Submitted:
21 February 2026
Posted:
26 February 2026
You are already at the latest version
Abstract
Obesity is one of the major global public health challenges. Pancreatic lipase (PL) is an important target for obesity management as it plays a key role in lipid absorption. However, the clinical application of synthetic PL inhibitors is limited by adverse effects. Previous studies showed that natural products from traditional Chinese medicines (TCMs) is a promising alternative for developing safe PL inhibitors. In this study, we integrated molecular docking, molecular dynamics (MD) simulations, MM/PBSA binding free energy calculations and in vitro enzymatic assays to systematically screen potential PL inhibitors from TCM constituents. Six compounds with docking scores ranging from −9.9 to −9.0 kcal/mol. MD simulations confirmed the structural stability of six ligand−PL complexes via RMSD, radius of gyration (Rg), hydrogen bond and conformational analysis. MM/PBSA calculations revealed the binding free energies of the six compounds to PL ranged from −21.38±0.40 to −13.33±0.58 kcal/mol. In vitro validation showed five of the six compounds exhibited PL inhibitory activity, among which Hydroxygenkwanin (HYD) was the most potent competitive inhibitor (IC₅₀= 0.128±0.009 mM), followed by Atractylenolide I (ATR-I) (IC50 = 0.584±0.031 mM) and Peiminine (PEI) (IC50 = 0.748±0.042 mM). This study validates the efficiency of the integrated in silico and in vitro strategy for screening natural PL inhibitors and provide valuable reference for the development of novel anti-obesity agents or functional food ingredients.
Keywords:
pancreatic lipase
; obesity
; traditional Chinese medicine
; molecular docking
; molecular dynamics simulation
; MM/PBSA
; inhibitor
1. Introduction
Obesity is a chronic metabolic disease caused by a severe imbalance between energy intake and expenditure, leading to excessive accumulation of body fat[1]. With shifting dietary patterns and increasingly sedentary lifestyles in modern society, the prevalence of obesity has risen dramatically, making it a major global public health challenge[2]. Obesity not only impedes daily activities but also elevates the risk of numerous serious diseases, such as stroke, diabetes, atherosclerosis, and cardiovascular diseases [3,4,5]. Therefore, the prevention and management of obesity carry substantial medical and societal importance.
PL is a key enzyme responsible for facilitating the absorption of lipids, and inhibition of its activity can effectively reduce fat absorption, making it an important therapeutic target for obesity management [6]. Orlistat is a clinically approved PL inhibitor; however, its clinical application is limited due to adverse effects such as steatorrhea and rectal pain [7]. Consequently, it is imperative to develop of new, safe, and effective PL inhibitors. Numerous studies have shown that many Traditional Chinese medicines (TCMs) possess lipid-lowering and anti-obesity properties [8,9]. Thus, natural products derived from TCMs hold great promise as a valuable resource for the developing of novel PL inhibitors.
Traditional drug screening methods suffer from several drawbacks, including intensive time and cost requirements as well as low efficiency. Molecular docking, as a structure-based drug discovery technique, can rapidly identify compounds that fit the target protein[10]. However, docking methods itself suffers from several drawbacks such as lack of solvation effects, poor scoring function, rigid protein conformation and so on [10,11]. MD simulation can incorporate protein flexibility, mimic the behavior of ligand−protein complex in the real biochemical environments [12]. Furthermore, binding free energy calculations with methods like Molecular Mechanics/Poisson-Boltzmann surface area (MM/PBSA) can provide more accurate ligand−protein binding energy than docking score [13]. So integrating molecular docking, MD simulation and MM/PBSA can effectively improve the efficiency and accuracy of drug screening [14,15].
In this study, potential PL inhibitors were screened from TCMs through the integration of molecular docking, molecular dynamics (MD) simulations, and in vitro validation. Three compounds were identified as potent PL inhibitors. This study may provide a valuable reference for the screening and development of novel PL inhibitors.
2. Materials and Methods
2.1. Screening with Molecular Docking
AutoDock Vina is one of the fastest and most widely used open-source docking engines [16]. Nguyen et al. demonstrated that AutoDock Vina 1.1.2 could redock the co−crystallized PL inhibitor (MUP) into the active site of PL, with an RMSD of 0.86 Å between the docked conformation and the crystal structure (PDB ID: 1LPB) , which is below the generally accepted threshold of 2.0 Å [17]. Similar docking protocol was also adopted to screen PL inhibitors from natural products [18,19]. Therefore, the docking protocol validated by Nguyen et al. [17] was employed in the present study, with detailed procedures described below. The PL crystal structure (PDB ID: 1LPB) was pretreated using AutoDock Tools 1.5.6 software [20], including removing water molecules, adding polar hydrogen atoms and assigning atomic charges. The grid box was centered on the centriod of the co-crystallized ligand at (8.4, 24.4, 52.6) Å with dimensions of 30 × 30 × 30 Å3. The molecular structures of chemical constituents of TCMs were downloaded from the TCMSP database [21] and were transformed from MOL2 to PDBQT format with Autodock Tools 1.5.6 software. The conformation with the lowest binding energy was selected as the optimal docking conformation. Molecular visualization of ligand−PL complexes was performed using Discovery Studio Visualizer (BIOVIA Discovery Studio 2019, Dassault Systèmes, San Diego, USA) and Pymol 2.5.5 (Schrödinger, LLC, 2025).
2.2. Molecular Dynamics Simulation
Molecular dynamics(MD) simulations were performed using the GROMACS 2024 software package [22]. The structure exhibiting the lowest binding energy from the preceding molecular docking simulation was selected as the initial conformation of the complex for MD simulation. Amber14SB−ILDN all-atom force field [23] and TIP3P solvent model were used to simulate the protein and water molecules respectively. Geometric optimization and electrostatic potential calculations for the ligands were performed using Gaussian 09 [24] software at the HF/6-31G(d) level. The antechamber module implemented in AmberTools23 [25] software package were employed to assign general AMBER force field (GAFF) [26] parameters to the ligands.
Each complex was solvated in an octahedral water box, with the minimum distance from the complex to box boundary set to 1.0 nm. Appropriate number of Na+ and Cl- ions were added to neutralize the overall charge of system. Before the simulation, energy minimization was performed with the steepest−gradient method to eliminate unreasonable intermolecular interactions in the system. Subsequently, a 300 ps equilibration simulation was performed under the constant temperature and volume (NVT) ensemble at 310 K, followed by a 500 ps simulation under the constant temperature and pressure (NPT) ensemble (310 K, 1 atm) to fully equilibrate the system. Finally, a 100 ns MD production simulation was performed on each system under the NPT condition at temperature of 310 K and 1 atm pressure with the time step of 2 fs. Subsequent trajectory analysis included the time−dependent variations in root mean square deviation (RMSD), radius of gyration (Rg) and the number of hydrogen bonds. MM/PBSA is a classical molecular mechanics−based approach used to quantify the binding affinity between receptors and ligands [27]. In this study, 80 conformations were extracted at 125 ps intervals from the last 10 ns of the MD trajectory for MM/PBSA calculations.
2.3. Materials and Reagents
Porcine pancreatic lipase purchased from Sigma−Aldrich Co. Orlistat, p−nitrophenyl palmitate (pNPP), Atractylenolide I, Linarin, Hydroxygenkwanin, Salvianolic acid B, Peiminine and Mulberroside A were all purchased from Yuanye Bio−Technology Co., Ltd. All the chemical reagents used in this study were of analytical grade.
2.4. Inhibition Rate and Inhibition Type on Pancreatic Lipase
The experiment was performed according to the method reported by Glisan et al. [28] with minor modifications. Briefly, 20 μL of PL solution (6.5 mg/mL), 10 μL of test compound solution, and 160 μL of PBS buffer (pH 8.0) were mixed and preincubated at 37 °C for 10 min to facilitate sufficient binding between the enzyme and the test compounds. The enzymatic reaction was initiated by the addition of 10 μL pNPP solution (3 mg/mL). Absorbance at 405 nm was recorded at 2−minute intervals over a 20−minute period. The linear region of the absorbance–time curve was used to determine the reaction rate K (i.e., the slope of the linear regression). The inhibition ratio was calculated using Eq. (1):
where Ka is the reaction rate with the presence of test compound, Kb is the blank reaction rate without enzyme and Kc is the reaction rate without test compound
The inhibition type was explored in a way similar to that described above. The reaction rate (v) was calculated according to the changes in absorbance over time. The reaction rates were measured under the condition that the concentration of inhibitor was held constant and and the concentration of pNPP ([S]) was varied. Then the reverse of v were fitted with the reverse of [S]. According to Michaelis−Menten equation (Eq.(2)), the inhibition type of the compounds could be determined by analyzing the slopes, intercepts and intersection points of the fitted curves.
