Submitted:
11 February 2026
Posted:
12 February 2026
You are already at the latest version
Abstract
Long‑acting injectables (LAIs) are widely used for chronic conditions such as schizophrenia, opioid use disorder and HIV. Their prolonged efficacy improves adherence and reduces dosing frequency. Among these systems, poly(lactide‑co‑glycolide) (PLGA)‑based formulations are commonly used to deliver drugs ranging from small molecules to peptides and proteins.
In vitro release (IVR) tests play a critical role in evaluating drug product performance for both immediate release and prolonged release dosage forms. However, there is a lack of standardized compendial IVR methods for the assessment of LAIs. This lack impedes the development of new drug products in this area and also complicates their regulatory approval process.
Considering the complexity of drug release mechanisms and the diversity of various formulation design approaches, it is not possible to devise a universal IVR method which would be applicable to all LAI products. The in vitro release test applied for quality control should be simple, robust, reproducible and discriminatory. On the other hand, more complex biorelevant media and methods are often used during development to better reflect the physiological conditions.
This article provides a comprehensive review of compendial and non-compendial methods used for in vitro release testing of PLGA-based LAIs (microspheres and in situ forming implants), with the goal of aiding the development and standardization of future methodologies.
Keywords:
long-acting injectables
; in vitro release test
; PLGA
; drug delivery systems
; biorelevance
1. Introduction
Long-acting injectables (LAIs) have emerged as valuable drug delivery systems because they provide sustained and consistent drug exposure over extended periods, thereby reducing the dosing burden associated with long-term oral therapy and improving adherence [1]. This advantage is particularly beneficial in chronic conditions where consistent therapeutic coverage is essential, including schizophrenia, HIV treatment and prevention, contraception, oncology, hormonal disorders, inflammatory and pain conditions, and ophthalmic diseases [2]. The main categories of long-acting injectables include aqueous suspensions or nanosuspensions, oil-based depot injections, polymer-based systems such as poly(lactide-co-glycolide) (PLGA) microspheres and in situ forming implants (ISFIs), and pre-formed implants that may be biodegradable or non-biodegradable [3]. These platforms enable different types of release control, allowing the formulation approach to be adjusted to the drug properties and therapeutic needs of the condition [4].
PLGA is the dominant polymer platform for long-acting injectable systems due to its biocompatibility, regulatory acceptance, and tunability through hydrophilic (glycolic)/hydrophobic (lactic) monomer ratio, molecular weight, and end-group chemistry [5,6]. Two major PLGA-based LAI delivery systems are microspheres and in situ forming implants (ISFIs). Microspheres, which can be manufactured via different technologies, exhibit distinct internal porosity, morphology, and drug distribution, all of which directly influence encapsulation efficiency and release behavior [7]. ISFIs, by contrast, consist of PLGA dissolved in water-miscible organic solvents (e.g., N-methyl-2-pyrrolidone, dimethyl sulfoxide) that undergo solvent exchange and polymer precipitation after injection, forming depots whose solidification and release characteristics depend on polymer composition and solvent hydrophilicity [8]. PLGA biodegrades through hydrolytic cleavage of ester bonds followed by autocatalytic erosion, with faster degradation observed for lower molecular-weight polymers, higher glycolide content, or more hydrophilic end groups [6]. The end products of PLGA degradation are lactic and glycolic acids, naturally occurring compounds that are eliminated through normal physiological pathways, including the tricarboxylic acid cycle and renal excretion [9].
Drug release from PLGA-based systems typically follows a triphasic profile comprising (i) an initial burst related to surface- or pore-associated drug, (ii) a lag phase, and (iii) a secondary release phase driven by polymer swelling and/or erosion. Furthermore, release from the PLGA LAIs can be controlled by at least three major mechanisms, or combinations thereof: diffusion through the polymer matrix, water-mediated transport processes and polymer erosion [10,11,12]. The balance among these mechanisms differs between microspheres and ISFIs. Microspheres, owing to their pre-formed microstructure, generally yield more predictable multiphasic profiles. In contrast, ISFIs often show a larger and more variable initial burst driven by rapid solvent efflux and crust formation, followed by diffusion- and erosion-controlled release due to the evolving depot morphology [8]. Release profiles further vary with particle size, porosity, glass transition temperature, polymer architecture (e.g., linear versus star-shaped PLGA), and drug–polymer interactions, including acylation or destabilization of sensitive molecules [13]. Overall, drug release from PLGA systems is inherently complex and governed by formulation attributes, polymer properties, and physiological conditions at the administration site.
The in vitro release (IVR) test is a key test of drug product performance which may serve several purposes. It is used as a tool for formulation development and process optimization, as well as for batch release, evaluation of drug product stability, life cycle management and for establishment of IVIVC [14,15,16]. Understanding the factors influencing drug release from both in vivo and in vitro perspectives is essential for development of meaningful in vitro release tests and specifications [17]. Considering the complexity of drug release mechanisms from long acting injectable products and the diversity of various formulation design approaches, it is not possible to devise a universal one-size-fits-all IVR method which would be applicable to all LAI products [4]. Currently, there are no standard compendial in vitro release methods for prolonged release parenteral dosage forms. Various IVR methods, using both compendial and noncompendial setups, have been explored for PLGA-based LAIs [4,18]. This article provides an overview of IVR methodologies for PLGA-based parenteral drug products from academic, industrial, and regulatory perspectives, with the aim of supporting future method development and standardization.
2. Experimental Setups for In Vitro Release Testing of PLGA LAIs
One of the major challenges in the development of long-acting parenteral drug products is the lack of compendial in vitro release testing methods [6]. As a general recommendation for IVR method development, existing compendial apparatuses should be used as a first approach, as this facilitates standardization and comparability between laboratories [17]. However, the standard pharmacopoeial apparatuses that are most commonly used for dissolution or in vitro release testing (i.e., basket and paddle apparatus according to Ph. Eur. and USP) were originally developed for oral dosage forms and, in many cases, may not be appropriate for testing of the modified release parenteral dosage forms. Therefore, for some parenteral dosage forms, modified compendial or non-compendial apparatuses may be required [16].
USP <1001> provides guidance on methods that have been demonstrated as useful for in vitro release testing of parenteral drug preparations. It provides examples of apparatuses that have been applied to various parenteral products, including microparticles (Apparatus 2 and 4, dialysis cell, incubation jar) and in situ forming preparations and implants (Apparatus 2, 4, and 7, incubation jar) [16]. Further insight into the types of apparatuses and methods currently applied to long-acting parenteral drug products can be obtained from the methods described in the USP Dissolution Methods Database (Table 1) and the FDA Dissolution Methods Database (Table 2) [19,20]. The recommendations from Ph. Eur. 5.17.1 and USP <1092>, although originally developed for oral dosage forms, may also be useful when developing an in vitro release test procedure for parenteral dosage forms [16,21,22].
Importantly, in vitro release kinetics are highly dependent on the selected test method and experimental conditions. For example, Garner et al. investigated the impact of in vitro testing conditions, such as vessel dimensions, agitation speed, the presence of solid beads in the vessel, and media exchange volume, on the in vitro release rate from PLGA microparticles. Their results demonstrated that significantly different release profiles can be obtained for the same formulation simply by modifying IVR testing conditions. In fact, the effects of these methodological differences were so pronounced that they outweighed differences between formulations or process variants [23]. Furthermore, the Schwendeman research group has systematically investigated the impact of release media composition on the release rate from PLGA microparticles. The effects of pH, buffering species, and the presence of plasticizer in the release medium were evaluated. These studies demonstrated that the composition of the release medium can be manipulated to alter the drug release rate from PLGA microparticles, but also to change the underlying release mechanism(s) [24,25].
A variety of different in vitro release methods for PLGA-based long-acting injectables have been described in the literature, using both compendial (pharmacopoeial) and non-compendial (non-pharmacopoeial) apparatuses. The most commonly used approaches can be grouped into three broad categories: sample-and-separate methods, dialysis methods, and flow-through methods. Each of these methods has its own advantages and limitations, as described in the following subsections.
2.1. Sample and Separate Method
Generally speaking, the sample and separate (SS) method is the simplest and most commonly used approach for dissolution or in vitro release testing. In this method, the sample is introduced into a container filled with release medium, and drug release is assessed over time (Figure 1).
The type of container varies widely between studies and may include vials, tubes, flasks, jars, or bottles [10,23,26,27]. The selection of the container is primarily guided by the required volume of release medium, which typically ranges from approximately 1 mL [28,29,30] to about 1000 mL [20]. It should be noted that the pharmacopoeial paddle and basket apparatuses also operate according to the sample and separate principle.
The release medium in the containers may be agitated using different techniques, including shaking water baths [31], orbital shakers [23,24,30], reciprocating shakers [32], magnetic stirrers [33], tube rotator systems [34], or paddle rotation in compendial paddle apparatuses [35]. Samples are withdrawn at predefined time points, followed by filtration or centrifugation to separate the release medium from undissolved material, and subsequent quantification of the released drug substance in the supernatant or filtrate. The selected filter should not adsorb the drug substance [22]. To maintain sink conditions, medium replacement is often performed after sampling.
Several PLGA-based systems that have an in vitro release method described in USP or in the FDA Dissolution Methods Database are tested using the sample and separate approach. For example, the USP monograph for Goserelin implants describes an IVR test performed using flat-bottom glass jars, whereas FDA-recommended methods for Triamcinolone acetonide intra-articular suspension and Triptorelin pamoate intramuscular suspension employ a paddle apparatus [19,20].
The main advantages of the SS method include its simplicity, wide availability of the experimental setup, and high flexibility with respect to release medium volume and equipment selection. Small containers and low media volumes are advantageous when low sample quantity is available or in case of limited analytical sensitivity. Additionally, the use of small-volume tubular containers may facilitate better control of PLGA implant formation [36]. On the other hand, larger containers and higher medium volumes may be employed to ensure sink conditions for poorly soluble drug substances. Medium evaporation can be minimized by using well-closed medium containers. Overall, the SS technique is cost-friendly, easy to set up, and suitable for long-term studies [4,23,35,37,38].
However, there are also several disadvantages of the SS method, including potential floating of microparticles [31,39] and microparticle aggregation [37,40]. Aggregation of the microparticles can be mitigated by the addition of surfactants [41,42] or by optimization of the agitation method [37,43]. Furthermore, the tendency for microparticle aggregation may be impacted by container geometry, as demonstrated by Garner et al. [23]. The experimental conditions should be optimized to eliminate aggregation as it may lead to poor reproducibility, slower release rates, and incomplete release profiles [23,31].
When the SS method is applied to microparticle systems, the sampling technique can become cumbersome with potential clogging of the filters [37] and unintended withdrawal of microspheres during sampling [40,44]. Microspheres removed from the system (e.g., adhering to filters or sampling probes) need to be reintroduced during medium replacement [31]. Sample loss or particle floating may further compromise reproducibility [31,39]. In addition, centrifugation-based separation requires subsequent resuspension of microparticles in the medium by shaking or vortexing, which may be difficult because of microparticle aggregation [33]. Finally, the wide variety of medium container dimensions and agitation methods limits inter-laboratory comparability of SS-based IVR data [18,23]. In order to standardize the experimental setup, the pharmacopoeial paddle apparatus (Apparatus 2) may be used in combination with standard vessels or mini-vessels.
2.2. Flow-Through Method
The setup for the flow-through method includes a medium reservoir, pump, flow-through cell, filtering system positioned on top of the flow-through cell, and a sample collector (Figure 2).
The tested LAI product is introduced into the flow-through cell, and the medium is continuously pumped through the cell, followed by analysis of the eluent. The flow-through apparatus mimics the injection site with respect to immobilized microparticles and the limited volume of medium inside the flow-through cell, while the continuously circulating medium simulates the dynamic in vivo environment [18,45].
Specifications related to flow-through cell size are described in pharmacopoeias (Apparatus 4 according to Ph. Eur. and USP) [46,47]. Apparatus 4 can be operated in open- or closed-loop mode, with different cell sizes, flow rates, and temperatures. In the open-loop configuration, fresh medium continuously flows through the cell and samples are collected using a fraction collector. In the closed-loop configuration, a fixed volume of medium is recirculated through the cell. In addition, the closed-loop setup minimizes medium evaporation during prolonged testing [48].
An example of the flow-through method using Apparatus 4 in closed-loop mode is described in the FDA Dissolution Methods Database for risperidone extended-release suspension (PLGA microparticles) [20].
Hydrodynamics within the flow-through cell may be modified by filling the cell with glass beads to achieve laminar flow or by removing them to allow turbulent flow. Microparticles are commonly layered between or mixed with glass beads to prevent aggregation and to achieve laminar flow within the cell [40,49,50]. For example, Rawat et al. described a sample preparation technique in which microspheres were divided into three approximately equal portions and layered between 1 mm glass beads within a 12 mm flow-through cell. An anti-static gun was used to neutralize static charge on glass beads, microspheres, and the spatula to facilitate sample preparation [51]. The ratio of microparticles to glass beads should not be excessive in order to avoid back-pressure issues [40].
