Submitted:
25 February 2026
Posted:
27 February 2026
You are already at the latest version
Abstract
Objectives: Mucopolysaccharidosis type VI is rare lysosomal storage disorder due to arylsulfatase B enzyme deficiency, leading to progressive multisystem disease and complex airway. Acute respiratory infections can precipitate airway embarrassment. A structured treatment guideline is currently lacking. We present 7 years old MPS VI male with respiratory distress highlighting the challenges in management. Methods: Case review focusing on clinical presentation, imaging findings, multidisciplinary deci-sion making during acute deterioration. Results: A child diagnosed with MPS VI at age of 3.5 years old, due to low arylsulfatase B enzyme activity and homozygous for c.275C>A p.(Thr92Lys) variant in ARSB gene. At 7 years, he presented with dyspnoea, increased respiratory effort with bilateral crepitations, noisy breathing. Initial man-agement included facemask oxygen, nebulised adrenaline, corticosteroids, bronchodi-lators. Computer tomography scan of the neck, chest showed complex upper airway, multiple tracheal narrowing, tortuosity, extra loop of truncus brachiocephalicus from arch of aorta. Potential interventions carried substantial risks due to abnormal airway and multisystem disease. Following extensive multidisciplinary discussion after careful consideration of the significant risks associated with invasive airway interventions, a shared decision was reached with the family to adopt a comfort focused palliative care approach. Despite the best supportive care, the child unfortunately passed away after 3 months. The family were involved in every decision process, were fully supported. Conclusion: MPS VI is associated with complex airways, multisystem disease. Multi-disciplinary decision making with family is critical to safe and appropriate care. The rarity of the disease, lack of guidelines, complex airways, multiple comorbidities make management challenging.
Keywords:
mucopolysaccharidosis VI
; airway management
; crisis
1. Introduction
Mucopolysaccharidoses (MPS) are rare, inherited, lysosomal storage diseases with a combined incidence of 1 in 22,000 [1]. They arise due to deficiency of lysosomal hydrolases leading to accumulation of glycosaminoglycans (GAGs) in various parts of the body. Depending on the type of enzyme deficiency, MPS can be classified into 8 different types [2]. Table 1, shows various types of MPS, with enzyme deficiency, incidence, inheritance and life expectancy [3].
MPS type VI or Maroteaux-Lamy syndrome, is one of the MPSs, it is a rare autosomal recessive inherited lysosomal disorder with birth prevalence between 1 in 43,261 and 1 in 1,505,160 live births [5]. According to epidemiological data Maroteaux-Lamy syndrome is the third most common MPS type in Kazakhstan with birth prevalence 0.12 per 100 000 live births [6]. The disease develops due to defect in Arylsulfatase B (ARSB) gene coding for the enzyme lysosomal hydrolase N-acetylgalactosamine 4-sulphatase leading to accumulation of GAGs such as dermatan sulphate and chondroitin sulphate in various parts of the body, causing multiorgan damage. MPS VI affects multiple systems including cardiovascular, respiratory and skeletal systems, but neurocognitive aspects are normally preserved in this type of MPS [7,8]. Respiratory system impairment is one of the leading causes of early mortality in MPS. Patients with MPS VI are known to have complex airways [7,9]. Abnormalities in the airway and ventilation can arise from lips to lungs. In the upper airways, these patients may have bulky upper airways and limited neck extension, mouth opening can make access to the airways difficult. Angulation in the trachea, stenosis, malacia and tortuosity can make navigation through the airway difficult. Often these patients also have reduced lung capacity, pulmonary changes, splinting of diaphragm due to hepatosplenomegaly making ventilation challenging. Musculoskeletal problems in the cervical, thoracolumbar spine, rib cage can add to the existing airway issues. Often these patients also have sleep disordered breathing, obstructive sleep apnoea [7]. Respiratory tract infections in this scenario may become challenging in an already compromised airway. We discuss an MPS VI diagnosed at the age of 3.5 years old, now 7 years of age presenting with airway embarrassment following respiratory tract infection. We discuss the various management methods, dilemmas and ethical issues in management of this complex situation by various health care professionals.
2. Materials and Methods
A detailed case review was undertaken, examining the child’s clinical presentation, progression of respiratory symptoms, and bedside assessment during acute deterioration. Diagnostic evaluation included analysis of cross-sectional imaging and three-dimensional airway reconstructions to characterise structural abnormalities contributing to respiratory compromise. The review also explored the multidisciplinary discussions involving respiratory, ENT (Ear, nose and Throat), anaesthetic, and palliative care teams, focusing on risk–benefit considerations and shared decision-making with the family.
