Submitted:
03 February 2026
Posted:
04 February 2026
You are already at the latest version
Abstract
This study reports the synthesis of four derivatives containing acridine (3a), quinoline (3b), indole (3c), and pyridine (3d) nuclei, as well as their interactions with DNA, physicochemical and pharmacokinetic predictions, antiproliferative activity, in silico and in vivo effects in zebrafish, and enzyme inhibition assays. UV–vis absorption studies with ssDNA revealed different spectroscopic effects, with binding constants (Kb) ranging from 1.41 × 10⁵ to 6.46 × 10⁴ M⁻¹. The fluorescence quenching constant (Ksv) with ethidium bromide (EB) varied between 0.53 and 0.67 × 10³ M⁻¹. The compounds intercalated into DNA base pairs, a mechanism confirmed by molecular docking, with 3b (quinoline) showing the most substantial interaction. All derivatives exhibited antitopoisomerase IIα activity at 100 μM and were cytotoxic against MCF-7 and T47-D breast tumor cells, particularly against the more aggressive T47-D lineage. No hemolytic activity was observed in human erythrocytes. In vivo assays in zebrafish embryos showed no toxicological or cardiotoxic effects. However, all compounds altered superoxide dismutase (SOD) and catalase (CAT) enzymatic activity, requiring further studies on reactive oxygen species (ROS) generation to assess potential adverse effects. Furthermore, good results were observed in the physicochemical and pharmacokinetic parameters of the synthesized compounds. These findings highlight the quinoline derivative (3b) as the most promising nitrogen heterocycle due to its antiproliferative activity and biomolecular interactions without adverse effects in zebrafish embryos, distinguishing it from clinically available agents.
Keywords:
acridine
; quinoline
; DNA binder
; Danio rerio
1. Introduction
Cancer continues to be a persistent public health challenge with increasing numbers on a global scale [1]. The resistance of tumor cells to available drugs and adverse effects drives the search for new antitumor medications, aiming to slow tumor growth, eliminate cancerous cells, and reduce unwanted side effects. In this context, organic compounds that contain nitrogen heterocyclic skeletons (N-heterocycles) are promising due to their favorable pharmacological properties [2,3,4,5,6].
Among the options for new drug candidates, nitrogen heterocycles based on acridine, quinoline, indole, and pyridine nuclei stand out because they exhibit cytotoxic activity against tumor cells, the ability to interact with DNA, and the inhibition of the topoisomerase enzyme [7]. These mechanisms of action are described by the works of Almeida et al. [8] with spiro-acridine derivatives, Ribeiro et al. [9] with quinoline derivatives, Alves et al. [10] with indole derivatives, and Narva et al. [11] with pyridine derivatives. These different nuclei have proven promising for cancer therapy by targeting key molecules and inducing the death of tumor cells.
Almeida et al. [8] produced new spiro acridines by introducing a cyano-N-acyl hydrazone group between the acridine and (non– and methoxy-substituted) phenyl rings followed by spontaneous cyclization. The compounds were specific for human topoisomerase IIα inhibition and retained the DNA interaction ability. Recently, Santos et al. [7] synthesized four new nitrogen heterocyclic derivatives containing the acridine, quinoline, indole, and pyridine nuclei and studied them by in vitro and in vivo tests, finding that the indole ring favored biological activity. Indole derivatives exhibited the highest DNA binding constant (Kb) of 9.54 × 10⁴ and fluorescence quenching (Ksv) of 0.64 × 10³ M⁻¹, the best topoisomerase enzyme inhibition profile with 82% inhibition of enzymatic activity, and cytotoxic activity against tumor cells, without side effects for zebrafish model [7].
For new anticancer molecule propositions, candidate drugs can be explored through interaction studies involving biomolecules such as DNA (the primary target for cancer therapy) [12]. Additionally, zebrafish (Danio rerio) have proven to be a valuable tool in anticancer research, providing crucial information on the efficacy and toxicity of new compounds to assess possible adverse effects and determine the best therapeutic dose of these compounds [13,14,15].
Based on the previous results for the indole derivative described by Santos et al. [7] and the data of Almeida et al. [8] for the spiro acridines, four nitrogen heterocyclic derivatives with different nuclei were tested here (Scheme 1): [5-oxo-1-phenyl-1,5-dihydro-10H-spiro(acridin-9,2-pyrro)-4-cya nitrile (3a - acridine)], 3-(quinoline-4yl)-2-cyano-N-phenylacrylamide (3b - quinoline), 2-cyano-3-(1H-indol-3yl)-N-phenyl acrylamide (3c - indole), and [3-(pyridine-4yl)-2-cyano-N-phenylacrylamide] (3d - pyridine). The molecules were obtained to improve biological activity and to reduce adverse effects on non-tumoral cells as well as on in vivo organisms, trying to contribute to the advancement of studies focused on cancer therapy. The compounds were characterized and evaluated for their ability to interact with DNA, physicochemical and pharmacokinetic predictions, in vitro and in silico methodologies, besides in vitro inhibition of the Topoisomerase IIa enzyme, antiproliferative activity in tumor and non-tumor cells, hemolytic activity with human erythrocytes, and in vivo toxicological studies with zebrafish embryos.
2. Results and Discussion
2.1. Nitrogen Heterocyclic Compounds
Studies on spiroacridine derivatives obtained through spontaneous cyclization conducted by Almeida et al. [8] raised interest in synthesizing new nitrogen heterocyclic derivatives with spirocyclization using the same side chemical portion, but with the cyano-acetamide spacer instead of the cyano-acylhydrazone. The present spacer spontaneously cyclized only in the acridine derivative (3a), as demonstrated in Scheme 1. The three other compounds were synthesized: quinoline (3b), indole (3c), and pyridine (3d). The synthesis of 3b was previously described Silva et al. [16].
As established by previous studies, it was observed that 9-acridinecarbaldehyde, 4-pyridecarbaldehyde, 4-quinolecarbaldehyde, and 3-indolecarbaldehyde reacted effectively in absolute ethanol in the presence of triethylamine with their respective intermediates, 2-cyano-N'-phenylacetamide, through a Knoevenagel condensation reaction [8,17]. This resulted in the formation of the compounds mentioned. The 9-acridinecarbaldehyde underwent spontaneous cyclization between the N1 group of the acetamide and the C-9 carbon of the acridine. Regarding the yields of the final compounds, they were found to be above 50%, except for the pyridine-ring compound 3d, which achieved a yield of 32%.
The structures of the four derivatives were confirmed by 1H NMR, 13C NMR, mass spectrometry, infrared spectroscopy, and high-resolution spectroscopy. The 1H NMR analysis allowed for the identification of distinct features, such as the singlets of the amide protons (NH) with chemical shifts ranging from δ 10.54 to 9.63 ppm. Additionally, the 1H NMR spectra of all derivatives revealed singlet (s), doublet (d), triplet (t), and multiplet (m) signals associated with the acridine aromatic groups, confirming the presence of a ring similar to that described in the literature [8,18]. Furthermore, the 13C NMR spectra corroborated the structures, as they presented signals corresponding to the carbon atoms. Exclusively in acridine compound (3a), a shift at 70.6 ppm was observed, indicating the formation of the quaternary carbon after the process of spontaneous cycling. As previously reported by Vilková et al. [19], cyclization occurs spontaneously, without the need for additional energy. Our theoretical results confirm that cyclization is energetically favored, as the cyclic isomer possesses lower energy (10 kJ mol⁻¹) compared to the open isomer, since molecules tend to adopt a lower energy form, rendering them more stable.
The infrared results also assisted in the structural characterization of the new compounds. The following bands were observed: 3314 – 3470 cm⁻¹, suggestive of axial deformation of NH; a band between 3019 and 3262 cm⁻¹, suggestive of axial deformation of CH; and a band between 1640 – 1695 cm⁻¹, suggestive of aromatic axial deformation of C=O [8,20]. Ultimately, mass spectrometry (MS) was useful in the structural characterization of the new nitrogenous derivatives. These data are presented in the Supplementary Material.
2.2. Physicochemical and Pharmacokinetic Characteristics of the Compounds
Previous the in vitro and in vivo evaluations, druglikeness studies generated by SwissADME and Multi-Parameter Optimization (MPO) obtained by Marvin, allowed to visualize the good physicochemical profile of the compounds (Table 1). Based on the results generated by SwissADME, it can be observed that all compounds have good characteristics, following Lipinski's rule of five [21], Ghose's rule [22], Veber's rule [23], Egan's rule [24], and Muegge's rule [25], indicating the potential of the compounds as drug candidates. Furthermore, regarding the multiparametric optimization (MPO) system that brings together drug candidates and active drugs, as indicated by Wager et al. [26], it is possible to detect that all have stable pharmacokinetics, with MPO above 4.0, which indicates good passive permeability (Papp), low efflux by P-glycoprotein (P-gp), and low metabolic clearance.
Although all compounds met druglikeness requirements and presented properties within the ideal range (Figure 1), compound 3d obtained the highest MPO value (4.67). This condition occurs because the LogP and LogD values (2.16) are close to the values considered ideal (LogP ≤ 3 and LogD ≤ 2), since 3d consists of the smallest structure (249.27 g/mol) and is composed of important polar groups. However, all molecules have good and similar TPSA results (Figure 2), mainly associated with carbonyl, nitrile, and N-substituted amide groups, which are present in all compounds [27]. Furthermore, 3c and 3d also have an imine nitrogen contributing to the polar area and have the same TPSA value (65.45 Å2), which is close to the result for 3b (64.98 Å2). However, because 3b and 3c have an indole and quinoline nucleus, respectively, they have a larger size (299.33 g/mol and 287.32 g/mol, respectively) and lipophilicity (3.54 and 3.48, respectively) than 3d, which has a pyridine group as a substituent.
Furthermore, compound 3a obtained the lowest MPO value (4.24), as it is the compound with the highest lipophilic character, presenting the highest LogD and LogP results (4.03 for both), which is associated with the presence of the acridine group, which also leads to an increase in molar mass (348.49 g/mol) and molar refractivity (MR) (110.26). In addition, it is evident that it is the compound with the lowest TPSA (Figure 2), due to the N-substituted amide in the structure binding to acridine to form a tertiary amine, which has a lower contribution to the polar area. It can also be seen that the formation of this bond also promotes a conformational restriction, causing 3a to have only one rotatable bond (RB), while the others have four [26,27].
Regarding the pKa values (Table 1), it is possible to classify all of them as low-potential bases, as they have strong conjugate acids, with pKa values below 5.0. This is because, despite having N-substituted amides and tertiary amine, which act as bases, in all molecules there is a displacement of the electronic density of these groups due to the mesomeric and/or steric effect, causing the base to have its electrons displaced throughout the structure. However, 3a and 3c have much lower pKa values because the free electrons of nitrogen in acridine and indole are actively participating in resonance, which promotes greater stabilization of the base. Meanwhile, in derivatives 3b and 3d, the free electrons are more available for protonation, making them weaker acids than 3a and 3c.
