
Submitted:
06 February 2026
Posted:
06 February 2026
You are already at the latest version
Abstract
Lung cancer is the leading cause of cancer-related mortality worldwide, accounting for more deaths than any other ma-lignancy. Despite advances in treatment, it remains highly lethal, with 5-year survival rates showing minimal improve-ment over the past several decades, highlighting a critical unmet clinical need. Macrophage Migration Inhibitory Factor (MIF) is a multifunctional cytokine that contributes to inflammation and cancer, promoting tumor growth, progression, and metastasis through modulation of the tumor microenvironment, stimulation of angiogenesis, and regulation of im-mune responses. Polymorphisms in the promoter region of MIF, such as high-expression CATT repeats, influence MIF expression and susceptibility to a range of inflammatory, autoimmune, and malignant disorders, yet their role in lung cancer remains largely unexplored. Therapeutic strategies targeting MIF, including small-molecule inhibitors, antibod-ies, and peptide-based agents, have shown promise in preclinical models, although their clinical translation is still lim-ited. This review discusses the dual role of MIF in inflammation and oncology, summarizes current therapeutic devel-opments, and emphasizes the potential of MIF-targeted interventions in lung cancer. It discusses the significance of ge-netic predisposition, particularly high-expression MIF alleles, in guiding personalized treatment strategies for lung can-cer and optimizing clinical outcomes in patients most likely to benefit from MIF inhibition.
Keywords:
tumor
; tautomerase
; MIF polymorphisms
; CATT repeats
; targeted therapy
; oxidized MIF (oxMIF)
; metastasis
; survival rate
1. Introduction
Lung cancer is one of the most aggressive and lethal malignancies worldwide, consistently ranking among the leading cancers in both incidence and mortality. It has remained the leading cause of cancer-related deaths worldwide since 1985, with both incidence and mortality steadily rising, with the high fatality rate causing mortality to mirror incidence [1]. Globally, in 2008, it accounted for 1.6 million new cases (13% of all cancers) and 1.4 million deaths (18% of cancer-related deaths), ranking first in incidence and mortality among men and fourth in incidence and second in mortality among women [2]. This upward trend continued with 1.8 million cases and 1.6 million deaths in 2012 [3] and 2.09 million cases and 1.76 million deaths in 2018 [4,5]. Although breast cancer briefly surpassed lung cancer in incidence in 2020, lung cancer remained the leading cause of cancer-related mortality with 1.8 million deaths [6]. By 2022, lung cancer again became the most commonly diagnosed cancer worldwide, with nearly 2.5 million new cases (12.4%) and remained the leading cause of cancer mortality with over 1.8 million deaths (18.7%) [7]. If current trends persist, global lung cancer burden is projected to reach 4.62 million cases and 3.55 million deaths by 2050 [8]. Overall, between 2008 and 2022, lung cancer accounted for approximately 12.4%–13% of all cancer cases and 18%–18.7% of cancer-related deaths worldwide. Thus, it remains the deadliest malignancy, causing more deaths than any other cancer; for example , in the United States, lung cancer mortality equaled the combined deaths from prostate, colon, breast, and pancreatic cancers in 2011 [9,10] and continued to exceed the combined deaths from breast, colorectal, and prostate cancers in 2022 [11].
Lung cancer is broadly categorized into two major histological types, small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC accounts for approximately 85–90% of all cases [12] and comprises several subtypes, including adenocarcinoma, squamous cell carcinoma, and large cell carcinoma [13,14]. Treatment strategies are stage-dependent, with early-stage NSCLC (I and II) typically managed through surgical resection, often followed by adjuvant therapy, while advanced or metastatic disease (III and IV) is primarily treated with chemotherapy and/or radiotherapy [15]. However, conventional chemotherapies are limited by non-specificity, low bioavailability, and drug resistance. To address these issues, newer approaches such as nanocarrier-based delivery systems, molecular targeted therapies, photothermal therapy, and immunotherapy have emerged [12]. Despite these advances, prognosis remains poor, with lung cancer still showing one of the lowest 5-year survival rates among all cancers [5] and survival rates have shown minimal improvement over the past several decades [16]. In this context, global data from 1995 to 2014 across approximately 70 countries showed that lung cancer remains highly lethal, with age-standardized 5-year net survival consistently low, fluctuating between 10–20% in both developed and developing regions [17,18]. During this period, survival rates remained largely unchanged, with improvements limited to 5–10% in 21 countries and exceeding 10% in only three countries,, highlighting the minimal improvement in lung cancer survival rates [17]. More recent analyses, covering data from 1990 to 2018, indicate that this pattern has changed very little, with global 5-year relative or net survival remaining in the range of 10–20% and remaining substantially lower than that observed for all other major cancers [19]. For instance, in the United States, the 5-year overall survival for lung cancer was only 15.6% between 2001 and 2007 and remained among the lowest at 19% during 2008–2014, compared with much higher rates for other cancers such as prostate (98%), melanoma (92%), and breast (90%) [20].
Given the substantial clinical burden of lung cancer, understanding the molecular mechanisms underlying its onset and progression is essential for developing novel and targeted therapies. The disease is highly complex and heterogeneous, driven by extensive genomic diversity, a broad mutation spectrum, and cumulative genetic and epigenetic changes that disrupt tumor suppressors and activate oncogenic pathways [21,22,23]. Among these, p53 is the most frequently mutated tumor suppressor, and its alterations are associated with poor prognosis, resistance to DNA-damaging therapies, and limited response to immunotherapy, highlighting the need for novel therapeutic strategies specifically targeting mutant p53 [24,25]. Other commonly affected tumor suppressors include retinoblastoma and the p16 pathway [26], while major oncogenic drivers include the rat sarcoma (RAS) and members of the human epidermal growth factor receptor (HER) family [27]. Tumor progression also relies on angiogenesis, driven mainly by vascular endothelial growth factor (VEGF), which enables growth beyond 3 millimeters by establishing a vascular network for nutrient and oxygen supply [23,28]. Notably, many of these pathways are influenced by macrophage migration inhibitory factor (MIF) (see Section 5 and Section 6).
Building on the evidence discussed above, lung cancer remains a major therapeutic challenge and a significant unmet clinical need, with persistently high incidence, mortality, and minimal improvements in survival despite advances in diagnostics and treatment. This review therefore focuses on MIF and its role in inflammatory diseases and cancer, with particular emphasis on its regulation of key molecular, inflammatory, and immune pathways in lung cancer, as well as emerging evidence linking MIF genetic polymorphisms to cancer susceptibility and its potential as a therapeutic target.
2. Overview of MIF
MIF is a multifunctional, pleiotropic cytokine that plays a central role in immune response and inflammation. It was first identified for its ability to inhibit the migration of immune cells, particularly macrophages and hence its name [29]. MIF is produced by a wide range of cell types, including macrophages, T cells, and endothelial cells, and participates in numerous physiological and pathological processes, such as leukocyte recruitment, immune responses, cellular proliferation, tumorigenesis, and the modulation of glucocorticoid activity [30].
MIF was first identified by Bloom and Bennett in 1966, when they described the phenomenon of migration inhibition during studies on delayed-type hypersensitivity, characterizing it as a soluble factor produced by sensitized lymphocytes that suppresses macrophage migration in vitro in response to specific antigens [29]. The cloning of human MIF cDNA in 1989 [31] enabled detailed exploration of its biological activities, revealing macrophages as both a source and target of MIF [32]. By 1993, MIF was recognized as a clinically relevant pro-inflammatory cytokine that potentiates lethal endotoxemia [33], sparking extensive research into its roles in immune regulation, inflammation, and receptor-mediated signaling, and leading to the identification of its molecular targets and disease-modifying effects [34].
2.1. Structure and Isoforms
MIF has a homotrimer structure composed of three identical 114–amino-acid subunits, arranged with three-fold rotational symmetry around a central solvent-accessible channel. Each monomer contains two antiparallel α-helices and a four-stranded β-sheet, and the central channel has been proposed as a binding site for small molecules such as glutathione, gangliosides, or dopachrome [35]. Its evolutionary conservation across plants, protozoans, nematodes, and other invertebrates underscores its fundamental biological importance [36].
Interestingly, increasing evidence indicates that MIF exists in two post-translational redox forms, oxidized and reduced [37]. This redox interconversion is regulated by redox-sensitive residues within its structure, particularly lysine-66 and cysteine-80 (the “switch cysteine”) [38]. The oxidized MIF (oxMIF) predominates in inflammatory and malignant conditions, while the reduced form is mainly present in healthy tissues, suggesting that oxMIF represents the active, disease-associated isoform [39,40] (see Section 4 and Section 5). Notably, oxidation at specific residues can also reduce MIF’s biological activity; for instance, oxidation of the N-terminal proline (Pro-1) impairs its tautomerase activity and ability to bind and activate CD74 signaling [41], suggesting that selective Pro-1 oxidation could represent a potential strategy to attenuate MIF’s pro-tumorigenic effects. Thus, the precise role of oxMIF in disease progression and its functional implications require further investigation. Clarifying how redox modifications influence MIF’s activity and signaling pathways may reveal new opportunities for therapeutic intervention in inflammatory and malignant diseases.
2.2. Signaling
Upon secretion, MIF exerts its effects through both autocrine and paracrine signaling, primarily via binding to its primary receptor CD74. This interaction forms a functional complex with the co-receptor CD44, which is essential for full activation of downstream pathways. The MIF–CD74–CD44 complex triggers both transient and sustained activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) mitogen-activated protein kinase (MAPK) cascade via Src family tyrosine kinase signaling [42]. This leads to nuclear factor kappa B (NF-κB) translocation to the nucleus, upregulation of phospholipase A2 and prostaglandin synthesis, and stimulation of the arachidonic acid pathway [43], which subsequently activates c-Jun N-terminal kinase (JNK) and enhances the translation of tumor necrosis factor-alpha (TNF-α) and other cytokine mRNAs [44]. In addition to CD74-dependent signaling, MIF can also interact with non-canonical C-X-C chemokine receptors (CXCR) including CXCR2 and CXCR4, thereby amplifying its role in inflammatory, autoimmune, and neoplastic processes [45]. Collectively, these receptor interactions highlight the complexity of MIF signaling and its broad implications in human pathophysiology.
2.3. Catalytic Activities
MIF exhibits unique enzymatic activities, functioning as a D-dopachrome tautomerase [46], a phenylpyruvate tautomerase [47], and a thiol-protein oxidoreductase [48], with the latter contributing to redox homeostasis, defense against oxidative stress and apoptosis inhibition [49]. MIF’s enzymatic activity may also exert a neuroprotective effect by detoxifying catecholamine breakdown products, converting toxic quinone derivatives into less harmful dihydroxyindoles, which serve as are precursors of neuromelanin [50]. A study using tautomerase-null MIF knock-in mice has shown that MIF’s catalytic Pro1 residue is essential for many of its biological effects and may play a role in mediating crucial protein–protein interactions [51,52]. The finding that MIF interact with its CD74 receptor near its tautomerase site suggested that small-molecule inhibitors targeting this region could MIF–CD74 interaction and thereby block the key protein–protein interaction essential to MIF’s function [53,54]. To facilitate the identification of such inhibitors, a biochemical assay was developed to evaluate compounds for inhibition of the MIF–CD74 protein–protein interaction [53]. A study by El-Turk et al. proposed that targeting MIF's C-terminal region could offer novel approaches for allosteric modulation of its enzymatic activity and aid in the development of new inhibitors of MIF tautomerase activity [55].
Many studies have shown that inhibition of MIF-tautomerase activity may represent a therapeutic approach for various inflammatory diseases and malignancies. For example, in a murine sepsis model, inhibition of MIF’s tautomerase activity with ISO-1, a widely used small-molecule MIF inhibitor commercially available for experimental studies, significantly reduced both lethality and TNF-α levels highlighting the MIF active site as a promising therapeutic target for treating sepsis [56]. In a murine model of chronic Pseudomonas aeruginosa infection, we found that mice lacking MIF tautomerase activity (mifP1G/P1G) exhibited significantly reduced lung inflammation, lower bacterial burden, and less tissue damage compared with wild-type animals [57]. SCD-19, a novel small-molecule tautomerase inhibitor identified by our group that completely abolishes MIF enzymatic activity and is more effective than ISO-1, markedly reduced pulmonary inflammation in the same model [58], highlighting a role of MIF’s tautomerase activity in driving pulmonary inflammation in cystic fibrosis. The small molecule 4-iodo-6-phenylpyrimidine (4-IPP) was shown to irreversibly bind to MIF’s catalytic N-terminal proline, thereby inhibiting its enzymatic activity and more effectively suppressing MIF-induced lung adenocarcinoma cell migration and growth than ISO-1 [59]. Our group further demonstrated that MIF’s tautomerase activity is critical for lung cancer progression as tumor growth was markedly reduced in MIF-knockout and enzymatically inactive MIFP1G/P1G mice [60]. In both a murine Lewis Lung Carcinoma (LLC) cell line and in tumor-bearing mice, SCD-19 more potently suppressed cell proliferation and tumor growth than ISO-1, reducing primary tumor volume in mice by approximately 90% [60].
Altogether, MIF’s tautomerase activity contributes to its biological functions and plays a role in mediating its pro-inflammatory and pathogenic effects, although its precise role in chronic inflammation and malignancy remains to be fully elucidated. Inhibition of this enzymatic function has been shown to markedly reduce inflammation, tumor progression, and infection severity in preclinical models. These findings highlight the pivotal role of MIF’s enzymatic activity in disease pathogenesis and support the development of novel, selective small-molecule inhibitors as a promising therapeutic approach for treatment of cancer and chronic inflammatory disorders.
3. MIF Promoter Polymorphisms and Their Clinical Implications
The human MIF gene is located on chromosome 22 (22q11.2) and is composed of three exons of 107, 172, and 66 base pairs (bp) and two introns of 188 and 94 bp (see Figure 1) [61,62]. Genetic variations within the MIF promoter region have been shown to influence its transcriptional regulation. Specifically, two functional polymorphisms have been identified, a functional tetranucleotide CATT5-8 repeat at −794 (rs5844572) and a G/C single-nucleotide polymorphism (SNP) at −173 (rs755622) [63]. Both variants are associated with differential MIF expressions. With respect to the −173 G/C SNP, the −173C allele enhances binding of the activator protein 4 (AP4) transcription factor, leading to increased transcriptional activity. Both in vitro and in vivo studies have demonstrated that this allele correlates with increased MIF gene and protein expression levels [63,64]. Similarly, the −794 CATT5-8 tetranucleotide repeat modulates MIF promoter activity in a length-dependent manner, with the CATT5 allele showing the lowest promoter activity [65], while longer repeats (CATT6-8) are associated with elevated MIF expression [66,67]. This is mediated by the transcription factor ICBP90, which binds to the CATT5-8 microsatellite region, where longer repeats strengthen the interaction and enhance MIF transcription [68], (see Figure 1).
Both −173 G/C and −794 CATT5-8 MIF polymorphisms, particularly alleles associated with higher MIF expression, have been linked to susceptibility and severity of various autoimmune, inflammatory, and neoplastic conditions. A comprehensive meta-analysis of 96 case-control studies involving thousands of participants showed that the MIF −173 G/C polymorphism is strongly associated with increased risk of autoimmune-inflammatory, infectious, and age-related diseases across diverse global populations, with significant associations in Asian, European, and Latin American cohorts [70]. A subsequent meta-analysis of 23 studies further confirmed this association with autoimmune disorders [71]. This -173 G/C variant has also been linked to cardiovascular diseases [72,73], increased susceptibility to pulmonary tuberculosis, particularly among Asians [74] and, based on a meta-analysis of ten studies involving 2,203 cancer cases and 2,805 controls, with overall cancer risk [75]. However, none of these studies specifically examined the potential link of the variant with lung cancer.
Likewise, accumulating evidence indicates that the CATT5–8 polymorphism contributes to the susceptibility and severity of inflammatory and autoimmune diseases, commonly alongside the −173 G/C variant. In rheumatoid arthritis (RA), the low-expressing CATT5 allele was associated with milder [65], while higher-expressing alleles, particularly CATT7, were correlated with elevated circulating MIF levels and more severe, erosive joint damage [64]. A more recent meta-analysis of 12 studies confirmed that the −173C allele, the CATT7 repeat, and the CATT7–MIF −173C haplotype were linked to increased RA susceptibility across multiple ethnicities [76]. Similarly, a meta-analysis of 13 studies revealed higher circulating MIF levels in systemic lupus erythematosus (SLE) patients, particularly among Asians, with the −173 C/G polymorphism, but not −794 CATT, linked to disease susceptibility [77]. Paradoxically, in a cohort of 1,369 SLE patients, the high-expression −794 CATT7/−173C haplotype was associated with lower SLE incidence, yet CATT5 alleles were less frequent in those with organ damage, indicating that high-expression CATT6–8 alleles contributed to more severe organ involvement [67]. In cystic fibrosis, CATT5 was associated with milder disease, lower rates of Pseudomonas colonisation [78], and a slower forced expiratory volume in 1 second (FEV1) decline [57].