2.5. Statistic Analysis
Data were expressed as mean±standard deviation of triplicate trials. Data analysis and plotting were performed with using the plotting module of scikit-learn [29] and software of GNU PSPP (https://www.gnu.org/software/pspp/).
3. Results
3.1. Molecular Docking
Molecular docking serves as an effective tool for screening large compound libraries and identifying potential bioactive hits [10]. 13132 chemical ingredients from TCMSP were screened with Autodock Vina. Analysis of molecular docking results revealed that the docking score representing the binding energies of known natural product PL inhibitors were generally below −7.0 kcal/mol, as shown in Table S1. To enhance the screening positive rate, this study defined the binding energy cutoff value as −9.0 kcal/mol (i.e., only compounds with binding energies below this value were selected). To ensure structural diversity of the candidates, compounds with high structural similarity were excluded. Ultimately, six compounds with no previously reported pancreatic lipase (PL) inhibitory activity were selected as candidates for further investigation, with docking scores ranging from −9.9 to −9.0 kcal/mol, as listed in Table 1. The chemical structures of these selected compounds were shown in Figure 1.
Binding conformation analysis revealed that all candidate compounds interacted with hydrophobic residues (e.g., Phe77, Tyr114, Phe215) which formed a hydrophobic cleft critical for substrate recognition [30]. Hydrophobic interactions have been previously identified as key drivers of inhibitor-PL binding [30,31]. Additionally, all compounds except PEI formed conventional hydrogen bonds with polar residues like Ser152, His263, Gly76 and His151 (Table 2). Figure 2A-F showed the detailed binding conformations of the compounds with PL. ATR-I established two hydrogen bonds with the Ser152 and His263 residues respectively, through its carbonyl group, along with π-π interactions with Phe215 and Tyr114 (Figure 2A). LIN formed four hydrogen bonds with Cys181 and Gln183, one π -π interaction and one π-σ interaction with Tyr114 and Ile209 respectively (Figure 2B). HYD binds to the active site of PL via multiple interactions, including three hydrogen bonds with Gly76, Phe77, and His151, a π-π interaction with Tyr114, and a π-σ interaction with Leu164 (Figure 2C). SAL-B forms a hydrogen bond with Asp79, two π-π stacking interactions with the side chains of Phe215 and Tyr114, and a π-cation interaction with Arg256 (Figure 2D). PEI formed two π-alkyl interactions with Ile78 and Trp252, along with additional hydrophobic interactions (Figure 2E). MUL-A established conventional hydrogen bonds with key residues including Arg256, Thr255, and Gly76, accompanied by multiple π-π and π-σ interactions with residues such as Tyr114, Phe215, Pro180, and Ala178 (Figure 2F). Although minor unfavorable interactions were observed for LIN and SAL-B, the overall synergistic effects of multiple non-covalent forces still favorable for the formation of ligand-PL complexes, as indicated by their negative binding energies.
3.2. Molecular Dynamics Simulation
To further evaluate the structural stability of the ligand-PL complexes, molecular dynamics (MD) simulations were carried out using the docked conformations as initial structures. For comparative analysis, MD simulations were also performed on apo-PL. The dynamic properties of these systems were characterized by analyzing the root mean square deviation (RMSD), radius of gyration (Rg), and the number of intermolecular hydrogen bonds.
3.2.1. Analysis of the RMSD and Rg
As illustrated in Figure 3A-B, the backbone RMSD of PL was maintained below ~0.25 nm throughout most of the simulation time in all systems. This result indicated that the structure of PL, whether in the ligand-bound or apo state, exhibited high stability under the employed simulation conditions. The RMSD of the six binding compounds varied significantly (Figure 3C-D). The RMSD of ATR-I (blue curve, Figure 3C) displayed only minor fluctuations within 0.05 nm throughout the entire simulation, owing to its rigid structure. HYD (green curve in Figure 3C) exhibits moderate fluctuations during the initial simulation phase, followed by a stabilization at a lower RMSD level (~0.15 nm) with only minor oscillations after approximately 70 ns, suggesting a gradual convergence toward a stable binding conformation. LIN (red curve in Figure 3C) exhibited the most pronounced and frequent fluctuations among all compounds, with RMSD values peaking at up to 0.4 nm, indicating substantial conformational variability and unstable binding state. The RMSDs of PEI (red curve in Figure 3D) kept below 0.2 nm throughout the simulation, and showed minor fluctuations in the last 10 nm, indicating a relative stable binding conformation. SAL-B and MUL-A exhibited several distinct RMSD fluctuations (blue and green curves in Figure 3D) , with peaks exceeding 0.3 nm during the 40-60 ns interval. Subsequently, their RMSD values gradually stabilized to a plateau over the final 30 ns of the simulation, indicating that their binding conformations likely evolved toward more stable states.
The radius of gyration (Rg) is usually used to assess the structural compactness of proteins. A smaller Rg value indicates a more compact and stable protein conformation. The Rg of the apo-form of PL and the six ligand-PL complexes were analyzed, with the resulting plots presented in Figure 3E-F. The apo-PL system (orange curve in Figure 3F) exhibited fluctuations within a narrow range of 2.65–2.70 nm and displayed a gradual decreasing trend over the course of the simulation, suggesting enhanced structural stability as the system approached equilibrium. The Rg values of all complexes ranged from 2.65 to 2.75 nm and exhibited a narrow fluctuation range. Collectively, these results indicated that PL maintained structural stability in all systems throughout the simulation, with ligand binding causing negligible disturbance to its global structure.
3.2.2. Binding Mode Variation During the Simulation
To explore the dynamic behavior of ligand binding in greater detail, the positions and conformations of the compounds at different simulation time points (0, 25, 50, 75, and 100 ns, colored red, green, blue, yellow, and orange, respectively) were visualized after alignment of the structure of PL (Figure 4). ATR-I departed from its initial binding site at 50 ns (blue, Figure 4A), then partially reverted to its original position and adopted an optimized binding conformation. LIN displayed obvious variations in its position and orientation throughout the simulation, accompanied by substantial conformational adaptation (Figure 4B), indicating an unstable binding mode. HYD maintained a relatively stable binding conformation throughout the simulation. Its core structure remained anchored within the active site of PL, with only minor adjustments observed in the orientation (Figure 4C). SAL-B and MUL-A showed comparable binding profiles (Figure 4D, 4E). Both exhibited substantial conformational fluctuations over the course of the simulation, indicative of their inherent flexibility, yet underwent no significant spatial displacement within the binding site. PEI stayed within the original binding pocket, optimizing its binding interactions with PL with merely minor conformational changes (Figure 4F).
3.2.3. Hydrogen Bond Analysis
Hydrogen bonds are crucial non-covalent interactions that contribute significantly to the binding affinity. Figure 5 showed the number of hydrogen bonds formed between the compounds and PL throughout the MD simulations. For ATR-I, HYD and PEI (Figure 5A, 5C and 5E, respectively), the number of hydrogen bonds generally are no more than three, with only one hydrogen bond observed in the majority of cases. In the case of LIN, SAL-B and MUL-A (Figure 4B, 4D and 4F), hydrogen bond counts spanned zero to seven or eight, a result indicative of a robust, dynamic hydrogen bond network with PL that stems from the multiple hydroxyl groups in these compounds.
3.2.4. MM/PBSA Calculation
The MM/PBSA method is a computational technique for evaluating the binding affinity of ligands with protein [13]. In this study, the binding free energies between the six compounds and the PL were calculated using the MM/PBSA method.The results were listed in Table 2. Among all the compounds, SAL-B and PEI exhibited the strongest binding affinity, with the total binding free energies (ΔGTOTAL) of −21.38±0.40 and −21.24±0.39 kcal/mol, followed by LIN and SAL-B with the ΔGTOTAL of −19.54±0.43 and−15.58±0.85 kcal/mol respectively. MUL-A and ATR-I showed similar binding affinity with ΔGTOTAL of −13.33±0.58 and −13.44±0.45 kcal/mol, respectively. Overall, all compounds exhibited negative binding free energies with PL, indicating that the formation of ligand-PL complexes was thermodynamically favorable. Although MM/PBSA generally provides more reliable results than docking methods, its performance in ligand ranking is still unsatisfactory[13]. Therefore, all six compounds were selected for subsequent in vitro validation.
