Submitted:
11 February 2026
Posted:
12 February 2026
You are already at the latest version
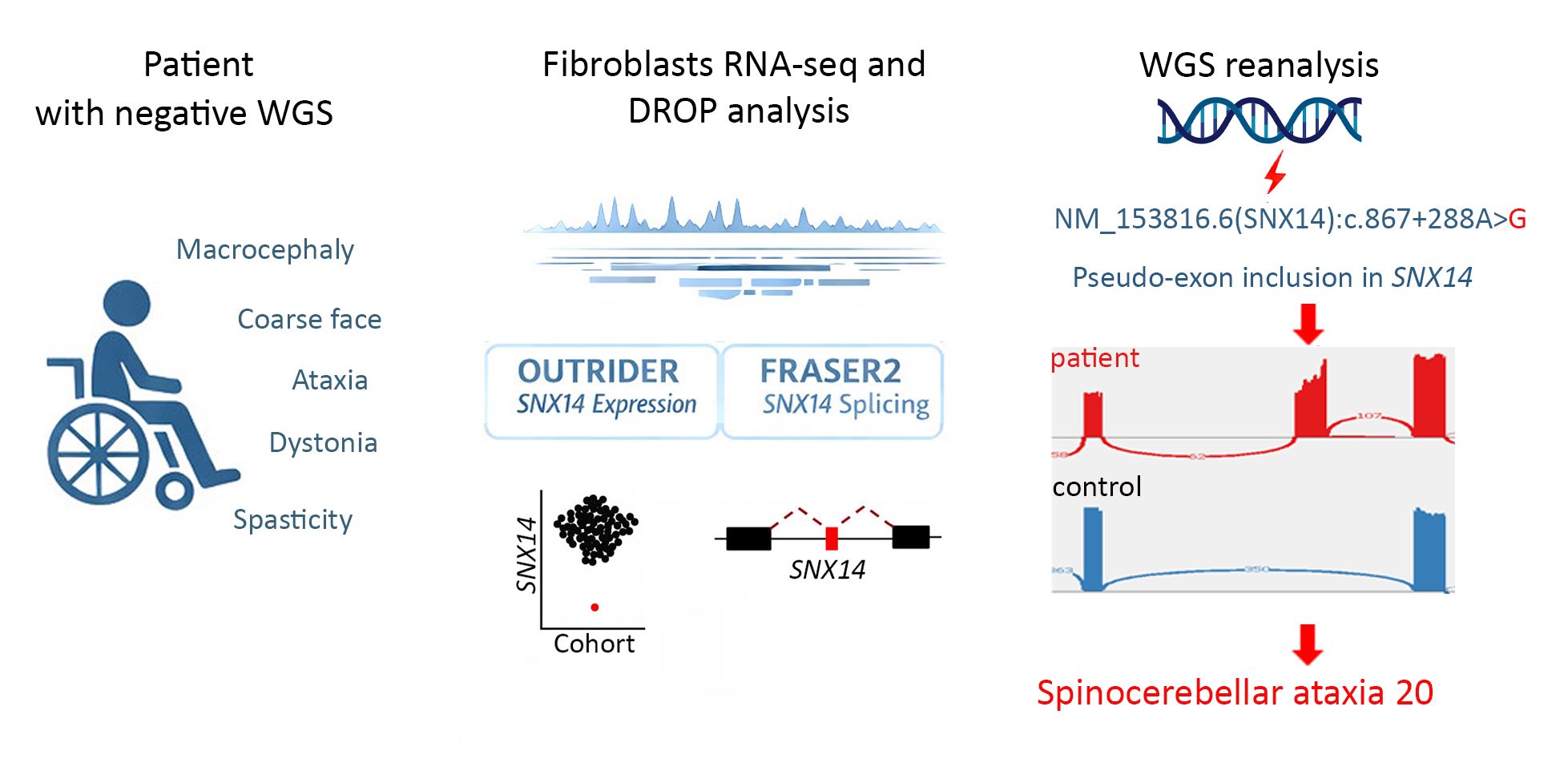
Abstract
Background: The contribution of intronic variants to the etiology of Mendelian diseases is still underrecognized, impacting the diagnostic yield. Whole genome sequencing (WGS) detects intronic variants, but beside canonical splice-sites, intronic variants are frequently excluded from the interpretation step or are classified as variants of uncertain significance (VUS). In fact, assessing their clinical significance often requires validation via RNA-sequencing or in vitro studies. Methods: We studied a 31-year-old patient with spinocerebellar ataxia who lacked a molecular diagnosis after WGS analysis. We applied the Detection of RNA Outliers Pipeline (DROP) to analyze RNA-sequencing (RNA-seq) data from patient fibroblasts. DROP integrates OUTRID-ER and FRASER 2.0 algorithms designed to identify aberrant gene expression and splicing, respectively. Results: DROP identified SNX14 as an outlier with significantly reduced gene expression and aberrant splicing. Retrospective WGS data inspection revealed a homozygous deep intronic variant NM_153816.6(SNX14):c.867+288A>G, which caused the splicing defect. Biallelic loss of function variants in SNX14 cause autosomal recessive spinocerebellar ataxia type 20 (SCAR20; OMIM 616354), and the patient’s clinical presentation was compatible with this disease. Conclusion: We present a deep intronic SNX14 variant as the cause of SCAR20. Integration of RNA-sequencing increases the diagnostic yield specifically by identifying and resolving the pathogenicity of deep intronic variants. Identification of aberrant splicing events can be therapeutically relevant, as these mechanisms are targets for antisense oligonucleotide (ASO) based interventions.