A child diagnosed with MPS VI at the age of 3.5 years old, due to low arylsulfatase B enzyme activity 2,7 μmol/L/h (normal reference ≥8,8 μmol/L/h). Genetic testing revealed pathogenic mutation c.275C>A p.(Thr92Lys) in homozygous condition in ARSB gene. According to the Human Gene Mutation Database (HGMD) Professional 2018.1, this variant has previously been described as disease causing for MPS VI by Karageorgos et al., 2007 [10]. At the time of MPS VI diagnosis child already had mild phenotypical changes as coarse facial features, short stature height z-score = -2.37, dysostosis multiplex, pectus carinatum, general muscle hypotonia. There were multiple mongoloid spots on the back. Difficulty breathing through the nose, snoring during night sleep was also noted. There was no history suggestive of obstructive sleep apnoea. Enzyme replacement therapy (ERT) was started immediately. He was carefully monitored during following 4 years with regular full system check-ups. At the age of 7 years old he was admitted to the hospital with main complains on shortness of breath, frequent respiratory infections, poor sleeping with snoring. His exercise tolerance was low and he had limited physical activity; he could mobilize inside home but outside the house he needed assistance. There were no acute ENT, ophthalmic, cervical spine or cognitive issues. At the moment of examination child was short stature for his age height 99cm (z-score= -4.76), weight 19kg (z-score= -1.85), BMI 19.4 (z-score=1.62). There was no history of sleep apnoea or sleep disordered breathing, however taking into account snoring during night sleep, we assume that child had obstructive sleep apnoea syndrome. His cardiac examination showed relative cardiac dullness borders +0.5 cm outside the left midclavicular line, right along the right edge of the sternum, upper at the third intercostal space on percussion. The auscultation revealed muffled heart sounds, systolic murmur at the cardiac apex and tachycardia with heart rate of 125 beats per minute. An echocardiogram revealed mitral valve cusp thickening with moderate regurgitation, left ventricular hypertrophy, aortic fibrous ring, ejection fraction 65%. Examination of respiratory system showed: tachypnoea respiratory rate of 24 breaths per minute, respiratory effort was high with use of accessory muscles. On auscultation there were bilateral chest crepitations, noisy breathing. The oxygen saturations on room air were fluctuating with lowest being 50%. On spirograph test there was severe restrictive type of pulmonary ventilation impairment, decreased FVC-20% (Forced vital capacity) and FEV1-23% (Forced expiratory volume in one second). As supportive treatment, face-mask oxygen, nebulised adrenaline, corticosteroids, bronchodilators and broadspectrum antibiotics were commenced. Computer tomography (CT) scan of the neck, chest, three- dimensional (3D) reconstructions showed, short neck, large head, small torso, broad spatulated ribs with reduced intercostal spaces seen in Figure 1 and Figure 2. The upper airway was bulky with high anterior larynx, large epiglottis, multiple tracheal narrowing’s with tortuosity shown in Figure 3. The lung parenchyma showed patchy bilateral opacities and narrowing in trachea, shown in Figure 4. In the root of the anterior neck there was an extra loop of truncus brachiocephalicus from arch of aorta over the anterior aspect of trachea at the level of thoracic inlet shown in Figure 5.
High flow nasal oxygen was commenced as this bypasses the oral cavity, this was better tolerated than face mask oxygen reducing the oxygen requirements marginally. Due to gradual worsening of clinical condition as evidenced by increasing oxygen requirements, exhaustion in the effort of breathing, escalation of care was considered by means of intubation and ventilation. The airway was planned as per the difficult airway society guidelines [11]; which is plan A: Endo tracheal intubation, plan B: Laryngeal mask airway, plan C: Bag and mask ventilation, plan D: Front of neck access. The challenges, consequence and methods to mitigate each step are described in Table 2.
The challenges in ventilation were ongoing infection, broad spatulate ribs with reduced inter costal space, contracted chest wall with reduced lung volumes evidenced by low FEV1 and FVC. Various methods of intubations as laid out in Table 2 were planned including fiberoptic and video laryngoscopy. Expertise from clinicians with special interest in MPS was also sought. The risks of every intervention were carefully discussed in as a multidisciplinary approach involving the family. final option would be palliation, as the risks outweighs the benefits. This would mean that the child will be kept comfortable and all supportive care will be offered but no active invasive interventions will be done. This would also involve not to actively cardiopulmonary resuscitation should there be a deterioration. With these options in hand, the family were invited for a discussion. All the risks were explained. Input from specialist nurses were done to make sure the family felt well supported. The family took their time and refused any invasive interventions despite worsening breathing. A collective multidisciplinary decision to keep the child comfortable and provide palliative care was made. High flow nasal oxygen was better tolerated than face mask oxygen and this reduced the oxygen requirements marginally. The child unfortunately passed away in sleep peacefully after 3 months of hospital admission despite the supportive care.
3. Discussion
Maroteaux–Lamy syndrome (MPS VI) is a rare lysosomal storage disorder characterised by arylsulfatase deficiency. This leads to dermatan sulphate accumulation in multiple tissues with significant clinical heterogeneity. Limited awareness among healthcare professionals contributes to delays in recognition and challenges in management. This is a progressive multisystem disease [12] making management complex. The clinical features are short stature, motor dysfunction [13], ocular, oro dental problems, organomegaly, cardio-respiratory insufficiency, coarse features, umbilical/inguinal hernias, sleep disorder, spinal cord compression, carpal tunnel syndrome [7]. The ENT problems include adeno tonsillar hypertrophy, hearing loss, otitis media, chronic sinusitis and airway problems [7,9]. The radiological features include dysostosis multiplex, cervical instability, compressive myelopathy, central vertebral beaking, thoracolumbar junctional kyphosis, cervicothoracic kyphosis [13] These radiological abnormalities further complicate clinical care. Garcia et al. [14] investigated the role of very early and continuous ERT. The have shown that ERT can slow disease progression, preserve endurance and motor function. The authors have also noted reduction in growth impairment when initiated early. However, ERT does not prevent long-term progression of skeletal or ocular disease. With respect to cardiac disease, the study has shown that cardiac valve involvement generally remained mild, although progression occurred in one patient. These limitations highlight the need for additional therapeutic strategies and continued research into disease-modifying treatments. Airway management remains one of the most challenging aspects of MPS VI care. Anatomical distortion, tracheal narrowing, vascular anomalies, and multisystem comorbidities significantly increase the risks associated with anaesthesia, intubation, extubation, and ventilatory support. Problems with anaesthesia have been noted in case reports as extremely challenging [15]. Although international recommendations on MPS VI published in 2019 [12] provide peri-operative guidance, there are no clear protocols for managing acute airway crises. Expert anaesthetic involvement, support from ENT, cardiology, and radiology, and strict avoidance of neck hyperextension have been recommended to minimise the risk of neurological injury. Tracheostomy may be needed when intubation, ventilation is not possible. The other indications of tracheostomy in MPS VI includes severe upper airway obstruction, severe sleep apnoea not treatable by CPAP (continuous positive airway pressure) or adenotonsillectomy [12]. Common causes as in non-MPS patients needing prolonged ventilation and failure to extubate are also indications for tracheostomy. Performing this procedure is technically difficult in MPS patients due to large head, short neck. Obtaining the appropriately sized tracheostomy tubes may also be challenging. The procedure may be potentially hazardous due to abnormal vascular anatomy such as in our patient and possibility of a catastrophic haemorrhage. Following successful intubation, extubation may also be equally challenging and is better performed in a staged fashion. The risks of any intervention need to be evaluated through a multidisciplinary approach that draws on local expertise and, when necessary, input from centres with greater experience. Given the high risk of respiratory deterioration, particularly during infections, personalised management plans and clearly defined ceilings of care with involvement of the family are crucial. Preventive strategies including infection control measures and immunisation against respiratory infections may reduce the frequency of acute episodes. Regular monitoring and anticipatory planning support safer elective and emergency interventions. In end stage of the disease, advance care planning and palliative approaches ensures that management aligns with the patient’s best interests.
