Submitted:
05 February 2026
Posted:
09 February 2026
You are already at the latest version
Abstract
Background/Objectives:
Tafamidis, a transthyretin kinetic stabilizer, increases circulating transthyretin levels in treated patients. While this effect is well documented, its underlying mechanism remains incompletely understood. This study aimed to evaluate the performance of a Physiologically Based Pharmacokinetic (PBPK) model performance and to calibrate a hypothesis-consistent Quantitative Systems Pharmacology (QSP) model of tafamidis and transthyretin dynamics, to explore mechanistic hypotheses underlying the clinically observed increase in circulating transthyretin and the associated dose–response relationship.
Methods:
A PBPK–QSP model was constructed in Simcyp (V23) using LUA-based modules. The PBPK part was parameterized from literature and validated against data from therapeutic single-dose, therapeutic multiple-dose, and supratherapeutic dose clinical studies. The QSP part of the model describes tafamidis–TTR binding kinetics, stabilization, and clearance of bound complexes. Simulations were performed in thirty virtual healthy male subjects aged 30–40 years, incorporating physiological variability in baseline TTR concentrations.
Results:
Mean predicted versus observed ratios of tafamidis AUC and C_max values were within a 1.3-fold range across validation studies. The integrated model reproduced the clinically reported 33% increase in TTR concentration through a calibrated clearance-scaling factor. It supports the hypothesis that reduced clearance of tafamidis-bound TTR may explain the observed effect without modifying TTR synthesis. Dose-sensitivity simulations indicated that patients with low baseline TTR may achieve adequate stabilization at reduced doses, while those with higher baseline TTR concentration may require higher doses.
Conclusions:
The developed PBPK–QSP model does not only reproduce tafamidis pharmacokinetics and TTR responses but also proposes a plausible mechanistic hypothesis implicating clearance modulation of stabilized TTR as a key driver of the clinical effect. This may potentially support rational dose individualization and inform future experimental research.
Keywords:
transthyretin
; tafamidis
; quantitative systems pharmacology
; physiologically based pharmacokinetics
1. Introduction
Transthyretin amyloidosis (ATTR) is a progressive, life-threatening disorder caused by destabilization of transthyretin (TTR), a homotetrameric protein primarily synthesized in the liver [1,2]. Tetramer dissociation into monomers is the rate-limiting step of amyloid formation. Liberated monomers can misfold, aggregate, and deposit in tissues such as the myocardium and peripheral nerves, giving rise to ATTR cardiomyopathy (ATTR-CM) or polyneuropathy. The disease occurs in hereditary (ATTRv) or wild-type (ATTRwt) forms and is associated with substantial mortality. The introduction of kinetic stabilizers that bind to the thyroxine-binding sites of TTR and inhibit tetramer dissociation, such as tafamidis, has significantly improved outcomes in ATTR cardiomyopathy [3,4,5].
A consistent pharmacodynamic (PD) signature of tafamidis therapy is an increase in circulating TTR concentrations, typically around 33% above baseline in both ATTRv and ATTRwt. Although this rise is clinically reproducible, its mechanistic basis remains unresolved [6,7]. Stabilization reduces tetramer dissociation, but it is unclear whether, under physiological, non–mass-conserved conditions, this effect can fully account for the magnitude of the observed increase [8,9,10]. Therefore, mechanistic understanding of the origin of the TTR concentration rise requires models that incorporate both drug–protein binding and TTR turnover physiology.
Several TTR-focused modeling approaches have been proposed over the past decade. Subunit-exchange kinetic models accurately quantify tetramer dissociation under physiological conditions using in vitro or ex vivo plasma experiments [8]. Minimal turnover models couple synthesis, dissociation, reassociation, and degradation of TTR and show that slowed dissociation alone cannot reproduce the >30% clinical rise in circulating TTR without invoking additional mechanisms, such as altered clearance or changes in monomer disposition [9,10]. Population PK–PD models, most notably Tess et al. (2023), integrate in vitro binding, TTR concentrations, and clinical endpoints from the Transthyretin Amyloidosis Cardiomyopathy Clinical Trial (ATTR-ACT) to link tafamidis exposure with TTR stabilization and disease progression [5,11]. These analyses confirm near-complete stabilization at the approved 80 mg tafamidis meglumine dose and provide insight into the relationship between binding-site occupancy and clinical biomarkers.
Physiologically based pharmacokinetic (PBPK) modeling provides a mechanistic representation of drug disposition across organs and tissues, accounting for absorption, distribution, metabolism, and excretion based on anatomical and physiological principles [12]. Quantitative systems pharmacology (QSP) extends this to molecular and cellular processes underlying drug action (here, ligand–protein binding, tetramer stabilization, monomer generation and loss, and TTR turnover) [13]. An integrated PBPK–QSP model is therefore uniquely suited to translate tafamidis pharmacokinetics into mechanistic predictions of TTR homeostasis, offering the ability to formulate and test biological hypotheses and identify parameters that drive uncertainty.
As mentioned above, the study by Tess et al. established an important link between tafamidis binding and disease progression by modeling TTR concentrations and clinical endpoints as a function of steady-state tafamidis exposure [11]. While valuable, this approach relies on population PK rather than multiorgan PBPK, and thus does not capture tissue-level distribution, dynamic binding competition, or physiological constraints on tafamidis disposition. Additionally, the model proposed by Tess et al. explicitly assumes that monomer degradation greatly exceeds reassociation and fits several kinetic parameters simultaneously to reproduce the ~30% TTR increase observed after treatment initiation.
In contrast, the PBPK–QSP framework proposed in this manuscript incorporates experimentally supported parameters wherever possible, including human in vivo TTR turnover data and in vitro kinetic constraints from subunit exchange assays [8,14,15]. This reflects a key conceptual stance: mechanistic models must respect the origin and quality of parameter estimates, rather than expanding the parameter set to force agreement with a single clinical time point. Any sufficiently flexible model can be tuned to match sparse observations; explanatory value arises predominantly when a model reproduces data based on a physiological description, and not because its degrees of freedom are abundant. Our integration therefore makes explicit which physiological measurements, such as organ-level clearance, monomer internalization, and elimination pathways, remain critical gaps for better mechanistic understanding of tafamidis-modified TTR dynamics.
The objective of this study was to develop and validate an integrated PBPK–QSP model of tafamidis kinetics and TTR turnover. The developed model: (1) captures tafamidis pharmacokinetics across organs and tissues; (2) quantitatively describes drug binding, stabilization, and TTR turnover under physiological, non–mass-conserved conditions; (3) enables mechanistic hypothesis testing to evaluate which biological processes can plausibly account for the clinically observed increase in circulating TTR; and (4) identifies physiological and kinetic parameters that constrain our mechanistic understanding of TTR homeostasis under stabilizer therapy, thereby mapping explicit blind spots, i.e., parameters and processes we know we do not yet know well enough, to guide future experimental and clinical data collection.
2. Materials and Methods
2.1. Clinical Trials Data Used for Model Development and Validation
Clinical pharmacokinetic data from selected tafamidis studies were used to develop and evaluate model performance against observed human exposure across a range of dosing conditions. Study characteristics and data sources are summarized in Table 1.
Model accuracy was assessed using fold error, defined as the ratio of the predicted to the observed pharmacokinetic summary statistic (), and reported for the corresponding mean, median, or geometric mean values, as applicable. To provide a global assessment of model bias and precision, Average Fold Error (AFE) and Absolute Average Fold Error (AAFE) were additionally calculated using fold errors derived from exposure metrics (AUC and ) only, consistent with standard PBPK model evaluation practice. AFE was defined as the geometric mean of the Predicted/Observed ratios, calculated as , while AAFE was defined as the geometric mean of the absolute fold deviations, calculated as . Model performance was interpreted against commonly applied PBPK acceptance criteria, with fold error values within 0.5–2.0 considered acceptable and values within 0.8–1.25 considered indicative of strong predictive performance.
2.2. PBPK Model Development
The general structure of the PBPK model was previously proposed and implemented in Simcyp Simulator (version 23) [20]. The model follows a whole-body, perfusion-limited PBPK framework in which all anatomical compartments are represented as well-stirred, kinetically homogeneous units, assuming instantaneous distribution within each tissue relative to inter-compartmental blood flow. Drug transport between compartments is governed by organ-specific blood flows and tissue-to-plasma partition coefficients.
The PBPK model-simulated plasma concentration of tafamidis is then used as an external, dynamic effect driving variable for the downstream QSP model. The simulated plasma concentration–time profile of tafamidis serves as a dynamic forcing function for the downstream QSP model, with no feedback from the QSP system to the PBPK component.
2.1.1. Drug Physicochemical Properties and Blood Proteins Binding
Tafamidis is a monoprotic acid with a pKa of 3.73, molecular weight of 308.12 g/mol, with the molecular weight of tafamidis meglumine salt being 503.33 g/mol [21]. A logP value (3.91), calculated using the ALOGPS model, was used to parametrize the PBPK model [22]. The plasma unbound fraction of the drug has a value of 0.008 (0.8%). Apart from its affinity for TTR, which is a key element of its mechanism of action, tafamidis also exhibits high affinity for plasma albumin, consistent with interactions between a weakly acidic small molecule and proteins with basic binding domains [23]. Binding to plasma proteins is part of the developed QSP model, as described further in the QSP model development and Results sections.
2.1.2. Dissolution and Absorption
Tafamidis has been reported as having low solubility and variable permeability; however, in vitro-in vivo extrapolation of Caco-2 permeability results suggests that it is actually a well-permeable drug, since its apparent permeability () equals from the apical to basolateral side and from basolateral to apical side [24]. The above-mentioned value was used to estimate effective permeability () using linear scaling, as described by Sun et al. [25].
According to regulatory data, tafamidis meglumine solubility at pH 6.8 buffer is 3.121 . It is also known that in an alkaline environment (0.1 M NaOH), solubility is higher than 4.19 , while solubility in water is higher than 4.63 [26]. The presence of meglumine, an alkalising agent, explains the higher solubility in pure water compared to the pH 6.8 buffer. In the developed PBPK model, tafamidis meglumine solubility was described as a pH-dependent parameter, incorporating the adjusted Henderson-Hasselbalch equation for prediction, based on the measured value at pH 6.8 [27]. Predicted intrinsic solubility, So = 0.0025 mg/mL, seems reasonable considering low solubility in low pH. The predicted pH dependent solubility is presented in Figure S1. The commercially available tafamidis meglumine is manufactured as gelatine-encapsulated powder; thus, it is assumed that it is released rapidly once it enters the duodenum, allowing for the immediate start of the dissolution process and absorption. For description of the absorption process, the ADAM (Advanced Dissolution, Absorption, and Metabolism) model was used, with the stomach serving as the dose-accepting compartment and upstream driver of intestinal absorption. Fed and fasted conditions were handled implicitly within the Simcyp ADAM framework by applying condition-specific gastrointestinal physiology (including gastric emptying time, luminal fluid volumes, bile salt concentrations, and intestinal transit), as implemented in the Simcyp Healthy Volunteer population. The above-reported Caco-2 data were used to estimate as [28]. It was assumed that permeability is the same for each of the seven intestinal subcompartments, whereas local pH, fluid dynamics, GIT transit time follow inter-individual variability as implemented in the Simcyp Healthy Volunteer population [20]. Additionally, the gastric emptying time varies between virtual individuals [29].
2.1.3. Volume of Distribution and Tissue-to-Plasma Partitioning
The apparent volume of distribution after oral administration of tafamidis meglumine to healthy volunteers was reported as 16 L (0.2 L/kg for an 80 kg patient) [30]. For a full PBPK distribution model, the volume of distribution at steady state (), accounting for distribution in the following tissues: adipose, bone, brain, gut, pancreas, heart, kidney, liver, lung, muscle, skin, spleen, was calculated following the Sawada equation [31].
Partition coefficients between each tissue and plasma () – except for bone, pancreas, muscle, and skin – were obtained from rat PK study on tafamidis distribution, following the assumption that drugs are distributed in rats similarly to humans [24,32]. For prediction of for bone, pancreas, muscle, and skin, the Rodgers and Rowland model was incorporated as described previously [33]. The model estimated as 13.53 L for an 80 kg patient (0.17 L/kg).
