Submitted:
17 March 2026
Posted:
18 March 2026
You are already at the latest version
Abstract
Molecular typing of enteroviruses (EVs) is essential for surveillance of hand, foot, and mouth disease (HFMD). Conventional reverse-transcription polymerase chain reaction (RT-PCR) targeting the VP4–VP2 region can be insufficiently sensitive, reducing the detectability of Enterovirus A (EV-A). We developed a single-round RT-PCR assay using a modified reverse primer design (C3R) for rapid EV detection and genotyping. Sensitivity was evaluated using EV-A71 and poliovirus type 1 reference strains and 60 EV-positive clinical specimens. The C3R-based assay showed ~1,000-fold higher sensitivity for EV-A71 than conventional assays (limit of detection: 6.6 copies/reaction). It detected 100% (60/60) of clinical specimens, whereas the conventional assay detected only 45.0% (27/60) and failed at cycle threshold (Ct) values >30. Our assay maintained 100% sensitivity even at low viral loads (Ct > 35; P < 0.0001). All amplicons yielded high-quality sequences for definitive genotyping. This C3R-based RT-PCR overcomes sensitivity limitations of existing protocols and provides reliable genotyping from low-copy specimens, supporting its use in routine diagnostics and large-scale HFMD surveillance.
Keywords:
hand
; foot
; and mouth disease
; herpangina
; enterovirus
; reverse-transcription polymerase chain reaction
1. Introduction
Over 116 types of enteroviruses (EVs) infect humans, although some have unknown pathogenicity. Among them, polioviruses (PVs) and many non-PVs circulate globally [1,2]. The major types that circulate in Japan include Coxsackievirus A (CVA), Coxsackievirus B (CVB), echoviruses, enterovirus D68(EV-D68), and EV-A71 [3]. EV-A types such as CVA6, CVA10, CVA16, and EV-A71 commonly cause hand, foot, and mouth disease (HFMD), whereas several CVA types cause herpangina. The EV-B types, including CVB and echoviruses, can also contribute to both [4,5]. These diseases predominantly affect children and significantly impact public health [6].
Epidemic patterns shift annually in Japan, where EV-A71 outbreaks are associated with neurological complications such as aseptic meningitis and encephalitis [6,7,8]. Severe cases involving CVA2 and CVA4 have also been reported in the country [9,10], highlighting the importance of timely and accurate typing to support diagnoses, outbreak responses, and surveillance.
Semi-nested reverse-transcription polymerase chain reaction (RT-PCR) of the VP4–VP2 region (hereinafter referred to as conventional VP4–VP2 RT-PCR) has been widely used since the 1990s as a reliable and simple method for detecting and typing EVs [11,12]. Conventional VP4-VP2 RT-PCR, along with VP1-based assays such as Consensus-DEgenerate Hybrid Oligonucleotide Primer PCR (CODEHOP-PCR) [8], are commonly used in clinical and surveillance settings. VP1 sequencing remains the gold standard for definitive typing and genotyping. Although nested VP1 assays have high sensitivity, their high costs and long turnaround times also limit their utility for rapid or large-scale screening. Notably, recent evidence has indicated that, in most instances, EV types can be assigned directly from conventional VP4–VP2 RT-PCR products via sequence-based analysis, providing a practical complement to VP1-based typing [12]. Nevertheless, conventional VP4–VP2 RT-PCR can underperform in low-copy specimens. In one case of EV-A71 infection in a cerebrospinal fluid (CSF) sample, conventional VP4–VP2 RT-PCR based on the EVP-4/OL68 primer returned negative results, while both 5’-untranslated region (UTR) nested RT-PCR and real-time PCR assays showed positive detection, underscoring a sensitivity gap under low-titer conditions [13]. In this context, more sensitive VP4–VP2 RT-PCR may serve as a rapid screening tool to triage specimens for downstream VP1 or whole-genome sequencing, and as a complementary fallback test when VP1 fails [14], thereby maintaining the continuity of EV typing in real-world clinical and public health workflows.
In Japan, conventional VP4–VP2 RT-PCR is incorporated into the official HFMD pathogen-detection manual [15] used by public health laboratories (National Institute of Infectious Diseases, 2023). Accordingly, improving the sensitivity of this widely adopted assay has direct operational relevance for routine diagnostics and national surveillance workflows [12]. Recent evidence indicates that EV types can be determined from VP4–VP2 sequences with high concordance in most cases [14].
To address this limitation [13], we designed a novel reverse primer, C3R, for VP4–VP4-VP2-RT-PCR to improve its detection of EV-A and other EVs associated with HFMD and herpangina. By focusing on a shorter, more conserved target region, our alternative RT-PCR assay enhances analytical sensitivity while preserving type-level resolution via direct Sanger sequencing, offers a faster turnaround time, and reduces contamination risk when compared with nested protocols. Herein, we report the performance of this optimized assay, referred to as C3R-based RT-PCR, compared with that of conventional OL68-1-based RT-PCR [11,12], using a specimen set enriched in HFMD and herpangina cases.
2. Materials and Methods
2.1. Primers and Probes
All primers and probes are listed in Table 1, and their corresponding genomic binding sites are shown schematically in Figure 1. For the VP4–VP2 single-round and semi-nested PCR assays, the forward primers EVP2 [16] and EVP4 [12] and the reverse primers OL68-1 [11] and C3R were used (the latter being designed by our team to target a conserved VP2 coding region).
For VP1 CODEHOP PCR [17], SO224/SO222 primers were used in the first round, followed by AN89/AN88 ones in the second round.
Pan-EV real-time RT-PCR (i.e., the reference assay) was performed using Primers F and R1, together with the corresponding probe, according to a previously described protocol [18].
Left: Workflow of the C3R-based VP4–VP2 RT-PCR protocol, including RNA extraction, cDNA synthesis using random hexamers, single-round VP4–VP2 PCR with the EVP2/C3R primer set, and the optional C3R-based semi-nested PCR using EVP4/C3R.
Right: Genomic locations of primers and probes used in the Pan-enterovirus one-step RT-qPCR [18], conventional VP4–VP2 RT-PCR, semi-nested VP4–VP2 PCR [12], and VP1 CODEHOP assays [17]. The primer and probe binding sites are mapped across the 5‘-NC, VP4, VP2, VP3, and VP1 regions to illustrate the target regions for each assay.
