Submitted:
18 February 2026
Posted:
26 February 2026
You are already at the latest version
Abstract
Detecting highly divergent or previously unknown viruses remains a major challenge with important clinical implications. In human sequencing datasets, alignment-based approaches frequently classify many assembled contigs as “unknown” because they lack detectable similarity to reference genomes. In this work, we introduce an ensemble of deep neural networks designed to identify viral sequences across diverse human biospecimens. The proposed framework operates on metagenomic sequence data derived from next-generation sequencing platforms, where biochemical nucleotide incorporation events are transduced into fluorescence-based signals and captured by optical sensor arrays, subsequently processed into digital sequence information.
The proposed ensemble integrates complementary convolutional architectures capable of capturing both local patterns and broader compositional signals directly from raw metagenomic contigs. The training data consisted of sequences from 19 metagenomic experiments. On this dataset, our approach achieves state-of-the-art performance, outperforming previous machine/deep learning methods for viral genome classification. Using 300 base pairs (bp) contigs, the model achieves an area under the ROC curve of 0.939, outperforming previously reported state-of-the-art methods evaluated on the same dataset under an identical testing protocol. If our system is combined with other recent methods for representing the DNA sequence as a feature vector, we can achieve an even higher area under the curve, approximately 0.94.
All developed code and dataset will be made publicly available to ensure full reproducibility of our results.
Keywords:
neural networks
; viroma
; DNA sequence
; ensemble
1. Introduction
DNA sequences lie at the core of all living systems, shaping virtually every biological process in both eukaryotes and prokaryotes. Understanding how information is encoded in DNA, and how sequence properties give rise to biological function, is fundamental not only for advancing basic biology but also for engineering new biological systems. However, the immense diversity and complexity of possible DNA sequences make such tasks inherently challenging. Many coding and noncoding regions remain poorly characterized, and traditional analytical methods often struggle to capture the full breadth of patterns embedded in biological data [1].
Machine learning has therefore become indispensable in bioinformatics, enabling the extraction of meaningful insights from rapidly expanding datasets. Classical methods such as random forests, support vector machines, hidden Markov models, and Bayesian networks have long contributed to fields including genomics, proteomics, systems biology, and structural biology [2]. More recently, deep learning has surpassed these traditional approaches by scaling to massive datasets and uncovering complex, nonlinear relationships. The transformative impact of deep learning is exemplified by breakthroughs such as AlphaFold 3 [3], and by the growing use of natural language processing inspired models to analyze biological sequences, which resemble linguistic data in their structure and statistical properties [4].
These advances are particularly relevant to the study of the human virome, the diverse community of viruses that inhabit the human body. Virome composition varies across individuals and has been linked to disease, yet its full influence on human health remains incompletely understood [5,6]. Metagenomic studies continue to reveal large numbers of previously unknown viruses, suggesting that only a fraction of human associated viruses have been discovered. Emerging evidence further indicates that yet unidentified viral pathogens may contribute to complex conditions [7], including autoimmune diseases such as diabetes [8] and multiple sclerosis [5,9].
Next-generation sequencing (NGS) has become a powerful means of directly capturing all genetic material present in clinical samples, enabling metagenomic analyses that bypass the need for prior knowledge of pathogens [10]. However, current computational approaches face significant limitations. Alignment-based tools such as BLAST often fail to classify highly divergent sequences, leaving many reads labeled as “unknown”. Profile Hidden Markov Model approaches like HMMER3 [11] improve sensitivity to distant homologs but still depend on existing reference databases, restricting their ability to detect highly novel viral genomes [9].
These challenges highlight the need for alternative computational strategies capable of generalizing beyond known viral diversity. Machine/Deep learning offers a promising framework for classifying viral sequences from metagenomic data without relying solely on sequence homology. Integrating modern machine learning and deep learning techniques with metagenomic sequencing presents a crucial opportunity to advance viral discovery and deepen our understanding of the human virome [2,9].
The main contributions of this work can be summarized as follows:
- We introduce an ensemble of neural networks that leverages heterogeneous feature representations for training, improving robustness and generalization.
- The proposed approach is built upon a well-established, for this topic, neural network architecture, extending it through the exploration of different network hyperparameters and pooling strategies to construct the ensemble.
- We provide a publicly available implementation of the proposed system to ensure reproducibility https://github.com/LorisNanni/Ensemble-Deep-Learning-Models-on-Raw-DNA-Sequences-for-Viral-Genome-Identification-in-Human-Samples.
- Extensive experimental evaluations demonstrate that the proposed method achieves state-of-the-art performance when compared to existing approaches on the same dataset and under the same evaluation protocol.
We acknowledge that a wide range of neural network architectures exists, as well as numerous methods for representing DNA sequences as matrices suitable for neural network processing [12]. However, providing an exhaustive survey or comparison of these innumerable approaches is not the objective of this work. Instead, our goal is to demonstrate that a simple ensemble of neural networks, composed of small models and therefore characterized by low computational cost on modern GPUs, is sufficient to achieve state-of-the-art performance on the evaluated dataset and task. Importantly, this is accomplished without making any a priori hyperparameter choices based on the full dataset, thereby avoiding potential sources of overfitting. All design decisions involved in constructing the ensemble are derived exclusively from the training/validation set, ensuring a principled and unbiased evaluation protocol.
2. Materials and Methods
In this section, we describe the neural network architectures adopted in this study, with particular emphasis on the ViraMiner architecture, which serves as the baseline model for viral sequence identification. We also detail the preprocessing strategy used to transform raw DNA sequences into a numerical representation suitable for convolutional neural networks.
All DNA sequences are encoded using the well-established one-hot encoding scheme for nucleotide sequences [13]. Let
denote the alphabet of possible characters, where (A), (C), (G), and (T) represent the four canonical nucleotides, and (N) is used as a fifth symbol to encode any ambiguous or non-canonical nucleotide. Each nucleotide () is mapped to a binary vector (), defined as
Given a DNA sequence of fixed length (L = 300), S = , the one-hot encoding transforms (S) into a binary matrix where each row corresponds to the one-hot representation of a single nucleotide in the sequence. This matrix constitutes the input to the neural networks described in the following. See Figure 1 for an example of one hot encoding.
Our baseline is ViraMiner, a convolutional neural network (CNN) based method designed for the identification of viral sequences integrated into the human genome. This choice is well motivated, as CNNs have already proven effective in analyzing biological sequences, such as DNA and amino acid sequences [14,15,16]. The architectures tested in this paper are shown in Figure 3 and Figure 4. Input DNA sequences are first encoded using a fixed-length numerical representation and processed by multiple parallel convolutional branches that operate at different receptive field sizes, enabling the model to capture both short-range and long-range sequence patterns. The convolutional layers perform hierarchical feature extraction, while pooling layers progressively reduce the dimensionality and improve translation invariance. The extracted features are then flattened and passed to fully connected layers that act as the final classifier. The network outputs a single sigmoid-activated probability score, indicating the likelihood that the input sequence contains viral genomic material. Moreover, with respect to ViraMiner architecture (i.e. the one shown in Figure 3), we added a batch normalization step after the convolutional layer and after the first fully connected layer.
