Submitted:
16 March 2026
Posted:
17 March 2026
You are already at the latest version
Abstract
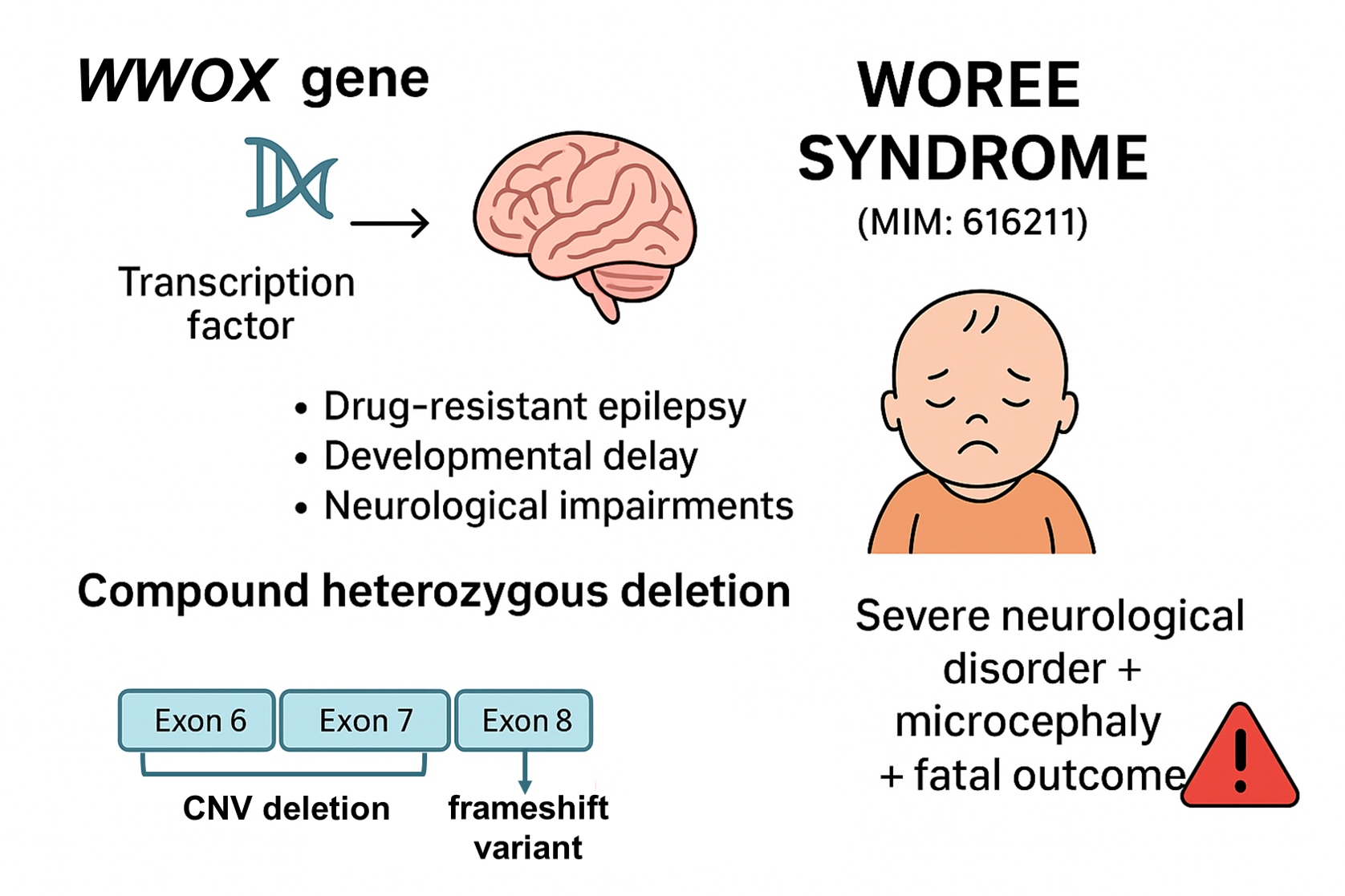
The WWOX gene, well-known as tumor suppressor, has also a crucial role as transcrip-tion factor in the developing brain. The bi-allelic loss of WWOX gene causes a condition characterized by drug-resistant epilepsy, developmental delay, and neurological impairments, often resulting in mortality within the first year of life, known as WOREE syndrome (MIM: 616211). Whole Exome Sequencing (WES) analysis was performed on a female patient who died within three months of birth and was diagnosed with mi-crocephaly, severe early-onset refractory seizures, and drug-resistant epileptic encephalopathy. WES revealed a 38 kb CNV deletion spanning WWOX exons 6-7, and a known frameshift variant in exon 8, impairing a highly clinically significant region of the encoded protein. Clinical and genetic features of reported WOREE patients with WWOX gene deletions similar to our patient were analyzed. Our case highlights the clinical heterogeneity of WWOX variants in WOREE syndrome and suggests that the novel compound heterozygous deletion may contribute to poor prognosis. A "WOREE syndrome plus" phenotype can be defined by severe neurological disorder, microcephaly, and a fatal outcome within the first year of life. Further researches need to elucidate WWOX pathophysiology and improve diagnostic and therapeutic strategies.