3.3. In Vitro Validation
The inhibitory activities of the six compounds were evaluated via an in vitro enzyme assay. The positive control, Orlistat, exhibited an IC₅₀ value of 0.481 ± 0.023 μM. For the candidate compounds, the IC₅₀ values were determined as follows: HYD, 0.128 ± 0.009 mM; ATR-I, 0.584 ± 0.031 mM; PEI, 0.748 ± 0.042 mM; SAL-B, 1.147 ± 0.065 mM; and MUL-A, 13.410 ± 0.724 mM. LIN showed no detectable inhibitory activity at concentrations up to 50 mM. Subsequently, inhibition kinetics were performed for the four most potent compounds (HYD, ATR-I, PEI, and SAL-B) by measuring enzyme activity at fixed inhibitor concentrations and varying substrate concentrations. The resulting Lineweaver-Burk plots (Figure 6) demonstrated that the slopes of the fitted lines increased progressively with rising inhibitor concentration, while the y-axis intercepts remained essentially unchanged. These kinetic features indicated that all four compounds act as competitive inhibitors of PL.
4. Discussion
In the present study, we integrated in silico screening with in vitro enzymatic assays to systematically identify potential PL inhibitors from TCM-derived compounds. Molecular docking using AutoDock Vina identified six candidates with binding energies below −9.0 kcal/mol. MM/PBSA calculations showed that the binding affinity of the candidates were in the range from −21.38±0.40 to −13.33±0.58 kcal/mol. In vitro enzymatic analysis demonstrated that five of the six selected compounds displayed inhibitory activity against PL, with IC₅₀ values ranging from 0.128± 0.009 mM (HYD) to 13.410 ± 0.724 mM (MUL-A). These values were markedly higher than that of the positive control orlistat (IC₅₀ = 0.481 ± 0.023 μM). This is because orlistat binds to PL via covalent interactions, whereas most natural products interact with PL primarily through non-covalent forces, thus generally exhibiting weaker inhibitory activity against PL than orlistat [32]. So these results support the reliability and efficiency of the in silico strategy used for screening potential PL inhibitors.
However, the binding free energy rankings derived from MM/PBSA calculations were not fully consistent with the in vitro inhibitory activities. For instance, SAL-B showed the most favorable predicted binding affinity, with a binding free energy of −21.38±0.40 kcal/mol, but displayed relatively weak inhibitory activity (IC50 = 1.147 ± 0.065 mM). In contrast, HYD exhibited a moderate binding free energy (−15.58±0.85 kcal/mol) yet the strongest in vitro potency (IC50 = 0.128 ± 0.009 mM). A plausible explanation for this discrepancy is that the entropic penalty was neglected in the MM/PBSA calculations, which may lead to substantial overestimation of the binding affinities of flexible ligands [13,32]. Previous studies have indicated that flexible molecules with 5 or more rotatable bonds may incur a total entropic penalty of 5-7 kcal/mol [33]. In the present work, SAL-B, MUL-A, and LIN possess 14, 8, and 7 rotatable bonds, respectively; their binding affinities were thus overestimated by at least 5-7 kcal/mol. By contrast, HYD, ATR-I, and PEI are rigid compounds with no more than two rotatable bonds, so the entropic contribution has a negligible effect on their calculated binding energies [13]. Thus, as demonstrated by in vitro experiments, the pancreatic lipase (PL) inhibitory activities of HYD, ATR-I and PEI were superior to those of the other three molecules with greater conformational flexibility.
In the present study, HYD exhibited the most potent inhibitory activity against porcine pancreatic lipase (PL), with an IC₅₀ value of 0.128±0.009 mM (128 μM). Structurally, HYD is a luteolin derivative generated by the methylation of the 7-OH on the A-ring of luteolin. Previous studies have demonstrated that the free hydroxyl groups on the A and B rings of flavones are essential for their PL inhibitory activity [34], while methylation [34,35] or glycosylation [35,36] of these hydroxyl moieties can significantly attenuate such activity. For example, methylation of the 4’-hydroxyl group of luteolin increased its IC50 from 259μM to 3033μM for the methylated product hesperetin [35]. Consistently, luteolin has been reported to inhibit PL with a lower IC₅₀ of 99 μM [35], showing stronger potency than HYD, indicated that methylation of the 7-hydroxyl group in luteolin impairs its PL inhibitory activity. This structure–activity relationship (SAR) also provides a clear explanation for the negligible PL inhibitory activity of linarin (LIN), a flavone glycoside substituted with 5-OH, 7-O-rutinoside, and 4’-OMe. LIN can be regarded as a derivative of apigenin (5,7,4’-OH), modified by 4’-O-methylation on the B-ring and 7-O-glycosylation on the A-ring. In contrast, apigenin has been reported to inhibit PL with an IC₅₀ of 256 μM [34]. For LIN, however, the loss of free hydroxyl groups caused by 4’-O-methylation may impair its capacity to form hydrogen bonds, while 7-O-glycosylation introduces a bulky hydrophilic moiety. Together, these modifications render it unable to form favorable interaction with PL, as reflected by its vibrant fluctuations in RMSD during the MD simulation (Figure 2C), thus leading to the nearly complete abolition of PL inhibitory activity.
5. Conclusions
In conclusion, this study demonstrates the utility of integrating in silico and in vitro approaches for identifying natural PL inhibitors from TCMs. Among the six test compounds, HYD, ATR-I and PEI were identified as potent inhibitors with IC₅₀ of 0.128 ± 0.009, 0.584 ± 0.031 and 0.748 ± 0.042 mM, respectively. Though their potency is lower than the positive control, orlista, they may used as promising leads for the development of novel anti-obesity drugs or functional food ingredients. Future in vivo and structural modification studies are needed further advance the translation of these findings into practical applications for obesity management.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Autodock Vina-based ocking cores of Traditional Chinese Medicine-derived pancreatic lipase inhibitors.
Author Contributions
Conceptualization, M.Z. and M.C.; methodology, N.X. and Y.W.; software, Z.Z.; validation, J.L. and T.L.; formal analysis, Z.X.; data curation, J.L.; writing—original draft preparation, Z.Z.; writing—review and editing, N.X.; visualization, J.L.; supervision, M.Z.; project administration, M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Beijing Life Science Academy (BLSA), No: 2024500CB0030, 2023000CA0040]; the National Natural Science Foundation of China [grant number 81603119]; the Natural Science Foundation of Beijing Municipality [grant number 7174316]; the Peking University Medicine Seed Fund for Interdisciplinary Research supported by “the Fundamental Research Funds for the Central Universities”[grant number No. BMU2022MX017, No. BMU2022MX003]; High Education Basic Research Project supported by Department of Education of Liaoning Province [grant number No. LJ212510162007].