Based on the determination of pharmacokinetic parameters by ADMETLab 3.0, it is possible to understand the results obtained from the evaluation of Table 2. In which, when highlighting the permeability process, all compounds have greater permeability compared to etoposide, which has a large number of polar groups and a large molecular mass of 588.55 g/mol. Furthermore, 3a, 3b, and 3c have high permeability via Caco-2 (6.60x10-5 cm/s, 1.11x10-5 cm/s, and 1.15x10-5 cm/s, respectively) and MDCK (2.23x10-5 cm/s, 1.48x10-5 cm/s, and 1.45x10-5 cm/s), while 3d has high permeability only for MDCK (1.72x10-5 cm/s), being moderate for Caco-2 (8.57x10-6 cm/s) [28]. It should be noted that this prediction is associated with the superior lipophilic potential of compounds 3a, 3b, and 3c in relation to 3d, which is also considered the smallest structure in relation to the others, having the lowest molar mass.
Furthermore, by understanding the distribution processes, it can be highlighted that derivative 3a has the greatest ability to penetrate tissues (1.08 L/kg), presenting the largest volume of distribution, in addition to having one of the most significant probabilities of crossing the blood-brain barrier (5.7%). However, in general, all have a strong tendency to remain in plasma, as they present distribution volume results lower than 2 L/kg, with 3b (0.59 L/kg), 3c (0.50 L/kg), and 3d (0.56 L/kg) presenting very low values in relation to 3a. This condition may be associated with a strong tendency for molecules to bind to plasma proteins, as they all have results above 90%. Regarding BBB permeability, with the exception of 3a, all have results below 1%, which may be associated with the greater hydrophilic character of the compounds [29,30].
Regarding excretion, there is an increase in plasma clearance from 3a to 3d, due to an increase in the polarity of the molecule and a decrease in lipophilicity and molar mass. Thus, 3c (0.30972 L/h/K) and 3d (0.30222 L/h/K) have moderate clearance, while 3a (0.2208 L/h/Kg) and 3b (0.2772 L/h/K) have low clearance. However, among the four compounds, only 3a has a relatively high half-life (3h 23min), while the others vary between 1 and 2 hours. This is because, in addition to having low clearance, compound 3a has the largest volume of distribution, which makes it the least available for metabolism and excretion [31].
According to the first-phase metabolism process, based on the results generated by ADMETlab 3.0 and Xenosite evaluated in Table 3, CYP1A2, CYP2C19, and CYP3A4 can be identified as important enzymes involved in the metabolism of the compounds. CYP1A2, a priori, proves to be effective in catalyzing reactions in 3a (82.1%), 3c (63.2%), and 3d (97.9%), promoting hydroxylation primarily in aromatic structures of neutral lipophilic planar substrates. Similarly, CYP2C19, which is presented as being capable of metabolizing only 3a (100%), also performs hydroxylations in aromatic and aliphatic structures of lipophilic, neutral, and intermediate-sized substrates. Finally, CYP3A4 promotes the metabolism of 3a (100%) and 3d (59.5%) in greater proportions, performing the oxidation of aliphatic structures, with a greater preference for large, lipophilic molecules [32,33].
This explains the strong tendency for oxidation reactions to form with the compounds, as shown in Figure 3. However, it is understood that compound 3b has a low probability of being metabolized by any of the enzymes mentioned, despite its structural similarity to the other compounds. Furthermore, it can also be seen that the amide group in compounds 3b, 3c, and 3d may be targeted by hepatic peptidases, resulting in a phase I hydrolysis reaction that leads to the formation of carboxylic acids and amines. In addition, among the phase II reactions, the predictions generated by Xenosite point to a strong tendency for the formation of conjugates with glucuronic acid, forming glucuronide complexes. For this, it is pointed out that the nitrogens involved in the cyclic structures consist of an important nucleophile for the reaction. However, only in 3a and 3c is the presence of secondary amines verified, which have greater reactivity for conjugation to occur, while in 3b and 3d tertiary amines stand out, which are less reactive, requiring prior oxidation steps to undergo glucuronidation [34].
Finally, among the toxicity criteria evaluated by ProTox 3.0, it is possible to predict that only compound 3a showed cardiotoxic potential, with a probability of 92%, while for other predictions, such as nephrotoxicity, carcinogenicity, mutagenicity, and cytotoxicity, it was inactive. For the other compounds, a good safety profile is understood. Only a slight potential for hepatotoxicity is presented, but no compound has a probability of greater than 60%, as shown in Table 4. Comprehensively, with the detection of toxicity points (Figure 4), based on the predictions generated by STopTox, it is possible to identify amine groups and aromatic portions associated with the toxic potential of the compounds. While carbonyl and nitrile nuclei are generally less toxic regions, for 3a, the less toxic potential of acridine substitution is understood. It can be observed that compound 3a has the most intense toxicity points, associated with the N-phenyl portion, which may be related to its higher probability of cardiotoxicity. In relation to hepatotoxicity, among the main associated aspects, it is possible to predict the ability of molecules to be metabolized by enzymes that make up CYP450, which has been observed to occur, but also the daily dose administered. Another important factor commonly associated with hepatotoxicity is lipophilicity, but the relationship between the LogP of the compounds and the results obtained is not understood [35].
2.3. DNA Interactions
Biological activities and toxic effects of nitrogen heterocyclic derivatives can be caused by the ability to bind [36]. Therefore, the compounds 3a, 3b, 3c and 3d were subjected to absorption analysis in the absence and presence of ssDNA. Spectroscopic analyses revealed absorption bands ranging from 271 to 420 nm. The maximum absorption wavelengths of the compounds in their free and bound forms, along with other spectrophotometric data of the derivatives, are available in Table 5. In the presence of increasing amounts of ssDNA (0 to 100 μM), changes in the absorption spectra were observed, exhibiting hypochromic and hyperchromic effects, with shifts in the maximum absorption peaks towards the blue (hypochromic) and red (bathochromic) regions (Table 5 and Supplementary Material S17). When it comes to the interaction of drug candidates with biomolecules, spectral changes and shifts in the maximum absorption peaks indicate the formation of ligand-DNA complexes [37,38,39,40].
The compounds 3a, 3b, and 3d demonstrated hypochromic effects in the presence of ssDNA, with varying degrees of absorption reduction. The highest hypochromic effect was 91.63%, observed for the quinoline derivative (3b). The presence of the derivatives contributed to the spectral changes, stabilizing the DNA structure and resulting in hypochromism, indicating an intercalative type of interaction [41]. In general, the combination of hypochromism and bathochromism is due to the insertion of the molecules between the DNA base pairs, establishing hydrogen bonds [41,42]. The characteristics were particularly notable for the compounds 3a (spiroacridine) and 3b (quinoline), demonstrating a shift of the absorption peak towards the red region (bathochromic effect), with values of 71 nm and 78 nm, respectively. Regarding the binding affinity constant (Kb), values ranged from 5.26 × 10⁴ to 6.46 × 10⁵ M⁻¹ (Table 5). The highest Kb value was observed for 3a (acridine). The Kb values found for the derivatives are characteristic of intercalators, which are often in the range between 10⁴ and 10⁶ M⁻¹ [36].
a Absorption of ssDNA scanned in the range of 200 and 600 nm. b Hyperchromicity for complexes formed derivatives and 100 mM of ssDNA in comparison to free compounds. c Hypochromicity for complexes formed derivatives and 100 mM of ssDNA in comparison to free compounds. d Binding constant (Kb) obtained from complexes formed derivatives-ssDNA.
In the development of new antitumor drugs, understanding and elucidating the mechanism of interaction of molecules with DNA is essential [43]. Absorption studies provide valuable information about the binding mechanism of compounds to DNA. However, for a better understanding, competition studies with DNA intercalating probes, such as ethidium bromide (EB), are necessary, as they already have their binding mode well established [41, (Rehman et al.; 2015; Thirunavukkarasu et al.; 2018). In this competition assay, we used EB as the DNA intercalating probe: EB-ssDNA, given that the fluorescence intensity of isolated DNA is weak. The assay was conducted both in the absence and presence of the derivatives 3a, 3b, 3c and 3d with emission detailed in Table 6.
EB is a fluorophore when intercalated with DNA serving as probe for by DNA complex formation [44]. As presented in Table 6, the fluorescence intensity of the EB-ssDNA complex exhibited its maximum emission peak at 599 nm. After the addition of the compounds, the fluorescence signal of the EB-ssDNA complexes decreased as the concentrations of the compounds increased, where the fluorescence suppression of the probe occurs due to the probe's fluorophore being replaced by another intercalating agent, resulting in the displacement of the probe and suggesting a more intense interaction between the compounds and DNA [42,44,45].
The fluorescence quenching values of the EB-ssDNA complex were calculated, with the lowest fluorescence quenching percentage being 53.29% for 3c (indole) and the highest percentage of 73.14% for 3b (quinoline). This indicates that the compounds compete with the probe and intercalate the DNA base pairs. Regarding the fluorescence quenching profile of the compounds, the Stern-Volmer equation was used to calculate the fluorescence quenching constant (Ksv) (Table 6). The highest Ksv values for EB-ssDNA were observed for 3a (acridine) with 6.7 × 10² M⁻¹. These data confirm the findings in the UV–vis absorption assay, as these same derivatives were the ones that presented the highest binding affinity constants (Kb), with acridine being particularly notable (Figure 5).
Competition studies using DNA intercalating probes such as EB were conducted by Santos et al. [7]. Their findings indicated that derivatives with different nuclei (acridine, quinoline, indole, and pyridine) caused fluorescence quenching of EB. Similarly to our findings, the fluorescence quenching values ranged from 0.34 to 0.64 × 10³ M⁻¹ for EB. In other words, the findings of the present study consolidate the data previously obtained by the research group, although they present different side groups. Furthermore, the data regarding intercalation between DNA base pairs corroborate the findings [20,45,46,47], who studied compounds with acridine, spiroacridine, indole, and pyridine nuclei, respectively, and attributed the ability to intercalate DNA base pairs to the planarity of polyaromatic compounds.
2.4. Human Topoisomerase Inhibition Assay
Considering that topoisomerase enzymes are involved in vital processes for DNA regulation, such as proliferation, differentiation, cell survival, and maintenance of genome integrity, various studies have conducted antitumor therapeutic investigations due to their functions [20,48]. Topoisomerase IIα (topo) is one of the main therapeutic targets due to its overexpression during the proliferation process of tumor cells [49]. In its mechanism of action, topoisomerase inhibitors cause damage to tumor cells by promoting DNA cleavages while simultaneously inhibiting the formation of new DNA strands, thereby activating the cell death process (apoptosis) [50,51,52].