On the other hand, Wang et al. investigated in vitro release from in situ forming risperidone and naproxen depots using flow-through cells operated with or without glass beads. The presence of glass beads caused mechanical compression and fragmentation of the PLGA matrix, resulting in accelerated degradation-driven release at later stages of the release profile while producing a lower initial release compared to bead-free conditions. Conversely, bead-free conditions preserved an intact, disk-like implant morphology and resulted in a more gradual release profile [52].
Compared with various non-compendial setups, IVR methods employing pharmacopoeial flow-through cells offer improved reproducibility, as they are based on a standardized apparatus with defined geometry and hydrodynamics, enabling meaningful inter-laboratory comparison [42]. Furthermore, the drug formulation is contained in the flow-through cell, making sampling and media replacement easy (i.e. there is no need for additional separation of the formulation from the medium containing the dissolved drug substance). Sampling convenience may be further enhanced by the use of commercially available automated systems [37]. Medium volume can be readily adjusted to maintain sink conditions for poorly soluble drug substances or reduced when analytical sensitivity is limited [40,51]. Furthermore, flow-through cells may be coupled with in situ fiber-optic UV probes, since the dispersed microparticles are enclosed inside the flow-through cell and therefore do not interfere with UV analysis. This is particularly advantageous for characterization of the burst release phase, as in situ measurement allows for easy collection of multiple frequent data points [40].
The main disadvantages of flow-through methods include the complexity of the apparatus [38] and the cumbersome setup procedure [53]. This method requires expensive and maintenance-intensive equipment that may not be readily available in many laboratories [23,38,54]. In addition, potential filter blockage may cause variations in flow rate and pressure buildup within the system [16,37,48]. Failures of O-rings or filters may occur during prolonged release testing [18]. Furthermore, adsorption of the analyte onto hydrophobic surfaces of the flow-through apparatus may occur; this effect can be mitigated by incorporating surfactants into the release medium [44].
2.3. Dialysis Method
In the dialysis method, the tested sample is introduced into a dialysis bag (donor compartment), which is sealed and placed into a vessel containing the release medium (acceptor compartment) (Figure 3).
The dialysis bag is composed of a semipermeable membrane that allows diffusion of the drug substance from the donor compartment into the acceptor compartment. Drug release is monitored by sampling from the acceptor compartment.
The membrane material and pore size must be carefully selected. The drug substance should not bind to the selected membrane [37]. The pore size of the dialysis membrane is characterized by its molecular weight cut-off (MWCO). Dialysis membranes with sufficiently high MWCO values are used for in vitro release studies to ensure that they are not a limiting factor for drug diffusion [37,54]. It has been reported that the MWCO should be approximately 100-fold higher than the molecular weight of the investigated drug substance [57]. Dialysis membranes should be pretreated according to the manufacturer’s instructions prior to use.
The volume of the release medium in the acceptor compartment should be at least 6–10 times greater than the volume inside the dialysis bag to maintain a sufficient concentration gradient and to provide a driving force for drug transport across the membrane [37]. Agitation of the outer medium is commonly applied to minimize the unstirred water layer effect. Various modes of agitation may be used, including a magnetic stirrer, shaker or a compendial paddle apparatus [33,37].
Several commercially available dialysis devices have been employed for in vitro release testing of PLGA-based systems, including Float-A-Lyzer [58], Slide-A-Lyzer MINI [59], Tube-O-DIALYZER [60], and the Dispersion Releaser [61]. Float-A-Lyzer (Repligen) is a ready-to-use dialysis device equipped with a flotation ring and cellulose ester membrane, available in a wide range of MWCO values (0.1–1000 kDa) and volumes (1–10 mL). These devices may be used in combination with pharmacopoeial Apparatus 1 or 2. Slide-A-Lyzer MINI (Thermo Fisher Scientific) is a disposable polypropylene cup (0.1–2 mL) with regenerated cellulose membrane of various MWCOs (2–20 kDa) and can be placed in conical or microcentrifuge tubes or alternatively used with a floating unit and a larger beaker. Tube-O-DIALYZER (G-BioSciences) utilizes regenerated cellulose membrane with MWCOs ranging from 1 to 50 kDa and accommodates sample volumes of 20 μL to 2.5 mL.
The Dispersion Releaser (Pharma Test) is a dialysis-based device designed for use with pharmacopoeial Apparatus 2 and standard or mini dissolution vessels. In this setup, the dissolution vessel represents the acceptor compartment, while the donor compartment consists of a dialysis membrane mounted around a cylindrical housing and sealed with two O-rings. The donor compartment is continuously agitated by a paddle stirrer equipped with a permanent magnet and the propulsion is transmitted to a magnetic stirring device in the acceptor compartment [38,61,62].
It is important to note that there are two kinetics involved in the total drug release rate determined using dialysis methods, namely the rate of the drug substance released from the carrier and the permeation rate through the dialysis membrane. Consequently, the dialysis membrane may limit the sensitivity of the method due to a delay in the membrane transport [38,63]. For this reason, it is recommended to conduct membrane permeability experiments during membrane selection, in order to yield an optimized dialysis method which enables rapid transport of the released drug into the acceptor compartment [38,64]. Membrane permeation rate is determined in a reference experiment performed with the drug substance solution. Subsequently, this membrane permeation rate may be used for normalization of release profiles through mathematical modeling approaches, such as the four-step model [41,62].
Compared with sample-and-separate methods, dialysis techniques do not require physical separation of the released drug substance from microparticles. Sampling and medium replacement are simplified due to physical separation of microparticles by the dialysis membrane [37]. In addition, dialysis setups more closely simulate in vivo conditions following subcutaneous or intramuscular administration, where microspheres are immobilized within the tissue rather than freely dispersed in the medium [37,48]. In case of in situ forming implants, dialysis methods may lead to standardized implant geometries and reduced variability in release profiles [65].
The disadvantages of the dialysis method include a cumbersome setup procedure for dialysis bags and the use of non-standardized apparatus which makes comparison between laboratories difficult. These issues may be overcome by using a commercially available dialyzer, which in turn simplifies and standardizes the setup and facilitates inter-laboratory comparison [37,53,59,64]. In addition, the lack of agitation in the donor compartment may lead to microparticle aggregation and poor reproducibility of the release data [39,66]. This issue has been addressed in the commercially available Dispersion Releaser device, where continuous agitation of the donor compartment is applied [61,62].
The small volume of medium within the dialysis bag and the limited membrane surface area may potentially lead to violation of the sink conditions in the inner compartment if the drug release from the carrier is faster than drug diffusion through the membrane [18,65]. To address this problem, reverse dialysis configuration has been proposed, in which the dialysis bag serves as the acceptor compartment while microparticles are suspended in the outer medium [18,67].
Furthermore, equilibration between the donor and acceptor compartments is slow in setups with small membrane surface area (e.g. membrane at one end of the tube), which may hinder accurate characterization of the initial burst effect. This issue may be addressed by using a dialysis setup with a large surface area to facilitate drug transport [33,37].
Finally, the dialysis method is not suitable for drug substances that bind to the dialysis membranes [37].
2.4. Implant Formation During IVR Test for In-Situ Forming Implants (ISFI)
PLGA-based ISFIs are typically composed of PLGA polymer dissolved in organic solvent (e.g. NMP, DMSO, triacetin) and the drug substance which may be suspended or dissolved in the drug product. Upon injection at the administration site, these systems form a drug releasing implant via phase inversion. In this process, sol to gel transition is obtained by solvent exchange, whereby organic solvent diffuses out of the polymeric system, while aqueous medium enters the system, causing water insoluble polymers to precipitate and entrap drug substances within the formed implant.
Importantly, unlike the evaluation of pre-formed implants, ISFI testing involves in situ formation of the implant within the IVR test. The in vitro formation of the implant, including factors such as injection parameters, resulting depot geometry, surface to volume ratio (S/V), and initial water uptake, critically determines its microstructure and consequently has a significant impact on the release profile. Several recent FDA-funded studies have emphasized that controlling depot formation during IVRT is the key determinant for achieving reproducible, discriminating, and potentially biopredictive IVR methods [8,65,68,69].
An early study recognized the importance of uniform implant preparation and tested IVR of thin layer implants prepared using a homemade holding cell. This manner of implant preparation was sufficient to discriminate among PLGA ISFIs containing leuprolide acetate prepared with different molecular weights [70]. In this setup, a mesh separated the holding cell from the release medium, allowing diffusion and phase inversion processes to occur while preserving the physical integrity of the forming implants and resulting in low IVR variability [70].
Zhang and Fassihi systematically investigated the impact of the implant formation method on in vitro release rate of the PLGA-based ISFIs containing naltrexone [60]. Three different formation methods were used and the formed implants were tested using a sample and separate method. Conventional injection of PLGA/NMP formulations into aqueous media led to unpredictable and non-uniform implant shapes due to rapid phase inversion. Injection through a needle produced thin, fragile, helix-like filaments with highly variable S/Vs which resulted in poor reproducibility. Injections without a needle yielded irregular spheres prone to entrapping air bubbles. These voids altered local density, caused flotation, and generated microenvironmental differences that accelerated or disrupted diffusion. Both of these methods of implant formation led to considerable variability in early burst release, overall release kinetics, and long-term depot integrity. To overcome geometry-related variability, the authors introduced a mold-based method for creating disk-shaped implants (1.4 cm diameter cylindrical cavity with 1.5 cm depth). The key characteristics of this procedure included defined dimensions of the depot, controlled injection force to simulate physiological resistance during subcutaneous injection without distorting the implant and immediate solidification since the polymer solution formed a uniform “gel disk” upon contact with aqueous buffer. This controlled depot preparation eliminated air entrapment, standardized the S/V, and resulted in reproducible release profiles across replicates. This study showed that standardizing depot geometry is a prerequisite for increasing reproducibility of the IVR method [60].
In situ forming implants prepared using different formation methods, which resulted in significantly different implant geometries, internal microstructures, and drug-release kinetics, were tested by Suh et al. [65]. Using leuprolide acetate in PLGA/NMP systems, the study evaluated five depot formation approaches, with in vitro release testing performed using a sample and separate method. The authors noted that smaller injection volumes (approximately 50 µL) are more manageable when administered directly into the IVR medium, as they tend to form relatively uniform depots. In contrast, larger volumes of ISFIs, as tested in this study (250 µL), result in greater variability due to increased implant size. Therefore, the depot preparation step was found to be more critical for achieving consistently shaped implants in case of higher ISFI dosing volumes [65].
In vitro formation methods resulting in irregular implant geometries (medium injection, flash freezing, gelatin capsule) substantially elevated the initial burst release due to uncontrolled expansion or structural breakage. In contrast, controlled-geometry methods (dialysis sacs and water-soluble PVA thin film sacs) maintained reproducible implant size and surface area, resulting in significantly reduced and more consistent burst release. Furthermore, microstructural imaging confirmed that controlled-geometry methods generated more uniform solid/semi-solid phase distributions, while irregular-geometry methods produced heterogeneous architectures related to variable release [65].
In a notable study, Wang et al. developed a new adapter-based method using water-dissolvable polyvinyl alcohol (PVA) films combined with a Teflon frame as a PVA carrier to improve in vitro release testing for in situ forming implants. Upon contact with aqueous medium, uniform depots are formed whereby PVA film dissolves. To evaluate the proposed adapter, two widely different implant types were selected: a risperidone formulation, where the drug remains suspended in PLGA/NMP matrix, and a naproxen formulation, where the drug fully dissolves in the same polymer/solvent system [52].
For risperidone formulations, the adapter-based method formed uniform, disk-like depots that resembled the implants obtained in in vivo testing on rabbits, improving the variability that was seen with direct injection. Adapter preparation captured three distinct release phases for risperidone implants—burst, diffusion-controlled release, and degradation-controlled release. In contrast, direct injections produced irregular implant geometry and resulted in apparent zero-order release. The IVR method with the proposed adapter showed discriminatory ability, differing between risperidone formulations with different PLGA ratios and molecular weights. Release profiles were reproducible across different sample injection volumes (0.1–0.6 mL), which was attributed to a constant diffusional path length and surface area per unit mass [52].
In the same study, naproxen formulations were prepared as solutions in PLGA/NMP systems. Upon formation within the adapter, naproxen depots exhibited consistent shape and minimized variability compared with direct injection. Naproxen release followed first-order kinetics regardless of the applied IVR method, indicating diffusion-dominated behavior with rapid solvent exchange. Release profiles were reproducible across different sample injection volumes (0.1–0.6 mL). The adapter reduced variability in both drug and NMP release profiles relative to the direct injection method. Differences between naproxen formulations with different PLGA grades were also observed, confirming adequate discriminatory power. Reproducibility was satisfactory across repeated studies and different adapter volumes, confirming robustness of the method for solution-type ISFIs [52].
A follow-up study from the same group compared different implant formation methods applied to ISFIs with distinct drug release mechanisms, namely PLGA/NMP suspensions containing meloxicam or risperidone and PLGA/NMP solution containing naproxen [35]. The study evaluated three depot formation methods: direct injection, the PVA film combined with a Teflon adapter, and the GF/F glass fiber membrane combined with a Teflon adapter. The PVA–Teflon adapters performed well in combination with naproxen and risperidone depots, which follow bulk-erosion driven release and remain structurally intact during the IVR test, maintaining consistent depot geometry and producing low-variability release profiles. In contrast, meloxicam depots underwent surface erosion and progressively thinned and fragmented when formed with the PVA–Teflon adapter, causing variability due to uncontrolled detachment of fragments. Using the GF/F–Teflon adapter prevented fragmentation and stabilized depot geometry, resulting in more reproducible meloxicam release despite late-stage GF/F membrane degradation [35].
