and Sudden Cardiac Death
Malignant ventricular arrhythmias are common in patients with EDMD, irrespective of left ventricular ejection fraction, and are associated with an increased risk of sudden cardiac death [44].
The risk of ventricular arrhythmias (VA) in this condition has been recognized for a long time [23,48]. The incidence of VA appears to be higher in the presence of significant left ventricular systolic dysfunction, similar to other forms of heart disease [49]. However, the true progression to sudden death in EDMD patients receiving prophylactic device implantation despite preserved left ventricular function remains unknown [27].
In EDMD (both EMD/emerin and LMNA/lamin A/C phenotypes), ventricular arrhythmias may occur as part of a broader “conduction–cardiomyopathy” continuum and can emerge even before advanced LV dysfunction is evident. From an ECG standpoint, the most reproducible correlations are not with a single lead “location,” but with conduction disease burden and its progression over time: marked PR prolongation/AV conduction delay and QRS widening (bundle-branch block or non-specific intraventricular conduction delay), particularly when these parameters worsen on serial ECGs, are associated with a more arrhythmogenic phase in cardiac laminopathies [16,25,50]. Cardiac MRI adds anatomical specificity: septal late gadolinium enhancement (LGE), a typical pattern in LMNA-related disease, has been linked to a higher incidence of major ventricular arrhythmias and helps refine risk beyond surface ECG alone [51].
In EDMD, LMNA-related cardiomyopathy appears to follow a particularly malignant course compared with other DCM aetiologies, with malignant ventricular arrhythmias and progressive end-stage, medically refractory heart failure being more frequently observed in LMNA mutation carriers [40,52,53]. A similar trend has also been reported in LMNA mutation carriers with a left ventricular ejection fraction >35% [52]. Patients with LMNA mutations have a high risk of premature sudden death, seemingly driven by ventricular tachyarrhythmias; this risk does not parallel the onset of heart failure or bradyarrhythmias and, accordingly, is not prevented by pacemaker implantation alone [45,54]. In a multicentre study including 59 patients with EDMD type 1 (EMD variant-carriers), a 50-year-old man with a permanent pacemaker died suddenly. Post-mortem device interrogation documented rapid sustained ventricular tachycardia coinciding with collapse, and an echocardiogram performed two weeks earlier showed only mildly reduced LVEF (43%), supporting the concept that malignant ventricular arrhythmias may occur despite the absence of severe LV systolic dysfunction [16].
In LMNA mutation carriers, the use of a recently validated risk score has been suggested. It is an interesting and highly useful tool developed based on the results of a multicenter study involving 839 patients, recruited from two different registries: 660 from the French Nationwide Registry and 179 from other countries. The aim of the study was to validate a model to estimate the 5-year absolute risk of developing life- threatening ventricular tachyarrhythmias (LTVTA) in patients with dilated cardiomyopathy (DCM) caused by LMNA mutations (i.e., patients with laminopathies in general) with adult-onset disease. The study population included 55 patients with Emery–Dreifuss muscular dystrophy (EDMD) from the French registries and 6 patients with EDMD from other registries (such as London, the United States, Bern, and Melbourne). The term LTVTA refers to the occurrence of:
- Sudden cardiac death
- Appropriate ICD shock for the termination of ventricular tachyarrhythmias (VT)
- Ventricular tachyarrhythmias associated with hemodynamic instability and other clinical manifestations
The exclusion criteria were: history of tachyarrhythmias before baseline, age < 16 years, development of neuromuscular or systemic disease before the age of 16, cardiomyopathies associated with other genetic mutations, lack of clinical data, first cardiological evaluation performed before January 2000.
The risk score is based on the presence or absence of several factors considered adverse in patients with laminopathies, as they are associated with an increased risk of malignant tachyarrhythmias:
- Male sex
- Baseline left ventricular ejection fraction <45%
- Non-missense genetic mutations
- Non-sustained ventricular tachycardia
- AVB
The score can be easily calculated in these patients, including those with EDMD due to LMNA mutations and adult-onset DCM, using the dedicated online calculator (https://lmna-risk-vta.fr/). Risk estimation is crucial for identifying patients who may benefit from implantable cardioverter-defibrillator (ICD) implantation, helping to avoid both underestimation and overestimation of risk [55].
3.2.7. Cardiac Implantable Electronic Device and Ablation Therapy
In EDMD normal myocardium is gradually replaced by the fibrous and adipose tissue, process that usually starts in the atria, then involving AV node and ventricles. This electro-anatomical substrate is responsible for frequent bradi- and tachyarrhythmias, usually predominant feature of disease. Conduction abnormalities, such as “sick sinus syndrome” (often associated with atrial standstill phenomenon) and various degrees of atrio-ventricular blocks, are common and can be associated with the development of cardiomyopathy. In according to ESC guidelines, in cases of any second or third degree AVB or HV ≥70ms, with or without symptoms, permanent pacing is indicated (class of recommendation I), while may be considered with PR ≥240msec or QRS duration ≥120ms (class of recommendation IIb) [56]. In a cases series by Steckiewicz et al. on 21 EMD patients followed for a mean period of 11 ± 8 years after pacemaker (PM) implantation, pacing mode was often changed from DDD to VVI during follow-up, because of atrial electrical silence, which is responsible of considerable atrial spike-to-P-wave delay and loss-of-capture and high incidence of atrial fibrillation/flutter [24]. Whenever pacing is indicated in this disease, an implantable cardioverter defibrillator (ICD) should be considered according to guidelines indications. In the report by Golzio et al., ventricular lead extraction was required only 18 months after pacemaker implantation. The patient subsequently underwent device upgrade to an ICD for ventricular tachycardia; nine months later, multiple appropriate ICD therapies successfully averted sudden cardiac death [57].
Sudden death is not uncommon in EDMD, so risk stratification is necessary [58]. As indicated in the 2022 ESC guidelines for the management of ventricular arrhythmias, should be considered not only in patients with heart failure and ejection fraction <35%, but also in cases that don’t meet the usual criteria, if they are factors, such as age, CTG expansion, sudden death in family history, ECG conduction abnormalities, PR prolongation, left bundle branch block, atrial arrhythmias, non-sustained VT, LV dysfunction with an ejection fraction of less than than 45%, structural abnormalities in CMR and lamin A/C mutation [59]. Cardiac resynchronization therapy may also find a place in the management of patients with EDMD, given the high incidence of heart failure in dilated cardiomyopathy, particularly in LMNA-mutated carriers, and intraventricular conduction disturbances. In 1999, Walker et al. published the first anecdotal case of biventricular ICD [60]. To date, there are no systematic studies on the outcomes and responsiveness of resynchronization therapy in Emery-Dreifuss disease. Moreover, no cases have been published on emerging selective pacing techniques, such as left bundle branch area pacing and His bundle pacing in this setting.
The prevalence of atrial arrhythmias ranged from 12.5% to 63% in the LMNA patient cohort, and from 10% to 73.3% in the EMD patients’ cohort, while malignant ventricular arrhythmias can be common, especially in LMNA-mutated patients. In a systematic review the ICD implantation resulted in device activation in 24 to 52.4% of ICD carriers [26]. Ablation therapy can be challenging, because of regional and transmural repolarization heterogeneity and electrical instability linked to diffuse fibrosis. Only three reported cases of ablation can be identified in EDMD patients. Blagova described a case of atrial flutter ablation in this setting, initially successful, but with recurrence within 4 months, complicated by worsening LV systolic function necessitating a heart transplant [29]. Carvalho reported a case involving an EDMD patient who underwent multiple ablations for atrial and ventricular tachyarrhythmias [61]. In 2020, Butt showed the only successful case of atrial flutter treatment through cavotricuspid isthmus ablation with arrhythmias freedom after thirty-five months’ follow-up [62]. Anecdotal cases demonstrate the complexity of electrical substrate in these patients. In addition, there is a lack of data concerning ventricular arrhythmias ablation in EDMD patients which could depend on reduced accessibility to ablative therapy or because it may be considered futile or ineffective. Valverde Soria described an Emery-Dreifuss dilated cardiomyopathy patient (EMD-related phenotype) with an ICD who developed recurrent ventricular fibrillation (VF) episodes consistently triggered by a short-coupled, monomorphic ventricular ectopic beat compatible with a Purkinje-related focus, in the context of an intraseptal arrhythmogenic substrate. The authors adopted a pragmatic, mechanism-oriented strategy combining substrate ablation with ICD-guided pace mapping to reproduce and localize the initiating ectopy, achieving successful elimination of the VF trigger and control of recurrences. Collectively, the case supports catheter ablation of Purkinje triggers as a potential adjunctive option in selected dilated cardiomyopathy patients suffering recurrent ICD shocks due to VF initiated by monomorphic ectopy [63]. Zeppenfeld’s review provides a practical, mechanism-based framework for VT ablation in non-ischemic cardiomyopathy, but it does not report data specifically addressing EDMD and it is not discussed as a distinct cardiomyopathy subtype. Still, the concepts can be reasonably extrapolated to EDMD when patients develop sustained monomorphic VT or recurrent ICD shocks. Non-ischemic VT is often driven by scar-related re-entry with heterogeneous, sometimes intramural/epicardial substrate distribution, features that can limit lesion effectiveness and contribute to recurrence. It is important to highlight the recognition of His-Purkinje system-dependent VTs (including bundle branch re-entry), because these may be comparatively “high-yield” targets for ablation in selected non-ischemic phenotypes [64].