The experimental assay was conducted to evaluate the effects of the derivatives against human topo IIα. In Figure 6, alterations in the migration of DNA bands (pUC19) are observed, representing different structural arrangements such as supercoiled DNA, circular and relaxed DNA, and linear DNA [46,53]. The inhibition of Topo IIα was analyzed based on the bands corresponding to supercoiled DNA (pUC19) in relation to the positive control. The derivatives: 3a (acridine), 3b (quinoline), 3c (indole), and 3d (pyridine) inhibited the activity of Topo IIα at 100 μM. Quantitatively, the positive control m-AMSA showed 100% inhibition, while the derivatives 3a, 3b, 3c, and 3d exhibited inhibitions of 77%, 82%, 93%, and 98%, respectively.
The appearance of linear DNA bands indicates that the compounds acted as Topo IIα poisons [20]. In addition to the formation of linear DNA, the formation of topoisomers was observed, suggesting the formation of a cleavage complex between Topo IIα, pUC19, and the tested compounds. The formation of this complex indicates that the compounds were effective in inhibiting the enzyme and can maintain DNA cleavage, thereby preventing cellular replication in tumor cells [46].
Previous to our study, Gouveia et al. [20] demonstrated with spiroacridine derivatives similar inhibition percentage of 61.70% at 100 μM, a result similar to our findings for 3a, which inhibited by 77% at 100 μM. In accordance with our findings, Silva Filho et al. [53] conducted enzymatic inhibition tests of Topo IIα using two acridine-thiosemicarbazone derivatives and observed that those substituted with phenyl and naphthyl groups promoted inhibition of enzymatic activity at 100 μM, with the best result for the derivative bearing the phenyl substituent on the side chain. Furthermore, in line with the findings of Santos et al. [7], the derivatives with indole and pyridine rings exhibited 82% and 80% inhibition of the topoisomerase enzyme at the same concentration tested here. However, our inhibition values were even higher, at 93% and 98%, respectively, indicating that the substitution of the amide nitrogen with a phenyl group in the different nuclei has enhanced their biological activity.
In addition to the spectroscopic studies of the in vitro interaction between the derivatives and DNA at the topoisomerase action site, computational studies of molecular docking and molecular dynamics were also conducted to corroborate the in vitro studies. Molecular docking studies have the capability to predict the binding conformation of molecules and assist in the discovery of new molecules capable of binding and acting on biological targets, such as DNA and topoisomerase IIα [9,54,55]. The binding energy data of the derivatives studied are presented in Table 7, and the docking poses are depicted in Figure 7.
As presented in Table 7, all derivatives exhibited negative binding energy values, with these values being lower than that of the ligand 9-acridine, which showed a binding energy of -6.99 kcal mol⁻¹. The binding energy values of the derivatives with the target 1G3X, in ascending order, were: 3d (-7.35), 3a (-8.07), 3c (-8.75), and 3b (-9.23) kcal mol⁻¹. The same sequence was observed for the target 5GWK: 3d < 3a < 3c < 3b, with 3b (quinoline) having the lowest binding energy value (-8.84 kcal mol⁻¹) for target 5GWK and (-9.23 kcal mol⁻¹) for target 1G3X. The binding pose of the ligand with the lowest binding energy, 3b, indicates that there was intercalative interaction between the DNA nitrogenous base pairs and that the ligands interact with the amino acid Arg-487 in a manner like etoposide (a co-crystallized drug present in the topo IIα structure), as observed in Figure 7. Additionally, 3a and 3b exhibited π-stacking interactions between the DNA base pairs and their aromatic rings; π-stacking interaction is essential in the intercalative interaction process [10].
The binding energy values confirm the findings from the in vitro assays, as 3b exhibited the second highest Kb and Ksv values in the competition tests with EB, and the compounds demonstrate a higher binding affinity within the ligand-receptor complex, indicating a stronger and more stable interaction.
2.5. Antiproliferative and Hemolytic Evaluations
In this study, using the sulforhodamine assay, the antiproliferative activity of the compounds 3a, 3b, 3c, and 3d was evaluated in tumor and non-tumor cells, with MCF-7 and T47-D being the tumor cell lines and HaCaT the non-tumor cell line. The compounds were tested at different concentrations, specifically 0.25; 2.5; 25; and 250 µM, with m-AMSA used as positive control. The GI₅₀ values (the molar concentration of the compound that inhibits 50% of cell growth) for both cell lines are presented in Table 8.
As presented in Table 8, the GI₅₀ values varied among the compounds and the cell lines. The GI₅₀ values for the tumor cell lines ranged between 3.42 and 34.41 µM. The compounds 3a and 3b exhibited higher cytotoxicity for the T47-D tumor cell line with GI₅₀ values of 3.42 and 8.07 µM, respectively. Acridine derivative was cytotoxic in MCF-7 (34.41 μM) and T47-D (3.42 μM), showing greater inhibition of cell growth for T47-D. This same compound showed the highest GI₅₀ in HaCaT (>100 μM), indicating low cytotoxicity in non-tumor cells and higher antiproliferative activity in tumor cells, especially T47-D.
Antiproliferative effects of phenyl-substituted acridine-thiosemicarbazone derivatives against the MCF-7 and HaCaT cell lines were described by Almeida et al. [36] . The authors reported that the most active compound inhibited the growth of all cell lines with GI₅₀ values below 10 µM for all tumor cell lines, including the non-tumor HaCaT line. In contrast, our compound 3a (acridine) with a substituted N-phenyl amide was not as active against HaCaT cells and was cytotoxic to MCF-7 with a GI₅₀ of 34.41 μM.
In turn, the quinoline derivative 3b exhibited GI₅₀ values of 33.62 μM for the MCF-7 cell line and 2.98 μM for HaCaT, showing low selectivity between tumor cells and greater selectivity for non-tumor cells and the T47-D line. However, in the HaCaT line, our compounds showed higher GI₅₀ values compared to the m-AMSA control. Perković et al. [39] and Santos et al. [7] investigated the antiproliferative activity of compounds containing pyridine, indole, and quinoline heterocyclic structures. Perković et al. [39] reported the results of the compounds against the MCF-7 tumor cell line and determined GI₅₀ values (μM) ranging from 2.0 ± 0.1 to 11.0 ± 0.3, a result like that of 3c (indole). Santos et al. [7] studied different nitrogen heterocyclic nuclei (acridine, quinoline, indole, and pyridine) and conducted studies with tumor cell lines including MCF-7, T47-D, and the non-tumor HaCaT. In the assays, the compounds demonstrated cytotoxic activity against tumor cells with different TGI values for the different nuclei studied. In the MCF-7 line, the highest TGI value (13.39 μM) was observed for the CAPA derivative (pyridine), while in our study with the same line, the GI₅₀ value was 33.27 μM. It is worth noting that the TGI values found in the study by Santos et al. [7] for the HaCaT line ranged between 1.64-6.17 μM across the different nuclei studied.
Meanwhile, the compounds 3c and 3d were not active against both breast tumor cell lines used (T47-D and MCF-7). Indole derivative exhibited a GI₅₀ of 9.44 μM and in the HaCaT tumor line a GI₅₀ of 4.29 μM. As previously described, some compounds showed proximity in GI₅₀ values between tumor and non-tumor cell lines, indicating lower selectivity and, consequently, potential toxicity to healthy cells. However, the results demonstrate that the GI₅₀ values of all derivatives were higher than those observed for the reference drug (positive control) m-AMSA.
Similar results to our findings were reported by Kim et al. [38] , who studied a series of analogs based on the indole nucleus. The results demonstrated that compounds (3a-b and 3d-e) were more cytotoxic against MCF-7 cells than gefitinib, with GI₅₀ values of 19.3, 9.6, 4.6, and 8.3 µM, respectively. Specifically, the inhibitory activity of compound 3d (GI₅₀: 4.6 µM) was approximately 4.5 times stronger than that of gefitinib (GI₅₀: 20.8 µM). These results confirm the cytotoxicity of indole compounds such as ICMD studied here, which presented a GI₅₀ of 9.44 µM for the MCF-7 cell line.
As previously described, some compounds exhibited similar GI₅₀ values between tumor and non-tumor cell lines, which may indicate potential toxicity to healthy cells. However, the results demonstrate that the GI₅₀ values of all derivatives were higher than those observed for the reference drug (positive control) m-AMSA, which already has reported toxicity against non-tumor cells.
Cancer therapy faces a constant therapeutic challenge, as drugs often do not act specifically against tumor cells, being cytotoxic also to healthy cells, including human erythrocytes, because cytotoxicity can cause damage to the cell membrane and immediate cell death [37,40]. For this reason, a hemolytic assay was conducted to evaluate possible damage to the cell membranes of human erythrocytes. According to the results in Table 8, the four compounds with different nuclei presented hemolysis rates below 7%, meeting the toxicity criterion (<10%). In other words, none of the compounds caused significant hemolysis in human erythrocytes, even at the highest tested concentration of 100 µM [56]. The highest hemolytic activity of 6.66% was exhibited by the acridine derivative (3a) at the highest concentration (100 µM), still being considered non-hemolytic. In turn, the reference drug m-AMSA showed a hemolytic activity of 19% at the highest concentration.
2.6. Teratogenic Potential and In Vivo Toxicity
Several anticancer drugs have their use limited due to the lack of safety data regarding toxicity in preclinical phase studies [43]. Such a problem can be addressed through in vivo analyses of the potential toxicity of a drug candidate [57]. For studies on toxicity, teratogenicity, and cardiotoxicity, zebrafish is widely recognized due to its genetic similarity to humans [58]. Despite its popularity, studies on zebrafish embryos with promising nuclei for cancer treatment are still few. Recently, Santos et al. [7] studied nitrogen heterocyclic derivatives and evaluated the teratogenic and toxicological profiles against zebrafish embryos, observing that the derivatives did not present toxicity or teratogenic activity.
In the study conducted here, zebrafish embryos were exposed to the compounds 3a, 3b, 3c, and 3d at concentrations of 0.5 μM and 1 μM, with their embryonic development evaluated over 72 hours post-fertilization (hpf). The data are presented in Table 9. During the assay, zebrafish embryos in the first 24 hpf showed 10% embryo coagulation in the negative control (E3 medium) and 100% embryo coagulation for the positive control, 3.4-dichloroaniline. Regarding the exposure to the derivatives, at 24 hpf, 10% coagulation was observed for the compound ACMD (acridine) at 0.5 μM, the same percentage of coagulation observed in the negative control. The toxicity of spiroacridine derivatives in zebrafish embryos was reported by Duarte et al. [59]. The authors describe that the compound AMTAC-06 did not induce lethal effects or morphological alterations during embryonic and larval development, with a LC50 greater than 126.2 μM after 96 hours of exposure, confirming our findings for our acridine derivative (3a).