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
During the preparation of this paper, the author utilized Doubao for proofreading and grammatical correction of certain sentences. The author has carefully reviewed, edited, and corrected all AI-generated content and assumes full responsibility for the integrity, accuracy, and viewpoints presented in this work.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| PL | Pancreatic lipase |
| TCM | Traditional Chinese medicine |
| MD | Molecular dynamics |
| MM/PBSA | Mechanics/Poisson-Boltzmann surface area |
| RMSD | Root Mean Square Deviation |
| Rg | Radius of Gyration |
| pNPP | p-nitrophenyl palmitate |
| ATR-I | Atractylenolide I |
| LIN | Linarin |
| HYD | Hydroxygenkwanin |
| SAL-B | Salvianolic Acid B |
| PEI | Peiminine |
| MUL-A | Mulberroside A |
References
- Ji, T.; Li, Y.; Ma, L. Sarcopenic obesity: An emerging public health problem. Aging Dis. 2022, 13, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, H. Obesity: Epidemiology, pathophysiology, and therapeutics. Front. Endocrinol. 2021, 12, 706978. [Google Scholar] [CrossRef] [PubMed]
- Swinburn, B.A.; Sacks, G.; Hall, K.D.; McPherson, K.; Finegood, D.T.; Moodie, M.L.; Gortmaker, S.L. The global obesity pandemic: shaped by global drivers and local environments. Lancet 2011, 378(9793), 804–814. [Google Scholar] [CrossRef]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [PubMed]
- Lopez-Jimenez, F.; Almahmeed, W.; Bays, H.; Cuevas, A.; Di Angelantonio, E.; le Roux, C.W.; Sattar, N.; Sun, M.C.; Wittert, G.; Pinto, F.J.; Wilding, J.P.H. Obesity and cardiovascular disease: Mechanistic insights and management strategies. Eur. J. Prev. Cardiol. 2022, 29, 2218–2237. [Google Scholar] [CrossRef]
- Subramaniyan, V.; Hanim, Y.U. Role of pancreatic lipase inhibition in obesity treatment: mechanisms and challenges towards current insights and future directions. Int J Obes. 2025, 49, 492–506. [Google Scholar] [CrossRef]
- McClendon, K.S.; Riche, D.M.; Uwaifo, G.I. Orlistat: Current status in clinical therapeutics. Expert Opin. Drug Saf. 2009, 8, 727–744. [Google Scholar] [CrossRef]
- Fan, Q.; Xu, F.; Liang, B.; Zou, X. The anti-obesity effect of traditional Chinese medicine on lipid metabolism. Front. Pharmacol. 2021, 12, 696603. [Google Scholar] [CrossRef]
- Li, C.; Zhang, H.; Li, X. The mechanism of traditional Chinese medicine for the treatment of obesity. Diabetes Metab. Syndr. Obes. 2020, 13, 3371–3381. [Google Scholar] [CrossRef]
- Kontoyianni, M. Docking and virtual screening in drug discovery. Methods Mol. Biol. 2017, 1647, 255–266. [Google Scholar]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23(8), 1899. [Google Scholar] [CrossRef] [PubMed]
- Menchon, G.; Maveyraud, L.; Czaplicki, G. Molecular dynamics as a tool for virtual ligand screening. Methods Mol. Biol. 2018, 1762, 145–178. [Google Scholar] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015, 10(5), 449–61. [Google Scholar] [CrossRef] [PubMed]
- Dankwa, B.; Broni, E.; Enninful, K.S.; Kwofie, S.K.; Wilson, M.D. Consensus docking and MM-PBSA computations identify putative furin protease inhibitors for developing potential therapeutics against COVID-19. Struct. Chem. 2022, 33, 2221–2241. [Google Scholar] [CrossRef]
- Al-Nema, M.; Gaurav, A.; Lee, V.S. Docking based screening and molecular dynamics simulations to identify potential selective PDE4B inhibitor. Heliyon. 2020, 6(9), e04856. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Nguyen, P.T.V.; Huynh, H.A.; Truong, D.V.; Tran, T.D.; Vo, C.T. Exploring Aurone Derivatives as Potential Human Pancreatic Lipase Inhibitors through Molecular Docking and Molecular Dynamics Simulations. Molecules 2020, 25(20), 4657. [Google Scholar] [CrossRef]
- Vulichi, S.R.; Runthala, A.; Rachamreddy, S.K.; Yaramanedi, R.S.P.; Sahoo, P.S.; Burra, P.V.L.S.; Kaur, N.; Akkiraju, S.; Kanala, S.R.; Chippada, A.R.; Murthy, S.D.S. Appraisal of pancreatic lipase inhibitory potential of Ziziphus oenoplia (L.)Mill. leaves by in vitro and in Silico Approaches. ACS Omega. 2023, 8, 16630–16646. [Google Scholar] [CrossRef]
- Prabhakar, L.; Dicky John, D.G.; Singh, S.R.; Murali, A. Computational analysis of marine algal compounds for obesity management against pancreatic lipase. J. Biomol. Struct. Dyn. 2023, 41, 4863–4872. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Ru, J.; Li, P.; Wang, J.; Zhou, W.; Li, B.; Huang, C.; Li, P.; Guo, Z.; Tao, W.; Yang, Y.; Xu, X.; Li, Y.; Wang, Y.; Yang, L. TCMSP: A database of systems pharmacology for drug discovery from herbal medicines. J. Cheminform. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Páll, S.; Zhmurov, A.; Bauer, P.; Abraham, M.; Lundborg, M.; Gray, A.; Hess, B.; Lindahl, E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153, 134110. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory. Comput. 2015, 11(8), 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford CT, USA, 2016. [Google Scholar]
- Case, DA; Aktulga, HM; Belfon, K; Cerutti, DS; Cisneros, GA; et al. AmberTools. J Chem Inf Model. 2023, 63(20), 6183–6191. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25(9), 1157–74. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Glisan, S.L.; Grove, K.A.; Yennawar, N.H.; Lambert, J.D. Inhibition of pancreatic lipase by black tea theaflavins: Comparative enzymology and in silico modeling studies. Food. Chem. 2017, 216, 296–300. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; Vanderplas, J.; Passos, A.; Cournapeau, D.; Brucher, M.; Perrot, M.; Duchesnay, E. Scikit-learn: Machine Learning in Python. J. Mach. Learn Res. 2011, 12, 2825–2830. [Google Scholar]
- Winkler, F.K.; D'Arcy, A.; Hunziker, W. Structure of human pancreatic lipase. Nature 1990, 343, 771–774. [Google Scholar] [CrossRef]
- Chen, T.; Tong, M.; Ma, K. Synthesis of Oleanolic Acid Derivatives and Their Inhibitory Effects on Pancreatic Lipase. Nat. Prod. Res. Dev. 2016, 28, 11–16. [Google Scholar]
- Hou, T.; et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 2. Entropy calculations and their influence on the prediction of binding affinities. J Chem Theory Comput. 2011, 7, 27–39. [Google Scholar]
- Kongsted, J.; Ryde, U. An improved method to predict the entropy term with the MM/PBSA approach. J Comput-Aided Mol Des. 2009, 23, 63–71. [Google Scholar] [CrossRef]
- Cardullo, N.; Muccilli, V.; Pulvirenti, L.; Tringali, C. Natural Isoflavones and Semisynthetic Derivatives as Pancreatic Lipase Inhibitors. J. Nat. Prod. 2021, 84, 654–665. [Google Scholar] [CrossRef]
- Li, M.; Chen, Y.T.; Ruan, J.C.; Wang, W.J.; Chen, J.G.; Zhang, Q.F. Structure-activity relationship of dietary flavonoids on pancreatic lipase. Curr. Res. Food. Sci. 2023, 6, 100424. [Google Scholar] [CrossRef]
- Tran, T.H.; Mai, T.T.; Ho, T.T.; Le, T.N.; Cao, T.C.; Thai, K.M.; Tran, T.S. Inhibition of pancreatic lipase by flavonoid derivatives: in vitro and in silico investigations. Adv Pharmacol Pharm Sci. 2024, 6655996. [Google Scholar]
Figure 1.
Chemical structure of the selected candidates from molecular docking.
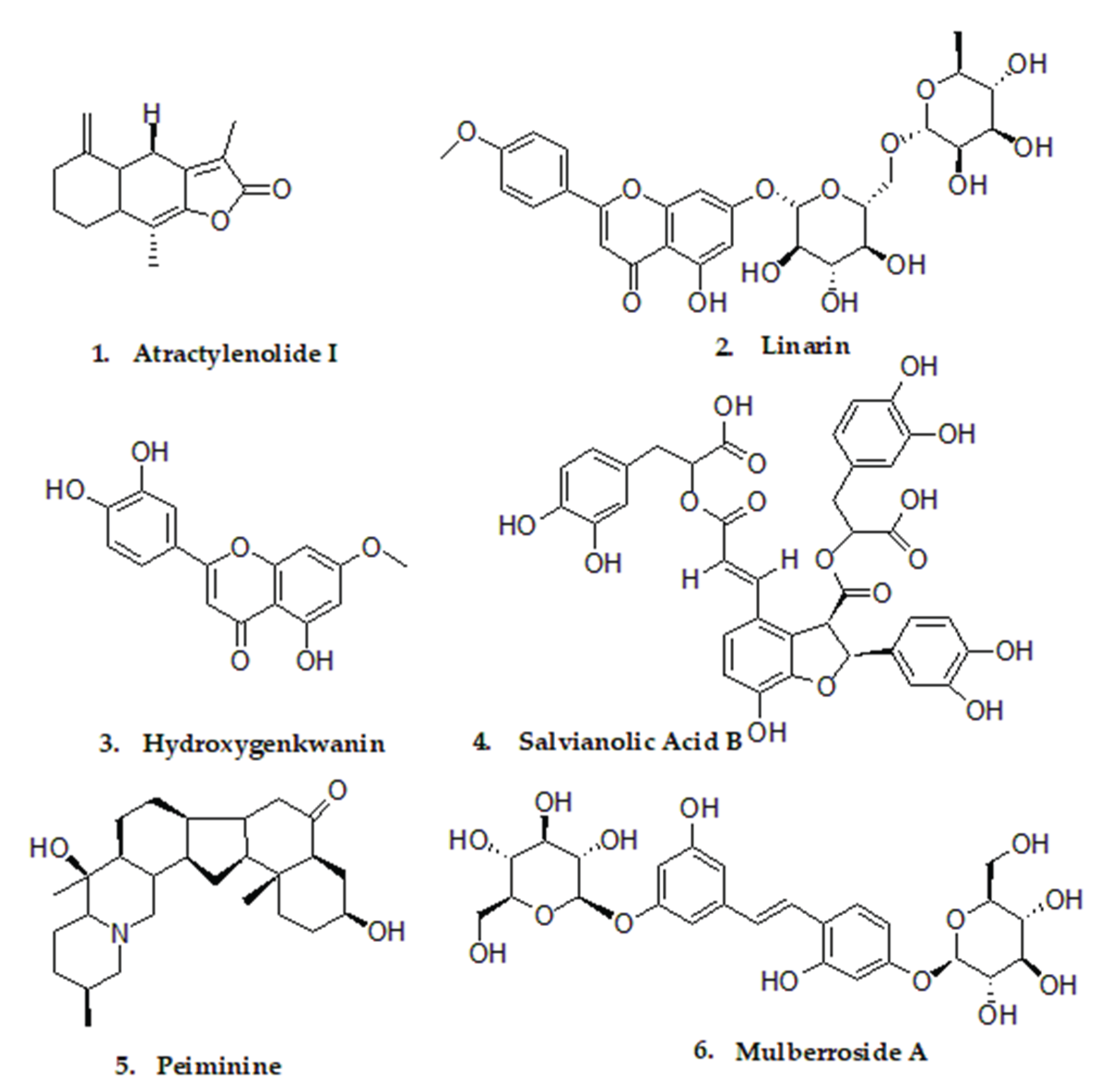
Figure 2.
