Submitted:
26 February 2026
Posted:
27 February 2026
You are already at the latest version
Abstract
Aminochrome, an endogenous neurotoxin, has been suggested that it is implicated in the loss of dopaminergic neurons that contain neuromelanin in the nigrostriatal system in Parkinson's disease. While aminochrome-induced oxidative stress, as well as its inhibitory effects on microtubule polymerization in neuronal models, are well documented, its impact on autophagosome–lysosome fusion and protein aggregation remains unexplored. The aim of this research was to evaluate the effects of aminochrome on the fusion between autophagosomes and lysosomes, and the accumulation of protein aggregated using SH-SY5Y cells differentiated into dopaminergic neurons. Our findings reveal that aminochrome like vinblastine, delays the fusion of autophagosomes with lysosomes, as evidenced by decreased colocalization of LC3β-LAMP1. This effect was also observed after the addition of autophagy inductors such as rapamycin and trehalose. Moreover, rapamycin and trehalose mitigate aminochrome-induced cell death, whereas vinblastine exacerbates it. Bafilomycin A1, despite reducing LC3β-LAMP1 colocalization, offers protection against aminochrome-induced cytotoxicity. These disruptions in autophagosome-lysosome fusion are associated with accumulation of perinuclear vimentin and ubiquitin aggregates together with increased colocalization between vimentin and ubiquitin. Interestingly, the presence of ubiquitin aggregates at the nuclear level were also observed. It is postulated that the effect of aminochrome on the microtubule network, particularly inhibition of the autophagosome-lysosome fusion and the accumulation of protein aggregates, could be one of the critical events that lead to cell death. Our findings underscore the therapeutic potential of targeting microtubule stabilization and autophagy/UPS induction in Parkinson's disease, an energy-dependent process, highlighting the need for further research into nuclear proteotoxicity mechanisms.
Keywords:
aminochrome
; autophagy
; microtubules
; Parkinson's disease
; protein aggregation
; ubiquitin
; vimentin
1. Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc). A common feature observed in postmortem brains of individuals with PD is the presence of abnormal protein aggregates in soma and neurites of neurons, known as Lewy’s bodies and Lewy’s neurites, respectively [1]. The accumulation of these protein aggregates is recognized as a contributing factor to the neurodegenerative process in both, sporadic and hereditary forms of PD [2].
The clearance of protein aggregates is a normal process in neurons, involving the ubiquitin-proteasome system (UPS) and the autophagy pathway. The latter relies on the formation of autophagosomes and their subsequent fusion with lysosomes for the recycling. Aggregated and misfolded proteins are ubiquitinated and recognized by proteosome for degradation [3]. Under prolonged stress conditions, the accumulation of these protein aggregates can lead to the formation of perinuclear inclusions known as aggresomes. These structures are composed of aggregated, ubiquitinated proteins and intermediate filament among other proteins. Vimentin, an intermediate filament, forms a cage surrounding aggregated proteins and is described as a marker of aggresomes [4,5,6]. An altered fusion process between autophagosomes and lysosomes is known to impact in the clearance of protein aggregates. Deficient protein clearance mechanisms and the accumulation of protein aggregates are common features of several neurodegenerative disorders, including PD [7].
Autophagy inductors may facilitate the clearance of protein aggregates. For instance, rapamycin and trehalose are compounds that stimulate the autophagosome-lysosomes fusion and are known to exert neuroprotective effects. Rapamycin functions as an inhibitor of the PI3K/AKT/mTOR pathway, whereas trehalose can act in both mTOR- dependent or -independent manners [8,9]. Both compounds modulate autophagy and promote mechanisms for clearing protein aggregates in neurodegenerative processes [10,11,12]. In contrast, bafilomycin A1, a vacuolar-type H+-ATPases inhibitor, prevents the fusion of autophagosomes with lysosomes by either disrupting V-ATPase-mediated acidification or Ca2+ depletion SERCA pump. It not only provides protection but also induces toxicity in PD models [13,14,15,16,17].
Although protein aggregation is considered an early event in the development of the PD, it does not appear to be the initial event. The disruption of the microtubule cytoskeleton network may precede it [18,19,20]. Microtubules, functioning as molecular motors that control vesicle trafficking within cells, are crucial for the vesicle fusion necessary for the clearance of the protein aggregates. Indeed, studies in models of PD have identified several neurotoxins that disrupt the microtubules network, [21,22,23,24]. Similarly, drugs that depolymerizes microtubules, such as vinblastine, are known to be neurotoxic [25]. In contrast, drugs that stabilize microtubules network act as neuroprotectors [23]. Nevertheless, the mechanism by which neurotoxins that disrupt the microtubule network impact neuronal degeneration remain to be elucidated.
It has been proposed that aminochrome is the endogenous neurotoxin that triggers neurodegeneration of dopaminergic neurons of the nigrostriatal system under a single-neuron degeneration model [26,27]. The synthesis of neuromelanin, which requires the formation of the neurotoxin aminochrome, is a normal and harmless process due to the presence of DT-diaphorase and glutathione transferase M2-2, which prevent the neurotoxic effects of aminochrome [26]. Aminochrome neurotoxicity is associated with the formation of aggregates of α- and β-tubulin [28,29] and the inhibition of microtubules polymerization through the formation of adducts with tubulin. Interestingly, the aminochrome effects on the microtubule network are event prior to cell death [23]. Aminochrome can disrupt microtubule-network-dependent processes, including mitochondrial transport, vesicular trafficking, and autophagy. Although some studies have reported the effects of aminochrome on the clearance mechanisms of misfolded or altered proteins [30], the specific mechanisms by which aminochrome-induced microtubule alterations contribute to neuronal death are not yet fully understood.
The aim of this study was to determine whether aminochrome inhibits the fusion of autophagosomes with lysosomes in a neuronal cell line by impairing the clearance of protein aggregates. To address this, we first examined whether aminochrome disrupts autophagosome–lysosome fusion and induces cell death in the presence of autophagy inducers and microtubule polymerization inhibitors. We then assessed whether aminochrome leads to the accumulation of intracellular protein aggregates.
2. Materials and Methods
2.1. Reagents
Dopamine and tyrosinase were purchased from Sigma-Aldrich (H8502 and T3824-50KU, respectively). For viability/cytotoxicity analysis, calcein AM (Invitrogen, L3224) and propidium iodide (Sigma-Aldrich, P4864) were used. For QRT-PCR analysis, a Brilliant III Ultra Fast SYBR Green QRT-PCR Master Mix from Agilent Technologies (Agilent Technologies, 600886) was used. For immunofluorescent analysis, the following were used: paraformaldehyde (PFA) (Winkler, 1.04005.1000) and DAKO fluorescent mounting medium (Dako, S3023). The following primary antibodies: anti-LAMP-1 (Santa Cruz Biotechnology, sc-17768), anti-ubiquitin (Santa Cruz Biotechnology, sc-271289) and anti-vimentin (Santa Cruz Biotechnology, sc-5565), purchased from Santa Cruz Biotechnology; and anti-LC3β (Abcam, ab51520) and anti-vimentin (Abcam, ab8978), from Abcam, were used. The following secondary antibodies: Alexa Fluor 488 donkey anti-rabbit (Jackson Immunoresearch, 711-545-152), Alexa Fluor 488 goat anti-mouse IgG (Jackson Immunoresearch, 115-545-003) and Cy3™ Goat anti-mouse IgG (Jackson Immunoresearch, 115-165-166) purchased from Jackson Immunoresearch, and Alexa Fluor 546 goat anti-rabbit (Life Technologies, A11035) and Alexa Fluor 546 goat anti-mouse (Life Technologies, A11030) from Life Technologies, were also used. To mark the nucleus, Hoechst (Invitrogen, H3569) was used. For the differentiation procedure, retinoic acid (Santa Cruz Biotechnology, sc-200898) and TPA (12-O-tetradecanoylphorbol-13-acetate) (Santa Cruz Biotechnology, sc-202021) from Santa Cruz Biotechnology were used. For cell treatment, vinblastine acquired from Laboratorio Chile under the name of Lemblastine, trehalose (Sigma-Aldrich, T0167) from Sigma-Aldrich and rapamycin (Santa Cruz Biotechnology, sc-3504A) and bafilomycin A1 (Santa Cruz Biotechnology, sc-201550A) both purchased from Santa Cruz Biotechnology were used.
2.2. Cell Culture and Differentiation
The cell culture and differentiation protocol were followed as described in Briceño [23]. The SH-SY5Y cell line was purchased from ATCC (ATCC, CRL-2266). The growth medium used was DMEM/F12 (Dulbecco’s Modified Eagle medium/F12 1:1) (Hyclone, sh30004.04) supplemented with 7.5% adult bovine serum (Biological industries, 04-003-1B), 1.5% fetal bovine serum (Biological industries, 04-001-1A), 1X of non-essential amino acids solution (MEM NEAA 100X) (Hyclone, sh30238.01), and 2X penicillin–streptomycin-amphotericin B solution (Biological industries, 03-033-1B) at a concentration of 200 U/mL of penicillin G sodium salt, 0.2 mg/mL of streptomycin sulfate, and 0.5 µg/mL of amphotericin B [31]. Cell cultures were kept in an incubator at 37ºC with an atmosphere of 5% CO2. When cells reached at 80% confluence, the differentiation of SH-SY5Y cells into dopaminergic neurons was initiated. Differentiation was carried out in two steps: in the first step, the cells were cultured for 3 days in DMEM/F12 media (supplemented with 4% bovine serum, 1X MEM, 2X penicillin–streptomycin-amphotericin B solution) containing 10 μM retinoic acid. In the second stage, the cells were cultured for 3 more days in the same media but instead of retinoic acid, 80 nM TPA (12-O-tetradecanoylphorbol-13-acetate) was used [32,33]. After 6 days of differentiation, the treatments were carried out for 0.5, 4, 9, 14 or 16 h with 50 μM aminochrome. Also, the cells were incubated with 10 μM rapamycin, 5 nM bafilomycin A1, 100 mM trehalose or 10 μM vinblastine in the presence or absence of 50 μM aminochrome for 16 h. Cells were treated with aminochrome in serum-free media for 1.5 h. After this time, the media was removed and replaced with fresh growth media, maintaining the other treatments until incubation time was completed. For treatments with bafilomycin (5 nM), rapamycin (10 µM), or trehalose (100 mM), cells were preincubated for 24 h.
2.3. Synthesis and Purification of Aminochrome
The synthesis and purification protocol for aminochrome was followed as described by Briceño et al. [23]. Dopamine (7.5 mmol) and 10 ng of tyrosinase were incubated in 25 mM potassium phosphate buffer pH 6 for 15–20 min at room temperature. To purify aminochrome, the incubation solution was loaded on a CM-Sephadex C50-1000 (18 x 0.7 cm) column (Sigma-Aldrich, C25120). The red–orange solution corresponding to aminochrome was collected and detected spectrophotometrically by measuring the absorbance at 480 nm. Aminochrome concentration was determined by the molar extinction coefficient of 3058 M-1 cm-1 [34].
2.4. Assessment of Cell Viability and Death
The protocol for assessing cell death was followed as described in Briceno et al. [23]. For live/dead experiments, cells were cultured in 24 well dishes according to culture and differentiation protocol previously described. Cell death was determined by using calcein AM and propidium iodide, two fluorescent reagents that discriminate the population of live cells from the dead cell population. After aminochrome treatment for 16 h at 37 °C, SH-SY5Y cells were washed with PBS (Dulbecco’s Phosphate Buffered Saline) (Biological Industries, 02-023-5A) 3 times for 5 min. Then, cells were incubated for 30 min in the dark with 1.5 μM of calcein AM and 1 μM of propidium iodide, both prepared in PBS. Live and dead cell counting was performed in a Leica fluorescence microscope (DM1L). One hundred cells per replicate were counted.