Keywords:
cerebellar ataxia
; deep intronic variant
; FRASER 2.0
; OUTRIDER
; RNA-sequencing
; SCAR20
; SNX14
1. Introduction
Next-generation sequencing has transformed Mendelian disease diagnostics, about 50% of cases remain genetically unsolved after whole exome and whole genome sequencing (WES and WGS) [1]. One challenge is posed by the interpretation of intronic variants outside canonical splice-sites, for example deep intronic variants, defined as located more than 100 bp away from the closest exon-intron boundary [2]. These variants can disrupt normal splicing and can create frameshifts and/or premature stop codons and thus being pathogenic [3]. Intronic variants are robustly identified by WGS, but are difficult to interpret in silico, and as a result many remain VUS, limiting the diagnostic yield [2,4]. RNA-seq has emerged as a powerful complementary tool, enabling the detection of aberrant gene expression and aberrant splicing. By pinpointing the affected gene, RNA-seq allows retrospective examination of WGS data to identify pathogenic variants [5,6]. The Detection of RNA Outliers Pipeline (DROP) is an integrative workflow to detect aberrant expression and aberrant splicing from raw RNA-seq data [7]. DROP integrates two statistical algorithms to prioritize disease-relevant genes: OUTRIDER, for the detection of expression outliers, and FRASER 2.0, for splicing outliers [8,9]. We applied DROP to RNA-seq data from skin fibroblasts of a 31-year-old patient with a neurodegenerative disease course featuring spinocerebellar ataxia and profound intellectual disability. The cause remained unknown despite WGS data analysis.
The pipeline identified aberrant expression and splicing of SNX14 in the patient fibroblasts. Retrospective analysis of WGS data revealed a homozygous deep intronic variant, NM_153816.6(SNX14):c.867+288A>G, GRCh37/hg19: chr6:g.86257731T>C, p?., responsible for pseudo-exon activation and reduced transcript levels. Biallelic pathogenic variants in Sorting nexin 14 (SNX14) cause SCAR20 (Spinocerebellar Ataxia, Autosomal Recessive 20; OMIM 616354), a condition overlapping with the clinical features of the patient. This study documents the pathogenicity of a deep intronic SNX14 variant and highlights the diagnostic value of RNA-seq analysis as an important tool to identify and assess pathogenic deep intronic variants which would otherwise remain VUS.
2. Materials and Methods
2.1. Whole Genome Sequencing (WGS) and Data Analysis
Genomic DNA was extracted from peripheral blood of the patient. Sample preparation for WGS was done using TruSeqTM PCR-free prep. WGS was performed using the Illumina HiSeq X instrument (Illumina, San Diego, CA) with 150 bp paired-end reads. The data were analyzed using the nf-core/raredisease pipeline, version 2.2.0 [10]. The reads were aligned to the GRCh38 human reference genome with BWA-MEM2 version 2.2.1 [11]. Duplicate reads were marked using Picard MarkDuplicates (version 3.3.0) (https://broadinstitute.github.io/picard/). Variant calling of each sample was performed with DeepVariant (version 1.6.1, gVCF mode) [12]. Joint genotyping of the trio was then conducted using GLnexus (version 1.4.1) [13]. Functional annotation was performed by the Ensembl Variant Effect Predictor (VEP, version 110) [14], and variant deleteriousness scores were computed by the Combined Annotation Dependent Depletion tool (CADD, version 1.6) [15]. The tools vcfanno (version 0.3.5) [16] and BCFtools (version 1.20) [17] were used to annotate variants with population allele frequencies from gnomAD (version 4.1) [18], clinical significance from ClinVar (downloaded on September 19, 2024) [19] and splice junction prediction scores from SpliceAI [20]. The final variant calling file (VCF) was analyzed using the FILTUS program [21]. We discarded variants with allelic frequency >0.01 in gnomAD v4.1. We also discarded variants with CADD phred score <10. We focused on single nucleotide variants (SNVs) small insertion/deletion (Indels) variants causing missense, nonsense, frameshift, or affected splice-sites.
2.2. RNA-seq and Data Analysis
Fibroblasts from skin biopsies of the patient and a cohort of 40 additional samples were cultivated in DMEM, high glucose (Gibco) with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5% CO2. The cohort consisted of skin biopsies from children with a genetic disease of known or unknown etiology and from healthy adults. The individuals, 47% female and 53% male were of varied ancestry.
RNA was extracted from cultured fibroblasts using the Ambion PARIS™ system (Thermo Fisher). Samples were prepared for RNA-seq with the Illumina Strand-specific TruSeq mRNA-seq library prep (Illumina Inc.). The libraries were indexed, pooled, and sequenced on an Illumina Novaseq X (Illumina Inc.) with 150 bp paired-end reads.