Binding conformations of the compounds with PL. (A) ATR-I; (B) LIN; (C) HYD; (D) SAL-B; (E)PEI; (F) MUL-A.
Figure 2.
Binding conformations of the compounds with PL. (A) ATR-I; (B) LIN; (C) HYD; (D) SAL-B; (E)PEI; (F) MUL-A.
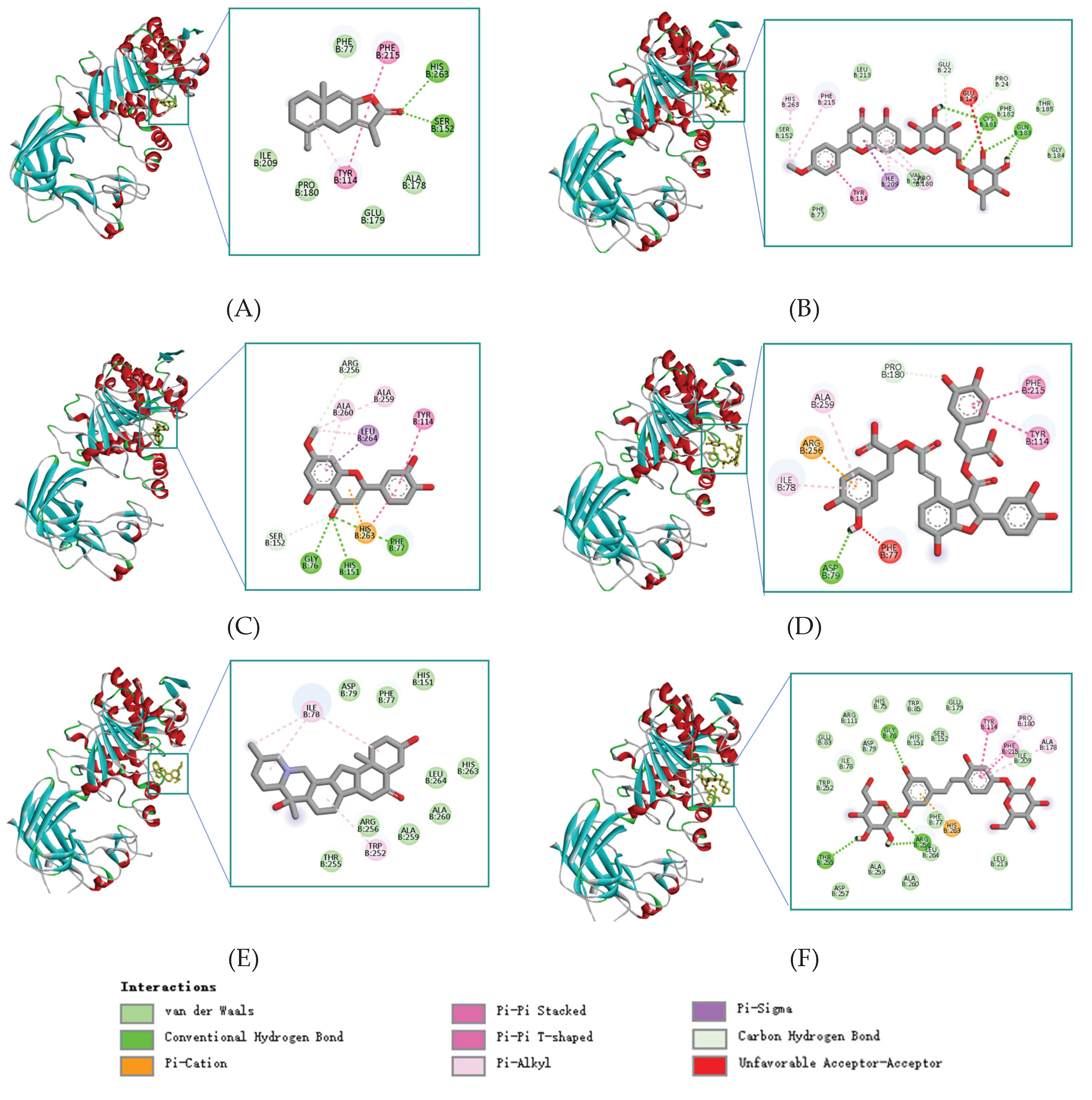
Figure 3.
The backbone RMSD plots of PL in ligand-bound and apo states (A-B) and the RMSD plots of the compounds (C-D). Radius of gyration (Rg) plots of ligand-PL complexes compared with the apo-PL (E-F).
Figure 3.
The backbone RMSD plots of PL in ligand-bound and apo states (A-B) and the RMSD plots of the compounds (C-D). Radius of gyration (Rg) plots of ligand-PL complexes compared with the apo-PL (E-F).