A complementary study from the same group systematically examined how in vitro implant formation conditions affect risperidone release from PLGA/NMP ISFIs and aimed to establish an IVIVC with a rabbit subcutaneous model [68]. Three methods of implant formation in the IVR test were developed to independently modulate depot morphology, exposed surface area, mechanical confinement, water uptake, and phase inversion kinetics. These methods used a PVA-film/Teflon adapter, a GF/F-membrane/Teflon adapter (sealed and half-open), and a sandblasted glass slide as molds for controlled implant formation.
The PVA-film method enabled unrestricted swelling and generated reproducible but non-biopredictive release characterized by a minimal burst, a prolonged lag phase, and accelerated degradation-driven release. Introducing GF/F glass fiber membranes allowed controlled confinement and tunable surface-to-volume ratios through sealed and half-open formats; however, membrane integrity was occasionally compromised at high loading, and mechanical pressure introduced artificial constraints not reflective of physiological conditions. In contrast, the sandblasted glass-slide ring method provided precise shape control without imposing mechanical stress, maintained depot adhesion during agitation, and ensured consistent geometry with high water accessibility. This setup enabled accurate adjustment of surface-to-volume ratios (20–60 cm⁻¹) and avoided confounding effects from swelling limitations or structural failure. In vivo, the risperidone implant formed a thin disk-shaped depot with a high surface-to-volume ratio (~50–60 cm⁻¹), rapidly released approximately 90% of NMP within 24 h, and exhibited a biphasic profile driven initially by diffusion and later by polymer degradation. Among the tested systems, the glass-slide ring method most closely reproduced these physiological conditions by matching the in vivo surface-to-volume ratio and permitting unhindered water uptake and phase inversion, thereby providing the best in vitro–in vivo alignment [68].
In the study by Karp et al., in vitro implants were produced using a custom flat holding-cell mold that generated uniform, thin, disk-shaped PLGA–progesterone implants [71]. The authors investigated release mechanisms including diffusion, dissolution, and swelling across formulations differing in solvents, PLGA concentration, and drug load. A defined amount of formulation was applied into the mold, and controlled exposure to a buffer triggered phase inversion, yielding reproducible implants with consistent geometry. Phase inversion took place for 20 h, after which the implants were considered fully formed. Because of this, the initial burst that occurred during the implant formation period was not captured in the in vitro release test. Instead, early drug release was captured indirectly through drug entrapment measurements. This standardized setup minimized variability and allowed clear discrimination among formulations in the post-burst phase [71]
Overall, these studies demonstrate that the method of implant formation is a primary determinant of IVR performance for ISFIs. The choice of the formation adapter, the degree of mechanical confinement, and the ability to control surface-to-volume ratio all play central roles in achieving reproducible release profiles. Importantly, adapting the depot formation method to match the physical characteristics of the depot formed in vivo may aid mimicking of in vivo release and guide early formulation development.
2.5. Release Medium
Selection of the release medium is generally governed by drug solubility and stability over the duration of the IVR study [37]. For PLGA-based LAIs, the most commonly used medium for in vitro release testing is pH 7.4 phosphate-buffered saline (PBS), corresponding to the physiological pH at subcutaneous and intramuscular administration sites. In addition, pH 7.4 HEPES-buffered saline (HBS) has been reported as an alternative buffer system [24,25]. In some studies, water has been used as the release medium [30,72]. However, as water quality and pH may vary, its use is recommended only when it has been demonstrated that pH fluctuations do not influence in vitro release characteristics [21,22]. In addition, buffers with acidic or alkaline pH may be used as release media for accelerated IVR testing.
To ensure complete drug release for poorly soluble drug substances, release media may require supplementation with solubilizing agents, including surfactants or organic solvents [20,73]. Furthermore, when microparticles exhibit poor wettability, a small amount of surfactant may be added into the medium to minimize microparticle aggregation or floating [16,42]. The nonionic surfactant polysorbate (Tween 20 or Tween 80) is frequently used for IVR testing of PLGA-based microparticles, typically at concentrations ranging from 0.02 % to 0.05 % [10,26,27,42]. Beyond the simple buffer systems such as PBS and HBS, more complex biorelevant release media are being developed in an attempt to simulate physiological conditions at the administration site.
Considering the usual duration of a long-term IVR test for LAI products (often weeks to months), special attention must be given to potential medium evaporation, microbial growth, and chemical degradation of the released drug substance in the medium [16]. Medium evaporation may be prevented by using sealed containers. When a poorly sealed apparatus is employed (e.g. pharmacopoeial paddle apparatus), an internal standard may be used to correct for changes in medium volume due to evaporation. In any case, it should be considered whether the decreased medium volume will affect the sink conditions [16].
Microbial growth can be limited by adding preservatives to the release medium, such as sodium azide. Concentrations ranging from 0.01 % to 0.15 % sodium azide have been reported in the literature [11,16,23,26,35,74].
In cases where chemical degradation of the drug substance occurs in the release medium, use of an alternative pH (where the drug substance is more stable) or addition of antioxidants (e.g. sodium ascorbate) may be beneficial. Complete medium replacement or use of flow-through apparatus in open-loop configuration may also be helpful, as the dissolved drug does not remain in the system for the entire duration of the test [16,31]. Drug degradation in the release medium may further be addressed by nonspecific analytical techniques, such as UV–Vis spectroscopy or HPLC with class separation, or by summation of peaks corresponding to the drug substance and its degradation products [16,56,75]. Alternatively, residual drug content in the microspheres may be quantified when instability in the release medium precludes direct measurement; however, this destructive approach requires a substantial amount of sample and is therefore not generally preferred [26,37,75].
IVR testing for quality control purposes is generally performed under sink conditions, meaning that the material already in solution does not exert a significant modifying effect on the dissolution rate of the remainder. Sink conditions normally occur when the volume of release medium is at least three- to ten-fold greater than the saturation volume [21]. Under sink conditions, IVR results are more likely to reflect properties of the dosage form [22]. Although the volume of fluid at subcutaneous and intramuscular administration sites is small, there is constant fluid movement and replacement, as well as drug diffusion away from the site [42]. In other words, the tissues surrounding the injected drug delivery system may function as a sink [76]. Nevertheless, when conducting biorelevant release testing and simulating the physiological environment, the sink conditions cannot be assumed in vivo. Estimation of drug distribution between the administration site and systemic circulation should therefore be performed on a product-specific basis, and for some products, IVR testing under non-sink conditions may be warranted [38,48].
2.6. Temperature
In vitro release testing for LAIs intended for intramuscular or subcutaneous administration is generally performed at 37 °C. Elevated temperatures may be used for accelerated IVR testing. According to pharmacopoeial requirements, the permissible temperature range for in vitro release testing is ±0.5 °C [46,47]. However, for certain PLGA-based systems, even tighter temperature control may be needed during IVR testing. For example, Rawat et al. demonstrated that temperature variations of ± 0.5 °C resulted in significant differences in release profiles of commercial risperidone PLGA microspheres and this was attributed to temperature-dependent changes in risperidone-catalyzed PLGA degradation [51].
2.7. Accelerated Methods
Long-acting injectable products are typically designed to deliver the drug substance over periods of weeks or months. Consequently, real-time in vitro release testing for these products also requires extended periods of time. Accelerated or short-term IVR testing is therefore considered a practical alternative, as it enables completion of release studies within days rather than months.
Development of an appropriate accelerated IVR method should be based on understanding of the in vitro drug-release mechanism. In addition, the impact of the applied accelerating conditions on the release mechanism should also be understood [45,48]. For example, drug release may be accelerated by addition of organic solvents to the release medium, which can solubilize the PLGA polymer and shift the release mechanism from erosion/diffusion-controlled kinetics to diffusion-controlled kinetics. Furthermore, for PLGA-based systems exhibiting multiphasic release profiles (e.g., burst release, lag phase, and secondary release), some of the release phases may be diminished or lost entirely under accelerated conditions [49].
Dosage form attributes that should be considered during accelerated method development include matrix glass transition temperature, solubility of formulation components in the release medium, polymer degradation rate, and stability of the dosage form under accelerated conditions [48].
Accelerated IVR methods represent an attractive tool for rapid assessment of formulation and process changes during drug product development, as well as for batch release and quality-control purposes [37,53]. However, a relationship between the real-time and accelerated release methods should be established in order to demonstrate suitability of the accelerated method as a routine quality control test. Optimally, a 1:1 correlation is achieved between the real-time and accelerated release profiles [45]. The scaling factor between the real-time and accelerated release profile may be determined as the ratio of the time required to reach 50 % drug release under real-time and accelerated conditions [51].
Ideally, an accelerated IVR test should increase the drug-release rate without altering the underlying release mechanism. However, a change in release mechanism may be acceptable if the accelerated method demonstrates adequate discriminatory power and preserves the rank-order relationship between formulations when compared with the real-time method [4,51].
Accelerated release is typically achieved by modifying one or more experimental conditions applied in real-time IVR testing, including temperature, pH, agitation, and addition of surfactants or organic solvents [45].
Elevation of temperature is the most frequently applied strategy to accelerate drug release from PLGA-based systems. At elevated temperatures, polymer chain mobility increases and this leads to accelerated drug release via diffusion. In addition, elevated temperature also increases the polymer hydration and polymer degradation rate, thereby accelerating the drug release via erosion [45,77]. However, it is generally recommended that the applied temperature should not go above the glass transition temperature (Tg), as temperatures above Tg may alter the release mechanism [18].
In a study by Zolnik et al. accelerated testing at elevated temperatures has demonstrated linear correlation with real-time data for erosion-controlled PLGA systems containing dexamethasone. On the other hand, the same accelerated release test appeared not to be suitable for formulations with dominantly diffusion-controlled release. Namely, the increased polymer mobility at elevated temperatures may lead to morphological changes, such as surface pore closure and microparticle aggregation, which in turn may hinder drug diffusion and decrease the drug release rate. Moreover, the applied accelerated test did not give an accurate prediction of the burst release phase, which is also diffusion-controlled. It was therefore recommended that the accelerated test should be supplemented by a real-time study to adequately assess the burst release phase [78].
Andhariya et al. developed an accelerated IVR test at 45 °C for naltrexone-loaded PLGA microparticles. The testing duration was reduced from 35-40 days (real-time test) to 6 days (accelerated test). A linear correlation between real-time and accelerated release profiles was observed, and the method was shown to be discriminatory toward manufacturing differences which lead to different particle size and porosity of the studied microparticle formulations [31].
Li et al. investigated accelerated IVR methods for PLGA-based ISFIs containing leuprolide acetate formulated with NMP, DMSO, or triacetin as cosolvents. Acceleration was achieved by increasing the test temperature to 50, 55, and 60 °C, resulting in substantial shortening of the release duration from approximately 50 days at 37 °C to 7–14 days at elevated temperatures. An Arrhenius relationship between real-time and accelerated release data was established for NMP- and DMSO-based formulations, enabling prediction of zero-order release at 37 °C. Among the evaluated temperatures, 50 °C provided the best discriminatory ability, whereas higher temperatures diminished differentiation between formulations. In contrast, accelerated testing at elevated temperatures was not suitable for triacetin-based formulations, since triacetin hydrolysis to acetic acid caused a pronounced pH reduction, accelerating the PLGA degradation, altering the release mechanism and preventing accurate real-time correlation [79].
In a study by Wang et al., increasing the temperature to 40 °C shortened the release duration of risperidone from PLGA/NMP ISFIs from approximately 45 to 25 days while preserving the real-time release mechanism, as demonstrated by the linear time-scaled correlation between the 37 °C and 40 °C profiles. However, addition of 0.1 % Triton X-100 at 40 °C accelerated the water uptake and altered PLGA phase separation, which changed the relative contributions of diffusion and degradation to the overall release kinetics. As a result, the kinetics no longer matched real-time release and no linear IVR correlation could be established [52].
Tipnis et al. explored the impact of temperature on in vitro release of triamcinolone acetonide from PLGA microparticles. Surprisingly, it was found that drug release is slower at 39 °C than at 35 °C and 37 °C and this was attributed to polymer plasticization at 39 °C, which in turn impacted the microparticle morphology. It was concluded that elevated temperature may not be a suitable parameter to accelerate drug release for all PLGA-based microparticle systems [74].
Modifications of release medium composition, including changes in pH or addition of organic solvents, have also been employed to accelerate drug release from PLGA-based systems. A change in medium pH can accelerate the hydrolytic degradation of PLGA. Both the acidic and alkaline conditions catalyze ester hydrolysis and polymer degradation and consequently may accelerate drug release. However, compared with temperature elevation, pH changes typically result in only moderate acceleration of the release rate [45,49]. Importantly, polymer erosion mechanisms differ under acidic and alkaline conditions: acidic media promote bulk erosion similar to that observed at pH 7.4, whereas strongly alkaline conditions (pH > 13) induce surface erosion [80].