Reads were aligned to the ENSEMBL reference GRCh37 release 87 (Homo_sapiens.GRCh37.dna.primary_assembly.fa and Homo_sapiens.GRCh37.87.gtf) with HISAT2 2.1.0 [22]. RNA sequencing data were processed with DROP [7], which integrates the statistical algorithm OUTRIDER and FRASER 2.0 for detection of aberrant expression and aberrant splicing respectively [8,9]. In OUTRIDER we used p value ≤ 0.05 as the cut-off for calling significant expression outliers [8]. In FRASER 2.0, split reads spanning the exon-exon junction and non-split reads spanning the splice sites were counted for splicing event calling [9]. The Intron Jaccard Index is computed using split and non-split reads for capturing several types of aberrant splicing. Significant aberrant splicing events were defined with FDR ≤ 0.1 [9]. The analysis cohort included 40 additional fibroblast samples to model background variation. Aberrant events were visually inspected using Integrative Genomics Viewer IGV [23].
3. Results
3.1. Clinical Presentation
The patient was a male born in 1995. His parents were consanguineous of Moroccan Berber de-scent and had two other children who were healthy (Figure 1A).
Pregnancy and delivery at term were uneventful. Early development was initially interpreted as unremarkable. In retrospect, however, there were early signs of developmental delay right from the beginning with poor head control, delayed visual tracking and eye contact and absence of consonant babbling.
Psychomotor delay was recognized at 7 months, when he was hospitalized for a respiratory tract infection. At 2 years, he experienced a seizure and was treated with antiepileptic medication for four years. Epilepsy recurred three years ago requiring antiepileptic medication.
He sat with support at 1 year, and without support around age 7-8 years, though this acquired skill was later lost. He has not achieved independent walking.
From 8 years of age, he developed dystonic posturing of the feet, later confirmed as equinovarus deformity, which was bilaterally surgically corrected (Figure 1C). He developed scoliosis, man-aged with orthotic bracing. Alopecia began around the age of 16.
Hearing impairment was suspected at age 2 years. A response threshold above 80 dB was found on brainstem auditory evoked response examination at 3 years, demonstrating neurogenic deaf-ness. He was fitted with hearing aids, which he refused to wear. His parents believe he has at least partial hearing.
At 22 years, he was wheelchair-bound with reduced mobility, particularly in his legs. No targeted or spontaneous movements were observed. Muscle tone was increased and there was dystonic posturing, particularly in the lower extremities. The resulting equinovarus foot deformities appeared more pronounced when he was awake. Deep tendon reflexes were difficult to elicit, but plantar responses were bilaterally extensor with fanning of the toes. There were no definite cranial nerve deficits, although visual tracking was limited. Sensory function appeared preserved.
Cognitive development was profoundly impaired. He did not have expressive verbal language, but only some vocalizations. His parents reported that he could visually engage with television and seemingly enjoy watching football, although he does not appear to understand the game. He displayed a range of stereotypic facial and hand movements (Supplementary file Video 1). Dys-morphic features included coarse facial traits, a prominent forehead, deep-set eyes, thick bushy eyebrows, thick lips, and a broad chin (Figure 1B). He was macrocephalic with head circumference 62 cm (2.5 cm >97th centile at 21 years) [23]. The patient was recently scheduled for clinical examination but was unable to attend due to technical problems related to wheelchair transportation. However, his guardian mother stated the that clinical condition had been stable since the last visit, except for recurrence of epilepsy.
Brain MRI performed at 11 years and at 21 years revealed atrophy of both cerebellar hemispheres and vermis (Figure 1 D-E) with bilateral symmetric dentate nucleus hyperintensity (Figure 1 D). There were no abnormal signal changes in the basal ganglia. MR spectroscopy (MRS) from the right cerebellar hemisphere showed elevated glutamate and myoinositol, with preserved peaks for choline, creatine, and N-acetyl aspartate (NAA), whereas lactate was normal. The MRS find-ings were concluded as nonspecific, but compatible with a degenerative disease [24].