The variants have also been implicated in cancer, with the high-expression −173 C allele and longer CATT6-8 repeats associated with increased risk of rectal [79] and non-cardia gastric cancer, particularly among Helicobacter pylori–infected individuals [80]. The CATT7 repeat was further linked to early-stage cervical cancer [81] and to higher prostate cancer incidence and recurrence [82]. In cutaneous squamous cell carcinoma, the MIF 5C (−794 CATT5/−173 C) and 7G (−794 CATT7/−173 G) haplotypes conferred higher disease susceptibility, accompanied by elevated circulating MIF levels [83].
To further investigate the functional impact of MIF promoter polymorphisms, our group developed novel humanized mouse models carrying the low-expressing CATT5 and high-expressing CATT7 human MIF alleles [84], providing an important experimental platform to elucidate how MIF genetic variation influences disease susceptibility and pathogenesis. The high-expression −794 CATT7 allele was linked to more severe COVID-19, with hospitalized patients exhibiting elevated serum MIF levels, and mice carrying this allele developing more severe disease than CATT5 carriers [84]. In preclinical asthma models, CATT7 mice showed pronounced airway inflammation than CATT5 or wild-type mice, which was effectively attenuated by the MIF inhibitor SCD-19 [85]. Moreover, mesenchymal stromal cells and SCD-19 treatment effectively attenuated airway inflammation and MIF-dependent macrophage priming in high-MIF CATT7 mice, further supporting the role of high-expression (CATT6-8) MIF alleles in asthma severity [86,87]. Similarly, a recent study using the YUMMER1.7 murine melanoma model demonstrated that tumors grew faster in high-MIF CATT7 mice, whereas CATT5 carriers exhibited a one week delay in tumor establishment and significantly reduced tumor burden by day 15 [88]. A comprehensive summary of diseases linked to MIF promoter polymorphisms, highlighting their associations with susceptibility and severity, is provided in Table 1.
Overall, high-expression promoter variants of MIF (i.e., the −173C allele and longer −794 CATT6-8 repeats) are frequently associated with greater disease susceptibility and/or severity across a variety of inflammatory, autoimmune, infectious and malignant disorders. These findings underscore not only the broad mechanistic importance of MIF in disease pathogenesis through immune activation, tissue injury, and tumor-promoting effects; but also highlight the translational potential of MIF-targeted therapies, particularly for individuals with high-expression alleles who may derive the greatest benefit from anti-MIF interventions. While studies have yet to examine MIF promoter polymorphisms in lung cancer, existing evidence suggests these variants may similarly influence lung tumor development and represent a target for further investigations.
4. MIF and Inflammatory Diseases
MIF is a key proinflammatory mediator that amplifies inflammatory responses both directly, by stimulating the release of proinflammatory cytokines, and indirectly, by counteracting the anti-inflammatory effects of glucocorticoids [98,99,100]. Administration of anti-MIF antibodies has been shown to completely protect adrenalectomised rodents from lethal arthritis, highlighting MIF’s role in amplifying inflammatory pathways when not counterbalanced by endogenous glucocorticoids [101]. As discussed previously in Section 3, the identification of functional polymorphisms in the MIF promoter region, which are associated with several inflammatory disorders, has further underscored MIF’s significance in the pathogenesis of inflammatory diseases [102]. Indeed, MIF has been implicated in a wide number of immune and inflammatory diseases. For example, in acute respiratory distress syndrome, MIF has been shown to be a key driver of the harmful inflammatory response which can be reduced by anti-MIF treatment [99]. Likewise, elevated MIF levels were detected in the bronchoalveolar lavage fluid of asthmatic patients compared to non-atopic individuals [103] and MIF-deficient mice exhibited reduced pulmonary inflammation and airway hyperresponsiveness following ovalbumin challenge compared to wild-type animals [104]. Recently, MIF has also been implicated in severe neutrophilic asthma, which is often resistant to glucocorticoid therapy, by promoting neutrophil recruitment to the lungs and reducing glucocorticoid effectiveness through inhibition of annexin-A1 [105], and to promote airway remodeling in addition to its established role in inflammation [106].
In cystic fibrosis, MIF and its tautomerase activity, which drive pulmonary inflammation and earlier Pseudomonas colonisation, have been linked to disease severity [57,78]. Furthermore, P. aeruginosa has been shown to exploit recombinant human MIF (rhMIF) to enhance biofilm formation, and rhMIF was found to impair the antibacterial efficacy of tobramycin in vitro [58].
Further evidence of MIF’s role in inflammatory pathogenesis comes from its involvement in autoimmune and systemic inflammatory diseases. In RA, MIF is associated with increased susceptibility [89], disease severity [64,65,107], and a higher risk of developing inflammatory polyarthritis [90]. In SLE, MIF and its oxidized form are elevated [77], with higher circulating [108], and urinary [109] concentrations correlating with disease activity and inflammatory organ involvement [67,110]. Plasma oxMIF rises during systemic flares, whereas urinary oxMIF increases specifically in lupus nephritis, reflecting local renal inflammation and disease severity [40]. Elevated MIF and oxMIF have also been detected in inflamed tissues of patients with Crohn’s disease and ulcerative colitis as well as in the plasma of individuals with severe sepsis and septicemia [40,111], further underscoring MIF’s central role in systemic inflammation.
Taken together, these findings highlight MIF as a central mediator of inflammatory and immune-driven pathology across diverse diseases. By promoting tissue inflammation, impairing glucocorticoid responsiveness, and enhancing immune cell recruitment, MIF contributes to disease progression in asthma, chronic pulmonary infections such as cystic fibrosis, and autoimmune conditions including SLE and RA. Importantly, its enzymatic and signaling functions position MIF as a promising therapeutic target, with anti-MIF strategies offering potential benefits for steroid-resistant, treatment-refractory, or genetically high-risk patient populations.
5. MIF and Cancer
MIF has emerged as a key regulator of tumor biology, promoting cancer progression through diverse mechanisms, including tumor growth, angiogenesis, and modulation of the immune microenvironment. The following subsections summarize the molecular mechanisms of MIF, its impact on anti-tumor immunity, and its clinical associations across cancer types.
5.1. Molecular Mechanisms of MIF-Driven Tumorigenesis
The link between inflammation and cancer has long been recognized, with chronic inflammatory diseases associated with increased risk of various malignancies [46,112]. While inflammatory pathways normally protect against infection and injury, they can also create a tumor-promoting microenvironment that supports growth and metastasis [113]. In this context, MIF acts as a crucial molecular bridge between inflammation and tumorigenesis by sustaining chronic inflammatory signaling and promoting tumor-supportive conditions [46]. MIF's interaction with its receptors CD74 and chemokine receptors CXCR2 and CXCR4 initiates downstream signaling pathways that enhance tumor growth and survival. As a downstream effect,, MIF is known to downregulate p53 and inhibit its nuclear localization, impairing its transcriptional activity [114,115,116]. As a key tumor suppressor, p53 normally triggers apoptosis in response to DNA damage, so its inhibition compromises genomic stability and allows potentially cancerous cells to survive and proliferate [117].
MIF further contributes to tumor progression by promoting angiogenesis. It stimulates endothelial cell migration and differentiation into vascular structures through activation of the MAPK and phosphatidylinositol-3-kinase/protein kinase B (PI3K/Akt) signaling [118], and upregulation of VEGF via MAPK-dependent pathways [119], thereby facilitating neovascularization and tumor growth. Within hypoxic tumor environments, MIF expression is upregulated in a hypoxia-inducible factor 1α (HIF-1α)–dependent manner [120]. MIF in turn stabilises and increases HIF-1α expression via the PI3K/Akt pathway and by inhibiting HIF-1α degradation [121]. This positive feedback loop enhances the transcription of HIF-1α–regulated genes that drive angiogenesis e.g., VEGF, glycolysis, invasion, and cell survival, processes that collectively support tumor progression and adaptation to hypoxia [122]. MIF also drives cancer progression and metastasis by inducing epithelial-to-mesenchymal transition (EMT) [123], and upregulating matrix metalloproteinase 9 [124], thereby facilitating extracellular matrix remodeling and tumor invasion.
The tautomerase active site of MIF is also critical for its oncogenic function, as tumor growth was markedly reduced in tautomerase-null in tautomerase-null (MIFP1G/P1G) mice in both skin [51] and lung cancer [60] models, highlighting the importance of this structural region in mediating protein–protein interactions essential for tumor progression [51,117]. Consistently, pharmacological inhibition of this site with the small-molecule inhibitor SCD-19 significantly suppressed lung cancer growth in vitro and in vivo [60]
Collectively, these findings highlight MIF as a multifunctional driver of tumor progression, linking inflammatory and stress-associated signaling to cancer cell survival, proliferation, and tumor–microenvironment interactions. Through its structural domains and downstream signaling pathways, MIF promotes key hallmarks of cancer, including enhanced growth, invasion, and resistance to cell death, underscoring its role as a central regulator of oncogenic processes. The major molecular pathways through which MIF contributes to cancer progression are summarized in Figure 2.
5.2. MIF–CD74 Signaling and Resistance to Immune Checkpoint Blockade
The MIF–CD74 axis plays a crucial role in promoting immunosuppression across multiple cancers by shaping the tumor microenvironment. MIF-CD74 interaction activates downstream pathways that polarize tumor-associated macrophages toward an M2 immunosuppressive phenotype and limit infiltration and activity of cytotoxic CD8+ T cells [125,126]. In Ewing’s sarcoma, this leads to impaired antitumor immunity, while inhibition of MIF–CD74 reprograms the microenvironment, enhances T cell activity, and suppresses tumor growth [127]. Similarly, in metastatic melanoma, blocking MIF–CD74 signaling on macrophages and dendritic cells reduces immunosuppressive factor expression and enhances cytotoxic T cell activation, restoring antitumor immune responses [125].
Mechanistically, MIF–CD74 interaction directly regulates programmed death-ligand 1 (PD-L1) expression on tumor cells, a key immune checkpoint molecule targeted by programmed cell death protein 1 (PD-1)/PD-L1 inhibitors. High CD74/PD-L1 expression correlates with immune evasion in melanoma models, providing a mechanistic basis for resistance to checkpoint blockade [128]. Inhibiting the MIF–CD74 axis increases T-cell infiltration, shifts macrophages toward a pro-inflammatory phenotype, and reduces PD-L1 expression in tumor cells, effectively enhancing innate immune activation and sensitizing tumors to immunotherapy [126]. In addition to its role in immune checkpoint regulation, MIF–CD74 signaling also influences responses to other therapies through remodeling of the tumor microenvironment. In NSCLC brain metastases, inhibition of the MIF–CD74 axis enhanced radiotherapy efficacy by reversing radiation-induced Akt phosphorylation and promoting polarization of microglia toward a pro-inflammatory M1 phenotype [129]. Although this study did not directly assess immune checkpoint inhibitors, it highlights the broader impact of MIF–CD74–mediated innate immune reprogramming on therapeutic responses.
Consistent with these observations, combining MIF-targeted therapies with PD-1/PD-L1 blockade potentially represents a promising strategy to overcome resistance in cancers characterized by MIF-driven immunosuppressive microenvironments. For example, in preclinical YUMMER1.7 melanoma and MC38 colorectal carcinoma mouse models, dual targeting of MIF and PD-1 enhanced antitumor responses, reduced tumor growth, prolonged survival, and promoted more complete tumor regression with enhanced intratumoral immune activation compared with anti–PD-1 or anti-MIF monotherapy [88].
Taken together, these studies identify the MIF–CD74 axis as a key regulator of tumor immune suppression and therapeutic resistance. By modulating macrophage polarization, T cell infiltration, and PD-L1 expression, MIF–CD74 signaling limits the efficacy of immune checkpoint blockade. PD-1/PD-L1 inhibitors have become an important component of cancer immunotherapy, with clinical activity reported across numerous solid tumors, highlighting the broad relevance of this therapeutic axis in oncology [130,131,132]. These results support further investigation of combination strategies that integrate MIF-targeted therapies with PD-1/PD-L1 blockade in additional preclinical models, and if proven effective, could subsequently be advanced to clinical trials.
5.3. MIF Overexpression and Clinical Associations Across Cancer Types
MIF is overexpressed in many types of cancer, and elevated expression has been linked to aggressive tumor phenotypes and poorer clinical outcomes across multiple malignancies [133,134]. A systematic meta-analysis of multiple solid tumors demonstrated that high MIF expression correlates with poorer overall survival and disease-free survival in cancer patients, supporting its prognostic relevance across diverse cancer types [135]. In line with these findings, pan-cancer analyses show that MIF is upregulated across numerous malignancies and is associated with genomic instability, immune suppression, and other features of aggressive disease [136]. While lung cancer is the primary focus of this review, selected evidence from other malignancies is mentioned below to demonstrate the broader clinical implications of MIF overexpression. Comprehensive pan-cancer and prognostic analyses are provided in other systematic reviews and meta-analyses [135,136].
Elevated MIF levels have been reported in colorectal cancer, where increased expression is observed in primary tumors, metastatic lesions, and patient serum, with higher circulating MIF concentrations correlating with disease severity and metastatic burden [124]. Similarly, MIF has been shown to be involved in the initiation and progression of head and neck cancers, where it suppresses apoptosis and facilitates both local and distant metastases and thereby contribute to poor prognosis [137]. Epithelial-derived MIF was shown to drive colorectal cancer progression and maintenance, highlighting its potential as a therapeutic target, with inhibition potentially enhancing anticancer therapy [138].
In addition to total MIF, the disease-associated oxMIF has been detected at elevated levels in several malignancies, including colorectal, pancreatic, ovarian, and lung cancers [40,139]. Within the tumor microenvironment, MIF oxidation appears to promote tumor growth and invasion [39,139]. Supporting the clinical relevance of oxMIF, a phase I clinical trial of the oxMIF-targeting antibody imalumab achieved disease stabilization in approximately one-quarter of heavily pretreated patients with advanced solid tumors [140]. Consistently, preclinical studies demonstrate that targeting oxMIF with monoclonal antibodies induces apoptosis, suppresses tumor cell proliferation, and enhances the efficacy of chemotherapeutic agents in both in vitro and in vivo cancer models [139,141].
Overall, MIF overexpression is a common feature across diverse malignancies and is closely associated with aggressive tumor behavior and poor clinical outcomes, highlighting its potential as both a biomarker and a therapeutic target in cancer.
6. MIF and Lung Cancer
As mentioned previously in Section 1 and Section 5, MIF is involved in many of the signaling pathways that are involved in lung cancer pathogenesis. Although MIF has been implicated in several inflammatory and malignant diseases, its specific therapeutic potential in lung cancer remains insufficiently explored. In vitro studies consistently demonstrate that MIF overexpression promotes NSCLC progression through autocrine and paracrine mechanisms, activating multiple oncogenic pathways such as MAPK and PI3K/Akt [115,142,143]. Elevated MIF mRNA and protein levels have been observed in several lung adenocarcinoma cell lines compared with normal primary alveolar epithelial cells [144]. Functionally, MIF overexpression enhances proliferation, migration, invasion, adhesion, and spreading of lung cancer cells, whereas MIF knockdown or pharmacological inhibition markedly suppresses these processes [142,145,146]. Mechanistically, MIF overexpression promoted lung cancer cell proliferation and the Warburg effect via NF-κB–dependent HIF-1α upregulation [147], while MIF silencing in H460 cells markedly reduced proliferation and triggered apoptosis through activation of caspases-3 and -4 [148]. Moreover, MIF promoted angiogenesis in NSCLC by inducing C-X-C motif chemokine ligand 8 (CXCL8) and VEGF expression through a CD74-dependent JNK/activator protein-1 (AP-1) signaling pathway [149].
In silico analyses have further supported these findings, identifying MIF as part of a 4-gene signature that stratifies lung adenocarcinoma patients into high- and low-risk groups for overall and recurrence-free survival, with markedly lower 5-year recurrence-free survival in the high-risk group (53% vs 90%) [150]. These observations highlight the potential prognostic significance of MIF in lung adenocarcinoma; however, the predictive value of such signatures for lung adenocarcinoma recurrence requires validation in experimental and clinical studies.