4. Conclusions
Maroteaux–Lamy syndrome (MPS VI) is a rare, progressive disorder with multisystem involvement, and respiratory infections can trigger rapid clinical deterioration. Such episodes may result in significant airway compromise and pose substantial risks to health. Careful airway evaluation and coordinated multidisciplinary management are essential when responding to acute crises. Key decisions often involve whether to proceed with interventions such as anaesthesia, intubation, ventilation, extubation, or tracheostomy, each of which must be considered in the context of the patient’s comorbidities. Any intervention should be planned collaboratively by a multidisciplinary team, taking a holistic view of all organ systems and weighing potential risks and benefits. Families should be supported and actively involved in shared decision-making. Personalised care plans should incorporate preventive strategies and outline approaches for both elective and emergency scenarios. Establishing advance care directives and defining ceilings of care are important components of anticipatory planning. In advanced disease, or when the risks of intervention outweigh potential benefits, palliative care should be considered to ensure that management remains aligned with the patient’s best interests.
Author Contributions
Conceptualization, MS, CG and AT.; methodology, AT.; formal analysis, investigation, resources, AT, MS, CG.; data curation, AT.; writing—AT, CG, MS; writing—review and editing, MS, AT, CG.; supervision CG, MS.; project administration, MS, AT. All authors have read and agreed to the published version of the manuscript.
Funding
No funding was received to produce this report.
Institutional Review Board Statement
This case report was prepared in accordance with ethical standards for clinical case reporting. As this is a single retrospective case report with no experimental intervention, formal ethical approval was not required according to local regulations. The authors affirm that the report adheres to the principles of the Declaration of Helsinki.
Informed Consent Statement
Informed consent for publication was obtained from the patient’s family, including consent to publish clinical details and imaging findings. All identifying information has been removed to ensure full confidentiality.
Data Availability Statement
All the relevant clinical information regarding this patient has been presented any additional information may be provided upon request.
Acknowledgments
We would like to thank the family for allowing us to submit the case report. The authors would like to thank all the healthcare professionals who were involved in the care of this patient providing multidisciplinary care. We also would like to than Dr Stuart Watson and team from 3DSPIN in Salford Care Organisation, UK for helping with 3D reconstruction.
Conflicts of Interest
The authors have no conflicts of interests to declare.
Abbreviations
The following abbreviations are used in this manuscript:
| MPS | Mucopolysaccharidosis |
| ARSB | Arylsulfatase B |
| HGMD | Human Gene Mutation Database |
| ENT | Ear, Nose and Throat |
| GAG | Glycos Amino Glycans |
| ERT | Enzyme Replacement Therapy |
| BMI | Body Mass Index |
| FVC | Forced Vital Capacity |
| FEV1 | Forced Expiratory Volume in 1 second |
| CT | Computerised Tomography |
| 3D | Three Dimensions |
| LMA | Laryngeal Mask Airway |
| CPAP | Continuous Positive Airway Pressure |
References
- Mehta, A.B.; Winchester, B. Lysosomal storage disorders: a practical guide; Wiley-Blackwell Chicester, 2012. [Google Scholar]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 2011, 50, v4–v12. [Google Scholar] [CrossRef] [PubMed]