Finally, a pragmatic management framework for the principal cardiac scenarios encountered in EDMD is provided in Table 5, emphasizing the need for proactive surveillance, early recognition of thromboembolic risk, and individualized device-based decision-making.
3.3. Current and Future Directions
The 2023 ESC cardiomyopathy guidance supports earlier primary-prevention ICD consideration in genotype-positive dilated/NDLVC settings, including high-risk genotypes with additional markers (e.g., syncope or LGE) even without overt LV dysfunction, and allows that an ICD may be considered in genotype-positive patients with LVEF >35% even in the absence of additional risk factors (Class IIb) [9]. In EDMD, these principles are biologically plausible but remain insufficiently validated; future studies should therefore test genotype-substrate-event links prospectively, define actionable thresholds, and explore precision therapeutics (including pathway-targeted strategies and emerging gene/RNA-based approaches) within multidisciplinary cardio-neuromuscular programmes.
Future research in EDMD should become genotype-driven, integrating systematic panel testing (EMD/LMNA/SYNE1-2 and related nuclear-envelope/LINC genes) with deep phenotyping and cascade screening, to refine prognostication and enable earlier, mechanism-based interventions. Patient-derived cellular models now provide actionable biological hypotheses: across EDMD1/2/5, dermal fibroblasts show a convergent profibrotic programme with increased TGFβ signalling and miR-21 upregulation, alongside α-SMA-positive myofibroblast over-representation. Importantly, CRISPR/Cas correction (or allele-specific disruption) rescues nuclear-envelope abnormalities and largely normalizes the disease miRNA signature, supporting proof-of-principle precision gene correction and biomarker discovery pipelines using isogenic lines and iPSC-based platforms [4].
Future directions in EDMD should pivot from a predominantly phenotype approach to a genotype-driven pathway, integrating systematic molecular diagnosis (EMD/LMNA-first, with expanded cardiomyopathy/arrhythmia panels where appropriate), variant reclassification over time, and cascade testing to refine penetrance, sex-specific expressivity, and age-related risk. This will require EDMD-specific longitudinal cohorts combining genomics with deep phenotyping (ECG/Holter burden, atrial myopathy metrics, CMR tissue characterization, biomarkers, and digital surveillance), and the development/validation of EDMD-tailored risk models for malignant ventricular arrhythmias and end-stage heart failure.
A pragmatic surveillance and management pathway for EDMD is proposed in Figure 4, integrating baseline phenotyping with subsequent decisions on anticoagulation, pacing or defibrillator therapy, arrhythmia management and advanced heart failure referral.
4. Conclusions
EDMD is a paradigmatic nuclear-envelope disorder in which cardiac disease is not a late “complication” of skeletal myopathy but often an early and independent driver of morbidity and mortality. Across genotypes, the dominant clinical trajectory is characterized by a progressive atriomyopathy-conduction disease axis (early atrial arrhythmias evolving toward atrial standstill and advanced bradyarrhythmias), variably accompanied by ventricular arrhythmias, cardiomyopathy, and heart failure. Critically, malignant ventricular tachyarrhythmias and sudden cardiac death may occur out of proportion to left ventricular dysfunction and are not neutralized by pacemaker therapy alone, supporting an earlier and more individualized consideration of ICD therapy, particularly in LMNA-related disease, within an aetiology-driven framework aligned with contemporary ESC cardiomyopathy and ventricular arrhythmia guidance.
From a clinical standpoint, EDMD mandates lifelong, structured cardiac surveillance that integrates serial ECG and extended rhythm monitoring, device diagnostics when present, and imaging (echocardiography and, when available, CMR tissue characterization) to capture evolving substrate and refine risk. Management should be explicitly proactive regarding thromboembolic prevention, given the high burden of atrial disease and the recurrent sequence of AF/AFL progressing to atrial standstill and stroke. While pharmacological and device strategies remain the backbone of care, evidence for catheter ablation in EDMD is sparse, yet selected cases suggest that mechanism-targeted approaches may be feasible in highly selected scenarios.
Finally, the field is poised to shift from descriptive phenotyping to genotype-driven precision care, leveraging deep longitudinal cohorts, EDMD-tailored risk models, and translational pipelines (isogenic systems and emerging gene/RNA-based strategies) aimed at disease modification. Until such data mature, optimal outcomes will depend on early recognition, multidisciplinary cardio-neuromuscular management, and anticipatory prevention of stroke, heart failure progression, and sudden death.
Supplementary Materials
none.
Author Contributions
Conceptualization, LG.G. and M.C.L.N.; methodology, L.G.G., M.M.; resources, L.G.G., M.C.L.N., F.C., N.F., A.N., S.G., P.C., G.D., O.T., G.A., C.d.G.; data curation, L.G.G; writing—original draft preparation, L.G.G., M.C.L.N., F.C., N.F.; writing—review and editing, L.G.G. and M.C.L.N.; visualization, S.G., G.A., C.d.G.; supervision, G.A., C.d.G., G.M.F.; project administration, L.G.G. All authors have read and agreed to the published version of the manuscript.”.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Acknowledgments
During the preparation of this manuscript, the authors used AI exclusively for grammar/syntax refinement and for assisting in the generation of figures. No AI system was involved in generating the scientific content or drafting the manuscript. The authors have reviewed and edited the output and take full responsibility for the content of this publication. The views and opinions expressed in this manuscript are those of the authors and do not necessarily reflect those of their affiliated istitutions.
Permissions/Copyright
No third-party copyrighted material has been used in the manuscript figures. All images/figures were entirely created by the authors specifically for this submission; therefore, no permissions are required and no copyright attribution is applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| A | late transmitral diastolic filling velocity |
| AF | atrial fibrillation |
| AFL | atrial flutter |
| AI | artificial intelligence |
| APBs | atrial premature beats |
| AT | atrial tachycardia |
| AV | atrioventricular |
| AVB | atrioventricular block |
| BMD | Becker muscular dystrophy |
| CK | creatine kinase |
| CMR | cardiac magnetic resonance |
| CRISPR/Cas | clustered regularly interspaced short palindromic repeats/CRISPR-associated system |
| CRT | cardiac resynchronization therapy |
| CTG | cytosine-thymine-guanine repeat expansion |
| DCM | dilated cardiomyopathy |
| DDD | dual-chamber paced, dual-chamber sensed, dual response to sensing |
| DMD | Duchenne muscular dystrophy |
| E | early transmitral diastolic filling velocity |
| ECG | electrocardiogram/electrocardiography |