2.5. Immunofluorescence LC3β-LAMP1 Double Staining
SH-SY5Y cells were cultured and differentiated on 12 mm gelatin-coated coverslips placed in 24-well plates. Cells were treated with 50 µM aminochrome for 16 h at 37 °C, washed with PBS, and fixed with 4% paraformaldehyde (PFA) for 30 min. To permeabilize the membrane and facilitate LC3β-I exposure, cells were incubated with 0.5% saponin for 15 min in the dark. Blocking was performed for 1 h using 6% bovine serum albumin (BSA). For LC3β and LAMP1 colocalization studies, cells were incubated separately overnight at 4 °C with rabbit anti-LC3β antibody (1:2,000) and mouse anti-LAMP1 antibody (1:100). Secondary antibodies, Alexa Fluor 488 donkey anti-rabbit and Alexa Fluor 546 goat anti-mouse (both at 1:200 dilution), were applied separately for 1.5 h. Nuclear staining was performed with Hoechst reagent (1:10,000) for 5 min. Coverslips were mounted on slides using DAKO fluorescent mounting medium. Images were acquired using a Carl Zeiss Observer.Z1 Axio fluorescence microscope (Göttingen, Germany) and analyzed with AxioVision Rel 4.8 software for deconvolution.
2.6. Immunofluorescence Vimentin Staining
SH-SY5Y cells were cultured and differentiated in 24 well plates on 12 mm coverslips previously gelatinized. Treatments were carried out in the presence and absence of 50 μM aminochrome for 16 h at 37 °C. Cells were washed with 1X Tris-buffered saline (TBS) and fixed with 4% PFA for 30 min. The cells were blocked for 1 h with 6% BSA 0.5% Triton X-100 1X TBS. The cells were incubated with primary antibody 1:100 dilution of rabbit anti vimentin overnight at 4 °C. After washing the cells with TBS, a 1:200 dilution of secondary antibody (AF-546 goat anti-rabbit IgG) was incubated for 1.5 h. For nucleus staining, Hoechst reagent was used at a 1:10,000 dilution in buffer Tris-EDTA for 5 min. Finally, coverslips were mounted onto slides using DAKO fluorescent mounting medium. Images were acquired using a Carl Zeiss Observer.Z1 Axio fluorescence microscope (Göttingen, Germany) and analyzed with AxioVision Rel 4.8 software for deconvolution.
2.7. Immunofluorescence Vimentin-Ubiquitin Double Staining
SH-SY5Y cells were cultured and differentiated on 12 mm gelatin-coated coverslips in 24-well plates. Treatments with 50 μM aminochrome were performed for 16 h at 37 °C, with untreated controls included. Cells were washed with 1X TBS and fixed with 4% PFA for 30 min. Blocking was carried out for 1 h in 6% bovine serum albumin (BSA) and 0.5% Triton X-100 in 1X TBS. Cells were incubated overnight at 4 °C with mouse anti-ubiquitin primary antibody (1:250 dilution) in 6% BSA and 0.5% Triton X-100 in 1X TBS. After washing, cells were incubated for 1.5 h in the dark with Alexa Fluor 488 goat anti-mouse IgG secondary antibody (1:200 dilution) in 3% BSA in 1X TBS. Subsequently, cells were incubated overnight at 4 °C in the dark with mouse anti-vimentin primary antibody (1:1,000 dilution) in 6% BSA and 0.5% Triton X-100 in 1X TBS. Following washes, cells were incubated for 1.5 h in the dark with Cy3™ goat anti-mouse IgG secondary antibody (1:800 dilution) in 3% BSA in 1X TBS. Nuclear staining was performed using Hoechst reagent (1:10,000 dilution) in Tris-EDTA buffer for 5 min. Coverslips were mounted on slides using Dako fluorescent mounting medium and stored at 4 °C in the dark. Images were acquired using a Carl Zeiss Observer.Z1 Axio fluorescence microscope (Göttingen, Germany) and analyzed with AxioVision Rel 4.8 software for deconvolution, including extended focus.
2.8. Quantitative Real-Time PCR
The total RNA from SH-SY5Y cells was extracted with TRIZOL reagent (Invitrogen, 15596-026) according to the manufacturer’s protocol and was quantified using a Nanodrop 3300 (Thermo). The cDNA was synthesized using oligo-dT (IDT) and Epicenter RT reagents according to the manufacturer’s instructions. Comparative quantitation real-time PCR for the UBI gene (Fw: 5’- CGCACCCTGTCTGACTACAA -3’; Rv: 5’AGGGATGCCTTCCTTGTCTT -3’) and LC3 gene (Fw: 5’-CAGTACTTGCATGGGGTTCA-3’; Rv: 5’-TCTGGTTTTCCCCGTTACAG-3’) were performed in triplicate using Brilliant III Ultra Fast SYBR Green QRT-PCR Master Mix in a Stratagene Mx3000p Detection system. The mRNA levels were normalized through the housekeeping gene GAPDH (Fw: 5’-GCCAAAAGGGTCATCATCTC-3’; Rv: 5’-TGTGGTCATGAGTCCTTCCA-3’). The following experimental run protocol was used: 25 °C for 1 s, 50 °C for 10 min, 95 °C for 3 min, followed by 40 cycles of 95 °C for 20 s, 60 °C for 20 s and a last segment that includes 95 °C for 1 min, 55 °C for 30 s and 95 °C for 30 s. Mx3300P Software was used to analyze real-time data.
2.9. Statistical Analysis
All data were expressed as mean ± SEM. Statistical significance was assessed using analysis of variance (ANOVA) for multiple comparisons followed by the Newman–Keuls post hoc test, while an unpaired t-test was used for two-group comparisons. In all cases, the significance level was set at α = 0.05. Normality was assessed using the Kolmogorov–Smirnov test.
3. Results
3.1. Inhibition of Autophagosome-Lysosome Fusion by Aminochrome in SH-SY5Y Cells
In order to determine whether aminochrome affects the fusion of autophagosome with lysosomes in SH-SY5Y cells, immunofluorescence double staining for microtubules-associated proteins light chains (LC3β) and Lysosomal-associated membrane protein 1 (LAMP1) (Figure 1A) was performed. In these experiments, image analysis for colocalization was performed, and the Pearson coefficient was quantified (Figure 1B) [35]. A significant decrease in the Pearson coefficient was observed in presence of 50 µM of aminochrome compared with control without aminochrome (26% decrease, P ˂ 0.01). Moreover, when the cells were treated with rapamycin, an inducer of autophagy, a significant increase in the Pearson coefficient was observed (27% increase, P ˂ 0.01) compared with control which was similarly inhibited by aminochrome (61% decrease, P ˂ 0.001). Likewise, when the cells were treated with bafilomycin A1, an inhibitor of autophagosome-lysosomes fusion [15,36,37], a significant decrease in the Pearson coefficient was observed (51% decrease, P ˂ 0.001) compared with control. However, no further decrease in the Pearson coefficient was observed when the cells were also incubated with 50 µM aminochrome. These results suggest that aminochrome inhibits the autophagosome-lysosomes fusion in a manner like bafilomycin A1, as no differences were observed between the effects of bafilomycin alone and in the presence of aminochrome.
3.2. Bafilomycin Inhibits the Autophagosome-Lysosome Fusion But Protects Against Aminochrome Toxicity in SH-SY5Y Cells
It was next investigated whether inhibition of autophagosome-lysosome fusion leads to toxic effects in SH-SY5Y cells. Cell death induced by aminochrome in the presence of rapamycin, an autophagy inducer, or bafilomycin A1, an inhibitor of autophagosome-lysosomes fusion was measured using calcein AM and propidium iodide in SH-SY5Y cells (Figure 1C). A significant increase (30-fold, P ˂ 0.001) in cell death was observed in presence of 50 µM of aminochrome compared with control. Moreover, when the cells were treated with aminochrome in the presence of rapamycin, a significant decrease (17%, P ˂ 0.01) in cell death was observed compared with aminochrome alone. Similarly, when the cells were treated with bafilomycin A1, a significant decrease (21%, P ˂ 0.001) in cell death was observed compared with aminochrome. Taken together, these results suggest that the inhibition of autophagosome-lysosomes fusion induced by aminochrome is toxic to cells, but not in the presence of bafilomycin A1 which provides protection against aminochrome toxicity, similar to the protective effect induced by rapamycin.
3.3. Aminochrome and Vinblastine Inhibit the Fusion of Autophagosome with Lysosomes in the Presence of Autophagy Inducers in SH-SY5Y Cells
To determine whether alterations in the microtubule network are primarily responsible for the inhibition of autophagosome-lysosome fusion in SH-SY5Y cells, immunofluorescence double staining for microtubules-associated proteins light chains (LC3β) and lysosomal-associated membrane protein 1 (LAMP1) (Figure 2A) was performed. The cells were incubated with aminochrome in the presence of vinblastine, a microtubules depolymerizing-agent [38,39], and rapamycin and trehalose, the latter autophagy inducers. In these experiments, image analysis for colocalization was conducted, and the Pearson coefficient was quantified (Figure 2B). A significant decrease (27%, P ˂ 0.001) in the Pearson coefficient was observed in the presence of 50 µM of aminochrome compared with control, which was further decreased (63%, P ˂ 0.001) in the presence of vinblastine. Moreover, when the cells were treated with rapamycin, an inductor of autophagy, a significant increase (22%, P ˂ 0.01) in the Pearson coefficient was observed compared with control which was similarly inhibited by aminochrome and vinblastine, (58% and 64%, P ˂ 0.001) respectively. Similarly, when the cells were treated with trehalose, an inductor of autophagy [40], a significant increase (25%, P ˂ 0.01) in the Pearson coefficient was observed compared with control which was similarly inhibited by aminochrome and vinblastine, (46% and 68%, P ˂ 0.001, respectively). These results suggest that microtubules-depolymerizing agents, such as aminochrome and vinblastine, inhibit the fusion of autophagosome with lysosomes even in the presence of autophagy inducers in SH-SY5Y cells.
3.4. Aminochrome Toxicity Is Enhanced in the Presence of Vinblastine But Inhibited by Autophagy Inductors
In order to evaluate whether the microtubules disruption is one of the primary events responsible for cell death is through the inhibition of autophagosome-lysosomes fusion in SH-SY5Y cells, cell death induced by aminochrome in the presence of vinblastine, a well-known microtubules-depolymerizing agent, was measured using calcein AM and propidium iodide in SH-SY5Y cells. Similarly, cell death induced by aminochrome in the presence or absence of rapamycin and trehalose, both autophagy inducers, was also measured (Figure 2C). A significant increase in the cell death (37 -fold, P ˂ 0.001) was observed in the presence of 50 µM of aminochrome compared with control, which was enhanced (21%, where P ˂ 0.001) in the presence of vinblastine. Moreover, when the cells were treated with rapamycin, in the presence of aminochrome, a significant decrease in the cell death (22%, P ˂ 0.001) was observed compared with aminochrome alone. Similarly, when the cells were treated with trehalose in the presence of aminochrome, a significant decrease in the cell death (53%, P ˂ 0.001) was observed compared with aminochrome alone. Additionally, a significant increase (10-fold, P ˂ 0.001) in cell death was observed in the presence of vinblastine compared with control. In contrast, when the cells were treated with trehalose or rapamycin, in the presence of vinblastine, no significant differences were observed compared to vinblastine alone. Consequently, aminochrome toxicity increases in the presence of vinblastine, but this is reduced by autophagy inducers (Figure 2C).