Organic solvents such as ethanol or acetonitrile have been used to accelerate drug release from PLGA-based systems [81,82,83]. Kamberi et al. demonstrated that acetonitrile increases the porosity of PLGA stent coating, leading to accelerated release [83]. Xie et al. developed an accelerated IVR method for thymopentin-loaded PLGA microparticles using a release medium containing 20 % ethanol in combination with a gradient heating program. The temperature of the release medium increased in three steps, starting with 2 hours at 40 °C, followed by 1 hour at 45 °C and finally 27 hours at 50 °C. The testing duration was reduced from 30 days (real-time test) to 30 hours (accelerated test). The developed accelerated method correlated well with the real-time release at 37 °C. In addition, it was found that the accelerated method with the gradient heating program (starting with temperature 40 °C) simulated well the burst release phase of the real-time test and that this approach may in some cases eliminate the need for an additional real-time study to assess the burst phase. Furthermore, the applied accelerated method was able to discriminate between formulations with different PLGA molecular weights and lactide-to-glycolide molar ratios [81].
Finally, it should be emphasized that elevated temperatures and extreme pH conditions may induce degradation of the drug substance [31,45,77]. Furthermore, stability of the release medium components, as well as robustness of the test apparatus under the applied accelerated conditions, should also be considered [45].
3. Method Validation and Discriminatory Power
In order to be used as a routine quality control test, the in vitro release method should be validated in accordance with ICH Q2(R2) guidelines [84].
Rawat et al. described validation of an accelerated IVR method for a commercial PLGA microsphere product (Risperdal Consta) using a flow-through apparatus [51] The method was validated for robustness (with respect to small variations in method parameters) and reproducibility (with respect to different analysts and apparatuses). The authors demonstrated robustness of the method toward variations in flow rate, size of the flow-through cell, size of the glass beads, cell-preparation technique, and amount of microspheres loaded into the cell. In contrast, tight control of temperature and pH of the release medium was identified as critical for achieving reproducible IVR profiles from the tested drug product. In addition, the authors highlight the importance of medium deaeration at the beginning of the IVR test, as well as throughout the test, as re-aeration of the medium may occur over time and affect release results [51]. It should be noted that complete validation of an IVR method for regulatory purposes would also need to cover validation of the analytical quantification step, including specificity/selectivity, accuracy, reportable range, and precision, in accordance with ICH Q2(R2) [84].
Furthermore, the discriminatory power of an IVR method intended for quality control purposes must be demonstrated. Discriminatory power refers to the ability of the test procedure to differentiate between batches manufactured with different critical process parameters and/or critical material attributes and/or critical process parameters, which may have an impact on bioavailability. In other words, the IVR test conditions should be chosen which, in combination with appropriately defined IVR specification limits, allow discrimination between acceptable and non-acceptable batches [16,22,85,86]. Additional guidance related to method validation and assessment of discriminatory power is available in the ICH Q2(R2)/ICH Q14 training materials [87].
The Burgess group has evaluated the discriminatory power of the IVR methods developed for PLGA microspheres containing various drug substances, including risperidone (poorly soluble small molecule), naltrexone (highly soluble small molecule), and leuprolide acetate (peptide). In these studies, IVR testing of risperidone- and naltrexone-loaded microspheres was performed using flow-through cells, whereas leuprolide-loaded microspheres were tested using a sample-and-separate method. In all cases, the IVR methods successfully discriminated between compositionally equivalent microspheres manufactured with deliberate process variations. Moreover, the in vivo pharmacokinetic profiles of the same microsphere formulations were determined following intramuscular administration in rabbits. In each of these studies, the in vivo release profiles of the process variants in the investigated animal model correlated well with in vitro release profiles, and the authors reported that a Level A IVIVC was established [28,42,88].
Zolnik and Burgess further evaluated the discriminatory power of the IVR test method towards dexamethasone-loaded PLGA microspheres prepared with different polymer molecular weights. The proposed in vitro method was able to differentiate between the two tested PLGA microsphere formulations exhibiting distinct in vivo release characteristics [78].
Furthermore, the potential of an in vitro release method to be used as a stability-indicating test has been described in a study with risperidone-loaded PLGA microspheres which were exposed to various storage conditions. After exposure of the microspheres to accelerated storage conditions, significantly faster in vitro release and a marked decrease in PLGA molecular weight were observed, confirming the discriminatory power of the applied IVR method towards potential stability-related changes for the tested drug delivery system [50].
4. Biorelevant Considerations
Subcutaneous (SC), intramuscular (IM), subgingival, and intra-articular sites are the primary routes explored for administering PLGA-based systems, with SC and IM being the most widely utilized. Biorelevant IVR methods for LAIs aim to better reflect the physiological conditions at the site of administration. Incorporating site-specific characteristics into IVR method design enhances the approximation of in vivo drug release profiles, and potentially enables more effective formulation screening in early development.
Subcutaneous (SC) administration
SC route of administration is characterized by the presence of adipocytes, an extracellular matrix (ECM) containing glycosaminoglycans such as hyaluronic acid (HA), limited blood capillaries, and interstitial fluid. Due to reduced vascularization and a denser ECM, drug absorption in subcutaneous tissue typically occurs at a slower rate compared to IM administration. Interstitial fluid (ISF) in SC tissue is the first fluid to interact with injected drugs [89]. ISF is constantly exchanged with plasma and lymph, driven by hydrostatic and osmotic pressures. Lymphatic drainage is crucial for the clearance of large molecules and particles, and local tissue movement (e.g. muscle contractions, injection trauma) further influences fluid dynamics [73,90]. Moreover, proteases in the SC tissue can degrade peptides and proteins, affecting their bioavailability [73]. ISF composition is similar to plasma but with lower protein and lipid content due to the selective permeability of capillary walls [89,91]. Major electrolytes include Na⁺ (132–146 mM), K⁺ (10–21 mM), Ca²⁺ (0.7–3.2 mM), Mg²⁺ (0.7–1.8 mM), Cl⁻ (104–158 mM), and HCO₃⁻ (20–28 mM). Protein levels range from 22–57 g/L, with albumin at 14–35 g/L, making up about 60% of total protein. Phosphatidylcholine and sphingomyelin are the main lipids in the ISF [89]. Gao et al. developed a Simulated Subcutaneous Interstitial Fluid (SSIF), a biorelevant medium which reflects the subcutaneous tissue by including major ions, buffers, and protein. SSIF consists of sodium (136.0 mM), potassium (3.9 mM), calcium (1.3 mM), magnesium (0.5 mM), chloride (114.9 mM), bicarbonate (20.6 mM), phosphate (21.0 mM), sulfate (20.5 mM), acetate (5.0 mM), Tris buffer (50.0 mM) and is further supplemented with 55% v/v fetal bovine serum to achieve a final protein concentration of 25 g/L [92].
In addition to using media compositions that replicate the characteristics of subcutaneous ISF, agarose gels are used as media for IVR testing to simulate the architecture of subcutaneous tissue in vitro [79,93,94]. These agarose-based systems act as diffusion-dominated barriers, minimizing convective transport and more closely simulating the gel-like extracellular matrix of SC tissue [94]. Typical agarose concentrations range from 0.5% to 2% w/v, prepared in phosphate-buffered saline (PBS) or phosphate buffer at physiological pH 7.4 [79,93,94]. The physical constraint imposed by the gel restricts swelling and promotes agglomeration of the dosage form. These are important contributing factors to the drug release kinetics which are not often encompassed in conventional (buffer) methods. For instance, Kožák et al. demonstrated that microspheres embedded in agarose gel merged into agglomerates, reducing effective surface area and slowing drug release, a phenomenon also observed in vivo but not in conventional IVR tests [94]. Agarose-based IVR systems thus could provide a more discriminative early assessment of formulation performance, enabling differentiation between formulations with varying polymer compositions, solvent systems, and device geometries that may otherwise appear similar in conventional tests [79,93,94]. Use of bromothymol blue in the gel showed acidic microenvironments at the depot surface early on, which is consistent with PLGA autocatalysis and acid accumulation reported in vivo [79]. Based on the available data, these systems appear to facilitate mechanistic investigations of processes occurring in vivo, thereby possibly enhancing the physiological relevance of the IVR test and predictiveness of in vivo performance [79,93].
However, there are also some limitations related to agarose gel based IVR systems. Firstly, agarose is chemically inert and lacks the complex biochemical components of the real extracellular matrix such as proteins, hyaluronic acid, lipids, enzymes, and cells. As a result, these gels do not reproduce specific binding interactions, enzymatic degradation, or immune responses that can impact solvent exchange in case of ISFIs, porosity, and drug–matrix interactions in vivo [94]. Secondly, although subcutaneous drug transport is primarily diffusion driven, some degree of convection still occurs in vivo due to the lymphatic flow or tissue movements. This may lead to an overestimation of diffusion barriers and potentially underestimate drug release rates compared to the in vivo situation [93]. These factors should be considered when interpreting IVR data from agarose-based systems, as they may not fully capture the dynamic and biochemical complexity of the subcutaneous environment.
Within the context of biorelevance and drug release, significant progress has been made in the characterization of peptides and proteins. Two systems in particular have emerged in this area that may be applicable to LAI formulations: the Pion SCISSOR platform and the BioJect system.
The Pion SCISSOR (Subcutaneous Injection Site Simulator) system, first described in 2015 and used for testing of human insulins and monoclonal antibodies, provides a mechanistic in vitro platform to simulate the fate of biopharmaceuticals following subcutaneous injection. The system employs a dialysis-based injection chamber, which can be loaded with defined concentrations of extracellular matrix (ECM) components such as HA, enabling the study of ECM influence on drug release. This chamber is immersed in a bicarbonate-based physiological buffer, maintained at 34°C and pH 7.4, to closely mimic the ionic composition and homeostatic conditions of the subcutaneous tissue. Hydrodynamics and interstitial pressure are mimicked by controlling the fluid volume and pressure within the chamber, while a modified dialysis membrane with micropores mimics the uptake of drugs into blood and lymphatic capillaries. Real-time monitoring of pH, pressure, and drug release allows the SCISSOR system to provide valuable insights into the physicochemical transitions and interactions that govern the absorption and bioavailability of subcutaneously administered biopharmaceuticals [95].
Recently a novel BioJect platform was developed to closely simulate the SC physiological microenvironment [96] for different insulin formulations. It integrates a compendial flow-through cell (Apparatus 4) with a perfusion system and simulates the extracellular matrix (ECM) by incorporating a customizable biomatrix within its flow-through cell, using hydrogels such as agarose as a base and supplementing them with physiologically relevant biopolymers like collagen and hyaluronic acid. This design mimics the structural, charge, and hydration properties of the native ECM, thereby replicating tissue retention, diffusion barriers, and the dynamic interactions that influence drug release and absorption in subcutaneous tissue. The modified simulated subcutaneous interstitial fluid (mSSIF) used in this setup consisted of 138.5 mM sodium, 10 mM potassium, 1.8 mM calcium, 0.8 mM magnesium, 111.3 mM chloride, 28 mM bicarbonate, 0.5 mM sulfate, 5 mM acetate, 4.2 mM phosphate, and 30 g/L total protein (bovine serum albumin), formulated to closely mimic the ionic and protein composition of human subcutaneous interstitial fluid [96].
For more details on biorelevant subcutaneous conditions and corresponding media compositions, the reader is kindly referred to a comprehensive review by Li et al. [73].
Intramuscular (IM) Administration
Unlike the SC environment, muscle tissue is highly vascularized and represents a dynamic site shaped by its extracellular matrix (ECM), interstitial fluid (ISF) composition, and local blood flow, all of which influence drug fate after injection. The ECM is organized into the endomysium, perimysium, and epimysium, composed mainly of type I collagen and hyaluronic acid (HA), with proteins such as fibronectin contributing to structural strength and viscoelasticity. HA acts as a hydration reservoir and provides elasticity, supporting smooth muscle movement and reducing friction [97]. A dense capillary network enables efficient nutrient, oxygen, and drug exchange. Because muscle has greater blood flow than SC tissue, intramuscularly injected drugs are typically absorbed more rapidly [97,98]. Electrolyte and protein levels are similar to those in SC tissue but fluctuate with muscle activity and perfusion changes [91]. ISF pH is around 7.4 but may vary under physiological or pathological conditions. Ultimately, the absorption and systemic availability of IM-injected drugs are dictated by the interplay of these ECM and ISF properties, as well as the physicochemical characteristics of the drug and formulation [97].
