Submitted:
26 February 2026
Posted:
02 March 2026
You are already at the latest version
Abstract
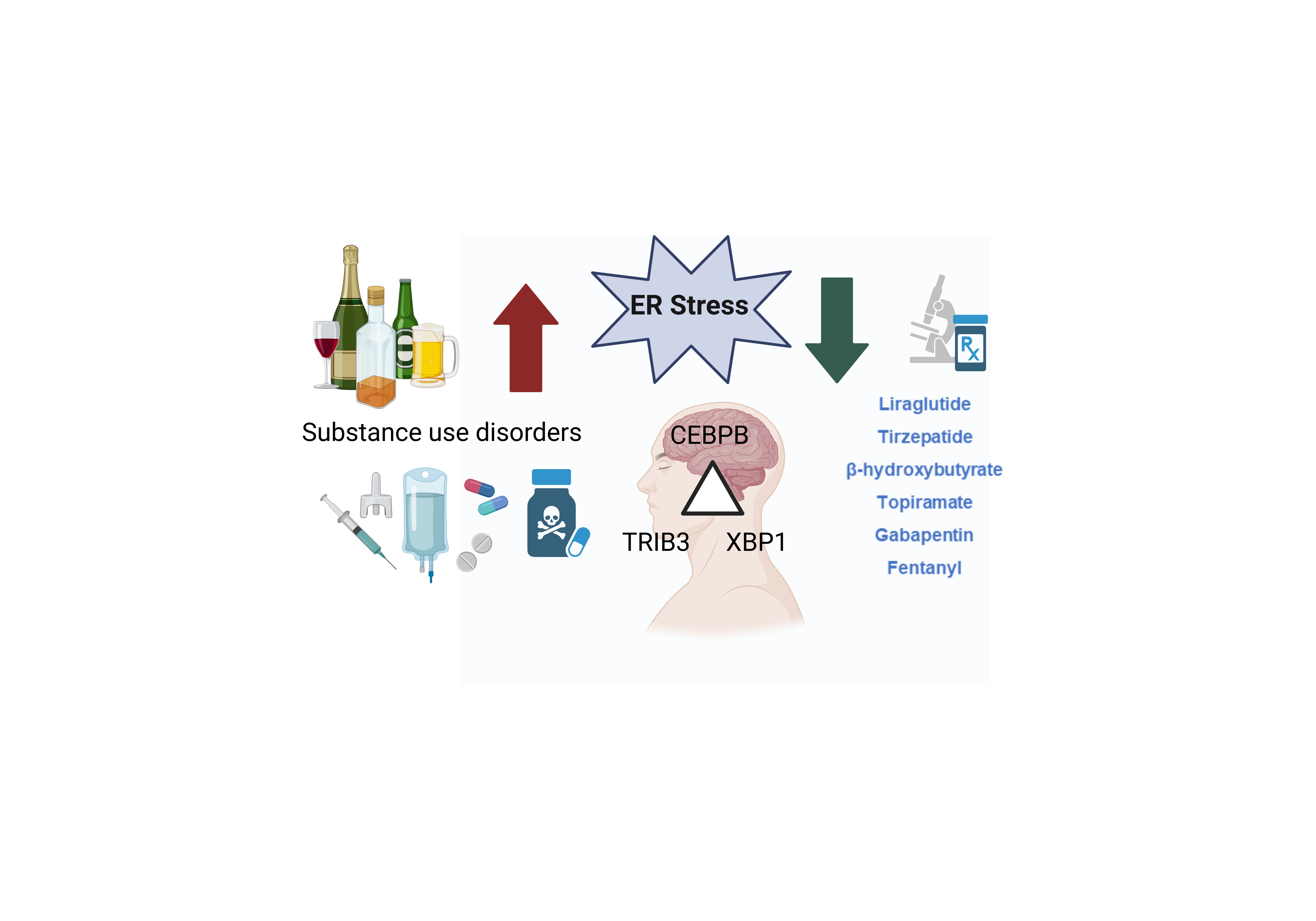
Opioid use disorder is a national crisis in the United States, with 3 US FDA-approved pharmacotherapies available and rapidly rising overdose deaths driven by synthetic opioids, i.e. fentanyl. Recent population-level evidence suggests that GLP-1 receptor agonists (GLP-1RA) may reduce the risk of opioid overdose, yet underlying mechanisms remain unclear. This study investigated molecular mechanisms of fentanyl and GLP-1RA. We performed RNA-seq in human iPSC-derived forebrain organoids treated with fentanyl, liraglutide, or exenatide. We then extended this analysis to iPSC-derived forebrain neurons exposed to additional therapeutic candidates: anticonvulsants (topiramate, gabapentin) and a metabolic modulator (β-hydroxybutyrate). We performed RNA-seq and functional genomic assays using iPSC-derived cell models. All drugs were tested at clinically relevant concentrations. Our results showed modulation of endoplastic reticulum (ER) stress signaling as a shared molecular mechanism across fentanyl, GLP-1RA, and other drug classes with therapeutic potential for substance use disorders (SUDs). Fentanyl, liraglutide, and exenatide consistently down-regulated ER stress–related genes, with TRIB3 emerging as the most strongly suppressed target in brain organoids. Additional stress-response genes, including DDIT3, ATF4, and PPP1R15A, were similarly reduced, indicating broad attenuation of ER stress pathways. We further identified CEBPB as a key upstream driver of these transcriptional changes. Finally, we confirmed that diverse compounds, including anticonvulsants and metabolic modulators, suppressed ER stress genes and reduced CEBPB DNA-binding activity in neurons. In summary, these findings reveal ER stress modulation, mediated in part through CEBPB, as a convergent mechanism across multiple drug classes and highlight potential relevance to therapeutic action in SUD.
Keywords:
ER stress
; opioid addiction
; pharmacotherapy
; iPSC
; brain organoids
1. Introduction
In recent years, synthetic opioid-involved overdose deaths have increased sharply [1]. Over 70,000 deaths due to drug overdose occurred in the US in 2019, and more than 50% involved synthetic opioids [1]. The number of opioid prescription medications, like oxycodone, and fentanyl, continues to be a considerable public health concern [2]. The US Center for Disease Control and Prevention (CDC) indicates that the US is currently in a “third wave” of opioid overdose deaths driven primarily by synthetic opioids like fentanyl [3,4,5]. The age-adjusted synthetic opioid death rate increased sharply by 1040% from 2013 to 2019 [1]. There are currently only three medications approved by the US Food and Drug Administration (FDA) for opioid use disorder (OUD): methadone, a full agonist of the µ opioid receptor; buprenorphine, a high-affinity partial agonist of the μ and κ opioid receptors; and naltrexone, a μ-opioid receptor antagonist [6]. Notably, the medications are underused. Only 1 in 5 US adults with OUD received medications to treat it in 2021 [7]. In contrast, type 2 diabetes (T2D), which is a prevalent medical condition worldwide, currently has over 37 FDA-approved medications that comprise 10 drug classes available for treatment [8]. The overwhelming scale and cost of the opioid epidemic, along with the fact that there are so few medications for OUD, have increased the urgency to discover new therapeutic options.
GLP-1 receptor agonists (GLP-1RA) such as semaglutide have been linked to a reduced risk of opioid overdose among patients with comorbid T2D and OUD [9]. Consistent with this finding, a recent study reported that individuals with OUD who received glucose-dependent insulinotropic polypeptide (GIP)/GLP-1 receptor agonist prescriptions experienced lower rates of opioid overdose compared with those who did not have these prescriptions [10]. Preclinical evidence further supports this potential relationship: a recent study demonstrated that exendin-4 can cross the blood-brain barrier and attenuate opioid reinforcement and drug-seeking behavior without impairing opioid-induced analgesia in rats being treated with oxycodone [11]. Collectively, this body of work highlights the potential therapeutic value of GLP-1 receptor agonists in OUD. Despite these observations, the mechanisms underlying this protective association remain unclear, prompting us to investigate the molecular signatures of fentanyl and GLP-1 receptor agonists in induced pluripotent stem cell (iPSC)-derived brain-like cells.
A central challenge in neuropsychopharmacology is the limited access to living human brain tissue, the primary site of pathology, limiting our ability to explore mechanisms and identify therapeutic targets. Patient-derived iPSC models help overcome this barrier by providing cells that retain individual’s unique genetic background by enabling the generation of human brain-like tissue. This system also allows controlled experimental manipulation of living, brain-relevant cells. Gaining insight into how opioids and GLP-1RAs function at cellular and molecular levels may reveal new therapeutic targets for OUD.
The present study was designed to investigate the molecular mechanisms of fentanyl and GLP-1 receptor agonists in iPSC-derived brain organoids. Notably, we found that the most significant differentially expressed gene across all treatment conditions was Tribbles pseudokinase 3 (TRIB3), a gene that is highly expressed in neurons and associated with endoplasmic reticulum (ER) stress. We then conducted functional genomics studies using iPSC-derived forebrain neurons and discovered that CCAAT/enhancer-binding protein beta (CEBPB) functions as a transcription factor that could alter the ER stress signaling pathway. Interestingly, in addition to GLP-1 receptor agonists, several medications with potential therapeutic benefits for substance use disorders (SUDs) were found to suppress the ER stress pathway via similar mechanisms.