3.5. Increased Perinuclear Vimentin Aggregates in SH-SY5Y Cells Is Induced by Aminochrome
In this study, it was observed that alterations in the microtubule network induced by aminochrome are a critical event affecting the autophagosome-lysosomes fusion, leading to cell death. Our objective was to investigate whether, under these conditions, aminochrome also induces the formation of perinuclear vimentin aggregates. Immunofluorescence staining for vimentin (Figure 3A) was performed, and the quantification of vimentin aggregates in the presence of aminochrome was conducted (Figure 3B). A significant increase (6-fold, P ˂ 0.001) of perinuclear vimentin aggregates were observed in the presence of 50 µM of aminochrome compared with control without aminochrome (Arrowhead, Figure 3A).
3.6. Increased Colocalization of Vimentin and Ubiquitin and Changes in Their Cellular Distribution Induced by Aminochrome
Perinuclear inclusions are composed of aggregated, ubiquitinated proteins and intermediate filaments. Vimentin sorrounds aggregated proteins, such as misfolded proteins, which are ubiquitinated and recognized by the proteosome for clearance [3,4,5,6]. It was previously shown that aminochrome induces the accumulation of perinuclear vimentin aggregates. Our objective was to investigate the effect of aminochrome on cellular distribution and the colocalization of ubiquitin and vimentin. Immunofluorescence double staining for vimentin and ubiquitin was performed (Figure 3C and 3E), image analysis for colocalization and quantification of the Pearson coefficient were conducted (Figure 3D). Similar to the results observed in Figure 3A, a homogeneous cytoplasmic vimentin labeling was noted, which changed to a perinuclear cellular distribution in the presence of aminochrome compared with control (Arrow). In the case of ubiquitin staining, it appears in the cytoplasm and nucleus, but was mainly concentrated in perinuclear (arrow) and nuclear (arrowhead) regions in the presence of aminochrome compared with control (Figure 3C and 3E). Ubiquitin in the perinuclear region colocalized with vimentin (Red arrow, Figure 3C), where a significant increase (12%, P ˂ 0.05) in the Pearson coefficient was observed in the presence of 50 µM of aminochrome compared with control (Figure 3D). The extended focus analysis of the deconvolved images in Z-stack provided more information about the cellular distribution of small vimentin aggregations. These aggregations were observed not only in perinuclear region but also in regions proximal or distal to the perinuclear region within cellular processes when the cells were treated with aminochrome (Figure 3E).
3.7. Increased LC3β and Ubiquitin Expression in SH-SY5Y Cells Induced by Aminochrome
In this study, it was observed that aminochrome affects the autophagosomes-lysosomes fusion and alters the clearance of proteins aggregates. Our objective was to investigate whether aminochrome also induces changes in the expression of genes related to autophagy and ubiquitin-proteasome system, such as LC3β and ubiquitin. Previously, it was reported that aminochrome affects the TUBB3 expression, where this effect possibly being a compensatory mechanism to protect against cellular damage [23]. In this context, it was studied whether early changes in the expression of LC3β and ubiquitin are observed. Gene expression was measured using qRT-PCR. It was observed that when cells were incubated in the presence of 50 µM aminochrome, a significant increase (5-fold, P ˂ 0.001) in the LC3β gene expression at 4 h compared with control was noted (Figure 4A). Similarly, a significant increase (9-fold, P ˂ 0.001) in the ubiquitin expression at 4 h compared to control was observed (Figure 4B).
A possible mechanism by which aminochrome affects autophagosome-lysosome fusion and promotes protein aggregates formation is illustrated in the following figure (Figure 5).
4. Discussion
4.1. Aminochrome Neurotoxicity and Microtubules Dysfunction
Microtubules, composed of α- and β-tubulin heterodimers, are the largest components of the cytoskeleton and play critical roles in cellular processes such as protein aggregate clearance, axonal transport, and organelle distribution and formation. Dysfunction of the microtubule network appears to be an early event in PD pathogenesis. Abnormal protein aggregation in the substantia nigra has been observed during the pre-motor stages of PD [41]. Microtubule destabilization has been observed in fibroblasts derived from both genetic and idiopathic PD patients as well as in animal models of PD [19,42,43]. Furthermore, early microtubule alterations have been replicated in animal models of PD. For instance, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induces early microtubule instability, which can be attenuated by the microtubule stabilizer Epothilone D, reducing nigrostriatal degeneration in dopaminergic neurons of C57BL/6 mice [20]. These findings suggest that modifications in the microtubule network may represent reversible early events that signal the onset of PD.
Several exogenous neurotoxins have been used to model PD; however, few models successfully replicate the slow and progressive nature of the disorder [44]. Notably, premotor symptoms can manifest decades before the onset of motor symptoms, a period during which significant degeneration of dopaminergic neurons has already occurred [45]. Aminochrome has been proposed as an endogenous neurotoxin responsible for nigrostriatal degeneration in PD [26,27]. It is implicated in multiple pathogenic mechanisms associated with PD, including the formation of neurotoxic α-synuclein oligomers, cytoskeletal dysfunction, neuroinflammation, oxidative stress through reactive oxygen species (ROS) production, mitochondrial impairment, dysfunction of proteasomal and lysosomal degradation systems, and endoplasmic reticulum stress [26]. Importantly, many of these mechanisms depend on an intact cytoskeletal network, which is disrupted early by aminochrome, prior to neuronal death [23]. Specifically, aminochrome induces aggregation of α- and β-tubulin [28,29] and inhibits microtubule polymerization via the formation of adducts with tubulin [23].
In this study, we investigated how aminochrome-induced microtubule alterations contribute to neuronal death. Our data indicate that aminochrome toxicity is mediated, in part, by inhibition of autophagosome–lysosome fusion, an effect that is potentiated by vinblastine, a microtubule-depolymerizing agent. Several mechanisms could underlie this fusion impairment, including microtubule instability and dysregulation of cytosolic calcium levels [15]. It is possible that the inhibition of microtubule polymerization induced by aminochrome is responsible for the observed decrease in autophagosome-lysosome fusion. The fact that vinblastine enhances this inhibition, evidenced by a further reduction in the Pearson correlation coefficient, and simultaneously increases cell death suggests that microtubule destabilization may be a key event triggering neuronal death. Previous studies have demonstrated that microtubule instability impairs autophagosome-lysosome fusion. For instance, vinblastine has been shown to reduce this fusion in HeLa cells and to promote cell death in RCSN-3 cells [25,39]. However, the extent to which inhibition of autophagosome-lysosome fusion contributes directly to cell death remains unclear. Some evidence suggests that impaired autophagic flux promotes the accumulation of protein aggregates [46]. In support of this, earlier studies have shown that aminochrome induces the formation of cytoskeletal protein aggregates [28]. Based on our findings, we propose that the disruption of the microtubule network by aminochrome particularly its effect on autophagosome-lysosome fusion may represent a critical mechanism contributing to cell death, though likely not the only one.
4.2. Bafilomycin A1 Protects Against Aminochrome Toxicity
Bafilomycin A1, a potent inhibitor of vacuolar-type H+-ATPases (V-ATPase), prevents the acidification of endosomes and lysosomes and disrupts autophagic flux [47]. Whether bafilomycin A1 inhibits autophagic flux by preventing autophagosome-lysosome fusion or by blocking lysosomal degradation remains under debate [15,36,48,49]. Increasing lysosomal pH has been proposed as responsible for autophagosome-lysosome fusion block and autophagy pathway inhibition. However, new studies demonstrated that bafilomycin A1-mediated fusion block is attributed to the depletion of the Ca2+ SERCA pump, rather than solely to loss of V-ATPase-mediated acidification [15,16]. In general, the mechanisms of action of bafilomycin A1 are varied and complex. This diversity of effects for bafilomycin A1 was described for a wide range of concentrations, some of them associated with toxic and protective effects and depending on treatment time [29,47,50]. To investigate the involvement of fusion inhibition in aminochrome-induced neuronal death, the SH-SY5Y cells were preincubated with 5 nM bafilomycin A1 and subsequently exposed to aminochrome in the presence of bafilomycin A1. Data show that aminochrome induces significant neuronal death in SH-SY5Y cells and significantly decreases autophagosome-lysosome fusion. While the inhibition of autophagosome-lysosome fusion is lower with aminochrome than with bafilomycin A1, reflected with a higher Pearson coefficient, aminochrome exerts a stronger effect on cell death compared with bafilomycin A1. These findings suggest that inhibition of autophagosome-lysosome fusion alone cannot fully account for aminochrome-induced neuronal death. In this study, it was shown that bafilomycin A1 protects against aminochrome toxicity without increasing its effect on fusion. How bafilomycin A1 exerts a neuroprotective effect against aminochrome-induced toxicity is unknown. Cytoprotective effects at low doses of bafilomycin A1 (≤10 nM or as high as 100nM) were reported [50]. Pivtoraiko et al. [50] reported that cytoprotection was associated with preserving cathepsin maduration and maintenance of autophagic flux under chloroquine-induced toxicity. On the other hand, prior studies suggested that bafilomycin-induced protection could involve competition between bafilomycin and aminochrome for the same binding site [29,51]. One potential explanation is that fusion inhibition may be linked to dysregulated calcium signaling. Increased calcium cytosolic is not only involved in the fusion inhibition, but also in autophagy activation [52]. The reversible binding of bafilomycin to V-ATPase may be displaced by aminochrome, potentially enhancing bafilomycin’s effect on SERCA by increasing cytosolic calcium levels and activating additional autophagy-related pathways. Further research is required to clarify the precise role of bafilomycin A1 in this context.
4.3. Autophagy Inducers Protect Against Aminochrome Toxicity
Rapamycin blocks the mTORC1 complex, thereby inducing autophagy [53]. The mTORC1 complex plays a central role in autophagy initiation by permitting phagophore formation [54]. In contrast, trehalose can induce autophagy via mTOR-dependent or -independent pathways, or through activation of TFEB (transcription factor EB) [9,40,55]. Although, the precise neuroprotection mechanism induced by trehalose remains unclear, evidence suggests that it not only enhances autophagy, but also inhibits misfolded proteins aggregation and exerts anti-inflammatory effects [12,56,57]. Both, rapamycin and trehalose have been shown to mitigate toxicity induced by various neurotoxic agents [25,58]. To determine how autophagic inducers protect against aminochrome-induced neuronal death, SH-SY5Y cells were preincubated with rapamycin and subsequently exposed to aminochrome and rapamycin. Data presented here indicate that rapamycin significantly reduces aminochrome-induced neuronal death, despite impairing autophagosome–lysosome fusion. Under these conditions, how can rapamycin exert a neuroprotective effect against aminochrome-induced toxicity? Possibly, previous activation of autophagy by rapamycin may facilitate the early clearance of protein aggregates induced by aminochrome, preventing their accumulation and subsequent engagement of alternative toxic pathways. In parallel experiments, the SH-SY5Y cells were preincubated with trehalose and then exposed to aminochrome and trehalose. Data presented here show that trehalose significantly decreases aminochrome-induced neuronal death, exhibiting a stronger protective effect than rapamycin. Trehalose-mediated neuroprotection likely involves multiple mechanisms, autophagy activation, inhibition of proteins aggregation, and anti-inflammatory effects. Similar to rapamycin, trehalose may enhance the early removal of aminochrome-induced protein aggregates, thereby preventing their accumulation and attenuating downstream toxic processes.
