Submitted:
26 March 2026
Posted:
27 March 2026
You are already at the latest version
Abstract
Background: Cancer remains a major global health challenge, with treatment efficacy limited by drug resistance and adverse effects. Drug repurposing offers opportunities for novel anticancer strategies. This study evaluated the cytotoxic, antiproliferative, and pro-apoptotic effects of metformin and caffeine, alone and in combination, in human cancer cell lines, and their potentialinteraction mechanisms. Methods: Human cervical carcinoma (HeLa), lung adenocarcinoma (A549), and colorectal carcinoma (HT29) cell lines were treated with metformin (0.05–50 mM) and caffeine (0.5–5 mM), alone or combined, for 24 and 48 h. Cell viability and proliferation were assessed using Trypan Blue and sulforhodamine B (SRB) assays. Apoptosis was analyzed by Annexin V/propidium iodide flow cytometry, and p53 expression in HeLa cells was determined by ELISA. Statistical analysis was performed using one-way ANOVA with Tukey’s post hoc test. Results: Metformin induced dose- and time-dependent cytotoxicity in all cell lines, with the lowest IC₅₀ values in HeLa and A549 cells after 48 h (2.28 and 3.30 mM; p < 0.05). Caffeine showed moderate antiproliferative activity, with strongest effects at 2.03 mM in HeLa and 2.01 mM in HT29 cells (p < 0.05). Combined treatment demonstrated variable effects depending on the cell line and treatment duration, with limited synergistic interaction observed only under specific conditions, while predominantly antagonistic effects were detected overall. Increased apoptosis and elevated p53 expression suggest activation of tumor-suppressive pathways. Conclusions: Metformin exhibits significant anticancer activity in vitro, supporting metformin repurposing in oncology. However,the addition of caffeine does not uniformly enhance its efficacy and appears to exert context-dependent effects.Further in vivo studies are required to confirm its clinical relevance.
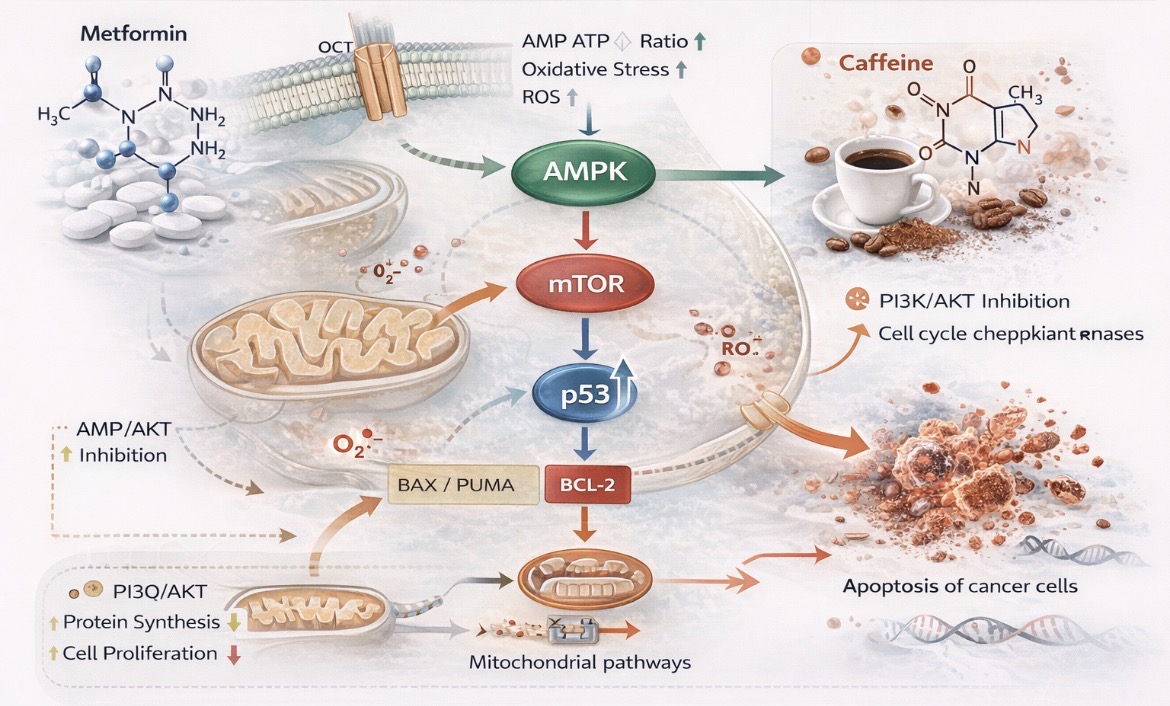
Keywords:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Cell Viability Assayand Proliferation Assays
2.3.1. Trypan Blue Exclusion Assay:
2.3.2. Sulforhodamine B (SRB) Assay