3.2. Variant Identification by DROP Pipeline on RNA-seq Data
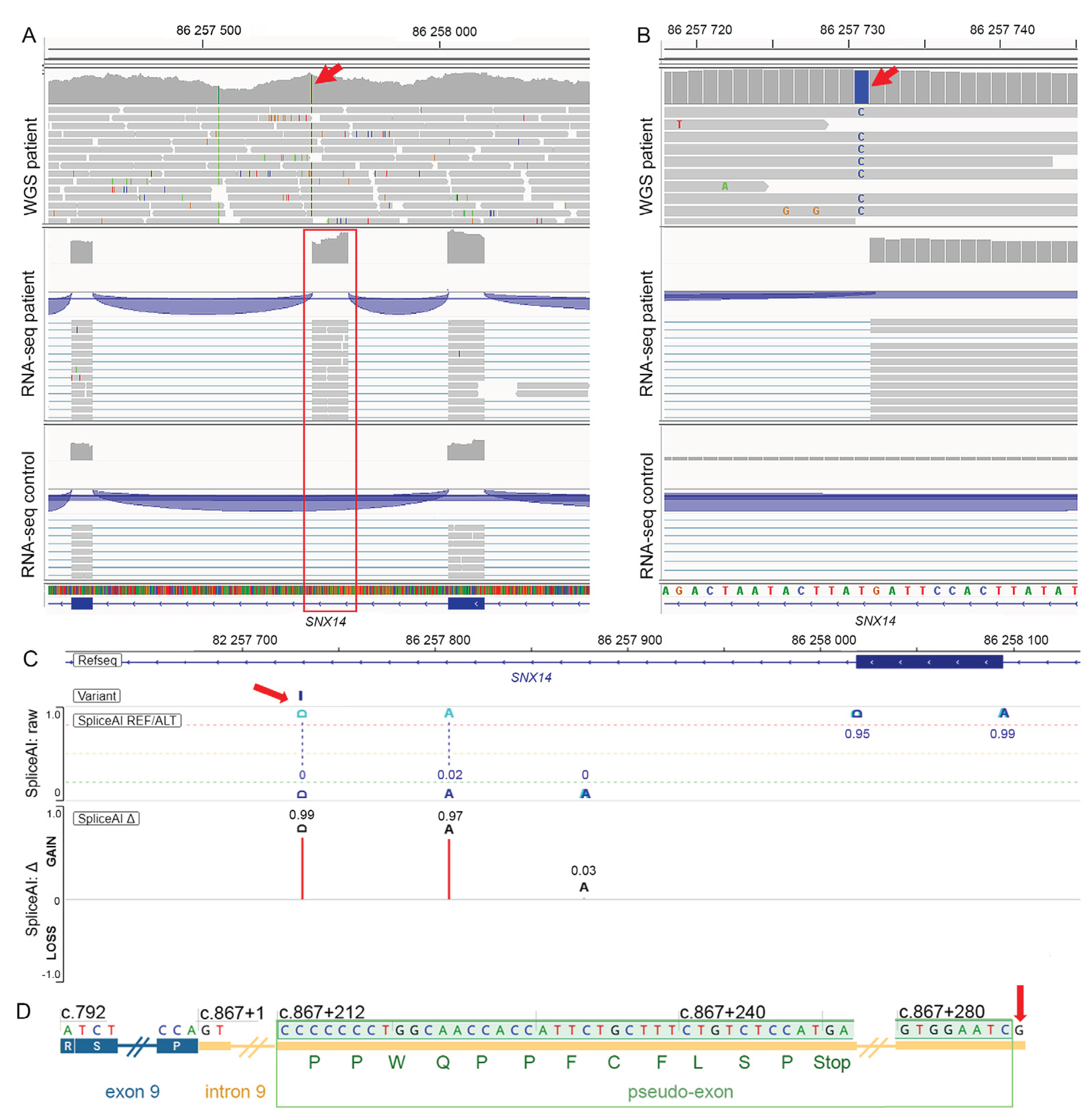
RNA-seq analysis using DROP revealed SNX14 as an outlier for both expression and splicing (Table S1 A-B). In OUTRIDER, SNX14 was identified as under-expressed (p adjusted value 5.35X10-4, log₂ fold change ≈ –2.17). FRASER 2.0 identified aberrant inclusion from SNX14 (NM_153816.6) intron 9 (chr6:86257269-86258018 bp) (p adjusted value 1.82 X10-8), consistent with a pseudo-exon activation event. Inspection of SNX14 in IGV demonstrated transcription of a pseudo-exon at chr6:86257732-86257807 bp, this event was not observed in any other samples in the cohort (Figure 2A). RNA-seq data from patient fibroblasts showed no SNX14 transcripts that support normal splicing.
3.3. Retrospective WGS Data Analysis
Initial WGS data analysis of the patient did not reveal any putative pathogenic variants, because deep intronic variants were not interpreted. After the pseudo-exon detection by DROP, retrospective WGS analysis of SNX14 identified the following variant: NM_153816.6(SNX14):c.867+288A>G, GRCh37/hg19: chr6:g.86257731T>C, p.? (Figure 2B). The SpliceAI algorithm predicted the variant to create a splice donor site (Δ= 0.99) at chr6:86257732 bp, and an acceptor site (Δ= 0.97) at chr6:86257807 bp (Figure 2C). This was consistent with RNA-seq data identifying a pseudo-exon at: chr6:86257732-86257807 bp. The inclusion of the pseudo-exon resulted in a frameshift and introduced a premature stop codon: NP_722523.1 (SNX14):p.(Asp290Profs*12) (Figure 2D). The premature stop codon likely triggered nonsense-mediated decay (NMD), explaining the reduced gene expression detected by OUTRIDER. The variant was not reported in gnomAD and had CADD PHRED 12.99 in (GRCh37-v1.6) and 24 in GRCh37-v1.7.