| ECV | extracellular volume |
| EDMD | Emery-Dreifuss muscular dystrophy |
| EMD | emerin gene |
| EMG | electromyography |
| ESC | European Society of Cardiology |
| HF | heart failure |
| HRS | Heart Rhythm Society |
| HV | His-ventricular interval |
| ICD | implantable cardioverter-defibrillator |
| ICMJE | International Committee of Medical Journal Editors |
| iPSC | induced pluripotent stem cell |
| LGE | late gadolinium enhancement |
| LINC | linker of nucleoskeleton and cytoskeleton |
| LMNA | lamin A/C gene |
| LV | left ventricle/left ventricular |
| LVEF | left ventricular ejection fraction |
| LTVTA | life-threatening ventricular tachyarrhythmias |
| MACE | major adverse cardiovascular events |
| miRNA | microRNA |
| NDLVC | non-dilated left ventricular cardiomyopathy |
| NSVT | non-sustained ventricular tachycardia |
| NYHA | New York Heart Association |
| PM | pacemaker |
| RNA | ribonucleic acid |
| SCD | sudden cardiac death |
| SVT | supraventricular tachyarrhythmias |
| SYNE1 | spectrin repeat containing nuclear envelope protein 1 gene |
| SYNE2 | spectrin repeat containing nuclear envelope protein 2 gene |
| TGFβ | transforming growth factor beta |
| VA | ventricular arrhythmias |
| VF | ventricular fibrillation |
| VVI | ventricular paced, ventricular sensed, inhibited response |
| VT | ventricular tachycardia |
References
- Emery, A. E.; Dreifuss, F. E. Unusual Type of Benign X-Linked Muscular Dystrophy. Journal of Neurology, Neurosurgery & Psychiatry 1966, 29(4), 338–342. [Google Scholar] [CrossRef]
- Wang, S.; Peng, D. Cardiac Involvement in Emery-Dreifuss Muscular Dystrophy and Related Management Strategies. Int. Heart J. 2019, 60(1), 12–18. [Google Scholar] [CrossRef] [PubMed]
- Pillers, DA; Von Bergen, NH. Emery-Dreifuss muscular dystrophy: a test case for precision medicine. Appl Clin Genet. 2016, 9, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Cattin, E.; Schena, E.; Mattioli, E.; Marcuzzo, S.; Bonanno, S.; Cavalcante, P.; Corradi, F.; Benati, D.; Farinazzo, G.; Cattaneo, M.; et al. Profibrotic Molecules Are Reduced in CRISPR-Edited Emery–Dreifuss Muscular Dystrophy Fibroblasts. Cells 2025, 14(17), 1321. [Google Scholar] [CrossRef]
- Heller, S. A.; Shih, R.; Kalra, R.; Kang, P. B. Emery-Dreifuss Muscular Dystrophy. Muscle and Nerve 2020, 61(4), 436–448. [Google Scholar] [CrossRef] [PubMed]
- Sanna, T. Cardiac Features of Emery–Dreifuss Muscular Dystrophy Caused by Lamin A/C Gene Mutations. European Heart Journal 2003, 24(24), 2227–2236. [Google Scholar] [CrossRef]
- Merlini, L.; Granata, C.; Dominici, P.; Bonfiglioli, S. Emery—Dreifuss Muscular Dystrophy: Report of Five Cases in a Family and Review of the Literature. Muscle and Nerve 1986, 9(6), 481–485. [Google Scholar] [CrossRef]
- Ishikawa, T.; Mishima, H.; Barc, J.; Takahashi, M. P.; Hirono, K.; Terada, S.; Kowase, S.; Sato, T.; Mukai, Y.; Yui, Y.; et al. N. Cardiac Emerinopathy: A Nonsyndromic Nuclear Envelopathy With Increased Risk of Thromboembolic Stroke Due to Progressive Atrial Standstill and Left Ventricular Noncompaction. Circ: Arrhythmia and Electrophysiology 2020, 13(10). [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J. R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C. R.; Biagini, E.; Blom, N. A.; De Boer, R. A.; et al. 2023 ESC Guidelines for the Management of Cardiomyopathies. European Heart Journal 2023, 44(37), 3503–3626. [Google Scholar] [CrossRef]
- Mah, J. K.; Korngut, L.; Fiest, K. M.; Dykeman, J.; Day, L. J.; Pringsheim, T.; Jette, N. A Systematic Review and Meta-Analysis on the Epidemiology of the Muscular Dystrophies. Can. J. Neurol. Sci. 2016, 43(1), 163–177. [Google Scholar] [CrossRef]
- Norwood, F. L. M.; Harling, C.; Chinnery, P. F.; Eagle, M.; Bushby, K.; Straub, V. Prevalence of Genetic Muscle Disease in Northern England: In-Depth Analysis of a Muscle Clinic Population. Brain 2009, 132(11), 3175–3186. [Google Scholar] [CrossRef]
- Viggiano, E.; Madej-Pilarczyk, A.; Carboni, N.; Picillo, E.; Ergoli, M.; Del Gaudio, S.; Marchel, M.; Nigro, G.; Palladino, A.; Politano, L. X-Linked Emery–Dreifuss Muscular Dystrophy: Study Of X-Chromosome Inactivation and Its Relation with Clinical Phenotypes in Female Carriers. Genes 2019, 10(11), 919. [Google Scholar] [CrossRef]
- Ben Yaou, R.; Leturcq, F.; Bonne, G. Emery-Dreifuss Muscular Dystrophy GeneReviews; University of Washington: Seattle: Seattle (WA), 2004. [Google Scholar]
- Orphanet. Emery-Dreifuss Muscular Dystrophy (ORPHA:261) [Internet]. Orphanet. 20 Mar 2026. Available online: Https://Www.Orpha.Net/En/Disease/Detail/261.
- Hershberger, R. E.; Givertz, M. M.; Ho, C. Y.; Judge, D. P.; Kantor, P. F.; McBride, K. L.; Morales, A.; Taylor, M. R. G.; Vatta, M.; Ware, S. M. Genetic Evaluation of Cardiomyopathy—A Heart Failure Society of America Practice Guideline. Journal of Cardiac Failure 2018, 24(5), 281–302. [Google Scholar] [CrossRef]
- Cannie, D. E.; Syrris, P.; Protonotarios, A.; Bakalakos, A.; Pruny, J.-F.; Ditaranto, R.; Martinez-Veira, C.; Larrañaga-Moreira, J. M.; Medo, K.; Bermúdez-Jiménez, F. J.; et al. Emery–Dreifuss Muscular Dystrophy Type 1 Is Associated with a High Risk of Malignant Ventricular Arrhythmias and End-Stage Heart Failure. European Heart Journal 2023, 44(48), 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Captur, G.; Arbustini, E.; Bonne, G.; Syrris, P.; Mills, K.; Wahbi, K.; Mohiddin, S. A.; McKenna, W. J.; Pettit, S.; Ho, C. Y.; et al. Lamin and the Heart. Heart 2018, 104(6), 468–479. [Google Scholar] [CrossRef] [PubMed]
- Kramarenko, D.; Walsh, R. Emery–Dreifuss Muscular Dystrophy: A Closer Look at Cardiac Complications. European Heart Journal 2023, 44(48), 5074–5076. [Google Scholar] [CrossRef]
- Madej-Pilarczyk, A. Clinical Aspects of Emery-Dreifuss Muscular Dystrophy. Nucleus 2018, 9(1), 268–274. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Carboni, N.; Bernasconi, P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells 2016, 5(3), 33. [Google Scholar] [CrossRef] [PubMed]
- 21 Yunisova, G.; Ceylaner, S.; Oflazer, P.; Deymeer, F.; Parman, Y. G.; Durmus, H. Clinical and Genetic Characteristics of Emery-Dreifuss Muscular Dystrophy Patients from Turkey: 30 Years Longitudinal Follow-up Study. Neuromuscular Disorders 2022, 32(9), 718–727. [Google Scholar] [CrossRef]
- Emery, AEH. Diagnostic Criteria for Neuromuscular Disorders, 2nd Ed. ed; Royal Society of Medicine Press: London, 1997. [Google Scholar]
- Bécane, H. M.; Bonne, G.; Varnous, S.; Muchir, A.; Ortega, V.; Hammouda, E. H.; Urtizberea, J. A.; Lavergne, T.; Fardeau, M.; Eymard, B.; et al. High Incidence of Sudden Death with Conduction System and Myocardial Disease Due to Lamins A and C Gene Mutation. Pacing Clin Electrophysiol 2000, 23 11 Pt 1, 1661–1666. [Google Scholar] [CrossRef]
- Steckiewicz, R.; Stolarz, P.; Świętoń, E.; Madej-Pilarczyk, A.; Grabowski, M.; Marchel, M.; Pieniak, M.; Filipiak, K. J.; Hausmanowa-Petrusewicz, I.; Opolski, G. Cardiac Pacing in 21 Patients with Emery-Dreifuss Muscular Dystrophy: A Single-Centre Study with a 39-Year Follow-Up. Kardiol Pol 2016, 74(6), 576–583. [Google Scholar] [CrossRef] [PubMed]