- Braunlin, E.A.; Harmatz, P.R.; Scarpa, M.; Furlanetto, B.; Kampmann, C.; Loehr, J.P.; Ponder, K.P.; Roberts, W.C.; Rosenfeld, H.M.; Giugliani, R. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. Journal of inherited metabolic disease 2011, 34, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.; Muenzer, J.; Scriver, CR; Beaudet, AL; Sly, WS; Valle, D; Childs, R; Kinzler, KW. The mucopolysaccharidoses, In: The Metabolic and Molecular Bases of Inherited Diseases, 8 ed.; McGraw-Hill: New York, 2001; pp. 3421–3452. [Google Scholar]
- Valayannopoulos, V.; Nicely, H.; Harmatz, P.; Turbeville, S. Mucopolysaccharidosis vi. Orphanet journal of rare diseases 2010, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Tulebayeva, A.; Mukhambetova, G.; Sharipova, M.; Tylki-Szymanska, A. The birth prevalence of mucopolysaccharidosis types I, II, III, IVA, VI, and VII in the Republic of Kazakhstan between 1984 and 2023. Diagnostics 2025, 15, 679. [Google Scholar] [CrossRef] [PubMed]
- D’Avanzo, F.; Zanetti, A.; De Filippis, C.; Tomanin, R. Mucopolysaccharidosis type VI, an updated overview of the disease. International Journal of Molecular Sciences 2021, 22, 13456. [Google Scholar] [CrossRef] [PubMed]
- Harmatz, P.; Shediac, R. Mucopolysaccharidosis VI: pathophysiology, diagnosis and treatment. Front Biosci (Landmark Ed) 2017, 22, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Gadepalli, C.; Stepien, K.M.; Sharma, R.; Jovanovic, A.; Tol, G.; Bentley, A. Airway Abnormalities in Adult Mucopolysaccharidosis and Development of Salford Mucopolysaccharidosis Airway Score. Journal of Clinical Medicine 2021, 10, 3275. [Google Scholar] [CrossRef] [PubMed]
- Karageorgos, L.; Brooks, D.A.; Pollard, A.; Melville, E.L.; Hein, L.K.; Clements, P.R.; Ketteridge, D.; Swiedler, S.J.; Beck, M.; Giugliani, R. Mutational analysis of 105 mucopolysaccharidosis type VI patients. Human mutation 2007, 28, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; El-Boghdadly, K.; Iliff, H.; Dua, G.; Higgs, A.; Huntington, M.; Mir, F.; Nouraei, S.R.; O’Sullivan, E.P.; Patel, A. Difficult Airway Society 2025 guidelines for management of unanticipated difficult tracheal intubation in adults. British Journal of Anaesthesia 2025. [Google Scholar] [CrossRef] [PubMed]
- Akyol, M.U.; Alden, T.D.; Amartino, H.; Ashworth, J.; Belani, K.; Berger, K.I.; Borgo, A.; Braunlin, E.; Eto, Y.; Gold, J.I. Recommendations for the management of MPS IVA: systematic evidence-and consensus-based guidance. Orphanet journal of rare diseases 2019, 14, 137. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Jilwan, M.; AlSayed, M. Mucopolysaccharidoses: overview of neuroimaging manifestations. Pediatric Radiology 2018, 48, 1503–1520. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.; Phillips, D.; Johnson, J.; Martin, K.; Randolph, L.M.; Rosenfeld, H.; Harmatz, P. Long-term outcomes of patients with mucopolysaccharidosis VI treated with galsulfase enzyme replacement therapy since infancy. Molecular Genetics and Metabolism 2021, 133, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Demis, A.A.; Oikonomidou, S.; Daglis, F.; Polymenakos, S.; Panagiotou, M. Double valve replacement in a patient with Maroteaux - Lamy syndrome as an ultimate team challenge. Journal of cardiothoracic surgery 2021, 16, 141–141. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Three-dimensional reconstruction anterio posterior view of the lower head, chest showing disproportionately large skull in relation to chest, short neck, broad ribs, large heart and reduced space for lungs.
Figure 1.
Three-dimensional reconstruction anterio posterior view of the lower head, chest showing disproportionately large skull in relation to chest, short neck, broad ribs, large heart and reduced space for lungs.
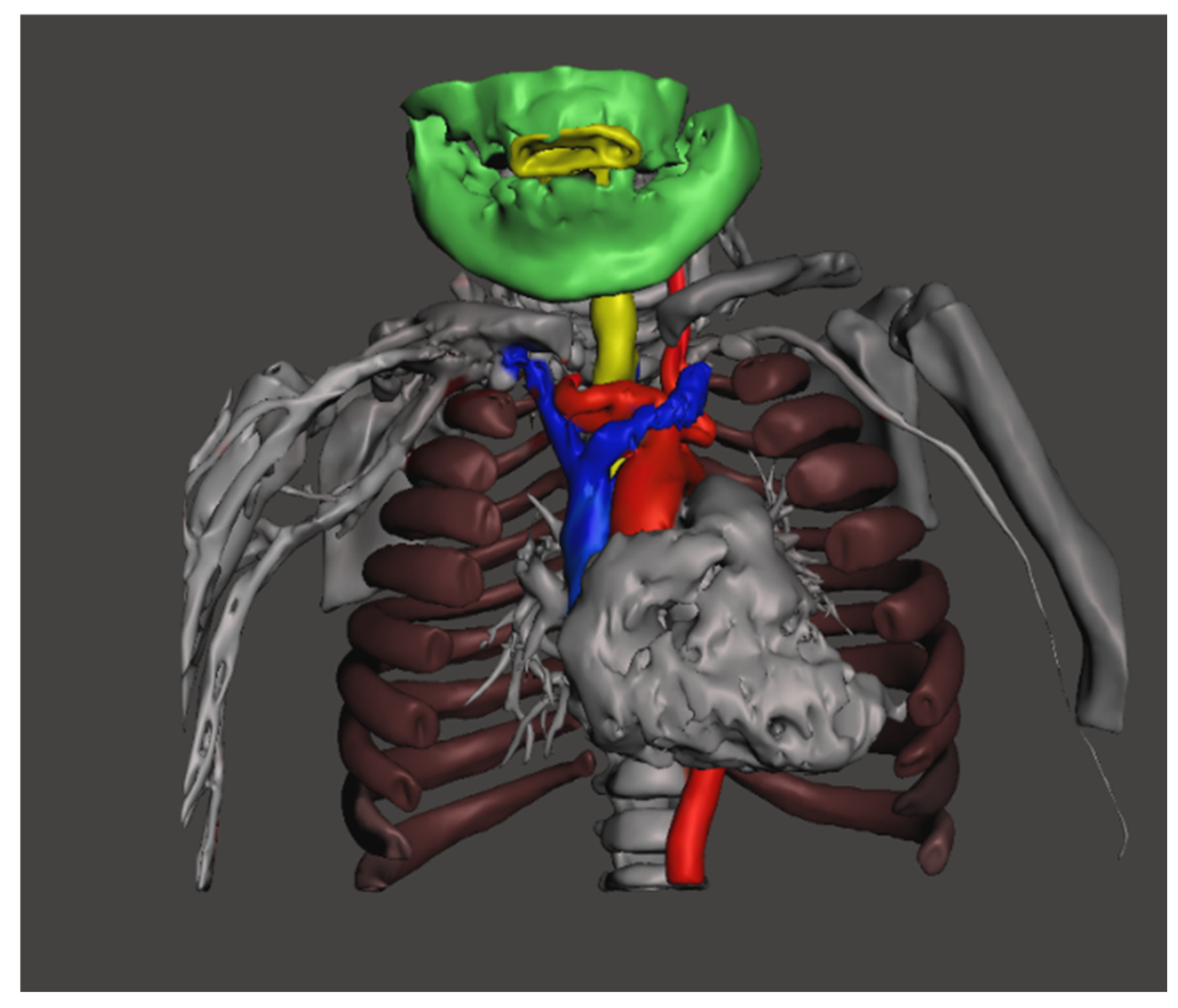
Figure 2.