Consistent with the in vitro and in silico findings, analyses of patient-derived lung tissues have shown that MIF was markedly upregulated in NSCLC compared with normal lung epithelium [151], with particularly high expression observed in patients with squamous cell carcinoma, where it correlated with poorer prognosis [152]. Elevated levels of oxMIF were also detected in lung tumor tissues [40,139]. Moreover, MIF expression was higher in multiple primary lung adenocarcinomas than in single primary tumors, with both exceeding normal lung tissue levels, implicating MIF in the development of multifocal disease [144]. In contrast, a pilot study of 25 patients with newly diagnosed lung carcinoma versus 25 matched controls found no difference in serum MIF levels [153], highlighting the limited utility of circulating MIF as a biomarker for lung cancer when compared with tissue expression.
Elevated MIF expression has also been associated with aggressive features in lung cancers. For instance, in lung squamous cell carcinoma, a high proportion of MIF-positive tumor cells correlated with lymph node metastasis, shorter disease-free survival, and poorer cancer-specific outcomes [145]. Analysis of 370 NSCLC tumor samples revealed an inverse correlation between MIF and miR-451 expression, with elevated MIF linked to advanced disease and poorer prognosis [154]. In NSCLC tissues from 87 patients, MIF exhibited a bimodal pattern, with one group comparable to normal tissue and another markedly elevated [155]. In this cohort, high MIF levels were associated with increased angiogenic chemokines and VEGF, enhanced tumor growth and vessel density, poorer survival, and higher risk of recurrence after tumor resection [155]. This bimodal expression pattern could potentially be driven by underlying MIF polymorphisms in the patients, which influence MIF expression profile; however, this possibility was not investigated in the study.
Increased MIF has also been demonstrated to confer resistance to chemotherapeutic agents. Cisplatin-resistant NSCLC tumors, as well as cisplatin-resistant A549 and H460 cells, exhibited elevated MIF expression, enhanced self-renewal, and M2 macrophage polarization, suggesting that MIF, together with markers such as proto-oncogene tyrosine-protein kinase Src and CD155, contributes to chemoresistance and tumor progression [156]. Similarly, in gefitinib-resistant lung adenocarcinoma patients, serum exosomal MIF levels were higher than the sensitive ones and positively correlated with tissue inhibitor of metalloproteinase-1 (TIMP1) expression [157]. Further in vitro studies showed that MIF mRNA was markedly upregulated in gefitinib-resistant A549 and PC9 cells, and M2 macrophage–derived exosomal TIMP1 was shown to promote gefitinib resistance by enhancing MIF–CD74 binding and activating the PI3K/Akt pathway [157]. Conversely, a retrospective study reported no association between serum MIF levels and chemotherapy response in NSCLC and SCLC patients [158], demonstrating that circulating MIF is less informative for patient identification and prediction of treatment response than tissue or exosome-derived MIF.
Beyond MIF, its receptor CD74 was strongly expressed in NSCLC specimens and frequently co-localized with MIF, a pattern associated with elevated angiogenic chemokines and increased tumor vascularity [159]. In lung adenocarcinoma tissues, CD74 expression was higher in tumors compared with adjacent normal tissue and correlated with lymph node metastasis and advanced TNM (tumor–node–metastasis) stage [160]. In vivo, TNF-α–driven inflammation in a urethane-induced lung adenocarcinoma mouse model upregulated MIF in macrophages and CD74 in tumor cells, while TNF-α blockade reduced their expression and tumor nodules, underscoring the TNF-α–MIF–CD74 axis in tumor progression [160].
Other in vivo studies further support the role of MIF and its receptors in promoting lung tumor growth and survival under experimental conditions. Jäger et al. demonstrated that A549 cells transduced to overexpress the MIF receptor CXCR4 exhibited increased proliferation and MIF production in vitro [161]. Intratracheal injection of these CXCR4-overexpressing cells into mice led to the highest lung tumor burden compared with mice receiving control or CXCR4-KO A549 cells. Bioluminescence and histological analyses revealed that treatment of mice with the MIF inhibitor ISO-1 significantly reduced tumor burden, highlighting the CXCR4–MIF axis as a driver of NSCLC progression in vivo [161]. While bioluminescence imaging provides a non-invasive method to monitor tumor growth, its accuracy can be limited by tissue depth and signal attenuation. High-resolution imaging techniques, such as micro-CT or MRI, could provide more precise tumor volume measurements, enabling better assessment of MIF-driven tumor progression and therapeutic responses.
Earlier in vivo work demonstrated that lung injury followed by intratracheal injection of LLC cells increased tumor growth in wild-type mice, with enhanced proliferation and reduced apoptosis, whereas MIF knockout mice exhibited neither injury-induced tumor growth nor alterations in proliferation or apoptosis, highlighting MIF’s essential role in linking tissue injury and repair to tumor progression [162]. Our previous work, as mentioned in Section 2.3, demonstrated that MIF’s tautomerase activity contributes to lung tumor progression in the LLC mouse model, with tumor growth markedly reduced in MIF-knockout and tautomerase-null mice, and that the novel small-molecule inhibitor SCD-19 effectively suppresses lung cancer growth in vitro and in vivo [60]. However, one limitation of this study was the use of a subcutaneous (ectopic) lung tumor model, where tumors develop outside their native lung environment, failing to replicate the lung’s unique stromal interactions and biomechanical forces, such as ventilation and perfusion dynamics, [163,164]. These factors may influence treatment responses, highlighting the need for further studies to investigate MIF’s role, its tautomerase activity, and polymorphisms in orthotopic primary lung cancer models that more accurately reflect lung cancer biology.
Collectively, evidence from in vitro, in silico, clinical, and in vivo studies identify MIF as a multifunctional oncogenic factor that promotes lung cancer growth and angiogenic signaling through multiple, interrelated mechanisms. Elevated MIF expression in patient tissues with disease severity and histological differentiation, supporting its potential as a biomarker for NSCLC staging and prognosis. Furthermore, elevated tissue MIF levels and their association with aggressive tumor phenotypes and chemotherapy resistance underscores the multifaceted role of MIF in lung cancer, positioning it as a potential prognostic biomarker and mediator of treatment response. In contrast, circulating serum MIF has shown limited predictive value of lung cancer, highlighting the need to investigate MIF genotyping of CATT polymorphisms as a more robust biomarker for patient stratification. These findings highlight the translational significance of MIF in lung cancer pathogenesis and emphasize the need for further studies to confirm its value as a biomarker and therapeutic target, particularly in patients with high MIF expression or MIF-related polymorphisms, thereby supporting a more personalised approach to lung cancer management.
7. Therapeutic Targeting of MIF
MIF is a key regulator of various cellular processes, and its dysregulation has been linked to a wide range of inflammatory and proliferative diseases. Thus, MIF is a promising therapeutic target, particularly in individuals with high baseline or inducible MIF expression, such as carriers of CATT6–8 alleles. MIF inhibitors can be generally divided into three main classes: small-molecule compounds, monoclonal antibodies, and peptide-based inhibitors [165].
7.1. Small-Molecule MIF Inhibitors
Small molecules targeting the MIF tautomerase active site or otherwise disrupting its function have demonstrated encouraging preclinical activity. Several such inhibitors have been identified using virtual screening techniques [53,166,167], and high-throughput activity-based assays [168,169]. Representative examples include ISO-1, the most widely used and commercially available research compound [170], 4-IPP [52,59], and SCD-19 [58,60]. These compounds primarily target the tautomerase site, which mediates MIF’s interaction with CD74, thereby attenuating downstream signaling. Beyond tautomerase-site inhibitors, several compounds have been reported to modulate MIF activity through alternative mechanisms. For instance, ebselen, synthetic organoselenium compound that mimics the antioxidant enzyme glutathione peroxidase, disrupts the trimeric structure of MIF by promoting its dissociation into monomers, thereby impairing MIF–CD74 signaling [169]. Another compound, p425, a small-molecule allosteric inhibitor, binds to the surface of the MIF trimer, blocking its tautomerase activity, interaction with CD74, and thereby its pro-inflammatory effects [171].
Ongoing studies have expanded the chemical diversity of small molecule MIF inhibitors to include oxadiazole, triazole, benzoxazole, benzopyran, and benzophenone derivatives, with improved potency and mechanistic diversity in preclinical models. For a detailed overview of these inhibitors, including their structure–activity relationships and mechanistic insights, readers are referred to a recent comprehensive review by Guo et al. [172]. Notable examples include the oxadiazole derivative IPG1576 which selectively inhibited MIF tautomerase activity and demonstrated oral bioavailability, safety, and in vivo efficacy in a murine pancreatic cancer model, reducing tumor growth and enhancing antitumor immunity [173]. In addition, a benzopyran (7-hydroxycoumarin) derivative compound inhibited MIF tautomerase activity at the nanomolar level and disrupted MIF–CD74 binding, resulting in reduced A549 cell proliferation in vitro [174]. However, this study did not include pharmacokinetic or toxicity assessments; therefore, comprehensive in vivo evaluation of metabolic stability, bioavailability, and safety profile will be necessary to advance this scaffold toward the development of potent, clinically viable MIF inhibitors. Another remarkable example of genotype-selective inhibition is the phenazine derivative 1-methoxy-5-formyl-4,6,8-trihydroxyphenazine (CMFT), which blocked ICBP90 binding to the −794 CATT5-8 MIF promoter microsatellite. CMFT inhibited MIF transcription in a promoter length–dependent manner, selectively reducing MIF mRNA and protein in macrophages isolated from high-expression CATT7 mice, with minimal effect in CATT5 macrophages [175]. Notably, CMFT belongs to a class of compounds with poor in vivo metabolic profiles [176], which could limit its further clinical development. Nonetheless, its selective mechanism of MIF inhibition offers a valuable framework for refining this compound class and developing future precision-based MIF modulators for individuals with genetically high MIF expression.
Among many small-molecule MIF inhibitors developed, only a few have advanced to clinical evaluation. Ibudilast, which binds near, but not directly at, the N-terminal proline [177,178], has completed Phase I/II trials for alcohol use disorder, demonstrating acceptable safety but modest efficacy [179,180]. It was also evaluated in combination with temozolomide in a Phase 1b/2a study for glioblastoma (NCT03782415), where safety, tolerability, and preliminary efficacy were assessed, although detailed results have not yet been posted [181]. IPG1094, another small-molecule MIF antagonist, is undergoing a Phase 1/2 open-label, dose-escalation trial in patients with advanced solid tumors, including NSCLC, head and neck cancer, and glioma (NCT06212076), evaluating safety, tolerability, pharmacokinetics, pharmacodynamics, and initial anti-tumor activity, with results still pending [182]. These trials highlight the potential of translating MIF-targeted small molecules into safe and effective human therapies.
In summary, although promising results have been observed in cellular and animal models, most small-molecule MIF inhibitors remain in remain in early developmental stages. The broad range of MIF’s biological activities means that its inhibition may lead to adverse effects or toxicity, highlighting the need for thorough safety evaluation. Additional challenges include limited pharmacokinetic and toxicological data, as well as species differences in preclinical models, which hinder clinical translation. Continued efforts to optimize selectivity, metabolic stability, and safety, together with mode-selective assays targeting specific MIF–receptor interactions, will be essential to fully realize the therapeutic potential of these compounds in inflammatory, immune, and cancer-related diseases.
7.2. Monoclonal Antibodies and Nanobodies
Monoclonal antibodies targeting MIF have been developed to neutralize its pro-inflammatory and pro-tumorigenic functions. These antibodies bind either native or oxidized form of MIF, thereby preventing interaction with its receptor CD74 and downstream signaling. In parallel, antibodies directed against CD74 itself have also been explored to disrupt MIF–CD74 signaling and inhibit MIF-mediated immune and tumor-promoting effects. Preclinical studies demonstrated therapeutic efficacy of anti-MIF antibodies in multiple disease models. In mice, it delayed the arthritis onset, reduced its frequency [183], lowered inflammatory cytokine levels and synovial inflammation [184]. In a rat model of anti-glomerular basement membrane disease, a neutralizing anti-MIF monoclonal antibody reduced proteinuria, preserved renal function, and attenuated histological signs of inflammation [185].
The human anti-oxMIF monoclonal antibody, imalumab, has undergone Phase I and early Phase IIa clinical trials in patients with advanced solid tumors, either alone or in combination with standard treatments [140,186]. The antibody was generally well tolerated, with allergic alveolitis as the only dose-limiting toxicity, and target engagement was confirmed, although objective tumor responses were modest, with stable disease seen in a subset of patients [140,186]. These results underscore the need for further studies to optimize dosing, assess combination strategies, and clarify the therapeutic potential of MIF-targeted antibodies in oncology. Similarly, the humanized anti-CD74 antibody milatuzumab was evaluated in a Phase 1b trial in patients with moderate SLE, where it demonstrated reductions in disease activity and acceptable tolerability, with only mild injection-site or flu-like reactions observed in the initial cohort [187,188]. However, broader randomized studies have not yet been conducted, and advancement into late-stage clinical development remains outstanding. Milatuzumab has also received FDA orphan drug designation for the treatment of multiple myeloma, but it has not yet been approved for this indication [189]. Notably, preclinical studies combining milatuzumab with the anti-CD20 antibody rituximab showed promising efficacy in mantle cell lymphoma model [190], highlighting potential for combination therapies in malignancies.
Nanobodies, single-domain antibody fragments, represent an emerging class of biologics with high solubility, stability, and superior tissue penetration compared to conventional antibodies [191]. Anti-MIF nanobodies with nanomolar affinity for murine and human MIF have been developed and shown therapeutic potential in models of septic shock and inflammatory organ damage [192]. Additional variants, including constructs with extended half-life, have provided more options for targeting MIF and evaluating improved stability and efficacy in preclinical studies [192]. To our knowledge, however, these anti-MIF nanobodies have so far been tested only in acute inflammatory disease models.
In conclusion, while antibodies and nanobodies offer high selectivity and potent engagement of extracellular MIF or its receptor interactions, challenges such as high production costs, and limited intracellular access hinder broader therapeutic development. Ongoing efforts to improve delivery and optimize these molecules may help address these limitations and make MIF-targeted biologics more feasible for clinical use.
7.3. Peptide Inhibitors
Peptide inhibitors represent a third class of therapeutics that may expand strategies for targeting MIF. For example, the novel chimeric peptide DRα1-MOG-35–55, which combines the HLA-DRα1 domain with the myelin oligodendrocyte glycoprotein 35–55 (MOG) peptide, binds to CD74 on monocytes and blocks MIF activity. This results in reduced tissue damage and improved symptoms of autoimmune encephalomyelitis in a mouse model [193]. The related inhibitor RTL1000, a fusion construct of DRα1 and DRβ1 domains with MOG-35–55 peptide (DRα1β1-MOG-35–55), also blocks MIF binding to CD74 and was well tolerated in a Phase 1 clinical trial for multiple sclerosis, highlighting its potential to promote targeted immunoregulation and central nervous system repair without inducing widespread immunosuppression [194,195]. The 17-amino acid peptide C36L1 also disrupts MIF–CD74 interactions on monocytes and dendritic cells, restoring antitumor immune responses by enhancing CD8+ T cell activity in a metastatic melanoma model [125]. Finally, the synthetic peptides MIF-(40–49) and MIF-(47–56), which mimic the N-like loop of parent MIF, competitively inhibit MIF binding to CXCR2 resulting in reduced monocyte arrest on aortic endothelial cells in vitro and MIF-dependent monocyte adhesion to atherosclerotic carotid arteries in vivo [196], highlighting their potential to limit vascular inflammation and atherogenesis.
8. Future Perspectives
Although numerous small-molecule MIF inhibitors have been developed, many demonstrate greater efficacy than ISO-1, the widely used and commercially available MIF inhibitor, none has yet reached clinical use. Similarly, anti-MIF antibodies and peptide-based inhibitors have been primarily tested in preclinical models, with only a few advancing into early-phase clinical trials, showing tolerability and preliminary biological activity, yet none have progressed to later-stage studies or regulatory approval. This underscores a substantial gap in translating these promising strategies into therapeutic applications and emphasizes the need to prioritize the development of clinically viable MIF inhibitors. Rigorous safety evaluation will be essential to translate preclinical potency into clinically viable therapies and fully realize the therapeutic potential of MIF-targeted interventions across inflammatory, immune, and oncologic diseases. Future research should focus on optimizing selectivity and pharmacokinetic profiles, including the exploration of novel derivatives with improved safety and tolerability, and on evaluating these inhibitors broadly in cancer, and lung cancer specifically, either as monotherapies or in combination with immunotherapies or standard chemotherapies.