2.4. Apoptosis Analysis by Flow Cytometry
2.5. ELISA Assay: Determination of Tumor-Suppressor Genes for p53
2.6. Statistics
3. Results
3.1. Metformin
3.1.1. Cytotoxic Activity
3.1.2. Flow Cytometry Analysis
3.1.3. Apoptosis and Necrosis Detection by Immunofluorescence Microscopy
3.1.4. Modulation of Tumor Suppressor p53 Expression Following Metformin and Combined Metformin–Caffeine Treatment in Cervical Cancer Cells (HeLa Cells)
3.2. Caffeine
3.3. Combination of metformin and Caffeine
3.4. Figures, Tables and Schemes











|
24h Recorded (450nm) |
Concentration (U/mL) p53 |
48h Recorded (450nm) |
Concentration (U/mL) p53 |
|
| MRC 5 | 0.412 | 23.04 | 0.511 | 28.58 |
| MRC 5 + Met1 | 0.385 | 21.53 | 0.415 | 23.21 |
| MRC 5 + Caff1 | 0.365 | 20.41 | 0.395 | 22.09 |
| HeLa | 1.371 | 76.67 | 0.518 | 28.57 |
| HeLa + Met | 0.755 | 42.23 | 0.583 | 32.60 |
| HeLa + Caff | 0.572 | 31.99 | 0.464 | 25.95 |
| Standard (S) | 0.3 | 5.51 | 0.504 | 28.19 |
| Control (C) | 1.344 | 75.16 | 0.623 | 34.84 |
| Cell line | Metformin 24h | Metformin 48h |
| HeLa | 6.04 | 2.28 |
| A549 | 14.79 | 3.30 |
| HT 29 | 26.53 | 10.54 |
| MRC 5 | 33.461 | 33.461 |
| Cell line | Caffeine24h | Caffeine48h |
| HeLa | 2.44 | 2.03 |
| A549 | 3.38 | 2.44 |
| HT 29 | 3.41 | 2.01 |
| MRC 5 | 3.55 1 | 2.621 |
| Cell line | Met + Caff 2 (24 h) | Met + Caff 2 (48h) |
| HeLa | 2.23 | 2.4 |
| A 549 | 12.39 | 6.36 |
| HT 29 | 18.71 | 13.46 |
| MRC 51 | 22.60 | 38.301 |
|
24h CI1 |
48h CI1 |
|
| MRC 5 0.96 | 1.53 | |
| HeLa 0.78 | 1.55 | |
| A549 1.13 | 2.34 | |
| HT 29 0.99 | 1.78 |
3.5. Drug Interaction Analysis by the Chou–Talalay Method
Isobologram Analysis and Theoretical Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Health Statistics 2024: Monitoring Health for the SDGs; WHO Press: Geneva, Switzerland, 2024; Available online: https://www.who.int/data/gho/publications/world-health-statistics (accessed on 2 September 2024).
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Institute of Public Health of Serbia. Cancer Incidence and Mortality in Serbia 2023. Available online: https://www.batut.org.rs/download/publikacije/pub2023v1.pdf (accessed on 2 September 2024).