A comprehensive study by Kozak and Lamprecht [99] explored several biorelevant IVR setups using the commercial LAI Risperdal® Consta®, including a previously developed muscle tissue based in vitro system containing native muscle lipids [100]. Conventional in vitro testing was performed in 0.1 M phosphate buffer pH 7.4 containing 0.02% polysorbate 20 and 0.05% sodium azide under mild horizontal agitation to represent a conventional IVR environment with freely suspended microspheres. It was demonstrated that neither the addition of enzymes nor albumin produced meaningful changes in risperidone release or PLGA degradation compared with buffer alone. Subsequently, release media incorporating isolated muscle lipids and defined lipid components were developed to examine the role of physiological fatty acids. Media rich with fatty acid, particularly those containing oleic acid, significantly accelerated risperidone release by shortening the lag phase and inducing earlier microsphere swelling. Furthermore, an agarose gel envelope method was employed to mimic the mechanical confinement of tissue surrounding the depot after intramuscular administration. In this gel system, risperidone release was slower due to restricted swelling and merging of softened microspheres, which reduced the effective surface area available for drug diffusion. A combined agarose-lipid setup partially restored the faster early release while maintaining slower release in the later phase, improving overall biorelevance relative to either approach alone. Finally, a simulated muscle tissue setup, incorporating microspheres directly into previously freeze-dried and then rehydrated porcine muscle tissue fixed with agarose, reproduced both the biochemical lipid environment and the mechanical constraints of intramuscular tissue. This muscle-based system produced a shorter lag phase and a slower post lag release, closely matching (published) human in vivo data. Removal of endogenous lipids from the muscle matrix stopped the early acceleration of drug release, indicating that tissue lipids drive the initial faster in vivo release. The study showed that fatty acid interactions dominate the early release phase by enhancing swelling and erosion of PLGA microspheres. It further showed that swelling restriction imposed by the muscle tissue governs the later phase slowing of release. The authors concluded that realistic IVIVR requires incorporating both biochemical (lipids) and biophysical (mechanical confinement) factors, with the simulated muscle tissue method providing the closest in vitro approximation of human in vivo release [99].
Intra-articular Administration
Synovial fluid (SF) is a viscous, viscoelastic (non-Newtonian) plasma dialysate rich in HA, proteins, primarily human serum albumin, and lubricin, supporting joint lubrication and shock absorption [101,102]. In healthy joints, the synovial fluid exhibits a pH of around 7.4 and a higher viscosity (up to 1 Pa·s), higher HA levels (1.5 - 4 mg/mL), and lower protein content (10 -30 mg/mL) compared with inflamed-joints SF, where HA decreases to 0.2 - 1.8 mg/mL and proteins rise to about 50 mg/mL. Pathological conditions such as arthritis degrade HA and reduce viscosity, impairing lubrication and increasing joint wear and pain [101,102] These changes in SF composition and rheology are critical for the development and evaluation of intraarticular drug delivery systems [101,102] Joint mechanics, high viscosity and viscoelastic nature of SF as well as its turnover affect drug fate, as movement enhances mixing and may promote clearance of small particles, while disease altered SF composition can modify drug distribution and retention [9,102]. Therefore, IVR method development for intra-articular delivery requires careful consideration of SF composition, rheology, and joint biomechanics in relation to the drug product of interest.
Magri et al. developed biorelevant synovial fluids (BSF) containing physiologically relevant HA, phospholipids, and proteins to represent healthy (H-BSF) and osteoarthritic (OA-BSF) environments. H-BSF consisted of 3.2 mg/mL HA, 1.9 mg/mL phosphatidylcholine, 19.6 mg/mL bovine serum albumin, and 2.9 mg/mL γ-globulin at pH 7.4. OA-BSF included the same HA concentration but higher phosphatidylcholine (5.4 mg/mL) and a basic pH 8.0. A protein-free variant, OAwp-BSF, maintained OA phospholipid and HA levels but excluded proteins which enabled isolating and studying the specific effects of proteins on drug solubility and release from PLGA microspheres and commercial methylprednisolone acetate suspension (DepoMedrone®). Protein-rich BSF increased methylprednisolone (MP) solubility and release from both PLGA microspheres and a DepoMedrone®. Solubility and drug release were highest in OA-BSF and lowest in OAwp-BSF, indicating that albumin and γ-globulin act as solubilizing components [103].
Subgingival Administration
The subgingival environment is a unique challenge for assessing biorelevance in vitro since it is characterized by complex biofilms, dynamic fluid composition, and host immune responses.
Development of biorelevant in vitro testing for subgingival administration should consider simulation of the periodontal pocket environment. The composition of simulated saliva or gingival crevicular fluid (GCF) should include physiologically relevant concentrations of mucins (typically 1–10 mg/mL), amylase (0.5–2 mg/mL), and total proteins (0.5–3 mg/mL), as these proteins influence drug release and biofilm interactions [104,105]. The media should have a pH between 6.8 and 7.4 to reflect the microenvironment of inflamed pockets [104]. The fluid volume should be based on realistic pocket geometry; however, these volumes vary significantly among patients. In healthy subgingival sites, GCF flow rates typically range from 3 to 8 μL/h per site, with a resting volume of about 0.05–0.06 μL, while intermediate pockets exhibit flow rates around 20 μL/h and the flow rates in deep pockets can reach up to 44 μL/h or higher, with resting volumes of up to 1.5 μL. Advanced periodontitis may present flow rates as high as 137 μL/h. The rapid turnover of GCF means that the pocket volume is replaced every 1–2 minutes in healthy conditions and as frequently as every 15–60 seconds in diseased pockets [106].
An FDA-funded science grant resulted in development of a biorelevant method using Small Volume Apparatus (SVA), which was designed specifically to reproduce the microenvironment of the periodontal pocket by integrating physiologically representative features [56]. This setup employed a custom-made polycarbonate chamber containing a slotted inner compartment wrapped in a 50 kDa dialysis membrane, which confined PLGA microspheres while allowing diffusion of the released drug substance into the surrounding medium. Approximately 10 mg of PLGA-based microspheres with minocycline hydrochloride (Arestin® and in-house formulations) were deposited into the inner chamber and immersed in 250 µL of simulated gingival crevicular fluid (sGCF). sGCF was prepared by dissolving citrate buffer salts, NaCl, KCl, CaCl₂, and low-level BSA (0.05 g/L) in water, adjusting the pH to 7.25. A syringe pump was employed to flush the sGCF through the outer chamber at 0.5 µL/min. This flow rate was chosen to reflect physiological GCF turnover while ensuring sustained concentration gradients across the membrane. This design enabled IVR testing under conditions of low volume and low flow rate that cannot be replicated by high volume compendial systems. Results of drug release from SVA were compared to the previously developed Apparatus 4 method which employs higher volume and flow of PBS based medium. Apparatus 4 method produced a rapid and complete release of Arestin® within 3 days, whereas the SVA method released less minocycline even over 14 days due to the very small medium volume and low flow rate that mimic periodontal pocket conditions. Although the rank order and discrimination of minocycline release was similar between Apparatus 4 and SVA, the potential to discriminate drug release in the initial times of release (<5 days) was greater for the biorelevant method. However, formulation screening potential as well as QC adaptability was better for the Apparatus 4 method [56].
To mimic the constrained geometry and slow fluid turnover of the periodontal pocket, another approach of a custom-made small-volume flow-through dissolution chamber (0.06 mL) was employed [107]. The method was based on a previously developed chamber by Ren et al. [108], originally designed for gelatin matrix systems and was adapted for PLGA-based products including Arestin microspheres and the Atridox in situ forming implant. The chamber simulated physiological conditions through temperature at 34 °C, a slow flow of simulated saliva at approximately 0.63 µL/min, and a medium composed of simulated saliva containing 0.137 M sodium chloride, 0.0014 M potassium phosphate monobasic, 0.017 M sodium phosphate dibasic, and 0.3% trypsin, adjusted to pH 8.0. Additional modifications, including filter paper to retain Arestin microspheres within the chamber and a blocking film to restrict the exposed surface area of the Atridox in situ forming implant, were implemented to reflect in vivo constraints. Under these conditions, the methods for both PLGA products indicated biorelevance by establishing a correlation with previously published in vivo data [107].
5. IVIVC
In vitro-in vivo correlation (IVIVC) is defined as a mathematical relationship between an in vitro property of a dosage form, usually drug release kinetics, and a relevant in vivo response [109,110]. A Level A IVIVC is the most informative, because it represents a point-to-point relationship in which the entire in vivo absorption profile or in vivo dissolution profile can be predicted from the in vitro release profile. Although Level A IVIVC is required for biowaivers, lower level IVIVCs may still support regulatory decision making when they are scientifically justified, validated, and fit for purpose [86,109,110]. The term in vitro–in vivo relationship (IVIVR) is also used to describe semiquantitative or rank order relationships between in vitro release and in vivo outcomes [76].
IVIVC can be used to set clinically relevant dissolution specifications. This implies the establishment of a link between drug product quality attributes (e.g. in vitro release), critical material attributes (CMAs), critical process parameters (CPPs) and in vivo performance (e.g. systemic exposure) [111].
For PLGA based LAIs, developing IVIVCs is particularly challenging because in vivo release depends on processes such as tissue hydration, enzymatic activity, polymer degradation, and dynamic changes in depot morphology, factors that cannot be fully replicated in standard in vitro tests. In addition, the long release duration of PLGA systems complicates alignment of in vitro and in vivo data [76]. A comprehensive discussion of these challenges and methodological considerations in development of IVIVC for PLGA based LAIs is provided in Wang et al. (2025), to which the reader is kindly referred for further guidance [76].
6. Conclusions and Future Considerations
Development of in vitro release methods for PLGA-based LAI products is challenging due to the lack of standardized methods, diversity of these drug delivery systems and complexity of the involved drug release mechanisms. Various in vitro release methods have been explored, which can broadly be grouped into sample-and-separate methods, dialysis methods and flow-through methods, each of them with their own advantages and shortcomings. IVR method development requires a careful assessment of experimental parameters including apparatus selection, medium composition, temperature and agitation, with controlled depot formation being particularly critical for in situ forming implants.
During early drug product development, the biorelevant IVR methods which closely mimic the physiological conditions at the administration site may be a valuable tool for formulation development and optimization. However, the complexity of these methods frequently limits their use for routine quality control. Development of QC methods often includes finding an optimum balance between biorelevance and practicality. The QC methods should be simple, robust, and reproducible while demonstrating adequate discriminatory power towards relevant critical material attributes, critical formulation variables and/or critical process parameters. Accelerated methods may be justified, provided they maintain the same release mechanism (or at least the same rank order between formulations) when compared with the real-time method. Ultimately, clinical relevance of the final method is desirable, implying that a link has been established between the in vitro release and in vivo performance.
Future progress in IVR methods for PLGA based LAIs will likely be related to a deeper mechanistic understanding of the involved release mechanisms both in vitro and in vivo, as well as a deeper understanding of the complex and dynamic physiological conditions present at the intended administration sites, enabled by the adoption of emerging technologies, such as in situ analytical monitoring and advanced computational modeling.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
Disclaimer
This article reflects the views of the authors and should not be construed to represent their
References
- Larrañeta, E.; Domínguez-Robles, J. Long-Acting Drug Delivery Systems: Current Landscape and Future Prospects. Drug Discov. Today 2025, 30, 104447. [Google Scholar] [CrossRef] [PubMed]
- Alidori, S.; Subramanian, R.; Holm, R. Patient-Centric Long-Acting Injectable and Implantable Platforms─An Industrial Perspective. Mol. Pharm. 2024, 21, 4238–4258. [Google Scholar] [CrossRef]
- Muddineti, O.S.; Omri, A. Current Trends in PLGA Based Long-Acting Injectable Products: The Industry Perspective. Expert Opin. Drug Deliv. 2022, 19, 559–576. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Berben, P.; Chakravarthi, S.S.; Chattorraj, S.; Garg, A.; Gourdon, B.; Heimbach, T.; Huang, Y.; Morrison, C.; Mundhra, D.; et al. Current State and Opportunities with Long-Acting Injectables: Industry Perspectives from the Innovation and Quality Consortium “Long-Acting Injectables” Working Group. Pharm Res 2023, 40, 1601–1631. [Google Scholar] [CrossRef] [PubMed]
- Park, K. PLGA-Based Long-Acting Injectable (LAI) Formulations. Journal of Controlled Release 2025, 382, 113758. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Tan, W.S.; Ho, K.L.; Mariatulqabtiah, A.R.; Kasim, N.H.A.; Abd. Rahman, N.; Wong, T.W.; Chee, C.F. Challenges and Complications of Poly(Lactic-Co-Glycolic Acid)-Based Long-Acting Drug Product Development. Pharmaceutics 2022, 14, 614. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Z.; Harris, A.; Yang, L. Bridging the Gap between Fundamental Research and Product Development of Long Acting Injectable PLGA Microspheres. Expert Opin. Drug Deliv. 2022, 19, 1247–1264. [Google Scholar] [CrossRef]
- Wang, X.; Burgess, D.J. Drug Release from in Situ Forming Implants and Advances in Release Testing. Adv. Drug Deliv. Rev. 2021, 178, 113912. [Google Scholar] [CrossRef]
- Lee, J.W.; Park, J.H.; Yu, G.W.; You, J.W.; Han, M.J.; Kang, M.J.; Ho, M.J. Sustained-Release Intra-Articular Drug Delivery: PLGA Systems in Clinical Context and Evolving Strategies. Pharmaceutics 2025, 17, 1350. [Google Scholar] [CrossRef]
- Otte, A.; Damen, F.; Goergen, C.; Park, K. Coupling the in Vivo Performance to the in Vitro Characterization of PLGA Microparticles. Int. J. Pharm. 2021, 604, 120738. [Google Scholar] [CrossRef]
- Doty, A.C.; Weinstein, D.G.; Hirota, K.; Olsen, K.F.; Ackermann, R.; Wang, Y.; Choi, S.; Schwendeman, S.P. Mechanisms of in Vivo Release of Triamcinolone Acetonide from PLGA Microspheres. Journal of Controlled Release 2017, 256, 19–25. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Release Mechanisms of PLGA-Based Drug Delivery Systems: A Review. Int. J. Pharm. X 2025, 10. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.N.; Jiang, W.; Wang, Y.; Loffredo, D.M. Challenges and Opportunities in the Development of Complex Generic Long-Acting Injectable Drug Products. Journal of Controlled Release 2021, 336, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Malavia, N.; Bao, Q.; Burgess, D.J. Novel Dissolution Methods for Drug Release Testing of Long-Acting Injectables. Int. J. Pharm. 2024, 664. [Google Scholar] [CrossRef] [PubMed]
- Anand, O.; Yu, L.X.; Conner, D.P.; Davit, B.M. Dissolution Testing for Generic Drugs: An FDA Perspective. AAPS Journal 2011, 13, 328–335. [Google Scholar] [CrossRef] [PubMed]
- United States Pharmacopeial Convention United States Pharmacopeia, USP NF 2026 Issue 1, 2026.