- Groh, W. J.; Bhakta, D.; Tomaselli, G. F.; Aleong, R. G.; Teixeira, R. A.; Amato, A.; Asirvatham, S. J.; Cha, Y.-M.; Corrado, D.; Duboc, D.; et al. 2022 HRS Expert Consensus Statement on Evaluation and Management of Arrhythmic Risk in Neuromuscular Disorders. Heart Rhythm 2022, 19(10), e61–e120. [Google Scholar] [CrossRef]
- Valenti, A. C.; Albini, A.; Imberti, J. F.; Vitolo, M.; Bonini, N.; Lattanzi, G.; Schnabel, R. B.; Boriani, G. Clinical Profile, Arrhythmias, and Adverse Cardiac Outcomes in Emery–Dreifuss Muscular Dystrophies: A Systematic Review of the Literature. Biology 2022, 11(4), 530. [Google Scholar] [CrossRef] [PubMed]
- Boriani, G.; Gallina, M.; Merlini, L.; Bonne, G.; Toniolo, D.; Amati, S.; Biffi, M.; Martignani, C.; Frabetti, L.; Bonvicini, M.; et al. Clinical Relevance of Atrial Fibrillation/Flutter, Stroke, Pacemaker Implant, and Heart Failure in Emery-Dreifuss Muscular Dystrophy: A Long-Term Longitudinal Study. Stroke 2003, 34(4), 901–908. [Google Scholar] [CrossRef] [PubMed]
- Marchel, M.; Madej-Pilarczyk, A.; Tymińska, A.; Steckiewicz, R.; Kochanowski, J.; Wysińska, J.; Ostrowska, E.; Balsam, P.; Grabowski, M.; Opolski, G. Echocardiographic Features of Cardiomyopathy in Emery-Dreifuss Muscular Dystrophy. Cardiology Research and Practice 2021, 2021, 1–7. [Google Scholar] [CrossRef]
- Blagova, O.; Nedostup, A.; Shumakov, D.; Poptsov, V.; Shestak, A.; Zaklyasminskaya, E. Dilated Cardiomyopathy with Severe Arrhythmias in Emery-Dreifuss Muscular Dystrophy: From Ablation to Heart Transplantation. J Atr Fibrillation 2016, 9(4), 1468. [Google Scholar] [CrossRef]
- Draminska, A.; Kuch-Wocial, A.; Szulc, M.; Zwolinska, A.; Styczynski, G.; Kostrubiec, M.; Hausmanowa-Petrusewicz, I.; Pruszczyk, P. Echocardiographic Assessment of Left Ventricular Morphology and Function in Patients with Emery–Dreifuss Muscular Dystrophy. International Journal of Cardiology 2005, 102(2), 207–210. [Google Scholar] [CrossRef]
- Smith, G.; Kinali, M.; Prasad, S.; Bonne, G.; Muntoni, F.; Pennell, D.; Nihoyannopoulos, P. Primary Myocardial Dysfunction in Autosomal Dominant EDMD. A Tissue Doppler and Cardiovascular Magnetic Resonance Study. Journal of Cardiovascular Magnetic Resonance 2006, 8(5), 723–730. [Google Scholar] [CrossRef]
- Haugaa, K. H.; Hasselberg, N. E.; Edvardsen, T. Mechanical Dispersion by Strain Echocardiography: A Predictor of Ventricular Arrhythmias in Subjects With Lamin A/C Mutations. JACC: Cardiovascular Imaging 2015, 8(1), 104–106. [Google Scholar] [CrossRef]
- Ditaranto, R.; Boriani, G.; Biffi, M.; Lorenzini, M.; Graziosi, M.; Ziacchi, M.; Pasquale, F.; Vitale, G.; Berardini, A.; Rinaldi, R.; et al. Differences in Cardiac Phenotype and Natural History of Laminopathies with and without Neuromuscular Onset. Orphanet J Rare Dis 2019, 14(1), 263. [Google Scholar] [CrossRef]
- Blaszczyk, E.; Gröschel, J.; Schulz-Menger, J. Role of CMR Imaging in Diagnostics and Evaluation of Cardiac Involvement in Muscle Dystrophies. Curr Heart Fail Rep 2021, 18(4), 211–224. [Google Scholar] [CrossRef]
- Bulmer, L.; Ljungman, C.; Hallin, J.; Dahlberg, P.; Polte, C. L.; Hedberg-Oldfors, C.; Oldfors, A.; Gummesson, A. EMD Missense Variant Causes X-Linked Isolated Dilated Cardiomyopathy with Myocardial Emerin Deficiency. Eur J Hum Genet 2025, 33(6), 775–783. [Google Scholar] [CrossRef]
- Russo, V.; Hudelo, J.; Marcel, M.; Florence, J.; Soulat, G.; Manka, R.; Pontana, F.; Dacher, J. N.; Toupin, S.; Nazarian, S.; et al. Role of Cardiovascular Magnetic Resonance in Diagnosis and Management of Muscular Dystrophies. J Cardiovasc Magn Reson 2026, 102693. [Google Scholar] [CrossRef]
- Holmström, M.; Kivistö, S.; Heliö, T.; Jurkko, R.; Kaartinen, M.; Antila, M.; Reissell, E.; Kuusisto, J.; Kärkkäinen, S.; Peuhkurinen, K.; et al. Late Gadolinium Enhanced Cardiovascular Magnetic Resonance of Lamin A/C Gene Mutation Related Dilated Cardiomyopathy. J Cardiovasc Magn Reson 2011, 13(1), 30. [Google Scholar] [CrossRef] [PubMed]
- Topriceanu, C.-C.; Al-Farih, M.; Joy, G.; Chan, F.; Webber, M.; Ilie-Ablachim, D. C.; Shiwani, H.; Tamang, M.; Banks, C.; Pettit, S.; et al. The Cardiovascular Magnetic Resonance Phenotype of Lamin Heart Disease. JACC Cardiovasc Imaging 2025, 18(6), 644–660. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G; Mercuri, E; Muchir, A; Urtizberea, A; Bécane, HM; Recan, D; Merlini, L; Wehnert, M; Boor, R; Reuner, U; et al. Clinical and Molecular Genetic Spectrum of Autosomal Dominant Emery-Dreifuss Muscular Dystrophy Due to Mutations of the Lamin A/C Gene. Ann Neurol 2000, 48(2), 170–80. [Google Scholar] [CrossRef]
- Boriani, G.; Biagini, E.; Ziacchi, M.; Malavasi, V. L.; Vitolo, M.; Talarico, M.; Mauro, E.; Gorlato, G.; Lattanzi, G. Cardiolaminopathies from Bench to Bedside: Challenges in Clinical Decision-Making with Focus on Arrhythmia-Related Outcomes. Nucleus 2018, 9(1), 442–459. [Google Scholar] [CrossRef]
- Feingold, B.; Mahle, W. T.; Auerbach, S.; Clemens, P.; Domenighetti, A. A.; Jefferies, J. L.; Judge, D. P.; Lal, A. K.; Markham, L. W.; Parks, W. J.; et al. Management of Cardiac Involvement Associated With Neuromuscular Diseases: A Scientific Statement From the American Heart Association. Circulation 2017, 136(13). [Google Scholar] [CrossRef]
- Finocchiaro, G.; Merlo, M.; Sheikh, N.; De Angelis, G.; Papadakis, M.; Olivotto, I.; Rapezzi, C.; Carr-White, G.; Sharma, S.; Mestroni, L.; et al. The Electrocardiogram in the Diagnosis and Management of Patients with Dilated Cardiomyopathy. European Journal of Heart Failure 2020, 22(7), 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Cesar, S.; Campuzano, O.; Cruzalegui, J.; Fiol, V.; Moll, I.; Martínez-Barrios, E.; Zschaeck, I.; Natera-de Benito, D.; Ortez, C.; Carrera, L.; et al. Characterization of Cardiac Involvement in Children with LMNA-Related Muscular Dystrophy. Front. Cell Dev. Biol. 2023, 11, 1142937. [Google Scholar] [CrossRef]
- Taylor, M. R. G.; Fain, P. R.; Sinagra, G.; Robinson, M. L.; Robertson, A. D.; Carniel, E.; Di Lenarda, A.; Bohlmeyer, T. J.; Ferguson, D. A.; Brodsky, G. L.; et al. Natural History of Dilated Cardiomyopathy Due to Lamin A/C Gene Mutations. Journal of the American College of Cardiology 2003, 41(5), 771–780. [Google Scholar] [CrossRef]
- Van Berlo, J. H.; De Voogt, W. G.; Van Der Kooi, A. J.; Van Tintelen, J. P.; Bonne, G.; Yaou, R. B.; Duboc, D.; Rossenbacker, T.; Heidbüchel, H.; De Visser, M.; et al. Meta-Analysis of Clinical Characteristics of 299 Carriers of LMNA Gene Mutations: Do Lamin A/C Mutations Portend a High Risk of Sudden Death? J Mol Med 2005, 83(1), 79–83. [Google Scholar] [CrossRef] [PubMed]
- Marchel, M.; Madej-Pilarczyk, A.; Tymińska, A.; Steckiewicz, R.; Ostrowska, E.; Wysińska, J.; Russo, V.; Grabowski, M.; Opolski, G. Cardiac Arrhythmias in Muscular Dystrophies Associated with Emerinopathy and Laminopathy: A Cohort Study. JCM 2021, 10(4), 732. [Google Scholar] [CrossRef]
- Bialer, M. G.; Mcdaniel, N. L.; Kelly, T. E. Progression of Cardiac Disease in Emery-dreifuss Muscular Dystrophy. Clinical Cardiology 1991, 14(5), 411–416. [Google Scholar] [CrossRef]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Bécane, H. M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and Molecular Genetic Spectrum of Autosomal Dominant Emery-Dreifuss Muscular Dystrophy Due to Mutations of the Lamin A/C Gene. Ann Neurol 2000, 48(2), 170–180. [Google Scholar] [CrossRef] [PubMed]