2. Materials and Methods
2.1. Study Subjects and Ethics Statements
This study was conducted in accordance with the protocol #20-00372, which was reviewed and approved by the Mayo Clinic Institutional Review Board. Participant confidentiality was strictly maintained throughout the study. Written consent was obtained from all individuals whose samples were used, including consent for study participation and publication of the results in a peer-reviewed scientific journal.
2.2. Generation of iPSCs, iPSC-Derived Forebrain Organoids and Neurons
We generated three iPSC lines for brain organoid differentiation (2 male and 1 female participants) using peripheral blood mononuclear cells (PBMCs) and the CytoTune™-iPS 2.0 Sendai Reprogramming Kit (A16517, Thermo Fisher, USA) as described previously [12,13]. All iPS cell lines showed normal karyotypes, and they all expressed pluripotency markers (Figure 1). Cell lines were regularly characterized and verified to be free from mycoplasma. We then generated iPSC-derived forebrain organoids and neurons as reported previously [12,14].
3-D iPSC-derived forebrain organoids were generated from 3 individuals. Briefly, iPS cells were cultured on Matrigel with mTeSR1 Plus media (STEMCELL technology, MA, USA). Embryoid bodies (EBs) were embedded in Matrigel and cultured with 1x N2, 1x NEAA and 1x Glutamax (Invitrogen, Grand Island, NY), 1 μM SB431542, and 1 μM CHIR99021 (Selleckchem) for a week. On day 14, organoids were cultured in a bioreactor [15]. Culture medium from days 14-70 consisted of DMEM/F12 medium supplemented with 1x N2, 1x B27, 1x NEAA and 1x Glutamax, 1x 2-metabptoethanol, 100x penicillin-streptomycin solution, and 2.5 μg/ml insulin (Sigma-Aldrich). The medium was changed every other day. From day 70 onward, Brainphy media supplemented with 20 ng/ml BDNF, 20 ng/ml GDNF (Peprotech), 0.2 mM L-ascorbic acid, and 0.5 mM cAMP (Sigma-Aldrich) was used [16,17]. Drug treatment for iPSC-derived forebrain organoids was initiated from day 83 to day 90 [14].
In addition, we generated iPSC-derived forebrain neurons for follow-up experiments. Neural progenitor cells were produced using the STEMdiff™ SMADi Neural Induction Kit (catalog #08581). These progenitors were then differentiated into a mixed population of forebrain-type (FOXG1-positive) neurons using the STEMdiff™ Forebrain Neuron Differentiation Kit in combination with the STEMdiff™ Forebrain Neuron Maturation Kit (catalog #08600).) Maturation medium was replaced every three days during continuous culture [18]. The iPSC-derived neurons were functionally mature by four weeks after differentiation from NPCs. These neurons formed synapses, fire action potentials, and had spontaneous synaptic activity [17,18]. Drug treatment for iPSC-derived forebrain neurons was initiated four weeks after differentiation.
2.3. Drug Treatment
Concentrations of fentanyl (3 nM) [19], liraglutide (50 nM) [20] and exenatide (150 pM) [21] were chosen to reflect blood drug levels observed in clinical studies. We treated iPSC-derived forebrain organoids with vehicle, fentanyl, liraglutide, or exenatide for seven days, with a daily medium change. Cells were collected for RNA extraction 24 hours after the final treatment. A similar experiment was conducted in iPSC-derived neurons that were treated with fentanyl (3 nM) [19], liraglutide (50 nM) [20], exenatide (150 pM), tirzepatide (100 nM) [22], topiramate (5 µM) [23], gabapentin (22 µM) [24], β-hydroxybutyrate (5 mM) [25] or thapsigargin (100 nM).
2.4. Immunostaining and Confocal Microscopy
Cells were fixed with 4% paraformaldehyde at room temperature for 15 minutes, followed by permeabilization using 0.2% Triton X-100 in PBS. Brain organoids were sectioned at a thickness of 10 μm. After blocking with 5% donkey serum in PBS for 30 minutes at room temperature, cells were incubated overnight with the primary antibody (see Supplementary Table S1) diluted in 3% BSA. Following washes, secondary antibodies were applied for 1 hour at room temperature. Nuclei were counterstained using antifade mounting medium containing DAPI (VECTOR Laboratories, Burlingame, CA, USA). Fluorescence images were acquired using an Olympus FV1200 confocal microscope.
2.5. RNA Sequencing (RNA-seq) and Data Analysis
Total RNA from iPSC-derived brain organoids and neurons was extracted using Trizol and the RNeasy mini kit (Qiagen, Valencia, CA, USA). The RNA integrity numbers (RIN) were between 9.6 and 9.9 for all RNA samples. RNA-seq in iPSC-derived forebrain organoids included two technical replicates for sequencing (n=4). RNA-seq experiments were conducted by Azenta using an Illumina HiSeq 6000 with eight samples in each lane using 100bp paired end index reads. Fastq files containing paired RNA-Seq reads were aligned with STAR [26] against the UCSC human reference genome (GRCh38.p14). Approximately 30 million reads were generated per sample, achieving an average mapping rate of ~90%. Duplication rates ranged from 25–35%, consistent with high library complexity and minimal technical (PCR) bias; low duplication rates are generally indicative of high-quality RNA-seq data. RNA-seq differential expression analysis was performed using the DESeq2 package with default parameters [27]. The Wald test was used to compare the beta estimate divided by its estimated standard error to a standard normal distribution to derive p-values. P values were then adjusted for multiple testing using the Benjamini and Hochberg method to produce false discovery rate (FDR) values. An FDR ≤0.05 was considered statistically significant. Gene Set Enrichment Analysis (GSEA) software [28,29], and the Database for Annotation, Visualization and Integrated Discovery (DAVID) [30,31] were used to perform pathway enrichment analysis. ChIP-X Enrichment Analysis 3 (ChEA3) was used to identify upstream transcription regulatory targets of differentially expressed genes [32].
2.6. Co-Immunoprecipitation (co-IP) and Western Blot Analysis
Co-immunoprecipitation was performed using iPSC-derived forebrain organoids. Protein samples were collected using 500 µL of IP lysis buffer supplemented with 5 µL of protease inhibitor cocktail and incubated on ice for 30 minutes. Immunoprecipitation was carried out using anti-CEBPB (abcam, catalog #: ab32358, USA), or control IgG (Cell Signaling Technology, Danvers, MA, USA). Magnetic protein G beads (ThermoFisher, Dynabeads® Protein G, catalog number: 10007D, USA) were added to the samples, which were rotated for 2 hours at 4°C. Immunoprecipitates were washed three times with ice-cold lysis buffer. CEBPB was immunoprecipitated using a CEBPB-specific antibody, with IgG serving as a negative control to assess non-specific binding. Approximately 1% of the input (~25 µg total protein) was loaded. The immunoprecipitated products were separated by SDS-PAGE gel and immunoblotted for XBP-1s, TRIB3, and CEBPB. A light-chain–specific secondary antibody was used to avoid interference from antibody heavy and light chains. Bound proteins were eluted using 50 µL of 4X Laemmli loading buffer and boiled at 95°C for 10 minutes. Proteins were then resolved on 4-20% SDS-PAGE gels and transferred to polyvinylidene fluoride membranes. The blocking buffer consisted of 5% skim milk in PBS. Membranes were incubated in blocking buffer for 1 hour at room temperature with gentle agitation. After blocking, membranes were incubated overnight at 4°C with primary antibodies against CEBPB, TRIB3 or X-box binding protein 1 (XBP1-s). Membranes were washed with TBST for 5 min on the agitator five times. Membranes were then incubated with Rabbit TrueBlot® ULTRA: Anti-Rabbit IgG HRP - 18-8816-31 for one hour at room temperature. Signal detection was performed using Clarity Western ECL Blotting Substrate (Bio-Rad, USA) and visualized with the GelDoc Go imaging system (Bio-Rad, USA). A complete list of antibodies used for co-IP and Western blotting is provided in Supplementary Table S1.
2.7. CEBPB Chromatin Immunoprecipitation (ChIP) Assay
ChIP assays were conducted on iPSC-derived forebrain neurons using the Pierce™ Magnetic ChIP Kit (Thermo Fisher Scientific, catalog #26157). DNA-CEBPB complexes were immunoprecipitated with anti-CEBPB antibodies, while normal rabbit IgG served as a negative control. Following purification, the recovered DNA was analyzed by qPCR using the primer sets listed in Supplementary Table S1. ChIP enrichment was calculated as the percentage of ChIP DNA relative to input DNA (% input). To correct for variability in chromatin preparation, Ct values from each IP fraction were normalized to the corresponding input Ct values. Data are presented as percent input.
2.8. Statistical Analysis
Sequencing analyses were performed using R Statistical Software (version 4.5.0; R Foundation for Statistical Computing, Vienna, Austria). Figure 5 was analyzed using one-way ANOVA (GraphPad Prism 10). P < 0.05 was considered statistically significant.
2.9. Data Availability
All data supporting our findings can be found on the main paper or in supplementary files. Sequencing data are available via the GEO database: GSE315126, and GSE315127.