4.4. Aminochrome Induces Nuclear and Perinuclear Aggregated-Protein Formation
Neurons are particularly vulnerable to oxidative damage, which promotes protein oxidation favoring misfolding and abnormal association [59]. When protein-folding capacity becomes overwhelmed, misfolded and aggregated proteins are primarily degraded through two major quality-control systems: the UPS and autophagy. These systems work in concert to maintain proteostasis through of cellular clearance and recycling. They remove damaged organelles, protein complexes, and lipid structures, including aggregates of misfolded proteins known as aggressomes [60]. These latter are typically formed when the production of misfolded proteins exceeds the degradative capacity of the UPS. Under prolonged stress, failure of these clearance pathways results in the accumulation of insoluble protein deposits [3]. Multiple proteins participate in proteostatic networks. Ubiquitin tags abnormal or misfolded proteins for degradation, while vimentin forms a cage-like structure surrounding ubiquitinated aggregates, thereby defining aggresomes. These inclusions usually contain various proteins, including α-synuclein, tubulin, ubiquitin, in addition to vimentin [61]. Previous studies demonstrated that aminochrome promotes the formation of perinuclear actin and tubulin aggregates, generates adducts with tubulin and destabilizes the microtubule network [23]. In the present study, accumulation of perinuclear vimentin-aggregates and cellular projections were induced by aminochrome. Furthermore, both perinuclear and nuclear ubiquitin labeling were detected in the presence of aminochrome. These findings suggest that aminochrome-induced microtubule destabilization and inhibition of autophagosome–lysosome fusion may contribute to protein aggregate accumulation. Consistent with previous reports showing that aminochrome toxicity involves the formation of synuclein oligomers [62] and that misfolded α-synuclein impairs UPS function [63], UPS inhibition may also occur under these conditions. Although the UPS was not directly evaluated in this study, previous evidence indicates that aminochrome disrupts the ubiquitin-protesome pathway [64], thereby promoting the agressomes formation [65]. Here, the results showed that aminochrome increased ubiquitin-vimentin colocalization which could indicate the accumulation of abnormal proteins that are not efficiently eliminated by UPS. While the perinuclear vimentin staining pattern supports the formation of aggresome-like structures, further studies are needed to discriminate whether misfolded protein inclusion bodies and insoluble protein deposits are also generated.
Interestingly, the proportion of cells displaying aggregates is higher than of dead cells under the same conditions, suggesting that not all cells harboring aggregates are committed to cell death. In Parkinson’s disease, surviving neurons frequently contain cytoplasmic inclusions known as Lewy bodies [66,67], suggesting that aggregate formation may represent a protective adaptation or an intermediate stage preceding cell death. Additional investigations are required to clarify this relationship. Similarly, it is known that aggresomes require the formation of autophagosomes for their elimination by autophagy [68]. Poly-ubiquitination of misfolded proteins favors both the formation of aggresomes and subsequent elimination by autophagy [69,70]. Since aminochrome toxicity also compromises autophagic-lysosomal dysfunction [29,62], this could further contribute to the accumulation of these aggregates. Indeed, the accumulation of ubiquitinylated protein aggregates in the post-mortem brain of patients with PD suggests a dysfunction of the aggresome-autophagy pathway [69,71]. Possibly, the simultaneous action of several of these mechanisms may act synergistically to promote the formation and accumulation of protein aggregates and, consequently, to cell death where destabilized microtubules determine the initiation of several irreversible processes.
Ubiquitin participates in the clearance of nuclear aggregates through the nuclear proteasomal system [72]. Under conditions of prolonged cellular stress, the formation of nucleolar aggresomes has also been documented [73,74]. The study shows the presence of aminochrome-induced nuclear ubiquitin aggregates, providing new insights into the nuclear effects of aminochrome. Whether aminochrome is actively transported into the nucleus to exert its effects directly or indirectly remains to be determined. The results shown in this study suggest the accumulation of altered proteins that are being ubiquitinated but not degraded by the nuclear proteasome, possibly due to impaired proteasomal activity or an overload that surpasses its degradative capacity. The presence of these aggregates likely reflects cumulative oxidative stress that may compromise nuclear proteins and other macromolecules. Consistent with this, previous studies have shown that aminochrome disrupts the cellular redox balance [75] and could generate oxidative damage to lipids and proteins. Aminochrome has also been observed to form adducts and generate oxidative damage to DNA [25,76]. Furthermore, early alterations in gene expression have been reported following aminochrome exposure [23]. Whether oxidative damage occurs early, generating changes in DNA and favoring epigenetic modifications, is unknown. The early upregulation of ubiquitin and LC3B suggests the activation of compensatory mechanisms. Nonetheless, it has been reported that oxidative stress triggers can disrupt normal epigenetic regulation, leading to the dysregulation of transcription factors such as nuclear factor erythroid 2–related factor 2 (NRF2) [77,78]. Interestingly, NRF2 not only regulates genes involved in antioxidant defense, such as glutathione S-transferase Mu 1 (GSTM1) and NAD(P)H quinone dehydrogenase 1 (NQO1) but also orchestrates the expression of autophagy- and proteasome-related genes [79,80,81,82,83].
5. Conclusions
Our findings suggest that the disruption of the microtubule network by aminochrome, particularly through inhibition of autophagosome-lysosome fusion and accumulation of protein aggregates, may represent a critical early event leading to neuronal death. Early microtubule destabilization may initiate a cascade of microtubule-dependent dysfunctions that contribute, directly or indirectly, to cell death.
Notably, the presence of nuclear protein aggregates raises the possibility that nuclear proteotoxic stress may play a significant role in aminochrome-induced neurotoxicity. Aminochrome-induced oxidative stress may be one of the primary triggers. Whether such nuclear changes affect proteins involved in epigenetic regulation remains unknown and represents an important avenue for future research.
In conclusion, therapeutic strategies aimed at stabilizing the microtubule network and enhancing autophagy or the UPS may hold promise for PD; particularly in the context of emerging research on nuclear proteotoxicity.
Author Contributions
Conceptualization, I.B.P. and J.S.A.; Methodology, A.B. and I.B.P.; Formal analysis, A.B. and I.B.P.; Investigation, A.B., C.N., K.C., P.P., N.S., and I.B.P.; Visualization, A.B. and I.B.P.; Resources, C.M., J.F.V., N.C., J.S.A., and I.B.P.; Writing—original draft preparation, I.B.P.; Writing—review and editing, C.M., J.F.V., N.C., J.S.A., and I.B.P.; Supervision, I.B.P.; Project administration, I.B.P.; Funding acquisition, I.B.P.; Validation, I.B.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by FONDECYT 1120337 and Universidad Santo Tomás projects 000012858 and 000016012. The APC was funded by Universidad Santo Tomás and Universidad Adolfo Ibáñez.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
No potential conflicts of interest are disclosed.
Abbreviations
The following abbreviations are used in this manuscript:
| PD | Parkinson’s disease |
| LC3β | Microtubule-associated protein 1 light chain 3 beta |
| LAMP1 | Lysosome-associated membrane glycoprotein 1 |
| PI3K | Phosphatidylinositol 3-kinase |
| AKT | RAC-gamma serine/threonine-protein kinase |
| mTOR | Serine/threonine-protein kinase mTOR |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| V-ATPases | Vacuolar-type H+-ATPases |
| SERCA | ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting |
| UPS | ubiquitin-proteasome system. |
References
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef]
- Reichmann, H.; Brandt, M.D.; Klingelhoefer, L. The nonmotor features of Parkinson’s disease: Pathophysiology and management advances. Curr. Opin. Neurol. 2016, 29, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Johnston, H.E.; Samant, R.S. Alternative systems for misfolded protein clearance: Life beyond the proteasome. FEBS J. 2021, 288, 4464–4487. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef]
- Morrow, C.S.; Porter, T.J.; Xu, N.; Arndt, Z.P.; Ako-Asare, K.; Heo, H.J.; Thompson, E.A.N.; Moore, D.L. Vimentin coordinates protein turnover at the aggresome during neural stem cell quiescence exit. Cell Stem Cell 2020, 26, 558–568.e9. [Google Scholar] [CrossRef] [PubMed]
- Morrow, C.S.; Moore, D.L. Vimentin’s side gig: Regulating cellular proteostasis in mammalian systems. Cytoskeleton 2020, 77, 515–523. [Google Scholar] [CrossRef]
- Bourdenx, M.; Koulakiotis, N.S.; Sanoudou, D.; Bezard, E.; Dehay, B.; Tsarbopoulos, A. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases. Prog. Neurobiol. 2017, 155, 171–193. [Google Scholar] [CrossRef]
- Hosseinpour-Moghaddam, K.; Caraglia, M.; Sahebkar, A. Autophagy induction by trehalose: Molecular mechanisms and therapeutic impacts. J. Cell Physiol. 2018, 233, 6524–6543. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, P.; Weng, R.; Wu, Z.; Qin, B.; Fang, J.; Wang, Y.; Qiu, S.; Yang, J.; Gu, L. Trehalose protects motor neurons after brachial plexus root avulsion by activating autophagy and inhibiting apoptosis mediated by the AMPK signaling pathway. Gene 2021, 768, 145307. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and clearance of protein aggregates in neurodegeneration. Cell Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Koprich, J.B.; Wang, Y.; Yu, W.B.; Xiao, B.G.; Brotchie, J.M.; Wang, J. Treatment with trehalose prevents behavioral and neurochemical deficits produced in an AAV α-synuclein rat model of Parkinson’s disease. Mol. Neurobiol. 2016, 53, 2258–2268. [Google Scholar] [CrossRef]
- Lee, H.J.; Yoon, Y.S.; Lee, S.J. Mechanism of neuroprotection by trehalose: Controversy surrounding autophagy induction. Cell Death Dis. 2018, 9, 712. [Google Scholar] [CrossRef]
- Shacka, J.J.; Klocke, B.J.; Shibata, M.; Uchiyama, Y.; Datta, G.; Schmidt, R.E.; Roth, K.A. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol. Pharmacol. 2006, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Klucken, J.; Poehler, A.M.; Ebrahimi-Fakhari, D.; Schneider, J.; Nuber, S.; Rockenstein, E.; Schlötzer-Schrehardt, U.; Hyman, B.T.; McLean, P.J.; Masliah, E.; Winkler, J. Alpha-synuclein aggregation involves a bafilomycin A1-sensitive autophagy pathway. Autophagy 2012, 8, 754–766. [Google Scholar] [CrossRef]
- Mauvezin, C.; Nagy, P.; Juhász, G.; Neufeld, T.P. Autophagosome–lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome–lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef]
- Kondo, M.; Hara, H.; Kamijo, F.; Kamiya, T.; Adachi, T. 6-Hydroxydopamine disrupts cellular copper homeostasis in human neuroblastoma SH-SY5Y cells. Metallomics 2021, 13, mfab041. [Google Scholar] [CrossRef]
- Cartelli, D.; Ronchi, C.; Maggioni, M.G.; Rodighiero, S.; Giavini, E.; Cappelletti, G. Microtubule dysfunction precedes transport impairment and mitochondria damage in MPP+-induced neurodegeneration. J. Neurochem. 2010, 115, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Cartelli, D.; Goldwurm, S.; Casagrande, F.; Pezzoli, G.; Cappelletti, G. Microtubule destabilization is shared by genetic and idiopathic Parkinson’s disease patient fibroblasts. PLoS ONE 2012, 7, e37467. [Google Scholar] [CrossRef]