4. Discussion
We report a patient with a homozygous deep intronic variant in SNX14 that activates a pseudo-exon inclusion and results in loss of functional SNX14 transcript, which likely caused the clinical presentation in line with autosomal recessive spinocerebellar ataxia type 20 (SCAR20). The clinical impact of deep intronic variants is poorly understood and are therefore possibly underreported as a cause of Mendelian disease in patients [2,4]. To date, more than 40 individuals with SCAR20 have been described, with the majority harboring SNX14 loss of function variants such as nonsense, frameshift, or canonical splice-site mutations [26,27,28,29,30,31,32,33,34,35,36,37]. In contrast, only one deep intronic variant (NM_153816.6:c.462-589A>G) has been described in two affected sisters [38]. We expand the SNX14 allelic spectrum beyond coding and canonical splice-site variants, highlighting the pathogenic relevance of non-coding variants in SCAR20.
The molecular and clinical features observed in the patient are consistent with the known biological role of SNX14. Constitutive loss of Snx14 cause embryonic lethality in mice and conditional Snx14 deletion in neurons and glia results in severe motor deficits and Purkinje cell degeneration, caused by disrupted microtubule organization and mitochondrial transport through destabilization of the spastin enzyme, leading to impaired axonal integrity and cerebellar ataxia [39]. SNX14 localizes to endoplasmic reticulum–lipid droplet contact sites and plays a key role in lipid homeostasis, autophagy, and organelle crosstalk, and its loss results in accumulation of lipid droplets, enlarged lysosomes, and impaired autophagosome clearance [26,35,40]. The patient phenotype including early onset early onset global developmental delay, macrocephaly, coarse facial features and cerebellar atrophy with ataxia and spasticity fall within the spectrum of congenital disorders of autophagy [41,42].
To our knowledge, alopecia is not a typical feature of SCAR20 and may be caused by a variant in another gene, which we were unable to identify.
Beyond its diagnostic relevance, the identification of cryptic splice events can be therapeutically relevant. Pseudo-exon activation represents a potentially targetable mechanism, as aberrant splice sites can be masked using ASOs, restoring normal splicing. Proof of concept for this strategy has already been demonstrated in several genetic disorders, including Leber congenital amaurosis type 10 caused by the deep intronic CEP290 variant NM_025114.4:c.2991+1655A>G, for which ASO-based therapies have advanced to clinical trials [43,44].
5. Conclusions
This study expands the mutational spectrum of SNX14, describing a novel pseudo-exon activating deep intronic variant. The variant was identified through RNA-seq analysis, highlighting a limitation of WGS-based diagnostics. It demonstrates how RNA-seq can provide a functional readout, turning challenging to interpret intronic variants into clinically relevant findings.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Video S1. The patient at the age of 22 years, wheelchair-bound and showing repetitive head and hand movements; Table S1. DROP analysis results showing SNX14 as gene expression outlier (panel A) and splicing outlier (panel B) in the patient compared to the cohort.
Author Contributions
Conceptualization DM, PS, EF; Clinical investigations: PS; Genetic data analyses: DM; Writing original draft preparation: DM, PS; MRI study: MSC; WGS and RNA-seq study: DM, AYMS, PMB, MES; Project administration EF; funding acquisition EF. All authors have read and agreed to the published version of the manuscript.
Funding
EF was supported by the Nasjonal kompetansetjeneste for sjeldne diagnoser (Norwegian National Advisory Unit on Rare Disorders).
Institutional Review Board Statement
The study was conducted in compliance with the Helsinki Declaration and with the ethical committee of the institutions involved. The study was approved by the Regional Committee for Medical Research Ethics – South-East Norway, REK 2010/1152a.
Informed Consent Statement
We obtained informed written consent from the family to perform genetic studies and publish photos and clinical and genetic information.
Data Availability Statement
The data will be available upon request. Distribution of sensitive data may be subject to restrictions.
Acknowledgments
We thank the patient and his family for participating to this study. WGS and RNA-seq were provided by the Norwegian High-Throughput Sequencing Centre, supported by the Research Council of Norway and Southeastern Health Authorities. We thank UNINETT Sigma2 for support with high-performance data storage and analysis.