Building on the need for more selective and clinically viable MIF inhibitors, selectively targeting oxMIF represents an emerging and therapeutically attractive strategy. Isoform-selective inhibition could attenuate pathological MIF signaling while preserving the homeostatic functions of reduced MIF in healthy tissues, thereby reducing the risk of side effects. The feasibility of this approach has been demonstrated by monoclonal antibody, imalumab that specifically binds the oxidized isoform and has shown acceptable safety and preliminary signs of activity in early clinical studies in patients with advanced solid tumors [140]. Second-generation bioengineered anti-oxMIF antibodies, such as ON203 and ON103, retain oxMIF specificity, exhibit improved biophysical properties, and show robust in vitro activity, with ON203 has demonstrated antitumor efficacy in a mouse preclinical model, significantly inhibiting the growth of human PC3 prostate cancer xenografts, highlighting the translational potential of isoform-selective oxMIF targeting [197]. However, to our knowledge, no small-molecule MIF inhibitors are oxMIF-specific, making the development of such compounds an unmet challenge and opportunity to improve the precision of MIF-targeted therapies.
Expanding on these molecular interventions, nanobody-based therapeutics have shown considerable promise in cancer due to their small size, high stability, strong target affinity, and superior penetration of solid tumors [198,199]. Their remarkable stability under proteolytic conditions and acidic pH, enable them to remain functional within the hostile tumor microenvironment, while their ability to recognize specific and cryptic epitopes allows precise targeting of tumor- and immune-associated factors [200,201]. Given the established role of MIF in cancer progression in general, and in lung cancer specifically, MIF may serve as a suitable epitope target for nanobody-based therapeutic strategies. However, anti-MIF nanobodies remain largely unexplored in oncology, including lung cancer. Further investigation of anti-MIF nanobodies could enable more precise modulation of the tumor microenvironment and open new avenues for targeted intervention in lung cancer, particularly in individuals with genetically high MIF expression.
Beyond molecular design, the use of nanocarriers to deliver targeted MIF therapies directly to affected organs represents an additional area for future studies. For example, inhalable or nebulized delivery systems could allow localized administration of non-encapsulated or nanocarrier-formulated anti-MIF therapies directly to the lungs in patients with lung cancer, potentially enhancing therapeutic efficacy while minimizing systemic exposure and off-target effects. Using nanomedicine approaches, these delivery systems could improve the distribution of MIF inhibitors, enable controlled release in response to tumor-specific signals such as acidic pH or enzymatic activity, and enhance their impact on cancer cells and the local tumor microenvironment, including immune and stromal components within lung tumors.
In addition, future studies should aim to validate MIF-related biomarkers to support patient stratification and therapeutic decision-making. Given the limited predictive value of circulating serum MIF, research should investigate tissue- or exosome-based MIF levels and MIF CATT genotyping as potential biomarkers to stratify patients with lung cancer and other MIF-driven diseases, and to guide therapeutic decisions, with further studies needed to confirm their clinical utility and ability to monitor treatment responses.
Another potential avenue for future investigation is the mechanistic interplay between MIF, high-expression CATT6–8 alleles, and key oncogenic pathways such as p53. Specifically, elucidating whether lung cancer patients carrying these high-expression CATT alleles and wild-type p53 exhibit poorer prognosis, and whether elevated MIF levels attenuate the tumor-suppressive activity of p53, could enhance our understanding of disease mechanisms and guide more effective therapies. Furthermore, assessing whether high-expression CATT alleles predict resistance to PD-1/PD-L1 checkpoint inhibitors could provide insights into the genetic basis of therapeutic response.
In a related context, the MIF CATT polymorphism has been extensively studied in inflammatory and autoimmune diseases, where individuals carrying high-expression alleles may benefit from MIF inhibition, including steroid-sparing strategies for treatment-resistant or glucocorticoid-dependent patients [165]. Extending these insights into cancer, particularly lung cancer, is a critical next step. Although MIF has been investigated in various cancers, the specific impact of high-expression MIF alleles on cancer biology remains poorly understood and has not yet been examined in lung cancer, representing a critical knowledge gap. These high-expression alleles are relatively common in the population, and understanding their influence on lung cancer initiation, progression, and metastasis could help identify patients genetically predisposed to elevated MIF levels who are most likely to benefit from MIF-targeted therapies. Future studies should assess the efficacy of MIF inhibition specifically in carriers of these alleles and explore the potential of small molecules, antibodies, peptide-based inhibitors, or nanobody-based strategies, with the goal of advancing personalized medicine approaches that tailor treatment to patients genetically primed for high MIF expression.
Author Contributions
Conceptualization of the review: MAS, OLG, IO, DF, MEA and SCD; writing—original draft preparation: MAS; critical reading and reviewing: OLG, IO, CO'C, DF, MEA and SCD; preparation of the figures: MAS. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
No new data were created or analyzed in this review. Data sharing is therefore not applicable.
Funding
This work was financially funded by a Research Ireland ARC Hub for Therapeutics grant.
Acknowledgments
The figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflicts of interest. The funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| 4-IPP | 4-iodo-6-phenylpyrimidine |
| AP1 | Activator Protein 1 |
| AP4 | Activator Protein 4 |
| CMFT | 1-methoxy-5-formyl-4,6,8-trihydroxyphenazine |
| cPLA2 | Cytosolic Phospholipase A2 |
| CXCL8 | C-X-C motif Chemokine Ligand 8 |
| CXCR | C-X-C Chemokine Receptor |
| EMT | epithelial-to-Mesenchymal Transition |
| ERK | Extracellular Signal-Regulated Kinase |
| FEV1 | Forced Expiratory Volume in 1 second |
| HIF-1α | Hypoxia-Inducible Factor 1α |
| JNK | c-Jun N-terminal Kinase |
| LLC | Lewis Lung Carcinoma |
| MAPK | Mitogen-Activated Protein Kinase |
| MIF | Macrophage Migration Inhibitory Factor |
| MOG | Myelin Oligodendrocyte Glycoprotein |
| NF-κB | Nuclear Factor kappa B |
| NSCLC | Non-Small Cell Lung Cancer |
| oxMIF | oxidized MIF |
| PD-1 | Programmed Cell Death Protein-1 |
| PD-L1 | Programmed Death-Ligand-1 |
| PI3K/Akt | Phosphatidylinositol-3-Kinase/Protein Kinase B |
| RA | Rheumatoid Arthritis |
| rhMIF | recombinant human MIF |
| SCLC | Small cell Lung Cancer |
| SLE | Systemic Lupus Erythematosus |
| SNP | Single-Nucleotide Polymorphism |
| TIMP1 | Tissue Inhibitor of Metalloproteinase-1 |
| TNF-α | Tumor Necrosis Factor-alpha |
| VEGF | Vascular Endothelial Growth Factor |
References
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. Adv Exp Med Biol 2016, 893, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J Clin 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J Clin 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Bade, B.C.; Dela Cruz, C.S. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin Chest Med 2020, 41, 1–24. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, Y.; Liu, J.; Feng, L.; Yu, J.; Chen, D. Global burden of lung cancer in 2022 and projections to 2050: Incidence and mortality estimates from GLOBOCAN. Cancer Epidemiol 2024, 93, 102693. [Google Scholar] [CrossRef]
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011, 61, 212–236. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin Chest Med 2011, 32, 605–644. [Google Scholar] [CrossRef]
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA Cancer J Clin 2025, 75, 10–45. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, B.; He, S. Advances and challenges in the treatment of lung cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2023, 169, 115891. [Google Scholar] [CrossRef]
- Lu, T.; Yang, X.; Huang, Y.; Zhao, M.; Li, M.; Ma, K.; Yin, J.; Zhan, C.; Wang, Q. Trends in the incidence, treatment, and survival of patients with lung cancer in the last four decades. Cancer Manag Res 2019, 11, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung cancer. Lancet (London, England) 2021, 398, 535–554. [Google Scholar] [CrossRef]
- Wathoni, N.; Puluhulawa, L.E.; Joni, I.M.; Muchtaridi, M.; Mohammed, A.F.A.; Elamin, K.M.; Milanda, T.; Gozali, D. Monoclonal antibody as a targeting mediator for nanoparticle targeted delivery system for lung cancer. Drug Deliv 2022, 29, 2959–2970. [Google Scholar] [CrossRef]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol Biomarkers Prev 2019, 28, 1563–1579. [Google Scholar] [CrossRef]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet (London, England) 2018, 391, 1023–1075. [Google Scholar] [CrossRef]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.S.; Bannon, F.; Ahn, J.V.; Johnson, C.J.; Bonaventure, A.; et al. Global surveillance of cancer survival 1995-2009: Analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet (London, England) 2015, 385, 977–1010. [Google Scholar] [CrossRef]
- Bi, J.H.; Tuo, J.Y.; Xiao, Y.X.; Tang, D.D.; Zhou, X.H.; Jiang, Y.F.; Ji, X.W.; Tan, Y.T.; Yuan, H.Y.; Xiang, Y.B. Observed and relative survival trends of lung cancer: A systematic review of population-based cancer registration data. Thorac Cancer 2024, 15, 142–151. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J Clin 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.C.; Shi, L.; Zhu, B.; Min, Z.; Jin, J. Genome analyses identify the genetic modification of lung cancer subtypes. Semin Cancer Biol 2017, 42, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Rina, A.; Maffeo, D.; Minnai, F.; Esposito, M.; Palmieri, M.; Serio, V.B.; Rosati, D.; Mari, F.; Frullanti, E.; Colombo, F. The Genetic Analysis and Clinical Therapy in Lung Cancer: Current Advances and Future Directions. Cancers (Basel) 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, B.; Łukasiewicz, H.; Samulak, D.; Piekarska, E.; Kołaciński, R.; Romanowicz, H. Lung Cancer-Epidemiology, Pathogenesis, Treatment and Molecular Aspect (Review of Literature). International journal of molecular sciences 2025, 26. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ashwood, L.M.; Kondrashova, O.; Strasser, A.; Kelly, G.; Sutherland, K.D. Breathing new insights into the role of mutant p53 in lung cancer. Oncogene 2025, 44, 115–129. [Google Scholar] [CrossRef]
- Saleh, M.M.; Scheffler, M.; Merkelbach-Bruse, S.; Scheel, A.H.; Ulmer, B.; Wolf, J.; Buettner, R. Comprehensive Analysis of TP53 and KEAP1 Mutations and Their Impact on Survival in Localized- and Advanced-Stage NSCLC. J Thorac Oncol 2022, 17, 76–88. [Google Scholar] [CrossRef]
- Kaye, F.J. RB and cyclin dependent kinase pathways: Defining a distinction between RB and p16 loss in lung cancer. Oncogene 2002, 21, 6908–6914. [Google Scholar] [CrossRef]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res 2020, 40, 6009–6015. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Bloom, B.R.; Bennett, B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science (New York, N.Y.) 1966, 153, 80–82. [Google Scholar] [CrossRef]
- Kasama, T.; Ohtsuka, K.; Sato, M.; Takahashi, R.; Wakabayashi, K.; Kobayashi, K. Macrophage migration inhibitory factor: A multifunctional cytokine in rheumatic diseases. Arthritis 2010, 2010, 106202. [Google Scholar] [CrossRef]
- Weiser, W.Y.; Temple, P.A.; Witek-Giannotti, J.S.; Remold, H.G.; Clark, S.C.; David, J.R. Molecular cloning of a cDNA encoding a human macrophage migration inhibitory factor. Proceedings of the National Academy of Sciences of the United States of America 1989, 86, 7522–7526. [Google Scholar] [CrossRef] [PubMed]
- Calandra, T.; Bernhagen, J.; Mitchell, R.A.; Bucala, R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. The Journal of experimental medicine 1994, 179, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Bernhagen, J.; Calandra, T.; Mitchell, R.; Martin, S.; Tracey, K.; Voelter, W.; Manogue, K.; Cerami, A.; Bucala, R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 1993, 365, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF signal transduction initiated by binding to CD74. The Journal of experimental medicine 2003, 197, 1467–1476. [Google Scholar] [CrossRef]
- Sugimoto, H.; Taniguchi, M.; Nakagawa, A.; Tanaka, I.; Suzuki, M.; Nishihira, J. Crystal structure of human D-dopachrome tautomerase, a homologue of macrophage migration inhibitory factor, at 1.54 A resolution. Biochemistry 1999, 38, 3268–3279. [Google Scholar] [CrossRef]
- Michelet, C.; Danchin, E.G.; Jaouannet, M.; Bernhagen, J.; Panstruga, R.; Kogel, K.-H.; Keller, H.; Coustau, C. Cross-kingdom analysis of diversity, evolutionary history, and site selection within the eukaryotic macrophage migration inhibitory factor superfamily. Genes 2019, 10, 740. [Google Scholar] [CrossRef]
- Schinagl, A.; Kerschbaumer, R.J.; Sabarth, N.; Douillard, P.; Scholz, P.; Voelkel, D.; Hollerweger, J.C.; Goettig, P.; Brandstetter, H.; Scheiflinger, F. Role of the cysteine 81 residue of macrophage migration inhibitory factor as a molecular redox switch. Biochemistry 2018, 57, 1523–1532. [Google Scholar] [CrossRef]
- Skeens, E.; Gadzuk-Shea, M.; Shah, D.; Bhandari, V.; Schweppe, D.K.; Berlow, R.B.; Lisi, G.P. Redox-dependent structure and dynamics of macrophage migration inhibitory factor reveal sites of latent allostery. Structure 2022, 30, 840–850. e846. [Google Scholar] [CrossRef]
- Thiele, M.; Donnelly, S.C.; Mitchell, R.A. OxMIF: A druggable isoform of macrophage migration inhibitory factor in cancer and inflammatory diseases. J Immunother Cancer 2022, 10. [Google Scholar] [CrossRef]
- Thiele, M.; Kerschbaumer, R.J.; Tam, F.W.; Völkel, D.; Douillard, P.; Schinagl, A.; Kühnel, H.; Smith, J.; McDaid, J.P.; Bhangal, G. Selective targeting of a disease-related conformational isoform of macrophage migration inhibitory factor ameliorates inflammatory conditions. The Journal of Immunology 2015, 195, 2343–2352. [Google Scholar] [CrossRef]
- Dickerhof, N.; Schindler, L.; Bernhagen, J.; Kettle, A.J.; Hampton, M.B. Macrophage migration inhibitory factor (MIF) is rendered enzymatically inactive by myeloperoxidase-derived oxidants but retains its immunomodulatory function. Free Radical Biology and Medicine 2015, 89, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.A.; Metz, C.N.; Peng, T.; Bucala, R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. The Journal of Biological Chemistry 1999, 274, 18100–18106. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.; Sun, S.; Al-Abed, Y. MIF, a controversial cytokine: A review of structural features, challenges, and opportunities for drug development. Expert Opin Ther Targets 2016, 20, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Swantek, J.L.; Cobb, M.H.; Geppert, T.D. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: Glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol Cell Biol 1997, 17, 6274–6282. [Google Scholar] [CrossRef]