- Cartelli, D.; Casagrande, F.; Busceti, C.L.; Bucci, D.; Molinaro, G.; Traficante, A.; Passarella, D.; Giavini, E.; Pezzoli, G.; Battaglia, G.; Cappelletti, G. Microtubule alterations occur early in experimental parkinsonism and the microtubule stabilizer epothilone D is neuroprotective. Sci. Rep. 2013, 3, 1837. [Google Scholar] [CrossRef]
- Cappelletti, G.; Surrey, T.; Maci, R. The parkinsonism-producing neurotoxin MPP+ affects microtubule dynamics by acting as a destabilizing factor. FEBS Lett. 2005, 579, 4781–4786. [Google Scholar] [CrossRef]
- Hongo, H.; Kihara, T.; Kume, T.; Izumi, Y.; Niidome, T.; Sugimoto, H.; Akaike, A. Glycogen synthase kinase-3β activation mediates rotenone-induced cytotoxicity with the involvement of microtubule destabilization. Biochem. Biophys. Res. Commun. 2012, 426, 94–99. [Google Scholar] [CrossRef]
- Briceño, A.; Muñoz, P.; Brito, P.; Huenchuguala, S.; Segura-Aguilar, J.; Paris, I.B. Aminochrome toxicity is mediated by inhibition of microtubule polymerization through the formation of adducts with tubulin. Neurotox. Res. 2016, 29, 381–393. [Google Scholar] [CrossRef]
- Stykel, M.G.; Humphries, K.; Kirby, M.P.; Czaniecki, C.; Wang, T.; Ryan, T.; Bamm, V.; Ryan, S.D. Nitration of microtubules blocks axonal mitochondrial transport in a human pluripotent stem cell model of Parkinson’s disease. FASEB J. 2018, 32, 5350–5364. [Google Scholar] [CrossRef] [PubMed]
- Paris, I.; Muñoz, P.; Huenchuguala, S.; Couve, E.; Sanders, L.H.; Greenamyre, J.T.; Caviedes, P.; Segura-Aguilar, J. Autophagy protects against aminochrome-induced cell death in substantia nigra-derived cell line. Toxicol. Sci. 2011, 121, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Muñoz, P.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Mannervik, B. Neuroprotection against aminochrome neurotoxicity: Glutathione transferase M2-2 and DT-diaphorase. Antioxidants 2022, 11, 296. [Google Scholar] [CrossRef]
- Huenchuguala, S.; Segura-Aguilar, J. Single-neuron neurodegeneration as a degenerative model for Parkinson’s disease. Neural Regen. Res. 2024, 19, 529–535. [Google Scholar] [CrossRef]
- Paris, I.; Perez-Pastene, C.; Cardenas, S.; Iturriaga-Vasquez, P.; Muñoz, P.; Couve, E.; Caviedes, P.; Segura-Aguilar, J. Aminochrome induces disruption of actin, alpha-, and beta-tubulin cytoskeleton networks in substantia nigra-derived cell line. Neurotox. Res. 2010, 18, 82–92. [Google Scholar] [CrossRef]
- Huenchuguala, S.; Muñoz, P.; Zavala, P.; Villa, M.; Cuevas, C.; Ahumada, U.; Graumann, R.; Nore, B.F.; Couve, E.; Mannervik, B.; Paris, I.; Segura-Aguilar, J. Glutathione transferase mu 2 protects glioblastoma cells against aminochrome toxicity by preventing autophagy and lysosome dysfunction. Autophagy 2014, 10, 618–630. [Google Scholar] [CrossRef]
- Xiong, R.; Siegel, D.; Ross, D. Quinone-induced protein handling changes: Implications for major protein handling systems in quinone-mediated toxicity. Toxicol. Appl. Pharmacol. 2014, 280, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.; Huenchuguala, S.; Muñoz, P.; Villa, M.; Paris, I.; Mannervik, B.; Segura-Aguilar, J. Glutathione transferase M2-2 secreted from glioblastoma cells protects SH-SY5Y cells from aminochrome neurotoxicity. Neurotox. Res. 2015, 27, 217–228. [Google Scholar] [CrossRef]
- Xie, H.R.; Hu, L.S.; Li, G.Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar] [PubMed]
- Ioghen, O.C.; Ceafalan, L.C.; Popescu, B.O. SH-SY5Y cell line in vitro models for Parkinson disease research—Old practice for new trends. J. Integr. Neurosci. 2023, 22, 20. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Lind, C. On the mechanism of the Mn3+-induced neurotoxicity of dopamine: Prevention of quinone-derived oxygen toxicity by DT-diaphorase and superoxide dismutase. Chem. Biol. Interact. 1989, 72, 309–324. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Elazar, Z.; Seglen, P.O.; Rubinsztein, D.C. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 2008, 4, 849–850. [Google Scholar] [CrossRef]
- Wilson, L.; Creswell, K.M.; Chin, D. The mechanism of action of vinblastine: Binding of [acetyl-3H]vinblastine to embryonic chick brain tubulin and tubulin from sea urchin sperm tail outer doublet microtubules. Biochemistry 1975, 14, 5586–5592. [Google Scholar] [CrossRef]
- Xie, R.; Nguyen, S.; McKeehan, W.L.; Liu, L. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol. 2010, 11, 89. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef]
- Ferrer, I.; Martinez, A.; Blanco, R.; Dalfó, E.; Carmona, M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: Preclinical Parkinson disease. J. Neural Transm. 2011, 118, 821–839. [Google Scholar] [CrossRef]
- Ren, Y.; Liu, W.; Jiang, H.; Jiang, Q.; Feng, J. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J. Biol. Chem. 2005, 280, 34105–34112. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Palmiter, R.D.; Xia, Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J. Cell Biol. 2011, 192, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Timoshina, Y.A.; Pavlova, A.K.; Voronkov, D.N.; Abaimov, D.A.; Latanov, A.V.; Fedorova, T.N. Assessment of the behavioral and neurochemical characteristics in a mouse model of the premotor stage of Parkinson’s disease induced by chronic low-dose MPTP. Int. J. Mol. Sci. 2025, 26, 8856. [Google Scholar] [CrossRef]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington: Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef]
- Zhang, S.; Shao, Z.; Liu, X.; Hou, M.; Cheng, F.; Lei, D.; Yuan, H. The E50K optineurin mutation impacts autophagy-mediated degradation of TDP-43 and leads to retinal ganglion cell apoptosis in vivo and in vitro. Cell Death Discov. 2021, 7, 49. [Google Scholar] [CrossRef]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome–lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Chu, H.Y.; Wang, W.; Chen, X.; Jiang, Y.E.; Cheng, R.; Qi, X.; Zhong, Z.M.; Zeng, M.S.; Zhu, X.F.; Sun, C.Z. Bafilomycin A1 increases the sensitivity of tongue squamous cell carcinoma cells to cisplatin by inhibiting lysosomal uptake of platinum ions but not autophagy. Cancer Lett. 2018, 423, 105–112. [Google Scholar] [CrossRef]
- Pivtoraiko, V.N.; Harrington, A.J.; Mader, B.J.; Luker, A.M.; Caldwell, G.A.; Caldwell, K.A.; Roth, K.A.; Shacka, J.J. Low-dose bafilomycin attenuates neuronal cell death associated with autophagy–lysosome pathway dysfunction. J. Neurochem. 2010, 114, 1193–1204. [Google Scholar] [CrossRef]
- Huenchuguala, S.; Muñoz, P.; Segura-Aguilar, J. The importance of mitophagy in maintaining mitochondrial function in U373MG cells: Bafilomycin A1 restores aminochrome-induced mitochondrial damage. ACS Chem. Neurosci. 2017, 8, 2247–2253. [Google Scholar] [CrossRef]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed]
- Deleyto-Seldas, N.; Efeyan, A. The mTOR–autophagy axis and the control of metabolism. Front. Cell Dev. Biol. 2021, 9, 655731. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; Piccolella, M.; Galbiati, M.; Garrè, M.; Morelli, E.; Vaccari, T.; Poletti, A. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef]
- Sevriev, B.; Dimitrova, S.; Kehayova, G.; Dragomanova, S. Trehalose: Neuroprotective effects and mechanisms—An updated review. NeuroSci. 2024, 5, 429–444. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Cho, E.D.; Jung Ahn, W.; Won Lee, K.; Lee, S.J.; Lee, H.J. Is trehalose an autophagic inducer? Unraveling the roles of non-reducing disaccharides on autophagic flux and alpha-synuclein aggregation. Cell Death Dis. 2017, 8, e3091. [Google Scholar] [CrossRef]
- Tao, D.; Xia, Y.; Liao, Q.; Yang, X.; Zhang, L.; Xie, C. Rapamycin mitigates neurotoxicity of fluoride and aluminum by activating autophagy through the AMPK/mTOR/ULK1 pathway in hippocampal neurons and NG108-15 cells. Sci. Rep. 2025, 15, 9801. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Fecto, F.; Esengul, Y.T.; Siddique, T. Protein recycling pathways in neurodegenerative diseases. Alzheimers Res. Ther. 2014, 6, 13. [Google Scholar] [CrossRef]
- Tanaka, M.; Kim, Y.M.; Lee, G.; Junn, E.; Iwatsubo, T.; Mouradian, M.M. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J. Biol. Chem. 2004, 279, 4625–4631. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Cardenas, S.; Huenchuguala, S.; Briceño, A.; Couve, E.; Paris, I.; Segura-Aguilar, J. DT-diaphorase prevents aminochrome-induced alpha-synuclein oligomer formation and neurotoxicity. Toxicol. Sci. 2015, 145, 37–47. [Google Scholar] [CrossRef]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; Kalia, L.V.; Kalia, S.K. Early-onset impairment of the ubiquitin–proteasome system in dopaminergic neurons caused by alpha-synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef]
- Zafar, K.S.; Siegel, D.; Ross, D. A potential role for cyclized quinones derived from dopamine, DOPA, and 3,4-dihydroxyphenylacetic acid in proteasomal inhibition. Mol. Pharmacol. 2006, 70, 1079–1086. [Google Scholar] [CrossRef]
- Chin, L.S.; Olzmann, J.A.; Li, L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 2010, 38, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, M.M.; Hill, W.D. Contribution of somal Lewy bodies to neuronal death. Brain Res. 1997, 775, 24–29. [Google Scholar] [CrossRef]
- Milber, J.M.; Noorigian, J.V.; Morley, J.F.; Petrovitch, H.; White, L.; Ross, G.W.; Duda, J.E. Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology 2012, 79, 2307–2314. [Google Scholar] [CrossRef]
- Fortun, J.; Dunn, W.A., Jr.; Joy, S.; Li, J.; Notterpek, L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 2003, 23, 10672–10680. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.S.; Olzmann, J.A.; Li, L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 2010, 38, 144–149. [Google Scholar] [CrossRef]
- Lee, J.Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef]
- Wenzel, H.J.; Hunsaker, M.R.; Greco, C.M.; Willemsen, R.; Berman, R.F. Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res. 2010, 1318, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L. Phase-to-phase with nucleoli—Stress responses, protein aggregation and novel roles of RNA. Front. Cell Neurosci. 2019, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Huna, A.; Inashkina, I.; Belyayev, A.; Krigerts, J.; Pastova, L.; Vazquez-Martin, A.; Erenpreisa, J. Nucleolar aggresomes mediate release of pericentric heterochromatin and nuclear destruction of genotoxically treated cancer cells. Nucleus 2017, 8, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Paris, I.; Perez-Pastene, C.; Couve, E.; Caviedes, P.; LeDoux, S.; Segura-Aguilar, J. Copper–dopamine complex induces mitochondrial autophagy preceding caspase-independent apoptotic cell death. J. Biol. Chem. 2009, 284, 13306–13315. [Google Scholar] [CrossRef]
- Zahid, M.; Saeed, M.; Yang, L.; Beseler, C.; Rogan, E.; Cavalieri, E.L. Formation of dopamine quinone–DNA adducts and their potential role in the etiology of Parkinson’s disease. IUBMB Life 2011, 63, 1087–1093. [Google Scholar] [CrossRef]
- Olawade, D.B.; Rashad, I.; Egbon, E.; Teke, J.; Ovsepian, S.V.; Boussios, S. Reversing epigenetic dysregulation in neurodegenerative diseases: Mechanistic and therapeutic considerations. Int. J. Mol. Sci. 2025, 26, 4929. [Google Scholar] [CrossRef]
- Rubio, K.; Hernández-Cruz, E.Y.; Rogel-Ayala, D.G.; Sarvari, P.; Isidoro, C.; Barreto, G.; Pedraza-Chaverri, J. Nutriepigenomics in environmental-associated oxidative stress. Antioxidants 2023, 12, 771. [Google Scholar] [CrossRef]
- Vauzour, D.; Buonfiglio, M.; Corona, G.; Chirafisi, J.; Vafeiadou, K.; Angeloni, C.; Hrelia, S.; Hrelia, P.; Spencer, J.P. Sulforaphane protects cortical neurons against 5-S-cysteinyl-dopamine-induced toxicity through activation of ERK1/2, Nrf2 and upregulation of detoxification enzymes. Mol. Nutr. Food Res. 2010, 54, 532–542. [Google Scholar] [CrossRef]
- Wong, M.; Ahmed, A.; Luo, W.; Bowman, A.B.; Tizabi, Y.; Aschner, M.; Ferrer, B. Combined manganese–iron exposure reduced oxidative stress is associated with the NRF2/NQO1 pathway in astrocytic C8-D1A cells. Biol. Trace Elem. Res. 2025. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, S.; Lin, Z.; Chen, X.; Jiao, Q.; Du, X.; Jiang, H. Targeting the interplay between autophagy and the Nrf2 pathway in Parkinson’s disease with potential therapeutic implications. Biomolecules 2025, 15, 149. [Google Scholar] [CrossRef]
Figure 1.