2.2. Viral Isolates and Analytical Sensitivity
Analytical sensitivity was evaluated using two reference isolates: EV-A71 (Nagoya; GenBank accession no.: AB482183.1), as a representative EV-A strain; and PV1 (Sabin 1; GenBank accession no.: V01150.1), as a representative non-EV-A strain. Viral RNA extracted from each isolate was serially diluted and tested side-by-side to compare the detection limits of our C3R-based single-round VP4-VP2 RT-PCR (using the C3R reverse primer we designed) and the single-round conventional VP4–VP2 RT-PCR (using the OL68-1 reverse primer). In this study, “single round” denotes a non-nested RT-PCR workflow.
2.3. Reference Assay for Defining EV Positivity
Inclusion as an EV-positive specimen was determined using a pan-EV one-step real-time RT-PCR targeting the 5’ non-coding (5’ NC/5’ UTR) region (primer sequences presented in Table 1) [18]. This real-time assay served as an eligibility-defining reference test for the subsequent cycle threshold (Ct)-based stratification.
For reactions in which a sigmoid amplification curve was observed but the fluorescence did not cross the assay-defined threshold, the reaction was not classified as negative for analytical purposes/ Instead, a conventional Ct value of 45 was assigned [19], and the data were retained in all Ct-based summaries and binning procedures.
2.4. Clinical Specimens
We retrospectively analyzed 60 clinical specimens collected in Japan between 2011 and 2022 from patients with suspected HFMD or herpangina. Specimens were included if they tested EV-positive by the reference 5′-NC real-time RT-PCR [18] assay described above.
The specimens included oropharyngeal swabs (n=45), saliva (n=8), stool (n=3), nasal secretions (n=2), one nasopharyngeal swab (n=1), and cerebrospinal fluid (n=1).
Co-detection of rhinovirus (RV) by multiplex or RV-specific assays performed at the submitting laboratories was recorded but not used for eligibility determination (i.e., the samples were included irrespective of RV status).
2.5. RNA Extraction
Viral RNA was extracted from 140 µL of each specimen using a QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) and eluted into 60 µL of Buffer AVE (RNase-free elution buffer). Carrier RNA was not used in this study. The Easy Dilution for real-time PCR kit (TaKaRa Bio Inc., Shiga, Japan) was used as the diluent to make the serial dilutions used for the assay’s sensitivity testing.
2.6. Pan-EV One-Step Real-Time RT-PCR
Pan-enterovirus real-time RT-PCR [18] was performed in a 20 µL reaction mixture using the One Step PrimeScript III RT-qPCR Mix with UNG (TaKaRa Bio). Each reaction contained 10 µL of the 2× reaction mix, 1.0 µL of Primer F (10 µM), 1.0 µL of Primer R1 (10 µM), 0.2 µL of Probe P (10 µM), 0.4 µL of ROX Reference Dye II (50×), 2.0 µL of extracted RNA, and 5.6 µL of RNase-free H₂O to reach a final volume of 20 µL.
Real-time RT-PCR was conducted on an ABI QuantStudio 3 instrument. The thermocycling conditions were as follows:
25 °C for 10 min, 52 °C for 5 min, and 95 °C for 10 s, followed by 45 cycles of 95 °C for 5 s and 58 °C for 30 s, with fluorescence detection in the FAM channel.
This assay served as the eligibility-defining reference test in this study, and its Ct values were recorded for downstream stratified analyses.
2.7. Complementary DNA Synthesis
All complementary DNA (cDNA) samples were synthesized using a PrimeScript™ RT reagent Kit (TaKaRa Bio Inc., Shiga, Japan). Each 20 µL reaction contained 4 µL of 5× PrimeScript buffer, 4 µL of Random 6-mer primer (100 µM), 1.0 µL of PrimeScript RT Enzyme Mix I, 1.0 µL of nuclease-free water, and 10 µL of RNA. Incubation was performed at 37 °C for 15 min, followed by enzyme inactivation at 85 °C for 5 s.
2.8. VP4–VP2 Single-Round PCR (C3R-Based and Conventional OL68-1-Based)
Amplification of the enterovirus VP4–VP2 region was performed using a single-round PCR with EmeraldAmp PCR Master Mix (TaKaRa, Shiga, Japan). To evaluate primer performance, the forward primer EVP2 [16] was paired with either the reverse primer C3R (this study) or OL68-1 [11], and results obtained with the two reverse primer sets were compared.
Each 25 µL reaction contained 25 µL of 2× EmeraldAmp PCR Master Mix, 0.5 µL of EVP2 (50 µM), 0.5 µL of either C3R or OL68-1 (50 µM), 21 µL of nuclease-free water, and 3 µL of cDNA.
Thermocycling was performed as follows: 95 °C for 5 min; 40 cycles of 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 45 s; followed by a final extension at 72 °C for 5 min. PCR products were analyzed by agarose gel electrophoresis.
2.9. VP4–VP2 Semi-Nested PCR
The 2nd PCR was performed as a conventional (non-real-time) semi-nested reaction using EmeraldAmp PCR Master Mix (TaKaRa, Shiga, Japan). The 1st-round products served as templates as follows:
1st PCR EVP2/C3R → 2nd PCR EVP4/C3R
1st PCR EVP2/OL68-1 → 2nd PCR EVP4/OL68-1
For each reaction (25 µL total volume), the mixture contained 25 µL of 2× EmeraldAmp PCR Master Mix, 0.5 µL of EVP4 [12] (50 µM), 0.5 µL of the appropriate reverse primer (C3R or OL68-1; 50 µM), 21 µL of nuclease-free water, and 3 µL of the 1st-round PCR product.
Thermal cycling conditions were: 95 °C for 5 min; 35 cycles of 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 45 s; followed by a final extension at 72 °C for 5 min. Amplicons were resolved by agarose gel electrophoresis. Primer sequences are provided in Table 1.
2.10. VP1 CODEHOP PCR [17]
For this assay, the first round (50 µL) used SO224/SO222 and cDNA with EmeraldAmp. The thermocycling conditions were: 95 °C for 5 min; 35 cycles of 95 °C for 30 s/42 °C for 30 s/72 °C for 30 s; final extension at 72 °C for 5 min.