2.1.4. Metabolism and Excretion
Tafamidis is metabolized primarily by UGT-mediated glucuronidation, predominantly by UGT1A1, UGT1A3, and UGT1A9 [34]. Unfortunately, no intrinsic clearance values or contributions of each of the enzymes can be found in the publicly available literature; thus, metabolism was described with a holistic parameter of oral clearance – 0.263 L/h – as stated in drug prescribing information [35]. Therefore, the model is not suitable for populations with altered UGT abundance (i.e. certain diseases including liver cirrhosis) or for assessing drug–drug interactions, as it lacks the mechanistic scalability offered by intrinsic clearance data. Based on documentation submitted to the FDA, a large fraction of pharmacologically inactive tafamidis metabolite is expected to be excreted with bile; the proposed model does not account for this route of elimination as there is not enough data to mechanistically describe such a process.
2.1.5. Enzyme- and Transporter-Mediated Drug–Drug Interactions
In vitro data presented in the regulatory documents do not exclude inhibition of intestinal UGT1A1 and induction of CYP2B6 and CYP3A4. It has been suggested that tafamidis might also inhibit OAT1, OAT3, and BCRP transporters [34]. Nevertheless, interaction with enzymes and transporters is not relevant for this model as drug-drug interactions (DDI) are not in scope of this study.
2.2. QSP Model Development
Tafamidis drug action was described using a basic ligand–protein binding model assuming two binding sites on the TTR molecule. The model assumes that after tafamidis binding, TTR undergoes conformational changes as previously observed [36,37]. Since structural modifications might alter molecular properties, the bound protein is hypothesized to exhibit distinct attributes compared to its unbound form, making it less susceptible to clearance or more susceptible to undergo tissue internalization [6,37,38]. In our analysis, we assumed that a kinetically stabilised TTR molecule does not dissociate to monomers at all. It is known that the dissociation rate of total TTR in plasma decreases as tafamidis concentration increases; however, it is not possible to specify quantitatively how various bound states of TTR contribute to this change [8]. The total TTR concentration remains at a dynamic steady-state level governed by its synthesis and degradation processes. These processes are quantified by rates and can be described using a zero-order synthesis and first-order degradation turnover framework as previously reported [9]. Under this formulation, total TTR dynamics can be expressed as:
yielding a steady-state concentration of:
Consequently, any change in the steady-state concentration of circulating TTR must arise from a change in the ratio between its synthesis and elimination rates. Let denote the baseline steady-state TTR concentration prior to tafamidis administration, and the steady-state concentration after drug saturation. An observed 33% increase corresponds to:
When monomer elimination is negligible, the steady-state tetramer concentration is governed by the balance between synthesis and degradation. In this framework, proportional scaling of either the synthesis rate or the degradation rate leads to mathematically identical steady-state solutions. Consequently, steady-state data alone cannot distinguish whether the observed increase in circulating TTR arises from enhanced synthesis or reduced elimination; these mechanisms bring structural non-identifiability of the model. This structural ambiguity is not specific to the present PBPK-QSP framework but reflects a general limitation of turnover models constrained solely by steady-state observations, as discussed previously in a phenomenological context [10].
To reproduce the observed population-average increase while keeping synthesis unchanged, we introduced a scaling factor on the effective degradation rate such that:
yielding the solution where .
Tafamidis binds directly to circulating TTR tetramers and induces stabilizing conformational changes, which in the model are assumed to alter internalization or clearance pathways of the protein; this assumption is supported by the high fraction of circulating TTR present in the drug-bound state.. In contrast, increased hepatic TTR synthesis is unlikely to underlie the observed rise in circulating TTR, as in vitro evidence supports post-translational modulation of secretion at the site of synthesis, requiring intracellular (endoplasmic reticulum) access; however, these findings do not necessarily translate quantitatively to the in vivo setting [39]. Accordingly, the QSP model is calibrated to a known population-level steady-state effect and is not intended to provide predictive discrimination between synthesis- and clearance-based mechanisms.
The main differences between the current and previously presented minimalistic QSP model include the application of two stable bound states of transthyretin and the introduction of a drug concentration variable as an input from the above-described PBPK model [9]. The previously presented minimalistic model was developed to provide a conceptual and mathematical framework for understanding TTR dynamics in vivo, with particular emphasis on disentangling the roles of tetramer stabilization, synthesis, and elimination. The model describes TTR as an open system with production, reversible tetramer–monomer interconversion, and irreversible elimination from the circulation. Under its core assumptions—rapid monomer–tetramer reassociation relative to monomer degradation—the model demonstrates analytically that changes in tetramer dissociation rate alone do not alter steady-state TTR concentration. Instead, clinically observed increases in circulating TTR can only be reproduced by concurrent changes in TTR production and/or elimination rates. Thus, the purpose of the minimal model development was mechanistic understanding to rationalize clinical observations and identify which physiological processes must be affected by tetramer stabilizers to explain increased serum TTR levels. The current work builds upon this framework by extending it to a mechanistic, state-resolved QSP model that explicitly represents multiple drug-bound TTR states, albumin binding, and dynamic tafamidis exposure provided by a PBPK model, thereby enabling quantitative simulation and prediction of clinically observed TTR responses. The general structure of the proposed model can be seen in Figure 1.
The analysis of drug effect relies on precise description of total TTR concentration () in plasma, as it is the only surrogate of drug action that can be quantitatively tracked. To characterize the sensitivity of model-predicted TTR responses to tafamidis dose, a dose-sensitivity analysis was performed under steady-state conditions by systematically varying the administered tafamidis meglumine dose around the recommended clinical regimen (4 × 20 mg, corresponding to 48.8 mg tafamidis). Simulations were conducted for fractional and multiple doses equal to twofold, three-quarters, one-half, and one-quarter of the standard dose, selected to span a clinically relevant and practically divisible range. For each dose level, steady-state values of total, bound, free, and monomeric TTR species were extracted, with particular emphasis on the unbound-to-total TTR ratio as an integrative, model-derived quantity summarizing dose-dependent changes in TTR binding at the population level.
We suggested a mechanism combining TTR degradation rate modification with molecular-level TTR binding. However, it should be noted that in some cases, steady-state TTR levels may increase through stabilization alone, without affecting degradation, although such increases are typically modest, not exceeding, on average, 15% [10].
The full model code, in the form of a notebook, together with parameter descriptions, can be found in the Supplementary Materials. We provide results for models with two different association and dissociation constants, coming from separate experiments, described in two articles; the code can be easily adjusted to switch from one set of parameters to another [37,40]. Results of the simulations are available as Excel files attached as the Supplementary Materials.
2.2.1. QSP Model Parametrisation
While a static model would be sufficient for monitoring steady-state concentrations, incorporation of dynamics enhances the model. It allows tracking delays in effect and accounting for time-dependent variations in individual drug concentrations. A total of six ordinary differential equations (ODEs) were used in the model (see equations below). The QSP model parameters are categorised into two groups: drug-specific parameters and system (patient)-specific parameters. The latter, namely system parameters, were provided as individual values for virtual individuals as detailed in a subsequent section. After analysis of the binding affinities, two sets of association and dissociation rate constants were applied: one derived from the equilibrium binding constant reported in Nelson et al. and the other one directly adopted from Corazza et al. [37,40]. To account for the observed TTR dynamics, a fixed scaling factor was introduced to describe changes in the clearance of the drug–protein complex. Specifically, the model was calibrated using scaling factors of 0.75 for TTR1 and TTR2 clearances, which allows the model to predict an average increase of 33% in TTR plasma concentration in healthy male volunteers, as previously reported in the literature [6].
Identifiability of the synthesis and clearance scaling factors was evaluated using a local Fisher Information Matrix (FIM)–based analysis with a steady-state pseudo-observation reflecting the reported 33% increase in total TTR concentration. Results of the analysis demonstrated that synthesis and clearance scaling factors cannot be estimated independently using available data, exhibiting strong correlation and a rank-deficient FIM. Consequently, the attribution of the observed steady-state increase to reduced effective clearance represents a mechanistically motivated modeling assumption rather than a statistically identifiable fact. Full details of the identifiability analysis are provided in the Appendix (R scripts).
Generative AI support (GPT-5.2 large language model, OpenAI; accessed January 2026) was used in a limited technical-assistance role during development of the Fisher Information Matrix identifiability workflow. Specifically, the model was used to help for- malize the computational procedure for local FIM construction, including symbolic-to- numeric mapping of model parameters, structuring of finite-difference sensitivity calcu- lations, and implementation logic for the steady-state pseudo-observation approach in R. The AI tool did not generate primary scientific results, select model assumptions, or inter- pret identifiability outcomes; all equations, parameterizations, scripts, and conclusions were defined, verified, and validated by the authors.
All parameters used for building the QSP model are presented in the Supplementary Material (see Table A2).
2.2.2. Equations Describing Tafamidis-TTR Kinetics
The concentration of single tafamidis-TTR complex increases when free transthyretin () binds to a tafamidis molecule, initiating stabilization of TTR. The single tafamidis-TTR complex also rises when the double tafamidis-TTR complex dissociates, releasing one tafamidis molecule and reverting to the single-bound form. Conversely, the single tafamidis-TTR complex concentration decreases through three mechanisms: irreversible degradation of the complex, dissociation of tafamidis from TTR into their free forms, and the binding of an additional tafamidis molecule to the single complex, forming the double tafamidis-TTR complex.
In Equation (5), kon1 represents the primary association rate of tafamidis with TTR (), while denotes the primary dissociation rate of tafamidis from TTR (). The formation of the double complex is governed by , the secondary association rate of tafamidis with the single-bound complex (), and its dissociation is described by (). The irreversible elimination is the same for single- and double-bound complexes, assigned as (), which is scalar adjusted via .
The concentration of the double tafamidis-TTR complex increases when a second tafamidis molecule binds to the single tafamidis-TTR complex, however without accounting for enhanced resistance to dissociation. Conversely, this concentration decreases through two primary mechanisms: irreversible degradation of the double complex and dissociation of one tafamidis molecule, which converts the double complex back into the single-bound form.
In Equation (6), represents the secondary association rate of tafamidis with the single-bound TTR complex (), while denotes the secondary dissociation rate of Tafamidis from the double complex ().
The concentration of free TTR increases through its natural synthesis in the body and the dissociation of tafamidis from the single tafamidis-TTR complex, releasing TTR back into its free form, as well as due to reassociation of monomers back to tetramers with rate ka (). Conversely, free TTR is reduced by three key processes: natural degradation of TTR, dissociation into monomers, which reflects destabilization of its native tetrameric structure and increases the risk of aggregation, and binding with tafamidis to form the single tafamidis-TTR complex.
In Equation (7), represents the rate of TTR synthesis (), while denotes the natural degradation rate of TTR (). The dissociation of TTR into monomers is described by (), and the binding of free TTR with tafamidis to form the single complex is governed by (). The reverse process, where tafamidis dissociates from the single complex, is described by ().
The concentration of monomers increases when free TTR dissociates from its tetrameric form, reflecting destabilization of the native structure. This dissociation is a critical step in the pathogenesis of amyloid diseases, as monomers are prone to misfolding and aggregation into amyloid fibrils. Conversely, the monomer concentration decreases when monomers re-associate to form stable tetramers, restoring TTR’s functional structure, undergo irreversible degradation, or clump to aggregates. For the proposed model the last two are indistinguishable, thus they are merged within single parameter.
In Equation (8), represents the dissociation rate of tetrameric TTR into monomers (), while denotes the association rate of unbound TTR monomers to reform tetramers (). The irreversible degradation of monomeric TTR is defined by (). Since no data exist on the concentration of dissociated TTR monomers in plasma, we assumed it results from the steady-state balance of TTR and its dissociation and association constants as presented in the Appendix A (Table A2, Equation (A1)).
Tafamidis can bind to albumin in the plasma, forming a tafamidis-albumin complex, thus reducing the availability of free tafamidis for transthyretin stabilisation. This interaction serves as a reservoir, modulating the pharmacokinetics of tafamidis, mainly by keeping the fraction unbound at a stable level. Conversely, the number of tafamidis-albumin complexes decreases when tafamidis dissociates from albumin, releasing free tafamidis back into the blood.
In Equation (9), represents the association rate of tafamidis with albumin (), while denotes the dissociation rate of tafamidis from the tafamidis-albumin complex ().