- Jankauskas, S.S.; Wong, D.W.; Bucala, R.; Djudjaj, S.; Boor, P. Evolving complexity of MIF signaling. Cellular signalling 2019, 57, 76–88. [Google Scholar] [CrossRef]
- Conroy, H.; Mawhinney, L.; Donnelly, S.C. Inflammation and cancer: Macrophage migration inhibitory factor (MIF)--the potential missing link. Qjm 2010, 103, 831–836. [Google Scholar] [CrossRef]
- Rosengren, E.; Aman, P.; Thelin, S.; Hansson, C.; Ahlfors, S.; Björk, P.; Jacobsson, L.; Rorsman, H. The macrophage migration inhibitory factor MIF is a phenylpyruvate tautomerase. FEBS letters 1997, 417, 85–88. [Google Scholar] [CrossRef]
- Kleemann, R.; Kapurniotu, A.; Frank, R.W.; Gessner, A.; Mischke, R.; Flieger, O.; Jüttner, S.; Brunner, H.; Bernhagen, J. Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol-protein oxidoreductase. J Mol Biol 1998, 280, 85–102. [Google Scholar] [CrossRef]
- Thiele, M.; Bernhagen, J. Link between macrophage migration inhibitory factor and cellular redox regulation. Antioxidants & redox signaling 2005, 7, 1234–1248. [Google Scholar] [CrossRef]
- Matsunaga, J.; Sinha, D.; Pannell, L.; Santis, C.; Solano, F.; Wistow, G.J.; Hearing, V.J. Enzyme activity of macrophage migration inhibitory factor toward oxidized catecholamines. The Journal of Biological Chemistry 1999, 274, 3268–3271. [Google Scholar] [CrossRef]
- Fingerle-Rowson, G.; Kaleswarapu, D.R.; Schlander, C.; Kabgani, N.; Brocks, T.; Reinart, N.; Busch, R.; Schütz, A.; Lue, H.; Du, X.; et al. A tautomerase-null macrophage migration-inhibitory factor (MIF) gene knock-in mouse model reveals that protein interactions and not enzymatic activity mediate MIF-dependent growth regulation. Mol Cell Biol 2009, 29, 1922–1932. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, D.; Zierow, S.; Syed, M.; Bucala, R.; Bhandari, V.; Lolis, E.J. Targeting distinct tautomerase sites of D-DT and MIF with a single molecule for inhibition of neutrophil lung recruitment. FASEB journal: Official publication of the Federation of American Societies for Experimental Biology 2014, 28, 4961–4971. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Leng, L.; Gandavadi, S.; Du, X.; Bucala, R.; Jorgensen, W.L. Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. Journal of medicinal chemistry 2009, 52, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Pantouris, G.; Syed, M.A.; Fan, C.; Rajasekaran, D.; Cho, T.Y.; Rosenberg, E.M., Jr.; Bucala, R.; Bhandari, V.; Lolis, E.J. An Analysis of MIF Structural Features that Control Functional Activation of CD74. Chem Biol 2015, 22, 1197–1205. [Google Scholar] [CrossRef]
- El-Turk, F.; Cascella, M.; Ouertatani-Sakouhi, H.; Narayanan, R.L.; Leng, L.; Bucala, R.; Zweckstetter, M.; Rothlisberger, U.; Lashuel, H.A. The conformational flexibility of the carboxy terminal residues 105-114 is a key modulator of the catalytic activity and stability of macrophage migration inhibitory factor. Biochemistry 2008, 47, 10740–10756. [Google Scholar] [CrossRef]
- Al-Abed, Y.; Dabideen, D.; Aljabari, B.; Valster, A.; Messmer, D.; Ochani, M.; Tanovic, M.; Ochani, K.; Bacher, M.; Nicoletti, F.; et al. ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. The Journal of Biological Chemistry 2005, 280, 36541–36544. [Google Scholar] [CrossRef]
- Adamali, H.; Armstrong, M.E.; McLaughlin, A.M.; Cooke, G.; McKone, E.; Costello, C.M.; Gallagher, C.G.; Leng, L.; Baugh, J.A.; Fingerle-Rowson, G.; et al. Macrophage migration inhibitory factor enzymatic activity, lung inflammation, and cystic fibrosis. American journal of respiratory and critical care medicine 2012, 186, 162–169. [Google Scholar] [CrossRef]
- Tynan, A.; Mawhinney, L.; Armstrong, M.E.; O'Reilly, C.; Kennedy, S.; Caraher, E.; Jülicher, K.; O'Dwyer, D.; Maher, L.; Schaffer, K.; et al. Macrophage migration inhibitory factor enhances Pseudomonas aeruginosa biofilm formation, potentially contributing to cystic fibrosis pathogenesis. FASEB journal: Official publication of the Federation of American Societies for Experimental Biology 2017, 31, 5102–5110. [Google Scholar] [CrossRef]
- Winner, M.; Meier, J.; Zierow, S.; Rendon, B.E.; Crichlow, G.V.; Riggs, R.; Bucala, R.; Leng, L.; Smith, N.; Lolis, E.; et al. A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer research 2008, 68, 7253–7257. [Google Scholar] [CrossRef]
- Mawhinney, L.; Armstrong, M.E.; C, O.R.; Bucala, R.; Leng, L.; Fingerle-Rowson, G.; Fayne, D.; Keane, M.P.; Tynan, A.; Maher, L.; et al. Macrophage migration inhibitory factor (MIF) enzymatic activity and lung cancer. Mol Med 2015, 20, 729–735. [Google Scholar] [CrossRef]
- Xu, L.; Li, Y.; Sun, H.; Zhen, X.; Qiao, C.; Tian, S.; Hou, T. Current developments of macrophage migration inhibitory factor (MIF) inhibitors. Drug discovery today 2013, 18, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Camacho Meza, G.; Avalos Navarro, G.; De La Cruz Mosso, U.; Ramírez Patiño, R.; Muñoz Valle, J.F.; Bautista Herrera, L.A. Macrophage migration inhibitory factor: Exploring physiological roles and comparing health benefits against oncogenic and autoimmune risks (Review). International journal of molecular medicine 2025, 56. [Google Scholar] [CrossRef] [PubMed]
- Donn, R.; Alourfi, Z.; De Benedetti, F.; Meazza, C.; Zeggini, E.; Lunt, M.; Stevens, A.; Shelley, E.; Lamb, R.; Ollier, W.E.; et al. Mutation screening of the macrophage migration inhibitory factor gene: Positive association of a functional polymorphism of macrophage migration inhibitory factor with juvenile idiopathic arthritis. Arthritis and rheumatism 2002, 46, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Radstake, T.R.; Sweep, F.C.; Welsing, P.; Franke, B.; Vermeulen, S.H.; Geurts-Moespot, A.; Calandra, T.; Donn, R.; van Riel, P.L. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis and rheumatism 2005, 52, 3020–3029. [Google Scholar] [CrossRef]
- Baugh, J.A.; Chitnis, S.; Donnelly, S.C.; Monteiro, J.; Lin, X.; Plant, B.J.; Wolfe, F.; Gregersen, P.K.; Bucala, R. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun 2002, 3, 170–176. [Google Scholar] [CrossRef]
- Llamas-Covarrubias, M.A.; Valle, Y.; Bucala, R.; Navarro-Hernández, R.E.; Palafox-Sánchez, C.A.; Padilla-Gutiérrez, J.R.; Parra-Rojas, I.; Bernard-Medina, A.G.; Reyes-Castillo, Z.; Muñoz-Valle, J.F. Macrophage migration inhibitory factor (MIF): Genetic evidence for participation in early onset and early stage rheumatoid arthritis. Cytokine 2013, 61, 759–765. [Google Scholar] [CrossRef]
- Sreih, A.; Ezzeddine, R.; Leng, L.; LaChance, A.; Yu, G.; Mizue, Y.; Subrahmanyan, L.; Pons-Estel, B.A.; Abelson, A.K.; Gunnarsson, I.; et al. Dual effect of the macrophage migration inhibitory factor gene on the development and severity of human systemic lupus erythematosus. Arthritis and rheumatism 2011, 63, 3942–3951. [Google Scholar] [CrossRef]
- Yao, J.; Leng, L.; Sauler, M.; Fu, W.; Zheng, J.; Zhang, Y.; Du, X.; Yu, X.; Lee, P.; Bucala, R. Transcription factor ICBP90 regulates the MIF promoter and immune susceptibility locus. The Journal of clinical investigation 2016, 126, 732–744. [Google Scholar] [CrossRef]
- Valdez, C.N.; Sánchez-Zuno, G.A.; Bucala, R.; Tran, T.T. Macrophage Migration Inhibitory Factor (MIF) and D-Dopachrome Tautomerase (DDT): Pathways to Tumorigenesis and Therapeutic Opportunities. International journal of molecular sciences 2024, 25. [Google Scholar] [CrossRef]
- Illescas, O.; Gomez-Verjan, J.C.; García-Velázquez, L.; Govezensky, T.; Rodriguez-Sosa, M. Macrophage Migration Inhibitory Factor -173 G/C Polymorphism: A Global Meta-Analysis across the Disease Spectrum. Frontiers in genetics 2018, 9, 55. [Google Scholar] [CrossRef]
- Du, X.; Li, R.; Song, S.; Ma, L.; Xue, H. The Role of MIF-173G/C Gene Polymorphism in the Susceptibility of Autoimmune Diseases. Mediators of inflammation 2020, 2020, 7825072. [Google Scholar] [CrossRef]
- Fouda, H.; Ibrahim, W.N.; Shi, Z.; Alahmadi, F.; Almohammadi, Y.; Al-Haidose, A.; Abdallah, A.M. Impact of the MIF -173G/C variant on cardiovascular disease risk: A meta-analysis of 9,047 participants. Frontiers in cardiovascular medicine 2024, 11, 1323423. [Google Scholar] [CrossRef]
- Shan, Z.X.; Fu, Y.H.; Yu, X.Y.; Deng, C.Y.; Tan, H.H.; Lin, Q.X.; Yu, H.M.; Lin, S.G. Association of the polymorphism of macrophage migration inhibitory factor gene with coronary heart disease in Chinese population. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics 2006, 23, 548–550. [Google Scholar]
- Tong, X.; Yan, Z.; Zhou, Q.; Liu, S.; Han, J.; Ma, Y.; Yang, X.; Fan, H. Association between the MIF-173G/C Polymorphism and Serum MIF levels with Pulmonary Tuberculosis: A Meta-analysis. Sci Rep 2017, 7, 234. [Google Scholar] [CrossRef]
- Zhang, X.; Weng, W.; Xu, W.; Wang, Y.; Yu, W.; Tang, X.; Ma, L.; Pan, Q.; Wang, J.; Sun, F. The association between the migration inhibitory factor -173G/C polymorphism and cancer risk: A meta-analysis. OncoTargets and therapy 2015, 8, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.C.; Lee, Y.H. Associations between circulating macrophage migration inhibitory factor (MIF) levels and rheumatoid arthritis, and between MIF gene polymorphisms and disease susceptibility: A meta-analysis. Postgraduate medical journal 2018, 94, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Sam, N.B.; Guan, S.Y.; Wang, P.; Li, X.M.; Wang, D.G.; Pan, H.F.; Ye, D.Q. Levels of the macrophage migration inhibitory factor and polymorphisms in systemic lupus erythematosus: A meta-analysis. Archives of medical science: AMS 2021, 17, 1232–1240. [Google Scholar] [CrossRef]
- Plant, B.J.; Gallagher, C.G.; Bucala, R.; Baugh, J.A.; Chappell, S.; Morgan, L.; O'Connor, C.M.; Morgan, K.; Donnelly, S.C. Cystic fibrosis, disease severity, and a macrophage migration inhibitory factor polymorphism. American journal of respiratory and critical care medicine 2005, 172, 1412–1415. [Google Scholar] [CrossRef]
- Chuo, D.; Lin, D.; Yin, M.; Chen, Y. Genetic Variants of the MIF Gene and Susceptibility of Rectal Cancer. Pharmgenomics Pers Med 2021, 14, 55–60. [Google Scholar] [CrossRef]
- Li, H.; Zang, J.; Wang, P.; Dai, L.; Zhang, J.; Wang, K. Gastric cancer susceptibility in gastric cancer relatives: Attributable risks of Macrophage migration inhibitory factor promoter polymorphism and Helicobacter pylori. Cytokine 2012, 60, 346–351. [Google Scholar] [CrossRef]
- Wu, S.; Sun, J.; Lian, J.; Shang, H.; Tao, H.; Xie, J.; Lin, W. Macrophage migration inhibitory factor promoter polymorphisms (-794CATT(5-7) ) as potential biomarker for early-stage cervical cancer. The journal of obstetrics and gynaecology research 2017, 43, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Siegler, K.L.; Vera, P.L.; Iczkowski, K.A.; Bifulco, C.; Lee, A.; Gregersen, P.K.; Leng, L.; Bucala, R. Macrophage migration inhibitory factor (MIF) gene polymorphisms are associated with increased prostate cancer incidence. Genes Immun 2007, 8, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Gutiérrez, E.; Ramos-Súarez, M.; Villalobos-Ayala, R.A.; Tlacuilo-Parra, A.; Muñoz-Valle, J.F.; Tarango-Martínez, V.; Valle, Y.; Padilla-Gutiérrez, J.R.; Rojas-Díaz, J.M.; Valdés-Alvarado, E. Haplotypes of. Mol Genet Genomic Med 2023, 11, e2252. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.J.; Fan, W.; Par-Young, J.; Piecychna, M.; Leng, L.; Israni-Winger, K.; Qing, H.; Gu, J.; Zhao, H.; Schulz, W.L.; et al. MIF is a common genetic determinant of COVID-19 symptomatic infection and severity. Qjm 2023, 116, 205–212. [Google Scholar] [CrossRef]
- Dunbar, H.; Hawthorne, I.J.; Tunstead, C.; Armstrong, M.E.; Donnelly, S.C.; English, K. Blockade of MIF biological activity ameliorates house dust mite-induced allergic airway inflammation in humanized MIF mice. FASEB journal: Official publication of the Federation of American Societies for Experimental Biology 2023, 37, e23072. [Google Scholar] [CrossRef]
- Hawthorne, I.J.; Dunbar, H.; Tunstead, C.; Schorpp, T.; Weiss, D.J.; Enes, S.R.; Dos Santos, C.C.; Armstrong, M.E.; Donnelly, S.C.; English, K. Human macrophage migration inhibitory factor potentiates mesenchymal stromal cell efficacy in a clinically relevant model of allergic asthma. Molecular therapy: The journal of the American Society of Gene Therapy 2023, 31, 3243–3258. [Google Scholar] [CrossRef]
- Dunbar, H.; Hawthorne, I.J.; McNamee, E.N.; Armstrong, M.E.; Donnelly, S.C.; English, K. The human MIF polymorphism CATT(7) enhances pro-inflammatory macrophage polarization in a clinically relevant model of allergic airway inflammation. FASEB journal: Official publication of the Federation of American Societies for Experimental Biology 2024, 38, e23576. [Google Scholar] [CrossRef]
- Tran, T.T.; Sánchez-Zuno, G.A.; Osmani, L.; Caulfield, J.; Valdez, C.N.; Piecychna, M.; Leng, L.; Armstrong, M.E.; Donnelly, S.C.; Bifulco, C.B.; et al. Improving immunotherapy responses by dual inhibition of macrophage migration inhibitory factor and PD-1. JCI insight 2025, 10. [Google Scholar] [CrossRef]
- Martínez, A.; Orozco, G.; Varadé, J.; Sánchez López, M.; Pascual, D.; Balsa, A.; García, A.; de la Concha, E.G.; Fernández-Gutiérrez, B.; Martín, J.; et al. Macrophage migration inhibitory factor gene: Influence on rheumatoid arthritis susceptibility. Hum Immunol 2007, 68, 744–747. [Google Scholar] [CrossRef]
- Barton, A.; Lamb, R.; Symmons, D.; Silman, A.; Thomson, W.; Worthington, J.; Donn, R. Macrophage migration inhibitory factor (MIF) gene polymorphism is associated with susceptibility to but not severity of inflammatory polyarthritis. Genes & Immunity 2003, 4, 487–491. [Google Scholar] [CrossRef]
- Sánchez, E.; Gómez, L.M.; Lopez-Nevot, M.A.; González-Gay, M.A.; Sabio, J.M.; Ortego-Centeno, N.; de Ramón, E.; Anaya, J.M.; González-Escribano, M.F.; Koeleman, B.P.; et al. Evidence of association of macrophage migration inhibitory factor gene polymorphisms with systemic lupus erythematosus. Genes Immun 2006, 7, 433–436. [Google Scholar] [CrossRef]
- Przybyłowska, K.; Mrowicki, J.; Sygut, A.; Narbutt, P.; Dziki, Ł.; Dziki, A.; Majsterek, I. Contribution of the -173 G/C polymorphism of macrophage migration inhibitory factor gene to the risk of inflammatory bowel diseases. Polski przeglad chirurgiczny 2011, 83, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Assis, D.N.; Takahashi, H.; Leng, L.; Zeniya, M.; Boyer, J.L.; Bucala, R. A Macrophage Migration Inhibitory Factor Polymorphism Is Associated with Autoimmune Hepatitis Severity in US and Japanese Patients. Digestive diseases and sciences 2016, 61, 3506–3512. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, F.; Zhang, X.; Li, Y.; Ma, H.; Zhou, Y.; Jin, Y.; Wang, H.; Bai, J.; Zhang, G.; et al. Association of MIF promoter polymorphisms with psoriasis in a Han population in northeastern China. Journal of dermatological science 2009, 53, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Savva, A.; Brouwer, M.C.; Roger, T.; Valls Serón, M.; Le Roy, D.; Ferwerda, B.; van der Ende, A.; Bochud, P.Y.; van de Beek, D.; Calandra, T. Functional polymorphisms of macrophage migration inhibitory factor as predictors of morbidity and mortality of pneumococcal meningitis. Proceedings of the National Academy of Sciences of the United States of America 2016, 113, 3597–3602. [Google Scholar] [CrossRef]