Effect of aminochrome on autophagosome-lysosome fusion and cell death in SH-SY5Y cells. (A) Immunofluorescence double labeling of microtubules-associated protein light chains (LC3β) (green) and lysosomal-associated membrane protein 1 (LAMP1) (red) in SH-SY5Y cells which were treated with 50 µM aminochrome (AM), 10 µM rapamycin (R), 5 nM bafilomycin (B), 50 µM aminochrome and 10 µM rapamycin (AM + R) or 50 µM aminochrome plus 5 nM bafilomycin (AM+ B) for 16 h at 37 °C. As a control, the cells were incubated only in culture medium (C). (B) Quantification of the Pearson coefficient of immunofluorescence double labeling in A. (C) Quantification of cell death. The images were obtained with a fluorescence microscope (Carl Zeiss, Göttingen, Germany; Observer.Z1 Axio model) and analyzed for deconvolution with the AxioVision Rel 4.8 software. The statistical significance was assessed using analysis of variance (ANOVA) for multiple comparisons and the Newman–Keuls test (**P < 0.01 and ***P < 0.001 compared with control; ♦P < 0.05, ♦♦P < 0.01 and ♦♦♦P < 0.001 compared among experimental groups differing from control). The experiment was performed with three replicates.
Figure 1.
Effect of aminochrome on autophagosome-lysosome fusion and cell death in SH-SY5Y cells. (A) Immunofluorescence double labeling of microtubules-associated protein light chains (LC3β) (green) and lysosomal-associated membrane protein 1 (LAMP1) (red) in SH-SY5Y cells which were treated with 50 µM aminochrome (AM), 10 µM rapamycin (R), 5 nM bafilomycin (B), 50 µM aminochrome and 10 µM rapamycin (AM + R) or 50 µM aminochrome plus 5 nM bafilomycin (AM+ B) for 16 h at 37 °C. As a control, the cells were incubated only in culture medium (C). (B) Quantification of the Pearson coefficient of immunofluorescence double labeling in A. (C) Quantification of cell death. The images were obtained with a fluorescence microscope (Carl Zeiss, Göttingen, Germany; Observer.Z1 Axio model) and analyzed for deconvolution with the AxioVision Rel 4.8 software. The statistical significance was assessed using analysis of variance (ANOVA) for multiple comparisons and the Newman–Keuls test (**P < 0.01 and ***P < 0.001 compared with control; ♦P < 0.05, ♦♦P < 0.01 and ♦♦♦P < 0.001 compared among experimental groups differing from control). The experiment was performed with three replicates.
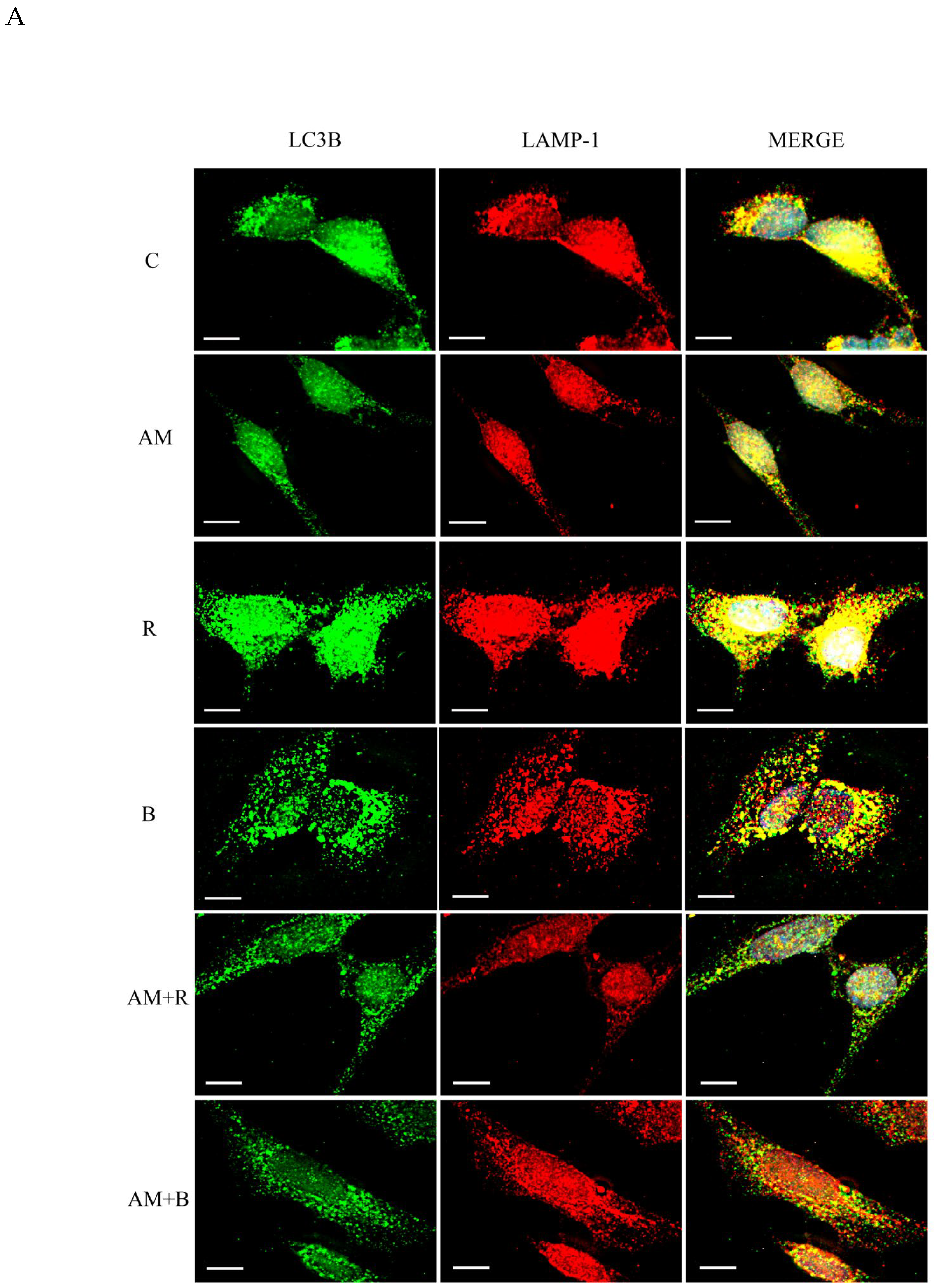
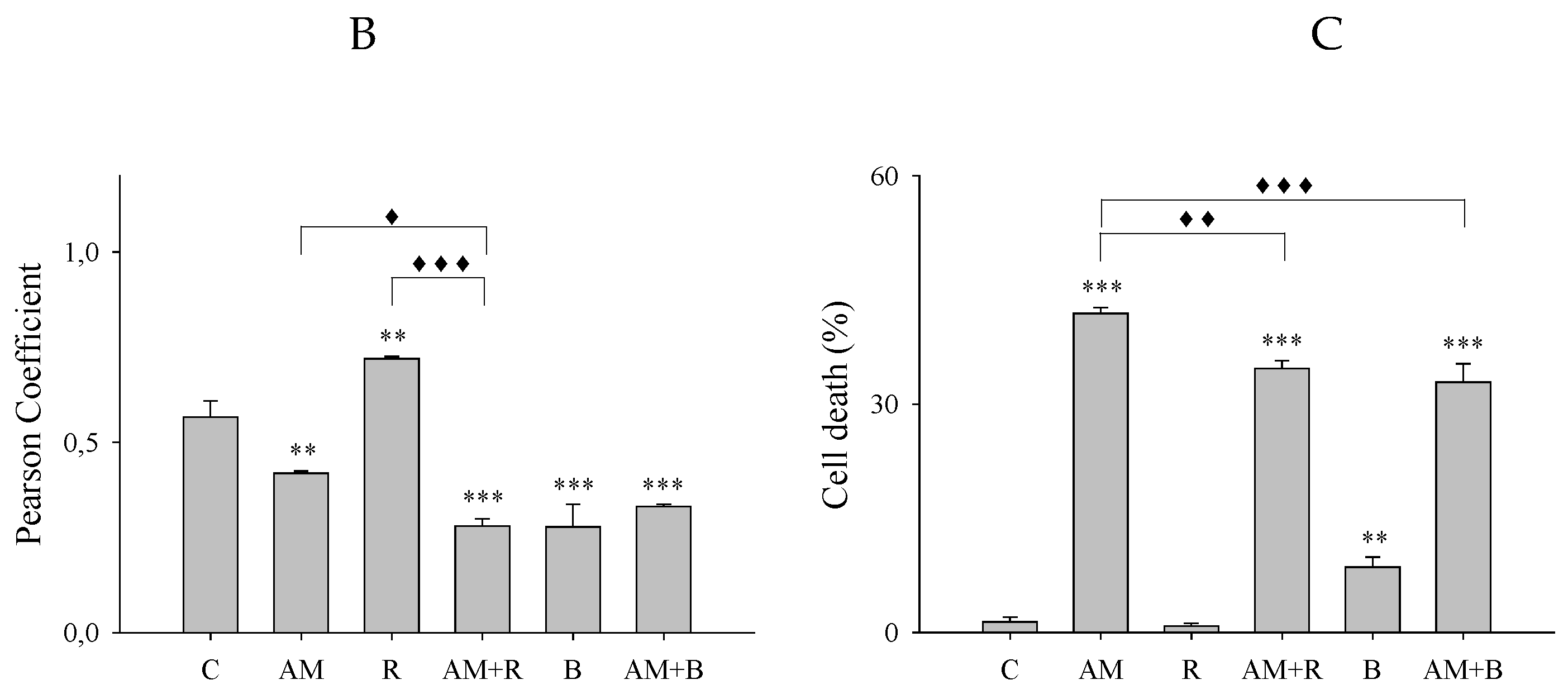
Figure 2.