- Priori, S. Task Force on Sudden Cardiac Death of the European Society of Cardiology. European Heart Journal 2001, 22(16), 1374–1450. [Google Scholar] [CrossRef]
- Rootwelt-Norberg, C.; Skjølsvik, E. T.; Chivulescu, M.; Bogsrud, M. P.; Ribe, M. P.; Aabel, E. W.; Beitnes, J. O.; Brekke, P. H.; Håland, T. F.; Hasselberg, N. E.; et al. Disease Progression Rate Is a Strong Predictor of Ventricular Arrhythmias in Patients with Cardiac Laminopathies: A Primary Prevention Cohort Study. EP Europace 2023, 25(2), 634–642. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Sala, S.; Lazzeroni, D.; Palmisano, A.; Gigli, L.; Esposito, A.; De Cobelli, F.; Camici, P. G.; Mazzone, P.; Basso, C.; et al. Septal Late Gadolinium Enhancement and Arrhythmic Risk in Genetic and Acquired Non-Ischaemic Cardiomyopathies. Heart, Lung and Circulation 2020, 29(9), 1356–1365. [Google Scholar] [CrossRef]
- Escobar-Lopez, L.; Ochoa, J. P.; Mirelis, J. G.; Espinosa, M. Á.; Navarro, M.; Gallego-Delgado, M.; Barriales-Villa, R.; Robles-Mezcua, A.; Basurte-Elorz, M. T.; Gutiérrez García-Moreno, L.; et al. Association of Genetic Variants With Outcomes in Patients With Nonischemic Dilated Cardiomyopathy. J Am Coll Cardiol 2021, 78(17), 1682–1699. [Google Scholar] [CrossRef]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D. B.; Frese, K. S.; Keller, A.; Jensen, K.; Katus, H. A.; Meder, B. Genotype-Phenotype Associations in Dilated Cardiomyopathy: Meta-Analysis on More than 8000 Individuals. Clin Res Cardiol 2017, 106(2), 127–139. [Google Scholar] [CrossRef]
- van Rijsingen, I. A. W.; Arbustini, E.; Elliott, P. M.; Mogensen, J.; Hermans-van Ast, J. F.; van der Kooi, A. J.; van Tintelen, J. P.; van den Berg, M. P.; Pilotto, A.; Pasotti, M.; et al. Risk Factors for Malignant Ventricular Arrhythmias in Lamin a/c Mutation Carriers a European Cohort Study. J Am Coll Cardiol 2012, 59(5), 493–500. [Google Scholar] [CrossRef]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N. K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.-N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140(4), 293–302. [Google Scholar] [CrossRef]
- Glikson, M.; Nielsen, J. C.; Kronborg, M. B.; Michowitz, Y.; Auricchio, A.; Barbash, I. M.; Barrabés, J. A.; Boriani, G.; Braunschweig, F.; Brignole, M.; et al. 2021 ESC Guidelines on Cardiac Pacing and Cardiac Resynchronization Therapy. European Heart Journal 2021, 42(35), 3427–3520. [Google Scholar] [CrossRef]
- Golzio, P. G.; Chiribiri, A.; Gaita, F. “Unexpected” Sudden Death Avoided by Implantable Cardioverter Defibrillator in Emery Dreifuss Patient. Europace 2007, 9(12), 1158–1160. [Google Scholar] [CrossRef]
- Wyse, D. G.; Nath, F. C.; Brownell, A. K. W. Benign X-Linked (Emery-Dreifuss) Muscular Dystrophy Is Not Benign. Pacing Clinical Electrophis 1987, 10(3), 533–536. [Google Scholar] [CrossRef]
- Zeppenfeld, K.; Tfelt-Hansen, J.; De Riva, M.; Winkel, B. G.; Behr, E. R.; Blom, N. A.; Charron, P.; Corrado, D.; Dagres, N.; De Chillou, C.A. 2022 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. European Heart Journal 2022, 43(40), 3997–4126. [Google Scholar] [CrossRef]
- Walker, S. Biventricular Implantable Cardioverter Defibrillator Use in a Patient with Heart Failure and Ventricular Tachycardia Secondary to Emery-Dreifuss Syndrome. Europace 1999, 1(3), 206–209. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A. A.; Levy, J. A.; Gutierrez, P. S.; Marie, S. K.; Sosa, E. A.; Scanavaca, M. Emery-Dreifuss Muscular Dystrophy: Anatomical-Clinical Correlation (Case Report). Arq Neuropsiquiatr 2000, 58(4), 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Butt, K.; Ambati, S. Atrial Arrhythmias in Emery-Dreifuss Muscular Dystrophy: Approach to Successful Ablation. HeartRhythm Case Rep 2020, 6(6), 318–321. [Google Scholar] [CrossRef] [PubMed]
- Valverde Soria, L.; Sanchez-Millan, P. J.; Fernandez-Sanchez, J. A.; Macías-Ruiz, R.; Jimenez-Jaimez, J.; Tercedor, L. Successful Ablation of Purkinje-Related Ventricular Ectopy Leading to Ventricular Fibrillation in Emery-Dreifuss Dilated Cardiomyopathy. J Interv Card Electrophysiol 2025, 68(5), 957–960. [Google Scholar] [CrossRef]
- Zeppenfeld, K. Ventricular Tachycardia Ablation in Nonischemic Cardiomyopathy. JACC Clin Electrophysiol 2018, 4(9), 1123–1140. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
From genetic defect to cardiovascular complications in Emery-Dreifuss muscular dystrophy. Schematic representation of the pathophysiological continuum underlying cardiac involvement in Emery-Dreifuss muscular dystrophy. Pathogenic variants in nuclear-envelope-related genes, classically including EMD and LMNA, lead to structural and functional disruption of the cardiomyocyte nucleus, with altered mechanotransduction, impaired nuclear stability, abnormal gene expression, and progressive fibro-fatty remodelling. These molecular and cellular abnormalities translate into a characteristic cardiac phenotype dominated by atrial myopathy, conduction system disease and arrhythmogenic cardiomyopathy. Clinically, this may manifest as supraventricular tachyarrhythmias, bradyarrhythmias, advanced atrioventricular block (AVB), ventricular arrhythmias, progressive ventricular dysfunction, heart failure, thromboembolic complications, and sudden cardiac death. The figure also summarizes the major therapeutic consequences of this disease trajectory, including pacemaker or implantable cardioverter–defibrillator implantation, anticoagulation in the setting of atrial disease and guideline-directed heart failure therapy when ventricular dysfunction develops. Abbreviations: EMD, emerin gene; ICD, implantable cardioverter–defibrillator; LMNA, lamin A/C gene.
Figure 2.
From genetic defect to cardiovascular complications in Emery-Dreifuss muscular dystrophy. Schematic representation of the pathophysiological continuum underlying cardiac involvement in Emery-Dreifuss muscular dystrophy. Pathogenic variants in nuclear-envelope-related genes, classically including EMD and LMNA, lead to structural and functional disruption of the cardiomyocyte nucleus, with altered mechanotransduction, impaired nuclear stability, abnormal gene expression, and progressive fibro-fatty remodelling. These molecular and cellular abnormalities translate into a characteristic cardiac phenotype dominated by atrial myopathy, conduction system disease and arrhythmogenic cardiomyopathy. Clinically, this may manifest as supraventricular tachyarrhythmias, bradyarrhythmias, advanced atrioventricular block (AVB), ventricular arrhythmias, progressive ventricular dysfunction, heart failure, thromboembolic complications, and sudden cardiac death. The figure also summarizes the major therapeutic consequences of this disease trajectory, including pacemaker or implantable cardioverter–defibrillator implantation, anticoagulation in the setting of atrial disease and guideline-directed heart failure therapy when ventricular dysfunction develops. Abbreviations: EMD, emerin gene; ICD, implantable cardioverter–defibrillator; LMNA, lamin A/C gene.

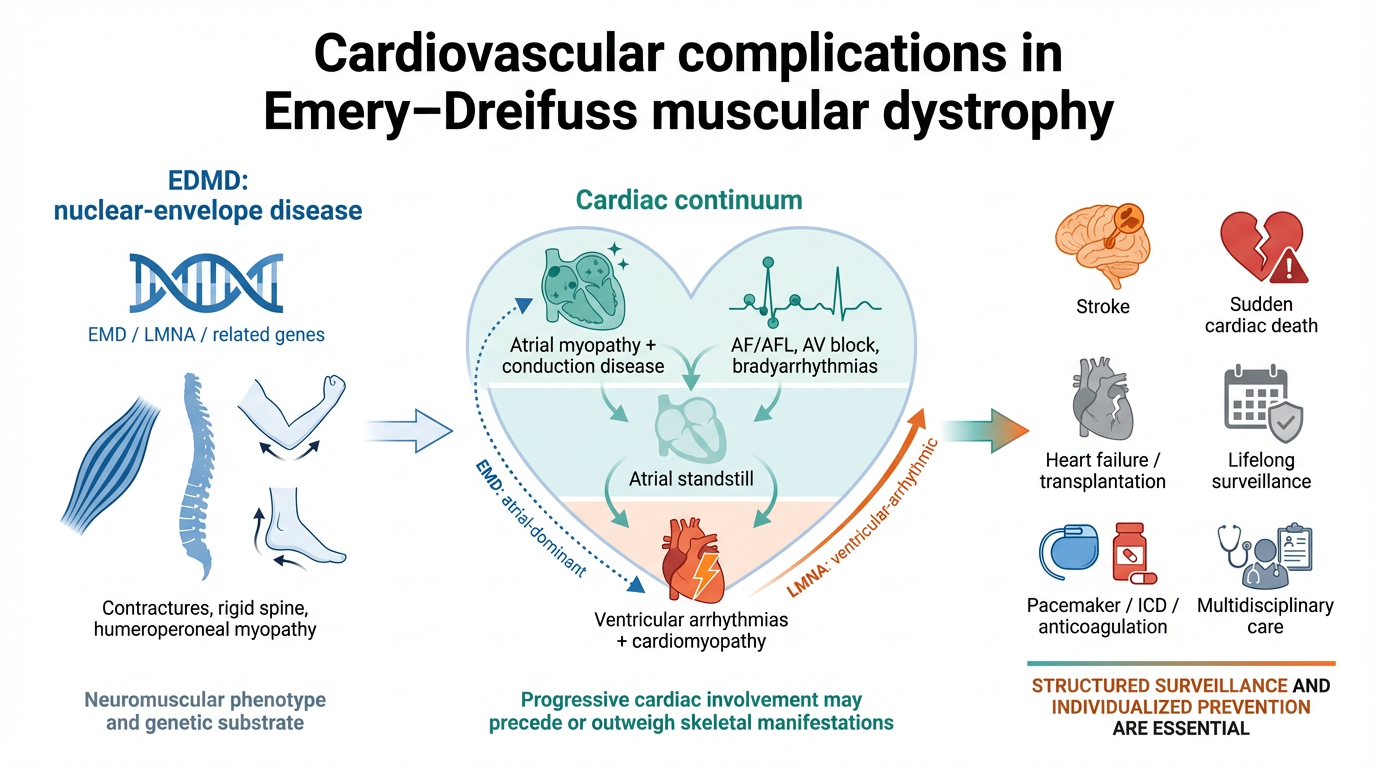
Figure 3.