3. Results
3.1. Transcriptomic Profiles in iPSC-Derived Forebrain Organoids
We generated iPSC-derived forebrain organoids from three individuals and all iPSC lines used in this study revealed normal karyotypes as determined by G-band karyotyping analysis (Figure 1A). Pluripotency markers including SRY-box transcription factor 2 (SOX2) and podocalyxin (TRA-1-81) were positive for all iPSC lines (Figure 1B). We then cultured iPSC-derived forebrain organoids in the bioreactors (Figure 1A). Brain organoids were treated with fentanyl, liraglutide, and exenatide for seven days (Figure 1C-D). RNA-seq data demonstrated that the most affected gene by fentanyl is TRIB3 (Figure 2A), which is a putative protein kinase that is induced by NF-κB and associated with type 2 diabetes [33,34]. TRIB3 is a regulator of the integrated stress response, and modulated DDIT3-depedent cell death during ER stress in neuronal cells [35,36]. However, previous studies suggested that it did not affect non-neuronal cells [35,36]. Notably, TRIB3 was also significantly down-regulated by both liraglutide and exenatide (Figure 2A). We subsequently explored genes involved in ER stress response, including DNA damage inducible transcript 3 (DDIT3), activating transcription factor 4 (ATF4), Protein phosphatase 1 regulatory subunit 15A (PPP1R15A) also known as growth arrest and DNA damage-inducible protein (GADD34), and hexokinase domain containing 1 (HKDC1), all of which were down-regulated in response to fentanyl, liraglutide, and exenatide (Figure 2A, and Supplementary Table S2). The ER stress pathway was down-regulated in iPSC-derived forebrain organoids treated with fentanyl or GLP-1 receptor agonists (Supplementary Tables S3–S5). These findings indicate that ER stress modulation may represent a common mechanism linking fentanyl and GLP-1 receptor agonists. Therefore, we set out to determine the underlying mechanisms.
3.1.1. CEBPB as Upstream Regulator of ER Stress Genes
We identified CEBPB as the most significant upstream regulator of down-regulated genes across all three treatment conditions (Figure 2B). Based on the STRING Database (Figure 2C) [37], CEBPB can physically interact with TRIB3 and several ER stress-related genes. We confirmed that CEBPB proteins interact with TRIB3 and XBP-1s by performing co-immunoprecipitation (Figure 2C). These findings suggest that ER stress regulation may represent a shared molecular mechanism across fentanyl and GLP-1 receptor agonists. Brain organoids contain various brain cell types, including neuronal and glial cells. As previously noted, TRIB3 regulates DDIT3-dependent cell death in neuronal cells under ER stress [35,36]. We then generated iPSC-derived forebrain neurons for follow-up mechanistic experiments.
3.1.2. Transcriptomic Profiles in iPSC-Derived Forebrain Neurons
We hypothesized that modulation of ER stress may represent a common molecular mechanism underlying the therapeutic effects of drugs for SUDs. To test this hypothesis, we treated iPSC-derived forebrain neurons with fentanyl, liraglutide, and exenatide, along with additional compounds, including tirzepatide, topiramate, gabapentin, and beta-hydroxybutyrate, all of which have shown therapeutic potential in the context of SUD. Specifically, tirzepatide is a dual incretin receptor agonist that targets both GLP-1 and GIP receptors, which may have a protective role in both OUD and alcohol use disorder (AUD) [10,38]. Topiramate and gabapentin are anticonvulsants with demonstrated therapeutic potential in treating opioid withdrawal [39,40]. β-hydroxybutyrate, a ketone body, was included due to emerging evidence suggesting that ketogenic diets may benefit some individuals with SUDs, such as OUD and AUD [41,42,43]. Therefore, we generated iPSC-derived forebrain neurons and treated them with fentanyl (3 nM) [19], liraglutide (50 nM) [20], exenatide (150 pM), tirzepatide (100 nM) [22], topiramate (5 µM) [23], gabapentin (22 µM) [24] or β-hydroxybutyrate (5 mM) [25] (Figure 3A–3B). The selected drug concentrations fell within clinically relevant ranges and were informed by prior pharmacokinetic studies. Exenatide was removed from the subsequent analysis due to library preparation failure. Remarkably, several ER stress genes, including XBP1, TRIB3, and DDIT3 were down-regulated (Figure 3B). XBP1 was one of the top hits across all treatment conditions. Normally, unspliced XBP1 mRNA is cleaved by the activated stress sensor IRE1α to generate spliced XBP1, a crucial transcription factor modulating ER stress response. Pathway enrichment analyses further demonstrated that down-regulated genes were most strongly enriched for ER stress-related pathways, including the unfolded protein response and IRE1α-mediated chaperone activation (Supplementary Figure S1). In contrast, up-regulated genes were linked to a broad range of pathways, including synaptic function, voltage-gated channel activity, and neurotransmission (Supplementary Figure S2). Collectively, these findings support the premise that modulation of the ER stress response represents a shared mechanism underlying the effects of these diverse compounds.
3.1.3. CEBPB Modulates ER Stress Related Genes by Decreasing Its DNA Binding
We identified 158 differentially expressed genes (DEGs) that overlapped all treatment conditions, the majority of which were down-regulated (Figure 4A). Strikingly, CEBPB again emerged as the top upstream regulator of these DEGs (FDR = 0.00168), with 17 genes predicted as CEBPB targets (Figure 4B). Even more striking, many of these targets are associated with ER stress, including DDIT3, TRIB3, XBP1, heat shock protein family A (Hsp70) member 9 (HSPA9), solute carrier family 7 member 11 (SLC7A11), fibronectin 1(FN1), fin bud initiation factor homolog (FIBIN), and homocysteine inducible ER protein with ubiquitin like domain 1 (HERPUD1) (Figure 4B. We validated these findings using our iPSC-derived cell model system. Specifically, iPSC-derived neurons were treated with thapsigargin, a pharmacological inducer of ER stress. As anticipated, DDIT3, TRIB3, HSPA9, XBP1, SLC7A11, FN1, FIBIN, and HERPUD1 mRNA levels were upregulated and the ER stress response emerged as the most significantly affected pathway (Figure 4C, and Supplementary Table S6).
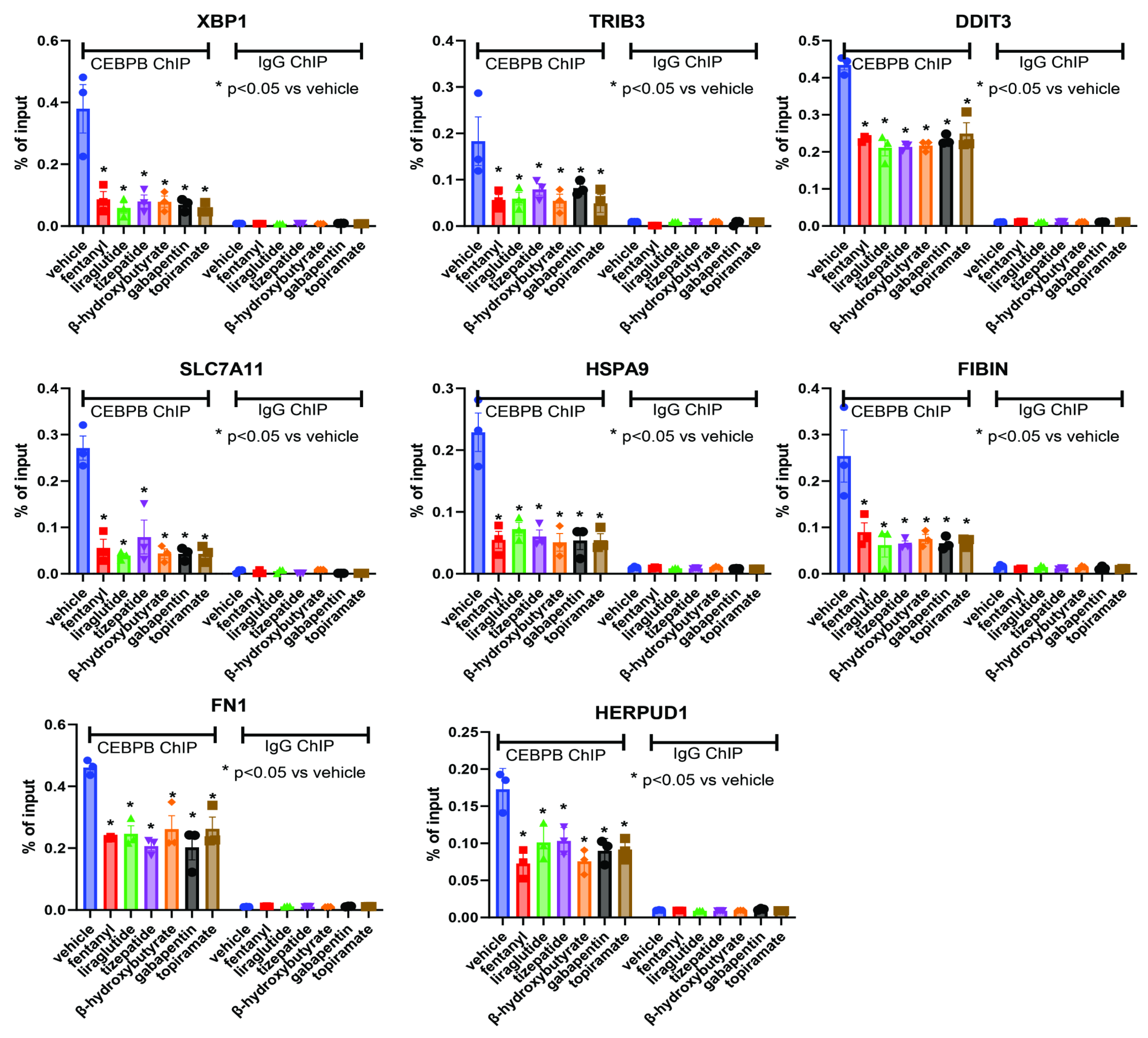
Although CEBPB acts as a transcription factor, its mRNA levels remained unchanged following treatment with fentanyl, liraglutide, tirzepatide, topiramate, gabapentin, or β-hydroxybutyrate in iPSC-derived forebrain neurons. We hypothesized that these treatments may modulate CEBPB’s DNA-binding activity rather than its expression, thereby influencing downstream gene regulation (Figure 4D). To test this hypothesis, we first queried the encyclopedia of DNA elements (ENCODE) database and found CEBPB binding sites in several of the above-mentioned ER stress-related genes, with strong signal intensities ranging from 500 to 1000. This prompted us to perform CEBPB ChIP assays to validate these interactions. As anticipated, CEBPB could bind to most of those ER-stress related genes in the absence of drug treatment (Figure 5). Remarkably, the DNA binding was significantly decreased in response to drug treatment (Figure 5). Together, these findings indicate that diverse SUD-relevant compounds converge on a shared mechanism of ER stress modulation, potentially mediated by reduced CEBPB binding to its target genes.