Albumin is present in abundance; however, it is necessary to track its concentration to capture all drug interactions. We assume that there is only one binding site of tafamidis to albumin and that only unbound albumin described with Equation 10 is available for binding [11].
The concentration of free tafamidis in plasma is determined by the total administered dose, excluding the amount bound to protein complexes. We assume that only the unbound drug is available to interact with TTR. Total concentration () mentioned in Equation 11 represents the total tafamidis concentration treated as the merging variable for the PBPK and QSP model. All concentrations are expressed in .
2.3. Population-Level Sampling of TTR Concentrations
To conduct simulations in a virtual population, baseline TTR concentrations were generated for thirty virtual healthy male subjects aged 30–40 years, using summary statistics reported by Ingenbleek et al. for age- and sex-stratified groups [41]. This age range was selected deliberately for methodological consistency. First, the only publicly available multiple-dose tafamidis pharmacokinetic data approaching steady state were generated in healthy male volunteers within this age range and were used to evaluate the PBPK component of the model [19]. Second, the most frequently cited human estimates of transthyretin elimination kinetics were derived from in vivo radiolabeled protein kinetic studies conducted in healthy males aged 30–40 years, as reported by Oppenheimer et al. and Socolow et al. [14,15]. These studies reported first-order degradation rate constants of approximately 0.363 day⁻¹ (Oppenheimer et al.) and 0.268 day⁻¹ (Socolow et al.). Ingenbleek et al. reported that baseline TTR concentrations within each age–sex stratum are approximately normally distributed [41]. Individual baseline TTR values were sampled randomly from a normal distribution parameterized using the reported mean and standard deviation for this group. The number of virtual subjects (n = 30) was selected to match the size of the multiple-dose clinical pharmacokinetic study used to evaluate the PBPK component, thereby aligning simulated between-subject variability with the available observed study cohort [19]. One baseline value was assigned per virtual subject and held constant across simulations, thereby preserving the reported between-subject variability in baseline TTR concentrations.
Female subjects were not simulated in the present work. Although baseline TTR distributions by sex are available, the data required to parameterize and validate sex-specific tafamidis exposure under a multiple dosing regime and to quantitatively support transferability of the observed TTR increase across sexes are limited [41]. To avoid introducing additional unverifiable assumptions, the present analysis was restricted to males, and generalization of the results to female populations is acknowledged as a limitation.
Initial values for optimisation were derived from Oppenheimer et al., as this study reports turnover estimates in a demographic group matching the selected virtual population [14]. Parameter fitting and population sampling were implemented using the R programming language. Full optimisation results, sampling scripts, and associated code are provided in the Supplementary Material.
3. Results
The developed PBPK-QSP model effectively describes the overall drug effect on TTR and successfully replicates tafamidis pharmacokinetic outcomes observed in multiple-dose studies conducted in a healthy population. The comparison of observed and predicted PK parameters (AUC, , ) is shown in Table 2.
The pharmacokinetic model’s performance in predicting tafamidis exposures after multiple-dose therapy in a 30–40-year-old male population is presented in Figure 2D. Moreover, simulated tafamidis plasma concentration–time profiles following a single supratherapeutic 400 mg dose were consistent with observed clinical data in a cohort of 42 healthy individuals (Figure 2A). The model also successfully captured the pharmacokinetics of single-dose administration in 16 healthy volunteers enrolled in trial B3461054, demonstrating robust predictive performance under both fasted and fed conditions (Figure 2B and 2C) [17]. Across all evaluated dosing regimens and nutritional conditions, predicted exposure metrics (AUC and ) showed good agreement with observations, with individual fold errors ranging from 0.74 to 1.12. Based on geometric mean AUC and Cmax values, the overall AFE was 0.93, indicating a slight underprediction, while the AAFE was 1.13, corresponding to a typical prediction error of approximately 13%.
Figure 3 illustrates the distribution of bound tafamidis across the single- and double-bound states of TTR, as well as its interaction with albumin, which acts as a buffering pool for circulating tafamidis and modulates its availability for TTR binding. Comparison of results obtained using the Nelson-based and Corazza-based binding parameter sets shows that the former yields a higher proportion of single-bound TTR–tafamidis complexes, whereas the latter shifts the distribution toward a higher abundance of double-bound complexes (Figure 3 and Supplementary Figure A2).
The steady-state unbound-to-total TTR ratio exhibited a clear dependence on tafamidis dose across the evaluated range. Relative to the recommended tafamidis meglumine regimen (4 × 20 mg; 48.8 mg tafamidis), stepwise dose reductions to three-quarters, one-half, and one-quarter of the standard dose were associated with progressively higher unbound-to-total TTR ratios, whereas a twofold dose increase resulted in a further reduction of the unbound fraction. The predicted dose–response relationship for the unbound-to-total TTR ratio is shown in Figure 4. Dose-dependent changes in total, bound, free, and monomeric TTR species are presented in the Supplementary Materials (Figure A3, Figure A4, Figure A5 and Figure A6).
4. Discussion
Tafamidis has been approved for the treatment of transthyretin amyloid cardiomyopathy based on demonstrated clinical benefit, including improved survival and reduced cardiovascular-related hospitalizations, together with strong experimental evidence showing its ability to kinetically stabilize transthyretin tetramers in vitro and ex vivo. The present modeling work does not aim to reproduce or predict clinical outcomes. Instead, it builds on the established mechanism of action of tafamidis—namely, stabilization of the transthyretin structure.
The presented PBPK-QSP model successfully replicates tafamidis pharmacokinetics in a healthy population, supporting its applicability for dose selection and clinical trial optimization. Tafamidis is a long-term therapy and accordingly the integrated PBPK–QSP model simulation was run over a sufficiently long time horizon to reach pharmacodynamic steady state. It should be noted that pivotal clinical evidence for tafamidis efficacy derives predominantly from elderly ATTR patients, often with multiple comorbidities. Age- and disease-related physiological changes may affect both tafamidis pharmacokinetics and TTR turnover; therefore, extrapolation of simulation results from young healthy males to the clinical ATTR population should be done cautiously. In the absence of age-matched longitudinal data suitable for parameterization and validation within this framework, the reported average ~33% increase in total TTR concentration was used as a calibration target, and this assumption is explicitly treated as a limitation of the model. Moreover, the model accurately predicts tafamidis plasma concentrations following a single supratherapeutic 400 mg dose (Figure 2A), supporting its applicability for dose selection.
On the PD side, the model captures trends in average TTR concentration changes; however, its predictive performance cannot currently be evaluated at the individual level. This is because the model was developed based on the data derived from population-level analysis. If the individual TTR trajectories were available, the model’s predictive accuracy for patient-specific responses could be assessed and further refined. The variability in TTR concentration appears to depend on the baseline protein levels and overall patient health, but the precise contributing factors remain unidentified. Addressing this limitation could potentially enhance the model’s predictive accuracy and increase its usefulness towards personalized medicine. Our analysis indicates that patients with low baseline TTR levels may achieve adequate stabilization at lower doses. For instance, a low-TTR patient (3.85 ) dosed with 60 mg tafamidis meglumine reached steady-state fractional stabilization of 2.55%, comparable to the population average of 2.19%. In contrast, a high-TTR patient (7.51 ) required 80 mg daily to reach 3.86% stabilization, a value close to that achieved by the average patient on 60 mg (4.59%). The dose levels in this analysis were chosen deliberately: while the standard regimen involves four 20 mg tablets, reducing the dose to three tablets in patients with low TTR could lower treatment costs without compromising therapeutic effect. Conversely, patients with high baseline TTR may require higher doses to achieve optimal stabilization. These simulations suggest that baseline TTR concentration may influence the exposure–stabilization relationship; however, any dose individualization based on baseline TTR would require prospective clinical validation and should be regarded as hypothesis-generating.
Dose sensitivity analysis has shown that there is no clear difference in the binding effect across the tested dose range (Figures S3–S6). However, the simulations suggest that baseline TTR may influence the exposure–stabilization relationship in extreme cases; this observation is hypothesis-generating and would require prospective clinical validation before informing dosing decisions. Patients with lower TTR levels could potentially decrease the dose or extend dosing gaps. A critical aspect of the model’s implementation is the necessity of applying a fixed scaling factor to describe changes in drug-protein complex clearance. Due to the lack of sufficient experimental data, this heuristic adjustment was required to align predictions with observed clinical data. Similarly, the rate of monomer degradation was set to a constant value in the absence of experimental data on its actual magnitude. This choice is justified by the fact that, under physiological conditions, the product of is expected to be substantially lower than , given that monomer concentrations are below 1% of tetramer concentrations (as reported by Sekijima et al. 2001) [42]. Consequently, variations in this parameter are unlikely to dominate system dynamics; however, this assumption should be revisited if future data indicate otherwise.
A key methodological point is that, in an open turnover system such as circulating transthyretin, a clinically observed steady-state increase in total TTR can generally be reproduced by multiple mechanistic hypotheses that act on different parts of the turnover balance. In particular, reduced tetramer dissociation, altered effective clearance/internalisation of stabilized species, and modulation of net production can lead to similar steady-state outcomes, rendering the underlying mechanism structurally ambiguous when only steady-state endpoints are available [10]. In this light, the present PBPK–QSP framework is intended not as a definitive mechanistic proof, but as a quantitative scaffold that preserves mechanistic structure where parameters are experimentally constrained (tafamidis exposure, binding stoichiometry and kinetics), while representing unresolved biology through a minimal number of calibrated terms. This modeling stance allows the integration of pharmacokinetic information with TTR turnover physiology and provides a principled way to constrain the space of plausible mechanisms consistent with observed PK and population-average TTR responses, thereby guiding which future measurements would be most informative for discriminating between competing hypotheses.
The proposed model does not uniquely identify the biological mechanism underlying the observed increase in TTR concentrations, but it constrains which turnover and clearance hypotheses are consistent with the available steady-state and pharmacokinetic data. Future research should focus on elucidating whether this phenomenon is driven by modified clearance, internalization of bound complexes, or potential effects of tafamidis on TTR synthesis rates. Additional studies are also needed to clarify albumin’s competitive role in tafamidis binding and its impact on pharmacokinetics. While the PBPK-QSP model demonstrates strong predictive capabilities, further refinements and validation studies are necessary to fully capture the complexities of tafamidis-TTR interplay. We acknowledge that the model relies on several assumptions without direct experimental validation, and further research is required to confirm its mechanistic foundations.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. PBPK-QSP results, QSP Lua code, Population sampling R script with necessary data, Figures S1–S6; Table S1; Equation (S1).
Author Contributions
Conceptualization, Seweryn Ulaszek. and Bartek Lisowski.; methodology, Seweryn Ulaszek.; software, Sebastian Polak.; validation, Bartek Lisowski, Seweryn Ulaszek.; investigation, Barbara Wiśniowska; resources, Sebastian Polak.; data curation, Seweryn Ulaszek.; writing—original draft preparation, Seweryn Ulaszek.; writing—review and editing, Sebastian Polak, Bartek Lisowski, Barbara Wiśniowska.; visualization, Seweryn Ulaszek and Sebastian Polak.; supervision, Sebastian Polak. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the European Union within the VITAL Horizon Europe project (Grant nr 101136728). The authors declared no competing interests for this work.
Acknowledgments
During the preparation of this manuscript, the authors used the GPT-5.2 large language model (OpenAI; accessed January 2026) for technical assistance in structuring and implementing the Fisher Information Matrix-based identifiability analysis workflow, including formulation of the computational procedure and R implementation logic. The authors reviewed and edited all generated material and take full responsibility for the content of this publication.