- Avalos-Navarro, G.; Del Toro-Arreola, A.; Daneri-Navarro, A.; Quintero-Ramos, A.; Bautista-Herrera, L.A.; Franco Topete, R.A.; Anaya Macias, B.U.; Javalera Castro, D.I.; Morán-Mendoza, A.J.; Oceguera-Villanueva, A.; et al. Association of the genetic variants (-794 CATT5-8 and -173 G > C) of macrophage migration inhibitory factor (MIF) with higher soluble levels of MIF and TNFα in women with breast cancer. Journal of clinical laboratory analysis 2020, 34, e23209. [Google Scholar] [CrossRef]
- Qin, L.; Qin, J.; Lv, X.; Yin, C.; Zhang, Q.; Zhang, J. MIF promoter polymorphism increases peripheral blood expression levels, contributing to increased susceptibility and poor prognosis in hepatocellular carcinoma. Oncol Lett 2021, 22, 549. [Google Scholar] [CrossRef]
- Calandra, T.; Echtenacher, B.; Roy, D.L.; Pugin, J.; Metz, C.N.; Hültner, L.; Heumann, D.; Männel, D.; Bucala, R.; Glauser, M.P. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nature medicine 2000, 6, 164–170. [Google Scholar] [CrossRef]
- Donnelly, S.C.; Haslett, C.; Reid, P.T.; Grant, I.S.; Wallace, W.A.; Metz, C.N.; Bruce, L.J.; Bucala, R. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nature medicine 1997, 3, 320–323. [Google Scholar] [CrossRef]
- Donnelly, S.C.; Bucala, R. Macrophage migration inhibitory factor: A regulator of glucocorticoid activity with a critical role in inflammatory disease. Mol Med Today 1997, 3, 502–507. [Google Scholar] [CrossRef]
- Leech, M.; Metz, C.; Bucala, R.; Morand, E.F. Regulation of macrophage migration inhibitory factor by endogenous glucocorticoids in rat adjuvant-induced arthritis. Arthritis and rheumatism 2000, 43, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K.; Bucala, R. Macrophage migration inhibitory factor, MIF alleles, and the genetics of inflammatory disorders: Incorporating disease outcome into the definition of phenotype. Arthritis and rheumatism 2003, 48, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.G.; Haslett, C.; Hirani, N.; Greening, A.P.; Rahman, I.; Metz, C.N.; Bucala, R.; Donnelly, S.C. Human circulating eosinophils secrete macrophage migration inhibitory factor (MIF). Potential role in asthma. The Journal of clinical investigation 1998, 101, 2869–2874. [Google Scholar] [CrossRef] [PubMed]
- Mizue, Y.; Ghani, S.; Leng, L.; McDonald, C.; Kong, P.; Baugh, J.; Lane, S.J.; Craft, J.; Nishihira, J.; Donnelly, S.C.; et al. Role for macrophage migration inhibitory factor in asthma. Proceedings of the National Academy of Sciences of the United States of America 2005, 102, 14410–14415. [Google Scholar] [CrossRef]
- Allam, V.; Pavlidis, S.; Liu, G.; Kermani, N.Z.; Simpson, J.; To, J.; Donnelly, S.; Guo, Y.K.; Hansbro, P.M.; Phipps, S.; et al. Macrophage migration inhibitory factor promotes glucocorticoid resistance of neutrophilic inflammation in a murine model of severe asthma. Thorax 2023, 78, 661–673. [Google Scholar] [CrossRef]
- Dunbar, H.; Hawthorne, I.J.; Tunstead, C.; Dunlop, M.; Volkova, E.; Weiss, D.J.; Santos, C.C.D.; Armstrong, M.E.; Donnelly, S.C.; English, K. The VEGF-Mediated Cytoprotective Ability of MIF-Licensed Mesenchymal Stromal Cells in House Dust Mite-Induced Epithelial Damage. Eur J Immunol 2025, 55, e202451205. [Google Scholar] [CrossRef]
- Morand, E.F.; Leech, M.; Weedon, H.; Metz, C.; Bucala, R.; Smith, M.D. Macrophage migration inhibitory factor in rheumatoid arthritis: Clinical correlations. Rheumatology (Oxford) 2002, 41, 558–562. [Google Scholar] [CrossRef]
- Connelly, K.L.; Kandane-Rathnayake, R.; Hoi, A.; Nikpour, M.; Morand, E.F. Association of MIF, but not type I interferon-induced chemokines, with increased disease activity in Asian patients with systemic lupus erythematosus. Sci Rep 2016, 6, 29909. [Google Scholar] [CrossRef]
- Vincent, F.B.; Slavin, L.; Hoi, A.Y.; Kitching, A.R.; Mackay, F.; Harris, J.; Kandane-Rathnayake, R.; Morand, E.F. Analysis of urinary macrophage migration inhibitory factor in systemic lupus erythematosus. Lupus Sci Med 2018, 5, e000277. [Google Scholar] [CrossRef]
- Foote, A.; Briganti, E.M.; Kipen, Y.; Santos, L.; Leech, M.; Morand, E.F. Macrophage migration inhibitory factor in systemic lupus erythematosus. The Journal of rheumatology 2004, 31, 268–273. [Google Scholar]
- Lehmann, L.E.; Novender, U.; Schroeder, S.; Pietsch, T.; von Spiegel, T.; Putensen, C.; Hoeft, A.; Stüber, F. Plasma levels of macrophage migration inhibitory factor are elevated in patients with severe sepsis. Intensive Care Med 2001, 27, 1412–1415. [Google Scholar] [CrossRef] [PubMed]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proceedings of the National Academy of Sciences of the United States of America 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.D.; Shoaibi, M.A.; Maestro, R.; Carnero, A.; Hannon, G.J.; Beach, D.H. A proinflammatory cytokine inhibits p53 tumor suppressor activity. The Journal of experimental medicine 1999, 190, 1375–1382. [Google Scholar] [CrossRef]
- Mitchell, R.A.; Liao, H.; Chesney, J.; Fingerle-Rowson, G.; Baugh, J.; David, J.; Bucala, R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: Regulatory role in the innate immune response. Proceedings of the National Academy of Sciences of the United States of America 2002, 99, 345–350. [Google Scholar] [CrossRef]
- Fingerle-Rowson, G.; Petrenko, O.; Metz, C.N.; Forsthuber, T.G.; Mitchell, R.; Huss, R.; Moll, U.; Müller, W.; Bucala, R. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proceedings of the National Academy of Sciences of the United States of America 2003, 100, 9354–9359. [Google Scholar] [CrossRef]
- O'Reilly, C.; Doroudian, M.; Mawhinney, L.; Donnelly, S.C. Targeting MIF in Cancer: Therapeutic Strategies, Current Developments, and Future Opportunities. Med Res Rev 2016, 36, 440–460. [Google Scholar] [CrossRef]
- Amin, M.A.; Volpert, O.V.; Woods, J.M.; Kumar, P.; Harlow, L.A.; Koch, A.E. Migration inhibitory factor mediates angiogenesis via mitogen-activated protein kinase and phosphatidylinositol kinase. Circ Res 2003, 93, 321–329. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, G.; Yang, S.; Qiao, J.; Li, T.; Yang, S.; Hong, Y. Macrophage migration inhibitory factor regulating the expression of VEGF-C through MAPK signal pathways in breast cancer MCF-7 cell. World journal of surgical oncology 2016, 14, 51. [Google Scholar] [CrossRef]
- Baugh, J.A.; Gantier, M.; Li, L.; Byrne, A.; Buckley, A.; Donnelly, S.C. Dual regulation of macrophage migration inhibitory factor (MIF) expression in hypoxia by CREB and HIF-1. Biochemical and biophysical research communications 2006, 347, 895–903. [Google Scholar] [CrossRef]
- Oda, S.; Oda, T.; Nishi, K.; Takabuchi, S.; Wakamatsu, T.; Tanaka, T.; Adachi, T.; Fukuda, K.; Semenza, G.L.; Hirota, K. Macrophage migration inhibitory factor activates hypoxia-inducible factor in a p53-dependent manner. PLoS ONE 2008, 3, e2215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xing, C.; Deng, Y.; Ye, C.; Peng, H. HIF-1α signaling: Essential roles in tumorigenesis and implications in targeted therapies. Genes & diseases 2024, 11, 234–251. [Google Scholar] [CrossRef]
- Yang, S.; He, P.; Wang, J.; Schetter, A.; Tang, W.; Funamizu, N.; Yanaga, K.; Uwagawa, T.; Satoskar, A.R.; Gaedcke, J.; et al. A Novel MIF Signaling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer research 2016, 76, 3838–3850. [Google Scholar] [CrossRef] [PubMed]
- He, X.X.; Chen, K.; Yang, J.; Li, X.Y.; Gan, H.Y.; Liu, C.Y.; Coleman, T.R.; Al-Abed, Y. Macrophage migration inhibitory factor promotes colorectal cancer. Mol Med 2009, 15, 1–10. [Google Scholar] [CrossRef]
- Figueiredo, C.R.; Azevedo, R.A.; Mousdell, S.; Resende-Lara, P.T.; Ireland, L.; Santos, A.; Girola, N.; Cunha, R.; Schmid, M.C.; Polonelli, L.; et al. Blockade of MIF-CD74 Signalling on Macrophages and Dendritic Cells Restores the Antitumour Immune Response Against Metastatic Melanoma. Frontiers in immunology 2018, 9, 1132. [Google Scholar] [CrossRef]
- de Azevedo, R.A.; Shoshan, E.; Whang, S.; Markel, G.; Jaiswal, A.R.; Liu, A.; Curran, M.A.; Travassos, L.R.; Bar-Eli, M. MIF inhibition as a strategy for overcoming resistance to immune checkpoint blockade therapy in melanoma. Oncoimmunology 2020, 9, 1846915. [Google Scholar] [CrossRef]
- He, F.; Xu, J.; Zeng, F.; Wang, B.; Yang, Y.; Xu, J.; Sun, X.; Ren, T.; Tang, X. Integrative analysis of Ewing's sarcoma reveals that the MIF-CD74 axis is a target for immunotherapy. Cell Commun Signal 2025, 23, 23. [Google Scholar] [CrossRef]
- Imaoka, M.; Tanese, K.; Masugi, Y.; Hayashi, M.; Sakamoto, M. Macrophage migration inhibitory factor-CD74 interaction regulates the expression of programmed cell death ligand 1 in melanoma cells. Cancer science 2019, 110, 2273–2283. [Google Scholar] [CrossRef]
- Liu, L.; Wang, J.; Wang, Y.; Chen, L.; Peng, L.; Bin, Y.; Ding, P.; Zhang, R.; Tong, F.; Dong, X. Blocking the MIF-CD74 axis augments radiotherapy efficacy for brain metastasis in NSCLC via synergistically promoting microglia M1 polarization. J Exp Clin Cancer Res 2024, 43, 128. [Google Scholar] [CrossRef]
- Lin, X.; Kang, K.; Chen, P.; Zeng, Z.; Li, G.; Xiong, W.; Yi, M.; Xiang, B. Regulatory mechanisms of PD-1/PD-L1 in cancers. Molecular cancer 2024, 23, 108. [Google Scholar] [CrossRef]
- Chang, E.; Pelosof, L.; Lemery, S.; Gong, Y.; Goldberg, K.B.; Farrell, A.T.; Keegan, P.; Veeraraghavan, J.; Wei, G.; Blumenthal, G.M.; et al. Systematic Review of PD-1/PD-L1 Inhibitors in Oncology: From Personalized Medicine to Public Health. The oncologist 2021, 26, e1786–e1799. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am J Cancer Res 2020, 10, 727–742. [Google Scholar]
- Guda, M.R.; Rashid, M.A.; Asuthkar, S.; Jalasutram, A.; Caniglia, J.L.; Tsung, A.J.; Velpula, K.K. Pleiotropic role of macrophage migration inhibitory factor in cancer. Am J Cancer Res 2019, 9, 2760–2773. [Google Scholar]
- Kindt, N.; Journe, F.; Laurent, G.; Saussez, S. Involvement of macrophage migration inhibitory factor in cancer and novel therapeutic targets. Oncol Lett 2016, 12, 2247–2253. [Google Scholar] [CrossRef]
- Koh, H.M.; Kim, D.C. Prognostic significance of macrophage migration inhibitory factor expression in cancer patients: A systematic review and meta-analysis. Medicine 2020, 99, e21575. [Google Scholar] [CrossRef]
- Pu, Y.; Yang, G.; Zhou, Y.; Pan, X.; Guo, T.; Chai, X. The Macrophage migration inhibitory factor is a vital player in Pan-Cancer by functioning as a M0 Macrophage biomarker. International immunopharmacology 2024, 134, 112198. [Google Scholar] [CrossRef]
- Lechien, J.R.; Nassri, A.; Kindt, N.; Brown, D.N.; Journe, F.; Saussez, S. Role of macrophage migration inhibitory factor in head and neck cancer and novel therapeutic targets: A systematic review. Head Neck 2017, 39, 2573–2584. [Google Scholar] [CrossRef]
- Schneider, K.L.; Claus, L.; Bucala, R.; Schulz-Heddergott, R. Targeting macrophage migration inhibitory factor as a potential therapeutic strategy in colorectal cancer. Oncogenesis 2025, 14, 30. [Google Scholar] [CrossRef]
- Schinagl, A.; Thiele, M.; Douillard, P.; Völkel, D.; Kenner, L.; Kazemi, Z.; Freissmuth, M.; Scheiflinger, F.; Kerschbaumer, R.J. Oxidized macrophage migration inhibitory factor is a potential new tissue marker and drug target in cancer. Oncotarget 2016, 7, 73486–73496. [Google Scholar] [CrossRef]
- Mahalingam, D.; Patel, M.R.; Sachdev, J.C.; Hart, L.L.; Halama, N.; Ramanathan, R.K.; Sarantopoulos, J.; Völkel, D.; Youssef, A.; de Jong, F.A.; et al. Phase I study of imalumab (BAX69), a fully human recombinant antioxidized macrophage migration inhibitory factor antibody in advanced solid tumours. British Journal of Clinical Pharmacology 2020, 86, 1836–1848. [Google Scholar] [CrossRef]
- Hussain, F.; Freissmuth, M.; Völkel, D.; Thiele, M.; Douillard, P.; Antoine, G.; Thurner, P.; Ehrlich, H.; Schwarz, H.P.; Scheiflinger, F.; et al. Human anti-macrophage migration inhibitory factor antibodies inhibit growth of human prostate cancer cells in vitro and in vivo. Molecular cancer therapeutics 2013, 12, 1223–1234. [Google Scholar] [CrossRef]
- Rendon, B.E.; Roger, T.; Teneng, I.; Zhao, M.; Al-Abed, Y.; Calandra, T.; Mitchell, R.A. Regulation of human lung adenocarcinoma cell migration and invasion by macrophage migration inhibitory factor. The Journal of Biological Chemistry 2007, 282, 29910–29918. [Google Scholar] [CrossRef]
- Nobre, C.C.; de Araújo, J.M.; Fernandes, T.A.; Cobucci, R.N.; Lanza, D.C.; Andrade, V.S.; Fernandes, J.V. Macrophage Migration Inhibitory Factor (MIF): Biological Activities and Relation with Cancer. Pathology oncology research: POR 2017, 23, 235–244. [Google Scholar] [CrossRef]
- Liu, W.; Yang, H.S.; Zhi, F.H.; Feng, Y.F.; Luo, H.H.; Zhu, Y.; Lei, Y.Y. Macrophage migration inhibitory factor may contribute to the occurrence of multiple primary lung adenocarcinomas. Clin Transl Med 2023, 13, e1368. [Google Scholar] [CrossRef]
- Koh, H.M.; Kim, D.C.; Kim, Y.M.; Song, D.H. Prognostic role of macrophage migration inhibitory factor expression in patients with squamous cell carcinoma of the lung. Thorac Cancer 2019, 10, 2209–2217. [Google Scholar] [CrossRef]
- Zhang, H.; Duan, J.; Wu, O. The expression of macrophage migration inhibitory factor in the non-small cell lung cancer. Saudi journal of biological sciences 2020, 27, 1527–1532. [Google Scholar] [CrossRef]
- Li, J.; Zhang, J.; Xie, F.; Peng, J.; Wu, X. Macrophage migration inhibitory factor promotes Warburg effect via activation of the NF-κB/HIF-1α pathway in lung cancer. International journal of molecular medicine 2018, 41, 1062–1068. [Google Scholar] [CrossRef]
- Guo, Y.; Hou, J.; Luo, Y.; Wang, D. Correction: Functional disruption of macrophage migration inhibitory factor (MIF) suppresses proliferation of human h460 lung cancer cells by caspase-dependent apoptosis. Cancer cell international 2013, 13, 84. [Google Scholar] [CrossRef]
- Coleman, A.M.; Rendon, B.E.; Zhao, M.; Qian, M.W.; Bucala, R.; Xin, D.; Mitchell, R.A. Cooperative regulation of non-small cell lung carcinoma angiogenic potential by macrophage migration inhibitory factor and its homolog, D-dopachrome tautomerase. Journal of immunology (Baltimore, Md.: 1950) 2008, 181, 2330–2337. [Google Scholar] [CrossRef]
- Carr, S.R.; Wang, H.; Hudlikar, R.; Lu, X.; Zhang, M.R.; Hoang, C.D.; Yan, F.; Schrump, D.S. A Unique Gene Signature Predicting Recurrence Free Survival in Stage IA Lung Adenocarcinoma. The Journal of thoracic and cardiovascular surgery 2023, 165, 1554–1564. [Google Scholar] [CrossRef]