- Martinez, M.N.; Rathbone, M.J.; Burgess, D.; Huynh, M. Breakout Session Summary from AAPS/CRS Joint Workshop on Critical Variables in the in Vitro and in Vivo Performance of Parenteral Sustained Release Products. Journal of Controlled Release 2010, 142, 2–7. [Google Scholar] [CrossRef]
- Andhariya, J. V.; Burgess, D.J. Recent Advances in Testing of Microsphere Drug Delivery Systems. Expert Opin. Drug Deliv. 2016, 13, 593–608. [Google Scholar] [CrossRef]
- United States Pharmacopoeia USP Dissolution Database. Available online: https://www.usp.org/resources/dissolution-methods-database (accessed on 4 February 2026).
- Food and Drug Administration FDA Dissolution Methods Database. Available online: https://www.accessdata.fda.gov/scripts/cder/dissolution/ (accessed on 4 February 2026).
- 5.17.1. Recommendations on dissolution testing. in: European Pharmacopeia 12.1, 2026; EDQM Council of Europe: Strasbourg, France.
- <1092> The dissolution procedure: development and validation, in: United States Pharmacopeia, USP NF 2026 Issue 1, The United States Pharmacopeial Convention, Rockville, MD, USA, 2026.
- Garner, J.; Skidmore, S.; Park, H.; Park, K.; Choi, S.; Wang, Y. Beyond Q1 / Q2 : The Impact of Manufacturing Conditions and Test Methods on Drug Release From PLGA-Based Microparticle Depot Formulations. J. Pharm. Sci. 2018, 107, 353–361. [Google Scholar] [CrossRef]
- Hirota, K.; Doty, A.C.; Ackermann, R.; Zhou, J.; Olsen, K.F.; Feng, M.R.; Wang, Y.; Choi, S.; Qu, W.; Schwendeman, A.S.; et al. Characterizing Release Mechanisms of Leuprolide Acetate-Loaded PLGA Microspheres for IVIVC Development I : In Vitro Evaluation. Journal of Controlled Release 2016, 244, 302–313. [Google Scholar] [CrossRef]
- Doty, A.C.; Zhang, Y.; Weinstein, D.G.; Wang, Y.; Choi, S.; Qu, W.; Mittal, S.; Schwendeman, S.P. Mechanistic Analysis of Triamcinolone Acetonide Release from PLGA Microspheres as a Function of Varying in Vitro Release Conditions. European Journal of Pharmaceutics and Biopharmaceutics 2017, 113, 24–33. [Google Scholar] [CrossRef]
- Li, T.; Chandrashekar, A.; Beig, A.; Walker, J.; Hong, J.K.Y.; Benet, A.; Kang, J.; Ackermann, R.; Wang, Y.; Qin, B.; et al. Characterization of Attributes and in Vitro Performance of Exenatide-Loaded PLGA Long-Acting Release Microspheres. European Journal of Pharmaceutics and Biopharmaceutics 2021, 158, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Wang, J.; Khaliq, N.U.; Meng, F.; Oucherif, K.A.; Xue, J.; Horava, S.D.; Cox, A.L.; Richard, C.A.; Swinney, M.R.; et al. Development of Poly(Lactide-Co-Glycolide) Microparticles for Sustained Delivery of Meloxicam. Journal of Controlled Release 2023, 353, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Andhariya, J. V; Jog, R.; Shen, J.; Choi, S.; Wang, Y.; Zou, Y.; Burgess, D.J. Development of Level A in Vitro-in Vivo Correlations for Peptide Loaded PLGA Microspheres. Journal of Controlled Release 2019, 308, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Doty, A.C.; Hirota, K.; Olsen, K.F.; Sakamoto, N.; Ackermann, R.; Feng, M.R.; Wang, Y.; Choi, S.; Qu, W.; Schwendeman, A.; et al. Validation of a Cage Implant System for Assessing in Vivo Performance of Long-Acting Release Microspheres. Biomaterials 2016, 109, 88–96. [Google Scholar] [CrossRef]
- Lizambard, M.; Menu, T.; Fossart, M.; Bassand, C.; Agossa, K.; Huck, O.; Neut, C.; Siepmann, F. In-Situ Forming Implants for the Treatment of Periodontal Diseases: Simultaneous Controlled Release of an Antiseptic and an Anti-Inflammatory Drug. Int. J. Pharm. 2019, 572, 118833. [Google Scholar] [CrossRef]
- Andhariya, J. V; Choi, S.; Wang, Y.; Zou, Y.; Burgess, D.J.; Shen, J. Accelerated in Vitro Release Testing Method for Naltrexone Loaded PLGA Microspheres. Int. J. Pharm. 2017, 520, 79–85. [Google Scholar] [CrossRef]
- Kamali, H.; Khodaverdi, E.; Hadizadeh, F.; Yazdian-Robati, R.; Haghbin, A.; Zohuri, G. An In-Situ Forming Implant Formulation of Naltrexone with Minimum Initial Burst Release Using Mixture of PLGA Copolymers and Ethyl Heptanoate as an Additive: In-Vitro, Ex-Vivo, and in-Vivo Release Evaluation. J. Drug Deliv. Sci. Technol. 2018, 47, 95–105. [Google Scholar] [CrossRef]
- D’Souza, S.S.; DeLuca, P.P. Development of a Dialysis in Vitro Release Method for Biodegradable Microspheres. AAPS PharmSciTech 2005, 6, 323–328. [Google Scholar] [CrossRef]
- Andhariya, J. V; Jog, R.; Shen, J.; Choi, S.; Wang, Y.; Zou, Y.; Burgess, D.J. In Vitro-in Vivo Correlation of Parenteral PLGA Microspheres : Effect of Variable Burst Release. 2019, 314, 25–37. [Google Scholar] [CrossRef]
- Wang, X.; Wang, R.; Roy, M.; Kwok, O.; Burgess, D.J. Long-Acting Injectable in Situ Forming Implants: Impact of Polymer Attributes and API. Int. J. Pharm. 2025, 670, 125080. [Google Scholar] [CrossRef]
- Yang, S.; Hu, M.; Liu, W.; Hou, N.; Yin, K.; Shen, C.; Shang, Q. Fabrication of PLGA in Situ Forming Implants and Study on Their Correlation of in Vitro Release Profiles with in Vivo Performances. J. Biomater. Sci. Polym. Ed. 2021, 32, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.S.; DeLuca, P.P. Methods to Assess in Vitro Drug Release from Injectable Polymeric Particulate Systems. Pharm. Res. 2006, 23, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Nothnagel, L.; Wacker, M.G. How to Measure Release from Nanosized Carriers? European Journal of Pharmaceutical Sciences 2018, 120, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Lee, K.; Choi, S.; Qu, W.; Wang, Y.; Burgess, D.J. A Reproducible Accelerated in Vitro Release Testing Method for PLGA Microspheres. Int. J. Pharm. 2016, 498, 274–282. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Raton, J.L.; Burgess, D.J. Application of USP Apparatus 4 and in Situ Fiber Optic Analysis to Microsphere Release Testing. Dissolut. Technol. 2005, 12, 11–14. [Google Scholar] [CrossRef]
- Xie, L.; Beyer, S.; Vogel, V.; Wacker, M.G.; Mäntele, W. Assessing the Drug Release from Nanoparticles: Overcoming the Shortcomings of Dialysis by Using Novel Optical Techniques and a Mathematical Model. Int. J. Pharm. 2015, 488, 108–119. [Google Scholar] [CrossRef]
- Shen, J.; Choi, S.; Qu, W.; Wang, Y.; Burgess, D.J. In Vitro-in Vivo Correlation of Parenteral Risperidone Polymeric Microspheres. Journal of Controlled Release 2015, 218, 2–12. [Google Scholar] [CrossRef]
- Bain, D.F.; Munday, D.L.; Smith, A. Modulation of Rifampicin Release from Spray-Dried Microspheres Using Combinations of Poly-(DL-Lactide). J. Microencapsul. 1999, 16, 369–385. [Google Scholar] [CrossRef]
- Rawat, A.; Burgess, D.J. USP Apparatus 4 Method for in Vitro Release Testing of Protein Loaded Microspheres. Int. J. Pharm. 2011, 409, 178–184. [Google Scholar] [CrossRef]
- Shen, J.; Burgess, D.J. Accelerated In-Vitro Release Testing Methods for Extended-Release Parenteral Dosage Forms. Journal of Pharmacy and Pharmacology 2012, 64, 986–996. [Google Scholar] [CrossRef]
- 2.9.3. Dissolution test for solid dosage forms. European Pharmacopeia 12.1, 2026; EDQM Council of Europe: Strasbourg, France.
- Dissolution, in: United States Pharmacopeia, USP NF 2026 Issue 1. Rockville, MD, USA, 2026; The United States Pharmacopeial Convention.
- D’Arcy, D.M.; Wacker, M.G.; Klein, S.; Shah, V.; Burke, M.D.; Hunter, D.G.; Xu, H. In-Vitro Product Performance of Parenteral Drug Products: View of the USP Expert Panel. Dissolut. Technol. 2022, 29, 204–218. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Burgess, D.J. Effect of Acidic PH on PLGA Microsphere Degradation and Release. Journal of Controlled Release 2007, 122, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Andhariya, J. V; Shen, J.; Wang, Y.; Choi, S.; Burgess, D.J. Effect of Minor Manufacturing Changes on Stability of Compositionally Equivalent PLGA Microspheres. Int. J. Pharm. 2019, 566, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Rawat, A.; Stippler, E.; Shah, V.P.; Burgess, D.J. Validation of USP Apparatus 4 Method for Microsphere in Vitro Release Testing Using Risperdal® Consta ®. Int. J. Pharm. 2011, 420, 198–205. [Google Scholar] [CrossRef]
- Wang, X.; Bao, Q.; Suh, M.S.; Kastellorizios, M.; Wang, R.; Burgess, D.J. Novel Adapter Method for in Vitro Release Testing of in Situ Forming Implants. Int. J. Pharm. 2022, 621, 121777. [Google Scholar] [CrossRef]
- D’Souza, S.; Faraj, J.A.; Dorati, R.; DeLuca, P.P. A Short Term Quality Control Tool for Biodegradable Microspheres. AAPS PharmSciTech 2014, 15, 530–541. [Google Scholar] [CrossRef]
- Kim, Y.; Park, E.J.; Kim, T.W.; Na, D.H. Recent Progress in Drug Release Testing Methods of Biopolymeric Particulate System. Pharmaceutics 2021, 13, 1313. [Google Scholar] [CrossRef]
- Peloso, C.; Yvorra, E.; Delamare, R.; Campana, M.; Grizot, S.; Lopez-Noriega, A. Improving in Vivo Release Prediction from in Situ Forming Depots with a Novel Flow-through in Vitro Dissolution Apparatus. Int. J. Pharm. 2025, 681. [Google Scholar] [CrossRef]
- Patel, S.K.; Greene, A.C.; Desai, S.M.; Rothstein, S.; Basha, I.T.; MacPherson, J.S.; Wang, Y.; Zou, Y.; Shehabeldin, M.; Sfeir, C.S.; et al. Biorelevant and Screening Dissolution Methods for Minocycline Hydrochloride Microspheres Intended for Periodontal Administration. Int. J. Pharm. 2021, 596. [Google Scholar] [CrossRef]
- Xu, X.; Khan, M.A.; Burgess, D.J. A Two-Stage Reverse Dialysis in Vitro Dissolution Testing Method for Passive Targeted Liposomes. Int. J. Pharm. 2012, 426, 211–218. [Google Scholar] [CrossRef]
- D’Souza, S.S.; Faraj, J.A.; DeLuca, P.P. A Model-Dependent Approach to Correlate Accelerated with Real-Time Release from Biodegradable Microspheres. AAPS PharmSciTech 2005, 6, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.B.; Lee, S.; Ju, H.J.; Kim, H.E.; Noh, J.H.; Choi, S.; Park, K.; Lee, H.B.; Kim, M.S. Preparation and Evaluation of Injectable Microsphere Formulation for Longer Sustained Release of Donepezil. Journal of Controlled Release 2023, 356, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fassihi, R. Release Rate Determination from in Situ Gel Forming PLGA Implant: A Novel ‘Shape-Controlled Basket in Tube’ Method. Journal of Pharmacy and Pharmacology 2020, 72, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.F.; Ashtikar, M.; Kojima, R.; Yoshida, T.; Kaihara, M.; Tajiri, T.; Shanehsazzadeh, S.; Modh, H.; Wacker, M.G. Predicting Drug Release and Degradation Kinetics of Long-Acting Microsphere Formulations of Tacrolimus for Subcutaneous Injection. Journal of Controlled Release 2021, 329, 372–384. [Google Scholar] [CrossRef]
- Janas, C.; Mast, M.P.; Kirsamer, L.; Angioni, C.; Gao, F.; Mäntele, W.; Dressman, J.; Wacker, M.G. The Dispersion Releaser Technology Is an Effective Method for Testing Drug Release from Nanosized Drug Carriers. European Journal of Pharmaceutics and Biopharmaceutics 2017, 115, 73–83. [Google Scholar] [CrossRef]
- Jug, M.; Hafner, A.; Lovrić, J.; Kregar, M.L.; Pepić, I.; Vanić, Ž.; Cetina-Čižmek, B.; Filipović-Grčić, J. An Overview of in Vitro Dissolution/Release Methods for Novel Mucosal Drug Delivery Systems. J. Pharm. Biomed. Anal. 2018. [Google Scholar] [CrossRef]
- Mast, M.P.; Modh, H.; Knoll, J.; Fecioru, E.; Wacker, M.G. An Update to Dialysis-Based Drug Release Testing—Data Analysis and Validation Using the Pharma Test Dispersion Releaser. Pharmaceutics 2021, 13. [Google Scholar] [CrossRef]
- Suh, M.S.; Kastellorizios, M.; Tipnis, N.; Zou, Y.; Wang, Y.; Choi, S.; Burgess, D.J. Effect of Implant Formation on Drug Release Kinetics of in Situ Forming Implants. Int. J. Pharm. 2021, 592, 120105. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Burgess, D.J. Evaluation of in Vivo-in Vitro Release of Dexamethasone from PLGA Microspheres. Journal of Controlled Release 2008, 127, 137–145. [Google Scholar] [CrossRef]
- Chidambaram, N.; Burgess, D.J. A Novel in Vitro Release Method for Submicron-Sized Dispersed Systems. AAPS PharmSci 1999, 1, 32–40. [Google Scholar] [CrossRef]
- Wang, X.; Roy, M.; Wang, R.; Kwok, O.; Wang, Y.; Wang, Y.; Qin, B.; Burgess, D.J. Towards in Vitro – In Vivo Correlation Models for in Situ Forming Drug Implants. Journal of Controlled Release 2024, 372, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration In Vitro Release Test (IVRT) for in Situ Gel/Depot-Forming Drug Products. Available online: https://www.fda.gov/media/184397/download?attachment (accessed on 6 February 2026).