Conflicts of Interest
Author Sebastian Polak is affiliated with Certara (Certara Predictive Technologies). The company was not involved in the design of the study, data collection, analysis, model development, interpretation of results, manuscript preparation, or the decision to publish. No financial support or other direct influence from the company was received for this work. The remaining authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AAFE | Absolute Average Fold Error |
| ADAM | Advanced Dissolution, Absorption, and Metabolism |
| AFE | Average Fold Error |
| AUC | Area Under the Curve |
| ATTR | Transthyretin Amyloidosis |
| BCRP | Breast Cancer Resistance Protein |
| BCS | Biopharmaceutics Classification System |
| Cmax | Maximum Plasma Concentration |
| FDA | Food and Drug Administration |
| GIT | Gastrointestinal Tract |
| ka | Association Rate Constant |
| kdiss | Dissociation Rate Constant |
| kon | Association Rate Constant |
| koff | Dissociation Rate Constant |
| Mdeg | Monomer Degradation Rate Constant |
| PBPK | Physiologically Based Pharmacokinetics |
| PD | Pharmacodynamics |
| Peff | Effective Permeability |
| Papp | Apparent Permeability |
| PK | Pharmacokinetics |
| QSP | Quantitative Systems Pharmacology |
| SD | Standard Deviation |
| So | Intrinsic Solubility |
| Tdeg | TTR Degradation Rate Constant |
| Tmax | Time to Maximum Concentration |
| TTR | Transthyretin |
| UGT | Uridine 5′-Diphospho-Glucuronosyltransferase |
| Vss | Volume of Distribution at Steady State |
Appendix A

Table A1.
Drug input parameters for PBPK model of tafamidis meglumine.
| Parameter | Value | Origin of data | Reference |
|---|---|---|---|
| Physicochemical properties and blood binding | |||
| Type of compound | Acid | Based on structure | - |
| MW, free acid (g/mol) | 308.12 | Based on structure | - |
| LogP | 3.91 | Predicted with ALOGPS | [43] |
| pKa | 3.7 | - | [21] |
| PSA (Å2) | 63.3 | Predicted | - |
| Blood and plasma binding | |||
| Blood to plasma ratio (B:P) | 0.55 | Methodological details unknown. Derived from regulatory documents. | [18] |
| Fraction unbound in plasma (fup) | 0.008 | Experimental | [18] |
|
Absorption (solubility and permeability) Advanced Dissolution, Absorption and Metabolism model (ADAM) | |||
| fugut | 0.67393 | Predicted | [33] |
| Caco-2 apparent permeability (Papp) (10-6 cm/s) | 30.9 | Experimental | [24] |
| Metoprolol permeability as a reference for Caco-2 experiment (Papp) (10-6 cm/s) | 15.5 | Experimental | [24] |
| Effective permeability in human intestine (10-4 cm/s) | 6.9706 | Predicted | [25] |
| Solubility at pH 6.8 (mg/mL) | 3.121 | Experimental | [26] |
| Intrinsic Solubility S0 (mg/mL) | 0.0024771 | Predicted with Henderson-Hasselbach equation using solubility at pH 6.8 as an input. | [44] |
| Distribution | |||
| Volume of distribution at steady state | 0.16909 L/kg | Predicted with Simcyp population data, B:P and tissue-to-plasma partition coefficients. | [31,45] |
| Tissue to plasma partition coefficients (Kp) | |||
| Adipose | 0.027 | Obtained from rats. Rats distribution of small molecules is expected to be similar to that of humans. | [24] |
| Brain | 0.022 | ||
| Gut | 0.254 | ||
| Heart | 0.113 | ||
| Kidney | 0.59 | ||
| Liver | 4.01 | ||
| Lung | 0.107 | ||
| Spleen | 0.063 | ||
| Bone | 0.10203 | Predicted using the Rodgers and Rowland method. | [45] |
| Pancreas | 0.06316 | ||
| Muscle | 0.036922 | ||
| Skin | 0.28047 | ||
| Additional Organ | 0.039244 | ||
| Elimination | |||
| Described as a total clearance = 0.262 L/hr. Tafamidis is eliminated mainly through glucuronidation in the liver, unfortunately there is not enough experimental data shared for bottom-up clearance prediction. | |||
Table A2.
Parameters used for building QSP model.
| Parameter Name | Value | Unit | Source |
|---|---|---|---|
| TTR Synthesis rate | Patient-specific | Based on Ingenbleek et al. dataset, as explained in “Population-level sampling of TTR concentrations” section [9,14,41]. | |
| TTR degradation rate | Patient-specific | Based on Ingenbleek et al. dataset, as explained in “Population-level sampling of TTR concentrations” section [9,14,41]. | |
| Dissociation rate of unbound TTR | 0.0024 | [8] | |
| Association rate of unbound TTR | 360000 | [46] | |
| Monomer degradation rate | 0.016 | There is no data on this parameter available in the literature. We assume that for healthy volunteers it is the same as average degradation rate of TTR [14]. | |
| Association rate constant for first tafamidis-TTR binding | 16200 | [37] | |
| Dissociation rate constant for first tafamidis-TTR binding | 50.2 OR 28.8 |
Two alternative values presented: one fitted using KD from Nelson et al. (2020) combined with kon from Corazza et al. (2019); the other directly using koff from Corazza et al. (2019) [37,40]. | |
| Association rate constant for second tafamidis-TTR binding | 12600 | [37] | |
| Dissociation rate constant for second tafamidis-TTR binding | 2998.8 OR 216 |
Two alternative values presented: one fitted using KD from Nelson et al. (2020) combined with kon from Corazza et al. (2019); the other directly using koff from Corazza et al. (2019) [37,40]. | |
| Association rate constant for tafamidis-albumin binding | 1000 | [40]. | |
| Dissociation rate constant for tafamidis-albumin binding | 1800 | Exact value unknown. Determined by simultaneous fitting with the albumin-tafamidis complex association rate to achieve KD = kdiss/ka [40]. | |
| Individual albumin concentration | Patient-specific | Albumin concentration calculated using a baseline (50.34 g/L) adjusted for age (coefficient = -0.0575) and BMI (coefficient = -0.0738), with an inter-individual coefficient of variation (CV) of 10%. | |
| Irreversible elimination rate of bound TTR-tafamidis complexes | Tdeg * 0.75 | Analytically solved for ~ 33% increase of total TTR concentration |
Equation (A1) defines baseline steady state of monomer concentration. Where is and are dissociation and association rates constants of TTR, is TTR synthesis rate and γ is degradation rate.
Figure A1.
Predicted pH dependent solubility profile.
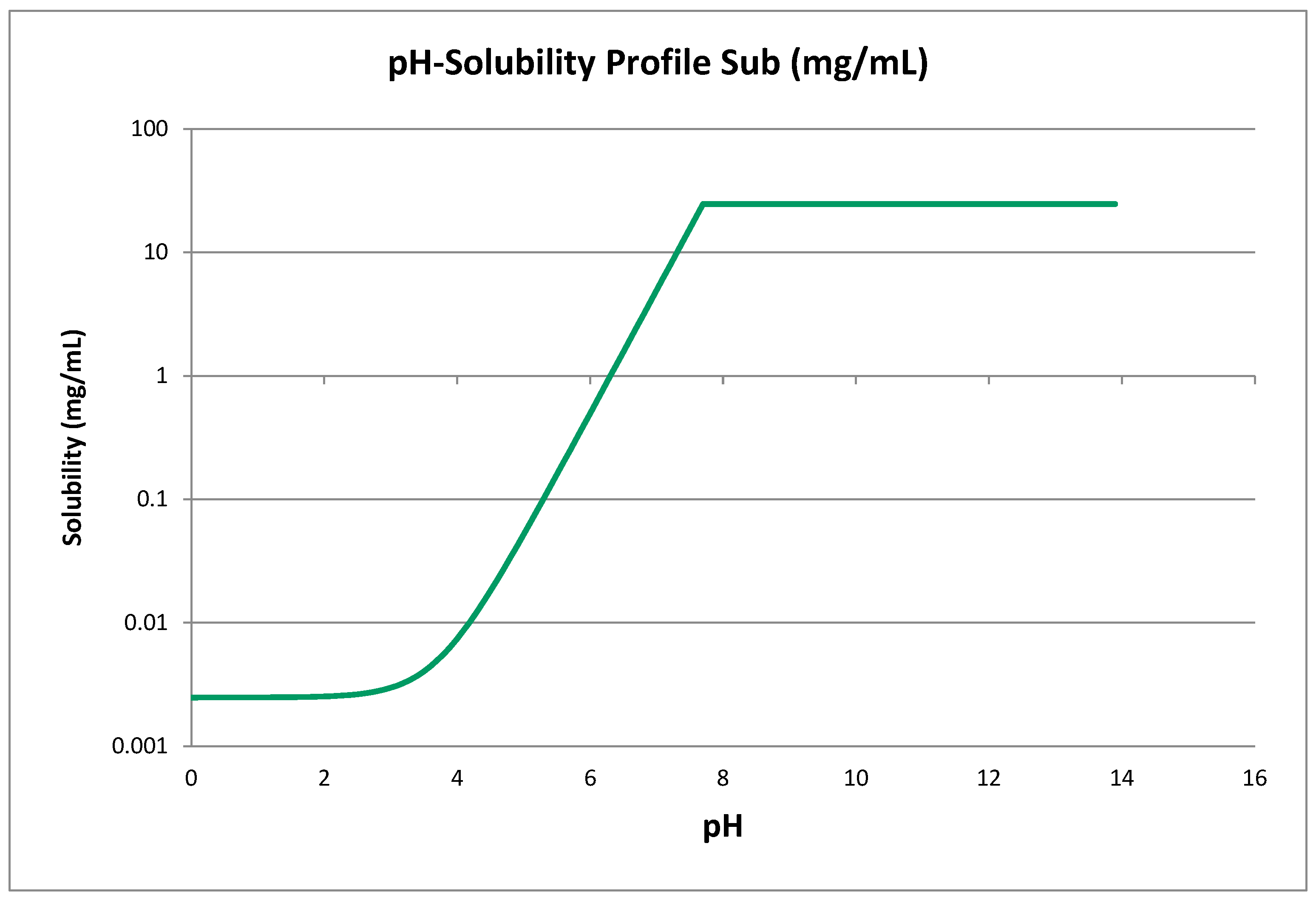
Figure A2.
Tafamidis binding when applied dissociation constants from Corrazza et al. 2019 [37]. In this scenario we observe much higher double bound tafamidis-TTR complex concentration, when compared to simulation with applied Nelson et.al. 2021 constants [40].
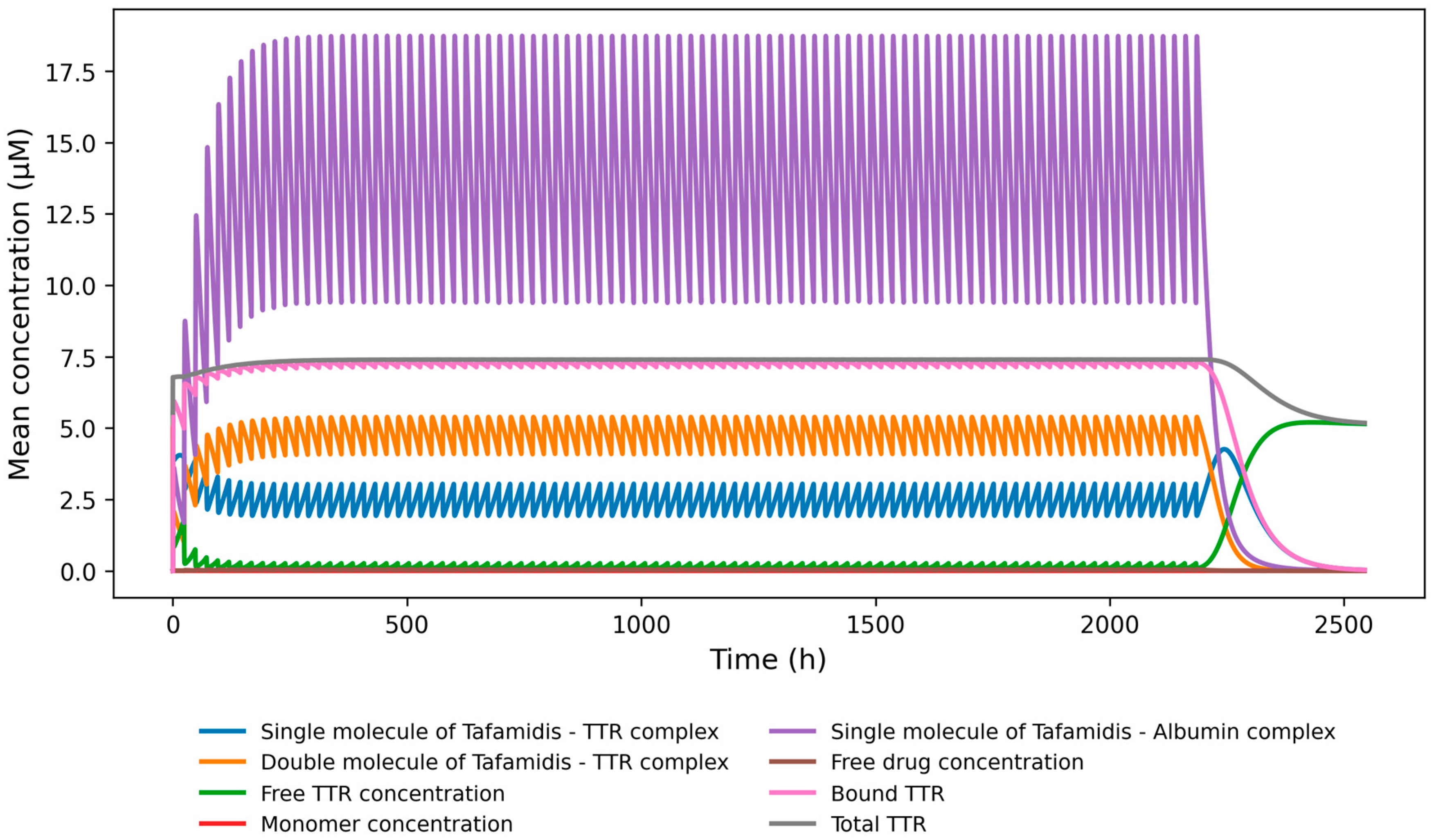
Figure A3.