- Kamimura, A.; Kamachi, M.; Nishihira, J.; Ogura, S.; Isobe, H.; Dosaka-Akita, H.; Ogata, A.; Shindoh, M.; Ohbuchi, T.; Kawakami, Y. Intracellular distribution of macrophage migration inhibitory factor predicts the prognosis of patients with adenocarcinoma of the lung. Cancer 2000, 89, 334–341. [Google Scholar] [CrossRef]
- Tomiyasu, M.; Yoshino, I.; Suemitsu, R.; Okamoto, T.; Sugimachi, K. Quantification of macrophage migration inhibitory factor mRNA expression in non-small cell lung cancer tissues and its clinical significance. Clinical cancer research: An official journal of the American Association for Cancer Research 2002, 8, 3755–3760. [Google Scholar] [PubMed]
- Khan, N.; Cromer, C.J.; Campa, M.; Patz, E.F., Jr. Clinical utility of serum amyloid A and macrophage migration inhibitory factor as serum biomarkers for the detection of nonsmall cell lung carcinoma. Cancer 2004, 101, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Tanaka, M.; Yoshida, M.; Umakoshi, M.; Nanjo, H.; Shiraishi, K.; Saito, M.; Kohno, T.; Kuriyama, S.; Konno, H.; et al. The low expression of miR-451 predicts a worse prognosis in non-small cell lung cancer cases. PLoS ONE 2017, 12, e0181270. [Google Scholar] [CrossRef] [PubMed]
- White, E.S.; Flaherty, K.R.; Carskadon, S.; Brant, A.; Iannettoni, M.D.; Yee, J.; Orringer, M.B.; Arenberg, D.A. Macrophage migration inhibitory factor and CXC chemokine expression in non-small cell lung cancer: Role in angiogenesis and prognosis. Clinical cancer research: An official journal of the American Association for Cancer Research 2003, 9, 853–860. [Google Scholar]
- Huang, W.C.; Kuo, K.T.; Wang, C.H.; Yeh, C.T.; Wang, Y. Cisplatin resistant lung cancer cells promoted M2 polarization of tumor-associated macrophages via the Src/CD155/MIF functional pathway. J Exp Clin Cancer Res 2019, 38, 180. [Google Scholar] [CrossRef]
- Peng, J.; Zhang, Y.; Li, B.; He, X.; Ding, C.; Hu, W. The inhibitory effect of M2 macrophage-derived exosomes on gefitinib resistant lung adenocarcinoma cells through the MIF/TIMP1/CD74 axis. Sci Rep 2025, 15, 28482. [Google Scholar] [CrossRef]
- Rupp, A.; Bahlmann, S.; Trimpop, N.; von Pawel, J.; Holdenrieder, S. Lack of clinical utility of serum macrophage migration inhibitory factor (MIF) for monitoring therapy response and estimating prognosis in advanced lung cancer. Tumour biology: The journal of the International Society for Oncodevelopmental Biology and Medicine 2024, 46, S341–S353. [Google Scholar] [CrossRef]
- McClelland, M.; Zhao, L.; Carskadon, S.; Arenberg, D. Expression of CD74, the receptor for macrophage migration inhibitory factor, in non-small cell lung cancer. The American journal of pathology 2009, 174, 638–646. [Google Scholar] [CrossRef]
- Cao, L.; Wang, X.; Liu, X.; Meng, W.; Guo, W.; Duan, C.; Liang, X.; Kang, L.; Lv, P.; Lin, Q.; et al. Tumor Necrosis Factor α-Dependent Lung Inflammation Promotes the Progression of Lung Adenocarcinoma Originating From Alveolar Type II Cells by Upregulating MIF-CD74. Laboratory investigation; a journal of technical methods and pathology 2023, 103, 100034. [Google Scholar] [CrossRef]
- Jäger, B.; Klatt, D.; Plappert, L.; Golpon, H.; Lienenklaus, S.; Barbosa, P.D.; Schambach, A.; Prasse, A. CXCR4/MIF axis amplifies tumor growth and epithelial-mesenchymal interaction in non-small cell lung cancer. Cell Signal 2020, 73, 109672. [Google Scholar] [CrossRef] [PubMed]
- Arenberg, D.; Luckhardt, T.R.; Carskadon, S.; Zhao, L.; Amin, M.A.; Koch, A.E. Macrophage migration inhibitory factor promotes tumor growth in the context of lung injury and repair. American journal of respiratory and critical care medicine 2010, 182, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhao, X.; Tan, L.; Que, P.; Zhao, T.; Huang, W.; Yao, D.; Tang, S. Animal models of lung cancer: Phenotypic comparison of different animal models of lung cancer and their application in the study of mechanisms. Animal Model Exp Med 2025. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.N.; Chen, B.; Liu, X.; Kurie, J.M.; Kim, J. A Method for Orthotopic Transplantation of Lung Cancer in Mice. Methods in molecular biology (Clifton, N.J.) 2022, 2374, 231–242. [Google Scholar] [CrossRef]
- Bilsborrow, J.B.; Doherty, E.; Tilstam, P.V.; Bucala, R. Macrophage migration inhibitory factor (MIF) as a therapeutic target for rheumatoid arthritis and systemic lupus erythematosus. Expert Opin Ther Targets 2019, 23, 733–744. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Trofimov, A.; Du, X.; Hare, A.A.; Leng, L.; Bucala, R. Benzisothiazolones as modulators of macrophage migration inhibitory factor. Bioorg Med Chem Lett 2011, 21, 4545–4549. [Google Scholar] [CrossRef]
- Trivedi-Parmar, V.; Jorgensen, W.L. Advances and Insights for Small Molecule Inhibition of Macrophage Migration Inhibitory Factor. Journal of medicinal chemistry 2018, 61, 8104–8119. [Google Scholar] [CrossRef]
- Ouertatani-Sakouhi, H.; Liu, M.; El-Turk, F.; Cuny, G.D.; Glicksman, M.A.; Lashuel, H.A. Kinetic-based high-throughput screening assay to discover novel classes of macrophage migration inhibitory factor inhibitors. J Biomol Screen 2010, 15, 347–358. [Google Scholar] [CrossRef]
- Ouertatani-Sakouhi, H.; El-Turk, F.; Fauvet, B.; Cho, M.K.; Pinar Karpinar, D.; Le Roy, D.; Dewor, M.; Roger, T.; Bernhagen, J.; Calandra, T.; et al. Identification and characterization of novel classes of macrophage migration inhibitory factor (MIF) inhibitors with distinct mechanisms of action. The Journal of Biological Chemistry 2010, 285, 26581–26598. [Google Scholar] [CrossRef]
- Lubetsky, J.B.; Dios, A.; Han, J.; Aljabari, B.; Ruzsicska, B.; Mitchell, R.; Lolis, E.; Al-Abed, Y. The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. The Journal of Biological Chemistry 2002, 277, 24976–24982. [Google Scholar] [CrossRef]
- Bai, F.; Asojo, O.A.; Cirillo, P.; Ciustea, M.; Ledizet, M.; Aristoff, P.A.; Leng, L.; Koski, R.A.; Powell, T.J.; Bucala, R.; et al. A novel allosteric inhibitor of macrophage migration inhibitory factor (MIF). The Journal of Biological Chemistry 2012, 287, 30653–30663. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhao, Y.; Yuan, Y.; Liao, Y.; Jiang, X.; Wang, L.; Lu, W.; Shi, J. Progress in the development of macrophage migration inhibitory factor small-molecule inhibitors. European journal of medicinal chemistry 2025, 286, 117280. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Xi, J.; Tian, B.; Zhang, Y.; Wang, Z.; Wang, F.; Li, Z.; Long, J.; Wang, J.; Fan, G.H.; et al. The Tautomerase Activity of Tumor Exosomal MIF Promotes Pancreatic Cancer Progression by Modulating MDSC Differentiation. Cancer immunology research 2024, 12, 72–90. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chen, D.; Song, S.; van der Vlag, R.; van der Wouden, P.E.; van Merkerk, R.; Cool, R.H.; Hirsch, A.K.H.; Melgert, B.N.; Quax, W.J.; et al. 7-Hydroxycoumarins Are Affinity-Based Fluorescent Probes for Competitive Binding Studies of Macrophage Migration Inhibitory Factor. Journal of medicinal chemistry 2020, 63, 11920–11933. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Leng, L.; Pantouris, G.; Manjula, R.; Piecychna, M.; Abriola, L.; Hu, B.; Lolis, E.; Armstrong, M.E.; Donnelly, S.C.; et al. A small-molecule allele-selective transcriptional inhibitor of the MIF immune susceptibility locus. The Journal of Biological Chemistry 2024, 300, 107443. [Google Scholar] [CrossRef]
- Lawther, R.P.; Phillips, S.L.; Cooper, T.G. Lomofungin inhibition of allophanate hydrolase synthesis in Saccharomyces cerevisiae. Molecular & general genetics: MGG 1975, 137, 89–99. [Google Scholar] [CrossRef]
- Cho, Y.; Crichlow, G.V.; Vermeire, J.J.; Leng, L.; Du, X.; Hodsdon, M.E.; Bucala, R.; Cappello, M.; Gross, M.; Gaeta, F.; et al. Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proceedings of the National Academy of Sciences of the United States of America 2010, 107, 11313–11318. [Google Scholar] [CrossRef]
- Günther, S.; Fagone, P.; Jalce, G.; Atanasov, A.G.; Guignabert, C.; Nicoletti, F. Role of MIF and D-DT in immune-inflammatory, autoimmune, and chronic respiratory diseases: From pathogenic factors to therapeutic targets. Drug discovery today 2019, 24, 428–439. [Google Scholar] [CrossRef]
- Burnette, E.M.; Ray, L.A.; Irwin, M.R.; Grodin, E.N. Ibudilast attenuates alcohol cue-elicited frontostriatal functional connectivity in alcohol use disorder. Alcoholism, clinical and experimental research 2021, 45, 2017–2028. [Google Scholar] [CrossRef]
- Ray, L.A.; Bujarski, S.; Shoptaw, S.; Roche, D.J.; Heinzerling, K.; Miotto, K. Development of the Neuroimmune Modulator Ibudilast for the Treatment of Alcoholism: A Randomized, Placebo-Controlled, Human Laboratory Trial. Neuropsychopharmacology: Official publication of the American College of Neuropsychopharmacology 2017, 42, 1776–1788. [Google Scholar] [CrossRef]
- Phase 1b/2a Single-center, Open-label, Dose Escalation Study to Evaluate the Safety, Tolerability, and Efficacy of MN-166 (Ibudilast) and Temozolomide Combination Treatment in Patients With Newly Diagnosed or Recurrent Glioblastoma. Available online: https://clinicaltrials.gov/study/NCT03782415 (accessed on 4 November 2025).
- Phase I/II Study of IPG1094 in Advanced Solid Tumors Patients. Available online: https://clinicaltrials.gov/study/NCT06212076 (accessed on 4 November 2025).
- Mikulowska, A.; Metz, C.N.; Bucala, R.; Holmdahl, R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. Journal of immunology (Baltimore, Md.: 1950) 1997, 158, 5514–5517. [Google Scholar] [CrossRef] [PubMed]
- Ichiyama, H.; Onodera, S.; Nishihira, J.; Ishibashi, T.; Nakayama, T.; Minami, A.; Yasuda, K.; Tohyama, H. Inhibition of joint inflammation and destruction induced by anti-type II collagen antibody/lipopolysaccharide (LPS)-induced arthritis in mice due to deletion of macrophage migration inhibitory factor (MIF). Cytokine 2004, 26, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y.; Bacher, M.; Yang, N.; Mu, W.; Nikolic-Paterson, D.J.; Metz, C.; Meinhardt, A.; Bucala, R.; Atkins, R.C. The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. The Journal of experimental medicine 1997, 185, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Patel, M.; Sachdev, J.; Hart, L.; Halama, N.; Ramanathan, R.; Sarantopoulos, J.; Liu, X.; Yazji, S.; Jäger, D.; et al. PD-011 Safety and efficacy analysis of imalumab, an anti-oxidized macrophage migration inhibitory factor (oxMIF) antibody, alone or in combination with 5-fluorouracil/leucovorin (5-FU/LV) or panitumumab, in patients with metastatic colorectal cancer (mCRC). Annals of Oncology 2016, 27, ii105. [Google Scholar] [CrossRef]
- Daniel, J.W.; William, A.W.; Heather, H.; David, M.G. CT-01 Phase IB study of IMMU-115 (humanised ANTI-CD74 antibody) targeting antigen presenting cells in patients with systemic lupus erythematosus (SLE). Lupus Science & Medicine 2016, 3, null. [Google Scholar] [CrossRef]
- Sciences, G. Phase Ib Study of SC Milatuzumab in SLE. Available online: https://trial.medpath.com/clinical-trial/7a7a5d14f71bbe7d/nct01845740-milatuzumab-systemic-lupus-erythematosus?utm_source=chatgpt.com (accessed on 04/11).
- Orphan Drug Designations and Approvals. Available online: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=255807 (accessed on 25 October 2025).
- Alinari, L.; Yu, B.; Christian, B.A.; Yan, F.; Shin, J.; Lapalombella, R.; Hertlein, E.; Lustberg, M.E.; Quinion, C.; Zhang, X.; et al. Combination anti-CD74 (milatuzumab) and anti-CD20 (rituximab) monoclonal antibody therapy has in vitro and in vivo activity in mantle cell lymphoma. Blood 2011, 117, 4530–4541. [Google Scholar] [CrossRef]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol 2007, 77, 13–22. [Google Scholar] [CrossRef]
- Sparkes, A.; De Baetselier, P.; Brys, L.; Cabrito, I.; Sterckx, Y.G.; Schoonooghe, S.; Muyldermans, S.; Raes, G.; Bucala, R.; Vanlandschoot, P.; et al. Novel half-life extended anti-MIF nanobodies protect against endotoxic shock. FASEB journal: Official publication of the Federation of American Societies for Experimental Biology 2018, 32, 3411–3422. [Google Scholar] [CrossRef]
- Meza-Romero, R.; Benedek, G.; Yu, X.; Mooney, J.L.; Dahan, R.; Duvshani, N.; Bucala, R.; Offner, H.; Reiter, Y.; Burrows, G.G.; et al. HLA-DRα1 constructs block CD74 expression and MIF effects in experimental autoimmune encephalomyelitis. Journal of immunology (Baltimore, Md.: 1950) 2014, 192, 4164–4173. [Google Scholar] [CrossRef]
- Meza-Romero, R.; Benedek, G.; Leng, L.; Bucala, R.; Vandenbark, A.A. Predicted structure of MIF/CD74 and RTL1000/CD74 complexes. Metab Brain Dis 2016, 31, 249–255. [Google Scholar] [CrossRef]
- Offner, H.; Sinha, S.; Burrows, G.G.; Ferro, A.J.; Vandenbark, A.A. RTL therapy for multiple sclerosis: A Phase I clinical study. Journal of neuroimmunology 2011, 231, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, S.; Lue, H.; Zernecke, A.; Kapurniotu, A.; Andreetto, E.; Frank, R.; Lennartz, B.; Weber, C.; Bernhagen, J. MIF-chemokine receptor interactions in atherogenesis are dependent on an N-loop-based 2-site binding mechanism. FASEB journal: Official publication of the Federation of American Societies for Experimental Biology 2011, 25, 894–906. [Google Scholar] [CrossRef]
- Rossmueller, G.; Mirkina, I.; Maurer, B.; Hoeld, V.; Mayer, J.; Thiele, M.; Kerschbaumer, R.J.; Schinagl, A. Preclinical Evaluation of ON203, A Novel Bioengineered mAb Targeting Oxidized Macrophage Migration Inhibitory Factor as an Anticancer Therapeutic. Molecular cancer therapeutics 2023, 22, 555–569. [Google Scholar] [CrossRef]
- Safarzadeh Kozani, P.; Naseri, A.; Mirarefin, S.M.J.; Salem, F.; Nikbakht, M.; Evazi Bakhshi, S.; Safarzadeh Kozani, P. Nanobody-based CAR-T cells for cancer immunotherapy. Biomarker research 2022, 10, 24. [Google Scholar] [CrossRef]
- Mustafa, M.I.; Mohammed, A. Nanobodies: A Game-Changer in Cell-Mediated Immunotherapy for Cancer. SLAS discovery: Advancing life sciences R & D 2023, 28, 358–364. [Google Scholar] [CrossRef]
- Maksymova, L.; Pilger, Y.A.; Nuhn, L.; Van Ginderachter, J.A. Nanobodies targeting the tumor microenvironment and their formulation as nanomedicines. Molecular cancer 2025, 24, 65. [Google Scholar] [CrossRef]
- Kunz, P.; Zinner, K.; Mücke, N.; Bartoschik, T.; Muyldermans, S.; Hoheisel, J.D. The structural basis of nanobody unfolding reversibility and thermoresistance. Sci Rep 2018, 8, 7934. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the human macrophage migration inhibitory factor (MIF) gene and its promoter polymorphisms. A) The human MIF gene is composed of three exons and two introns. Two functional polymorphisms in the promoter region, a tetranucleotide repeat (−794 CATT5-8) and a single-nucleotide variant (−173 G/C), modulate MIF transcriptional activity. Created with BioRender.com based on concepts described in Reference [69]. B) Longer CATT6-8 repeats and the −173C allele are associated with increased MIF expression, which promote inflammatory responses, angiogenesis and cell proliferation and predispose individuals to inflammatory, autoimmune and malignant disorders and likely influence the development and prognosis of lung cancer. Created in BioRender. Selo, M. (2026) https://BioRender.com/gqd3pdi, accessed on 05/12/2025.