4. Discussion
Opioid use disorder remains a national crisis, with only three FDA-approved pharmacotherapies available and a rapidly rising overdose death level driven by synthetic opioids, such as fentanyl. Our study identifies a unifying biologic mechanism, attenuation of ER stress signaling, that is shared across fentanyl, GLP-1 receptor agonists, anticonvulsants, and metabolic modulators in human iPSC-derived neural models. These diverse drug classes consistently suppress ER stress-related genes through a common transcriptional regulator, CEBPB. These findings highlight ER stress modulation as a previously unrecognized point of convergence in therapeutic responses and suggest new avenues for developing or repurposing treatments for opioid addiction.
Fentanyl and GLP-1 receptor agonists, despite acting on pharmacologically distinct primary targets, converge on a common downstream pathway involving attenuation of ER stress signaling in human iPSC-derived forebrain organoids. In the present study, the fentanyl concentration used was 3 nM, which is substantially lower than concentrations commonly used in most in vivo and in vitro studies (typically ranging from 100 nM to 250 µM) [44]. The recommended serum concentrations of fentanyl for analgesia and anesthesia are 1-2 ng/ml (~2.97-5.94 nM) and 10-20 ng/ml (~29.7-59.4 nM), respectively, with a lethal dose of fentanyl in humans estimated at 2 mg. Approximately 7 ng/ml or higher blood concentrations have been linked to deaths in cases involving polysubstance misuse. We emphasize that drug effects can vary markedly depending on treatment conditions, including dose, duration, and frequency of exposure. The concentration used here was selected to be clinically relevant based on human pharmacokinetic studies.
Liraglutide is a 31-amino acid polypeptide chain with the molecular formula C₁₇₂H₂₆₅N₄₃O₅₁ and a molecular weight of 3751.26 Da. Exenatide is a 39-amino acid synthetic peptide amide with the molecular formula C₁₈₄H₂₈₂N₅₀O₆₀S and a molecular weight of 4186.6 Da. Although these two GLP-1 receptor agonists belong to the same drug class, we observed a greater number of differentially expressed genes in iPSC-derived forebrain organoids treated with exenatide (Figure 2A). This observation aligns with our previous study, in which oxycodone and buprenorphine produced distinct transcriptional profiles in iPSC-derived forebrain organoids despite both targeting opioid receptors [14]. These findings indicate that individual drugs within the same class can produce unique molecular signatures, underscoring the importance of patient-derived models for evaluating drug effects in the human brain. It should be noted that several immune-related pathways were down-regulated across all three treatments (Supplementary Table S3–S5). Both drug classes produced a similar transcriptional signature in genes involved in ER stress. The most significant suppression of TRIB3 was observed after fentanyl exposure, and its expression was similarly reduced by liraglutide and exenatide in iPSC-derived forebrain organoids (Figure 2A). In line with this observation, TRIB3 mRNA expression was consistently down-regulated across all treatment conditions in iPSC-derived forebrain neurons (Figure 3B). Importantly, the drug concentrations used in the present study are clinically relevant, making these effects physiologically plausible and pointing to ER stress modulation as a potential shared mechanism of opioid and GLP-1 therapies. We identified CEBPB as the key transcriptional regulator orchestrating these shared responses.
CEBPB plays an important role in ER stress, inflammation, and metabolic disturbances [45]. A key insight from this work is the identification of CEBPB as a common upstream regulator whose activity, rather than expression, appears to be modulated by these compounds in iPSC-derived forebrain neurons. Although CEBPB mRNA levels remained unchanged, its predicted downstream targets, including numerous ER stress genes, were consistently down-regulated. ChIP assays confirmed direct CEBPB binding to multiple ER stress-related loci under basal conditions, and drug treatments significantly reduced this DNA-binding activity. These findings suggest that CEBPB acts as a regulatory hub through which pharmacologic interventions converge to attenuate ER stress signaling. Given previous reports linking CEBPB activity to cellular stress responses, neuroinflammation, and synaptic plasticity, diminished DNA binding of CEBPB may represent a crucial mechanism through which drugs exert neuroprotective effects [46].
Several anti-diabetic medications, including metformin and liraglutide, have been shown to exert indirect ER stress-modulating effects [47]. The convergence of fentanyl and GLP-1 receptor agonists on common ER stress pathways carries important implications for understanding therapeutic action in SUD. Extending this analysis to additional compounds, including anticonvulsants and metabolic modulators, further supports the concept that therapeutic efficacy in SUD may depend not only on receptor-specific effects but also on the restoration of cellular homeostasis via attenuation of ER stress. ER stress signaling is increasingly recognized as a critical contributor to addiction biology, as chronic drug exposure (e.g., opioids) activates cellular stress pathways that drive neuroplastic changes across mesocorticolimbic circuits [48]. ER stress influences synaptic remodeling, neuroinflammation, and vulnerability to neurotoxicity, all processes that influence reward learning and behavioral sensitization [49]. In this context, the convergence of fentanyl and GLP-1 receptor agonists on the down-regulation of ER stress-associated genes suggests that attenuation of ER stress may represent a shared protective or homeostatic mechanism that counteracts maladaptive plasticity induced by addictive substances. Recent real-world data further indicate that GLP-1 agonists may reduce drug-seeking behaviors and alter reward circuitry through mechanisms beyond their primary receptor-specific actions [50,51]. The convergence of GLP-1 receptor agonists, anticonvulsants, metabolic modulators, and opioid ligands on a shared ER stress signature underscores the possibility that therapeutic efficacy in SUD may depend not only on receptor-specific mechanisms but also on broader restoration of cellular homeostasis. Our results may help explain why certain drugs with distinct primary targets nonetheless show overlapping clinical benefits in treating opioid withdrawal, AUD, and other SUD-related conditions. While further work is needed to determine whether pharmacologic modulation of ER stress can be leveraged therapeutically for SUD, our results point to a promising avenue for drug repurposing and mechanism-guided treatment development.
Several limitations to this study should be noted. We initially conducted RNA-seq on iPSC-derived forebrain organoids, but the small sample size limits the broader applicability of our findings. Our iPSC-derived brain-like cells are region-specific. We investigated iPSC-derived forebrain organoids, a part of the brain that has been implicated in the pathophysiology of addiction [52,53]. Given the complexity of the human brain, focusing on a single region may not fully capture the intricacies of the entire central nervous system. However, the iPS cell model system, particularly in psychiatric research, represents a powerful research tool for generating and testing mechanistic hypotheses. Future research should expand to include additional brain regions and leverage single-cell sequencing in iPSC-derived brain organoids to explore cell-type-specific and drug-specific effects more comprehensively. Despite these limitations, our study provides novel mechanistic insights into drug actions in iPSC-derived brain-like cells. The consistent ER stress signature observed across treatments and platforms strongly supports a convergent mechanism. Further work will be needed to determine whether modulating CEBPB-dependent transcriptional networks yields therapeutic benefits and whether these ER stress-related mechanisms extend to in vivo or clinical settings.
5. Conclusions
This study identifies modulation of ER stress as a shared molecular mechanism underlying the effects of fentanyl, GLP-1 receptor agonists, and other therapeutic candidates for SUD. Fentanyl, liraglutide, and exenatide consistently down-regulated ER stress-related genes, with TRIB3 emerging as the most notably affected target across treatments in iPSC-derived forebrain organoids. Additional stress-response genes, including DDIT3, ATF4, PPP1R15A, and HKDC1, were similarly suppressed, indicating broad attenuation of ER stress signaling. We identified CEBPB as the key driver of these shared transcriptional changes. Functional studies in iPSC-derived forebrain neurons further revealed strong down-regulation of ER stress genes and reduced CEBPB DNA-binding activity across diverse drug classes, including anticonvulsants and metabolic modulators. Together, these findings highlight ER stress modulation, mediated through CEBPB, as a convergent mechanism with therapeutic relevance for SUD.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
M. Ho wrote the manuscript. M. Ho designed the research. M. Ho, and C. Zhang, performed the research; C. Zhang, and H. Li analyzed the data and contributed analytical tools. All authors have given final approval of this version of the manuscript.