Figure 2.
Viraminer branches.
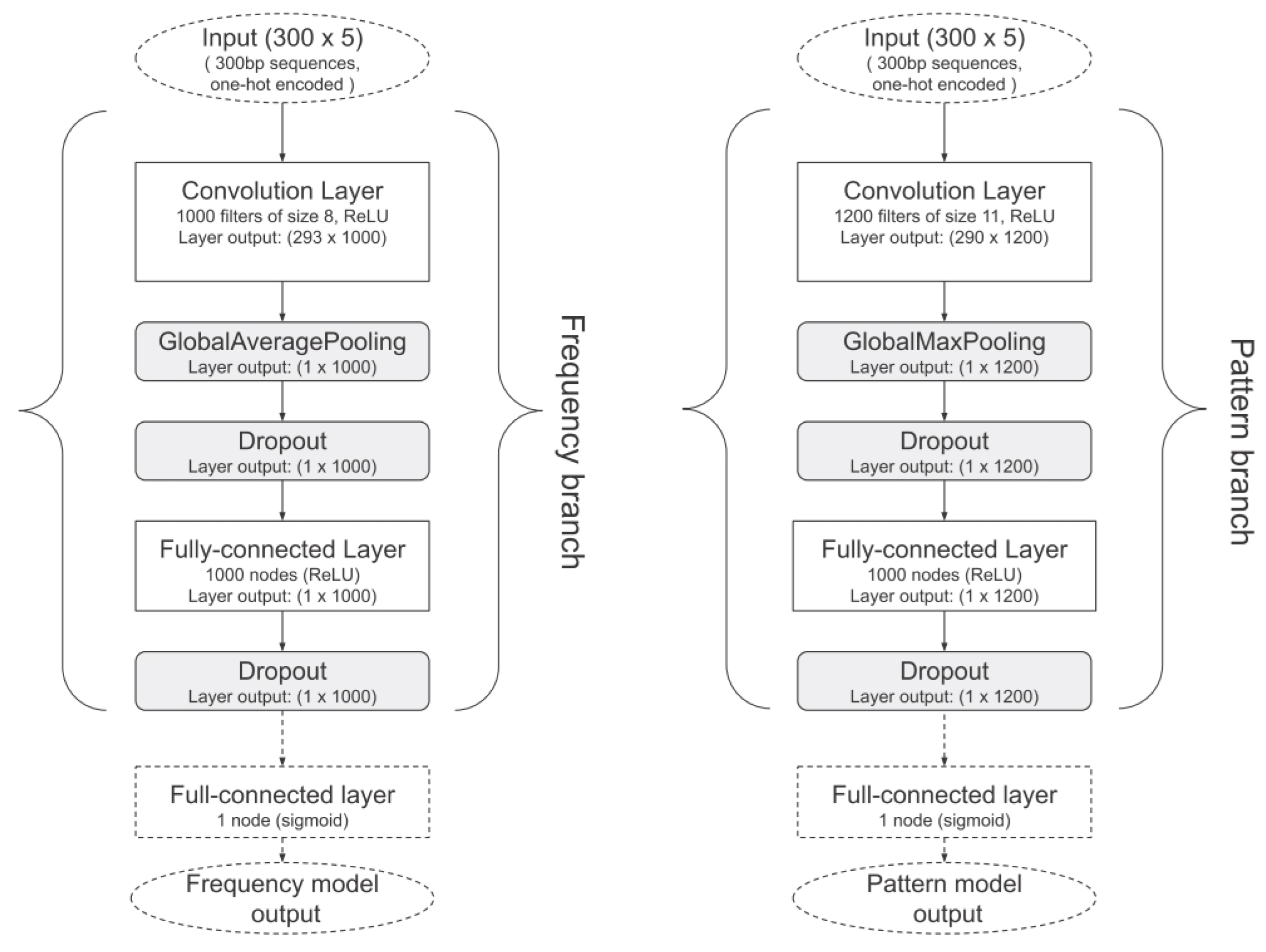
Figure 3.
Viraminer full architecture.
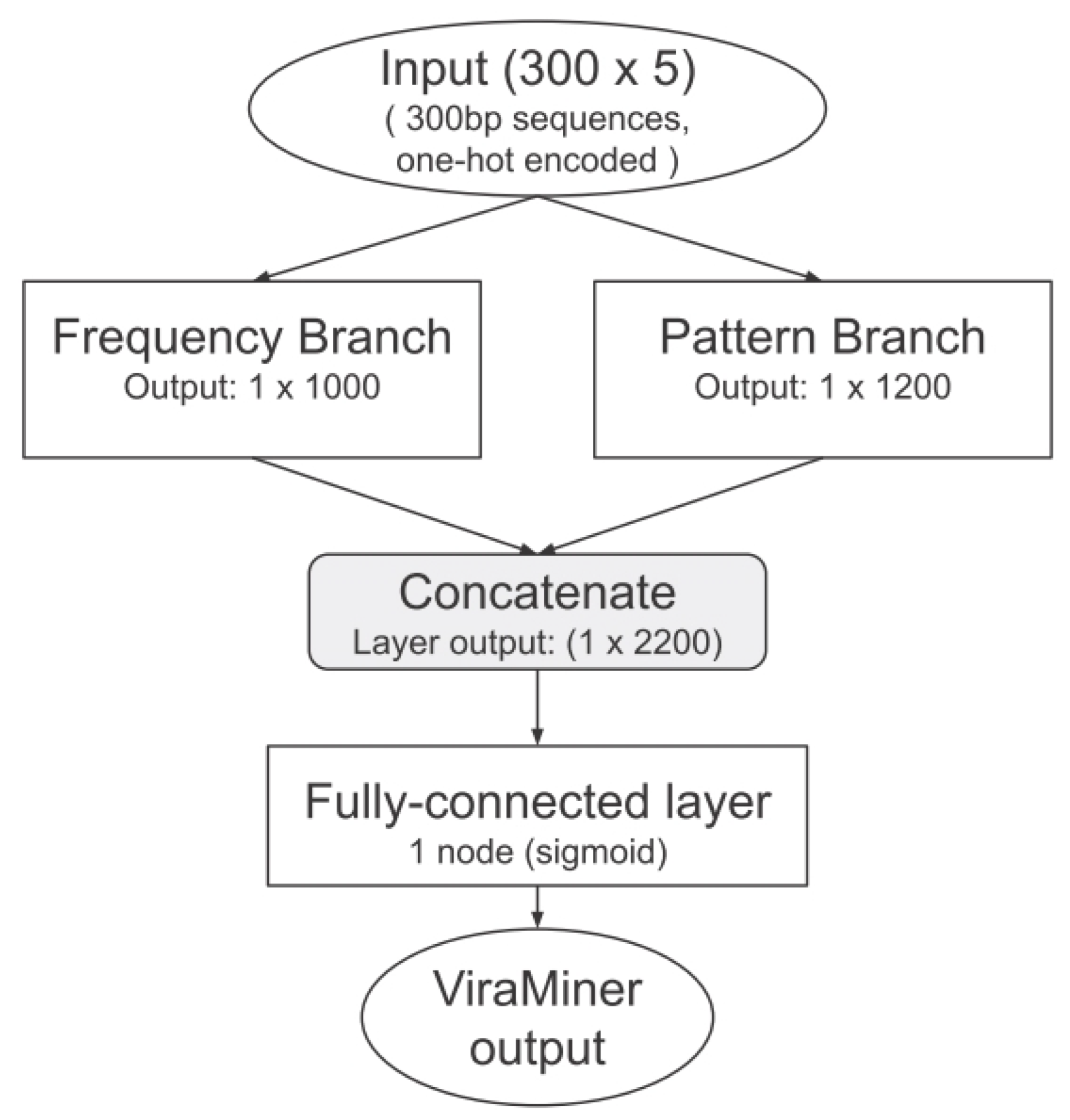
Figure 4.