Conflicts of Interest Statement: The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACMG/AMP | American College of Medical Genetics and Genomics/ Association for Molecular Pathology |
| CADD | Combined Annotation Dependent Depletion |
| DROP | Detection of RNA Outliers Pipeline |
| FDR | False discovery rate |
| IGV | Integrative Genomics Viewer |
| Indels | Small insertion/deletion |
| NAA | N-acetyl aspartate |
| NMD | Nonsense-mediated decay |
| RNA-seq | RNA-sequencing |
| SNVs | Single Nucleotide Variants |
| SNX14 | Sorting nexin 14 |
| SCAR20 | Spinocerebellar ataxia, autosomal recessive 20 |
| VUS | Variants of uncertain significance |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
References
- Pandey, R.; Brennan, N. F.; Trachana, K. A meta-analysis of diagnostic yield and clinical utility of genome and exome sequencing in pediatric rare and undiagnosed genetic diseases. Genet Med 2025, 27, pp. 101398. [Google Scholar] [CrossRef]
- Vaz-Drago, R.; Custodio, N.; Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum Genet 2017, 136, 1093–1111. [Google Scholar] [CrossRef]
- Park, E.; Pan, Z.; Zhang, Z. The Expanding Landscape of Alternative Splicing Variation in Human Populations. Am J Hum Genet 2018, 102, 11–26. [Google Scholar] [CrossRef]
- Lord, J.; Baralle, D. Splicing in the Diagnosis of Rare Disease: Advances and Challenges. Front Genet 2021, 12, pp. 689892. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Macakova, K.; Sinson, J. C. Clinical validation of RNA sequencing for Mendelian disorder diagnostics. Am J Hum Genet 2025, 112, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Kernohan, K. D.; Boycott, K. M. The expanding diagnostic toolbox for rare genetic diseases. Nat Rev Genet 2024, 25, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Yepez, V. A.; Mertes, C.; Muller, M. F. Detection of aberrant gene expression events in RNA sequencing data. Nat Protoc 2021, 16, 1276–1296. [Google Scholar] [CrossRef]
- Brechtmann, F.; Mertes, C.; Matuseviciute, A. OUTRIDER: A Statistical Method for Detecting Aberrantly Expressed Genes in RNA Sequencing Data. Am J Hum Genet 2018, 103, 907–917. [Google Scholar] [CrossRef]
- Scheller, I. F.; Lutz, K.; Mertes, C. Improved detection of aberrant splicing with FRASER 2.0 and the intron Jaccard index. Am J Hum Genet 2023, 110, 2056–2067. [Google Scholar] [CrossRef]
- Neethiraj, R.; Wm, J.; Jemt, A. nf-core/raredisease: 2.2.0 - Dogmatix. 2024, pp. [Google Scholar]
- Vasimuddin, M.; Misra, S.; Li, H. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), 2019. [Google Scholar]
- Poplin, R.; Chang, P. C.; Alexander, D. A universal SNP and small-indel variant caller using deep neural networks. Nature Biotechnology 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Yun, T.; Li, H.; Chang, P. C. Accurate, scalable cohort variant calls using DeepVariant and GLnexus. Bioinformatics 2021, 36, 5582–5589. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S. E. The Ensembl Variant Effect Predictor. Genome Biol 2016, 17, pp. 122. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med 2021, 13, pp. 31. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B. S.; Layer, R. M.; Quinlan, A. R. Vcfanno: fast, flexible annotation of genetic variants. Genome Biol 2016, 17, pp. 118. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J. K.; Liddle, J. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, pp. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Francioli, L. C.; Goodrich, J. K. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 2024, 625, 92–100. [Google Scholar] [CrossRef]
- Landrum, M. J.; Lee, J. M.; Benson, M. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016, 44, D862–868. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J. F. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548 e524. [Google Scholar] [CrossRef]
- Vigeland, M. D.; Gjotterud, K. S.; Selmer, K. K. FILTUS: a desktop GUI for fast and efficient detection of disease-causing variants, including a novel autozygosity detector. Bioinformatics 2016, 32, 1592–1594. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J. T.; Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Walker, L. C.; Hoya, M.; Wiggins, G. A. R. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am J Hum Genet 2023, 110, 1046–1067. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Akizu, N.; Cantagrel, V.; Zaki, M. S. Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome-autophagosome dysfunction. Nat Genet 2015, 47, 528–534. [Google Scholar] [CrossRef]
- Jazayeri, R.; Hu, H.; Fattahi, Z. Exome Sequencing and Linkage Analysis Identified Novel Candidate Genes in Recessive Intellectual Disability Associated with Ataxia. Arch Iran Med 2015, 18, 670–682. [Google Scholar]