3-dimensional reconstruction of the lower head, chest showing short neck, large jaw, hyoid bone is tucked under the jaw, board spatulate ribs and reduced intercostal spaces.
Figure 2.
3-dimensional reconstruction of the lower head, chest showing short neck, large jaw, hyoid bone is tucked under the jaw, board spatulate ribs and reduced intercostal spaces.
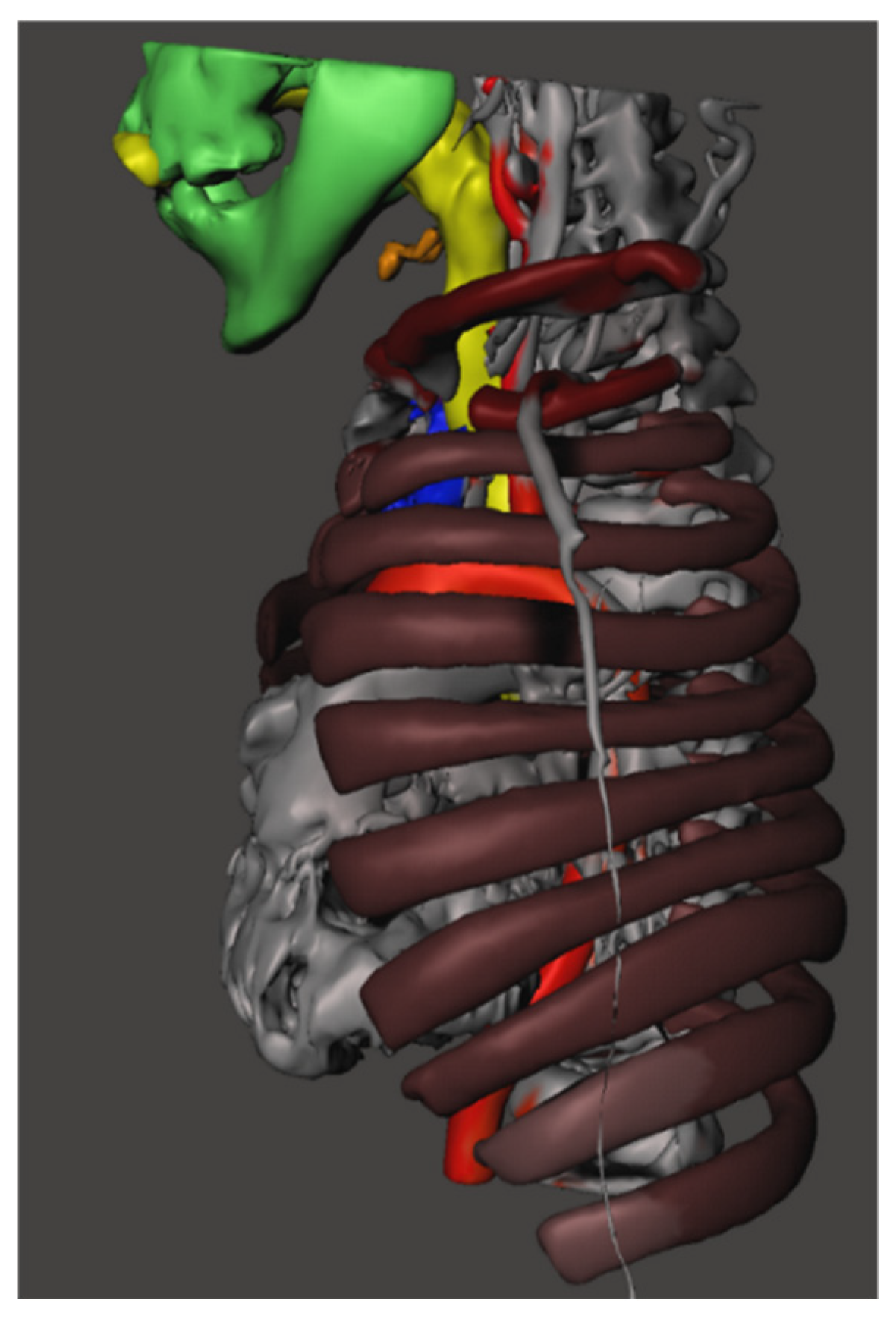
Figure 3.
3D reconstruction of anterio-posterior and left lateral cross section view of the oropharynx, larynx and trachea showing high larynx where the epiglottis marked with an arrow, multiple areas of tracheal narrowing’s marked with arrows.