Our best model architecture.
The model is trained on a labeled dataset of viral and human sequences, using the Adam algorithm [17] to optimize the binary cross-entropy loss function.
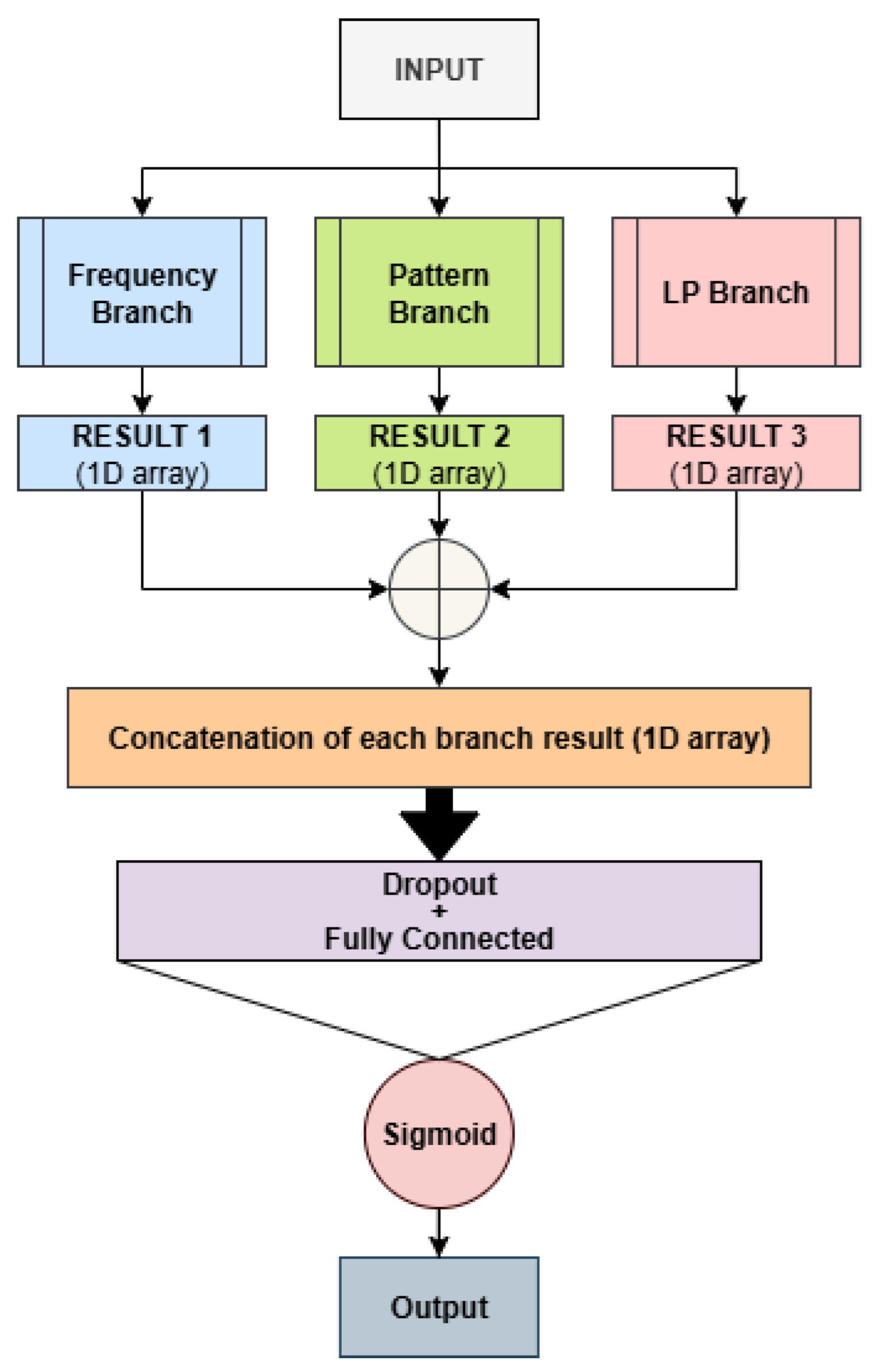
In addition to the pattern branch shown in Figure 2, we also tested another branch, called the LP branch, in which the pooling operator is a parameterized pooling, whose parameter is learned during training. The parametrized pooling, named “Power-Average” pooling layer [18], is defined as follows: on each window, the function computed is:
when: , this corresponds to Max Pooling; p = 1, corresponds to mean Pooling.
The value of p is a hyperparameter, see Table 1, in the context of Power-Average (Lp) pooling, ’norm type’ corresponds to the power parameter p.
The hyperparameters tested over all the models are reported in the following Table 1. It is important to emphasize that all hyperparameters were selected using only the validation set. The test set is completely blind and was never used for hyperparameter tuning, to avoid any form of overfitting on our data and to ensure that the reported results are as reliable as possible. As the learning-rate scheduler, we used a decay approach, which reduces the optimizer’s learning rate by a factor of 0.5 every 5 epochs.
Throughout the remainder of the paper, the term branch refers to the network associated with a specific backbone (e.g., columns in Figure 2). For instance, the branch "pattern" corresponds to the network shown in the last column of Figure 2.
The Merger combines multiple branches into a single system, as illustrated in Figure 4. Specifically, the outputs of the first fully connected layer of all branches are concatenated, followed by a dropout layer and a fully connected layer that produces the final model output. For the Merger models, only the best-performing branch of each type was included, using the hyperparameters selected for each branch when trained independently.
The ensemble proposed in this work consists of ten networks based on the previously presented architectures. When the architecture name starts with Bx, it denotes a single-branch network of type x, as illustrated in the previous figure, where corresponds to the frequency branch, to the pattern branch, and to the LP branch. Conversely, architectures labeled Mx+y indicate multi-branch models that combine branches x and y, as exemplified in Figure 4. The ensemble is constructed using a simple sum rule, where the outputs of the individual networks are fused by summation, without introducing any additional learnable parameters.