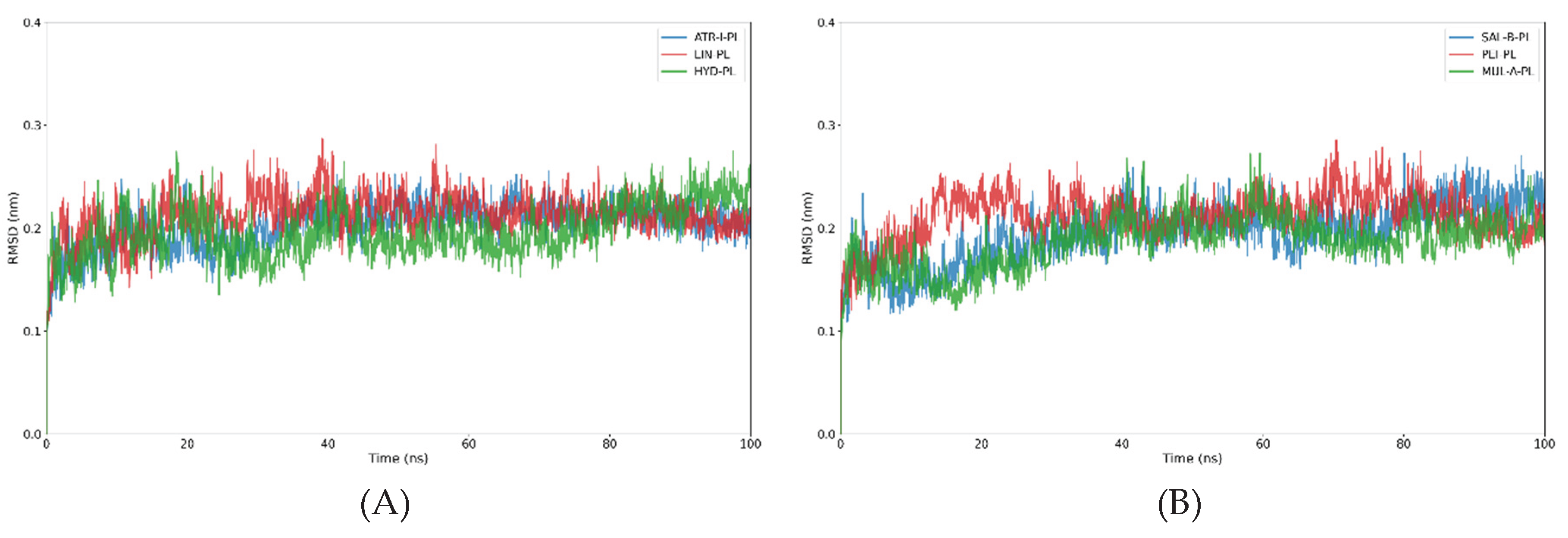
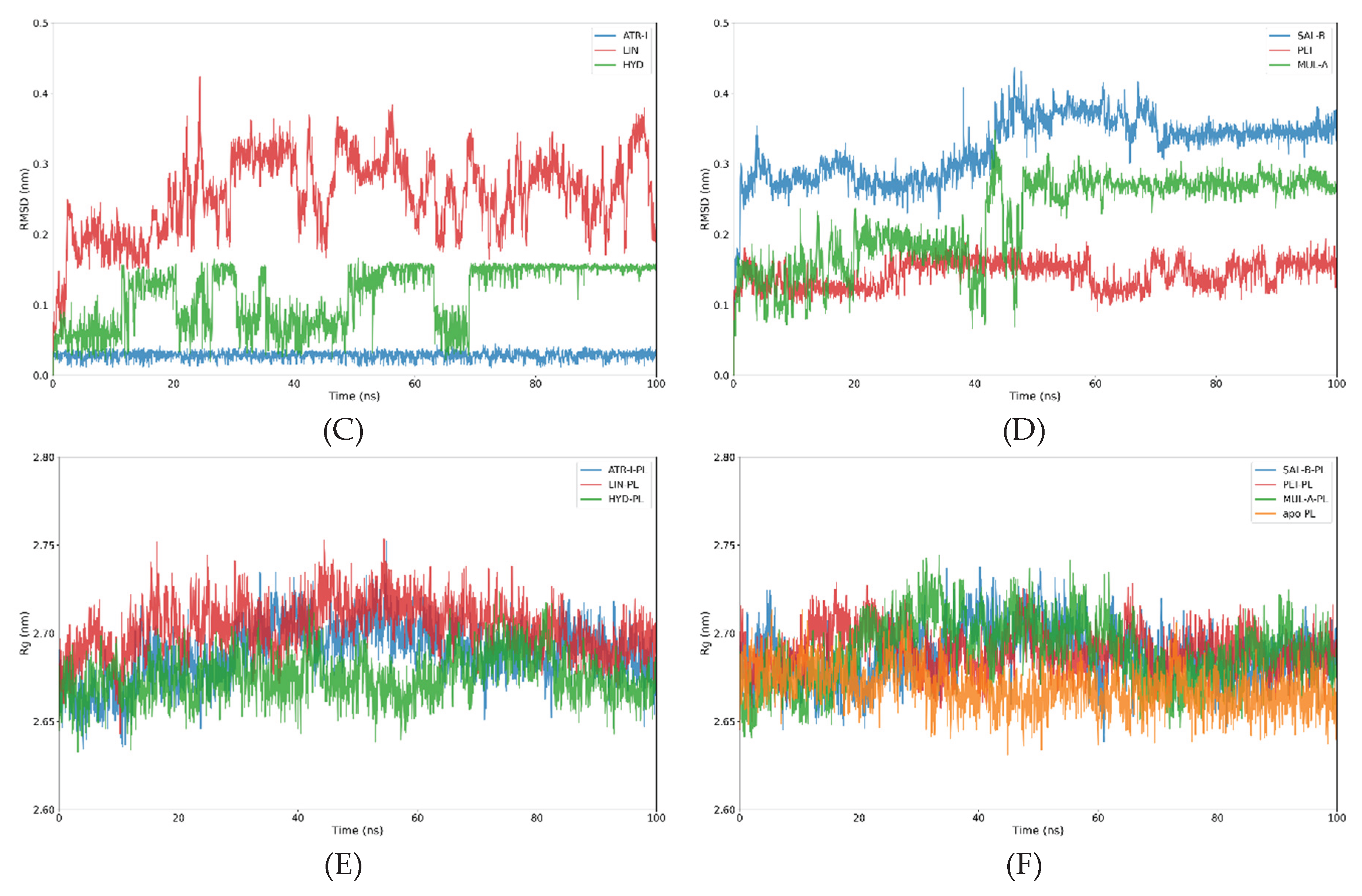
Figure 4.
The variation in binding mode of compounds after aligning the PL backbone, at 0 (red), 25 (green), 50 (blue), 75 (yellow), and 100 (orange) ns. (A)ATR-I; (B)LIN; (C)HYD; (D)SAL-B; (E) PEI; (F)MUL-A.
Figure 4.
The variation in binding mode of compounds after aligning the PL backbone, at 0 (red), 25 (green), 50 (blue), 75 (yellow), and 100 (orange) ns. (A)ATR-I; (B)LIN; (C)HYD; (D)SAL-B; (E) PEI; (F)MUL-A.
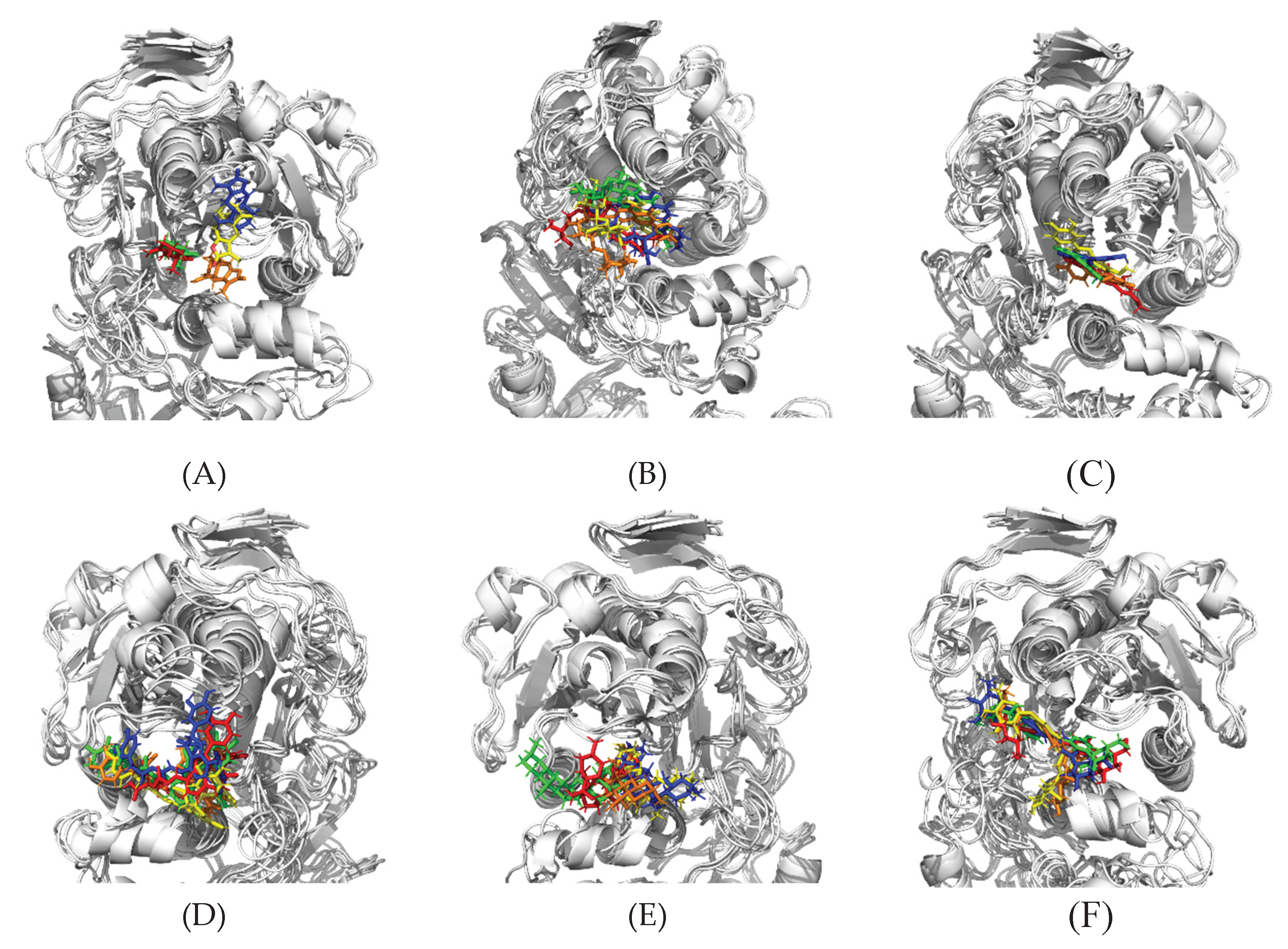
Figure 5.