Keywords:
autosomal recessive spinocerebellar ataxia 12 (SCAR12
; MIM:614322)
; infantile epileptic encephalopathy 28 (IEE28
; MIM:616211)
; WW domain-containing oxidoreductase (WWOX) gene (MIM: 605131)
; WWOX-related epileptic encephalopathy (WOREE) syndrome
; Whole Exome Sequencing (WES)
1. Introduction
The WW domain-containing oxidoreductase (WWOX) gene (MIM: 605131) is situated within the fragile site FRA16D on chromosome 16q23.3-q24.1, one of the common hot spots of genomic instability for germline and somatic copy number variations (CNVs) [1,2]. Germline mutations of WWOX are rare and primarily associated with severe developmental disorders, while somatic mutations causing the cancer predisposition [2]. Widely known as tumor suppressor, the WWOX gene encodes a 414-amino acid enzyme belonging to the short-chain dehydrogenases/reductases (ADH/SDR), which in the role of transcription factor exerts pleiotropic functions regulating various cellular pathways (e.g., cell proliferation, apoptosis and steroid metabolism) [1]. Even if less studied in the brain, WWOX seems to hold a pivotal position in the central nervous system’s biology, significantly contributing to neuronal development, migration, and proliferation [3]. Normally expressed in neurons, oligodendrocytes, and astrocytes, it serves as a scaffold protein due ist WW domain and interaction ability to counteract various proteins, e.g., cytoskeleton proteins, cellular trafficking and microtubule-based transport [3].
Bi-allelic or pathogenic variants in WWOX are implicated in autosomal recessive conditions, such as spinocerebellar ataxia type 12 (SCAR12; MIM:614322) and infantile epileptic encephalopathy 28 (IEE28; MIM:616211) [2]. These disorders often present with neurological manifestations including epilepsy, developmental delay (DD), and motor impairments [2]. Animal models have provided critical insights into WWOX deficiency, demonstrating phenotypes resembling epilepsy. The WWOX knockout (KO) mouse model mirrors human symptoms such as epilepsy, structural brain malformations, and learning impairments, alongside severe metabolic disorders and premature death within weeks of birth [4]. Currently, 88 disease-causing variants in WWOX are cataloged in the Human Gene Mutation Database (HGMD) database (www.hgmd.cf.ac.uk/), illustrating distinct genotype-phenotype correlations: (i) null genotypes: absent psychomotor development, poor spontaneous motility, absent eye contact from birth, drug-resistant epilepsy starting in the 1st weeks of life, as well as possible retinal degeneration, premature death, and acquired microcephaly; (ii) compound heterozygous genotypes with one null allele and one missense mutation: less severe encephalopathy combining IEE with delayed psychomotor development and increased life expectancy; (iii) genotypes with biallelic hypomorphic missense mutations: SCAR12 [5].
Homozygous and compound heterozygous variants in the WWOX gene have been linked to WWOX-related epileptic encephalopathy (WOREE) syndrome (more details in the official website of ‘WWOX foundation for WWOX-related syndromes’: www.wwox.org/what-is-wwox). WOREE has been characterized by severe early-onset epileptic encephalopathy, resistant seizures to conventional antiseizure medications (ASMs) and diverse brain anomalies including impaired myelination, thickened corpus callosum, white matter abnormalities, and/or cerebral atrophy [6,7], accompanied by DD, psychomotor impairments, spasticity with hyperreflexia and hypokinesia, and an inability to walk or communicate verbally. Microcephaly was also reported [8,9] and most affected of them succumb within the first year of life [2]. Despite advancements, the full spectrum and precise genotype-phenotype correlations of WWOX-related disorders remain areas of ongoing research.