The second round (50 µL) used AN89/AN88, under conditions of: 95 °C for 5 min; 35 cycles of 95 °C for 30 s/60 °C for 30 s/72 °C for 30 s; final extension at 72 °C for 5 min.
2.11. Gel Electrophoresis
Amplicons from all assays (including VP4–VP2 RT-PCR rounds 1 and 2) were separated on 1.5% agarose gels. After electrophoresis, gels were post-stained with MIDORI Green Advance (NIPPON Genetics EUROPE GmbH, Düren, Germany) or GelRed Nucleic Acid Gel Stain (Biotium, Inc., Fremont, CA, USA) and imaged. Samples were considered positive only when a discrete band was observed at the assay-specific target size. For the VP4–VP2 assays, the target sizes were ~750 bp (round 1) and ~650 bp (round 2). Rhinovirus was distinguished by its characteristically shorter products (~100 bp smaller). Negative and positive controls were processed in parallel.
2.12. Sanger Sequencing and EV Typing
The PCR products were either enzymatically cleaned or gel-purified, and subsequently sequenced in both directions using amplification primers. Their chromatograms were visually inspected for quality. Low-quality 5’ and 3’ ends were trimmed, and forward/reverse reads were assembled into a consensus sequence. Ambiguous bases were resolved by reexamining the electropherograms or through repeat sequencing when necessary.
EV typing was performed based on the VP4–VP2 region, following the criteria and thresholds established by Kitamura and Arita [14]. Type assignment was determined based on the percentage of amino acid identity relative to the reference prototype strains. A 95% identity threshold was used as the primary discriminator, while accounting for established exceptions (e.g., PV1–PV2, CVA6, and specific EV-C types). In cases where the identity values were borderline or fell within known exception ranges, the final assignment was supported by VP1 sequencing and phylogenetic analyses [17].
2.13. In Silico Primer Coverage and 3′-End-Weighted Suitability Scoring (C3R vs OL68-1)
2.13.1. Panel and Alignment
The reverse primers C3R (i.e., the experimental primers) and OL68-1 (i.e., the control primers) were assessed against a curated panel of 100 representative EV genotypes (National Center for Biotechnology Information (NCBI) taxonomy ID: 12059) spanning EV-A, EV-B, EV-C, and EV-D (accession numbers listed in the Supplementary Table S2). For each genotype, we extracted the 20 nt reverse primer-binding segment in reverse-complement orientation and aligned the primers accordingly, emphasizing extension from the primer 3′ end.
2.13.2. International Union of Pure and Applied Chemistry (IUPAC) handling
Ambiguous nucleotide symbols were interpreted according to the standard International Union of Pure and Applied Chemistry (IUPAC) definitions [20].
For each sequence position i, primer and template nucleotides were considered concordant when the intersection of their IUPAC-defined nucleotide sets was non-empty; otherwise, the position was scored as a mismatch.
Primer-side “N” was treated as universally permissive and therefore always scored as a match, whereas missing or blank template positions were treated as mismatches.
2.13.3. Primer–Template Fit Score Calculation
Primer–template suitability scores were calculated using the IUPAC-based matching rules described above, combined with a 3′-end–weighted positional scoring model.A 20-nt window at the 3′ end was evaluated using position-specific weights (positions 1–12 = 0.2; 13–16 = 0.5; 17–20 = 1.0).
The mismatch indicator was defined as 0 for matches and 1 for mismatches, and the final score normalized to a 0–100 scale as:
For pre-aligned primer–template pairs, the full alignment length was evaluated rather than restricting the analysis to the rightmost 20 nt.
The web-based scoring tool developed for this study is openly available at: https://fujimoto-t.github.io/primer-score/primer-fit-score-en.html (accessed 2026-03-04).
2.13.4. Outputs and Interpretation
For each primer × genotype, the final score was computed and summarized by species and genotype (full per-genotype tables presented in the Supplementary Table S2). As operational heuristics for single-round PCR, scores were interpreted as 90–100 (near-perfect 3′-proximal match; high likelihood of amplification), 70–89 (generally acceptable; review potential 3′-proximal issues and consider optimization), 50–69 (suboptimal; potentially reduced efficiency), and <50 (poor; amplification likely impaired). These thresholds reflect established evidence that primer–template mismatches, particularly at or near the 3′ end, disproportionately impair priming and extension, thereby affecting qPCR sensitivity and quantification [21,22]. Accordingly, we prioritized 3′-end integrity over global identity, consistent with common primer-design guidance and reporting standards [19].
3. Results
3.1. In Silico Evaluation of Reverse Primers (Summary)
To contextualize the wet- laboratory results, we first summarized the in silico evaluation of the reverse primers. Using 100 representative EV genotypes, we assessed 3′-end compatibility of the redesigned C3R and conventional OL68-1 primers. C3R maintains consistently high 3′-end fitness across EV-A, EV-B, EV-C, and EV-D, with only a few moderate drops in EV-B and EV-C. This pattern suggests broadly reliable single-round amplification, particularly in EV-A and a broad subset of EV-B. In contrast, OL68-1 exhibits multiple 3′-proximal mismatches in EV-A and scattered reductions in EV-B and EV-C, consistent with its reduced empirical sensitivity observed later in wet-lab validation.
3.2. Analytical Sensitivity with Reference Isolates
Using RNA from reference isolates, the C3R-based VP4–VP2 RT-PCR assay demonstrated higher analytical sensitivity for the EV-A types. For EV-A71 (Nagoya strain), the limit of detection (LOD) was 66 copies in the single-round reaction and 6.6 copies in the second-round (semi-nested) reaction. In contrast, the OL68-1-based VP4–VP2 RT-PCR did not yield a detectable amplicon at or above 6.6 × 10⁴ copies in the first round, and required 6,600 copies to produce a positive band in the second round.
For PV 1 (Sabin 1), both assays showed comparable performance, with detection of 24 copies in both the single-round (Figure 2) and second-round reactions.
These findings confirm that the redesigned C3R reverse primer substantially improves the analytical sensitivity for EV-A (e.g., EV-A71), while preserving the expected performance for EV-C viruses (e.g., PV1).