Effect of aminochrome and autophagy inducers on autophagosome-lysosome fusion in SH-SY5Y cells. (A) Immunofluorescence double labeling of microtubules-associated protein light chain (LC3β) (green) and lysosomal-associated membrane protein 1 (LAMP1) (red) in SH-SY5Y cells which were treated with 50 µM aminochrome (AM), 10 µM vinblastine (V), 50 µM aminochrome and 10 µM vinblastine (AM + V), 10 µM rapamycin (R), 50 µM aminochrome and 10 µM rapamycin (AM + R), 10 µM vinblastine and 10 µM rapamycin (V + R), 100 mM trehalose (T), 50 µM aminochrome plus 100 mM trehalose (AM + T), or 10 µM vinblastine plus 100 mM trehalose (V + T) for 16 h at 37 °C. As a control, the cells were incubated only in culture medium (C). (B) Quantification of the Pearson coefficient of immunofluorescence double labeling in A. (C) Quantification of cell death. The images were obtained with a fluorescence microscope (Carl Zeiss, Göttingen, Germany; Observer.Z1 Axio model) and analyzed for deconvolution with the AxioVision Rel 4.8 software. The statistical significance was assessed using analysis of variance (ANOVA) for multiple comparisons and the Newman–Keuls test (**P < 0.01 and ***P < 0.001 compared with control; ♦P < 0.05 and ♦♦♦P < 0.001 compared among experimental groups differing from control). The experiment was performed with three replicates.
Figure 2.
Effect of aminochrome and autophagy inducers on autophagosome-lysosome fusion in SH-SY5Y cells. (A) Immunofluorescence double labeling of microtubules-associated protein light chain (LC3β) (green) and lysosomal-associated membrane protein 1 (LAMP1) (red) in SH-SY5Y cells which were treated with 50 µM aminochrome (AM), 10 µM vinblastine (V), 50 µM aminochrome and 10 µM vinblastine (AM + V), 10 µM rapamycin (R), 50 µM aminochrome and 10 µM rapamycin (AM + R), 10 µM vinblastine and 10 µM rapamycin (V + R), 100 mM trehalose (T), 50 µM aminochrome plus 100 mM trehalose (AM + T), or 10 µM vinblastine plus 100 mM trehalose (V + T) for 16 h at 37 °C. As a control, the cells were incubated only in culture medium (C). (B) Quantification of the Pearson coefficient of immunofluorescence double labeling in A. (C) Quantification of cell death. The images were obtained with a fluorescence microscope (Carl Zeiss, Göttingen, Germany; Observer.Z1 Axio model) and analyzed for deconvolution with the AxioVision Rel 4.8 software. The statistical significance was assessed using analysis of variance (ANOVA) for multiple comparisons and the Newman–Keuls test (**P < 0.01 and ***P < 0.001 compared with control; ♦P < 0.05 and ♦♦♦P < 0.001 compared among experimental groups differing from control). The experiment was performed with three replicates.
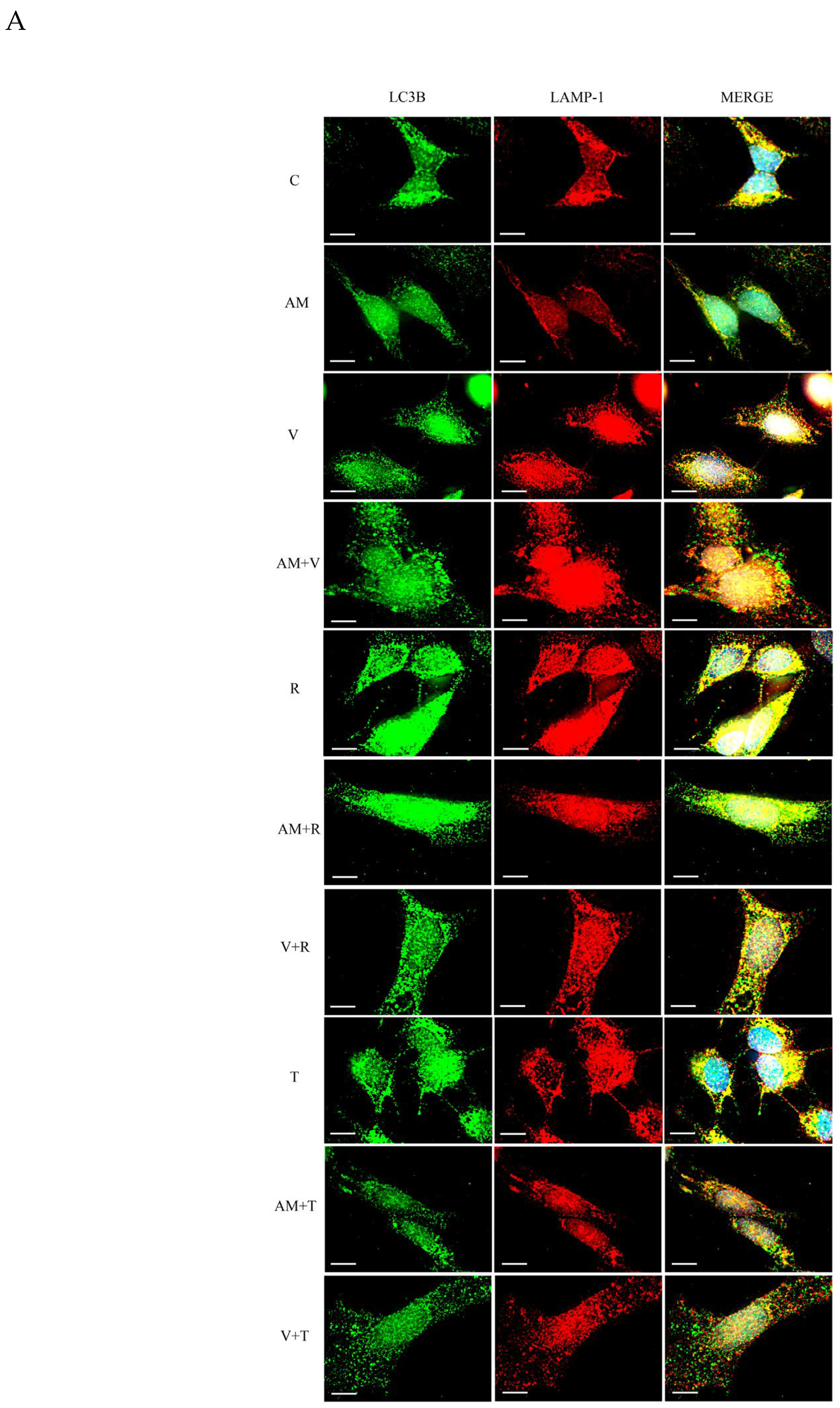
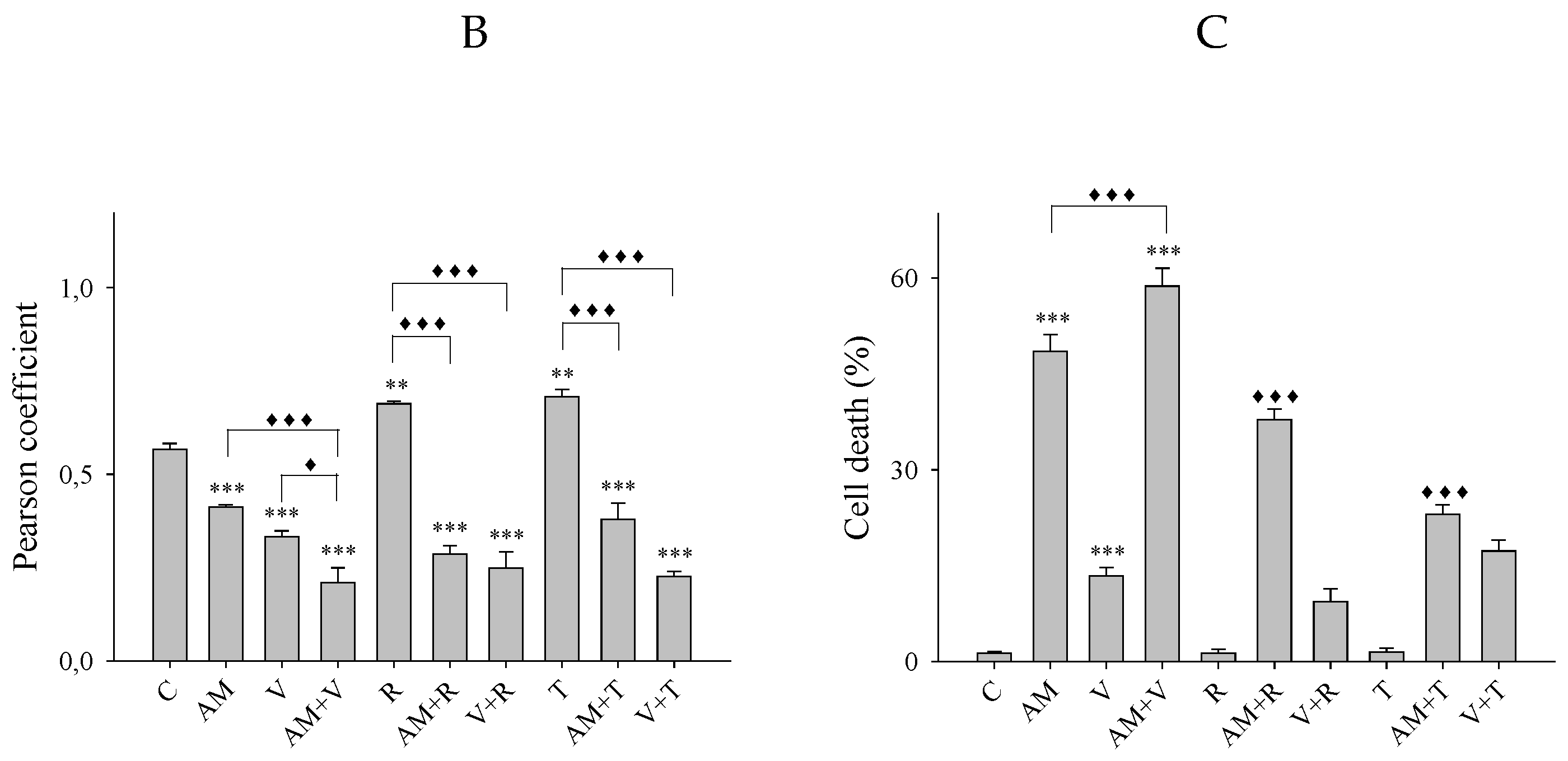
Figure 3.
Vimentin aggregates by aminochrome in SHSY5Y cells. (A) Immunofluorescence labeling of vimentin (red) in SH-SY5Y cells which were treated with 50 µM aminochrome (AM), for 14 h at 37 °C. As a control, the cells were incubated only in culture medium (C). (B) Quantification of vimentin aggregates. (C) Immunofluorescence double labeling of ubiquitin (green) and vimentin (red) in SH-SY5Y cells which were treated with 50 µM aminochrome (AM) for 16 h at 37 °C. As a control, the cells were incubated only in culture medium (C). (D) Quantification of the Pearson coefficient of immunofluorescence double labeling in C. (E) Immunofluorescence double labeling of ubiquitin (green) and vimentin (red) in cells SH-SY5Y which were treated with 50 µM aminochrome (AM) for 16 h at 37 °C. The images were obtained with a fluorescence microscope (Carl Zeiss, Göttingen, Germany; Observer.Z1 Axio model) and analyzed for deconvolution with the AxioVision Rel 4.8 software. Images in Z-stack were also obtained and analyzed for extended focus for figure E. The statistical significance was assessed using unpaired t-test for two-group comparisons (*P < 0.05 and ***P < 0.001 compared with control). The experiments were performed with three to four replicates.