- Karaca, E.; Harel, T.; Pehlivan, D. Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron 2015, 88, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Maia, N.; Soares, G.; Silva, C. Two Compound Heterozygous Variants in SNX14 Cause Stereotypies and Dystonia in Autosomal Recessive Spinocerebellar Ataxia 20. Front Genet 2020, 11, pp. 1038. [Google Scholar] [CrossRef] [PubMed]
- Raslan, I. R.; Silva, T. Y. T.; Kok, F. Clinical and Genetic Characterization of a Cohort of Brazilian Patients With Congenital Ataxia. Neurol Genet 2024, 10, pp. e200153. [Google Scholar] [CrossRef] [PubMed]
- Sait, H.; Moirangthem, A.; Agrawal, V. Autosomal recessive spinocerebellar ataxia-20 due to a novel SNX14 variant in an Indian girl. Am J Med Genet A 2022, 188, 1909–1914. [Google Scholar] [CrossRef]
- Shao, Y.; Yang, S.; Li, J. Compound heterozygous mutation of the SNX14 gene causes autosomal recessive spinocerebellar ataxia 20. Front Genet 2024, 15, pp. 1379366. [Google Scholar] [CrossRef]
- Shukla, A.; Upadhyai, P.; Shah, J. Autosomal recessive spinocerebellar ataxia 20: Report of a new patient and review of literature. Eur J Med Genet 2017, 60, 118–123. [Google Scholar] [CrossRef]
- Sousa, S. B.; Ramos, F.; Garcia, P. Intellectual disability, coarse face, relative macrocephaly, and cerebellar hypotrophy in two sisters. Am J Med Genet A 2014, 164A, 10–14. [Google Scholar] [CrossRef]
- Thomas, A. C.; Williams, H.; Seto-Salvia, N. Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am J Hum Genet 2014, 95, 611–621. [Google Scholar] [CrossRef]
- Trujillano, D.; Bertoli-Avella, A. M.; Kumar Kandaswamy, K. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet 2017, 25, 176–182. [Google Scholar] [CrossRef]
- Al Shamsi, B.; Al Maimani, A.; Al Hanaie, M. Exploring the Genetic Variations Underlying SNX14-Linked Autosomal Recessive Spinocerebellar Ataxia Type 20: A Case Series of 17 Patients From a Single Center in the Omani Population and Review of Literature. Am J Med Genet A 2025, pp. e70001. [CrossRef] [PubMed]
- Levchenko, O.; Filatova, A.; Mishina, I. Homozygous deep intronic variant in SNX14 cause autosomal recessive Spinocerebellar ataxia 20: a case report. Front Genet 2023, 14, pp. 1197681. [Google Scholar] [CrossRef]
- Zhang, H.; Hong, Y.; Yang, W. SNX14 deficiency-induced defective axonal mitochondrial transport in Purkinje cells underlies cerebellar ataxia and can be reversed by valproate. Natl Sci Rev 2021, 8, pp. nwab024. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.; Liu, Y.; Datta, S. SNX14 mutations affect endoplasmic reticulum-associated neutral lipid metabolism in autosomal recessive spinocerebellar ataxia 20. Hum Mol Genet 2018, 27, 1927–1940. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D. Congenital Disorders of Autophagy: What a Pediatric Neurologist Should Know. Neuropediatrics 2018, 49, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Saffari, A.; Wahlster, L. Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain 2016, 139, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Russell, S. R.; Drack, A. V.; Cideciyan, A. V. Intravitreal antisense oligonucleotide sepofarsen in Leber congenital amaurosis type 10: a phase 1b/2 trial. Nat Med 2022, 28, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, M. C.; van Roon-Mom, W.; Aartsma-Rus, A. Possibilities and limitations of antisense oligonucleotide therapies for the treatment of monogenic disorders. Commun Med (Lond) 2024, 4, pp. 6. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A) Pedigree of the patient’s family. The patient was homozygous for a single nucleotide variant in SNX14: Chr6(GRCh37):g.86257731T>C. (B-C) Photos of the patient at 22 years. (B) Lower extremities showing dystonic posture and talipes equinovarus. (C) The patient presented prominent forehead, deep-set eyes, bushy eyebrows, and thick lips. Alopecia was also evident. (D-E) Brain MRI of the patient at 21 years. (D) Coronal T2-weighted image shows both cerebellar hemispheres (asteristks) and vermis (V) atrophy with bilateral symmetric dentate nucleus hyperintensity (arrows). (E) Sagittal T1-weighted image shows pronounced cerebellar vermis atrophy.
Figure 1.
(A) Pedigree of the patient’s family. The patient was homozygous for a single nucleotide variant in SNX14: Chr6(GRCh37):g.86257731T>C. (B-C) Photos of the patient at 22 years. (B) Lower extremities showing dystonic posture and talipes equinovarus. (C) The patient presented prominent forehead, deep-set eyes, bushy eyebrows, and thick lips. Alopecia was also evident. (D-E) Brain MRI of the patient at 21 years. (D) Coronal T2-weighted image shows both cerebellar hemispheres (asteristks) and vermis (V) atrophy with bilateral symmetric dentate nucleus hyperintensity (arrows). (E) Sagittal T1-weighted image shows pronounced cerebellar vermis atrophy.