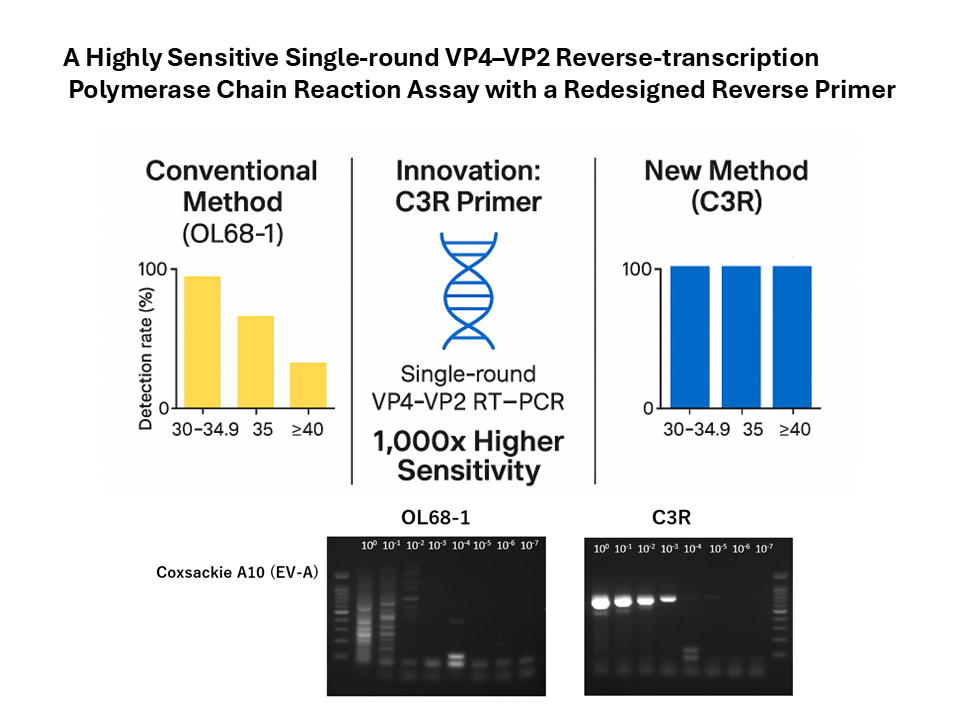
To illustrate these trends in single-round assays, representative amplicons from serial 10-fold dilutions are shown in Figure 2: C3R yields robust bands at low EV-A template inputs, where OL68-1 failed or produced only weak amplification, whereas EV-C (PV-1) exhibited comparable sensitivity between the two primers.
Serial 10-fold dilutions of enteroviral reference strains were amplified using single-round VP4–VP2 RT-PCR with either the conventional OL68-1 reverse primer or the redesigned C3R primer. For EV-A types, such as CVA10 and CVA5, the C3R primer markedly improved detection sensitivity, yielding clear amplicons at low template concentrations, whereas OL68-1 either failed to generate detectable bands or produced only weak amplification. As a control, EV-C (PV1) showed similar sensitivity between the two primers, with both assays detecting amplification down to 10⁻⁶ dilution (estimated LOD: ~19.5 copies).
Consistent with these trends, EV-A71 (Nagoya strain) showed a single-round LOD of 66 copies and a second-round (semi-nested) LOD of 6.6 copies with the C3R-based assay, whereas the OL68-1-based assay required substantially higher input to detect amplification (no detectable band at ≥6.6 × 10⁴ copies in the first round; 6,600 copies required in the second round).
3.3. Clinical Performance in HFMD/Herpangina Cohort
Across 60 EV/RV-positive specimens (oropharyngeal swabs 45; saliva 8; stool 3; nasal secretions 2; nasopharyngeal swab 1; cerebrospinal fluid 1), Ct values ranged from 22.17 to 45.00 (median 30.64)(Supplementary Table 1).
The C3R-based RT-PCR detected 60/60 (100%) specimens in the first round, whereas the OL68-1-based assay detected 27/60 (45%). Pairwise comparison on the same 60 specimens showed both positive = 27, C3R-based RT-PCR-only positive = 33, conventional VP4–VP2 RT-PCR-only positive = 0, both negative = 0; McNemar’s exact test [23] gave P=1.16×10⁻¹⁰, indicating a significant superiority of the C3R-based RT-PCR.
When stratified by Ct, specimens positive by the conventional VP4–VP2 RT-PCR had a lower median Ct (higher viral load) of 27.89 (n=27), whereas specimens negative by the conventional assay had a higher median Ct of 31.66 (n=33) (Mann–Whitney U, P=2.78 × 10⁻⁴), consistent with its reduced sensitivity at low template abundance. In contrast, the C3R-based RT-PCR remained positive across the entire Ct range analyzed (22.17–45.00), with Ct=45 representing reactions that showed a sigmoid amplification curve but did not cross the assay-defined threshold and were therefore retained in analysis as Ct=45 by convention.
These Ct-stratified trends are summarized in Figure 2, which shows preserved detection by the C3R-based assay across all Ct strata, in contrast to a sharp drop- in the conventional assay beyond Ct 30.
The detection rates of C3R-based RT-PCR (blue) and conventional OL68-1-based VP4–VP2 RT-PCR (orange) are shown across predefined Ct strata. The conventional assay demonstrates a pronounced decline in positivity beyond Ct 30, consistent with limited sensitivity at low template abundance, whereas the C3R-based RT-PCR maintains 100% detection across all Ct bands, including samples represented as Ct ≥ 40 (with Ct=45 assigned by convention for threshold-negative sigmoid reactions; see Methods). The numbers below each category denote the sample counts within each Ct stratum.
By grouping EV species, C3R-based RT-PCR achieved 100% detection of EV-A (53/53), EV-B (6/6), and EV-D (1/1). In contrast, conventional VP4–VP2 RT-PCR detected EV-A in 25/53 (47%), EV-B in 2/6 (33%), and EV-D in 0/1 (0%) patients in single-round testing.