Funding
This work was supported in part by the National Institutes of Health [grant numbers K01 AA28050, R01 AA27486, and R01 DA57928]; the Brain & Behavior Research Foundation [grant number 31329], the Mayo Clinic Research Pipeline K2R Program, the Terrance and Bette Noble Foundation, a generous gift from Donna Giordano, and the Mayo Clinic Center for Individualized Medicine. H.L. was supported by grants from the Mayo Clinic Center for Cell Signaling in Gastroenterology (NIH: P30DK084567), the Mayo Clinic Nutrition Obesity Research Program, the Glenn Foundation for Medical Research, Mayo Clinic Comprehensive Cancer Center (NIH; P30CA015083), Susan Morrow Legacy Foundation and Mr. and Mrs. Duca, the National Institutes of Health (NIH; U19 AG74879, P50 CA136393, U54 AG79779, P01 AG 62413, R03 OD038392).
Institutional Review Board Statement
This study was conducted in accordance with the protocol #20-00372, which was reviewed and approved by the Mayo Clinic Institutional Review Board.
Informed Consent Statement
Written consent was obtained from all individuals whose samples were used, including consent for study participation and publication of the results in a peer-reviewed scientific journal.
Data Availability Statement
All data supporting our findings can be found on the main paper or in supplementary files. Sequencing data are available via the GEO database: GSE315126, and GSE315127.
Acknowledgments
We extend our sincere gratitude to Dr. Leila Jones for providing administrative support for our addiction research program. The Scientific Publications staff at Mayo Clinic provided copyediting support.
Conflicts of Interest
All authors declare that they have no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AUD | alcohol use disorder (AUD) |
| CDC | Center for Disease Control (CDC) |
| ChIP | chromatin Immunoprecipitation (ChIP) |
| Co-IP | co-immunoprecipitation (co-IP) |
| DEGs | differentially expressed genes (DEGs) |
| EB | embryoid body (EB) |
| ER | endoplasmic reticulum (ER) |
| FDA | Food and Drug Administration (FDA) |
| GIP | glucose-dependent insulinotropic polypeptide (GIP) |
| GLP-1RA | GLP-1 receptor agonists |
| iPSC | Induced pluripotent stem cell |
| OUD | opioid use disorder (OUD) |
| SUD | substance use disorders (SUD) |
| TRIB3 | Tribbles Pseudokinase 3 |
| CEBPB | CCAAT/enhancer-binding protein beta |
| ATF4 | activating transcription factor 4 |
| DDIT3 | DNA damage inducible transcript 3 |
| XBP1 | X-box binding protein 1 |
| SOX2 | SRY-box transcription factor 2 |
| PPP1R15A | Protein phosphatase 1 regulatory subunit 15A |
| GADD34 | growth arrest and DNA damage-inducible protein |
| HKDC1 | hexokinase domain containing 1 |
| TRA-1-81 | podocalyxin |
| SLC7A11 | solute carrier family 7 member 11 |
| FIBIN | fin bud initiation factor homolog |
| FN1 | fibronectin 1 |
| HERPUD1 | homocysteine inducible ER protein with ubiquitin like domain 1 |
| ENCODE | The Encyclopedia of DNA Elements |
| T2D | type 2 diabetes (T2D) |
References
- Mattson, C.L.; Tanz, L.J.; Quinn, K.; Kariisa, M.; Patel, P.; Davis, N.L. Trends and Geographic Patterns in Drug and Synthetic Opioid Overdose Deaths - United States, 2013-2019. MMWR. Morbidity and mortality weekly report 2021, 70, 202–207. [Google Scholar] [CrossRef]
- Blanco, C.; Volkow, N.D. Management of opioid use disorder in the USA: Present status and future directions. The Lancet 2019, 393, 1760–1772. [Google Scholar] [CrossRef]
- Scherrer, J.F.; Tucker, J.; Salas, J.; Zhang, Z.; Grucza, R. Comparison of Opioids Prescribed for Patients at Risk for Opioid Misuse Before and After Publication of the Centers for Disease Control and Prevention’s Opioid Prescribing Guidelines. JAMA Network Open 2020, 3, e2027481. [Google Scholar] [CrossRef]
- Ling, W.; Nadipelli, V.R.; Aldridge, A.P.; Ronquest, N.A.; Solem, C.T.; Chilcoat, H.; Albright, V.; Johnson, C.; Learned, S.M.; Mehra, V.; et al. Recovery From Opioid Use Disorder (OUD) After Monthly Long-acting Buprenorphine Treatment: 12-Month Longitudinal Outcomes From RECOVER, an Observational Study. Journal of Addiction Medicine 2020, 14, e233–e240. [Google Scholar] [CrossRef] [PubMed]
- Kosten, T.R.; Petrakis, I.L. The Hidden Epidemic of Opioid Overdoses During the Coronavirus Disease 2019 Pandemic. JAMA Psychiatry 2021, 78, 585–586. [Google Scholar] [CrossRef]
- Oesterle, T.S.; Thusius, N.J.; Rummans, T.A.; Gold, M.S. Medication-Assisted Treatment for Opioid-Use Disorder. Mayo Clinic Proceedings 2019, 94, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- NIDA. Only 1 in 5 U.S. adults with opioid use disorder received medications to treat it in 2021. National Institute on Drug Abuse website. Available online: https://nida.nih.gov/news-events/news-releases/2023/08/only-1-in-5-us-adults-with-opioid-use-disorder-received-medications-to-treat-it-in-2021 (accessed on 16 October 2025).
- Feingold, K.R. Oral and Injectable (Non-Insulin) Pharmacological Agents for the Treatment of Type 2 Diabetes. In Endotext; Feingold, K.R., Ahmed, S.F., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., et al., Eds.; MDText.com, Inc. Copyright © 2000-2025, MDText.com, Inc.: South Dartmouth (MA), 2000. [Google Scholar]
- Wang, W.; Volkow, N.D.; Wang, Q.; Berger, N.A.; Davis, P.B.; Kaelber, D.C.; Xu, R. Semaglutide and Opioid Overdose Risk in Patients With Type 2 Diabetes and Opioid Use Disorder. JAMA Network Open 2024, 7, e2435247. [Google Scholar] [CrossRef] [PubMed]
- Qeadan, F.; McCunn, A.; Tingey, B. The association between glucose-dependent insulinotropic polypeptide and/or glucagon-like peptide-1 receptor agonist prescriptions and substance-related outcomes in patients with opioid and alcohol use disorders: A real-world data analysis. Addiction 2025, 120, 236–250. [Google Scholar] [CrossRef]
- Zhang, Y.; Kahng, M.W.; Elkind, J.A.; Weir, V.R.; Hernandez, N.S.; Stein, L.M.; Schmidt, H.D. Activation of GLP-1 receptors attenuates oxycodone taking and seeking without compromising the antinociceptive effects of oxycodone in rats. Neuropsychopharmacology 2020, 45, 451–461. [Google Scholar] [CrossRef]
- Ho, M.-F.; Zhang, C.; Zhang, L.; Wei, L.; Zhou, Y.; Moon, I.; Geske, J.R.; Choi, D.-S.; Biernacka, J.; Frye, M.; et al. TSPAN5 influences serotonin and kynurenine: Pharmacogenomic mechanisms related to alcohol use disorder and acamprosate treatment response. Molecular Psychiatry 2020, 26, 3122–3133. [Google Scholar] [CrossRef]
- Biernacka, J.M.; Coombes, B.J.; Batzler, A.; Ho, A.M.-C.; Geske, J.R.; Frank, J.; Hodgkinson, C.; Skime, M.; Colby, C.; Zillich, L.; et al. Genetic contributions to alcohol use disorder treatment outcomes: A genome-wide pharmacogenomics study. Neuropsychopharmacology 2021, 46, 2132–2139. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.-F.; Zhang, C.; Moon, I.; Zhu, X.; Coombes, B.J.; Biernacka, J.; Skime, M.; Oesterle, T.S.; Karpyak, V.M.; Schmidt, K.; et al. Single cell transcriptomics reveals distinct transcriptional responses to oxycodone and buprenorphine by iPSC-derived brain organoids from patients with opioid use disorder. Molecular Psychiatry 2022. [Google Scholar] [CrossRef]
- Qian, X.; Jacob, F.; Song, M.M.; Nguyen, H.N.; Song, H.; Ming, G.-l. Generation of human brain region–specific organoids using a miniaturized spinning bioreactor. Nature Protocols 2018, 13, 565. Available online: https://www.nature.com/articles/nprot.2017.152#supplementary-information. [CrossRef] [PubMed]
- Ho, M.-F.; Zhang, C.; Wei, L.; Zhang, L.; Moon, I.; Geske, J.R.; Skime, M.K.; Choi, D.-S.; Biernacka, J.M.; Oesterle, T.S.; et al. Genetic variants associated with acamprosate treatment response in alcohol use disorder patients: A multiple omics study. British journal of pharmacology 2022, 173, 16. [Google Scholar] [CrossRef]