When the term ’VM’ appears in parentheses after the network name, it indicates that we use exactly the same architectures as those reported in [9], i.e., identical layers and hyperparameters.
In conclusion, the proposed ensemble is composed of the following networks:
- Bf (VM)
- Bf
- B1
- Bp (VM)
- Bp
- Mf+p+1
- Mf+1
- Mp+1
- Mf+p
- Original ViraMiner architecture.
Please note that for the Merger architecture we use only the branches proposed here, and not the original ones from ViraMiner.
2.1. Dataset
The metagenomic data used in this study were obtained through Next Generation Sequencing technologies, including Illumina NextSeq, MiSeq, and HiSeq platforms, following the manufacturers’ standard protocols. The dataset originated from human-derived samples collected from multiple patient cohorts. These analyses aimed to identify viral genomes and other microorganisms in individuals affected by disease, as well as in matched control subjects.
All sequencing runs were processed using a validated bioinformatics pipeline. In brief, the workflow begins with read quality assessment, during which sequences are filtered according to their Phred quality scores. High-quality reads are then aligned against human, bacterial, phage, and vector reference genomes using BWA-MEM, and any reads showing more than 95% sequence identity over at least 75% of their length are excluded from subsequent analyses [19]. The remaining reads are normalized and assembled de novo using multiple assemblers, including IDBA-UD, Trinity, SOAPdenovo, and SOAPdenovo-Trans. The resulting contigs undergo taxonomic assignment via BLAST. The implementation of the pipeline is publicly available on GitHub (https://github.com/NGSeq/ViraPipe and https://github.com/NIASC/VirusMeta).
The training set comprised 19 independent NGS experiments. After de novo assembly, contigs were annotated using PCJ-BLAST [20] with the following configuration: Blastn algorithm, match reward of 1, mismatch penalty of 1, gap opening cost of 0, gap extension cost of 2, and an e-value threshold of . All contigs labeled through this workflow were aggregated to train multiple machine learning models. Notice that a contig is a continuous DNA sequence assembled from overlapping shorter sequencing reads. To prepare the input data for neural networks, labeled contigs were segmented into fixed-length fragments of 300 bp, any residual nucleotides that did not fit into a full-length segment were discarded. The dataset contains around 320,000 DNA sequences overall. Its primary difficulty lies in the significant class imbalance, as viral sequences make up only about 2% of the total. The data was divided into training, validation, and test subsets.
3. Results
The Area Under the Receiver Operating Characteristic curve (AUROC), Precision, and Recall were used as evaluation metrics to identify the best-performing models. For a binary classification problem with true positives (TP), false positives (FP), true negatives (TN), and false negatives (FN), the metrics are defined as follows:
-
The ROC curve plots the True Positive Rate (TPR) against the False Positive Rate (FPR):The AUROC is the area under this curve:
These performance indicators are also used in the literature, since accuracy is not informative when the dataset is highly imbalanced and, as is well known in the literature, accuracy is not a reliable metric for strongly imbalanced datasets.
In Table 2 we report the following approaches:
- FullE, the ensemble proposed here;
- Mf+p+1, our best stand-alone net;
- Viraminer, the original method proposed in [9];
- [2], the comparison of our method with this paper is very interesting, not only because it uses the same dataset and the same data split, but also because this method is based on self-attention and convolutional operations on nucleic acid sequences, leveraging two prominent deep learning strategies commonly used in computer vision and natural language processing.
The trade-off between precision and recall is a critical aspect of viral genome classification. A contig is labeled as viral when the model output exceeds a specified threshold. In [9], the authors report achieving a precision of 0.90 with a recall of 0.32. By contrast, when our ensemble model is tuned to the same precision level (0.90), it attains a higher recall of 0.4011.
In the following figures, we directly compare our ensemble (red curve) with the original ViraMiner model (black curve): in Figure 5 using the ROC curve, in Figure 6 using the precision–recall curve, and in Figure 7 using the DET-curve. In these cases, the performance difference between the two methods is clear. It is evident that this performance gain does not come for free, since the inference time of our approach is roughly nine times higher than that of the original ViraMiner. However, we consider this a secondary issue given the computational power currently available with modern GPUs.
To ensure that the effectiveness of our model was not influenced by unnoticed biases in the real datasets, as in [9], we additionally evaluated it using data generated with the ART sequencing read simulator [24] (see [9] for details on simulation settings). The simulated dataset contained slightly fewer reads than the real one (210,000) but included a higher proportion of viral sequences (around 10%). We first applied the models trained exclusively on the 19 real metagenomic datasets: this produced an AUROC of 0.751 for Viraminer and 0.782 for our approach. Next, we retrained ViraMiner and our ensemble directly on the simulated data, we reused the same hyperparameters and training setup as before. The model was trained on 80% of the simulated data, with 10% for validation and 10% for testing. Under this configuration, the ViraMiner achieved a test AUROC of 0.921, while our approach 0.932.
As further experiment, we evaluated a Foundation Model for DNA barcoding as a feature extractor. The extracted features were subsequently used to train a Support Vector Machine (SVM), LibSVM toolbox https://www.csie.ntu.edu.tw/~cjlin/libsvm/. This experiment is particularly interesting because the Foundation Model was pre-trained on DNA sequences that are completely different from those analyzed in this study. Specifically, we employed a masked language model (MLM), BarCodeMAE, as a foundation model to extract numerical vector representations (embeddings) from DNA sequences. BarCodeMAE was originally trained on arthropod DNA barcoding data, which is biologically unrelated to our target domain. Despite this domain mismatch, our goal was to investigate whether a model pretrained on large-scale, heterogeneous genomic data could still capture generalizable sequence features useful for downstream tasks. To obtain a fixed-length representation of each DNA sequence, we derived a single embedding for the entire DNA sequence by applying global average pooling over the sequence of 768-dimensional token-level output vectors produced by the model, excluding padding and special tokens. These pooled embeddings were then used as input features to train the SVM classifier. As expected, the standalone performance of this approach was not particularly strong. The SVM trained solely on the embeddings extracted from the pretrained MLM achieved an AUROC of 0.6818, indicating only moderate discriminative capability. Nevertheless, a more interesting finding emerged when combining this classifier with our main ensemble. By fusing the SVM outputs with the ensemble predictions using a simple weighted sum rule, where the weights are obtained using the validation data, we observed a slight improvement in overall performance, reaching an AUROC of 0.9395, which is marginally higher than that of the proposed ensemble alone. Although the gain is minimal, it is noteworthy, as it suggests that the foundation model still captures non-random and complementary information that can contribute to downstream classification.
The architecture of the Foundation Model is shown in Figure 8.