Total TTR sensitivity to dose.
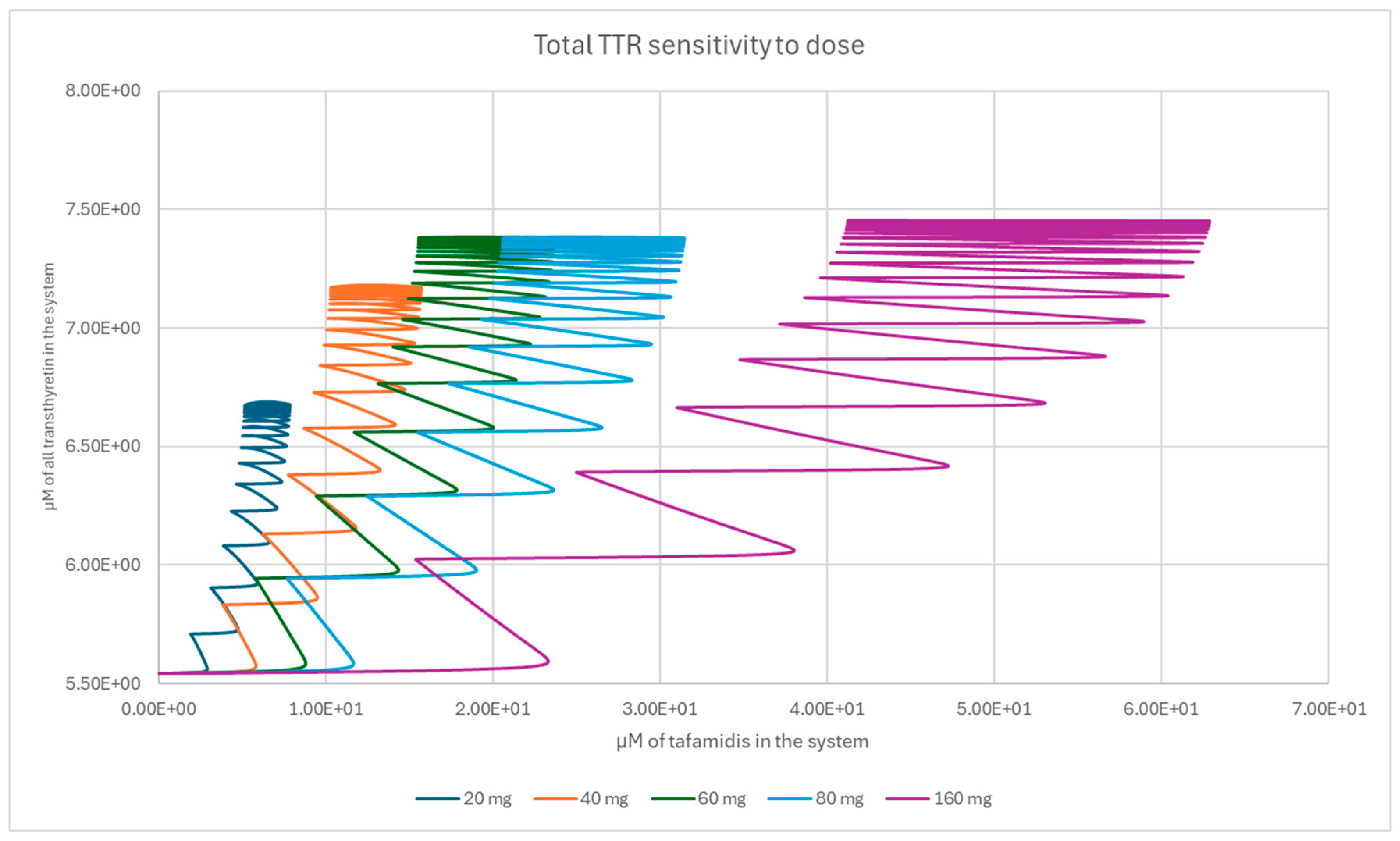
Figure A4.
Bound TTR sensitivity to dose.
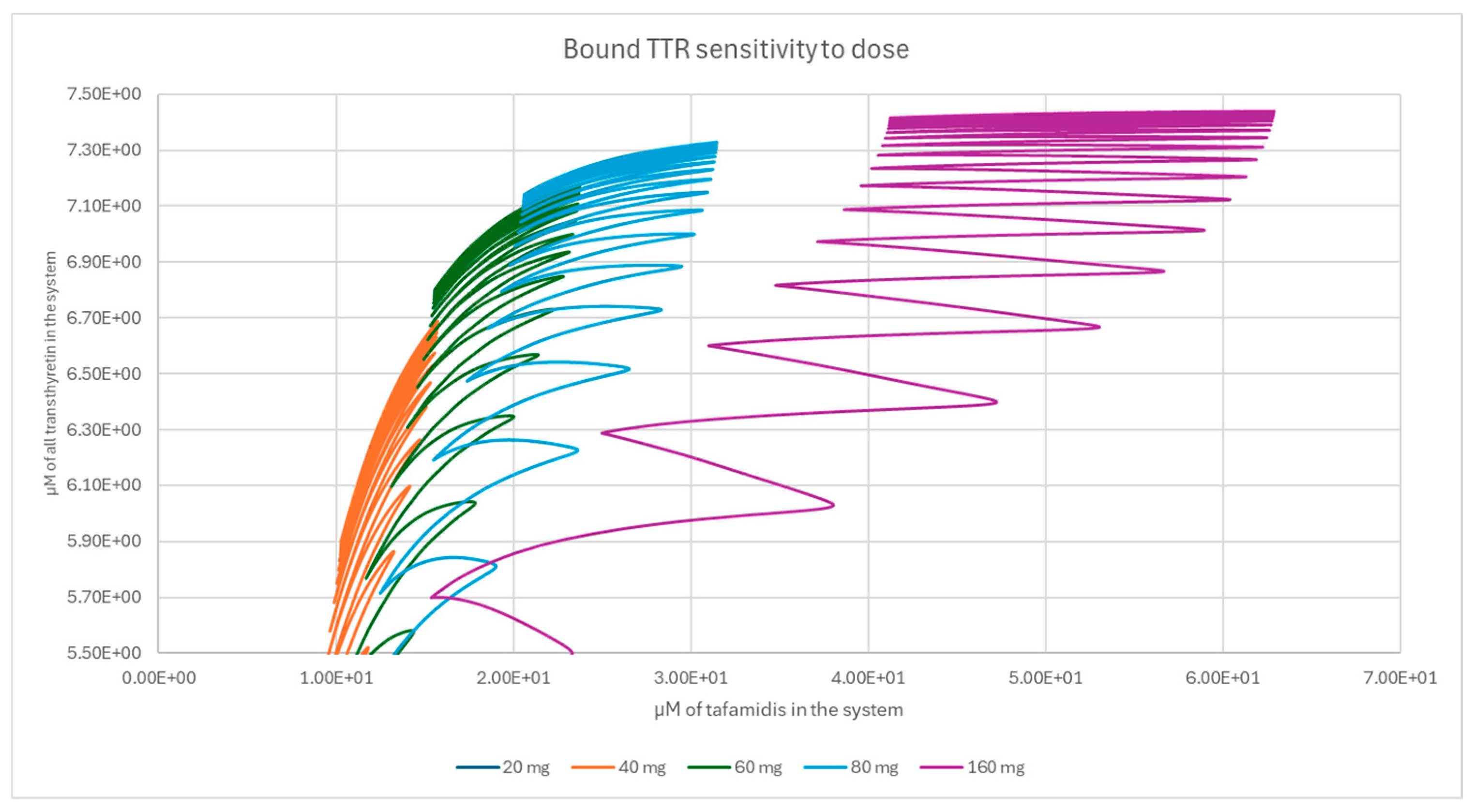
Figure A5.
Monomer sensitivity to dose.