At 24 hpf, coagulation was also observed in 2.5% and 5% of the embryos exposed to 3b (quinoline) and 3c (indole) at 1 μM, respectively. By the end of the 72 hpf exposure to the derivatives, no other embryos exhibited rates of mortality, malformations, or delays in embryonic development. These data indicate the absence of toxicity of the derivatives to the embryos, as the coagulation rate is within the prescribed parameters [60], which predicts a survival rate of 80% after 72 hpf [61,62]. Figure 8 shows the embryonic development at 72 hpf after exposure to the derivatives.
Quinoline derivative (3b), in addition do not exhibiting toxicity to the embryos, also demonstrated anticancer activity against the T47-D tumor cell line, diverging from the findings described by Sittaramane et al. [63]. The authors determined the toxicological and anticancer activity of derivatives with a quinoline nucleus, with these compounds inhibiting the development of zebrafish embryos. The most toxic to the embryos were the compounds 2-benzyl-1,1,1-trifluoro-3-(quinolin-2-yl)propan-2-ol (2) and trifluoro-3-(isoquinolin-1-yl)-2-(thiophen-2-yl)propan-2-ol (3), with an LC50 value of 14.14 μM. However, compound (2) possessed potent antiproliferative activity against tumor cells.
The toxicity and teratogenicity of nitrogen heterocyclic derivatives were described in the work of Santos et al. [7], where acridine, quinoline, indole, and pyridine compounds did not present significant mortality rates in embryos, with coagulation rates ranging from 2.5% to 20%, the highest rate (20%) being observed for the compound with a pyridine nucleus. In contrast to our findings for the compound 3d (pyridine), which did not exhibit any embryo coagulation during the 72 hpf. The authors also reported no tail detachment at 24 hpf for the acridine compound at 0.5 μM and 1 μM, and for the quinoline and indole derivatives, no tail detachment was also evidenced at the same concentrations and exposure interval, a parameter not observed in our study.
In addition to toxicity, cardiotoxicity is an increasing concern in cancer treatment. Chemotherapeutic treatment contributes to deleterious cardiac effects: additive or synergistic [64,65]. Cardiovascular events such as cardiomyopathy, myocardial fibrosis, and arrhythmias are secondary effects associated with oncological treatment [64,66,67]. For example, epirubicin, idarubicin, daunorubicin, and doxorubicin anthracyclines are used in cancer therapy, and the cardiotoxicity of these agents has been reported [68,69]. The treatment of cardiotoxicity in cancer patients requires therapeutic alternatives and ongoing preclinical phase studies to prevent unwanted cardiac effects [68,70]. Therefore, the analysis of cardio-oncology is necessary to prevent or mitigate cardiovascular damage from potential drug candidates.
The heart rate of the embryos was monitored, and both increases and decreases in heart rate were observed at the two tested concentrations, ranging between 130 and 143 bpm, within the expected range for zebrafish, which varies from 120–180 bpm [71,72]. Acridine compound (3a) at 0.5 μM, compared to the control, increased the heart rate by 5%, without significant statistical differences (Figure 9). Ramachandran and Nagarajan [73] evaluated the toxicity of new acridine-flavone hybrids in zebrafish larvae, in which the compounds did not show cardiac or hepatotoxic toxicity, corroborating our findings.
Quinoline derivative (3b) at 0.5 and 1 μM caused a greater increase in heart rate in zebrafish embryos. However, this increase was only 6% compared to the control. Although these data show variations in heartbeats, there are no statistically significant differences, indicating no cardiac toxicity. In contrast to previous studies, Han et al. [74] evaluated the toxicity of 12 different (fluoro)quinolones in zebrafish embryos and reported that all antibiotics at concentrations ranging from 0.1–20 mM caused different types of malformations such as head deformation, shortened tail, reduced pigmentation, pericardial edema, cardiac malformation, and death, with more than 80% of the mortality rate associated with cardiac dysfunction. These observations are distinct from the findings for the quinoline studied here.
Teratogenic and toxicological studies were conducted by Santos et al. [7]. In this study, under the same experimental conditions used here, zebrafish embryos exposed to nitrogen heterocyclic derivatives containing indole and pyridine nuclei showed an increase in the heart rate of the embryos. The indole derivative (CAIC) at 0.5 μM increased the heart rate of the embryos by 14.8% compared to the control, and this was statistically different compared to the control (p < 0.001). Pyridine compound (CAPA) increased by 11.3% (0.5 μM) and 10.5% (1 μM). One of the mechanisms that may be related to these findings is the generation of reactive oxygen or nitrogen species. Here, indole and pyridine derivatives promoted a decrease in heart rate of 2% to 3%; however, compared to the control, this reduction was not statistically different. Therefore, it is possible to assume that the compounds studied here appear more promising due to having lower mortality rates without statistically significant increases in heart rate, especially in quinoline derivative, which stood out in all interaction assays.
Finally, another useful biochemical parameter with zebrafish tissues for monitoring the embryos toxicity is by redox balance enzymes activities evaluations. The generation of reactive oxygen species (ROS) is a physiological process in cellular metabolism [75]. However, an excess of ROS can cause damage to cells, leading the organism to oxidative stress and the onset of diseases, including cardiological diseases and cancer [75,76]. The antioxidant defense system includes the enzymes: superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPX). These act especially against the superoxide radical anion (O₂⁻) and hydrogen peroxide (H₂O₂) [77].
Superoxide dismutase (SOD) and catalase (CAT) are antioxidant enzymes that catalyze the conversion of reactive species into non-reactive products. SOD catalyzes the conversion of the superoxide anion into hydrogen peroxide, and catalase catalyzes the conversion of hydrogen peroxide into oxygen and water [78]. In the present study, the enzymatic activity of CAT showed both an increase and a reduction in all studied nitrogen heterocycles (Figure 10). Acridine derivative (3a) reduced enzyme activity at both tested concentrations, with no statistically significant reduction at 0.5 μM. On the other hand, compared to the control, at the concentration of 1 μM, CAT activity was reduced by 13%, with statistical significance. Statistical differences were also observed for 3b at 0.5 and 1 μM and 3c at 0.5 μM. Enzyme activity increased by 35% at 0.5 μM and reduced by 62% at 1 μM of QAMD. Compared to the control, 3c (0.5 μM) reduced CAT enzymatic activity by 33% which may indicate a deficiency in enzyme activity or that the enzyme's function is compromised in the presence of the compounds, which can contribute to the emergence of ROS. After all, it has been reported in the literature that the decrease in antioxidant defenses causes the development of diseases such as Alzheimer's, Parkinson's, and cancer [79].
In the SOD activity no heterocyclic compound decreased enzymatic activity. However, an increase in catalytic activity and statistical differences compared to the control were observed. This represents an increase of 90% (3a 0.5 μM), 120% (3c 0.5 μM), 110% (3c 1 μM), 93% (3d 0.5 μM), and 90% (3d 1 μM). Although 3b increased the enzymatic activity of SOD, there was no statistical difference (p = 0.9). Both SOD and CAT, enzymatic activity was altered, except for 3b, which only altered the enzymatic activity of CAT. The increase in SOD and CAT activities can also be justified by the elevated production of ROS [80].
Considering the increase in heart rate in embryos and changes in the activity of antioxidant enzymes (CAT and SOD), it is possible to hypothesize a potential alteration in the redox state. However, this variation in enzymatic activity can also be considered a cellular response against oxidative stress. Therefore, more information is needed regarding the generation of radicals, considering that in some types of cancer, the downregulation (decrease) of catalase expression can contribute to a more oxidative cellular environment and aid in tumor progression. On the other hand, catalase can be expressed more abundantly in some types of cancer, which may be associated with tumor cell resistance to treatment [81].
3. Material and methods
3.1. Chemicals, Reagents and Equipment
The starting materials ethyl 2-cyanoacetate and aniline were acquired from Sigma Aldrich and used for the synthesis of the intermediate reagent 2-cyano-N’-phenylacetamide as well as the acridine, quinoline, indole, and pyridine aldehydes. Common solvents used for synthesis and analysis were provided by Sigma-Aldrich (Saint Louis, MO, USA), Merck (Darmstadt, Germany), and Fluka (Buchs, SG, Switzerland) and used without purification. Melting points were measured in capillary tubes using a Quimis Model 340 apparatus (Quimis, Diadema, Brazil). Thin-layer chromatography (TLC) was performed on Fluka Analytical silica gel 60 plates with a thickness of 0.25 mm and fluorescent indicator 254 nm and visualized under UV light (254 or 365 nm).
Infrared spectra were obtained using KBr discs on an IRPrestige-21 spectrometer, Shimadzu®. Data were processed using Origin software 8.0. Proton nuclear magnetic resonance (1H-NMR) and carbon-13 NMR experiments were conducted on a Varian Model Plus spectrophotometer (Varian, Santa Clara, CA, USA) operating at 300 MHz, using DMSO-d6 as the solvent. Spectra were plotted and interpreted using Mestre Nova 12.0 software. Chemical shifts (δ) were expressed in ppm and coupling constants (J) were given in Hertz (Hz). Signal multiplicities were designated as follows: singlet (s), doublet (d), multiplet (m). Mass spectra were recorded using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) on an Autoflex III mass spectrometer(Bruker Daltonics, Billerica, MA, USA).
The Salmon sperm DNA (ssDNA) and ethidium bromide (EB) used for interaction analysis were procured from Sigma Aldrich (Saint Louis, MO, USA). Human serum albumin (HSA) utilized in this study was obtained following the method described by Alves et al. (2021b) The compounds were prepared as solutions in dimethyl sulfoxide (DMSO) solvent, followed by further dilution in Tris-HCl buffer (0.1 M; pH 7.6). UV-visible absorption spectra and fluorescent emission spectra were recorded using the Thermo Scientific 51119200 spectrometer and the JASCO FP-6300 spectrofluorometer (Tokyo, Japan), respectively.
3.2. Procedure for Synthesis of 2-cyano-N-phenyl acrylamide Derivatives
The intermediate 2-cyano-N-phenylacetamide (2) was obtained through a nucleophilic addition reaction between ethyl 2-cyanoacetate (1) and aniline. The final isopropyl acrylamides (3a-3d) were obtained through a nucleophilic addition reaction between 2-cyano-N-phenylacrylamide (2) and different aromatic aldehydes in an ethanolic and basic medium at a temperature of 78 °C, with the reaction time varying from 3 to 96 hours, followed by the spontaneous cyclization of ACMD (3a) according to Scheme 1. The reaction was monitored by Analytical Thin-Layer Chromatography (TLC), determining the end of the reaction. The reaction was filtered, and the obtained crystals were washed with cold distilled water and then recrystallized in ethanol. The 1H and 13C NMR, IR spectroscopy, and mass spectrometry data are presented in Supplementary material S1–S14.