Figure 3.
Vimentin aggregates by aminochrome in SHSY5Y cells. (A) Immunofluorescence labeling of vimentin (red) in SH-SY5Y cells which were treated with 50 µM aminochrome (AM), for 14 h at 37 °C. As a control, the cells were incubated only in culture medium (C). (B) Quantification of vimentin aggregates. (C) Immunofluorescence double labeling of ubiquitin (green) and vimentin (red) in SH-SY5Y cells which were treated with 50 µM aminochrome (AM) for 16 h at 37 °C. As a control, the cells were incubated only in culture medium (C). (D) Quantification of the Pearson coefficient of immunofluorescence double labeling in C. (E) Immunofluorescence double labeling of ubiquitin (green) and vimentin (red) in cells SH-SY5Y which were treated with 50 µM aminochrome (AM) for 16 h at 37 °C. The images were obtained with a fluorescence microscope (Carl Zeiss, Göttingen, Germany; Observer.Z1 Axio model) and analyzed for deconvolution with the AxioVision Rel 4.8 software. Images in Z-stack were also obtained and analyzed for extended focus for figure E. The statistical significance was assessed using unpaired t-test for two-group comparisons (*P < 0.05 and ***P < 0.001 compared with control). The experiments were performed with three to four replicates.
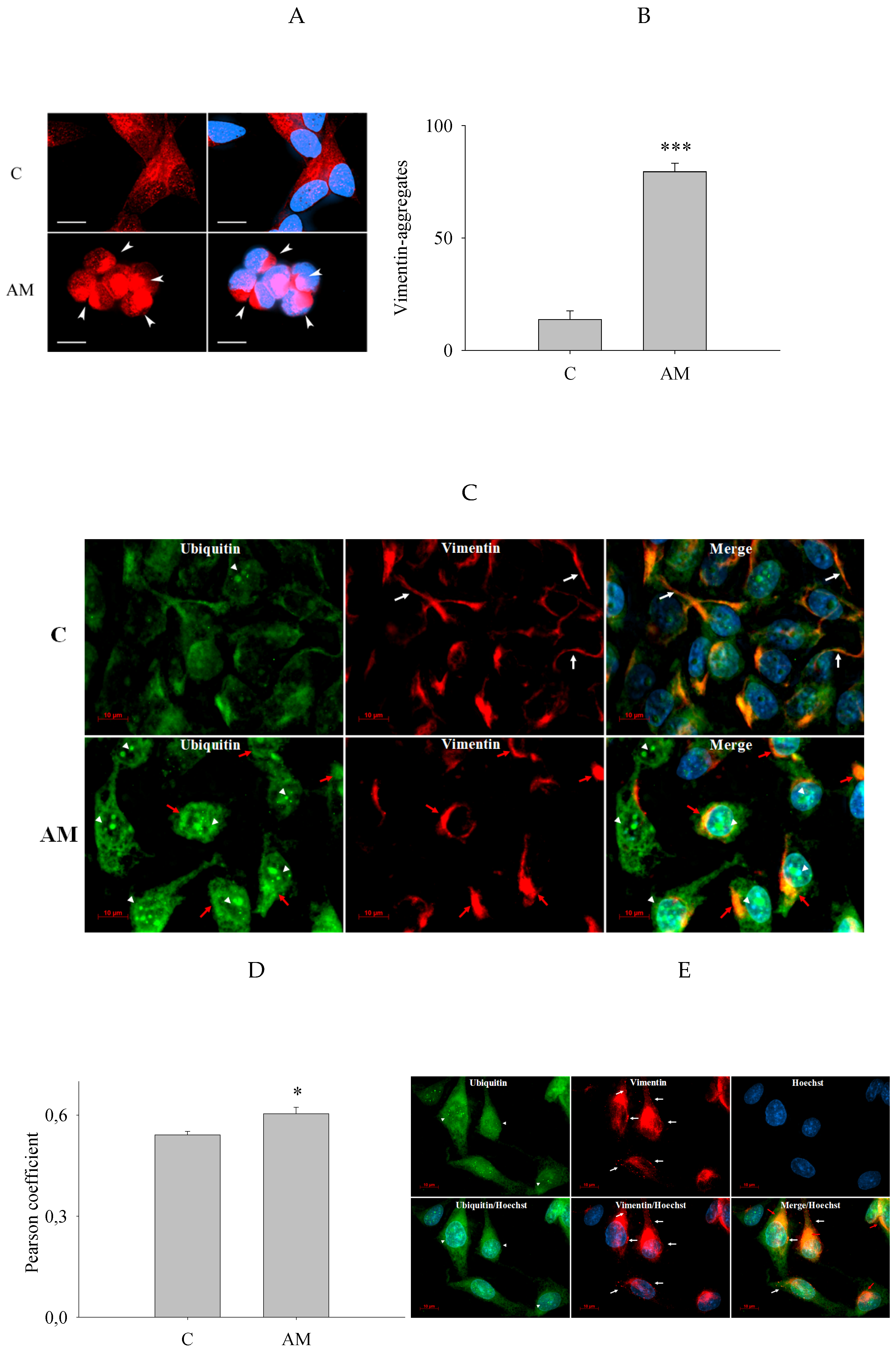
Figure 4.
LC3β and ubiquitin expression induced by aminochrome in SH-SY5Y cells. Quantitative real-time PCR. Total RNA from SH-SY5Y cells treated with 50 µM aminochrome for 0.5, 4, 9 and 14 h at 37 °C and 5% CO2 was extracted as described under experimental procedures. mRNA levels were normalized using the housekeeping gene GAPDH. The statistical significance was assessed using unpaired t-test for two-group comparisons (***P < 0.001 compared with control). The experiment was performed with three to six replicates.
Figure 4.
LC3β and ubiquitin expression induced by aminochrome in SH-SY5Y cells. Quantitative real-time PCR. Total RNA from SH-SY5Y cells treated with 50 µM aminochrome for 0.5, 4, 9 and 14 h at 37 °C and 5% CO2 was extracted as described under experimental procedures. mRNA levels were normalized using the housekeeping gene GAPDH. The statistical significance was assessed using unpaired t-test for two-group comparisons (***P < 0.001 compared with control). The experiment was performed with three to six replicates.
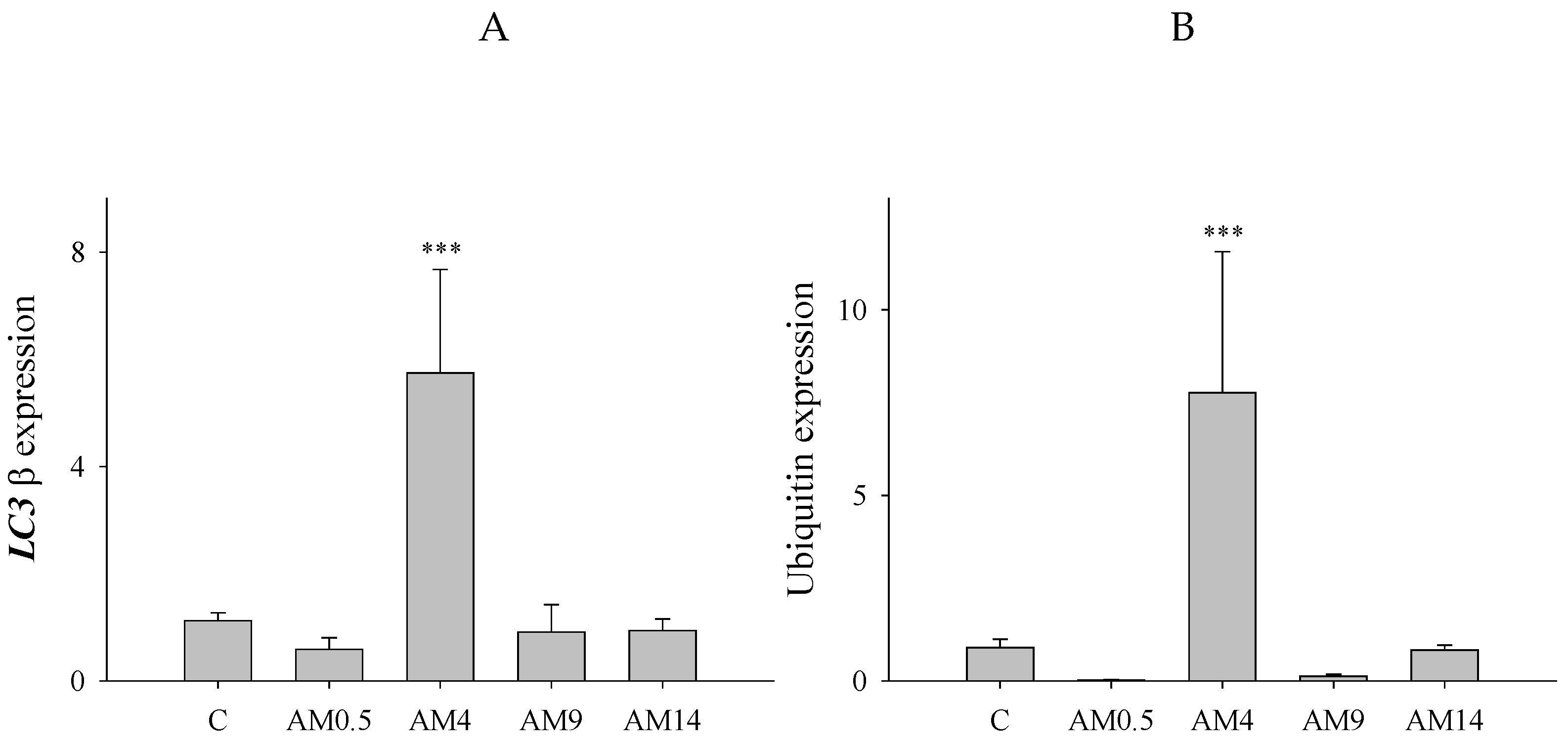
Figure 5.
Possible mechanism of action of aminochrome on autophagosome-lysosome fusion and the protein aggregates formation. Aminochrome forms adducts with tubulin and induces microtubule depolymerization (1), altering the fusion of autophagosomes with lysosomes (2). Decreased fusion between autophagosomes and lysosomes and disrupted UPS could favor the accumulation of protein aggregates (3). Aminochrome promotes the formation of vimentin aggregates and ubiquitin aggregates toward perinuclear regions that colocalize with ubiquitin (4). Vimentin aggregates in cell processes were also observed. Nuclear labeling of ubiquitin aggregates was induced by aminochrome (5). However, the initial effects of aminochrome on the microtubule network and possibly on autophagy and UPS mechanisms are not the only ones that could explain its toxic effects. A series of events could be triggered directly or indirectly by aminochrome where deregulation of calcium signaling, and mitochondrial disruption could play a significant role (6).
Figure 5.
Possible mechanism of action of aminochrome on autophagosome-lysosome fusion and the protein aggregates formation. Aminochrome forms adducts with tubulin and induces microtubule depolymerization (1), altering the fusion of autophagosomes with lysosomes (2). Decreased fusion between autophagosomes and lysosomes and disrupted UPS could favor the accumulation of protein aggregates (3). Aminochrome promotes the formation of vimentin aggregates and ubiquitin aggregates toward perinuclear regions that colocalize with ubiquitin (4). Vimentin aggregates in cell processes were also observed. Nuclear labeling of ubiquitin aggregates was induced by aminochrome (5). However, the initial effects of aminochrome on the microtubule network and possibly on autophagy and UPS mechanisms are not the only ones that could explain its toxic effects. A series of events could be triggered directly or indirectly by aminochrome where deregulation of calcium signaling, and mitochondrial disruption could play a significant role (6).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