In this study, we present a case report of a female patient died early at a few months of age, affected by microcephaly and severe early-onset refractory seizures, and exhibited a drug-resistant epileptic encephalopathy. A trio-based whole-exome sequencing (WES) identified two mutations in the baby girl, a 38 kb CNV deletion spanning WWOX exons 6–7, and a known frameshift variant c.1043del (p.Phe348Serfs*57) in exon 8 (ClinVar ID: 421407); both absent in her parents.
There appear to be no similar cases in the literature that report the same genotype associated with early death. Our study would shed light on the rare condition of WWOX-associated phenotype, investigating both her distinct clinical and genetic features.
2. Materials and Methods
2.1. Ethical Compliance
The study was conducted ethically in accordance with the World Medical Association Declaration of Helsinki and approved by the Ethics Committee of the University of Catania, Italy (Ethical Committee Catania 1 Clinical Registration n. 210/2023/PO). Written informed consent was obtained from the parents of the patient for publication of the details of their medical case and any accompanying images.
2.2. Genetic Testing and Genomic Data Analysis
The genomic Deoxyribonucleic Acid (DNA) of proband and her parents was extracted from a buccal swab to perform the clinical whole exome sequencing (WES) using the Blueprint Genetics Whole Exome Plus Test (version 3, February 2023). Variant data were annotated using tools like VcfAnno and Variant Effect Predictor (VEP), referencing public databases such as gnomAD SVs v2.1, ExAC, ClinVar, and HGMD. Variant classification followed Blueprint Genetics Variant Classification Schemes modified from the American College of Medical Genetics and Genomics (ACMG) guideline 2015 [10]. WES findings were also analyzed using the database tracks (OMIM Allelic Variants, ClinVar Variants, dbSNP, and Decipher v.21.11) available from UCSC genome browser (genome.ucsc.edu). The study was approved by the NICU ethics committee, and the proband’s parents provided written consent in accordance with the Declaration of Helsinki.
3. Results
3.1. Case Presentation
The patient, a female infant, was admitted to the neonatal intensive care unit (NICU) due to seizures after few hours from birth. She was born after an uneventful pregnancy and spontaneous vaginal delivery at full term, the birth weight was 3.100 g (-0.88 SDS—Standard Deviation Score), the length was 50 cm (-0.2 SDS), and the head circumference was 32 cm (-2.07 SDS), consistent with microcephaly. Apgar score was 9 and 10 after 1 and 5 min, respectively. The familiar medical history was unremarkable. Seizures, characterized by body stiffening, horizontal nystagmus, eyelid myoclonus and epileptic spasms, began shortly after birth and were accompanied by vomiting. There was no significant family history or dysmorphic features noted upon examination. The infant was started on levetiracetam for seizure management. The first electroencephalogram (EEG) was normal, but due to the persistence of seizures, treatment with levetiracetam as an ASM was initiated.
Neurological examination revealed several abnormalities, including lack of gaze fixation with horizontal nystagmus, axial hypotonia with spastic hypertonia of the limbs, reduced general movements, and reduced patellar reflexes. At 8 days of life, she presented multi-daily asymmetric tonic seizures with eye deviation to the left and left arm hyperextension. A second EEG performed during sleep, showed multifocal interictal epileptiform discharges (IED) with a focus on the bilateral centro-temporal areas. The ictal EEG revealed spikes on the right occipital derivations. Based on the electrical pattern and the persistence of critical episodes, carbamazepine was added as an adjunctive ASM. At this stage, the differential diagnosis included early infantile epileptic encephalopathy, neonatal encephalopathy secondary to hypoxic-ischemic encephalopathy, and encephalopathy due to structural brain abnormalities. The EEG pattern strongly indicated early infantile epileptic encephalopathy. Blood tests for metabolic disorders were performed, resulting within normal limits. Brain MRI at 10 days of age was unmarkable. Unfortunately, the infant passed away for cardiac arrest after three months of ages due to pulmonary infections and respiratory failure.
3.2. Mutational Analysis
WES analysis (hg19) identified two heterozygous pathogenic variants of WWOX gene (NM_016373.4) (Figure 1).