As a final experiment, we evaluate the EDEN method [25], a multiscale DNA sequence encoding framework based on kernel density estimation. EDEN represents nucleotide sequences as position-aware density profiles computed at multiple biologically relevant scales and processes them with a hybrid deep convolutional neural network. We consider two evaluation settings: (i) training the EDEN architecture from scratch using our training set, and (ii) fine-tuning an EDEN model pre-trained shared by the original authors of EDEN https://github.com/zabihis/EDEN. When training EDEN network from scratch, performance is very low. It improves when using a pre-trained version. We conducted several experiments, also varying the optimization methods. The best method, consistently selected based on the validation set, achieves an area under the ROC curve (AUC) of 0.8775 on the test set, which is still clearly lower than the performance of the methods we propose. If we include this method in our ensemble, through a weighted fusion, where the EDEN weight is selected using the validation set (with the ensemble weight fixed to 1), we obtain an AUC of 0.9395. If, within the same weighted combination, we also add the scores produced by an SVM trained on Barcodemae, with its weight again determined on the validation set, the AUC further increases to 0.9399. Although these improvements are modest and mainly refine the performance, it is interesting to note that feature extraction methods fully pre-trained on datasets different from the one used in our study can still contribute to enhancing overall performance.
4. Conclusion
In this study, we presented a convolutional neural network-based ensemble for the identification of viral sequences in heterogeneous human metagenomic samples. The proposed ensemble strategy combines complementary representations of DNA sequences, enabling the model to capture both local patterns and global distributional properties. Specifically, the architecture integrates multiple CNN configurations characterized by input features, different pooling mechanisms, hyperparameters selection, which contribute distinct and complementary information to the final prediction.
The system was trained and evaluated on assembled metagenomic contigs derived from 19 independent experiments. Experimental results show that the proposed method attains strong discriminative performance, achieving an AUROC of 0.939 on the test set. This represents a substantial improvement over earlier approaches trained on the same data, indicating that the proposed ensemble can exploit richer information for viral classification.
Overall, these findings reinforce the suitability of CNN-based models for viral genome analysis and highlight the importance of architectural diversity in achieving robust performance.
We will provide a publicly available implementation of the proposed system to facilitate reproducibility https://github.com/LorisNanni/Ensemble-Deep-Learning-Models-on-Raw-DNA-Sequences-for-Viral-Genome-Identification-in-Human-Samples, from the same link, the dataset is also available for download.
Author Contributions
Conceptualization, M.D.N., S.B., S.P.G., L.N. and D.F.; methodology, M.D.N., S.B., S.P.G., L.N. and D.F.; software, M.D.N., S.B., S.P.G., L.N. and D.F.; validation,M.D.N., S.B., S.P.G., L.N. and D.F.; formal analysis, M.D.N., S.B., S.P.G., L.N. and D.F.; investigation, M.D.N., S.B., S.P.G., L.N. and D.F.; data curation, M.D.N., S.B., S.P.G., L.N. and D.F.; writing—original draft preparation, M.D.N., S.B., S.P.G., L.N. and D.F.; writing—review and editing, M.D.N., S.B., S.P.G., L.N. and D.F.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
All datasets used in this study are publicly available on the GitHub repository associated with this paper.
Acknowledgments
We would like to acknowledge the support that NVIDIA provided us through the GPU Grant Program. We used a donated GPU to train deep networks used in this work.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- He, S.; Gao, B.; Sabnis, R.; Sun, Q. Nucleic Transformer: Classifying DNA Sequences with Self-Attention and Convolutions. ACS Synthetic Biology 2023, 12, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Do, D.T.; Le, T.Q.T.; Le, N.Q.K. Using Deep Neural Networks and Biological Subwords to Detect Protein S-sulfenylation Sites. Briefings in Bioinformatics 2020, 22, bbaa128. [Google Scholar] [CrossRef] [PubMed]
- Wylie, K.M.; Mihindukulasuriya, K.A.; Sodergren, E.; Weinstock, G.M.; Storch, G.A. Sequence analysis of the human virome in febrile and afebrile children. PLoS ONE 2012, 7, e27735. [Google Scholar] [CrossRef] [PubMed]
- Cesanelli, F.; Scarvaglieri, I.; De Francesco, M.A.; Alberti, M.; Salvi, M.; Tiecco, G.; Castelli, F.; Quiros-Roldan, E. The Human Virome in Health and Its Remodeling During HIV Infection and Antiretroviral Therapy: A Narrative Review. Microorganisms 2026, 14. [Google Scholar] [CrossRef]
- Zhang, D.; Cao, Y.; Dai, B.; Zhang, T.; Jin, X.; Lan, Q.; Qian, C.; He, Y.; Jiang, Y. The virome composition of respiratory tract changes in school-aged children with Mycoplasma pneumoniae infection. Virology Journal 2025, 22, 10. [Google Scholar] [CrossRef] [PubMed]
- Mercalli, A.; Lampasona, V.; Klingel, K.; Albarello, L.; Lombardoni, C.; Ekstrom, J.; et al. No evidence of enteroviruses in the intestine of patients with type 1 diabetes. Diabetologia 2012, 55, 2479–2488. [Google Scholar] [CrossRef]
- Vincente, A.T.Z.B.J.D.R. ViraMiner: Deep learning on raw DNA sequences for identifying viral genomes in human samples. PLOS ONE 2019, 1–17. [Google Scholar]
- Meiring, T.L.; Salimo, A.T.; Coetzee, B.; Maree, H.J.; Moodley, J.; Hitzeroth, I. Next-generation sequencing of cervical DNA detects human papillomavirus types not detected by commercial kits. Virology Journal 2012, 9, 164. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Research 2013, 41, e121. [Google Scholar] [CrossRef]
- Randhawa, G.S.; Soltysiak, M.P.M.; El Roz, H.; de Souza, C.P.E.; Hill, K.A.; Kari, L. Machine learning using intrinsic genomic signatures for rapid classification of novel pathogens: COVID-19 case study. PLOS ONE 2020, 15, e0232391. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nature Biotechnology 2015, 33, 831–838. [Google Scholar] [CrossRef]
- Kelley, D.R.; Snoek, J.; Rinn, J.L. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Research 2016, 26, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nature Methods 2015, 12, 931. [Google Scholar] [CrossRef]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Molecular Systems Biology 2016, 12, 878. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. In Proceedings of the Proceedings of the 3rd International Conference on Learning Representations (ICLR), San Diego, CA, USA, 2015. [Google Scholar]
- Radenović, F.; Tolias, G.; Chum, O. Fine-Tuning CNN Image Retrieval with No Human Annotation. IEEE Transactions on Pattern Analysis and Machine Intelligence;arXiv 2019, arXiv:1711.0251241, 1655–1668. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Nowicki, M.; Bzhalava, D.; Bała, P. Massively Parallel Implementation of Sequence Alignment with Basic Local Alignment Search Tool Using Parallel Computing in Java Library. Journal of Computational Biology 2018, 25, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Bzhalava, Z.; Tampuu, A.; Bała, P.; Vicente, R.; Dillner, J. Machine Learning for detection of viral sequences in human metagenomic datasets. BMC Bioinformatics 2018, 19, 336. [Google Scholar] [CrossRef]
- Bzhalava, Z.; Tampuu, A.; Bała, P.; Vicente, R.; Dillner, J. Machine Learning for detection of viral sequences in human metagenomic datasets. BMC Bioinformatics 2018, 19, 336. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: a next-generation sequencing read simulator. Bioinformatics 2011, 28, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Zabihi, S.; Hashemi, S.; Mansoori, E. EDEN: multiscale expected density of nucleotide encoding for enhanced DNA sequence classification with hybrid deep learning. BMC Bioinformatics 2026, 27, 40. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
One hot encoding.