At the genotype level within this 60-specimen set, C3R-based RT-PCR was uniformly positive for CVA6 (24/24), CVA16 (12/12), CVA10 (9/9), EV-A71 (6/6), CVB3 (1/1), CVB5 (1/1), CVA4/5/9 (each1/1), Echovirus 18/30/9 (each 1/1), and EV-D68 (1/1), whereas the OL68-1-based assay was positive for CVA6 (9/24), CVA16 (7/12), CVA10 (5/9), EV-A71 (2/6), CVB5 (1/1), CVA4 (1/1), CVA5 (1/1), CVA9 (1/1), and negative for CVB3, EV-D68, Echo 9/18/30 in the first round. These data demonstrate the broad inclusivity of C3R-based RT-PCR across EV species and genotypes in a single-round format.
All amplicons produced by C3R-based RT-PCR were within the expected size range (~750 bp in the 1st round; ~650 bp in the 2nd round) and were directly amenable to Sanger sequencing (Methods).
<25 / 25–29.9 / 30–34.9 / 35–39.9 / 45 (with 45 assigned by convention for sigmoid amplification curves that do not reach the threshold)
3.4. In Silico Coverage and Fitness of C3R (vs. OL68-1)
C3R:
Primer–template fitness across 100 EV genotypes showed that 68/100 scored 100 (68.0%) and 31/100 scored 94.1–97.6 (31.0%), indicating perfect or near-perfect matches at the primer 3′ end and predicting robust single-round amplification (Figure 3). The only notable outlier was EV-C104 (EU840733.1), which scored 83.33 due to multiple 3′-proximal mismatches, suggesting reduced amplification efficiency relative to other genotypes (full per-genotype scores are listed in Supplementary Table S2).
OL68-1:
OL68-1 exhibited a species-dependent pattern. In EV-C and EV-D, OL68-1 frequently achieved 97.6–100, occasionally 100, whereas C3R typically fell in the 94–98 range for these species. In EV-B, OL68-1 generally scored around 97.6 (with some at 88.1), but was often lower than C3R, which frequently reached 100 across Echo and CVB representatives; for EV-B81/EV-B87, OL68-1 scored 97.6 or 88.1, while C3R was 95.24. In EV-A, OL68-1 commonly scored ≈89.3, consistent with multiple 3′-proximal mismatches and the assay’s reduced empirical sensitivity for EV-A, while C3R was near-uniformly 100. Full per-genotype scores and alignments are provided in Supplementary Table S2.
Figure 4.
In silico primer–template fitness of C3R and OL68-1 across Enterovirus species A–D.

Fitness scores (0–100 scale) for C3R (blue) and OL68-1 (red) are presented using a subset of representative genotypes on the x-axis (labels are thinned for readability); trends reflect the full dataset for EV-A, EV-B, EV-C, and EV-D. C3R showed uniformly high fitness in EV-A (predominantly 100) and across most EV-B genotypes, with OL68-1 typically around 97.6 in EV-B (occasionally 88.1). In EV-C and EV-D, both primers generally exhibited high fitness; OL68-1 sometimes achieved perfect matches, whereas C3R typically scored in the mid-to-high 90s, with EV-C104 being the only notable low C3R outlier (83.33). Comprehensive per-genotype results and alignments are available in Supplementary Table S2.
4. Discussion
4.1. Major Findings
Our study demonstrates that the newly optimized single-round PCR assay utilizing the C3R reverse primer significantly enhances both analytical and diagnostic sensitivity for HFMD-associated EVs compared with conventional VP4–VP2 RT-PCR. For EV-A71, we observed an approximately 1,000-fold gain in analytical sensitivity (LOD, 6.6 copies/reaction), and in clinical specimens, the C3R-based RT-PCR assay achieved 100% detection (60/60), including low-viral-load samples (Ct ≥30 and up to 35), whereas the conventional assay detected 45% (27/60). This superiority was statistically significant in paired testing (McNemar’s exact test, P = 1.16 × 10⁻¹⁰), underscoring that the observed differences are unlikely to be due to chance.
4.2. Mechanistic Interpretation Anchored by Standard Strains
The superior performance of C3R is mechanistically consistent with its 3′-proximal concordance at the VP2 C3 site, which reduces the penalty incurred at the polymerase-critical end of the primer–template interface. The standard-strain experiments provide an orthogonal anchor for this interpretation: EV-A71 exemplifies the gain under 3′-end mismatch pressure that hampers OL68-1, whereas PV-1 (EV-C) demonstrates that the redesign does not trade sensitivity for breadth in a non-EV-A background. This alignment of the sequence-level rationale and reference-isolate behavior explains why the clinical gains are concentrated in EV-A-rich settings without implying cross-board disadvantages for EV-C and EV-D.
Notably, although C3R carries higher degeneracy than OL68-1 (96 vs. 8 possible sequence variants), its use resulted in less nonspecific amplification in our hands. This indicates that primer–template compatibility at the 3′ critical region exerts a stronger influence on amplification specificity than the nominal degree of degeneracy. Thus, the improved match at the VP2-C3 site appears to offset the theoretical increase in nonspecific binding expected from a more degenerate primer.
4.3. C3R-Based RT-PCR: Single-Round Alternative to Conventional VP4–VP2 RT-PCR
The poor single-round performance of conventional VP4–VP2 RT-PCR against EV-A can be mechanistically explained by sequence mismatches at critical sites in contemporary EV-A lineages (e.g., CVA6 clade D and recent EV-A71 strains), as shown by our in silico coverage analysis (Supplementary Table S2) and concordant external reports that OL68-1 frequently underperforms EV-A typing compared with alternative VP2 primer schemes. Conversely, the C3R primer was rationally redesigned to target the conserved VP2-coding region with minimal mismatches across EV-A, enabling stable amplification even when viral shedding is low (early/late infection and in the CSF) [13] or when RNA is partially degraded. Moreover, while nested VP1 protocols such as CODEHOP-PCR can often compensate for sensitivity in low-titer samples, their principal limitations include increased cost, longer turnaround times, added workflow complexity, and a higher risk of carryover contamination compared to single-round assays, which reduces their feasibility for rapid screening or surge-scale surveillance. Notably, the reference-isolate comparison (EV-A71 gain with PV-1 parity) provides direct experimental corroboration of the 3′-end–driven rationale, supporting a C3R-first, single-round strategy without implying a generalized disadvantage in EV-C/EV-D.