The first is a CNV deletion c.(516+1_517-1)_(791+1_792-1)del, which involves exons 6–7 (chr16:78420649-78459043; at least 38 Kb), resulting in the disruption of open reading frame. Coordinates of CNV represent an estimate of the alteration’s genomic position, since we are unable to determine the exact breakpoints using the current next-generation sequencing method. The second is a known frameshift variant c.1043del (p.Phe348Serfs*57) (ClinVar ID: 421407) in exon 8 of WWOX gene (chr16:78,466,634-78,466,634) (Figure 1). In detail, the present CNV loss, also confirmed by digital Polymerase Chain Reaction (PCR), results in an out-of-frame deletion of exons 6–7 of the WWOX gene, potentially leading to the disruption of the open reading frame and, consequently, protein truncation or nonsense-mediated mRNA decay. Deletions encompassing WWOX exons 6–7 have been reported in one individual in the ExAC control cohorts. In addition, overlapping deletions involving exon 6 or 7 or both have been detected in 17 individuals in the ExAC control cohorts and in 4 individuals in the gnomAD control cohorts (gnomAD SVs v2.1, ExAC data available in the gnomAD browser). Furthermore, the same rearrangement has not been found in Decipher database v.21.11. Instead, the variant c.1043del (p.Phe348Serfs*57) (ClinVar ID: 421407) appears to induce a frameshift in the penultimate exon of the gene, resulting in a premature stop codon in the final exon. No additional genetics findings were reported. Analysis of asymptomatic parental samples would have required for the inheritance pattern determination of variants but did not perform.
4. Discussion
WWOX genetic variants exhibit a certain degree of heterogeneity from a phenotypic perspective. The clinical wide spectrum ranges from a mild phenotype, SCAR12, to a severe condition known as WOREE syndrome, depending on the specific mutation and its impact on WWOX gene expression [7]. Consequently, the diagnosis and the treatment are challenging due to the different phenotypes observed among affected individuals. Diseases caused by WWOX variants is inherited in an autosomal recessive manner. WOREE syndrome typically manifests with early-onset epilepsy, growth impairment, delayed psychomotor development, and progressive microcephaly correlated with a high mortality rate in first years [3].
The first animal model, a spontaneous 13-bp deletion in exon 9 of the WWOX gene in the lde/lde (lethal dwarfism with epilepsy) rat, was reported associated with dwarfism, ataxic gait, a high incidence of epileptic seizures, and postnatal lethality, highlighting the WWOX’s role in developmental neurobiology [11]. This preclinical model showed a post-natal lethal phenotype, with severe neurological disorder, early mortality before the first year of age and possible association with microcephaly. This phenotipic picture could be defined as ‘WOREE syndrome plus’.
The WWOX gene would be linked to neuronal migration and therefore involved in the growth of cranial circumference, hence the microcephaly. However, the pathophysiological mechanism driving the development of WOREE syndrome due to the complete or partial loss of function of WWOX remains incompletely understood. A more comprehensive overview of phenotypic and genetic features of WOREE patients aligned with our case was provided in Figure 2.
According to our results, no cases with similar our CNV deletion on WWOX gene, reported the clinical sign of microcephaly (Figure 2). To note, evidence of microcephaly was associated with other genotypes [9,12]. As highlighted in Figure 2, Piard et al. reports another patient who experienced early death at 6 months of age, similar to our patient (who passed away at 3 months), with epileptic encephalopathy onset at 2 days of life in both cases [7]. In addition, there are other studies describing a clinical picture characterized by very early infantile-onset epileptic encephalopathy. Mallaret et al. reported a homozygous missense variant c.139C>A, p.(Pro47Thr), within the WW domain, in four siblings with childhood-onset cerebellar ataxia, generalized tonic-clonic epilepsy, and intellectual disability [4]. They also identified a homozygous missense variant c.1114G>C, p.Gly372Arg, within the C-terminal part of WWOX’s dehydrogenase/reductase domain, in another family with two affected siblings exhibiting generalized tonic-clonic epilepsy, intellectual disability, and ataxia. Moreover, early death was rarely reported [7]. Abdel-Salam et al. identified, through exome sequencing, a homozygous nonsense pathogenic variant c.160G>T, p.Arg54* in a girl from a consanguineous family with severe growth retardation, microcephaly, epileptic seizures, retinopathy, and early death [8], as in our patient. Additionally, Mignot et al. identified biallelic pathogenic variants in patients with infantile epileptic encephalopathy, including deletions, truncating and missense variants, and delineated the phenotypic spectrum of WWOX variants [13].