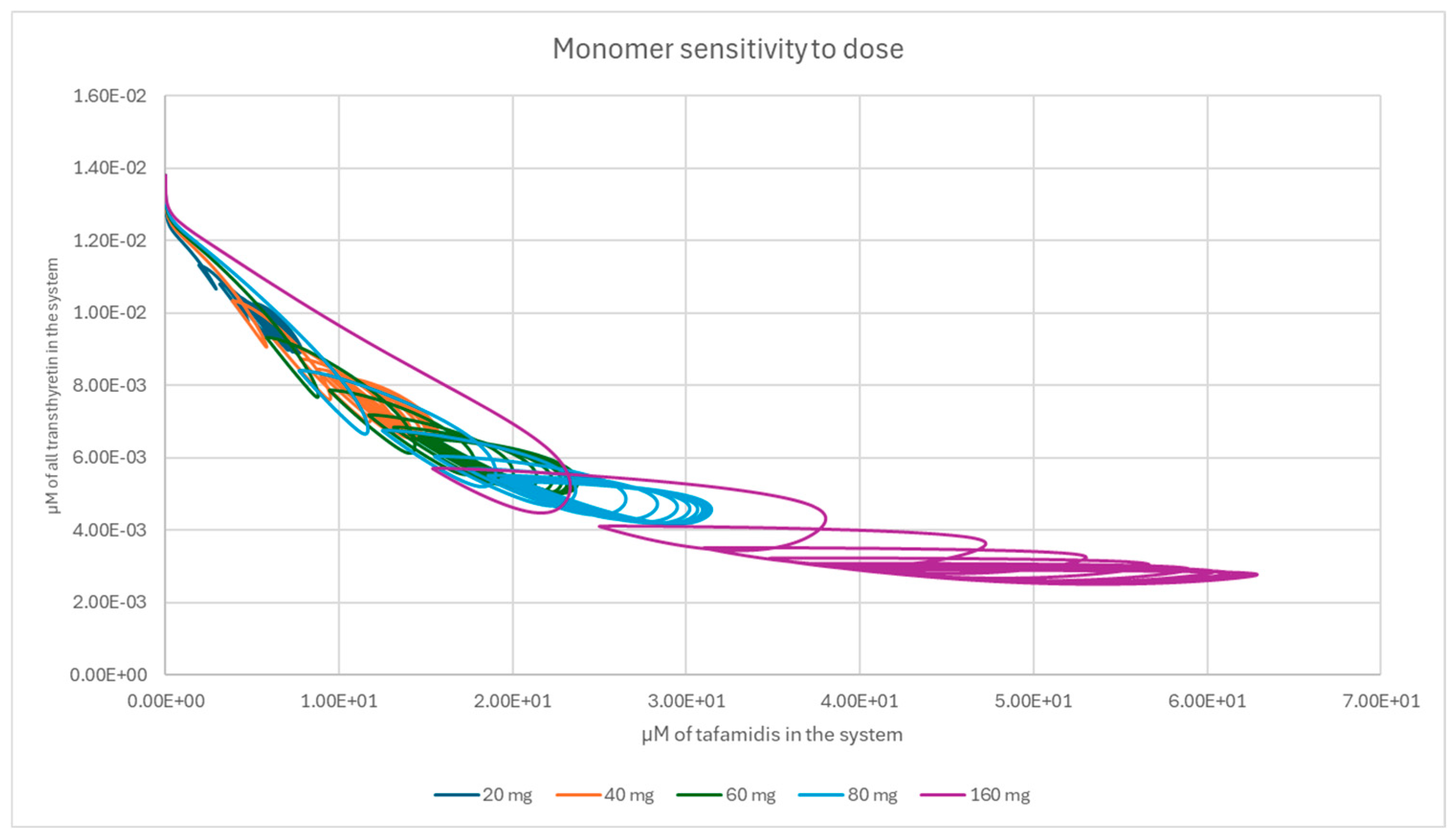
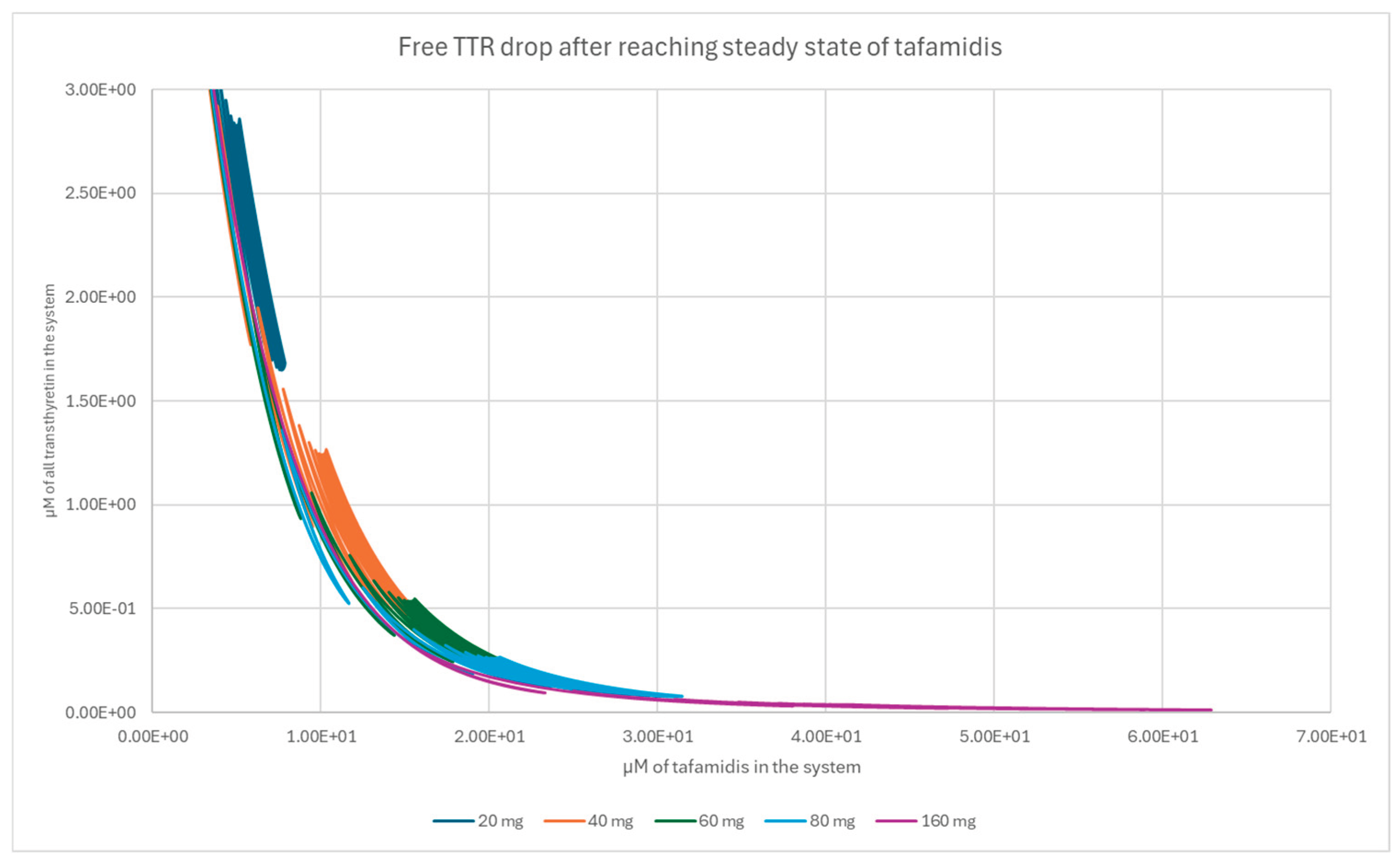
Figure A6.
Free TTR sensitivity to dose.
References
- Ruberg, F.L.; Berk, J.L. Transthyretin (TTR) Cardiac Amyloidosis. Circulation 2012, 126, 1286–1300. [Google Scholar] [CrossRef]
- Ueda, M. Transthyretin: Its function and amyloid formation. Neurochem. Int. 2022, 155, 105313. [Google Scholar] [CrossRef]
- Powers, E.T.; Amass, L.; Baylor, L.; Fernández-Arias, I.; Riley, S.; Kelly, J.W. Transthyretin Kinetic Stabilizers for ATTR Amyloidosis: A Narrative Review of Mechanisms and Therapeutic Benefits. Cardiol. Ther. 2025, 14, 333–350. [Google Scholar] [CrossRef]
- FDA approves new treatments for heart disease caused by a serious rare disease, transthyretin mediated amyloidosis. Jun 2019. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatments-heart-disease-caused-serious-rare-disease-transthyretin-mediated#:∼:text=On%20May%203%2C%20the%20U.S.,approved%20treatments%20for%20ATTR%2DCM.
- Maurer, M.S.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.H.; Haddad, M.; Walker, C.R.; Dorbala, S.; Cuddy, S.A.M. Effect of Tafamidis on Serum Transthyretin Levels in Non-Trial Patients With Transthyretin Amyloid Cardiomyopathy. JACC CardioOncology 2021, 3, 580–586. [Google Scholar] [CrossRef]
- Gamino, D.; Teruya, S.; De Los Santos, J.; Helmke, S.; Guadalupe, S.; Maurer, M. Tafamidis Increases Serum TTR (Prealbumin) Levels in both ATTRh and ATTRwt Cardiac Amyloidosis. J. Card. Fail. 2019, 25, S21. [Google Scholar] [CrossRef]
- Rappley, I.; et al. Quantification of Transthyretin Kinetic Stability in Human Plasma Using Subunit Exchange. Biochemistry 2014, 53, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Ulaszek, S.; Wiśniowska, B.; Lisowski, B. No body fits in the test tube – the case of transthyretin. Amyloid 2024, 31, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, B.; Ulaszek, S.; Wiśniowska, B.; Bernhauerová, V.; Polak, S. Phenomenological model of transthyretin stabilization. Sci. Rep. 2026. [Google Scholar] [CrossRef]
- Tess, D.A.; Maurer, T.S.; Li, Z.; Bulawa, C.; Fleming, J.; Moody, A.T. Relationship of binding-site occupancy, transthyretin stabilisation and disease modification in patients with tafamidis-treated transthyretin amyloid cardiomyopathy. Amyloid 2023, 30, 208–219. [Google Scholar] [CrossRef]
- Rowland, M.; Peck, C.; Tucker, G. Physiologically-Based Pharmacokinetics in Drug Development and Regulatory Science. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 45–73. [Google Scholar] [CrossRef] [PubMed]
- Danhof, M.; de Jongh, J.; De Lange, E.C.M.; Della Pasqua, O.; Et, A. Mechanism-Based Pharmacokinetic-Pharmacodynamic Modeling: Biophase Distribution, Receptor Theory, and Dynamical Systems Analysis. Annu. Rev. Pharmacol. Toxicol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, J.H.; Surks, M.I.; Bernstein, G.; Smith, J.C. Metabolism of Iodine-131-Labeled Thyroxine-Binding Prealbumin in Man. Science 1965, 149, 748–751. [Google Scholar] [CrossRef]
- Socolow, E.L.; Woeber, K.A.; Purdy, R.H.; Holloway, M.T.; Ingbar, S.H. Preparation of I-131-labeled human serum prealbumin and its metabolism in normal and sick patients. J. Clin. Invest. 1965, 44, 1600–1609. [Google Scholar] [CrossRef]
- Klamerus, K.J.; Watsky, E.; Moller, R.; Wang, R.; Riley, S. The effect of tafamidis on the QTc interval in healthy subjects. Br. J. Clin. Pharmacol. 2015, 79, 918–925. [Google Scholar] [CrossRef]
- Pfizer. “A Phase 1, Open-label, Randomized, Four-period, Four-sequence, Single-dose, Crossover Study In Healthy Volunteers, To Determine The Relative Bioavailability Of Pf-06291826 61 Mga Tafamidis Free Acid Soft Gelatin Capsules Compared To Commercial 4 × 20 Mg Tafamidis Meglumine Soft Gelatin Capsules Administered Under Fasted And Fed Conditions And The Effect Of Food On The Oral Bioavailability Of Pf-06291826 61 Mga Tafamidis Free Acid Soft Gelatin Capsules,” clinicaltrials.gov, Clinical trial registration NCT03280173, Apr. 2018. Available online: https://clinicaltrials.gov/study/NCT03280173 (accessed on Jan. 16, 2026).
- Center for drug evaluation and research. “Application number 211996Orig1s000 212161Orig1s000.” FDA Office of Clinical Pharmacology, Nov. 02, 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211996Orig1s000,%20212161Orig1s000ClinPharmR.pdf.
- Lockwood, P.A.; et al. The Bioequivalence of Tafamidis 61-mg Free Acid Capsules and Tafamidis Meglumine 4 × 20-mg Capsules in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 9, 849–854. [Google Scholar] [CrossRef]
- Jamei, M.; et al. Population-Based Mechanistic Prediction of Oral Drug Absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef]
- Sinha, U.; Rao, S. Methods of treating ttr amyloidosis using ag10. Available online: https://patents.google.com/patent/CA3094711A1/.
- Tetko, I.V.; et al. Virtual Computational Chemistry Laboratory – Design and Description. J. Comput. Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef]
- Tafamidis - NDA 212161. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211996Orig1s000,%20212161Orig1s000PharmR.pdf (accessed on Oct. 15, 2025).
- Lee, K.-R.; et al. Pharmacokinetics of tafamidis, a transthyretin amyloidosis drug, in rats. Xenobiotica 2018, 48, 831–838. [Google Scholar] [CrossRef]
- Sun, D.; et al. Comparison of Human Duodenum and Caco-2 Gene Expression Profiles for 12,000 Gene Sequences Tags and Correlation with Permeability of 26 Drugs. Pharm. Res. 2002, 19, 1400–1416. [Google Scholar] [CrossRef] [PubMed]
- “VYNDAQEL PRODUCT MONOGRAPH,” Pfizer, Oct. 2022. Available online: https://pdf.hres.ca/dpd_pm/00067760.PDF.
- Hansen, N.T.; Kouskoumvekaki, I.; Jørgensen, F.S.; Brunak, S.; Jónsdóttir, S.Ó. Prediction of pH-Dependent Aqueous Solubility of Druglike Molecules. J. Chem. Inf. Model. 2006, 46, 2601–2609. [Google Scholar] [CrossRef]
- Tchaparian, E.; Xu, G.; Huang, T.; Jin, L. Cell Based Experimental Models as Tools for the Prediction of Human Intestinal Absorption. Poster Presentation, 15th North American Regional International Society for the Study of Xenobiotics Meeting, 2008. [Google Scholar]
- Hens, B.; et al. Gastrointestinal transfer: In vivo evaluation and implementation in in vitro and in silico predictive tools. Eur. J. Pharm. Sci. 2014, 63, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Australian Public Assessment Report for Tafamidis and Tafamidis meglumine. Therapeutic Goods Administration. Sep 2020. Available online: https://www.tga.gov.au/sites/default/files/auspar-tafamidis-200903.pdf.