4.4. Future Application to Improve the Conventional VP4–VP2 Manual Protocol
Importantly, the insights gained from this redesign process provide a clear roadmap for upgrading existing manual VP4–VP2 RT-PCR workflows. Many laboratories continue to employ OL68-1–based protocols as part of legacy EV typing algorithms. Our findings indicate that simply replacing the reverse primer within these manual protocols—while keeping the remaining steps unchanged—would yield a substantial improvement in sensitivity, particularly for EV-A–dominant HFMD contexts. As several national EV surveillance systems still rely on VP4–VP2 as a front-line assay, we plan to extend our optimization framework to develop a manual version of the C3R-enhanced VP4–VP2 PCR protocol. This will allow institutions with limited access to real-time PCR platforms or automated extraction systems to benefit from the same sensitivity gains demonstrated in the present study, while preserving compatibility with existing sequencing and reporting pipelines.
4.5. Practical Implications for Typing Workflows.
Because the improvement is derived from primer–template compatibility rather than cycling heuristics or nested amplification, C3R can be positioned as a first-line, single-round VP4–VP2 RT-PCR that triages specimens to VP1 or whole-capsid sequencing only when identities are borderline or epidemiologically critical. In laboratories where OL68-1 has been retained for historical reasons, the EV-A71/PV-1 pair provides a simple commissioning panel to verify that the expected differential is reproduced locally before a broad rollout. This standard-anchored validation is particularly relevant during HFMD surges, where time-to-result and contamination control favor a single-round strategy.
Importantly, because C3R enables reliable typing using single-round PCR without the need for nested amplification, the workflow becomes both faster and more economical. Eliminating the second PCR reduces reagent use, labor, and hands-on time, while also shortening the overall turnaround by 2–3 h. In practical terms, laboratories can process higher sample volumes at a lower per-specimen cost, which is a critical advantage during seasonal HFMD peaks and public-health surge responses.
4.6. Applicability to One-Step and Real-Time Formats.
In addition, the VP2C3R primer was also compatible with a one-step RT-PCR workflow and yielded robust amplification in our hands (data not shown). This suggests that VP2C3R is not only advantageous for single-round conventional RT-PCR but is also a promising candidate for real-time PCR assays targeting the VP4–VP2 region. Moving forward, we plan to expand both the number and diversity of clinical specimens (e.g., respiratory samples, sterile-site fluids, and stool) to systematically benchmark VP2C3R performance across one-step and real-time formats and to define its optimal placement within diagnostic algorithms.
4.7. Rationale for Selecting the VP4–VP2 Region and Positioning in Diagnostic Algorithms
Although VP1 sequencing remains the gold standard for definitive typing and genotyping [17,23], supported by a long-standing correlation between the serotype and the VP1 sequence, its relatively large amplicon (~ 900 bp) and reliance on nested workflows can impair its performance and timeliness in routine clinical specimens.
Our data show that the VP4–VP2 segment, which is shorter and flanked by conserved primer sites, presents a pragmatic target for HFMD-associated EVs. In line with a recent evaluation of VP4–VP2 typing that demonstrated high RT-PCR sensitivity for this region and proposed a 95% amino acid identity threshold for type assignment (with limited exceptions such as PV1–PV2, CVA6/10, and some EV-C types) [14], our single-round PCR using C3R achieved species/genotype coverage across EV-A71, CVA6/10/16, EV-B types, and EV-D68.
4.8. Clinical and Public-Health Significance
In surge settings for HFMD and other EV activities [9,24], a single-round assay that reliably detects EV-A71 and related EVs can shorten reporting by ~2–3 h, support early risk stratification (e.g., neurologic complications), and improve throughput at a lower per-specimen cost. As a Tier-1 screen [14], frontline VP4–VP2 single-round RT-PCR can triage only borderline or epidemiologically critical specimens to VP1 or whole-capsid sequencing, conserving resources for “deep” characterization.
A sensitive front-end screen aligns with international surveillance frameworks that prioritize early detection of severe EV presentations. In Europe, WHO/ENPEN data demonstrate that non-polio enteroviruses are important causes of paralysis and recommend standardized reporting of severe cases, underscoring the value of a rapid assay before VP1 or whole-capsid sequencing [25]. In the United States, national AFM surveillance and NESS emphasize prompt identification of EV-positive specimens during EV-D68 activity [26], where a fast VP4–VP2 gate helps triage AFM-suspect or ICU cases for deeper genomic investigation. In Japan, notifiable acute encephalitis surveillance and nationwide AFP surveillance (<15 years) reveal large pediatric caseloads with substantial unknown-pathogen proportions, highlighting the importance of rapid EV detection to direct confirmatory sequencing—reinforced by Japan’s nationwide AFM investigation during the 2015 EV-D68 season [27].
4.9. Limitations and Future Directions
Our evaluation focused on the HFMD-related EV-A/EV-B isolates and a small number of EV-D isolates. Performance against less common EV-C/EV-D genotypes warrants broader testing, including in cohorts with CSF-rich meningitis and AFM presentations. Short amplicon typing can be affected by recombination and lineage turnover; thus, borderline identity results (~92–96% AA) should trigger escalation to VP1 or whole-capsid sequencing and be interpreted in conjunction with clinical/epidemiological metadata.
Abbreviations (Alphabetical Order)
The following abbreviations are used in this manuscript:
cDNA — complementary DNA
CODEHOP — consensus-degenerate hybrid oligonucleotide primer
CSF — cerebrospinal fluid
Ct — cycle threshold
CVA — Coxsackievirus A
CVB — Coxsackievirus B
EVs — enteroviruses
FAM — 6-carboxyfluorescein
HFMD — hand, foot, and mouth disease
IUPAC — International Union of Pure and Applied Chemistry
LOD — limit of detection
NC — non-coding
PCR — polymerase chain reaction
PV — poliovirus
RT — reverse-transcription
RV — rhinovirus
UTR — untranslated region
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: title, Ct-stratified clinical detection of enteroviruses in the HFMD/herpangina cohort by C3R- and OL68-1-based single-round RT-PCR.; Table S2: title, Primer‒template alignments and in silico fitness scores of C3R and OL68-1 across 100 representative Enterovirus genotypes.