Regarding the patient genotype, we detected a double pathogenic alteration of WWOX gene, most likely associated with inauspicious prognosis, the rapidity of progression until the exitus after a few months of life, incompatible with the postnatal life. WES data infact, showed that both pathogenic variants, the CNV and the frameshift, disrupt a clinically significant region of the protein regarding the C-terminus of the ADH/SDR and the mitochondrial binding region translated by exon 6-8 of the gene.
The frameshift variant has been already reported in trans with exon 7–8 out-of-frame deletion or in the homozygous state in at least two individuals with WWOX-related developmental and epileptic encephalopathy [9,14]. Instead, establishing a causative and contributory role of the CNV to the fatal for the child’s survival is more complex. Similar rearrangements to our CNV have been documented in the ExAC (17 individuals) and gnomAD SVs v2.1 (4 individuals) control cohort databases. However, WWOX exons 6–7 deletion has been previously described in trans with a WWOX missense variant in at least 3 individuals with WOREE syndrome [7]. In addition, out-of-frame deletion of exon 6 or in-frame deletion of exon 7 have been found in several individuals with WWOX-related phenotypes [7,12,15]. Currently, there are no available data or indications correlating the protein damage caused by the frameshift mutation in exon 8 with the CNV deletion in exons 6–7. In the absence of this information, the functional loss in ADH/SHR domains and mitochondria targeting sequence of the protein is to considered to be plausibly deleterious. Our patient exhibited microcephaly, which it has already been reported in the literature to be associated with deletions in exons 2, 6, and 7 [8,9], as well as with the very early onset of epileptic encephalopathy in the first days of life. However, the progressive clinical deterioration of our patient, leading to death within the first 3 months of life, demonstrates the “catastrophic” impact that the specific WWOX genotype might have on lifelong prognosis.
5. Conclusions
To date, only one other patient has been described with a genotype characterized by compound heterozygous deletion of the WWOX gene in WOREE syndrome [16]. Both this case and our case involve newborns who experienced seizures shortly after birth with refractoriness to ASMs. In the first case, MRI images showed hyperintensity of white matter and delayed myelination, while in our case, MRI findings were unremarkable. Literature reports other MRI anomalies such as hippocampal hypoplasia, corpus callosum abnormalities, or even enlargement of subarachnoid spaces and asymmetry of lateral ventricles. These alterations suggest a role of the WWOX gene in brain homeostasis and developmental processes. Early mortality was a common outcome in both cases, emphasizing the low life expectancy in these individuals.
Author Contributions
Conceptualization, R.F., A.S. and X.G.P.; methodology, A.S., S.M. and R. Rocca; software, R.Rizzo and G.F.; validation, R.F., M.R. and X.G.P.; formal analysis, V.S. and S.M.; investigation, R.Rizzo and G.F.; resources, R.Rizzo; data curation, R.Rocca, R.Rizzo and G.F.; writing—original draft preparation, A.S., X.G.P., R.F., R.Rocca and R.Rizzo; writing—review and editing, A.S., S.M. and X.G.P.; visualization, S.M., V.S. and G.F.; supervision, M.R.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of the University of Catania, Italy (Ethical Committee Catania 1 Clinical Registration n. 210/2023/PO).
Informed Consent Statement
Written informed consent was obtained from the parents of the patient for publication of the details of their medical case and any accompanying images.