- Sawada, Y.; Hanano, M.; Sugiyama, Y.; Harashima, H.; Iga, T. Prediction of the volumes of distribution of basic drugs in humans based on data from animals. J. Pharmacokinet. Biopharm. 1984, 12, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Mayumi, K.; Tachibana, M.; Yoshida, M.; Ohnishi, S.; Kanazu, T.; Hasegawa, H. The Novel In Vitro Method to Calculate Tissue-to-Plasma Partition Coefficient in Humans for Predicting Pharmacokinetic Profiles by Physiologically-Based Pharmacokinetic Model With High Predictability. J. Pharm. Sci. 2020, 109, 2345–2355. [Google Scholar] [CrossRef]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP), “EMA assessment report - Vyndaqel,” Assessment report - Vyndaqel. Available online: https://www.ema.europa.eu/en/documents/variation-report/vyndaqel-h-c-2294-x-0049-g-epar-assessment-report_en.pdf (accessed on Dec. 15, 2025).
- Vyndaqel and Vyndamax full prescribing information. Available online: https://www.fda.gov/media/126283/download.
- Ji, A.X.; Betz, A.; Sinha, U. Differential Binding Affinities and Kinetics of Transthyretin Stabilizers. J. Cardiovasc. Pharmacol. 2025, 86, 204. [Google Scholar] [CrossRef]
- Corazza, A.; et al. Binding of Monovalent and Bivalent Ligands by Transthyretin Causes Different Short- and Long-Distance Conformational Changes. J. Med. Chem. 2019, 62, 8274–8283. [Google Scholar] [CrossRef]
- Sousa, M.M.; Saraiva, M.J. Internalization of Transthyretin. J. Biol. Chem. 2001, 276, 14420–14425. [Google Scholar] [CrossRef] [PubMed]
- Romine, C.; Wiseman, R.L. Starting at the beginning: endoplasmic reticulum proteostasis and systemic amyloid disease. Biochem. J. 2020, 477, 1721–1732. [Google Scholar] [CrossRef]
- Nelson, L.T.; Paxman, R.J.; Xu, J.; Webb, B.; Powers, E.T.; Kelly, J.W. Blinded potency comparison of transthyretin kinetic stabilisers by subunit exchange in human plasma. Amyloid 2021, 28, 24–29. [Google Scholar] [CrossRef]
- Ingenbleek, Y. Plasma Transthyretin Reflects the Fluctuations of Lean Body Mass in Health and Disease. In Recent Advances in Transthyretin Evolution, Structure and Biological Functions; Richardson, S. J., Cody, V., Eds.; Springer: Berlin, Heidelberg, 2009; pp. 329–357. [Google Scholar] [CrossRef]
- Sekijima, Y.; Tokuda, T.; Kametani, F.; Tanaka, K.; Maruyama, K.; Ikeda, S.-I. Serum transthyretin monomer in patients with familial amyloid polyneuropathy. Amyloid 2001, 8, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Drug bank website. Available online: https://go.drugbank.com/drugs/DB11644.
- Avdeef, A. Solubility of sparingly-soluble ionizable drugs. Adv. Drug Deliv. Rev. 2007, 59, 568–590. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically Based Pharmacokinetic Modeling 1: Predicting the Tissue Distribution of Moderate-to-Strong Bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Wiseman, R.L.; Johnson, S.M.; Kelker, M.S.; Foss, T.; Wilson, I.A.; Kelly, J.W. Kinetic Stabilization of an Oligomeric Protein by a Single Ligand Binding Event. J. Am. Chem. Soc. 2005, 127, 5540–5551. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic diagram of the PBPK–QSP linkage and QSP structure. Tafamidis plasma concentrations simulated by the PBPK model enter the QSP module as the time-varying total drug concentration, from which free tafamidis is computed based on competitive binding to transthyretin and albumin.
Figure 1.
Schematic diagram of the PBPK–QSP linkage and QSP structure. Tafamidis plasma concentrations simulated by the PBPK model enter the QSP module as the time-varying total drug concentration, from which free tafamidis is computed based on competitive binding to transthyretin and albumin.
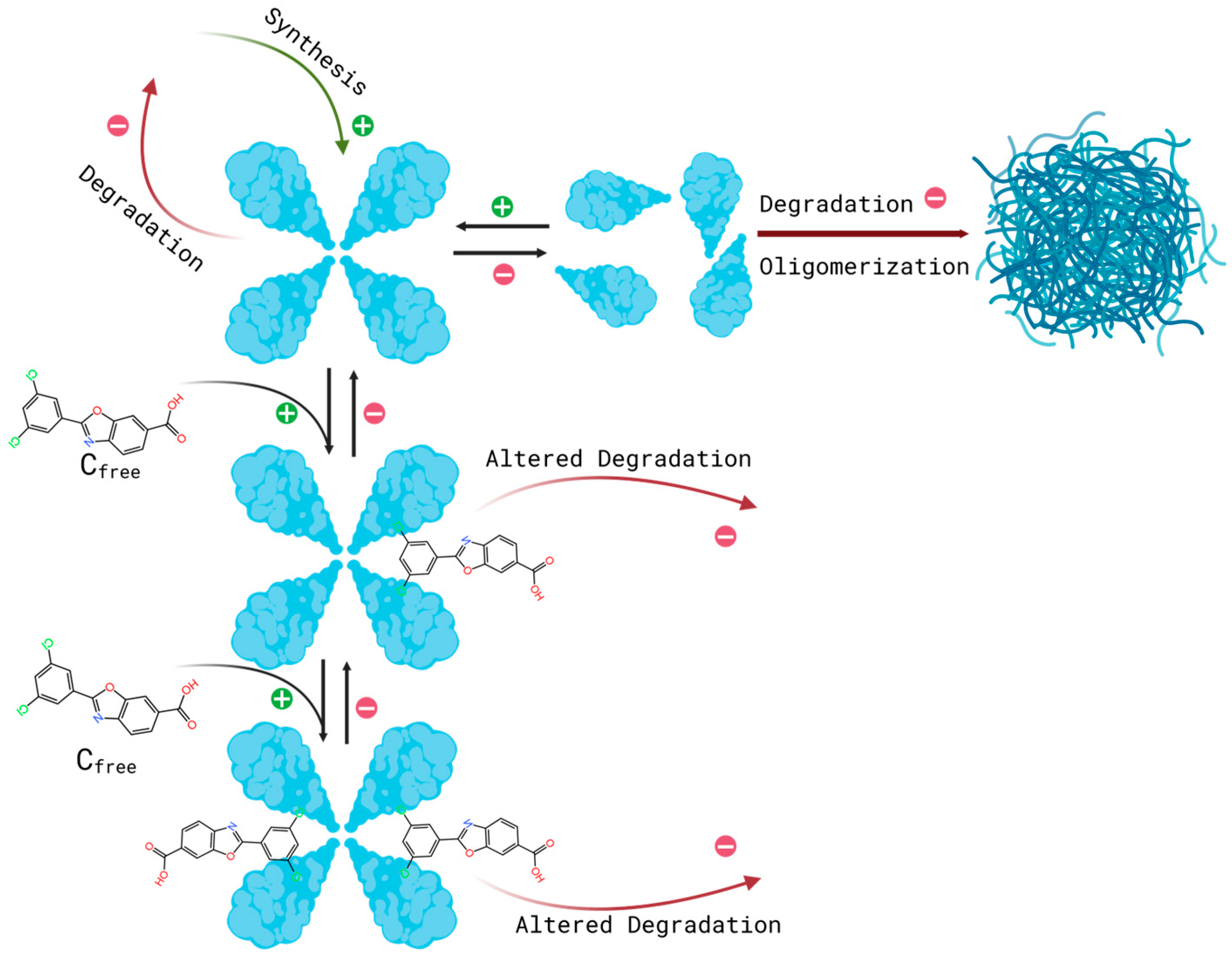
Figure 2.
Observed clinical data (open circles) versus simulated mean concentration across 10 virtual trials (solid line), with the 5th–95th percentile range across virtual trials shown as a shaded area, following (A) a single supratherapeutic dose of tafamidis meglumine (400 mg), (B) a single dose administered in the fasted state (4×20 mg), (C) a single dose administered in the fed state (4×20 mg), and (D) the seventh day of multiple dosing (4×20 mg).
Figure 2.
Observed clinical data (open circles) versus simulated mean concentration across 10 virtual trials (solid line), with the 5th–95th percentile range across virtual trials shown as a shaded area, following (A) a single supratherapeutic dose of tafamidis meglumine (400 mg), (B) a single dose administered in the fasted state (4×20 mg), (C) a single dose administered in the fed state (4×20 mg), and (D) the seventh day of multiple dosing (4×20 mg).
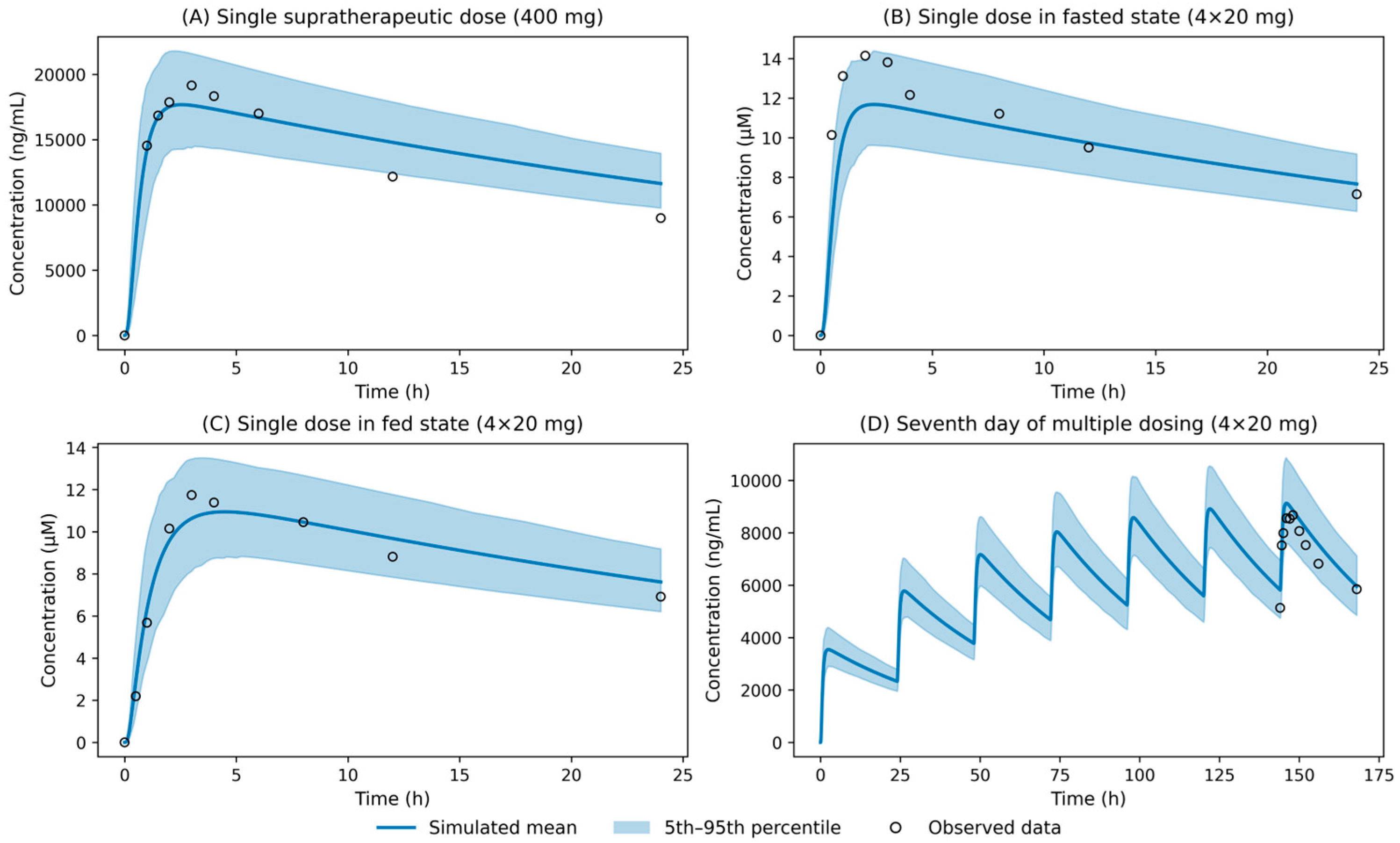
Figure 3.