Author Contributions
Conceptualization, T.F.; Methodology, T.F., M.O., K.Ko. and M.A.; Software, T.F.; Validation, M.O., K.Ka., T.S., Y.N., K.Ko., M.K.U. and T.F.; Formal analysis, T.F.; Investigation, M.O., K.Ka., T.S., Y.N., K.Ko. and M.K.U.; Resources, M.O., K.Ka., T.S., T.F. and M.A.; Data curation, T.F.; Writing—original draft, T.F., M.A., K.Ko. and M.O.; Writing—review and editing, all authors; Visualization, T.F.; Supervision, T.F. and M.A.; Project administration, T.F. and M.A.; Funding acquisition, M.A. All authors have read and agreed to the published version of the manuscript. Abbreviations: T.F. = Tsuguto Fujimoto; M.O. = Miki Ogi; K.Ka. = Kazuhiro Kitakawa; T.S. = Takako Sano; Y.N. = Yoshihiro Nishimura; K.Ko. = Kouichi Kitamura; M.K.U. = Minami Kikuchi Ueno; M.A. = Minetaro Arita.
Funding
This research was supported by the Japan Agency for Medical Research and Development (AMED) under Grant Numbers JP24fk0108627 and JP25fk0108716.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of the National Institute of Infectious Diseases (NIID), Japan (protocol No. 1533, approved on May 29, 2023).
Informed Consent Statement
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki. The requirement for individual informed consent was waived because the study used residual clinical specimens derived from administrative public health testing. Specimens and associated data were de-identified at the local public health institutes before transfer to the National Institute of Infectious Diseases (NIID). NIID and the Tokyo Metropolitan Institute of Public Health do not retain re-identification keys, whereas four collaborating institutes maintain linkage tables under restricted access. Information for opt-out (study purpose and contact details) is publicly available on the NIID website, enabling refusal; upon withdrawal, use of specimens is discontinued and any data already generated are excluded from analysis. The study protocol was approved by the NIID Ethics Review Committee (approval date: 29 May 2023).:
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in the section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
This research was supported by AMED under Grant Number JP25fk0108716.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Xie, Z.; Khamrin, P.; Maneekarn, N.; Kumthip, K. Epidemiology of Enterovirus Genotypes in Association with Human Diseases. Viruses 2024, 16, 1165. [Google Scholar] [CrossRef] [PubMed]
- Jartti, M.; Flodström-Tullberg, M.; Hankaniemi, M.M. Enteroviruses: Epidemic Potential, Challenges and Opportunities with Vaccines. J. Biomed. Sci. 2024, 31, 73. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.; Carr, M.J.; Kobayashi, M.; Hanaoka, N.; Fujimoto, T. Enterovirus-Associated Hand-Foot-and-Mouth Disease and Neurological Complications in Japan and the Rest of the World. Int. J. Mol. Sci. 2019, 20, 5201. [Google Scholar] [CrossRef] [PubMed]
- Aswathyraj, S.; Arunkumar, G.; Alidjinou, E.K.; Hober, D. Hand, Foot and Mouth Disease (HFMD): Emerging Epidemiology and the Need for a Vaccine Strategy. Med. Microbiol. Immunol. 2016, 205, 397–407. [Google Scholar] [CrossRef]
- Yu, H.; Li, X.W.; Liu, Q.B.; Deng, H.L.; Liu, G.; Jiang, R.M.; et al. Diagnosis and Treatment of Herpangina: Chinese Expert Consensus. World J. Pediatr. 2020, 16, 129–134. [Google Scholar] [CrossRef]
- Takahashi, S.; Metcalf, C.J.E.; Arima, Y.; Fujimoto, T.; Shimizu, H.; Van Doorn, H.R.; et al. Epidemic Dynamics, Interactions and Predictability of Enteroviruses Associated with Hand, Foot and Mouth Disease in Japan. J. R. Soc. Interface 2018, 15, 20180507. [Google Scholar] [CrossRef]
- Ooi, M.H.; Wong, S.C.; Lewthwaite, P.; Cardosa, M.J.; Solomon, T. Clinical Features, Diagnosis, and Management of Enterovirus 71. Lancet Neurol. 2010, 9, 1097–1105. [Google Scholar] [CrossRef]
- Takechi, M.; Fukushima, W.; Nakano, T.; Inui, M.; Ohfuji, S.; Kase, T.; et al. Nationwide Survey of Pediatric Inpatients with Hand, Foot, and Mouth Disease, Herpangina, and Associated Complications during an Epidemic Period in Japan: Estimated Number of Hospitalized Patients and Factors Associated With Severe Cases. J. Epidemiol. 2019, 29, 354–362. [Google Scholar] [CrossRef]
- Nagai, T.; Hanaoka, N.; Katano, H.; Konagaya, M.; Tanaka-Taya, K.; Shimizu, H.; et al. A Fatal Case of Acute Encephalopathy in a Child due to Coxsackievirus A2 Infection: A Case Report. BMC Infect. Dis. 2021, 21, 1167. [Google Scholar] [CrossRef]
- Ishii, M.; Hoshina, T.; Fujimoto, T.; Hanaoka, N.; Konagaya, M.; Shimbashi, R.; et al. A Pediatric Case of Encephalopathy with Hypoglycemia Induced by Coxsackievirus A4 Infection. Pediatr. Infect. Dis. J. 2024, 43, e124–e126. [Google Scholar] [CrossRef]
- Olive, D.M.; Al-Mufti, S.; Al-Mulla, W.; Khan, M.A.; Pasca, A.; Stanway, G.; Al-Nakib, W. Detection and Differentiation of Picornaviruses in Clinical Samples Following Genomic Amplification. J. Gen. Virol. 1990, 71, 2141–2147. [Google Scholar] [CrossRef]
- Ishiko, H.; Shimada, Y.; Yonaha, M.; Hashimoto, O.; Hayashi, A.; Sakae, K.; Takeda, N. Molecular Diagnosis of Human Enteroviruses by Phylogeny-Based Classification Using the VP4 Sequence. J. Infect. Dis. 2002, 185, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Yoshida, S.; Munemura, T.; Taniguchi, K.; Shinohara, M.; Nishio, O.; et al. Detection and Quantification of Enterovirus 71 Genome from Cerebrospinal Fluid of an Encephalitis Patient by PCR. Jpn. J. Infect. Dis. 2008, 61, 497–499. Available online: https://www.jstage.jst.go.jp/article/yoken/61/6/61_JJID.2008.497/_article. [CrossRef] [PubMed]
- Kitamura, K.; Arita, M. Evaluation of VP4-VP2 Sequencing for Molecular Typing of Human Enteroviruses. PLoS ONE 2024, 19, e0311806. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Infectious Diseases (NIID). Hand, Foot, and Mouth Disease Pathogen Detection Manual; NIID: Tokyo, Japan, 2023; Available online: https://id-info.jihs.go.jp/manuals/pathogen-detection/HFMdis20230704.pdf (accessed on 13 March 2026).