3.2. 1(E)5’-oxo-1’-phenyl-1’,5’-dihydro-10H-spiro{acridine-9,2’-pyrrole]-4’-carbonitrile (ACMD)- compound (3a)
Yellow powder. Formula: C23H15N3O; M.W.: 349.1215 g/mol; Yield: 75%; Melting Point: 240–242 °C; Rf: 0.57 (n-hexane/EtOAc 6:4). IR (KBr, cm−1): 3314 (NH), 3094 (CH), 2235 (CN), 1695 (C=O), 1450 e 1343 (aromatic CN). 1H NMR (DMSO-d6): δ 9.63 (s, 1H, NH), 8.57 (s, 1H, CH), 7.22 (m, 2H, J = 8.4 e J =1.6), 7.12 (m, 3H, aromatic), 7.07 (m, 2H, spiroacridine), 6.85 (m, 4H, spiroacridine), 6.76 (m, 2H, spiroacridine). 13C NMR: δ 70.6, 108.9, 112.2, 113.1, 115.5, 120.6, 125.4, 127.1, 128.1, 129.0, 130.5, 136.2, 138.7, 163.3, 164.1. HRMS m/z [M+H]+: calculated: 349.3847; found: 350.1168.
3.2.2. (E)-2-cyano-N-phenyl-3-(quinoline-4-yl)acrylamide (QAMD) - compound (3b)
Yellow powder. Formula: C19H13N3O; M.W.: 299.1059 g/mol; Yield: 55%; Melting Point: 195-197 °C; Rf: 0.50 (n-hexane/EtOAc 1:1). IR (KBr, cm−1): 3470 (NH), 3262 (CH), 2233 (CN), 1681 (C=O), 1550 e 1498 (aromatic C=C). 1H NMR (DMSO-d6): δ 10.34 (s, 1H, NH), 9.04 (d, 1H, J = 4.4 aromatic quinoline), 8.24 (d, 1H, J = 8.0 aromatic quinoline), 8.16 (d, 1H, J = 8.4 aromatic quinoline), 7.89 (t, 1H, J = 8.0, aromatic), 7.75 (m, 3H, aromatic), 7.54 (d, 1H, J = 4.4, aromatic quinoline), 7.39 (t, 2H, J = 7.6 aromatic), 7.17 (t, 1H, J = 7.6 aromatic quinoline), 5.73 (s, 1H, C=CH). 13C NMR: δ 114.4, 118.2, 121.4, 124.0, 125.0, 125.5, 128.2, 129.2, 130.2, 130.8, 137.5, 137.7, 147.8, 150.9, 160.0. HRMS m/z [M+H]+: calculated: 299.1059; found: 300.1570.
3.2.3. (E)-2-cyano-3-(1H-indol-3-yl)-N-phenylacrylamide (ICMD) - compound (3c)
Yellow powder. Formula: C18H13N3O; M.W.: 287.1059 g/mol; Yield: 60%; Melting Point: 276–277 °C; Rf: 0.55 (n-hexane/EtOAc 7:3). IR (KBr, cm−1): 3322 (NH), 3040 (CH), 2212 (CN), 1640 (C=O), 1570 e 1442 (aromatic C=C). 1H NMR (DMSO-d6): δ 12.43 (d, 1H, NH, indole), 10.17 (s, 1H, NH, amide), 8.62 (s, 1H, C=CH), 8.55 (s, 1H, C=CH, indole), 8.01 (m, 1H, aromatic indole), 7.70 (d, 2H, J = 7.9, aromatic), 7.59 (m, 1H, aromatic), 7.37 (t, 2H, J = 7.8, aromatic), 7.29 (m, 2H, aromatic), 7.13 (t, 1H, J = 7.4 aromatic). 13C NMR: δ 110.3, 113.2, 119.1, 121.1, 121.1, 123.8, 124.4, 128.1, 129.1, 131.1, 137.0, 140.4, 143.2, 161.7. HRMS m/z [M+H]+: calculated: 287.1059; found: 310.0880.
3.2.4. (E)-2-cyano-N-phenyl-3-(pyridin-4-yl)acrylamide (PAMD) - compound (3d)
Yellow powder. Formula: C15H11N3O; M.W.: 249.0902 g/mol; Yield: 32%; Melting Point: 220–222 °C; Rf: 0.40 (n-hexane/EtOAc 1:1). IR (KBr, cm−1): 3330 (NH), 3019 (CH), 2225 (CN), 1676 (C=O), 1598 e 1444 (aromatic C=C). 1H NMR (DMSO-d6): δ 10.54 (s, 1H, NH), 8.81 (s, 2H, aromatic pyridine), 8.27 (s, 1H, C=CH), 7.81 (d, 2H, J = 5.2 aromatic pyridine), 7.66 (d, 2H, J = 7.6, aromatic), 7.37 (t, 2H, J = 8.0, aromatic), 7.14 (t, 1H, J = 7.6, aromatic). 13C NMR: δ 112.5, 115.7, 121.0, 123.4, 125.1, 129.3, 138.4, 139.5, 148.8, 151.2, 160.1. HRMS m/z [M+H]+: calculated: 249.0902; found: 250.1020.
3.3. UV–Vis Absorption Spectroscopy Studies with DNA
Initially, the ssDNA solution was dissolved in Tris-HCl buffer (0.1 M, pH 7.6) and stored at 8 °C for 24 hours. The purity of the ssDNA solution was determined using an absorption spectroscopy assay at wavelengths of 260 nm and 280 nm. DMSO was used for compound dilution, with a final concentration of 1 mM. The test solutions of the compounds 3a, 3b, 3c and 3d were prepared at a concentration of 5 μM. These solutions were exposed to increasing concentrations of ssDNA (10, 20, 40, 60, 80, and 100 μM). The test solutions (ssDNA and compounds) were homogenized and left to rest at 25 °C for 10 minutes. The absorbance readings of the samples were then performed using a UV-vis Thermo Scientific 51119200 spectrophotometer in 96-well plates, at a wavelength range of 200-600 nm. The McGhee and Von Hippel [82] equation:
[Derivative] / (εa - εf) = [Derivative] / (εb - εf) + 1 / Kb (εb - εf) [DNA] / (εa - εf)
[DNA] / (εb - εf) + 1 / Kb (εb - εf) Where, Ea, Eb, and Ef are the apparent, bound, and free extinction coefficients, respectively. The graphs for [Compound]/ (Ea - Ef) versus [Compound] and [DNA]/ (Ea - Ef) versus [DNA] were used to obtain the Kbs values from the ratio of inclination and interception, using the software SigmaPlot 10.0. was used to determine the binding constant (Kb). The analyses were performed in triplicate.
3.3.1. Fluorescent DNA Probe Assay
The fluorescence emissions of EB were measured in the presence and absence of ssDNA. The investigation involved a concentration of 10 μM of EB and 100 μM of ssDNA. These measurements were conducted using a rectangular quartz cuvette with a 1 cm optical path and a JASCO FP-6300 spectrofluorometer (Tokyo, Japan). For the EB fluorescence spectra, excitation was set at 526 nm, and emission was recorded in the range of 550 to 100 nm. To evaluate the displacement of EB by the tested compounds, solutions containing EB-ssDNA were exposed to the derivatives at concentrations of 10, 20, 40, 60, 80, and 100 μM in Tris-HCl buffer (0.1 M, pH 7.6). The tests were conducted in triplicate. All solutions were carefully mixed and allowed to stabilize for 10 minutes in the mentioned buffer before undergoing fluorescence analysis. The concentrations used in the test solutions were determined after a scan of the absorption profile of the free test compounds was conducted. The emission suppression behavior was analyzed using the Stern-Volmer equation (Ksv) [83]:
where, F0 and F are the fluorescence intensities in the absence and in the presence of the derivatives, respectively. Ksv is the constant of linear suppression. [Q] represents the concentration of the compound. Binding data were obtained with the software SigmaPlot 10.0.
F0 / F = 1 + Ksv [Q]
3.4. Physicochemical and Pharmacokinetic Predictions
The MarvinSketch® academic license platform (https://chemaxon.com/products/marvin) was used for the two-dimensional representation of the chemical structure and to quantitatively estimate the similarity to drugs based on multiple parameter optimization (MPO), following the equation:
where w is the weighting factor assigned to the calculated value x of a property k, which varies from 0 to 1, staying within (xₖ ≤ xₐ) or outside (xb < xₖ) the desirability limit.
The MPO estimate covers parameters such as intrinsic lipophilicity (logP ≤ 3), pH (logD ≤ 2), molecular weight (200 < MW ≤ 500 g/mol), topological polar surface area (40 < TPSA ≤ 90 Ų), number of hydrogen bond donors (HBD ≤ 1), and pKa (≤ 8), which will result in a score ranging from 0 to 6 (M= 6), which is a pharmacokinetic feasibility score. In addition, drug similarity criteria from Lipinski's “rule of five” were also applied, and compatibility with the rules of Veber, Ghoshe, Egan, and Muegge was assessed using the SwissADME platform (http://www.swissadme.ch/).
In order to understand parameters related to physicochemical and pharmacokinetic aspects, different platforms were used, such as SwissADME and ADMETlab 3.0 (https://admetlab3.scbdd.com/) to predict factors related to absorption (Caco-2 permeability -2 and MDCK permeability), distribution (plasma protein binding, distribution volume, and blood-brain barrier permeability), metabolism (CYP1A2, CYP2C19, and CYP3A4), and excretion (clearance). The half-life was determined using the following formula:
In addition, other platforms such as Xenosite (https://xenosite.org/) were used to describe metabolization points. Finally, aspects related to the toxicity of the compounds were described using ProTox-3. 0 (https://tox.charite.de/protox3/index.php?site=compound_input) and TopTox (https://stoptox.mml.unc.edu/predict?smiles=CCc1ccc%28C%23N%29c%28F%29c1F).
3.5. Molecular Docking
AutoDock 4.2.6, combined with the Lamarckian genetic algorithm (Morris et al.; 2009), was used for the molecular docking analyses.
3.5.1. Preparation of Ligand Structure
The ChemDraw 15.0 software was used to construct the structure of the analyzed compounds, and the structures were optimized based on the MM2 semi-empirical theory. After optimization, the structures were saved in pdb format for docking studies.
3.5.2. Topoisomerase IIα and HSA in the Presence of Ligands
For topoisomerase IIα, the parameters for generating the three-dimensional grid (3D grids) were previously configured in AutoGrid 4.2.6. This intercalating agent contained 70 x 70 x 70 grid points with a spacing of 0.0375 nm. The grid was centered at the position of the co-crystallized ligand in the A chain of the protein. For HSA, the 3D grids were configured using AutoDockTools and AutoGrid 4.2.6. In both cases, the 3D grids had dimensions of 126 x 126 x 126 points with a spacing of 0.0375 nm. The center was defined as the first grid at the position of the co-crystallized ligand in site I of the A chain of the protein, while the second grid was centered at the position of the co-crystallized ligand in site II of the A chain of the protein. For both macromolecules, the Lamarckian genetic algorithm in AutoDock 4.2.6 was applied to search for the optimal conformation and orientation of the ligands. From the docking experiments, 100 conformations were generated and analyzed. The lowest energy conformation for each ligand was determined and examined using AutoDockTools and Discovery Studio Visualizer [7].