- Ho, M.-F.; Zhang, L.; Moon, I.; Skime, M.; Ho, A.M.-C.; Choi, D.-S.; Biernacka, J.; Kaddurah-Daouk, R.; Wen, Z.; Frye, M.A.; et al. TSPAN5, an alcohol responsive gene that alters serotonin levels in human induced pluripotent stem cells: Novel molecular links to alcohol use disorder. Alcoholism: Clinical and Experimental Research 2019, 43, S786. [Google Scholar]
- Wen, Z.; Nguyen, H.N.; Guo, Z.; Lalli, M.A.; Wang, X.; Su, Y.; Kim, N.-S.; Yoon, K.-J.; Shin, J.; Zhang, C.; et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014, 515, 414–418. [Google Scholar] [CrossRef]
- Lötsch, J.; Walter, C.; Parnham, M.J.; Oertel, B.G.; Geisslinger, G. Pharmacokinetics of Non-Intravenous Formulations of Fentanyl. Clinical Pharmacokinetics 2013, 52, 23–36. [Google Scholar] [CrossRef]
- Jacobsen, L.V.; Flint, A.; Olsen, A.K.; Ingwersen, S.H. Liraglutide in Type 2 Diabetes Mellitus: Clinical Pharmacokinetics and Pharmacodynamics. Clin Pharmacokinet 2016, 55, 657–672. [Google Scholar] [CrossRef]
- Cirincione, B.; Mager, D.E. Population pharmacokinetics of exenatide. Br J Clin Pharmacol 2017, 83, 517–526. [Google Scholar] [CrossRef]
- Schneck, K.; Urva, S. Population pharmacokinetics of the GIP/GLP receptor agonist tirzepatide. CPT Pharmacometrics Syst Pharmacol 2024, 13, 494–503. [Google Scholar] [CrossRef]
- Bae, E.-K.; Lee, J.; Shin, J.-W.; Moon, J.; Lee, K.-J.; Shin, Y.-W.; Kim, T.-J.; Shin, D.; Jang, I.-J.; Lee, S.K. Factors influencing topiramate clearance in adult patients with epilepsy: A population pharmacokinetic analysis. Seizure 2016, 37, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Swearingen, D.; Aronoff, G.M.; Ciric, S.; Lal, R. Pharmacokinetics of immediate release, extended release, and gastric retentive gabapentin formulations in healthy adults. Int J Clin Pharmacol Ther 2018, 56, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Shivva, V.; Cox, P.J.; Clarke, K.; Veech, R.L.; Tucker, I.G.; Duffull, S.B. The Population Pharmacokinetics of D-β-hydroxybutyrate Following Administration of (R)-3-Hydroxybutyl (R)-3-Hydroxybutyrate. Aaps j 2016, 18, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014, 15, 550. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics 2003, 34, 267. Available online: https://www.nature.com/articles/ng1180#supplementary-information. [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.; Ma’ayan, A. ChEA3: Transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Research 2019, 47, W212–W224. [Google Scholar] [CrossRef]
- Fang, N.; Zhang, W.; Xu, S.; Lin, H.; Wang, Z.; Liu, H.; Fang, Q.; Li, C.; Peng, L.; Lou, J. TRIB3 alters endoplasmic reticulum stress-induced β-cell apoptosis via the NF-κB pathway. Metabolism 2014, 63, 822–830. [Google Scholar] [CrossRef]
- Pitale, P.M.; Saltykova, I.V.; Adu-Agyeiwaah, Y.; Calzi, S.L.; Satoh, T.; Akira, S.; Gorbatyuk, O.; Boulton, M.E.; Pardue, M.T.; Garvey, W.T.; et al. Tribbles Homolog 3 Mediates the Development and Progression of Diabetic Retinopathy. Diabetes 2021, 70, 1738–1753. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. Embo j 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Örd, D.; Örd, T. Characterization of human NIPK (TRB3, SKIP3) gene activation in stressful conditions. Biochemical and Biophysical Research Communications 2005, 330, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Quddos, F.; Hubshman, Z.; Tegge, A.; Sane, D.; Marti, E.; Kablinger, A.S.; Gatchalian, K.M.; Kelly, A.L.; DiFeliceantonio, A.G.; Bickel, W.K. Semaglutide and Tirzepatide reduce alcohol consumption in individuals with obesity. Scientific Reports 2023, 13, 20998. [Google Scholar] [CrossRef]
- Salehi, M.; Kheirabadi, G.R.; Maracy, M.R.; Ranjkesh, M. Importance of gabapentin dose in treatment of opioid withdrawal. J Clin Psychopharmacol 2011, 31, 593–596. [Google Scholar] [CrossRef]
- Mokhber, N.; Soltanifar, A.; Talebi, M. The Effect of Topiramate in the Treatment of Opioid (Heroin) Withdrawal. European Psychiatry 2009, 24, E424–E443. [Google Scholar] [CrossRef]
- Kong, D.; Sun, J.X.; Yang, J.Q.; Li, Y.S.; Bi, K.; Zhang, Z.Y.; Wang, K.H.; Luo, H.Y.; Zhu, M.; Xu, Y. Ketogenic diet: A potential adjunctive treatment for substance use disorders. Front Nutr 2023, 10, 1191903. [Google Scholar] [CrossRef]
- Trinko, R.; Diaz, D.M.; Foscue, E.; Thompson, S.L.; Taylor, J.R.; DiLeone, R.J. Ketogenic diet enhances the effects of oxycodone in mice. Scientific Reports 2023, 13, 7507. [Google Scholar] [CrossRef]
- Wiers, C.E.; Vendruscolo, L.F.; van der Veen, J.-W.; Manza, P.; Shokri-Kojori, E.; Kroll, D.S.; Feldman, D.E.; McPherson, K.L.; Biesecker, C.L.; Zhang, R.; et al. Ketogenic diet reduces alcohol withdrawal symptoms in humans and alcohol intake in rodents. Science Advances 2021, 7, eabf6780. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.; Kermanizadeh, A. A Review of Toxicological Profile of Fentanyl—A 2024 Update. Toxics 2024, 12, 690. [Google Scholar] [CrossRef] [PubMed]
- van der Krieken, S.E.; Popeijus, H.E.; Mensink, R.P.; Plat, J. CCAAT/Enhancer Binding Protein β in relation to ER Stress, Inflammation, and Metabolic Disturbances. BioMed Research International 2015, 2015, 324815. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Long, C.; Yi, P.; Zhang, G.; Wan, W.; Rao, X.; Ying, J.; Liang, W.; Hua, F. C/EBPβ: A transcription factor associated with the irreversible progression of Alzheimer's disease. CNS Neurosci Ther 2024, 30, e14721. [Google Scholar] [CrossRef]
- Griffin, H.; Sullivan, S.C.; Barger, S.W.; Phelan, K.D.; Baldini, G. Liraglutide Counteracts Endoplasmic Reticulum Stress in Palmitate-Treated Hypothalamic Neurons without Restoring Mitochondrial Homeostasis. International Journal of Molecular Sciences 2023, 24, 629. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, R.; Leong Bin Abdullah, M.F.I. The association between neuropsychiatric effects of substance use and occurrence of endoplasmic reticulum and unfolded protein response: A systematic review. Toxicol Lett 2024, 391, 71–85. [Google Scholar] [CrossRef]
- Sprenkle, N.T.; Sims, S.G.; Sánchez, C.L.; Meares, G.P. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener 2017, 12, 42. [Google Scholar] [CrossRef]
- Wang, W.; Volkow, N.D.; Berger, N.A.; Davis, P.B.; Kaelber, D.C.; Xu, R. Association of semaglutide with risk of suicidal ideation in a real-world cohort. Nat Med 2024, 30, 168–176. [Google Scholar] [CrossRef]
- Wium-Andersen, I.K.; Wium-Andersen, M.K.; Fink-Jensen, A.; Rungby, J.; Jørgensen, M.B.; Osler, M. Use of GLP-1 receptor agonists and subsequent risk of alcohol-related events. A nationwide register-based cohort and self-controlled case series study. Basic Clin Pharmacol Toxicol 2022, 131, 372–379. [Google Scholar] [CrossRef]
- Goldstein, R.Z.; Volkow, N.D. Drug Addiction and Its Underlying Neurobiological Basis: Neuroimaging Evidence for the Involvement of the Frontal Cortex. American Journal of Psychiatry 2002, 159, 1642–1652. [Google Scholar] [CrossRef]
- Ceceli, A.O.; Bradberry, C.W.; Goldstein, R.Z. The neurobiology of drug addiction: Cross-species insights into the dysfunction and recovery of the prefrontal cortex. Neuropsychopharmacology 2022, 47, 276–291. [Google Scholar] [CrossRef]
Figure 1.
Generation and characterization of iPSC-derived forebrain organoids. (A) Induced pluripotent stem (iPS) cells were cultured on Matrigel-coated plates. G-band karyotyping confirmed that all iPS cell lines used in this study exhibited normal chromosomal integrity. A bioreactor system was employed to grow the iPSC-derived brain organoids. (B) All iPSC lines expressed key pluripotency markers. Panel B shows representative immunostaining for the pluripotency markers SOX2 and TRA-1-81. (C) Panel C presents representative immunostaining images of day-90 iPSC-derived forebrain organoids, highlighting SOX2-positive progenitor cells and TUJ1-positive neurons. (D) A schematic overview of the differentiation protocol used to generate the iPSC-derived forebrain organoids.
Figure 1.