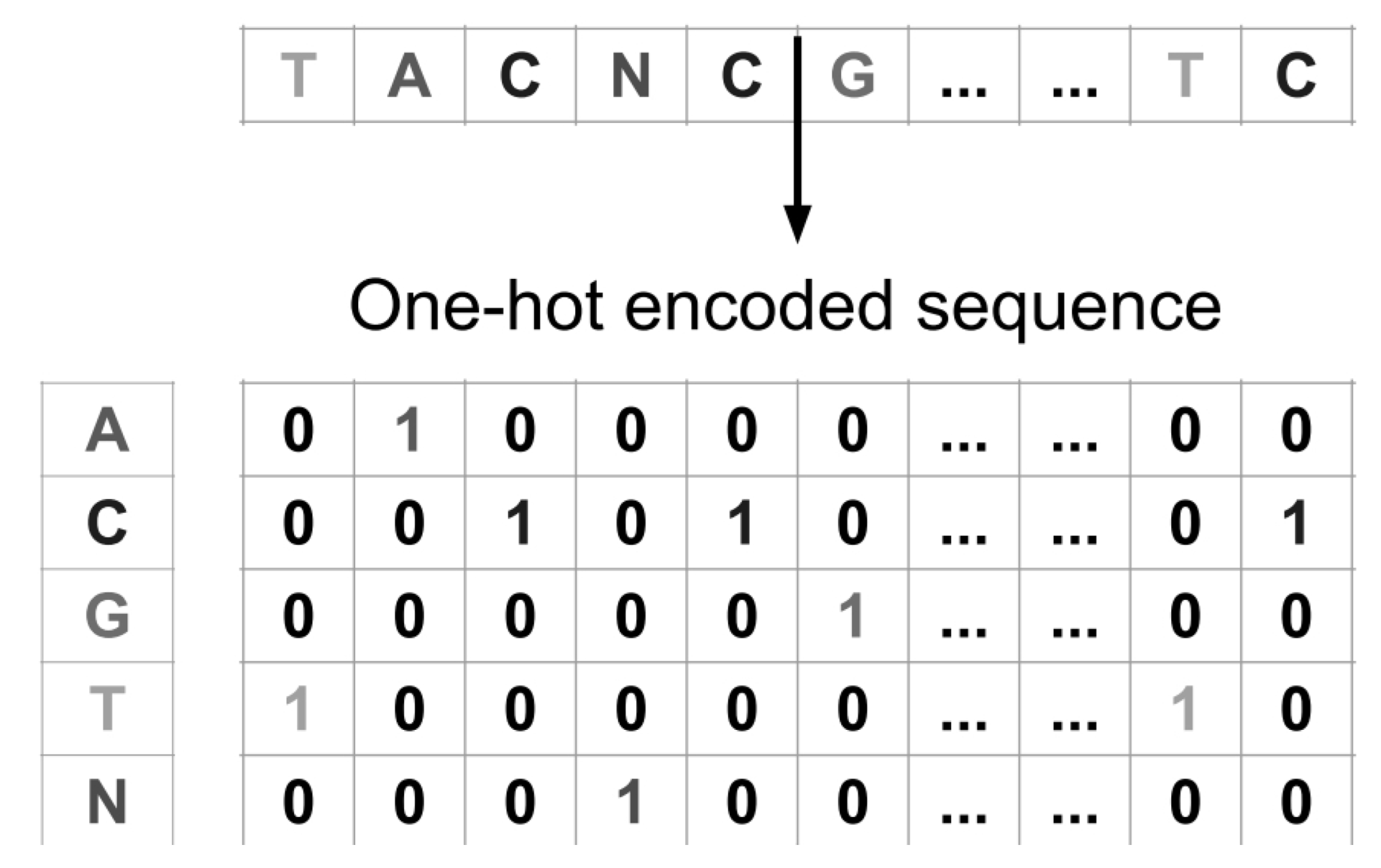
Figure 5.
ROC curves.
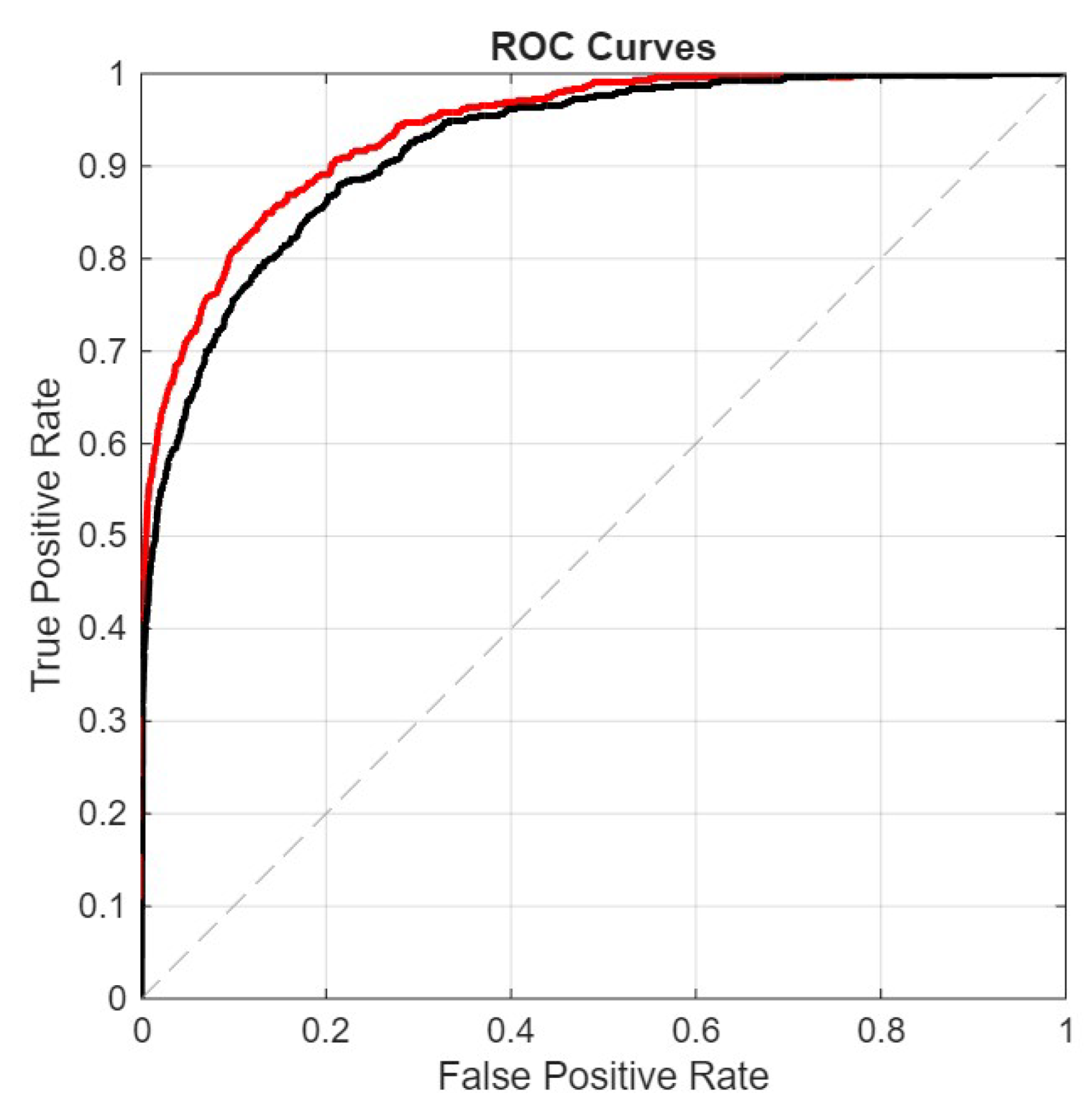
Figure 6.