Natural history of cardiac involvement in Emery-Dreifuss muscular dystrophy. EDMD is characterized by a progressive cardio-neuromuscular phenotype in which early atrial disease and conduction abnormalities often precede overt ventricular dysfunction. The disease trajectory may evolve from atrial ectopy, atrial fibrillation/flutter, and sinus node dysfunction toward atrial standstill, advanced AVB, ventricular arrhythmias, cardiomyopathy, and major adverse outcomes including stroke, sudden cardiac death, end-stage heart failure, and heart transplantation. Although overlap is common, EMD-related disease more often shows an early atrial-dominant course, whereas LMNA-related disease is generally associated with a more malignant ventricular-arrhythmic phenotype. Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; AVB, atrioventricular block; EDMD, Emery-Dreifuss muscular dystrophy; EMD, emerin gene; HF, heart failure; LMNA, lamin A/C gene; SCD, sudden cardiac death.
Figure 3.
Natural history of cardiac involvement in Emery-Dreifuss muscular dystrophy. EDMD is characterized by a progressive cardio-neuromuscular phenotype in which early atrial disease and conduction abnormalities often precede overt ventricular dysfunction. The disease trajectory may evolve from atrial ectopy, atrial fibrillation/flutter, and sinus node dysfunction toward atrial standstill, advanced AVB, ventricular arrhythmias, cardiomyopathy, and major adverse outcomes including stroke, sudden cardiac death, end-stage heart failure, and heart transplantation. Although overlap is common, EMD-related disease more often shows an early atrial-dominant course, whereas LMNA-related disease is generally associated with a more malignant ventricular-arrhythmic phenotype. Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; AVB, atrioventricular block; EDMD, Emery-Dreifuss muscular dystrophy; EMD, emerin gene; HF, heart failure; LMNA, lamin A/C gene; SCD, sudden cardiac death.

Figure 4.
Pragmatic workflow for cardiac surveillance and clinical decision-making in EDMD. Patients with suspected or confirmed EDMD, as well as genotype-positive relatives, require structured baseline cardiac assessment based on electrocardiography, prolonged rhythm monitoring, echocardiography, and cardiac magnetic resonance when available. Findings such as conduction disease, atrial arrhythmias or atrial standstill, ventricular arrhythmic markers, and progressive ventricular dysfunction should guide individualized decisions regarding pacing, anticoagulation, implantable cardioverter-defibrillator therapy, catheter ablation in selected cases, and referral for advanced heart failure therapies. Lifelong multidisciplinary follow-up is essential across all disease stages. Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; CMR, cardiac magnetic resonance; CRT, cardiac resynchronization therapy; ECG, electrocardiogram/electrocardiography; EDMD, Emery–Dreifuss muscular dystrophy; HF, heart failure; ICD, implantable cardioverter–defibrillator; LGE = late gadolinium enhancement; LMNA, lamin A/C gene; LV, left ventricle/left ventricular; NSVT, non-sustained ventricular tachycardia.
Figure 4.
Pragmatic workflow for cardiac surveillance and clinical decision-making in EDMD. Patients with suspected or confirmed EDMD, as well as genotype-positive relatives, require structured baseline cardiac assessment based on electrocardiography, prolonged rhythm monitoring, echocardiography, and cardiac magnetic resonance when available. Findings such as conduction disease, atrial arrhythmias or atrial standstill, ventricular arrhythmic markers, and progressive ventricular dysfunction should guide individualized decisions regarding pacing, anticoagulation, implantable cardioverter-defibrillator therapy, catheter ablation in selected cases, and referral for advanced heart failure therapies. Lifelong multidisciplinary follow-up is essential across all disease stages. Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; CMR, cardiac magnetic resonance; CRT, cardiac resynchronization therapy; ECG, electrocardiogram/electrocardiography; EDMD, Emery–Dreifuss muscular dystrophy; HF, heart failure; ICD, implantable cardioverter–defibrillator; LGE = late gadolinium enhancement; LMNA, lamin A/C gene; LV, left ventricle/left ventricular; NSVT, non-sustained ventricular tachycardia.


Table 1.
Integrated diagnostic framework for Emery-Dreifuss muscular dystrophy: from neuromuscular phenotype to genetic confirmation and cardiac screening. The table summarizes the contemporary diagnostic approach to EDMD, integrating the characteristic neuromuscular phenotype with cardiac manifestations, family history, and molecular confirmation. This framework reflects the current shift from a purely descriptive syndrome-based diagnosis to an aetiology-driven, multidisciplinary model aimed at early recognition, prognostic stratification, and cascade family screening. Abbreviations: CK, creatine kinase; CMR, cardiac magnetic resonance; ECG, electrocardiogram/electrocardiography; EDMD, Emery–Dreifuss muscular dystrophy; EMG, electromyography; EMD, emerin gene; LMNA, lamin A/C gene; LINC, linker of nucleoskeleton and cytoskeleton; SYNE1, spectrin repeat containing nuclear envelope protein 1 gene; SYNE2, spectrin repeat containing nuclear envelope protein 2 gene.
Table 1.
Integrated diagnostic framework for Emery-Dreifuss muscular dystrophy: from neuromuscular phenotype to genetic confirmation and cardiac screening. The table summarizes the contemporary diagnostic approach to EDMD, integrating the characteristic neuromuscular phenotype with cardiac manifestations, family history, and molecular confirmation. This framework reflects the current shift from a purely descriptive syndrome-based diagnosis to an aetiology-driven, multidisciplinary model aimed at early recognition, prognostic stratification, and cascade family screening. Abbreviations: CK, creatine kinase; CMR, cardiac magnetic resonance; ECG, electrocardiogram/electrocardiography; EDMD, Emery–Dreifuss muscular dystrophy; EMG, electromyography; EMD, emerin gene; LMNA, lamin A/C gene; LINC, linker of nucleoskeleton and cytoskeleton; SYNE1, spectrin repeat containing nuclear envelope protein 1 gene; SYNE2, spectrin repeat containing nuclear envelope protein 2 gene.
| Domain | Key features | Clinical relevance | Diagnostic tools | Implications for management |
| Neuromuscular phenotype | Early selective contractures involving elbows, Achilles tendons, and cervical/paraspinal muscles; humeroperoneal muscle weakness; rigid spine | Core phenotypic hallmark of classical EDMD and major clue for clinical suspicion | Neurological examination, musculoskeletal assessment, functional evaluation | Triggers targeted cardiological assessment and genetic testing |
| Skeletal muscle laboratory/electrophysiology | Serum CK often normal or only mildly elevated; EMG usually shows non-specific myopathic changes | Helpful as supportive but non-definitive findings; normal CK does not exclude EDMD | Serum CK, electromyography | Supports integrated work-up; should not delay molecular testing |
| Peripheral neuropathy / overlap phenotype | Occasional neurogenic features, mainly in selected LMNA-related cases; myopathy–neuropathy overlap may occur | Does not exclude EDMD when the typical contracture–myopathy pattern is present; helps refine phenotype | Nerve conduction studies, EMG, neurological re-evaluation | Broadens differential diagnosis and may justify extended genetic testing |
| Cardiac phenotype | Conduction disease, sinus node dysfunction, atrial fibrillation/flutter, atrial standstill, ventricular arrhythmias, cardiomyopathy, heart failure | Major determinant of morbidity and mortality; may precede overt neuromuscular manifestations | 12-lead ECG, prolonged Holter monitoring, echocardiography, CMR when available | Enables early rhythm surveillance, anticoagulation assessment, and device-based risk stratification |
| Family history | Sudden cardiac death, pacemaker implantation, cardiomyopathy, muscular dystrophy, stroke, unexplained syncope | Strengthens suspicion of inherited cardio-neuromuscular disease | Pedigree analysis, family interview | Supports cascade screening and anticipatory evaluation of relatives |
| Genetic confirmation | Pathogenic/likely pathogenic variants in EMD, LMNA, SYNE1, SYNE2, or related nuclear-envelope/LINC complex genes | Confirms diagnosis, refines prognosis, and supports genotype-oriented follow-up | Targeted gene testing, cardiomyopathy/neuromuscular gene panels, variant interpretation | Enables counselling, longitudinal risk stratification, and family screening |
| Female carrier / genotype-positive relative evaluation | Female EMD carriers or asymptomatic relatives may show delayed or isolated cardiac involvement | Clinically relevant because cardiac disease may emerge late and independently of skeletal manifestations | ECG, Holter, echocardiography, genetic counselling | Supports lifelong cardiac surveillance even in apparently asymptomatic carriers |
| Integrated final diagnosis | Combined recognition of typical neuromuscular pattern, compatible cardiac phenotype, and molecular confirmation | Contemporary diagnosis is multidisciplinary and aetiology-driven rather than purely descriptive | Joint neurology–cardiology–genetics assessment | Supports individualized monitoring and prevention of arrhythmic, thromboembolic, and heart failure complications |
Table 2.