- Astaneh, R.; Erfan, M.; Moghimi, H.; Mobedi, H. Pharmaceutics, Preformulation and Drug Delivery Changes in Morphology of in Situ Forming PLGA Implant Prepared by Different Polymer Molecular Weight and Its Effect on Release Behavior. J. Pharm. Sci. 2009, 98, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Karp, F.; Turino, L.N.; Mariano, R.N.; Luna, J.A.; Islan, G.A.; Mengatto, L.N. Progesterone-PLGA in Situ Formed Implants: Structural Parameters Correlations with in Vitro and in Vivo Mathematical Models. J. Pharm. Sci. 2025, 114, 104014. [Google Scholar] [CrossRef] [PubMed]
- Elder, S.H.; Ross, M.K.; Nicaise, A.J.; Miller, I.N.; Breland, A.N.; Hood, A.R.S. Development of in Situ Forming Implants for Controlled Delivery of Punicalagin. Int. J. Pharm. 2024, 652, 123842. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chow, P.Y.; Lin, T.P.; Cheow, C.; Li, Z.; Wacker, M.G. Simulate SubQ: The Methods and the Media. J. Pharm. Sci. 2023, 112, 1492–1508. [Google Scholar] [CrossRef] [PubMed]
- Tipnis, N.P.; Shen, J.; Jackson, D.; Leblanc, D.; Burgess, D.J. Flow-through Cell-Based in Vitro Release Method for Triamcinolone Acetonide Poly (Lactic-Co-Glycolic) Acid Microspheres. Int. J. Pharm. 2020, 579, 119130. [Google Scholar] [CrossRef]
- Giles, M.B.; Hong, J.K.Y.; Liu, Y.; Tang, J.; Li, T.; Beig, A.; Schwendeman, A.; Schwendeman, S.P. Efficient Aqueous Remote Loading of Peptides in Poly(Lactic-Co-Glycolic Acid). Nat. Commun. 2022, 13, 1–9. [Google Scholar] [CrossRef]
- Wang, Y.; Otte, A.; Park, H.; Park, K. In Vitro-in Vivo Correlation (IVIVC) Development for Long-Acting Injectable Drug Products Based on Poly(Lactide-Co-Glycolide). Journal of Controlled Release 2025, 377, 186–196. [Google Scholar] [CrossRef]
- Rawat, A.; Bhardwaj, U.; Burgess, D.J. Comparison of in Vitro-in Vivo Release of Risperdal® Consta® Microspheres. Int. J. Pharm. 2012, 434, 115–121. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Leary, P.E.; Burgess, D.J. Elevated Temperature Accelerated Release Testing of PLGA Microspheres. Journal of Controlled Release 2006, 112, 293–300. [Google Scholar] [CrossRef]
- Li, Z.; Mu, H.; Weng Larsen, S.; Jensen, H.; Østergaard, J. An in Vitro Gel-Based System for Characterizing and Predicting the Long-Term Performance of PLGA in Situ Forming Implants. Int. J. Pharm. 2021, 609. [Google Scholar] [CrossRef] [PubMed]
- Von Burkersroda, F.; Schedl, L.; Gopferich, A. Why Degradable Polymers Undergo Surface Erosion or Bulk Erosion. Biomaterials 2002, 23, 4221–4231. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Li, Z.; Zhang, L.; Chi, Q.; Yang, Y.; Zhang, H.; Yang, Y.; Mei, X. A Novel Accelerated in Vitro Release Method to Evaluate the Release of Thymopentin from PLGA Microspheres. Pharm. Dev. Technol. 2015, 20, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, J.; Tang, X.; Li, M.; Ma, S.; Liu, C.; Gao, Y.; Zhang, Y.; Liu, Y.; Yu, F.; et al. An Accelerated Release Method of Risperidone Loaded PLGA Microspheres with Good IVIVC. Curr. Drug Deliv. 2017, 15, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Kamberi, M.; Nayak, S.; Myo-Min, K.; Carter, T.P.; Hancock, L.; Feder, D. A Novel Accelerated in Vitro Release Method for Biodegradable Coating of Drug Eluting Stents: Insight to the Drug Release Mechanisms. European Journal of Pharmaceutical Sciences 2009, 37, 217–222. [Google Scholar] [CrossRef]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), Q2(R2) Validation of analytical procedures. 2023. Available online: https://www.ich.org/page/quality-guidelines.
- European Medicines Agency. EMA/CHMP/CVMP/QWP/336031/2017; Reflection paper on the dissolution specification for generic solid oral immediate release products with systemic action. 2017.
- European Medicines Agency. Guideline on quality of oral modified release products. EMA/CHMP/QWP/428693/2013, 2014. [Google Scholar]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), Q2(R2)/Q14 Training on Validation of analytical procedures and Q14: Analytical Procedure Development. 2025. Available online: https://www.ich.org/page/quality-guidelines.
- Andhariya, J. V; Shen, J.; Choi, S.; Wang, Y.; Zou, Y.; Burgess, D.J. Development of in Vitro-in Vivo Correlation of Parenteral Naltrexone Loaded Polymeric Microspheres. Journal of Controlled Release 2017, 255, 27–35. [Google Scholar] [CrossRef]
- Torres-Terán, I.; Venczel, M.; Stieler, T.; Parisi, L.; Kloss, A.; Klein, S. Prediction of Subcutaneous Drug Absorption - Characterization of Subcutaneous Interstitial Fluids as a Basis for Developing Biorelevant in Vitro Models. Int. J. Pharm. 2023, 638. [Google Scholar] [CrossRef]
- Kinnunen, H.M.; Mrsny, R.J. Improving the Outcomes of Biopharmaceutical Delivery via the Subcutaneous Route by Understanding the Chemical, Physical and Physiological Properties of the Subcutaneous Injection Site. Journal of Controlled Release 2014, 182, 22–32. [Google Scholar] [CrossRef]
- Torres-Terán, I.; Venczel, M.; Klein, S. Prediction of Subcutaneous Drug Absorption - Do We Have Reliable Data to Design a Simulated Interstitial Fluid? Int. J. Pharm. 2021, 610. [Google Scholar] [CrossRef]
- Gao, G.F.; Thurn, M.; Wendt, B.; Parnham, M.J.; Wacker, M.G. A Sensitive in Vitro Performance Assay Reveals the in Vivo Drug Release Mechanisms of Long-Acting Medroxyprogesterone Acetate Microparticles. Int. J. Pharm. 2020, 586. [Google Scholar] [CrossRef]
- Sun, Y.; Jensen, H.; Petersen, N.J.; Larsen, S.W.; Østergaard, J. Concomitant Monitoring of Implant Formation and Drug Release of in Situ Forming Poly (Lactide-Co-Glycolide Acid) Implants in a Hydrogel Matrix Mimicking the Subcutis Using UV–Vis Imaging. J. Pharm. Biomed. Anal. 2018, 150, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Kožák, J.; Rabišková, M.; Lamprecht, A. In-Vitro Drug Release Testing of Parenteral Formulations via an Agarose Gel Envelope to Closer Mimic Tissue Firmness. Int. J. Pharm. 2021, 594. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, H.M.; Sharma, V.; Contreras-Rojas, L.R.; Yu, Y.; Alleman, C.; Sreedhara, A.; Fischer, S.; Khawli, L.; Yohe, S.T.; Bumbaca, D.; et al. A Novel in Vitro Method to Model the Fate of Subcutaneously Administered Biopharmaceuticals and Associated Formulation Components. Journal of Controlled Release 2015, 214, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Qin, Q.; Benetti, A.A.; Kahouadji, L.; Wacker, M.G. BioJect: An in Vitro Platform to Explore Release Dynamics of Peptides in Subcutaneous Drug Delivery. Journal of Controlled Release 2025, 380, 1058–1079. [Google Scholar] [CrossRef]
- McCartan, A.J.S.; Mrsny, R.J. In Vitro Modelling of Intramuscular Injection Site Events. Expert Opin. Drug Deliv. 2024, 21, 1155–1173. [Google Scholar] [CrossRef]
- Villa Nova, M.; Gan, K.; Wacker, M.G. Biopredictive Tools for the Development of Injectable Drug Products. Expert Opin. Drug Deliv. 2022, 19, 671–684. [Google Scholar] [CrossRef]
- Kozak, J.; Lamprecht, A. Biorelevant in Vitro Drug Release Conditions Ameliorate In-Vitro-in-Vivo Relationship of Parenteral Risperidone Microspheres. J. Drug Deliv. Sci. Technol. 2023, 88, 104953. [Google Scholar] [CrossRef]
- Kozak, J.; Rabiskova, M.; Lamprecht, A. Muscle Tissue as a Surrogate for In Vitro Drug Release Testing of Parenteral Depot Microspheres. AAPS PharmSciTech 2021, 22. [Google Scholar] [CrossRef]
- Mertz, N.; Østergaard, J.; Yaghmur, A.; Larsen, S.W. Transport Characteristics in a Novel in Vitro Release Model for Testing the Performance of Intra-Articular Injectables. Int. J. Pharm. 2019, 566, 445–453. [Google Scholar] [CrossRef]
- Ouerfelli, N.; Vrinceanu, N.; Mliki, E.; Amin, K.A.; Snoussi, L.; Coman, D.; Mrabet, D. Rheological Behavior of the Synovial Fluid: A Mathematical Challenge. Front. Mater. 2024, 11, 1–13. [Google Scholar] [CrossRef]
- Magri, G.; Selmin, F.; Cilurzo, F.; Fotaki, N. Biorelevant Release Testing of Biodegradable Microspheres Intended for Intra-Articular Administration. European Journal of Pharmaceutics and Biopharmaceutics 2019, 139, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Bong Lee, J.; Gittings, S.; Iachelini, A.; Bennett, J.; Cram, A.; Garnett, M.; Roberts, C.J.; Gershkovich, P. Development and Optimisation of Simulated Salivary Fluid for Biorelevant Oral Cavity Dissolution. European Journal of Pharmaceutics and Biopharmaceutics 2021, 160, 125–133. [Google Scholar] [CrossRef]
- Acquier, A.B.; Pita, A.K.D.C.; Busch, L.; Sánchez, G.A. Comparison of Salivary Levels of Mucin and Amylase and Their Relation with Clinical Parameters Obtained from Patients with Aggressive and Chronic Periodontal Disease. Journal of Applied Oral Science 2015, 23, 288–294. [Google Scholar] [CrossRef]
- Goodson, J.M. Gingival Crevice Fluid Flow. Periodontol. 2000 2003, 31, 4–6. [Google Scholar] [CrossRef]
- Wanasathop, A.; Murawsky, M.; Li, S.K. Modification of Small Dissolution Chamber System for Long-Acting Periodontal Drug Product Evaluation. Int. J. Pharm. 2022, 618, 121646. [Google Scholar] [CrossRef]
- Ren, W.; Murawsky, M.; La Count, T.; Wanasathop, A.; Hao, X.; Kelm, G.R.; Kozak, D.; Qin, B.; Li, S.K. Dissolution Chamber for Small Drug Delivery System in the Periodontal Pocket. AAPS Journal 2019, 21. [Google Scholar] [CrossRef]
- Food and Drug Administration Guidance for Industry Guidance for Industry Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. 1997.