Time-dependent hydrogen bond profiles between the compounds and PL. (A)ATR-I; (B)LIN; (C)HYD; (D)SAL-B; (E) PEI; (F)MUL-A.
Figure 5.
Time-dependent hydrogen bond profiles between the compounds and PL. (A)ATR-I; (B)LIN; (C)HYD; (D)SAL-B; (E) PEI; (F)MUL-A.
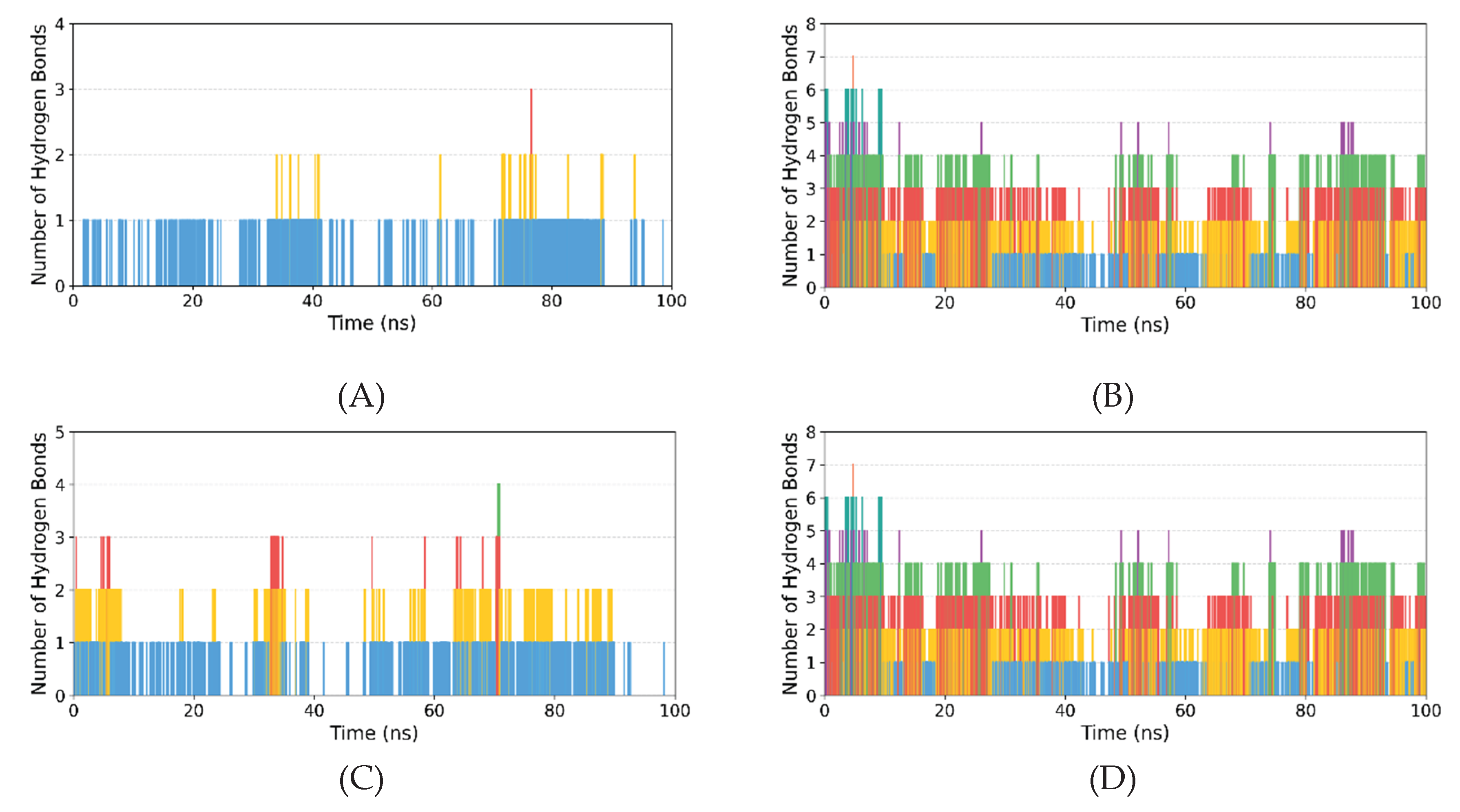
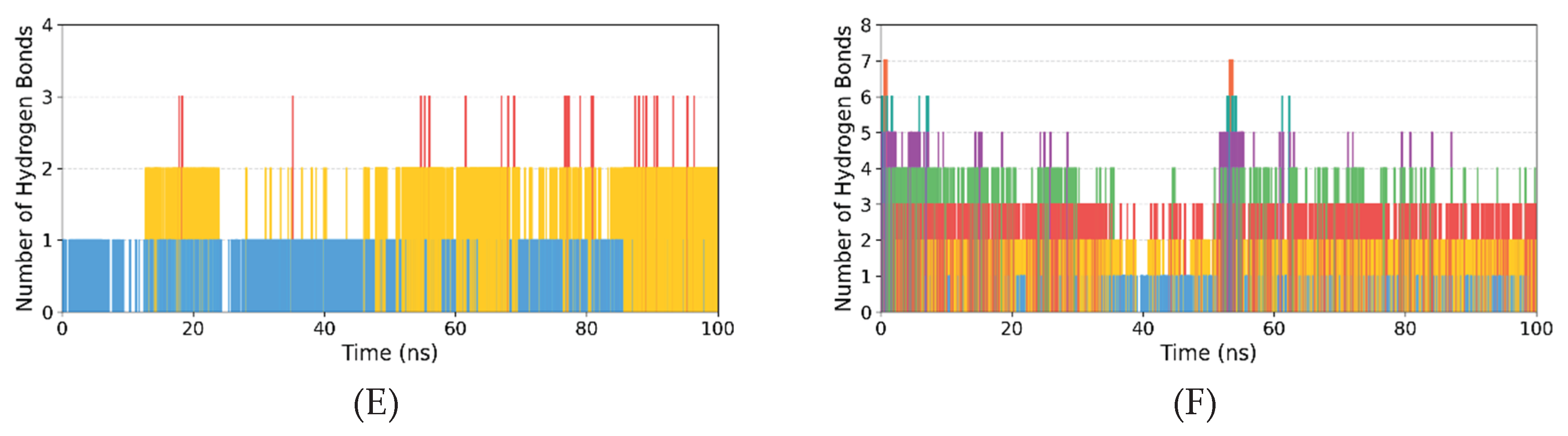
Figure 6.
Lineweaver–Burk plots for PL inhibition by four compounds (A) HYD; (B) PEI; (C) ATR-I and (D) SAL-B.
Figure 6.
Lineweaver–Burk plots for PL inhibition by four compounds (A) HYD; (B) PEI; (C) ATR-I and (D) SAL-B.