Genotype–phenotype correlations in Emery-Dreifuss muscular dystrophy. The table summarizes the main clinical differences across EDMD-related genetic subgroups, emphasizing how EMD-associated disease is often characterized by an earlier atrial-dominant course, whereas LMNA-related disease typically carries a more malignant ventricular-arrhythmic and heart failure trajectory. Less common nuclear-envelope/LINC-complex phenotypes remain less well defined and require individualized interpretation. Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; AV, atrioventricular; EDMD, Emery–Dreifuss muscular dystrophy; EMD, emerin gene; HF, heart failure; ICD, implantable cardioverter–defibrillator; LMNA, lamin A/C gene; LINC, linker of nucleoskeleton and cytoskeleton; NSVT, non-sustained ventricular tachycardia; VF, ventricular fibrillation; VT, ventricular tachycardia.
Table 2.
Genotype–phenotype correlations in Emery-Dreifuss muscular dystrophy. The table summarizes the main clinical differences across EDMD-related genetic subgroups, emphasizing how EMD-associated disease is often characterized by an earlier atrial-dominant course, whereas LMNA-related disease typically carries a more malignant ventricular-arrhythmic and heart failure trajectory. Less common nuclear-envelope/LINC-complex phenotypes remain less well defined and require individualized interpretation. Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; AV, atrioventricular; EDMD, Emery–Dreifuss muscular dystrophy; EMD, emerin gene; HF, heart failure; ICD, implantable cardioverter–defibrillator; LMNA, lamin A/C gene; LINC, linker of nucleoskeleton and cytoskeleton; NSVT, non-sustained ventricular tachycardia; VF, ventricular fibrillation; VT, ventricular tachycardia.
| Genotype / disease subgroup | Inheritance | Typical extra-cardiac phenotype | Predominant cardiac phenotype | Characteristic clinical course | Main management implications |
| EMD-related EDMD (EDMD1 / emerinopathy) | X-linked | Classical early contractures, humeroperoneal weakness, rigid spine; full phenotype mainly in males; female carriers often milder and later | Early atrial disease and conduction abnormalities; AF/AFL, atrial standstill, sinus node dysfunction, AV block; ventricular dysfunction may occur later | Often atrial-dominant in the early phase, with progression from atrial ectopy/tachyarrhythmias to atrial standstill and thromboembolic risk; malignant ventricular arrhythmias may still occur in selected patients | Lifelong rhythm surveillance; strong attention to atrial arrhythmias, atrial standstill, and thromboembolic prevention; pacing frequently required, but ICD may be needed in selected higher-risk cases |
| LMNA-related EDMD / laminopathy (EDMD2 and overlapping laminopathic cardiomyopathy phenotypes) | Usually autosomal dominant | Neuromuscular phenotype may be classical, subtle, delayed, or even overshadowed by cardiac manifestations; both sexes affected | Conduction disease, atrial arrhythmias, ventricular arrhythmias, dilated cardiomyopathy, progressive LV dysfunction, heart failure | Often more ventricular-arrhythmic and malignant, with higher risk of NSVT/VT/VF, sudden cardiac death, and end-stage heart failure; cardiac-first presentation is common | Early ICD-oriented risk stratification is crucial; CMR and LMNA-specific risk models may refine prognosis; surveillance must address both arrhythmias and HF progression |
| SYNE1 / SYNE2-related EDMD (EDMD4/5 and related LINC-complex phenotypes) | Usually autosomal dominant or recessive depending on variant/context | Variable contractures and myopathy; phenotype often heterogeneous and less classically defined than EMD/LMNA forms | Cardiac phenotype less well characterized; may include conduction abnormalities, arrhythmias, and cardiomyopathic features | Natural history remains less clearly defined because of limited cohort data; penetrance and severity appear variable | Requires individualized follow-up within inherited cardio-neuromuscular programs; management usually extrapolated from broader EDMD/laminopathy principles |
| Female EMD carriers / genotype-positive relatives with limited neuromuscular phenotype | X-linked carrier state | Often absent, subtle, or delayed skeletal manifestations | Late-onset conduction disease, atrial arrhythmias, and other isolated cardiac manifestations may occur | Cardiac disease may emerge independently of overt muscular phenotype, often later in life | Cardiac surveillance should not be omitted on the basis of a mild skeletal phenotype or female sex |
| “Cardiac emerinopathy” / isolated or cardiac-predominant EMD phenotype | X-linked | Minimal or absent clinically relevant skeletal muscle involvement | Progressive atrial arrhythmias, atrial standstill, conduction disease, stroke risk; possible ventricular involvement/non-compaction in selected cases | Demonstrates that cardiac disease can dominate the phenotype and may be the presenting manifestation | Highlights the need to suspect EDMD-spectrum disease even in apparently isolated inherited arrhythmic/conduction phenotypes |
Table 3.
Spectrum of cardiac involvement in Emery-Dreifuss muscular dystrophy: major manifestations, clinical consequences, and monitoring implications. Cardiac disease in EDMD spans a broad continuum including conduction system disease, atrial myopathy with tachyarrhythmias and atrial standstill, ventricular arrhythmias, cardiomyopathy, heart failure, and thromboembolic complications. The table emphasizes the prognostic impact of each domain and the monitoring tools most relevant to longitudinal follow-up. Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; APBs, atrial premature beats; AV, atrioventricular; CMR, cardiac magnetic resonance; EDMD, Emery–Dreifuss muscular dystrophy; ECG, electrocardiogram/electrocardiography; HF, heart failure; ICD, implantable cardioverter–defibrillator; LV, left ventricle/left ventricular; NSVT, non-sustained ventricular tachycardia; NYHA, New York Heart Association; VF, ventricular fibrillation; VT, ventricular tachycardia.
Table 3.
Spectrum of cardiac involvement in Emery-Dreifuss muscular dystrophy: major manifestations, clinical consequences, and monitoring implications. Cardiac disease in EDMD spans a broad continuum including conduction system disease, atrial myopathy with tachyarrhythmias and atrial standstill, ventricular arrhythmias, cardiomyopathy, heart failure, and thromboembolic complications. The table emphasizes the prognostic impact of each domain and the monitoring tools most relevant to longitudinal follow-up. Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; APBs, atrial premature beats; AV, atrioventricular; CMR, cardiac magnetic resonance; EDMD, Emery–Dreifuss muscular dystrophy; ECG, electrocardiogram/electrocardiography; HF, heart failure; ICD, implantable cardioverter–defibrillator; LV, left ventricle/left ventricular; NSVT, non-sustained ventricular tachycardia; NYHA, New York Heart Association; VF, ventricular fibrillation; VT, ventricular tachycardia.
| Cardiac domain | Typical manifestations | Main clinical consequences | Prognostic significance | Preferred monitoring tools |
| Conduction system disease / bradyarrhythmias | Sinus bradycardia, sinoatrial block, first-degree AV block, bundle branch block, Mobitz II AV block, complete AV block | Fatigue, presyncope, syncope, pacemaker requirement, low-output symptoms | Hallmark of EDMD; often progressive and may precede overt cardiomyopathy | Serial ECG, prolonged Holter monitoring, device interrogation |
| Atrial myopathy and atrial ectopy | Atrial premature beats, atrial tachycardia, electrical instability at a young age | Symptoms, progression to AF/AFL, marker of evolving atrial disease | Often an early manifestation, especially in EMD-related disease | ECG, extended rhythm monitoring, device diagnostics when available |
| Atrial fibrillation / atrial flutter | Paroxysmal or persistent AF/AFL, often occurring early in life | Palpitations, haemodynamic intolerance, thromboembolic risk, progression to atrial standstill | Major determinant of stroke risk and marker of advanced atrial remodelling | ECG, prolonged Holter, device diagnostics, echocardiographic atrial assessment |
| Atrial standstill | Absence of P waves, atrial electrical silence, lack of atrial contraction, loss of atrial capture | Severe bradycardia, ineffective atrial pacing, thromboembolism, stroke | End-stage expression of atrial disease with major pacing and anticoagulation implications | ECG, echocardiography, device interrogation |
| Ventricular arrhythmias | Ventricular ectopy, NSVT, sustained VT, VF | Sudden cardiac death, ICD therapies, recurrent hospitalizations | One of the main determinants of prognosis, particularly in LMNA-related disease | Holter monitoring, ICD diagnostics, CMR, risk score integration in LMNA |
| Structural cardiomyopathy | Atrial enlargement, LV systolic dysfunction, dilated phenotype, occasional regional dysfunction | Reduced exercise tolerance, progressive ventricular remodelling, arrhythmic substrate | Variable across genotypes but strongly linked to HF progression and arrhythmic burden | Echocardiography, CMR, serial imaging follow-up |
| Heart failure | LV dysfunction, NYHA III–IV symptoms, progressive congestion, end-stage disease | Hospitalization, reduced survival, transplantation in selected cases | Major cause of mortality together with sudden cardiac death | Clinical follow-up, echocardiography, CMR, device-based follow-up |
| Thromboembolism / stroke | Cerebral embolic events, often in association with AF/AFL or atrial standstill | Permanent disability, recurrent events, increased mortality | Disproportionately high risk relative to patient age; requires proactive prevention | Rhythm surveillance, atrial mechanical assessment, anticoagulation-oriented evaluation |
Table 4.