Figure 3.
3D reconstruction of anterio-posterior and left lateral cross section view of the oropharynx, larynx and trachea showing high larynx where the epiglottis marked with an arrow, multiple areas of tracheal narrowing’s marked with arrows.
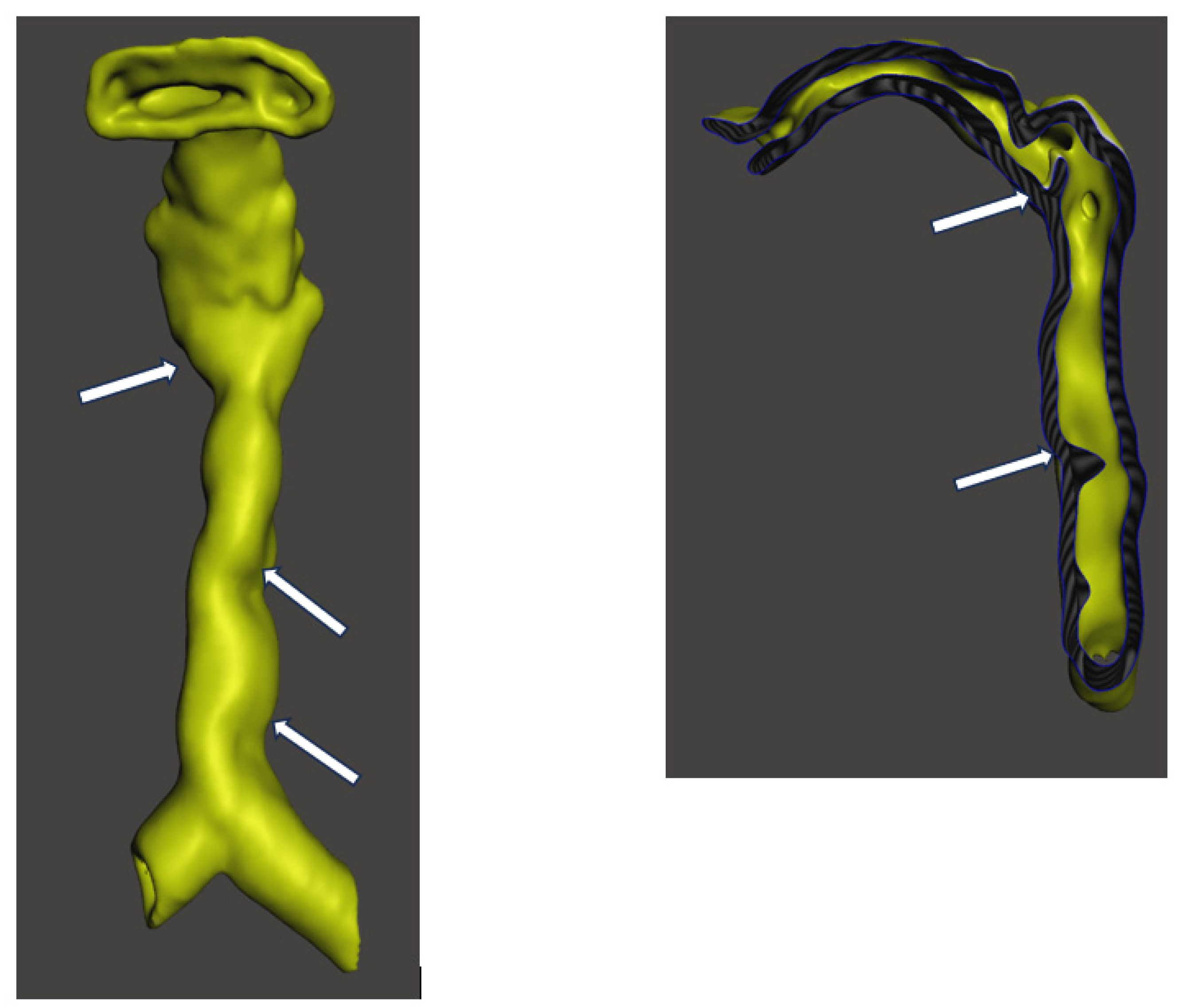
Figure 4.
СT scan, 3dimensional reconstruction shows tortuous and narrowed trachea and patchy infiltrates marked by arrows.
Figure 4.
СT scan, 3dimensional reconstruction shows tortuous and narrowed trachea and patchy infiltrates marked by arrows.