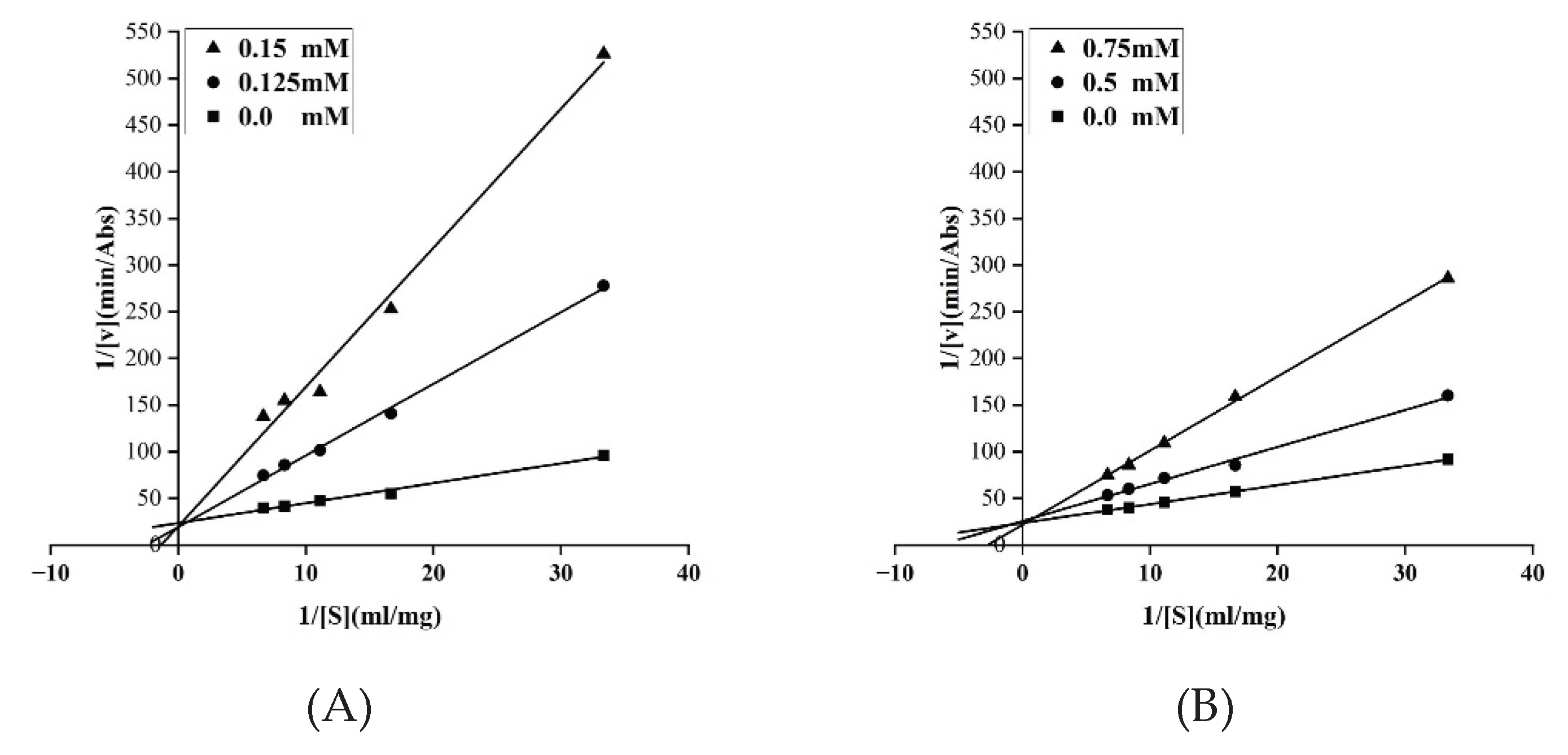
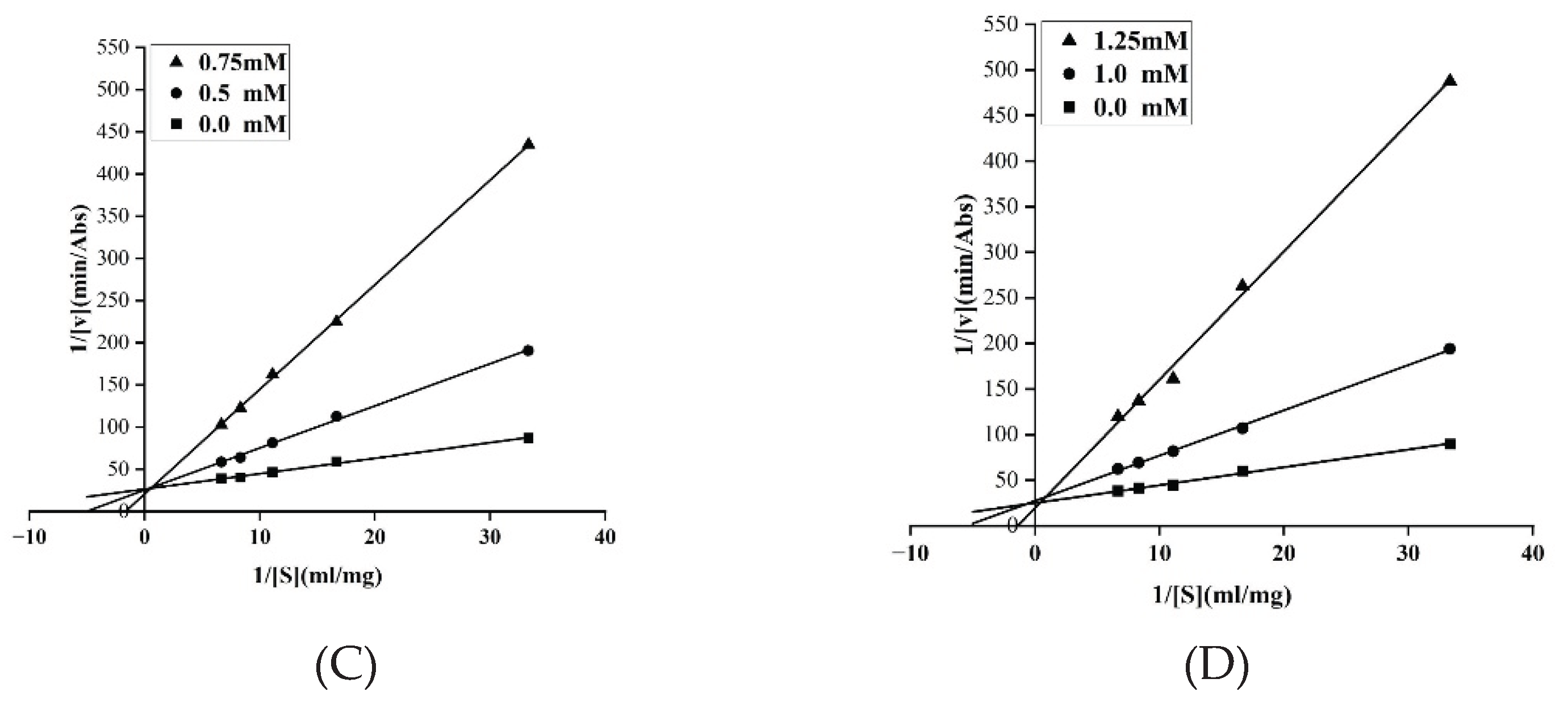

Table 1.
Docking scores (kcal/mol) of the selected candidates of PL inhibitor.
| Mol_ID | Mol_Name | Score | Hydrogen Bonds | Hydrophobic Interactions |
|---|---|---|---|---|
| MOL000043 | Atractylenolide I (ATR-I) |
−9.0 | Ser152,His263 | Phe77,Tyr114,Ala178,Phe215 |
| MOL001790 | Linarin (LIN) | −9.3 | Cys181,Glu183 | Phe182,Thr185,Val210,Leu213 |
| MOL005530 | Hydroxygenkwanin (HYD) |
−9.3 | Gly76,Phe77,His151 | Arg256,Tyr114,Ala260,Leu264 |
| MOL007074 | Salvianolic Acid B (SAL-B) | −9.1 | Asp79 | Pro180,Ile78,Tyr114,Phe215 |
| MOL009593 | Peiminine (PEI) | −9.4 | None | Phe77,Ile78,His151,Trp252, Thr255,Arg256,Ala259,Leu264 |
| MOL012733 | Mulberroside A (MUL-A) | −9.9 | Gly76,Thr255,Arg256 | Phe77,ILE78,Tyr114,Pro180, Ile209,Phe215,Ala259,Leu264 |
Table 2.
Binding free energies (kcal/mol) of PL with the six compounds calculated by MM/PBSA.
| Compound | ΔEVDW | ΔEELE | ΔEPB | ΔENPOLAR | ΔGGAS | ΔGSOLV | ΔGTOTAL |
|---|---|---|---|---|---|---|---|
| ATR-I | -17.08±0.51 | -5.35±0.50 | 11.09±0.60 | -2.11±0.04 | -22.43±0.90 | 8.99±0.56 | -13.44±0.45 |
| LIN | -26.47±0.72 | -43.49±1.61 | 53.71±1.39 | -3.29±0.05 | -69.96±1.62 | 50.42±1.37 | -19.54±0.43 |
| HYD | -35.35±0.39 | -6.88±0.60 | 29.99±0.93 | -3.34±0.02 | -42.23±0.82 | 26.65±0.92 | -15.58±0.85 |
| SAL-B | -28.27±0.47 | -63.91±1.09 | 75.13±0.83 | -4.33±0.03 | -92.18±0.88 | 70.80±0.82 | -21.38±0.40 |
| PEI | -38.97±0.29 | -13.26±0.65 | 34.85±0.50 | -3.87±0.04 | -52.23±0.62 | 30.99±0.50 | -21.24±0.39 |
| MUL-A | -48.58±0.43 | -28.76±0.76 | 68.96±1.07 | -4.95±0.03 | -77.34±0.84 | 64.01±1.06 | -13.33±0.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