Imaging phenotype in Emery-Dreifuss muscular dystrophy: echocardiographic and CMR findings with clinical interpretation. Echocardiography and cardiac magnetic resonance provide complementary information in EDMD, ranging from chamber remodelling and ventricular dysfunction to early myocardial deformation abnormalities and tissue characterization. The table highlights the incremental value of multimodality imaging for surveillance and risk stratification. Abbreviations: CMR, cardiac magnetic resonance; E/A ratio, ratio of early (E) to late (A) transmitral diastolic filling velocities; ECV, extracellular volume; EDMD, Emery-Dreifuss muscular dystrophy; HF, heart failure; LGE, late gadolinium enhancement; LV, left ventricle/left ventricular.
Table 4.
Imaging phenotype in Emery-Dreifuss muscular dystrophy: echocardiographic and CMR findings with clinical interpretation. Echocardiography and cardiac magnetic resonance provide complementary information in EDMD, ranging from chamber remodelling and ventricular dysfunction to early myocardial deformation abnormalities and tissue characterization. The table highlights the incremental value of multimodality imaging for surveillance and risk stratification. Abbreviations: CMR, cardiac magnetic resonance; E/A ratio, ratio of early (E) to late (A) transmitral diastolic filling velocities; ECV, extracellular volume; EDMD, Emery-Dreifuss muscular dystrophy; HF, heart failure; LGE, late gadolinium enhancement; LV, left ventricle/left ventricular.
| Imaging modality / parameter | Main findings in EDMD | Stage of disease in which useful | Potential clinical value | Main limitations |
| Standard transthoracic echocardiography | Atrial enlargement, variable LV systolic dysfunction, occasional dilated cardiomyopathy phenotype, chamber remodelling | Baseline and longitudinal follow-up across all stages | First-line structural and functional surveillance; identifies progression toward cardiomyopathy and HF | May underestimate early myocardial disease or subtle tissue abnormalities |
| Conventional Doppler / diastolic assessment | Impaired relaxation, altered E/A ratio, prolonged isovolumetric relaxation time, restrictive filling in advanced disease | Early subclinical dysfunction to overt cardiomyopathy | Detects early functional abnormalities beyond gross chamber dimensions | Interpretation may be affected by rhythm status, especially AF/AFL |
| Tissue Doppler imaging | Abnormal myocardial velocities despite preserved conventional indices in some patients | Early/subclinical stages | May reveal early myocardial dysfunction before overt LV remodelling | Operator-dependent and not fully standardized across centres |
| Speckle-tracking echocardiography / strain | Reduced deformation indices, impaired longitudinal mechanics, possible increased mechanical dispersion | Subclinical and intermediate disease stages | May refine early phenotyping and help identify patients at higher arrhythmic risk | Limited availability and limited EDMD-specific validation |
| Cardiac magnetic resonance: functional assessment | More accurate quantification of chamber volumes and ventricular function; detection of regional abnormalities | Particularly useful when echo findings are equivocal or disease progression is suspected | Improves phenotype definition and longitudinal characterization | Access and expertise may be limited |
| Cardiac magnetic resonance: tissue characterization | Mid-wall or septal LGE, increased extracellular volume, abnormal mapping indices, evidence of fibrosis substrate | Early to advanced disease, especially in LMNA-related phenotypes | Supports arrhythmic risk refinement and identification of subclinical myocardial involvement | Evidence in EDMD-specific cohorts remains limited and partly extrapolated from laminopathy series |
| Combined imaging approach | Echo for serial surveillance plus CMR for phenotypic clarification and tissue characterization | Across the disease continuum | Best overall strategy for integrated follow-up and risk assessment | Resource-dependent and not always feasible in routine practice |
Table 5.
Pragmatic management of cardiac complications in EDMD: surveillance, anticoagulation, device therapy and advanced options. The table proposes a clinically oriented framework for surveillance and treatment of the main cardiac scenarios encountered in EDMD, from early asymptomatic stages to advanced arrhythmic disease and heart failure. Particular emphasis is placed on proactive rhythm surveillance, anticoagulation when atrial disease emerges, and individualized selection of pacing versus defibrillator strategies. Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; AV, atrioventricular; CMR, cardiac magnetic resonance; CRT, cardiac resynchronization therapy; ECG, electrocardiogram/electrocardiography; EDMD, Emery–Dreifuss muscular dystrophy; HF, heart failure; ICD, implantable cardioverter–defibrillator; LMNA, lamin A/C gene; LV, left ventricle/left ventricular; NSVT, non-sustained ventricular tachycardia; VT, ventricular tachycardia; VF, ventricular fibrillation.
Table 5.
Pragmatic management of cardiac complications in EDMD: surveillance, anticoagulation, device therapy and advanced options. The table proposes a clinically oriented framework for surveillance and treatment of the main cardiac scenarios encountered in EDMD, from early asymptomatic stages to advanced arrhythmic disease and heart failure. Particular emphasis is placed on proactive rhythm surveillance, anticoagulation when atrial disease emerges, and individualized selection of pacing versus defibrillator strategies. Abbreviations: AF, atrial fibrillation; AFL, atrial flutter; AV, atrioventricular; CMR, cardiac magnetic resonance; CRT, cardiac resynchronization therapy; ECG, electrocardiogram/electrocardiography; EDMD, Emery–Dreifuss muscular dystrophy; HF, heart failure; ICD, implantable cardioverter–defibrillator; LMNA, lamin A/C gene; LV, left ventricle/left ventricular; NSVT, non-sustained ventricular tachycardia; VT, ventricular tachycardia; VF, ventricular fibrillation.
| Clinical scenario | Main problem | Suggested management approach | Key caveats in EDMD | Escalation options |
| Asymptomatic genotype-positive patient / early phenotype | Silent progression of conduction disease, atrial disease, or myocardial involvement | Lifelong structured surveillance with ECG, prolonged Holter, echocardiography, and CMR when available | Cardiac manifestations may precede or outweigh skeletal symptoms | Earlier referral to inherited cardiomyopathy / neuromuscular centre |
| Progressive conduction disease / bradyarrhythmias | Symptomatic bradycardia, advanced AV block, chronotropic incompetence | Permanent pacing according to guideline-based indications | Pacemaker therapy does not abolish the risk of malignant ventricular arrhythmias or sudden death | Consider ICD instead of pacing alone in selected high-risk patients |
| AF / AFL | Tachyarrhythmia, symptoms, stroke risk, progression of atrial disease | Rate/rhythm control strategy plus anticoagulation when indicated | Brady–tachy syndrome and coexisting conduction disease are common | Intensified rhythm monitoring, device-guided surveillance |
| Atrial standstill | Severe atrial myopathy, absent atrial contraction, embolic risk, atrial lead failure/loss of capture | Individualized pacing strategy and strong consideration of anticoagulation | Represents advanced atrial disease and may complicate device management | Device revision, specialist electrophysiology input |
| Ventricular arrhythmias / high-risk arrhythmic phenotype | NSVT, sustained VT/VF, sudden death risk | ICD-oriented risk stratification, especially in LMNA-related disease or when additional high-risk markers are present | Ventricular arrhythmias may occur despite only mild or moderate LV dysfunction | ICD implantation, tertiary arrhythmia referral |
| LV dysfunction / heart failure | Progressive cardiomyopathy with symptomatic HF | Guideline-directed medical therapy, serial imaging, clinical reassessment, device optimization when appropriate | EDMD-specific HF evidence is limited; progression may be genotype-dependent | CRT consideration, advanced HF referral |
| Recurrent arrhythmias despite drug/device therapy | Recurrent ICD therapies, refractory flutter/VT, electrical instability | Selected catheter ablation in expert centres | Diffuse fibrosis, intramural/atrial substrate, and disease progression may reduce success rates | Repeat ablation, combined advanced device/HF strategy |
| End-stage disease | Medically refractory HF and/or severe arrhythmic burden | Evaluation for advanced heart failure therapies | Rare but clinically relevant in advanced EDMD | Heart transplantation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