3.6. Human Topoisomerase IIα Inhibition
The topoisomerase IIα inhibition assay was conducted following the methodology described by Almeida et al. [8] . Subsequently, quantitative analysis was performed by densitometry to obtain the percentage of enzymatic inhibition by the derivatives, using image processing software (Scion Image, Beta 4.0.2.), according to Gouveia et al. [20] .
3.7. Evaluation of Antiproliferative Activity In Vitro
The sulforhodamine B method, as described by Monks et al. [84], was used in the antiproliferative assay with tumor and non-tumor cells. The breast cancer cell lines MCF-7 and T47D, as well as immortalized normal human keratinocytes (HaCaT), were utilized. In cell culture, the cells were grown and maintained in RPMI-1640 medium supplemented with 5% fetal bovine serum and 1% antibiotics, consisting of penicillin (1000 IU/mL) and streptomycin (1000 μg/mL). For the assay, 6 × 10³ cells were plated in 96-well plates and incubated at 37 °C, 5% CO2 for 24 hours. After this period, the cells were incubated with the compounds 3a, 3b, 3c and 3d at concentrations of 0.25, 2.5, 25, and 250 μM, which were previously dissolved in DMSO and diluted in RPMI-1640 medium. The same concentrations were used for positive control, amsacrine (m-AMSA). After 48 hours of incubation and exposure to the compounds, the cells were fixed with 50% trichloroacetic acid solution. Cell proliferation was quantified by absorbance reading at a wavelength of 540 nm, using sulforhodamine B stain. Non-linear regression analysis with Origin 8.0® software (OriginLab Corporation) was used to determine the concentration values that inhibited 50% of cell growth (GI50) for each cell line. Each experimental assay was performed in triplicate.
3.8. Hemolytic Activity Studies of the Derivatives
The human erythrocytes used were obtained from a voluntary donation by a healthy donor, with a volume of 5 mL. The collected blood sample was subjected to 3 washes with saline solution (0.9% NaCl) and centrifuged at 2500 rpm for 5 minutes. All plasma residue was removed from the sample, resulting in a concentrated red blood cell solution. The 0.5% stock solution was prepared by diluting 250 μL of the concentrated solution in 50 mL of saline solution. In the test solution, 2 mL of the red blood cell stock solution was used. For the assay, the compounds 3a, 3b, 3c and 3d were previously diluted in 1% DMSO, with a final concentration of 1 mM, and then test solutions were prepared. The red blood cell test solution was exposed to concentrations of 5, 10, 25, 50, and 100 μM of the compounds. The negative control used was 2 mL of red blood cells mixed with 10 μL of saline solution, while the positive control consisted of 2 mL of red blood cells supplemented with 10 μL of 1% Triton X-100. The negative control corresponded to 0% hemolysis and the positive control to 100% hemolysis. All samples were incubated for one hour at room temperature and then centrifuged at 2500 rpm for 5 minutes at 4°C. The supernatant was collected and transferred to a 96-well plate for reading on a spectrophotometer at a wavelength of 540 nm. The entire assay was conducted in triplicate. Equation: % Hemolysis = [(Sample Abs – Negative Control Abs) / Positive Control Absorbance)] x 100, where: Sample Abs is the absorbance of the test solution (Red Blood Cells + Derivatives), Negative Control Abs is the absorbance of the negative control (Red Blood Cells + Saline Solution), and Positive Control Abs is the absorbance of the positive control (Red Blood Cells + Triton), was used to calculate the hemolysis rate [85].
3.9. In Vivo Studies
3.9.1. Care and Obtention of Zebrafish Embryos
Zebrafish specimens were commercially obtained and maintained in a laboratory at the University of Pernambuco (UPE), Garanhuns Campus. The adult fish were separated into male and female groups in 16-liter tanks connected to a water recirculation and filtration system, with specific and appropriate conditions for the reproductive cycle, including a light/dark cycle of 14 hours of light and 10 hours of darkness, pH of 7.2, and a temperature of 26°C. The fish were fed daily ad libitum with Tropical® fish food (Poytara) and Artemia spp. All animal care and monitoring were conducted according to ethical standards and approved by the Ethics Committee on the Use of Animals of the University of Pernambuco (CEUA-UPE) under Process No. 008/2021, following the guidelines of CONCEA (National Council for the Control of Animal Experimentation).
After 2 months of monitoring, the fish were subjected to a reproductive event to obtain embryos, using one female and two males. The fish were separated and transferred to a breeding tank with a gridded bottom at around 4:00 PM, while maintaining the previously described conditions. The reproduction occurred in the early hours of the following day, and the embryos were collected and analyzed for viability using a Motic Phantera microscope. Embryos considered non-viable (coagulated or unfertilized) were discarded, and viable embryos were used for exposure.
3.9.2. Teratogenic Potential
The teratogenic studies followed the guidelines established by the OECD [60]. After analyzing the viable embryos, the fertilized eggs were washed with E3 medium (composed of 5 mM NaCl, 0.17 mM KCl, 0.33 mM MgSO4.7H2O, and 0.33 mM CaCl2 at pH 7.2) and then incubated at 25 °C. In 24-well plates, 20 embryos per well were exposed to the compounds 3a, 3b, 3c and 3d at concentrations of 1 μM and 0.5 μM, starting 6 hours post-fertilization (hpf), at a temperature of 26 ± 1°C. The positive control used was 3,4-dichloroaniline, and the negative control was E3 medium. The embryos were monitored at 24 hpf, 48 hpf, and 72 hpf, considering lethality parameters such as coagulated embryos, absence of somite formation, failure of tail detachment, and absence of heartbeat [60]. During the 72 hpf, the embryos were photographed using a Motic Phantera microscope.
3.9.3. Enzymatic Assays of Embryo Tissues
After 72 hpf of embryo exposure to the derivatives, enzymatic studies were conducted on the enzymes Superoxide dismutase (SOD) (EC 1.15.1.1) and catalase (CAT) (EC 1.11.1.6). For tissue homogenization, a tissue homogenizer (NT-136 – Nova Técnica) was used, along with 100 μL of ice-cold PBS buffer at 0.1 M, pH 7.2, supplemented with sodium orthovanadate (1 mM) and phenylmethylsulfonyl fluoride (PMSF, 200 μg/mL). After homogenization, this solution was centrifuged for 10 minutes at 800 x g (4°C) in a (Thermo Scientific – Heraeus megafuge 16R), the pellet was discarded, and the supernatant was used for the enzymatic assays. Protein quantification was previously determined using the bicinchoninic acid method [86]. Following Misra and Fridovich [18], adaptations were made, and the following conditions were established for SOD activity evaluation at 480 nm, epinephrine (60 mM) diluted in 0.05% acetic acid was used as a substrate, monitored for 9 minutes in the presence of 50 mM glycine buffer at pH 10.0. CAT activity was evaluated at 240 nm with 300 mM H2O2 as the substrate. The catalytic event was monitored for 3 minutes [87]. All assays were performed in triplicate.
3.9.4. Statistical Analysis
The data obtained from the assays were initially subjected to the Kolmogorov-Smirnov test to assess normality. Subsequently, one-way ANOVA followed by Tukey's post-hoc test was performed. Results with p < 0.05 were considered statistically significant. Statistical analyses were conducted using IBM® SPSS® software, and graphs were generated using OriginLab 8®.
4. Conclusion
The biological activity of the four studied molecules indicates their ability to interact with target biomolecules in cancer therapy (DNA). Among the analyzed compounds, those that exhibited the most significant interaction with DNA, as assessed by UV-vis spectroscopy and fluorescence probes, were 3a (acridine) and 3b (quinoline), with Kb values of 2.23 × 10⁵ and 1.41 × 10⁵, and Ksv values of 0.67 × 10³ and 0.63 × 10³ (EB), respectively. In the molecular docking study, 3b demonstrated the highest binding energy. All compounds inhibited topoisomerase activity and were cytotoxic against tumor cells while exhibiting minimal hemolytic activity, suggesting potential efficacy in cancer therapy. In the evaluation of teratogenicity, toxicity, and cardiotoxicity in zebrafish embryos, no toxic effects were observed at the tested concentrations. However, further investigations are necessary to assess their influence on redox balance, considering the observed changes in superoxide dismutase (SOD) and catalase (CAT) activity. Thus, based on the evidence presented in this study, the employed techniques demonstrate the value of this new class of nitrogen heterocycles and provide insights into the development of novel anticancer molecules. Reducing the acridine nucleus to a quinoline core while maintaining the N-phenyl-substituted amide side group appears to be a promising approach.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Maria Regina Silva; Methodology, Jéssica Celerino dos Santos, Josival Emanuel Alves, Josefa Gerlane da Silva, Maria Regina Silva, Jamire Muriel da Silva and Nabuêr Francieli da Silva; Software, Caio Victor Soares; Validation, Jéssica Celerino dos Santos, Josival Emanuel Alves and Caio Victor Soares; Formal analysis, Rafael David de Azevedo, Lucia Patrícia da Silva, Jamire Muriel da Silva and Nabuêr Francieli da Silva; Investigation, Jéssica Celerino dos Santos, Josival Emanuel Alves, Josefa Gerlane da Silva, Maria Regina Silva, Lucia Patrícia da Silva and Caio Victor Soares; Resources, Jéssica Celerino dos Santos, Maria Regina Silva and Jamire Muriel da Silva; Data curation, Rafael David de Azevedo and Josefa Gerlane da Silva; Writing – original draft, Jéssica Celerino dos Santos, Josival Emanuel Alves, Rafael David de Azevedo, Maria Regina Silva, Lucia Patrícia da Silva, Jamire Muriel da Silva and Nabuêr Francieli da Silva; Writing – review & editing, Rafael David de Azevedo, Jamerson Ferreira de Oliveira, Maria do Carmo de Lima, Ricardo Olimpio de Moura and Sinara Mônica de Almeida; Supervision, Jamerson Ferreira de Oliveira, Maria do Carmo de Lima, Ricardo Olimpio de Moura and Sinara Mônica de Almeida; Project administration, Ricardo Olimpio de Moura and Sinara Mônica de Almeida; Funding acquisition, Ricardo Olimpio de Moura and Sinara Mônica de Almeida. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fundação de Amparo à Ciência e Tecnologia de Pernambuco grant number APQ-1001-4.03/24, APQ-1508-4.03/24, APQ-1581-4.03/25, National Council for Scientific and Technological Development grant number DCR-0010-4.03/20.