Precision-Recall curves.
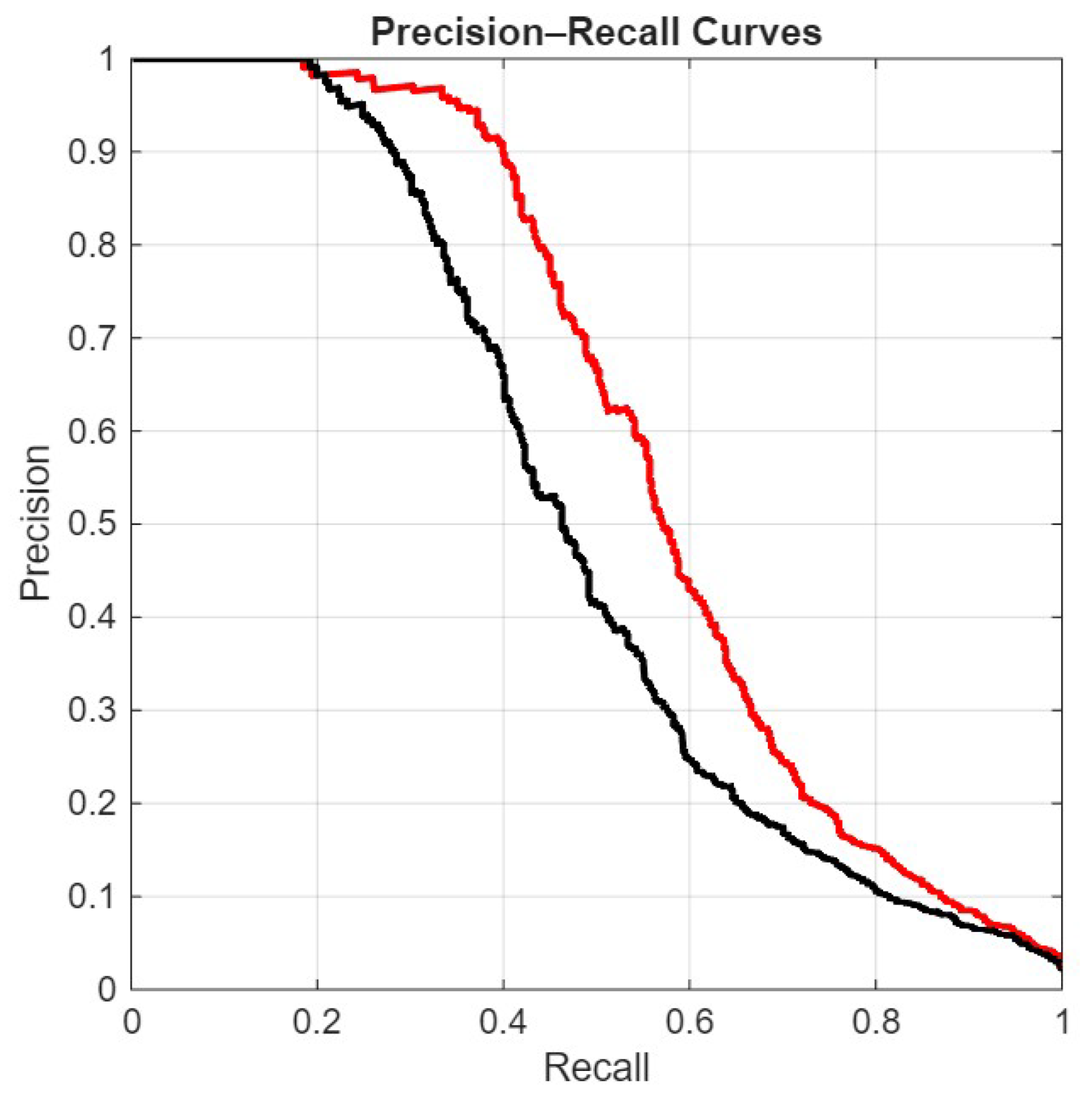
Figure 7.
DET curves.