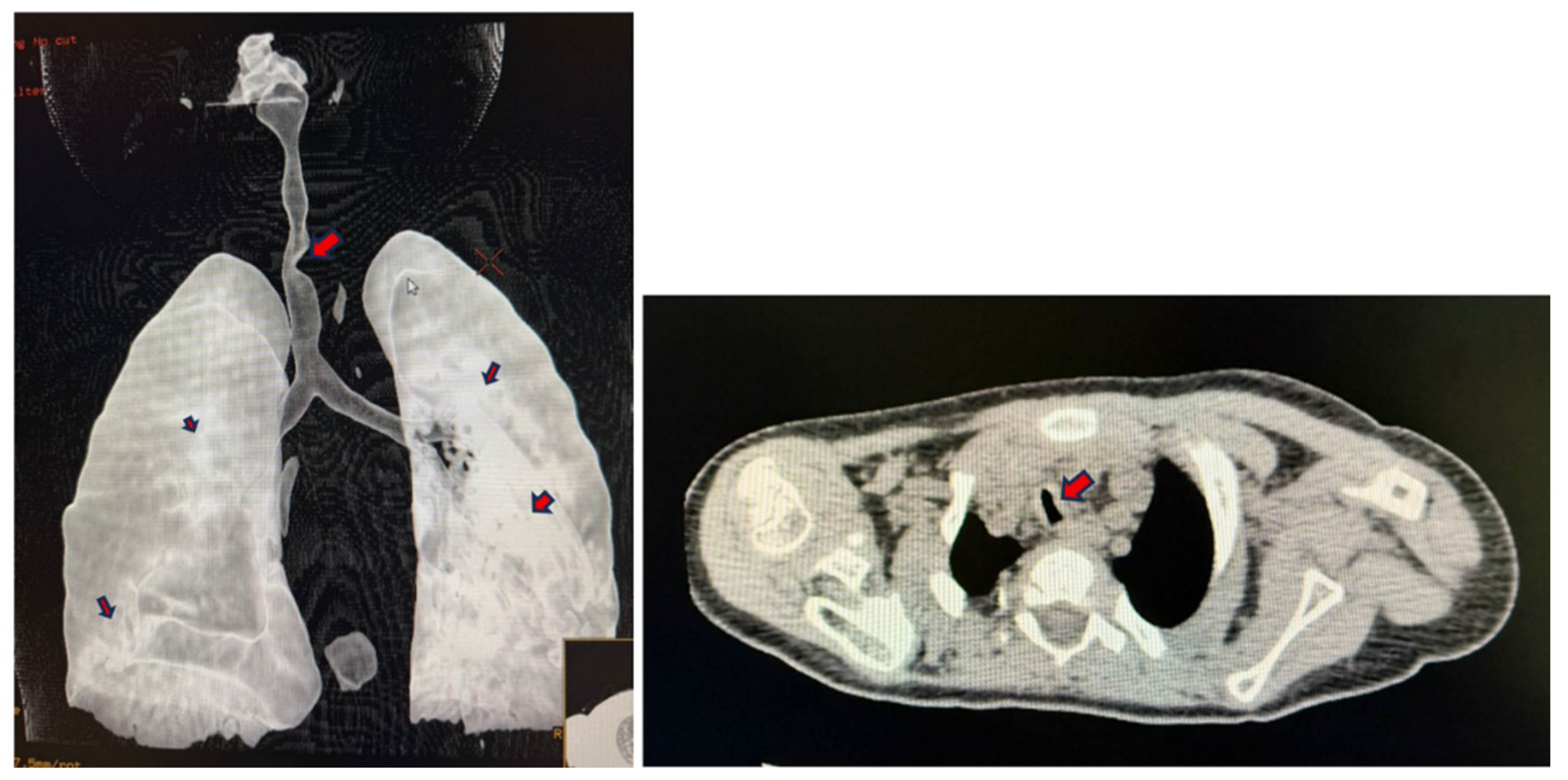
Figure 5.
3dimensional reconstruction of the root of the neck, thoracic inlet showing extra loop of truncus brachiocephalus in upper mediastinum marked with an arrow.
Figure 5.
3dimensional reconstruction of the root of the neck, thoracic inlet showing extra loop of truncus brachiocephalus in upper mediastinum marked with an arrow.
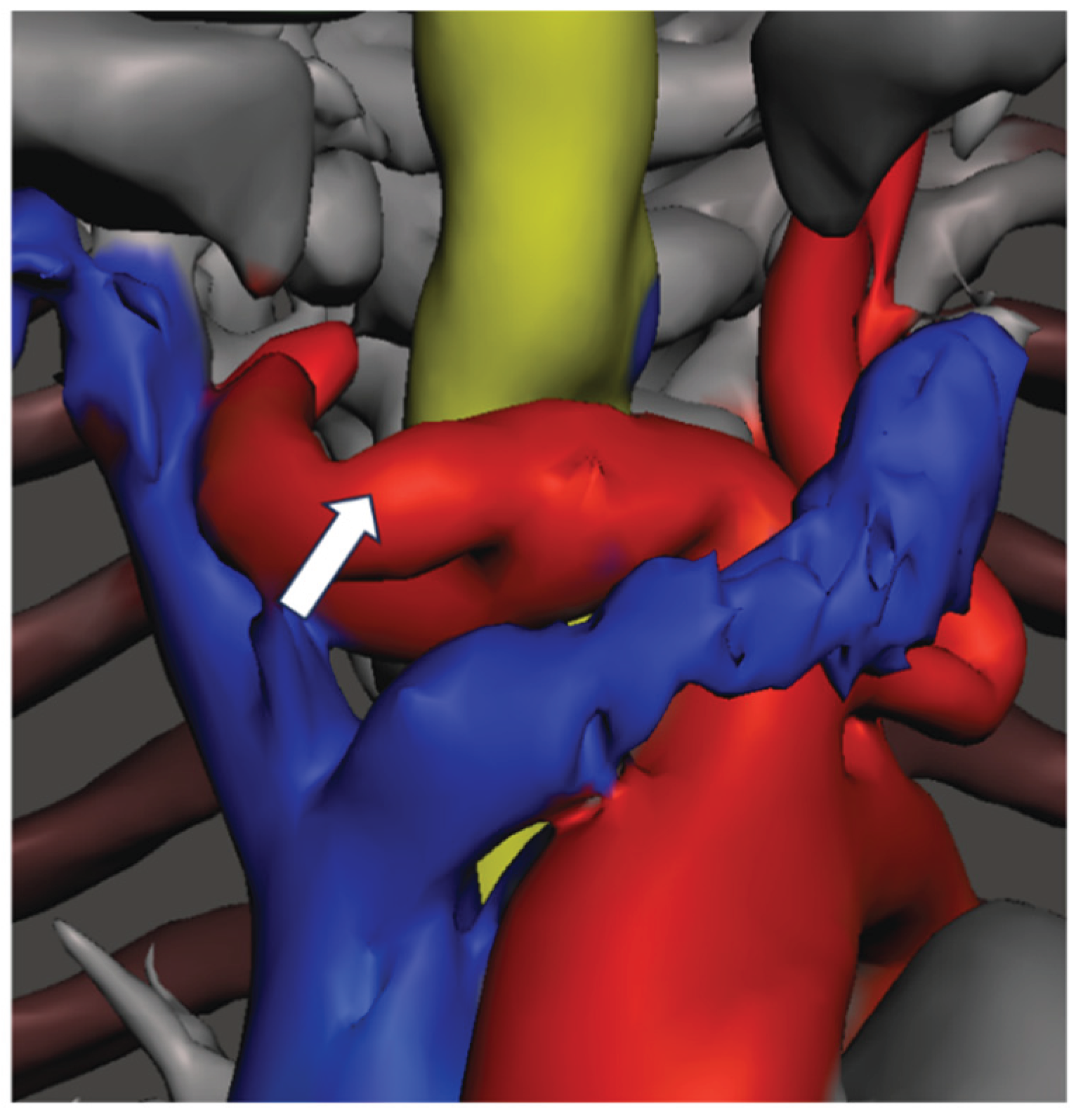

Table 1.