Generation and characterization of iPSC-derived forebrain organoids. (A) Induced pluripotent stem (iPS) cells were cultured on Matrigel-coated plates. G-band karyotyping confirmed that all iPS cell lines used in this study exhibited normal chromosomal integrity. A bioreactor system was employed to grow the iPSC-derived brain organoids. (B) All iPSC lines expressed key pluripotency markers. Panel B shows representative immunostaining for the pluripotency markers SOX2 and TRA-1-81. (C) Panel C presents representative immunostaining images of day-90 iPSC-derived forebrain organoids, highlighting SOX2-positive progenitor cells and TUJ1-positive neurons. (D) A schematic overview of the differentiation protocol used to generate the iPSC-derived forebrain organoids.
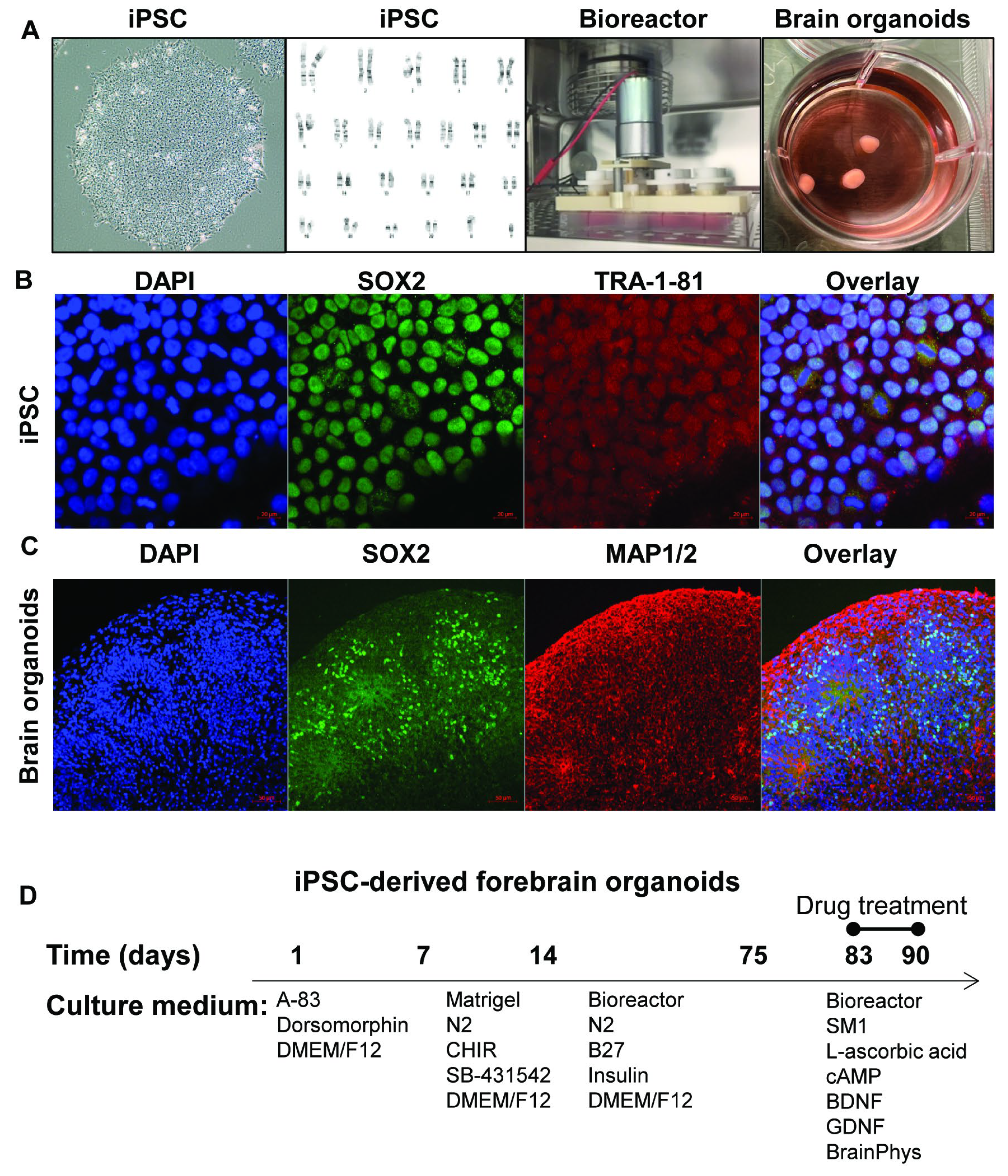
Figure 2.
RNA-seq analysis of iPSC-derived forebrain organoids treated with fentanyl, liraglutide, and exenatide. (A) Volcano plots depict the most significantly altered gene expression profiles following drug treatment, based on RNA-seq analysis (drug treatment vs. vehicle, FDR < 0.05). iPSC-derived forebrain organoids from three individuals were included in the analysis. Each treatment condition was analyzed using two technical RNA-seq replicates generated from independent library preparations. (B) Upstream regulator analysis was conducted using differentially expressed genes (FDR < 0.05). The top panel shows predicted upstream regulators associated with downregulated genes, while the bottom panel displays those linked to upregulated genes. (C) CEBPB emerged as the top upstream regulator of downregulated genes across all three drug-treatment conditions. STRING database indicated that CEBPB interacts with multiple proteins involved in ER stress signaling, including TRIB3 and XBP1. (D) Co-immunoprecipitation experiments confirmed a physical interaction between CEBPB and the spliced form of XBP1 (XBP1-s), and TRIB3 in iPSC-derived forebrain organoids.
Figure 2.
RNA-seq analysis of iPSC-derived forebrain organoids treated with fentanyl, liraglutide, and exenatide. (A) Volcano plots depict the most significantly altered gene expression profiles following drug treatment, based on RNA-seq analysis (drug treatment vs. vehicle, FDR < 0.05). iPSC-derived forebrain organoids from three individuals were included in the analysis. Each treatment condition was analyzed using two technical RNA-seq replicates generated from independent library preparations. (B) Upstream regulator analysis was conducted using differentially expressed genes (FDR < 0.05). The top panel shows predicted upstream regulators associated with downregulated genes, while the bottom panel displays those linked to upregulated genes. (C) CEBPB emerged as the top upstream regulator of downregulated genes across all three drug-treatment conditions. STRING database indicated that CEBPB interacts with multiple proteins involved in ER stress signaling, including TRIB3 and XBP1. (D) Co-immunoprecipitation experiments confirmed a physical interaction between CEBPB and the spliced form of XBP1 (XBP1-s), and TRIB3 in iPSC-derived forebrain organoids.
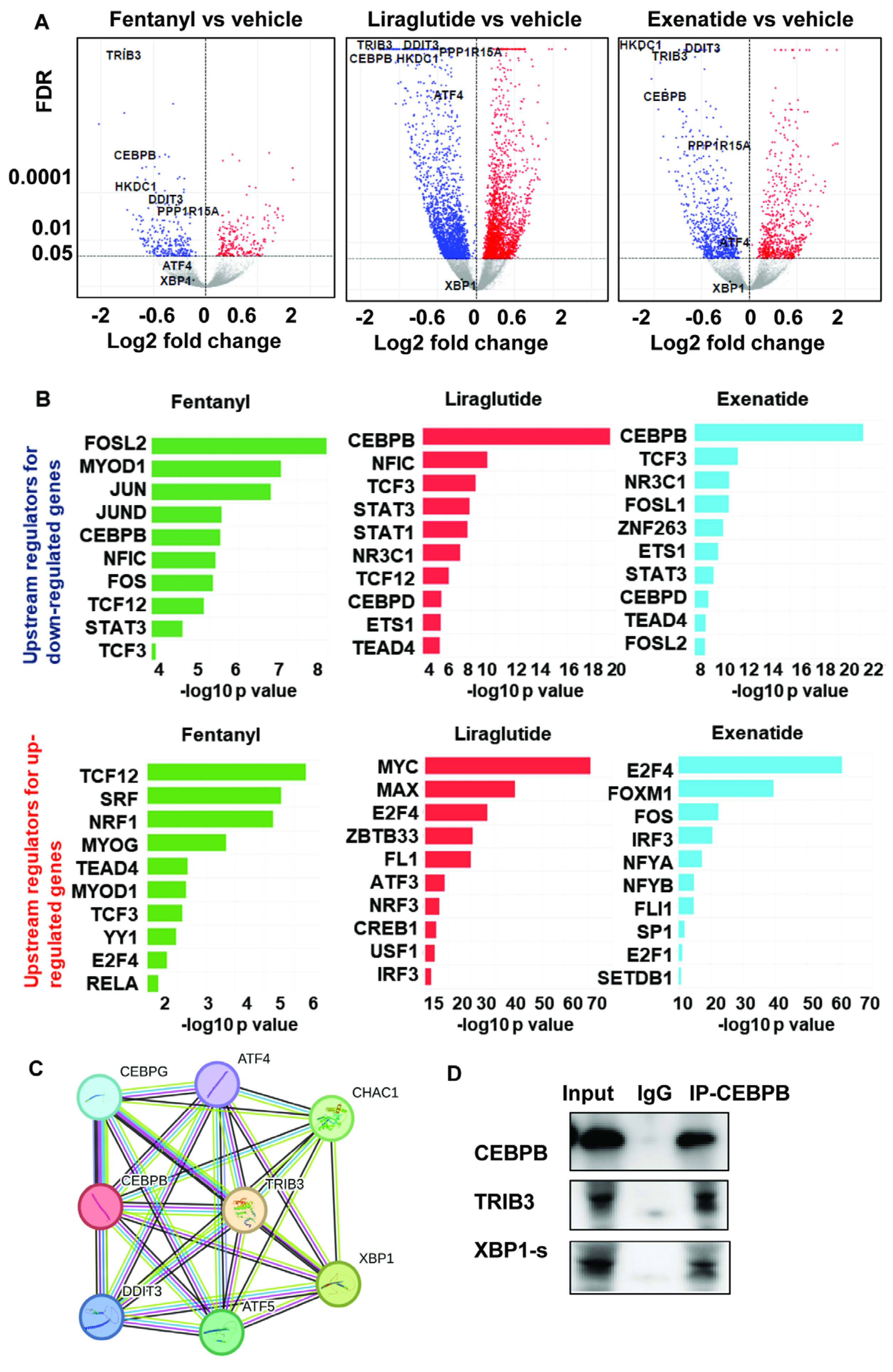
Figure 3.