- European Medicines Agency Guideline on the Pharmacokinetic and Clinical Evaluation of Modified Release Dosage Forms. 2015, 44, 1–46.
- Suarez-Sharp, S.; Cohen, M.; Kesisoglou, F.; Abend, A.; Marroum, P.; Delvadia, P.; Kotzagiorgis, E.; Li, M.; Nordmark, A.; Bandi, N.; et al. Applications of Clinically Relevant Dissolution Testing : Workshop Summary Report. AAPS J. 2018, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the experimental setup for sample and separate method (AI-assisted drawing using OpenAI, February 2026).
Figure 1.
Schematic representation of the experimental setup for sample and separate method (AI-assisted drawing using OpenAI, February 2026).

Figure 2.
Schematic representation of the experimental setup for flow-through method (AI-assisted drawing using OpenAI, February 2026).
Figure 2.
Schematic representation of the experimental setup for flow-through method (AI-assisted drawing using OpenAI, February 2026).
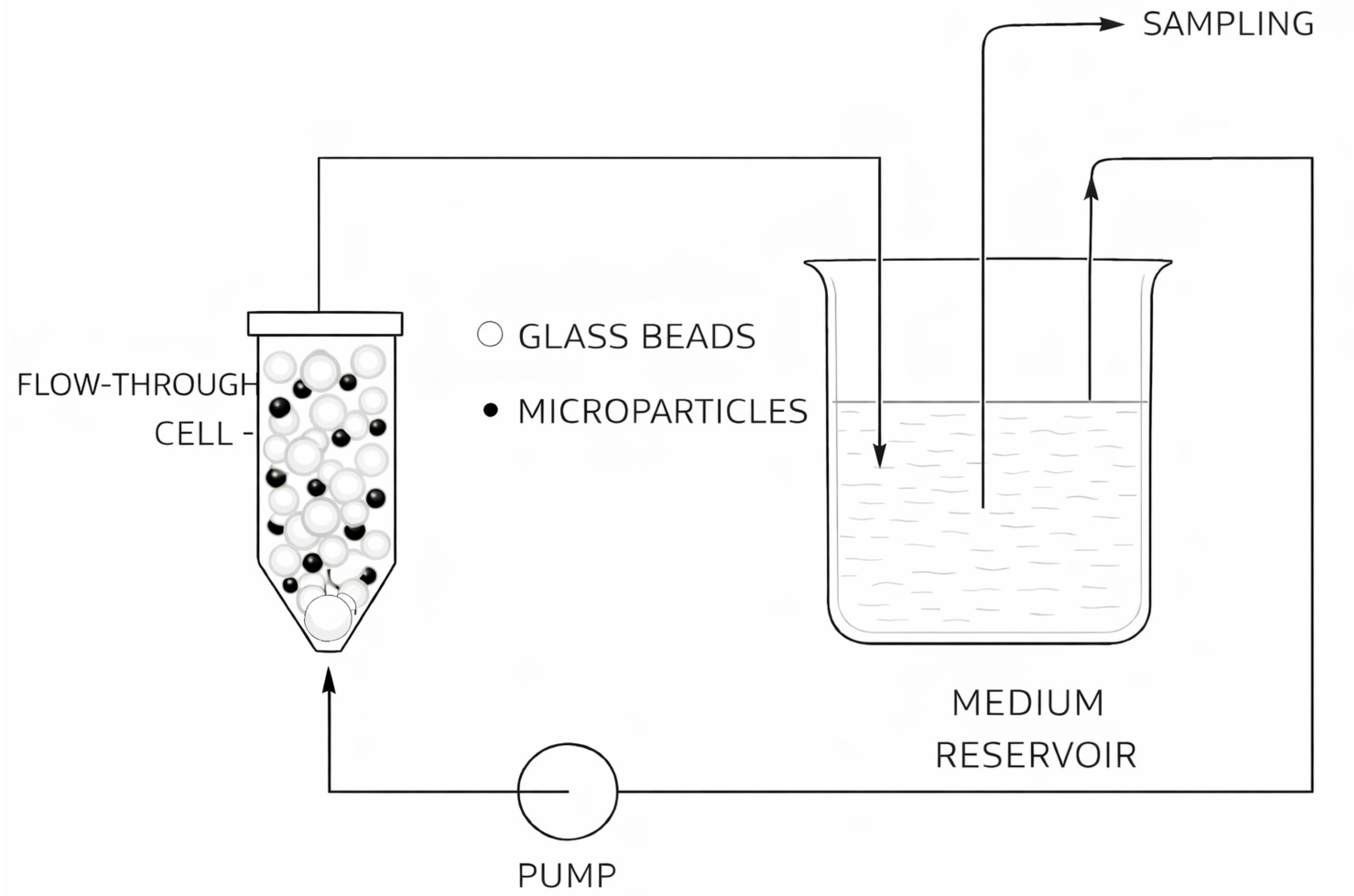
Figure 3.
Schematic representation of the experimental setup for dialysis method ((AI-assisted drawing using OpenAI, February 2026).
Figure 3.
Schematic representation of the experimental setup for dialysis method ((AI-assisted drawing using OpenAI, February 2026).
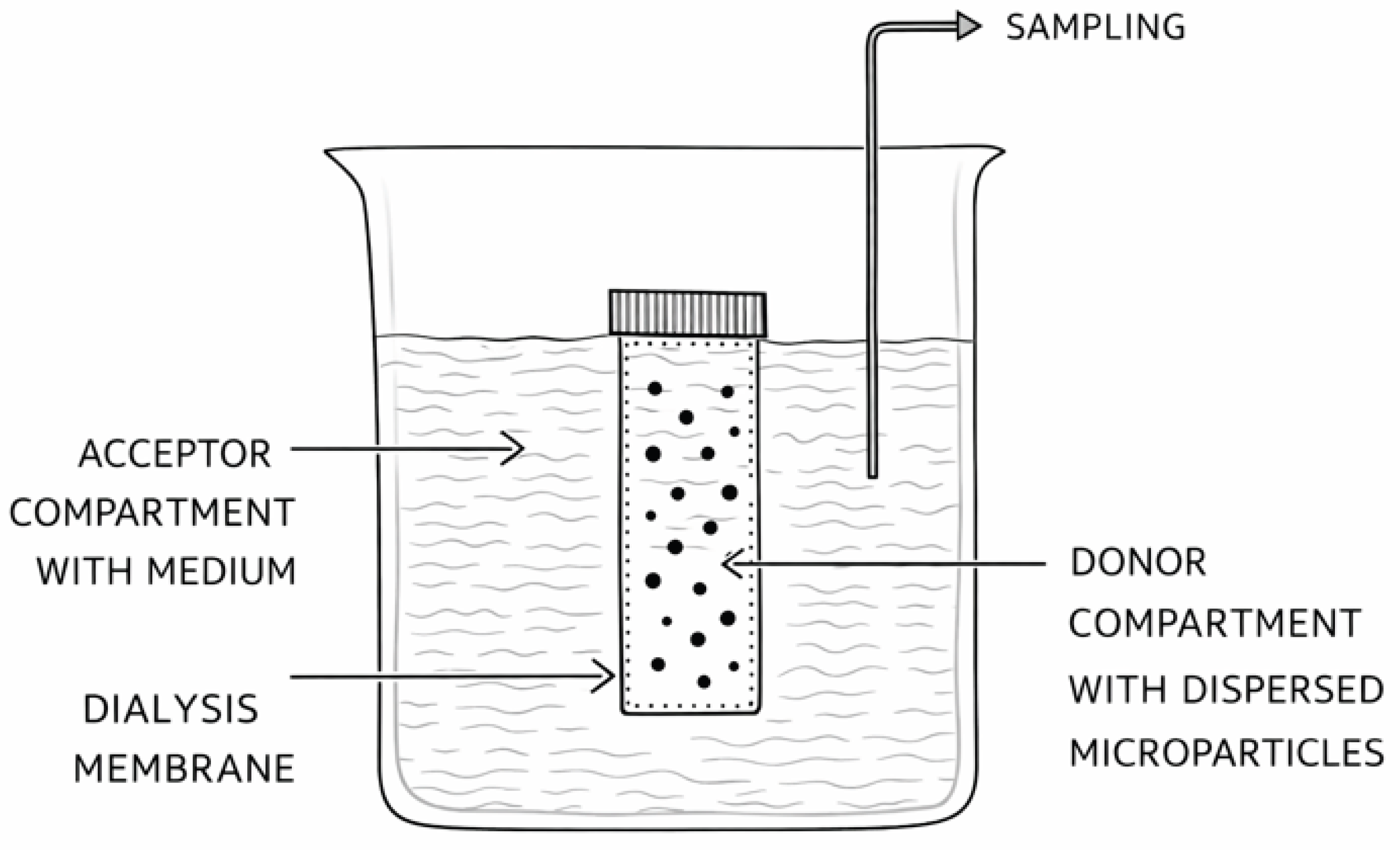

Table 1.
In vitro release methods used for PLGA LAI products according to the USP Dissolution Methods Database [19].
Table 1.
In vitro release methods used for PLGA LAI products according to the USP Dissolution Methods Database [19].
| Monograph | Dosage Form | Apparatus | Speed (rpm) | Medium | Volume (ml) | Test time | Exceptions |
| Goserelin Implants | Implants (3.6 mg) |
Flat bottom glass jar | / | Phosphate/citrate buffer, pH 7.4 | 50 | 168, 336, 408, 504, 672 h | 39 ± 0.5°C |
| Goserelin Implants | Implants (10.8 mg) |
Flat bottom glass jar | / | Phosphate buffered saline, pH 7.4 | 50 | 72, 336, 840, 1344, 2016 h | 39 ± 0.5°C |
| Minocycline Periodontal System | Periodontal system | Tube rotator | / | Phosphate buffer, pH 4.2 | 10 | 4, 24, 48, 72 h |
catalog # 400110 Labquake |
Table 2.
In vitro release methods used for PLGA LAI products according to the FDA Dissolution Methods Database [20].
Table 2.
In vitro release methods used for PLGA LAI products according to the FDA Dissolution Methods Database [20].
| Drug Name | Dosage Form | Apparatus | Speed (rpm) | Medium | Volume (ml) | Recommended sampling times |
| Dexamethasone | Implant (intravitreal) | VII (with reciprocating 50 mesh baskets) | 30 cycles per min | Phosphate buffered saline containing 0.05 g/L sodium dodecyl sulfate at 45 ± 0.5°C | 30 | 12, 24, 48, 72, 96, 120, 144, 168, 192, 216 and 240 hours |
| Goserelin acetate | Implant | 120-mL Wheaton jar | Swirl orbit of 50 mm at 205 rpm for 6 seconds (Prior to sampling, the jar is removed from incubation and mechanically swirled with digital orbital shaker) |
50 mL of phosphate buffered saline, pH 7.4, at 39°C (warmed overnight before the implants are added) | 50 | 3, 14, 35, 56 and 84 days (10.8 mg strength); 7, 14, 17, 21 and 28 days (3.6 mg strength) |
| Naltrexone | Injectable suspension | Develop an in vitro release method using Apparatus IV (flow-through cell), and, if applicable, Apparatus II (paddle) or any other appropriate method, for comparative evaluation by the Agency | / | Phosphate buffered saline with 0.02% Tween 20 and 0.02% Sodium azide, pH 7.4 (final osmolality should be 270 ± 20 mOsm), or any other appropriate medium, at 37°C. | / | / |
| Risperidone | For suspension (extended release) | IV (Flow thru cell-closed loop) | / | 50 mM potassium phosphate buffer, pH 7.4 | 1000 | 8, 24, 96, 120, 144, 168, 192, 216 and 264 hours |
| Triamcinolone Acetonide | Intra-articular, for suspension (extended release) | II (paddle) | 75 | 0.3% SDS in 10 mM phosphate buffer, pH 7.2 + 0.02% sodium azide @35°C | 1000 | 1, 2, 4, 8, 12, 16, 24, 36, 48, 72, 96 and 120 hours |
| Triptorelin Pamoate | For intramuscular suspension (extended release) | II (Paddle) | 75 | 50 mL of methanol to 950 mL of water | 950 | 1, 8, 24, 96, and 168 hours |
| Triptorelin Pamoate | Injectable suspension | II (Paddle) | 200 | Water-Methanol (95:5); Reconstitute vial in 2 mL Water for Injection, add to 500 mL medium at 37°C | 500 | 1, 6, 12, 24, 48 and 72 hours |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