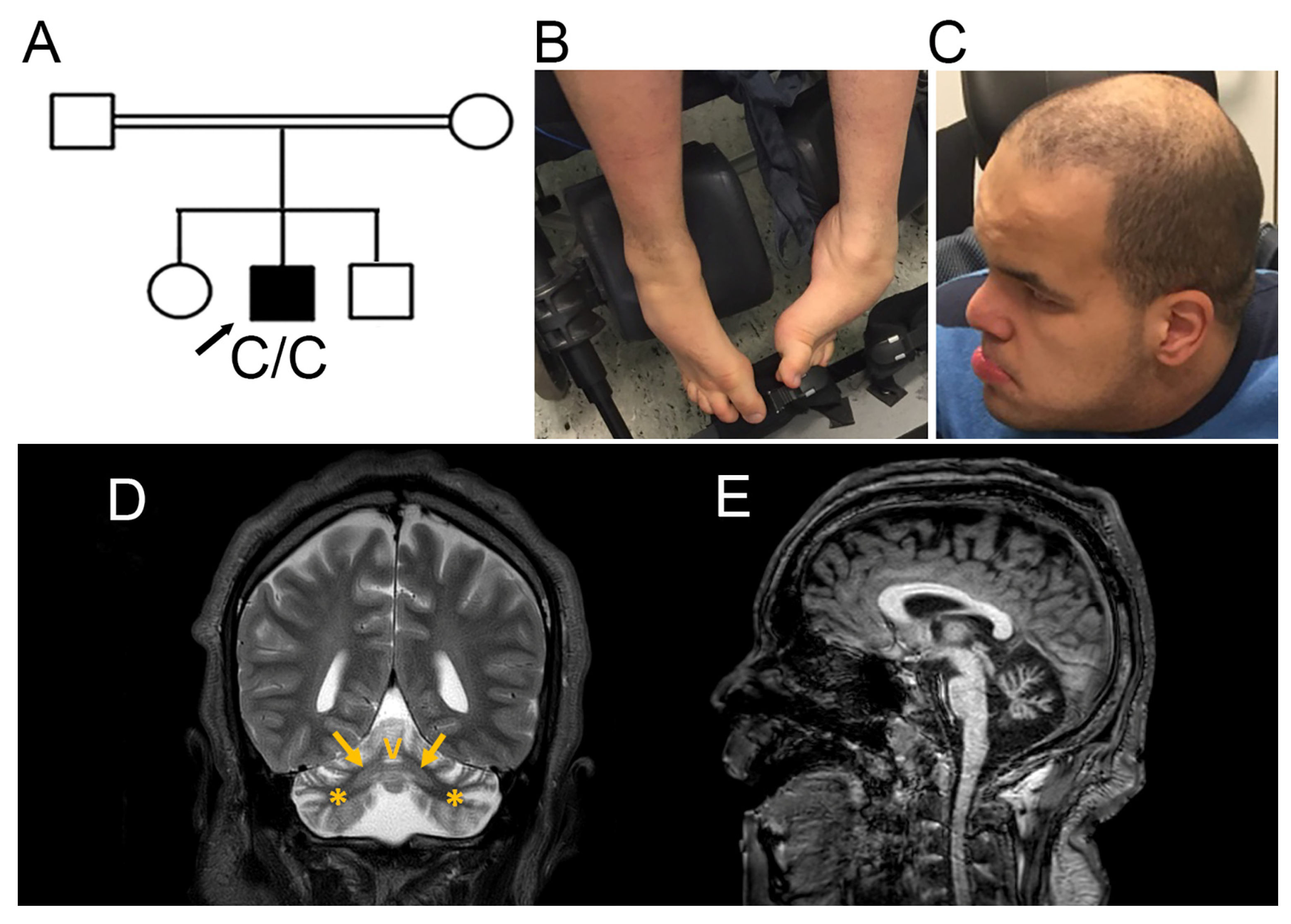
Figure 2.
(A) IGV screenshot of RNA-seq data showing the SNX14 homozygous variant chr6:86257731T>C (red arrow) in the patient (top panel) and pseudo-exon inclusion event at chr6:86257732–86257807 (red box in the middle panel). A representative control sample showinga no transcription of this intronic region (bottom panel). (B) Zoom-in of the intronic region surrounding the homozygous variant. Top panel: WGS of the patient. Middle panel: RNA-seq of the patient showing pseudo-exon inclusion. Bottom panel: RNA-seq of a representative control sample. The red arrow indicates the variant. (C) Visualization of the SpliceAI prediction of the SNX14: chr6:86257731T>C variant (red arrow). The SpliceAI algorithm predicted the intronic variant to create a splice donor site (Δ= 0.99) at chr6:86257732 bp, and an acceptor site (Δ= 0.97) at chr6:86257807 bp. (D) Schematic of the pseudo-exon activation caused by the NM_153816.6(SNX14):c.867+288A>G (red arrow). The variant leads to inclusion of a pseudo-exon (green) within intron 9. Translation of the pseudo-exon introduces a premature stop codon after 12 amino acids (green letters), likely triggering NMD. Exons are shown as blue boxes, introns as yellow lines, and the pseudo-exon is highlighted in green. All genomic positions refer to GRCh37 and are shown in bp.
Figure 2.
(A) IGV screenshot of RNA-seq data showing the SNX14 homozygous variant chr6:86257731T>C (red arrow) in the patient (top panel) and pseudo-exon inclusion event at chr6:86257732–86257807 (red box in the middle panel). A representative control sample showinga no transcription of this intronic region (bottom panel). (B) Zoom-in of the intronic region surrounding the homozygous variant. Top panel: WGS of the patient. Middle panel: RNA-seq of the patient showing pseudo-exon inclusion. Bottom panel: RNA-seq of a representative control sample. The red arrow indicates the variant. (C) Visualization of the SpliceAI prediction of the SNX14: chr6:86257731T>C variant (red arrow). The SpliceAI algorithm predicted the intronic variant to create a splice donor site (Δ= 0.99) at chr6:86257732 bp, and an acceptor site (Δ= 0.97) at chr6:86257807 bp. (D) Schematic of the pseudo-exon activation caused by the NM_153816.6(SNX14):c.867+288A>G (red arrow). The variant leads to inclusion of a pseudo-exon (green) within intron 9. Translation of the pseudo-exon introduces a premature stop codon after 12 amino acids (green letters), likely triggering NMD. Exons are shown as blue boxes, introns as yellow lines, and the pseudo-exon is highlighted in green. All genomic positions refer to GRCh37 and are shown in bp.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