Institutional Review Board Statement
The animal study protocol was approved by the Ethics Committee of the Use of Animals of the University of Pernambuco (CEUA-UPE) (protocol code No. 008/2021).
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Acknowledgments
The authors would like to thank the Foundation of Support to Sciences and Technology in Pernambuco [Fundação de Amparo à Ciência e Tecnologia de Pernambuco – FACEPE] – Grants nº APQ-1001-4.03/24; APQ-1508-4.03/24; APQ-1581-4.03/25. The authors also are grateful for financial support of Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (Grant nº DCR-0010-4.03/20), Brazil.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Geng, W.-C.; Jiang, H.; Wu, B. Recent advances in biocatalysis of nitrogen-containing heterocycles. Biotechnol. Adv. 2022, 54, 107813. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Kozurkova, M. Acridine derivatives as inhibitors/poisons of topoisomerase II. J. Appl. Toxicol. 2022, 42, 544–552. [Google Scholar] [CrossRef]
- Lang, D.K.; Kaur, R.; Arora, R.; Saini, B.; Arora, S. Nitrogen-Containing Heterocycles as Anticancer Agents: An Overview. Anticancer. Agents Med. Chem. 2020, 20, 2150–2168. [Google Scholar] [CrossRef]
- Mermer, A.; Keles, T.; Sirin, Y. Recent studies of nitrogen containing heterocyclic compounds as novel antiviral agents: A review. Bioorg. Chem. 2021, 114, 105076. [Google Scholar] [CrossRef]
- dos Santos, J.C.; Alves, J.E.F.; de Azevedo, R.D.S.; de Lima, M.L.; de Oliveira Silva, M.R.; da Silva, J.G.; da Silva, J.M.; de Carvalho Correia, A.C.; do Carmo Alves de Lima, M.; de Oliveira, J.F.; de Moura, R.O.; de Almeida, S.M.V. Study of nitrogen heterocycles as DNA/HSA binder, topoisomerase inhibitors and toxicological safety. Int. J. Biol. Macromol. 2024, 254, 127651. [Google Scholar] [CrossRef]
- Almeida, S.M.V.; Lafayette, E.A.; Silva, W.L.; Lima Serafim, V.; Menezes, T.M.; Neves, J.L.; Ruiz, A.L.T.G.; Carvalho, J.E.; Moura, R.O.; Beltrão, E.I.C.; Carvalho Júnior, L.B.; Lima, M. do C.A. New spiro-acridines: DNA interaction, antiproliferative activity and inhibition of human DNA topoisomerases. Int. J. Biol. Macromol. 2016, 92, 467–475. [Google Scholar] [CrossRef]
- Ribeiro, A.G.; Almeida, S.M.V.; Oliveira, J.F.; Souza, T.R.C. L.; Santos, K.L.; Albuquerque, A.P. de B.; Nogueira, M.C.B.L.; Carvalho Junior, L.B.; Moura, R.O.; Silva, A.C.; Pereira, V.R.A.; Castro, M.C.A.B.; Lima, M. do C.A. Novel 4-quinoline-thiosemicarbazone derivatives: Synthesis, antiproliferative activity, in vitro and in silico biomacromolecule interaction studies and topoisomerase inhibition. Eur. J. Med. Chem. 2019, 182, 111592. [Google Scholar] [CrossRef]
- Alves, J.E.F.; de Oliveira, J.F.; de Lima Souza, T.R.C.; de Moura, R.O.; de Carvalho Júnior, L.B.; Alves de Lima, M. do C.; de Almeida, S.M.V. Novel indole-thiazole and indole-thiazolidinone derivatives as DNA groove binders. Int. J. Biol. Macromol. 2021, 170, 622–635. [Google Scholar] [CrossRef]
- Narva, S.; Chitti, S.; Bala, B.R.; Alvala, M.; Jain, N.; Kondapalli, V.G.C.S. Synthesis and biological evaluation of pyrrolo[2,3- b ]pyridine analogues as antiproliferative agents and their interaction with calf thymus DNA. Eur. J. Med. Chem. 2016, 114, 220–231. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, P.-K. DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.; More, L.A.; Asnani, A. Zebrafish Models of Cancer Therapy-Induced Cardiovascular Toxicity. J. Cardiovasc. Dev. Dis. 2021, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Letrado, P.; de Miguel, I.; Lamberto, I.; Díez-Martínez, R.; Oyarzabal, J. Zebrafish: Speeding Up the Cancer Drug Discovery Process. Cancer Res. 2018, 78, 6048–6058. [Google Scholar] [CrossRef] [PubMed]
- Raby, L.; Völkel, P.; Le Bourhis, X.; Angrand, P.-O. Genetic Engineering of Zebrafish in Cancer Research. Cancers (Basel) 2020, 12, 2168. [Google Scholar] [CrossRef]
- Silva, P.; de Almeida, M.; Silva, J.; Albino, S.; Espírito-Santo, R.; Lima, M.; Santos, V. (E)-2-Cyano-3-(1 H-Indol-3-yl)-N-Phenylacrylamide, a Hybrid Compound Derived from Indomethacin and Paracetamol: Design, Synthesis and Evaluation of the Anti-Inflammatory Potential. International Journal of Molecular Sciences 2020, 21(7), 2591. [Google Scholar] [CrossRef]
- Lafayette, E.; Almeida, S.M.V.; Da Rocha Pitta, M.; Carneiro Beltrão, E.; Gonçalves da Silva, T.; Olímpio de Moura, R.; Da Rocha Pitta, I.; De Carvalho, L.; Do Carmo Alves de Lima, M. Synthesis, DNA Binding and Topoisomerase I Inhibition Activity of Thiazacridine and Imidazacridine Derivatives. Molecules 2013, 18, 15035–15050. [Google Scholar] [CrossRef]
- Misra, H.P.; Fridovich, I. The Role of Superoxide Anion in the Autoxidation of Epinephrine and a Simple Assay for Superoxide Dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [CrossRef]
- Vilková, M.; Prokaiová, M.; Imrich, J. Spontaneous cyclization of (acridin-9-ylmethyl)thioureas to spiro [dihydroacridine-9′(10′H),5-imidazolidine]-2-thiones, a novel type of acridine spirocycles. Tetrahedron 2014, 70, 944–961. [Google Scholar] [CrossRef]
- Gouveia, R.G.; Ribeiro, A.G.; Segundo, M.Â.S.P.; de Oliveira, J.F.; de Lima, M. do C.A.; de Lima Souza, T.R.C.; de Almeida, S.M.V.; de Moura, R.O. Synthesis, DNA and protein interactions and human topoisomerase inhibition of novel Spiroacridine derivatives. Bioorg. Med. Chem. 2018, 26, 5911–5921. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. Journal of medicinal chemistry 2002, 45(12), 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. Journal of Medicinal Chemistry 2001, 44, No. 12. [Google Scholar] [CrossRef]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Pettersson, M.; Hou, X.H.; Kuhn, M.; Wager, T.T.; Kauffman, G.W.; Verhoest, P.R. Quantitative Assessment of the Impact of Fluorine Substitution on P-Glycoprotein (P-gp) Mediated Efflux, Permeability, Lipophilicity, and Metabolic Stability. J. Med. Chem. 2016, 59, 5284−5296. [Google Scholar] [CrossRef]
- Ma, X.L.; Chen, C.; Yang, J. Predictive model of blood-brain barrier penetration of organic compounds. Acta Pharmacologica Sinica 2005, 26(4), 500–512. [Google Scholar] [CrossRef]
- Holt, K.; Nagar, S.; Korzekwa, K. Methods to Predict Volume of Distribution. Current Pharmacology Reports 2009, 5, 391–399. [Google Scholar] [CrossRef]
- Iwata, H.; Matsuo, T.; Mamada, H.; Motomura, T.; Matsushita, M.; Fujiwara, T.; Handa, K. Predicting Total Drug Clearance and Volumes of Distribution Using the Machine Learning-Mediated Multimodal Method through the Imputation of Various Non clinical Data. J. Chem. Inf. Model. 2022, 62, 4057−4065. [Google Scholar] [CrossRef]
- Zheng, M.; Luo, X.; Shen, Q.; Wang, Y.; Du, Y.; Zhu, W.; Jiang, H. Site of metabolism prediction for six biotransformations mediated by cytochromes P450. Bioinformatics 2009, 25(10), 1251–1258. [Google Scholar] [CrossRef]
- Barreiro, E.J.; Fraga, C.A.M. Química medicinal: as bases moleculares da ação dos fármacos, 3. ed.; Artmed: Porto Alegre, 2015; p. 608 p. ISBN 978-8582711170. [Google Scholar]
- Dang, N.L.; Hughes, T.B.; Krishnamurthy, V.; Swamidass, S.J. A simple model predicts UGT-mediated metabolism. Bioinformatics 2016, 32(20), 3183–3189. [Google Scholar] [CrossRef]
- Yu, K.; Geng, X.; Chen, M.; Zhang, J.; Wang, B.; Ilic, K.; Tong, W. High Daily Dose and Being a Substrate of Cytochrome P450 Enzymes Are Two Important Predictors of Drug-Induced Liver Injury. Drug Metab Dispos 2014, 42, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.M.V.; Lafayette, E.; Da Silva, L.; Amorim, C.; De Oliveira, T.; Ruiz, A.; De Carvalho, J.; De Moura, R.; Beltrão, E.; De Lima, M.; Carvalho Júnior, L. Synthesis, DNA Binding, and Antiproliferative Activity of Novel Acridine-Thiosemicarbazone Derivatives. Int. J. Mol. Sci. 2015, 16, 13023–13042. [Google Scholar] [CrossRef] [PubMed]
- Fortes, M.P.; da Silva, P.B.N.; da Silva, T.G.; Kaufman, T.S.; Militão, G.C.G.; Silveira, C.C. Synthesis and preliminary evaluation of 3-thiocyanato-1H-indoles as potential anticancer agents. Eur. J. Med. Chem. 2016, 118, 21–26. [Google Scholar] [CrossRef]
- Kim, Y.J.; Pyo, J.S.; Jung, Y.-S.; Kwak, J.-H. Design, synthesis, and biological evaluation of novel 1-oxo-1,2,3,4-tetrahydropyrazino[1,2-a]indole-3-carboxamide analogs in MCF-7 and MDA-MB-468 breast cancer cell lines. Bioorg. Med. Chem. Lett. 2017, 27, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Perković, I.; Beus, M.; Schols, D.; Persoons, L.; Zorc, B. Itaconic acid hybrids as potential anticancer agents. Mol. Divers. 2022, 26, 1–14. [Google Scholar] [CrossRef]