- Rotbart, H.A. Enzymatic RNA Amplification of the Enteroviruses. J. Clin. Microbiol. 1990, 28, 438–442. [Google Scholar] [CrossRef]
- Nix, W.A.; Oberste, M.S.; Pallansch, M.A. Sensitive, Seminested PCR Amplification of VP1 Sequences for Direct Identification of All Enterovirus Serotypes from Original Clinical Specimens. J. Clin. Microbiol. 2006, 44, 2698–2704. [Google Scholar] [CrossRef]
- Wolffs, P.F.; Bruggeman, C.A.; van Well, G.T.; van Loo, I.H. Replacing Traditional Diagnostics of Fecal Viral Pathogens by a Comprehensive Panel of Real-Time PCRs. J. Clin. Microbiol. 2011, 49, 1926–1931. [Google Scholar] [CrossRef]
- Bustin, S.A.; Ruijter, J.M.; van den Hoff, M.J.B.; Kubista, M.; Pfaffl, M.W.; Shipley, G.L.; et al. MIQE 2.0: Revision of the Minimum Information for Publication of Quantitative Real-Time PCR Experiments Guidelines. Clin. Chem. 2025, 71, 634–651. [Google Scholar] [CrossRef]
- Johnson, A.D. An Extended IUPAC Nomenclature Code for Polymorphic Nucleic Acids. Bioinformatics 2010, 26, 1386–1389. [Google Scholar] [CrossRef]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.; Schutten, M. The Effect of Primer-Template Mismatches on Nucleic Acid Detection Using the 5′ Nuclease Assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef]
- Rejali, N.A.; Moric, E.; Wittwer, C.T. The Effect of Single Mismatches on Primer Extension. Clin. Chem. 2018, 64, 801–809. [Google Scholar] [CrossRef]
- Thoelen, I.; Moes, E.; Lemey, P.; Mostmans, S.; Wollants, E.; Lindberg, A.M.; et al. Serotype and Genotype Correlation of VP1 and the 5′ NCR among Enterovirus B Species. J. Clin. Microbiol. 2004, 42, 963–971. [Google Scholar] [CrossRef]
- Fujimoto, T.; Chikahira, M.; Yoshida, S.; et al. Outbreak of Central Nervous System Disease Associated with Hand, Foot, and Mouth Disease in Japan during the Summer of 2000. Microbiol Immunol. 2002, 46, 621–627. [Google Scholar] [CrossRef]
- De Schrijver, S.; Vanhulle, E.; Ingenbleek, A.; Alexakis, L.; Johannesen, C.K.; Broberg, E.K.; et al. Epidemiological and Clinical Insights into Enterovirus Circulation in Europe, 2018–2023: A Multicenter Retrospective Surveillance Study. J. Infect. Dis. 2025, 232, e104–e115. [Google Scholar] [CrossRef]
- Whitehouse, E.R.; Lopez, A.; English, R.; Getachew, H.; Ng, T.F.F.; Emery, B.; et al. Surveillance for Acute Flaccid Myelitis, United States, 2018–2022. MMWR Morb. Mortal. Wkly. Rep. 2024, 73, 70–76. [Google Scholar] [CrossRef]
- Chong, P.F.; Kira, R.; Mori, H.; Okumura, A.; Torisu, H.; Yasumoto, S.; et al. Clinical Features of Acute Flaccid Myelitis Temporally Associated with an Enterovirus D68 Outbreak in Japan. Clin. Infect. Dis. 2018, 66, 653–664. [Google Scholar] [CrossRef]
Figure 1.
Schematic overview of the C3R-based VP4–VP2 RT-PCR workflow and primer/probe positions across the enterovirus genome.
Figure 1.
Schematic overview of the C3R-based VP4–VP2 RT-PCR workflow and primer/probe positions across the enterovirus genome.

Figure 2.
Enhanced VP4–VP2 single-round RT-PCR sensitivity using the C3R reverse primer compared with the conventional OL68-1 primer.
Figure 2.
Enhanced VP4–VP2 single-round RT-PCR sensitivity using the C3R reverse primer compared with the conventional OL68-1 primer.

Figure 3.
Detection rate by Ct band.


Table 1.
Primers used in this study.
| Primer/Probe name | Polarity | Sequence (5′→3′) | Position (V01149 PV1, Mahoney) | Primer length (bp) | Reference |
| EVP2 | + | CCTCCGGCCCCTGAATGCGGCTAAT | 444-468 | 25 | [16] |
| EVP4 | + | CTACTTTGGGTGTCCGTGTT | 541-560 | 20 | [12] |
| OL68-1 | − | GGTAAYTTCCACCACCANCC | 1178-1197 | 20 | [11] |
| C3R | - | TCNGGRAAYTTCCAVYACCA | 1181-1200 | 20 | This study |
| Primer F | + | CCCTGAATGCGGCTAATCC | 452-470 | 19 | [18] |
| Primer R1 | - | ATTGTCACCATAAGCAGCCA | 577-596 | 20 | [18] |
| Probe P(FAM) | + | AACCGACTACTTTGGGTGTCCGTGTTTC | 535-562 | 28 | [18] |
| SO224 | + | GCIATGYTIGGIACICAYRT | 1966-1977 | 20 | [17] |
| AN89 | + | CCAGCACTGACAGCAGYNGARAYNGG | 2603-2628 | 26 | [17] |
| SO222 | - | CICCIGGIGGIAYRWACAT | 2951-2969 | 19 | [17] |
| AN88 | - | TACTGGACCACCTGGNGGNAYRWACAT | 2951-2969 | 27 | [17] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