- Wagle, N.S.; Nogueira, L.; Devasia, T.P.; et al. Cancer treatment and survivorship statistics, 2025. CA Cancer J. Clin. 2025, 75, 308–340. [Google Scholar] [CrossRef]
- Mithoowani, H.; Febbraro, M. Non-small-cell lung cancer in 2022: A review for general practitioners in oncology. Curr. Oncol. 2022, 29, 1828–1839. [Google Scholar] [CrossRef]
- Han, X.; Robinson, L.A.; Jensen, R.E.; Smith, T.G.; Yabroff, K.R. Factors associated with health-related quality of life among cancer survivors in the United States. JNCI Cancer Spectr. 2021, 5, pkaa123. [Google Scholar] [CrossRef]
- Flory, J.; Lipska, K. Metformin in 2019. JAMA 2019, 321, 1926–1927. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef]
- Scheen, A.J. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 1996, 30, 359–371. [Google Scholar] [CrossRef]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.J.; Kitagawa, H.; Memmott, R.M.; Gills, J.J.; Dennis, P.A. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol. Metab. 2013, 24, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Vujovic, S.; Perovic, S.; Vlaovic, M.; Scepanovic, A.; Scepanovic, S. From metabolism to longevity: Molecular mechanisms underlying metformin’s anticancer and anti-aging effects. Curr. Issues Mol. Biol. 2026, 48, 286. [Google Scholar] [CrossRef]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef]
- Morales, D.R.; Morris, A.D. Metformin in cancer treatment and prevention. Annu. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.; Sonenberg, N.; Pollak, M. Metformin is an AMPK-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef]
- Dowling, R.J.O.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mTOR signaling in cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacology & Therapeutics 2015. [Google Scholar]
- Dai, Q.; et al. Caffeine sensitizes cancer cells to anticancer therapy. Cancer Letters 2012. [Google Scholar]
- Bode, A.M.; Dong, Z. The enigmatic effects of caffeine in cell cycle and cancer. Cancer Lett. 2007, 247, 26–39. [Google Scholar] [CrossRef]
- Fagundes, T.R.; Madeira, T.B.; Melo, G.P.; et al. Caffeine improves the cytotoxic effect of dacarbazine on melanoma cells. Bioorg. Chem. 2022, 120, 105576. [Google Scholar] [CrossRef]
- Strober, W. Trypan blue exclusion test of cell viability. CurrProtoc Immunol. 2001, 21(1), A.3B.1–A.3B.2. [Google Scholar]
- Shekan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990, 82(13), 1107–12. [Google Scholar]
- Bogdanović, G.; Raletić-Savić, J.; Marković, N. In vitro assays for antitumor-drug screening on human tumor cell lines: dye exclusion test and colorimetric cytotoxicity assay. Arch Oncol. 1994, 2(4), 181–4. [Google Scholar]
- Higuchi, K.; Mitsuhashi, N.; Saitoh, J.; Maebayashi, K.; Sakurai, H.; Akimoto, T.; Niibe, H. Caffeine enhanced radiosensitivity of rat tumor cells with a mutant-type p53 by inducing apoptosis in a p53-independent manner. Cancer Lett. 2000, 152(2), 157–62. [Google Scholar] [CrossRef]
- Chou, TC.; Talalay, P. Quantitative analysis of dose–effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chou, TC. Drug combination studies and their synergy quantification using the Chou–Talalay method. Cancer Res. 2010, 70(2), 440–446. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK: A key regulator of energy balance in cells and organisms. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B. Metformin inhibits mitochondrial complex I of cancer cells. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Heckman-Stoddard, B.; DeCensi, A.; Sahasrabuddhe, V.; Ford, L. Repurposing metformin for cancer prevention and treatment. Diabetologia 2017, 60, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Pernicova, I.; Korbonits, M. Metformin—Mode of action and implications for cancer therapy. Endocr. Rev. 2014, 35, 688–719. [Google Scholar]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in cancer prevention and therapy. Ann. Transl. Med. 2018, 6, 1–11. [Google Scholar]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Zhuang, Y; Chan, DK; Haugrud, AB; Miskimins, WK. Mechanisms by Which Low Glucose Enhances the Cytotoxicity of Metformin to Cancer Cells Both In Vitro and In Vivo. PLOS ONE 2014, 9, e108444. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nature Reviews Molecular Cell Biology 2007.Nat Rev Mol Cell Biol. 2007, 4, 275–83. [Google Scholar] [CrossRef] [PubMed]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; BrûléS; ViolletB; Kalender, A.; et al. Metformin inhibits mTOR signaling. Cell Metabolism 2010, 11(5), 390–401. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324(5930), 1029–1033. [Google Scholar] [CrossRef]
- Marciniak, M.; Torbicki, A.; Korpalski, M.; Pawluczyk, M.; Pawlikowski, K.; Żygłowicz, M.; Augustyn, D.; Gaworek, P.; Trybuła, A. Metformin in Oncology- Its Effect on Cancer Development and Progression. Medical Science 2025, 29, e132ms3665. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