Data Availability Statement
The data that support the findings of this study are not publicly available due to reasons of sensitivity, but are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ADH/SDR | Short-chain Dehydrogenases/Reductases |
| ACMG | American College of Medical Genetics and Genomics |
| ASMs | Anti-Seizure Medications |
| bp | Base pairs |
| CNVs | Copy Number Variations |
| DD | Developmental Delay |
| DNA | Deoxyribonucleic Acid |
| EEG | Electroencephalogram |
| ExAC | Exome Aggregation Consortium |
| HGMD | Human Gene Mutation Database |
| IEE | Infantile Epileptic Encephalopathy |
| IEE28 | Infantile Epileptic Encephalopathy 28 |
| IED | Interictal Epileptiform Discharges |
| KO | Knockout |
| MIM | Mendelian Inheritance in Man |
| MRI | Magnetic Resonance Imaging |
| NICU | Neonatal Intensive Care Unit |
| PCR | Polymerase Chain Reaction |
| SCAR12 | Autosomal Recessive Spinocerebellar Ataxia |
| SDS | Standard Deviation Score |
| VEP | Variant Effect Predictor |
| WES | Whole Exome Sequencing |
| WOREE | WWOX-Related Epileptic Encephalopathy |
| WES | Whole Exome Sequencing |
References
- Hussain, T.; Liu, B.; Shrock, M.S.; Williams, T.; Aldaz, C.M. WWOX, the FRA16D gene: A target of and a contributor to genomic instability. Genes Chromosomes Cancer 2019, 58, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Banne, E.; Abudiab, B.; Abu-Swai, S.; et al. Neurological Disorders Associated with WWOX Germline Mutations—A Comprehensive Overview. Cells 2021, 10, 824. [Google Scholar] [CrossRef]
- Aldaz, C.M.; Hussain, T. WWOX Loss of Function in Neurodevelopmental and Neurodegenerative Disorders. Int. J. Mol. Sci. 2020, 21, 8922. [Google Scholar] [CrossRef] [PubMed]
- Mallaret, M.; Synofzik, M.; Lee, J.; et al. The tumour suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Brain 2014, 137, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; et al. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, L.; Scorrano, G.; Spiaggia, R.; et al. Neuroimaging features of WOREE syndrome: A mini-review of the literature. Front. Pediatr. 2023, 11, 1301166. [Google Scholar] [CrossRef] [PubMed]
- Piard, J.; Hawkes, L.; Milh, M.; et al. The phenotypic spectrum of WWOX-related disorders: 20 additional cases of WOREE syndrome and review of the literature. Genet. Med. 2019, 21, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, G.; Thoenes, M.; Afifi, H.H.; Körber, F.; Swan, D.; Bolz, H.J. The supposed tumor suppressor gene WWOX is mutated in an early lethal microcephaly syndrome with epilepsy, growth retardation and retinal degeneration. Orphanet J. Rare Dis. 2014, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.L.; Trivisano, M.; Mandelstam, S.A.; et al. WWOX developmental and epileptic encephalopathy: Understanding the epileptology and the mortality risk. Epilepsia 2023, 64, 1351–1367. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Y.; Chou, Y.T.; Lai, F.J.; et al. WWOX deficiency leads to neurodevelopmental and degenerative neuropathies and glycogen synthase kinase 3β-mediated epileptic seizure activity in mice. Acta Neuropathol. Commun. 2020, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.C.; Cao, Y.; Fung, E.L.W.; et al. Expansion of the clinical and molecular spectrum of WWOX-related epileptic encephalopathy. Am. J. Med. Genet. A 2023, 191, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Mignot, C.; Lambert, L.; Pasquier, L.; et al. WWOX-related encephalopathies: Delineation of the phenotypical spectrum and emerging genotype-phenotype correlation. J. Med. Genet. 2015, 52, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.S.; Wen, X.J.; Zhang, S.; Wang, D.G.; Xiong, Y.; Li, Z.M. Identification of compound heterozygous deletion of the WWOX gene in WOREE syndrome. BMC Med. Genom. 2023, 16, 291. [Google Scholar] [CrossRef] [PubMed]
- Vetri, L.; Calì, F.; Saccone, S.; et al. Whole Exome Sequencing as a First-Line Molecular Genetic Test in Developmental and Epileptic Encephalopathies. Int. J. Mol. Sci. 2024, 25, 1146. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
UCSC Genome Browser illustration of pathogenic variants of WWOX (NM_016373.4) found in the proband. UCSC genome browser (genome.ucsc.edu) shows the 38 kb CNV deletion (chr16:78420649-78459043; hg19) shaded in blue, while in yellow the frameshift variant c.1043del (p.Phe348Serfs*57) (ClinVar ID:). Only for illustrative purpose, the two variants are represented within the same gene sequence, without indicating the location on specific allele and their cis/trans effects. Coding exons (ex) are represented by bars and introns with horizontal lines.
Figure 1.
UCSC Genome Browser illustration of pathogenic variants of WWOX (NM_016373.4) found in the proband. UCSC genome browser (genome.ucsc.edu) shows the 38 kb CNV deletion (chr16:78420649-78459043; hg19) shaded in blue, while in yellow the frameshift variant c.1043del (p.Phe348Serfs*57) (ClinVar ID:). Only for illustrative purpose, the two variants are represented within the same gene sequence, without indicating the location on specific allele and their cis/trans effects. Coding exons (ex) are represented by bars and introns with horizontal lines.

Figure 2.
A focused summary of main clinical and genetic features of the our proband and WOREE patients with 6–7 exon containing CNV variants.
Figure 2.
A focused summary of main clinical and genetic features of the our proband and WOREE patients with 6–7 exon containing CNV variants.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