Various types of MPS; reproduced from Braunlin et al. [3], who complied data from Neufeld et al. [4] and Valayannopoulos et al. [5].
| MPS type (eponym) | Incidence per 105 live births; inheritance pattern | Typical age at diagnosis | Typical life expectancy if untreated | Enzyme deficiency | GAG |
|---|---|---|---|---|---|
| MPS I Hurler (H) MPS I Hurler-Scheie (H-S) MPS I Scheie (S) | 0.11-1.67; AR | H: < 1 year H-S: 3–8 years S: 10–20 years | H: death in childhood H-S: death in teens or early adulthood S: normal to slightly reduced lifespan | α-L-iduronidase | DS, HS |
| MPS II (Hunter) | 0.1-1.07; XR | 1-2 years when rapidly progressing | rapidly progressing: death < 15 years slowly progressing: death in adulthood | iduronate-2-sulfatase | DS, HS |
| MPS III (Sanfilippo) A-B-C-D | 0.39-1.89; AR | 4-6 years | death in puberty or early adulthood | heparan sulfamidase (A) N-acetyl-α-D-glucosaminidase (B) acetyl-CoA-α-glucosaminidase N-acetyltransferase (C) N-acetylglucosamine-6-sulfatase (D) | HS |
| MPS IV (Morquio) A-B |
0.15-0.47; AR | 1-3 years | death in childhood- middle age | N-acetylgalactosamine-6-sulfatase (A) β-galactosidase (B) | CS, KS (A) KS (B) |
| MPS VI (Maroteaux-Lamy) |
0-0.38; AR | rapidly progressing: 1–9 years slowly progressing: > 5 years | rapidly progressing: death in 2nd-3rd decade slowly progressing: death in 4-5th decade | N-acetylgalactosamine-4-sulfatase | DS |
| MPS VII (Sly) | 0-0.29; AR | neonatal to adulthood | death in infancy- 4th decade** | β-D-glucuronidase | CS, DS, HS |
| MPS IX (Natowicz)* | unknown | adolescence | unknown | hyaluronidase | CS |
| MPS X*** | unknown AR | childhood-onset | unknown | Arylsulfatase K | DS |
AR: autosomal recessive; CS: chondroitin sulfate; DS: dermatan sulfate; GAG: glycosaminoglycan; H: Hurler syndrome; HS: heparan sulfate; H-S: Hurler-Scheie syndrome; KS: keratan sulfate; S: Scheie syndrome; XR: X-linked recessive. *only 1 patient reported in literature (Natowicz et al. 1996); **death can occur in utero with hydrops fetalis, ***10 patients reported in literature (Al Fahdi I et al.2025).
Table 2.
Airway plan as per the difficult airway society guidelines [11] of plan A, B, C and D; the challenges, consequences and methods to mitigate these challenges.
Table 2.
Airway plan as per the difficult airway society guidelines [11] of plan A, B, C and D; the challenges, consequences and methods to mitigate these challenges.
| Airway plan | Challenges | Consequence | Methods to mitigate |
|---|---|---|---|
| Plan A Endo tracheal intubation |
Reduced mouth opening, large tongue, high anterior larynx, large tongue, limited neck extension. Tortuous trachea with multiple narrowing’s can make passage of the endo tracheal tube difficult. | Access to the larynx and passage of the endo tracheal tube into the trachea will be difficult | Using small endo tracheal tube 1.Nasal intubation 2.Oral intubation with video laryngoscope or Hopkins rod telescope 3.Awake nasal or oral fiberoptic using an airway conduit |
| Plan B Laryngeal Mask airway (LMA) |
Limited mouth opening, large tongue, high anterior larynx, bulky supraglottic | Inserting the LMA and securing a seal will be difficult | Using re-enforced LMA, which are more flexible to reach anterior larynx |
| Plan C Bag and mask ventilation |
Limited mouth opening, large tongue which can fall posteriorly occluding the airway, bulky oropharynx, bulky supra glottis | Inability to pass oxygen beyond the oropharynx due to obstruction | 1.Guedel’s airway to by-pass the tongue base 2.Nasopharyngeal airway to bypass the epiglottis |
|
Plan D Front of neck access |
Short neck, limited extension, large head, small torso Large vessels in the thoracic inlet |
Accessing the cervical trachea will be difficult, large vessel catastrophic haemorrhage, inserting the right sized tracheostomy tube | Avoid tracheostomy, if attempted perform high tracheostomy. Planning the right tracheostomy tube before the surgery |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