Results of simulation of tafamidis binding accounting for dissociation rates of drug from Nelson et al. 2021 [40].
Figure 3.
Results of simulation of tafamidis binding accounting for dissociation rates of drug from Nelson et al. 2021 [40].
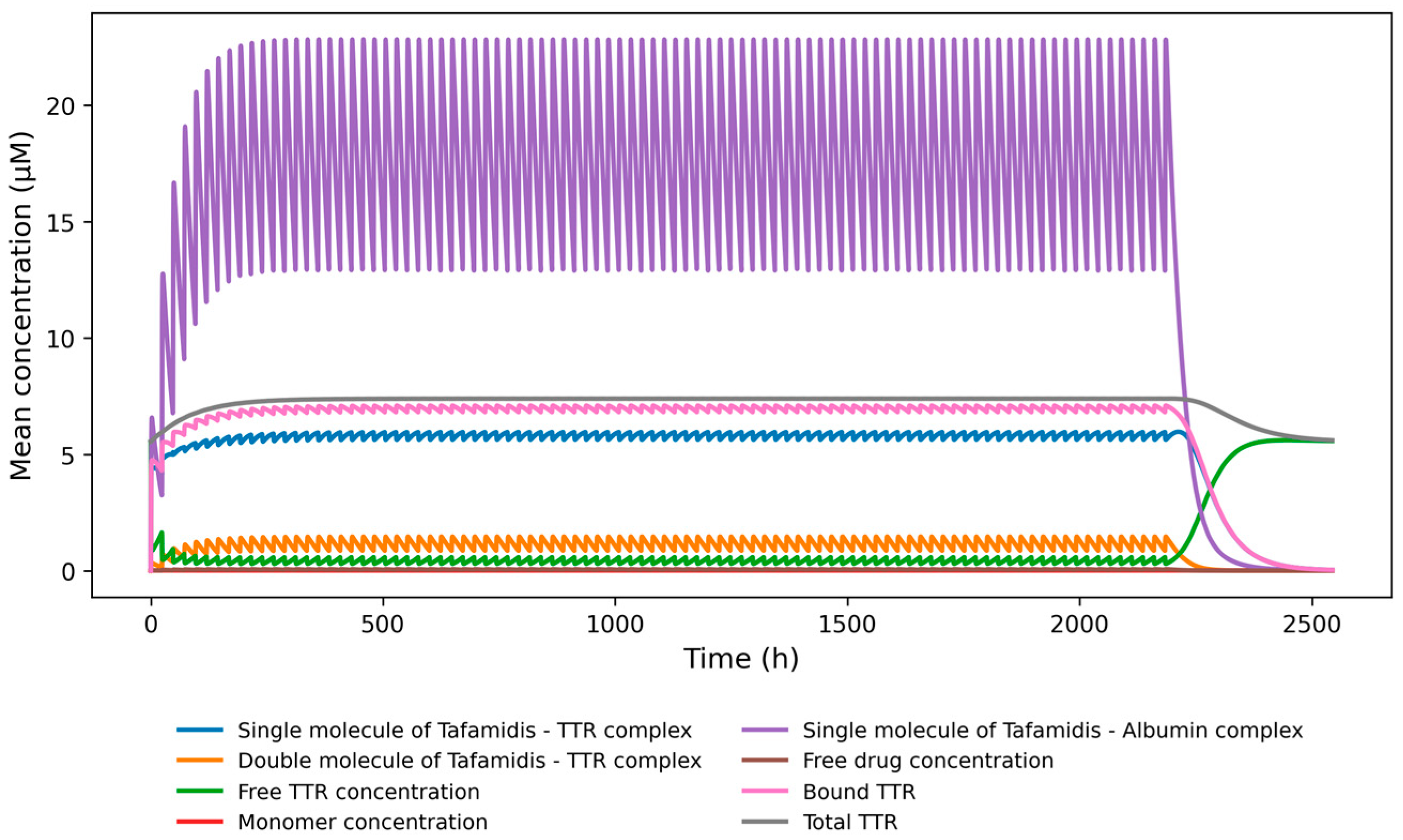
Figure 4.
Unbound-to-total TTR dose sensitivity analysis. Whiskers represent standard deviation.
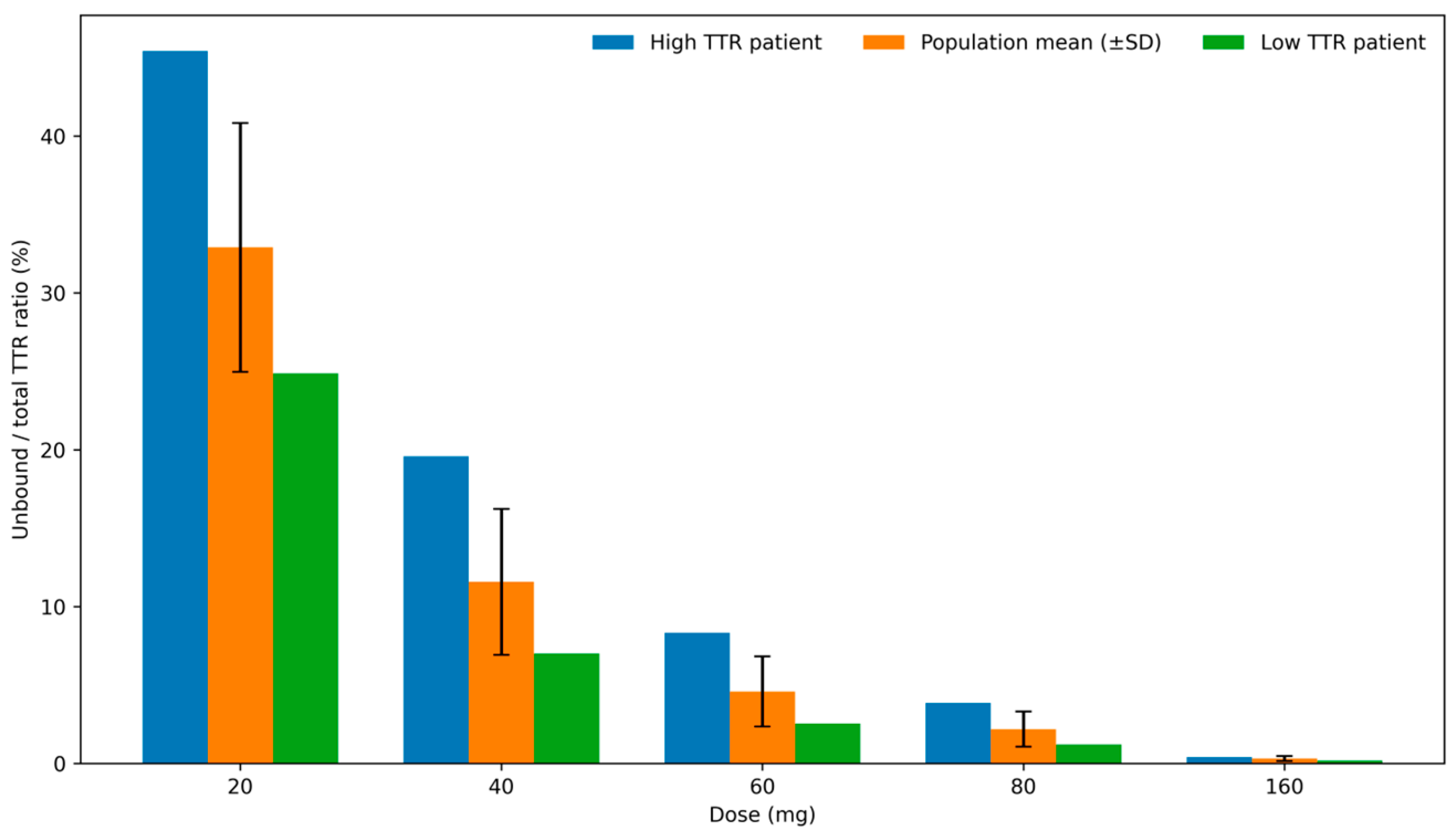
Table 1.
Tafamidis clinical data and studies characterization.
| Study / Trial identifier | Study purpose and design | Dose regimen and PK data used in this work |
|---|---|---|
| Thorough QT study (NCT01775761) [16]. | Phase I; randomized crossover; healthy volunteers (n = 42); fasted; QTc evaluation with PK as secondary endpoint. | Single oral supratherapeutic dose of tafamidis meglumine (400 mg); observed plasma PK (0–24 h); mean concentration–time data used to evaluate high-exposure behavior (Figure 2A). |
| Single-dose relative bioavailability and food-effect study (B3461054) [17,18]. | Phase I; open-label; randomized 4-period, 4-sequence crossover; healthy volunteers (n = 16); comparison of free acid and meglumine formulation; comparison of fasted and fed conditions. | Single oral dose of tafamidis meglumine 80 mg (4×20 mg); observed plasma PK under fasted and fed conditions; mean concentration–time profiles used to evaluate food effect (Figure 2B,C). |
| Phase Ib bioequivalence study (B3461056 - Lockwood et al., 2020) [19]. |
Phase I; open-label; randomized crossover; healthy male volunteers (n = 30); fasted; bioequivalence study. | Tafamidis meglumine 80 mg (4×20 mg) administered once daily for 7 days; observed steady-state plasma PK; mean concentration–time profiles on day 7 used to evaluate multiple-dose behavior (Figure 2D). |
Table 2.
Observed and PBPK-predicted pharmacokinetic parameters of tafamidis meglumine across multiple dosing regimens and nutritional conditions, with corresponding fold errors.
Table 2.
Observed and PBPK-predicted pharmacokinetic parameters of tafamidis meglumine across multiple dosing regimens and nutritional conditions, with corresponding fold errors.
| Study / scenario | Dosing regimen | State | PK parameter | Statistic | Observed | Predicted | Fold error (Pred/Obs) |
|---|---|---|---|---|---|---|---|
| B3461056 | Multiple dose, 4 × 20 mg, Day 7 | Fasted | AUC144–168h (ng x h/mL) | Mean (SD) | 169600 (35637) | 180557 (20998) | 1.06 |
| AUC144–168h (ng x h/mL) | Median (range) | 171000 (125000–258000) | 179721 (127744–254519) | 1.05 | |||
| AUC144–168h (ng x h/mL) | Geometric mean (%CV) | 166200 (20) | 179362 (12) | 1.08 | |||
| Cmax (ng/mL) | Mean (SD) | 9241 (1796) | 9160 (1039.26) | 0.99 | |||
| Cmax (ng/mL) | Median (range) | 8950 (6580–14600) | 9085 (6819.28–12840.15) | 1.02 | |||
| Cmax (ng/mL) | Geometric mean (%CV) | 9087 (18) | 9102 (11) | 1.00 | |||
| Tmax (h) | Median (range) | 2.0 (0.5–6.0) | 1.90 (1.18–3.84) | 0.95 | |||
| B3461054 | Single dose, 80 mg (4 × 20 mg) | Fasted | AUC0–inf (ng x h/mL) | Geometric mean (%CV) | 203400 (18) | 188274 (12) | 0.93 |
| Fasted | Cmax (ng/mL) | Geometric mean (%CV) | 4835 (20) | 3586 (12) | 0.74 | ||
| Fasted | Tmax (h) | Median (range) | 1.5 (0.5–4.05) | 2.26 (1.33–4.53) | 1.51 | ||
| Fed | AUC0–inf (ng x h/mL) | Geometric mean (%CV) | 208100 (23) | 188505 (12) | 0.91 | ||
| Fed | Cmax (ng/mL) | Geometric mean (%CV) | 4132 (15) | 3446.93 (12) | 0.83 | ||
| NCT01775761 | Single dose, 400 mg (supratherapeutic dose study) | Fasted | Cmax (ng/mL) | Geometric mean (%CV) | 20360 (18) | 17659 (13) | 0.87 |
| Fasted | AUC0–24h (ng x h/mL) | Geometric mean (%CV) | 305400 (15) | 342313 (13) | 1.12 | ||
| Fasted | Tmax (h) | Median (range) | 2.0 (1–6) | 2.3 (1.37–5.07) | 1.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