Figure 1.
Schematic representation of the human macrophage migration inhibitory factor (MIF) gene and its promoter polymorphisms. A) The human MIF gene is composed of three exons and two introns. Two functional polymorphisms in the promoter region, a tetranucleotide repeat (−794 CATT5-8) and a single-nucleotide variant (−173 G/C), modulate MIF transcriptional activity. Created with BioRender.com based on concepts described in Reference [69]. B) Longer CATT6-8 repeats and the −173C allele are associated with increased MIF expression, which promote inflammatory responses, angiogenesis and cell proliferation and predispose individuals to inflammatory, autoimmune and malignant disorders and likely influence the development and prognosis of lung cancer. Created in BioRender. Selo, M. (2026) https://BioRender.com/gqd3pdi, accessed on 05/12/2025.
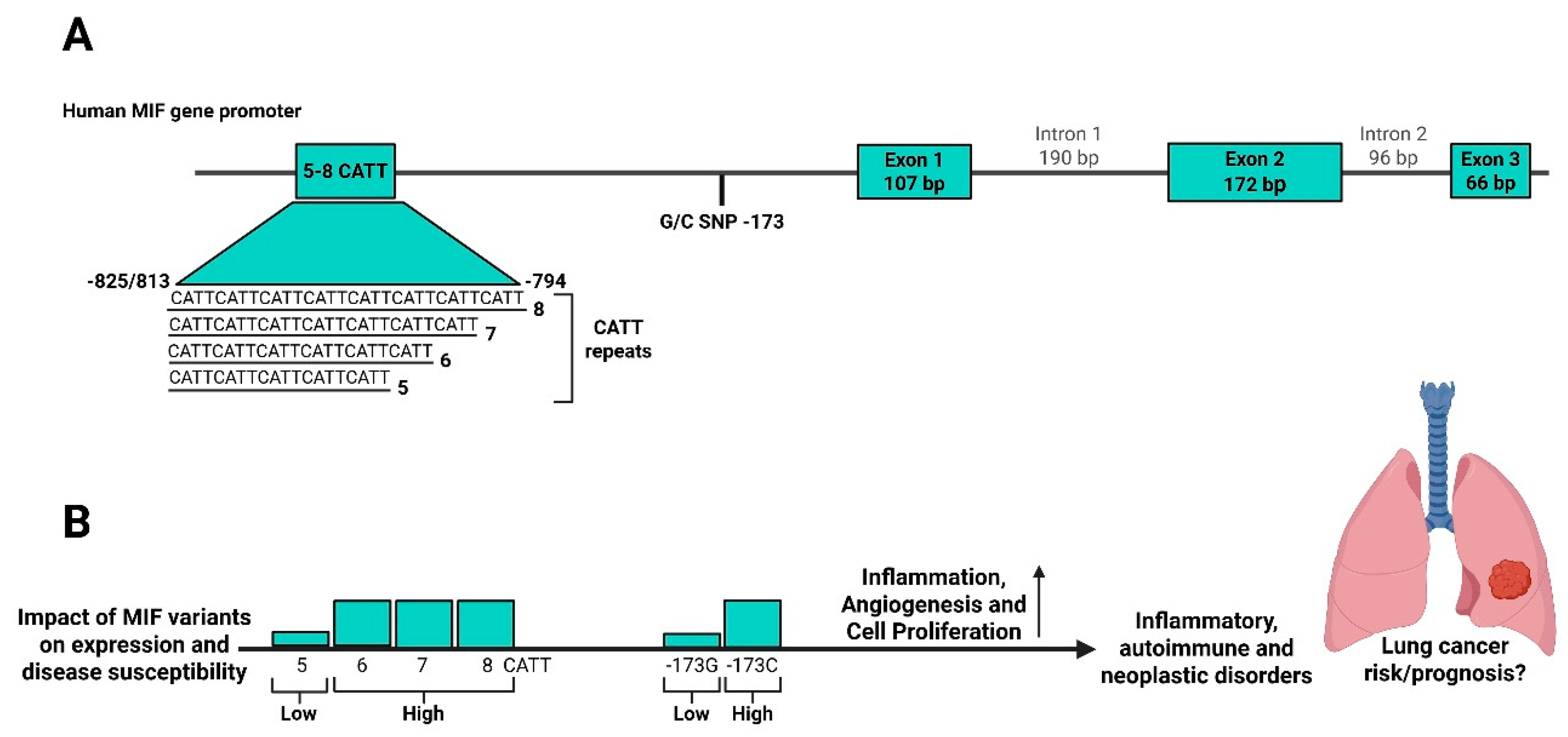
Figure 2.
Schematic representation of Macrophage migration inhibitory factor (MIF)-mediated signaling in tumor progression. MIF binds to the CD74/CD44 complex or CXC chemokine receptors 2 and 4 (CXCR2/4), thereby activating downstream mitogen-activated protein kinase–extracellular signal-regulated kinase 1/2 (MAPK–ERK1/2) and phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signaling pathways. ERK1/2 signaling stimulates cytosolic phospholipase A2 (cPLA2) and inhibits nuclear translocation of p53, promoting tumor cell survival and proliferation. Concurrently, PI3K/Akt activation induces nuclear factor kappa B (NF-κB) –mediated transcription of proinflammatory cytokines and vascular endothelial growth factor (VEGF), driving inflammation and angiogenesis. MIF also promotes epithelial–mesenchymal transition (EMT) and stabilises hypoxia-inducible factor 1α (HIF-1α), which further enhances MIF and VEGF expression and drives a metabolic shift toward aerobic glycolysis (the Warburg effect). This establishes a positive feedback loop that supports tumor growth, angiogenesis, invasion, and metastatic progression. Created in BioRender. Selo, M. (2026) https://BioRender.com/f0f538a, accessed on 02/12/2025.
Figure 2.
Schematic representation of Macrophage migration inhibitory factor (MIF)-mediated signaling in tumor progression. MIF binds to the CD74/CD44 complex or CXC chemokine receptors 2 and 4 (CXCR2/4), thereby activating downstream mitogen-activated protein kinase–extracellular signal-regulated kinase 1/2 (MAPK–ERK1/2) and phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signaling pathways. ERK1/2 signaling stimulates cytosolic phospholipase A2 (cPLA2) and inhibits nuclear translocation of p53, promoting tumor cell survival and proliferation. Concurrently, PI3K/Akt activation induces nuclear factor kappa B (NF-κB) –mediated transcription of proinflammatory cytokines and vascular endothelial growth factor (VEGF), driving inflammation and angiogenesis. MIF also promotes epithelial–mesenchymal transition (EMT) and stabilises hypoxia-inducible factor 1α (HIF-1α), which further enhances MIF and VEGF expression and drives a metabolic shift toward aerobic glycolysis (the Warburg effect). This establishes a positive feedback loop that supports tumor growth, angiogenesis, invasion, and metastatic progression. Created in BioRender. Selo, M. (2026) https://BioRender.com/f0f538a, accessed on 02/12/2025.
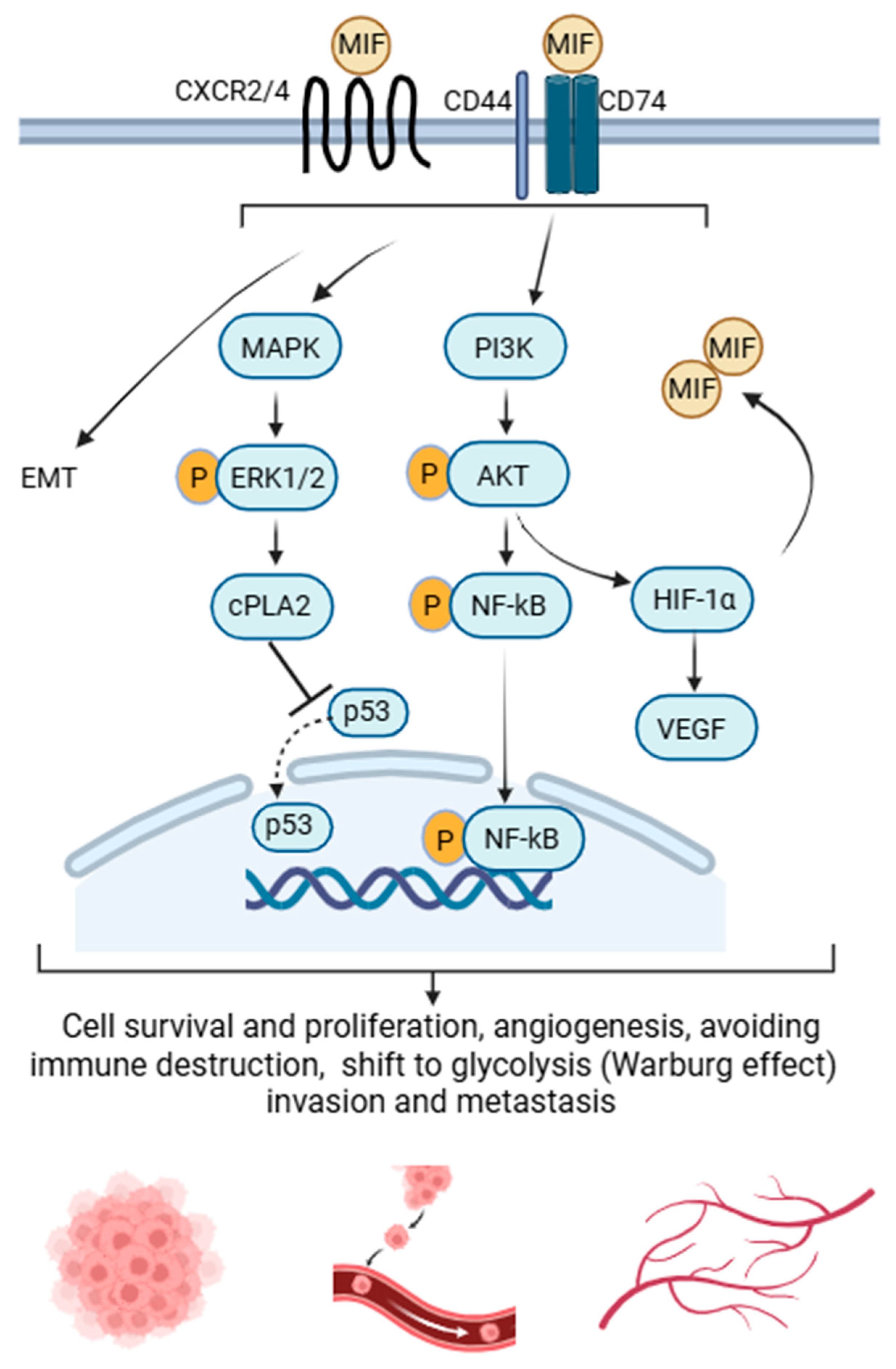

Table 1.
Associations of MIF promoter polymorphisms (−173 G/C and −794 CATT5–8) with disease susceptibility and severity in inflammatory, autoimmune, and malignant disorders.
Table 1.
Associations of MIF promoter polymorphisms (−173 G/C and −794 CATT5–8) with disease susceptibility and severity in inflammatory, autoimmune, and malignant disorders.
|
Disease or Condition |
Polymorphism(s) |
Associated Allele |
Reported Association | Reference(s) |
| Rheumatoid arthritis | −173 G/C and −794 CATT5-8 |
−173C and CATT7 alleles |
Increased susceptibility and Increased severity |
[89] [64] [76] |
| CATT5 allele | Low disease severity | [65]. | ||
| Inflammatory polyarthritis | −173 G/C and −794 CATT5-8 |
-173C CATT7 alleles |
Increased susceptibility | [90] |
| Systemic lupus erythematosus | −173 G/C and −794 CATT5-8 |
−173C and CATT7 alleles |
−173C and CATT7 are associated with increased end-organ damage, but their relationship with disease incidence is controversial, as studies report both increased and decreased risk. | [77,91] [67] |
| Juvenile idiopathic arthritis | −173 G/C | −173C allele | Increased susceptibility | [63] |
| Ulcerative colitis | −173 G/C | −173C allele | Increased risk | [92] |
| Autoimmune hepatitis | −173 G/C | -173 CC/GC genotypes | Higher alanine aminotransferase levels and greater steroid requirements were observed compared with the GG genotype. | [93] |
| Psoriasis | −173 G/C | −173C allele | Associated with male and late-onset psoriasis | [94] |
| Pulmonary tuberculosis | −173 G/C | −173C allele | Increased susceptibility, particularly in Asians. | [74] |
| Coronary heart disease | −173G/C | −173C allele | Increased risk in Arab and Asian populations | [72,73] |
| Pneumococcal meningitis | −173 G/C and −794 CATT5-8 |
−173C and CATT7 alleles | Increased morbidity and mortality | [95] |
| Cystic fibrosis | −794 CATT5-8 | CATT5 allele | Milder disease, reduced Pseudomonas colonisation, and slower FEV1 decline in CATT5 than CATT6-8 carriers. | [78] [57]. |
| COVID-19 patients and mouse model | −794 CATT5-8 | CATT7 allele | Increased disease severity but reduced susceptibility in comparison with CATT5 | [84]. |
| Asthma (preclinical mouse model) | −794 CATT5-8 | CATT7 allele | More pronounced airways inflammation in CATT7 than in CATT5 | [85,87]. |
| Cancer (various types) | −173G/C | −173C (G/C + C/C genotypes) | Increased overall cancer risk | [75] |
| Breast cancer | -173 G/C and −794 CATT5-8 | -173C and CATT7 alleles | Higher circulating MIF levels without increased susceptibility | [96] |
| Cutaneous squamous cell carcinoma | -173 G/C and −794 CATT5-8 | 5C (CATT5/ -173C) and 7G (CATT7/ -173G) haplotypes | Linked to increased susceptibility and higher circulating MIF levels | [83] |
| Rectal cancer | -173 G/C and −794 CATT5-8 | -173C and longer (CATT6-8) repeats | Elevated serum MIF levels and increased susceptibility | [79] |
| Non-cardia gastric cancer | -173 G/C) and −794 CATT5-8 | -173C and longer (CATT6-8) repeats | Increased susceptibility to gastric cancer, synergistic effect with H. pylori infection. | [80] |
| Hepatocellular carcinoma | -173 G/C | −173C (CC and GC) genotypes | Associated with increased susceptibility, poor prognosis and metastasis | [97] |
| Early-stage cervical cancer | −794 CATT5-8 | CATT7 allele | Associated with increased susceptibility. | [81] |
| Prostate cancer | -173 G/C and −794 CATT5-8 | G/C and C/C genotypes and CATT7 allele | G/C and C/C genotypes associated with increased incidence; CATT7 allele associated with increased incidence and higher risk of recurrence | [82]. |
| Melanoma | -173 G/C | −173C (C/C) genotype | More frequent in patients than controls, suggesting increased susceptibility | [88] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