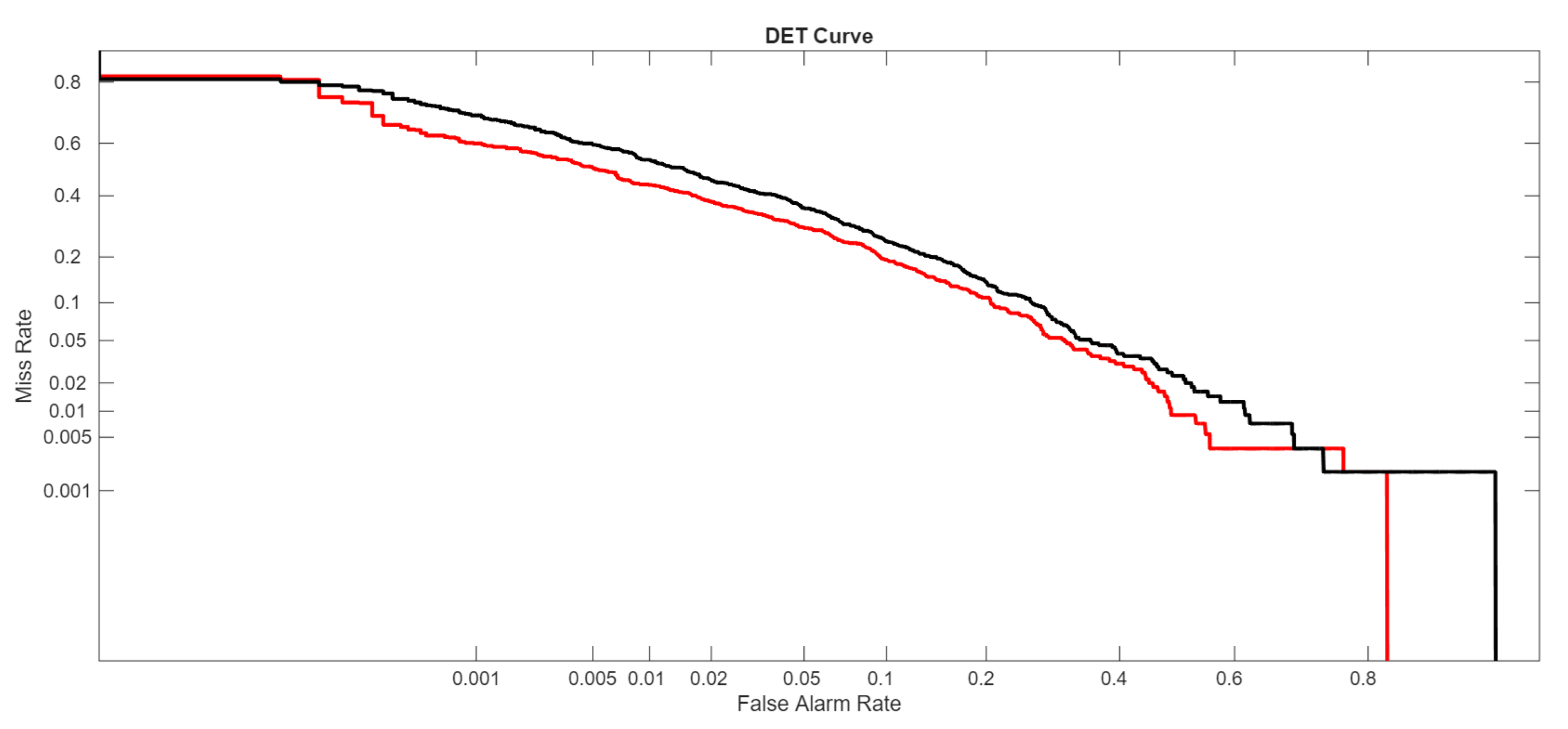
Figure 8.
BarcodeMAE Architectures
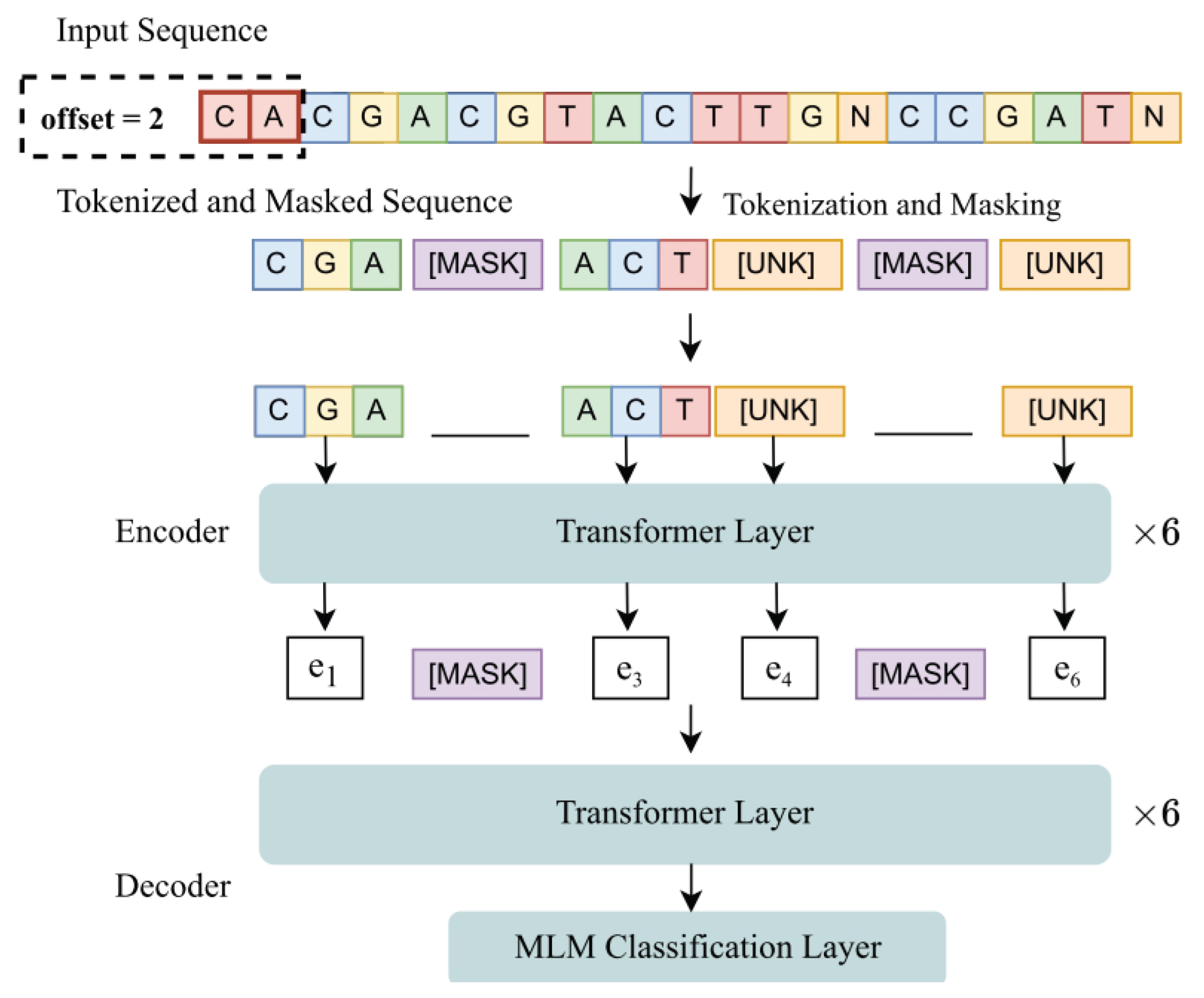

Table 1.
Tested hyperparameter configurations.
| Hyperparameter | Tested Configurations |
|---|---|
| Learning rate | 0.1, 0.01, 0.001. |
| Optimizer | Adam |
| Epochs | 30 |
| Batch size | 128 |
| Loss function | Binary Cross-Entropy |
| Dropout | 0.1, 0.2, 0.5. |
| Number of filters | 1000, 1200, 1500 |
| Kernel size | from 5 to 18 |
| Norm type (only LP) | from 1 to 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