RNA-seq analysis of iPSC-derived forebrain neurons treated with fentanyl, liraglutide, Tirzepatide, topiramate, gabapentin, and β-hydroxybutyrate. (A) A schematic overview of the differentiation protocol used to generate the iPSC-derived forebrain neurons. (B) Volcano plots depict the most significantly altered gene expression profiles following drug treatment, based on RNA-seq analysis (drug treatment vs. vehicle, FDR < 0.05). iPSC-derived forebrain neurons from a single individual were used for this RNA-seq analysis. Each treatment condition was analyzed with three technical replicates generated from independent library preparations.
Figure 3.
RNA-seq analysis of iPSC-derived forebrain neurons treated with fentanyl, liraglutide, Tirzepatide, topiramate, gabapentin, and β-hydroxybutyrate. (A) A schematic overview of the differentiation protocol used to generate the iPSC-derived forebrain neurons. (B) Volcano plots depict the most significantly altered gene expression profiles following drug treatment, based on RNA-seq analysis (drug treatment vs. vehicle, FDR < 0.05). iPSC-derived forebrain neurons from a single individual were used for this RNA-seq analysis. Each treatment condition was analyzed with three technical replicates generated from independent library preparations.

Figure 4.
CEBPB DNA-binding activity may contribute to modulation of the ER stress signaling pathway in response to drug treatment. (A) A total of 158 differentially expressed genes (DEGs) overlapped across all treatment conditions, with most showing downregulation (FDR < 0.05). (B) Of these 158 DEGs, 17 were identified as CEBPB target genes using the ChEA3 database. All 17 targets were downregulated following drug treatment (FDR < 0.05). (C) Volcano plots depict the most significantly altered gene expression profiles following drug treatment, based on RNA-seq analysis (thapsigargin vs. vehicle, FDR < 0.05). iPSC-derived forebrain neurons from a single individual were used for this RNA-seq analysis. Each treatment condition was analyzed with three technical replicates generated from independent library preparations. (D) The proposed study model demonstrates that these drug treatments may alter CEBPB DNA-binding activity, thereby affecting downstream gene regulation involved in ER stress signaling.
Figure 4.
CEBPB DNA-binding activity may contribute to modulation of the ER stress signaling pathway in response to drug treatment. (A) A total of 158 differentially expressed genes (DEGs) overlapped across all treatment conditions, with most showing downregulation (FDR < 0.05). (B) Of these 158 DEGs, 17 were identified as CEBPB target genes using the ChEA3 database. All 17 targets were downregulated following drug treatment (FDR < 0.05). (C) Volcano plots depict the most significantly altered gene expression profiles following drug treatment, based on RNA-seq analysis (thapsigargin vs. vehicle, FDR < 0.05). iPSC-derived forebrain neurons from a single individual were used for this RNA-seq analysis. Each treatment condition was analyzed with three technical replicates generated from independent library preparations. (D) The proposed study model demonstrates that these drug treatments may alter CEBPB DNA-binding activity, thereby affecting downstream gene regulation involved in ER stress signaling.
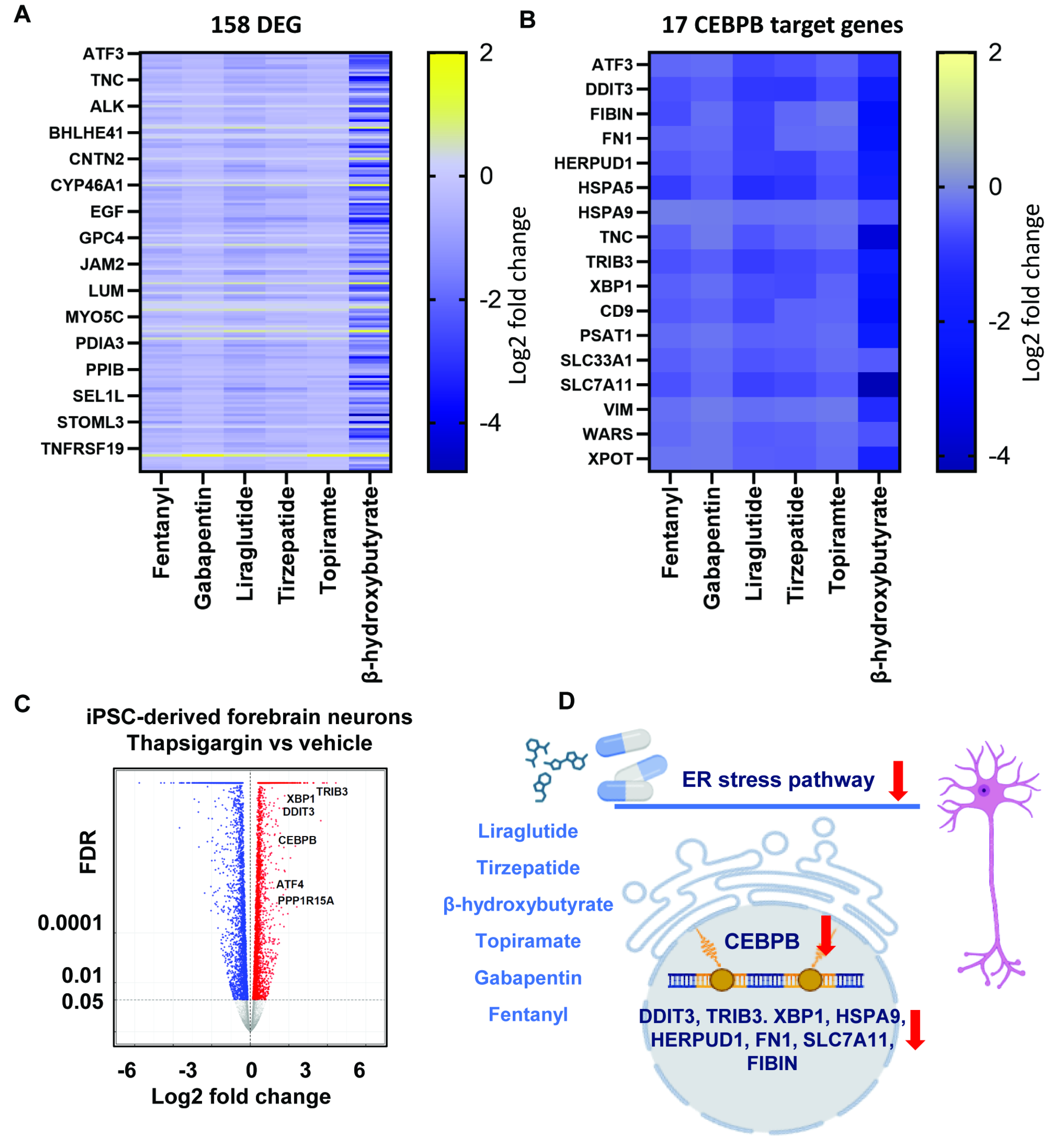
Figure 5.
CEBPB-driven ER Stress attenuation unifies the actions of fentanyl, GLP-1 receptor agonists, and candidate SUD therapeutics. ChIP assays were conducted in iPSC-derived forebrain neurons, confirming that changes in CEBPB binding correlated with altered mRNA expression of eight ER stress-related genes. ChIP DNA enrichment was quantified by qPCR and expressed as % input (n=3). ChIP-qPCR was performed in triplicate. Drug treatments were compared with vehicle controls using one-way ANOVA followed by Tukey-Kramer post hoc analysis. CEBPB ChIP and IgG control samples were analyzed separately. *P < 0.05 vs vehicle. Three independent experiments were performed.
Figure 5.
CEBPB-driven ER Stress attenuation unifies the actions of fentanyl, GLP-1 receptor agonists, and candidate SUD therapeutics. ChIP assays were conducted in iPSC-derived forebrain neurons, confirming that changes in CEBPB binding correlated with altered mRNA expression of eight ER stress-related genes. ChIP DNA enrichment was quantified by qPCR and expressed as % input (n=3). ChIP-qPCR was performed in triplicate. Drug treatments were compared with vehicle controls using one-way ANOVA followed by Tukey-Kramer post hoc analysis. CEBPB ChIP and IgG control samples were analyzed separately. *P < 0.05 vs vehicle. Three independent experiments were performed.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