- Zhao, M.; Cui, Y.; Zhao, L.; Zhu, T.; Lee, R.J.; Liao, W.; Sun, F.; Li, Y.; Teng, L. Thiophene Derivatives as New Anticancer Agents and Their Therapeutic Delivery Using Folate Receptor-Targeting Nanocarriers. ACS Omega 2019, 4, 8874–8880. [Google Scholar] [CrossRef]
- Rehman, S.U.; Sarwar, T.; Husain, M.A.; Ishqi, H.M.; Tabish, M. Studying non-covalent drug–DNA interactions. Arch. Biochem. Biophys. 2015, 576, 49–60. [Google Scholar] [CrossRef]
- Ma, F.-F.; Cai, Z.-B.; Li, S.-L.; Tian, Y.-P. Synthesis, photophysical properties, and DNA-binding of novel A-π-D-π-A’ two-photon absorption chromophores. J. Photochem. Photobiol. A Chem. 2018, 364, 705–714. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Zhou, J.; Wu, S.; Wei, Y.; Chang, A.; Liu, X.; Shangguan, D. DNA interaction, cellular localization and cytotoxicity of quinacridone derivatives. Dye. Pigment. 2015, 121, 328–335. [Google Scholar] [CrossRef]
- Singh, K.; Srivastava, P.; Patra, A.K. Binding interactions with biological targets and DNA photocleavage activity of Pr(III) and Nd(III) complexes of dipyridoquinoxaline. Inorganica Chim. Acta 2016, 451, 73–81. [Google Scholar] [CrossRef]
- Lafayette, E.A.; de Almeida, S.M.V.; Cavalcanti Santos, R.V.; de Oliveira, J.F.; Amorim, C.A. da C.; da Silva, R.M.F.; Pitta, M.G. da R.; Pitta, I. da R.; de Moura, R.O.; de Carvalho Júnior, L.B.; de Melo Rêgo, M.J.B.; de Lima, M.C.A. Synthesis of novel indole derivatives as promising DNA-binding agents and evaluation of antitumor and antitopoisomerase I activities. Eur. J. Med. Chem. 2017, 136, 511–522. [Google Scholar] [CrossRef]
- Oliveira, J.F.; Lima, T.S.; Vendramini-Costa, D.B.; de Lacerda Pedrosa, S.C.B.; Lafayette, E.A.; da Silva, R.M.F.; de Almeida, S.M.V.; de Moura, R.O.; Ruiz, A.L.T.G.; de Carvalho, J.E.; de Lima, M. do C.A. Thiosemicarbazones and 4-thiazolidinones indole-based derivatives: Synthesis, evaluation of antiproliferative activity, cell death mechanisms and topoisomerase inhibition assay. Eur. J. Med. Chem. 2017, 136, 305–314. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, J.; Zhuang, R.; He, R.; Xi, J.; Pan, X.; Shao, Y.; Pan, J.; Sun, J.; Cai, Z.; Liu, S.; Huang, W.; Lv, X. Synthesis and evaluation of a series of pyridine and pyrimidine derivatives as type II c-Met inhibitors. Bioorg. Med. Chem. 2017, 25, 3195–3205. [Google Scholar] [CrossRef]
- Singh, V.; Afshan, T.; Tyagi, P.; Varadwaj, P.K.; Sahoo, A.K. Recent development of multi-targeted inhibitors of human topoisomerase II enzyme as potent cancer therapeutics. Int. J. Biol. Macromol. 2023, 226, 473–484. [Google Scholar] [CrossRef]
- Shinde, Y.; Patil, R.; Badireenath, K.V.; Merugu, S.B.; Mokashi, V.; Harihar, S.; Marrot, J.; Butcher, R.J.; Salunke-Gawali, S. Keto-enol tautomerism of hydroxynaphthoquinoneoxime ligands: Copper complexes and topoisomerase inhibition activity. J. Mol. Struct. 2022, 1262, 133081. [Google Scholar] [CrossRef]
- Anurag, W.; Amol, D. Topoisomerase: An Overview. Int. J. Adv. Res. Sci. Commun. Technol. 2022, 145–155. [Google Scholar] [CrossRef]
- Ibrahim, M.K.; Taghour, M.S.; Metwaly, A.M.; Belal, A.; Mehany, A.B.M.; Elhendawy, M.A.; Radwan, M.M.; Yassin, A.M.; El-Deeb, N.M.; Hafez, E.E.; ElSohly, M.A.; Eissa, I.H. Design, synthesis, molecular modeling and anti-proliferative evaluation of novel quinoxaline derivatives as potential DNA intercalators and topoisomerase II inhibitors. Eur. J. Med. Chem. 2018, 155, 117–134. [Google Scholar] [CrossRef]
- Murugavel, S.; Ravikumar, C.; Jaabil, G.; Alagusundaram, P. Synthesis, crystal structure analysis, spectral investigations (NMR, FT-IR, UV), DFT calculations, ADMET studies, molecular docking and anticancer activity of 2-(1-benzyl-5-methyl-1H-1,2,3-triazol-4-yl)-4-(2-chlorophenyl)-6-methoxypyridine – A novel poten. J. Mol. Struct. 2019, 1176, 729–742. [Google Scholar] [CrossRef]
- da Silva Filho, F.A.; de Freitas Souza, T.; Ribeiro, A.G.; Alves, J.E.F.; de Oliveira, J.F.; de Lima Souza, T.R.C.; de Moura, R.O.; do Carmo Alves de Lima, M.; de Carvalho Junior, L.B.; de Almeida, S.M.V. Topoisomerase inhibition and albumin interaction studies of acridine-thiosemicarbazone derivatives. Int. J. Biol. Macromol. 2019, 138, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xing, J.; Xu, Y.; Zhou, N.; Peng, J.; Xiong, Z.; Liu, X.; Luo, X.; Luo, C.; Chen, K.; Zheng, M.; Jiang, H. In silico ADME/T modelling for rational drug design. Q. Rev. Biophys. 2015, 48, 488–515. [Google Scholar] [CrossRef]
- Queiroz, C.M.; de Oliveira Filho, G.B.; Espíndola, J.W.P.; do Nascimento, A.V.; Aliança, A.S.S.; de Lorena, V.M.B.; Feitosa, A.P.S.; da Silva, P.R.; Alves, L.C.; Leite, A.C.L.; Brayner, F.A. Thiosemicarbazone and thiazole: in vitro evaluation of leishmanicidal and ultrastructural activity on Leishmania infantum. Med. Chem. Res. 2020, 29, 2050–2065. [Google Scholar] [CrossRef]
- Cassar, S.; Adatto, I.; Freeman, J.L.; Gamse, J.T.; Iturria, I.; Lawrence, C.; Muriana, A.; Peterson, R.T.; Van Cruchten, S.; Zon, L.I. Use of Zebrafish in Drug Discovery Toxicology. Chem. Res. Toxicol. 2020, 33, 95–118. [Google Scholar] [CrossRef]
- Azevedo, R.D.S.; Falcão, K.V.G.; Amaral, I.P.G.; Leite, A.C.R.; Bezerra, R.S. Mitochondria as targets for toxicity and metabolism research using zebrafish. Biochim. Biophys. Acta - Gen. Subj. 2020, 1864, 129634. [Google Scholar] [CrossRef]
- Duarte, S.S.; Silva, D.K.F.; Lisboa, T.M.H.; Gouveia, R.G.; Ferreira, R.C.; De Moura, R.O.; Da Silva, J.M.; De Almeida, É.; Rodrigues-Mascarenhas, S.; Da Silva, P.M.; Farias, D.F.; Da Costa Ribeiro Souza, J.A.; De Paula Medeiros, K.C.; Gonçalves, J.C.R.; Sobral, M.V. Anticancer Effect of a Spiro-acridine Compound Involves Immunomodulatory and Anti-angiogenic Actions. Anticancer Res. 2020, 40, 5049–5057. [Google Scholar] [CrossRef]
- OECD. Test No. 236: Fish Embryo Acute Toxicity (FET) Test, OECD Guidelines for the Testing of Chemicals, Section 2; OECD, 2013. [Google Scholar] [CrossRef]
- Busquet, F.; Strecker, R.; Rawlings, J.M.; Belanger, S.E.; Braunbeck, T.; Carr, G.J.; Cenijn, P.; Fochtman, P.; Gourmelon, A.; Hübler, N.; Kleensang, A.; Knöbel, M.; Kussatz, C.; Legler, J.; Lillicrap, A.; Martínez-Jerónimo, F.; Polleichtner, C.; Rzodeczko, H.; Salinas, E.; Schneider, K.E.; Scholz, S.; van den Brandhof, E.-J.; van der Ven, L.T.M.; Walter-Rohde, S.; Weigt, S.; Witters, H.; Halder, M. OECD validation study to assess intra- and inter-laboratory reproducibility of the zebrafish embryo toxicity test for acute aquatic toxicity testing. Regul. Toxicol. Pharmacol. 2014, 69, 496–511. [Google Scholar] [CrossRef]
- Pogorelcnik, B.; Perdih, A.; Solmajer, T. Recent Developments of DNA Poisons - Human DNA Topoisomerase IIα Inhibitors - as Anticancer Agents. Curr. Pharm. Des. 2013, 19, 2474–2488. [Google Scholar] [CrossRef]
- Sittaramane, V.; Padgett, J.; Salter, P.; Williams, A.; Luke, S.; McCall, R.; Arambula, J.F.; Graves, V.B.; Blocker, M.; Van Leuven, D.; Bowe, K.; Heimberger, J.; Cade, H.C.; Immaneni, S.; Shaikh, A. Discovery of Quinoline-Derived Trifluoromethyl Alcohols, Determination of Their in vivo Toxicity and Anticancer Activity in a Zebrafish Embryo Model. Chem Med Chem 2015, 10, 1802–1807. [Google Scholar] [CrossRef]
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of anticancer treatments: Epidemiology, detection, and management. CA. Cancer J. Clin. 2016, 66, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Rosa, G.M.; Gigli, L.; Tagliasacchi, M.I.; Di Iorio, C.; Carbone, F.; Nencioni, A.; Montecucco, F.; Brunelli, C. Update on cardiotoxicity of anti-cancer treatments. Eur. J. Clin. Invest. 2016, 46, 264–284. [Google Scholar] [CrossRef] [PubMed]
- Essa, H.; Wright, D.J.; Dobson, R.; Lip, G.Y.H. Chemotherapy-Induced Arrhythmia – Underrecognized and Undertreated. Am. J. Med. 2021, 134, 1224–1231.e1. [Google Scholar] [CrossRef] [PubMed]
- Ewer, M.S.; Ewer, S.M. Cardiotoxicity of anticancer treatments. Nat. Rev. Cardiol. 2015, 12, 547–558. [Google Scholar] [CrossRef]
- Cruz, M.; Duarte-Rodrigues, J.; Campelo, M. Cardiotoxicidade na terapêutica com antraciclinas: estratégias de prevenção. Rev. Port. Cardiol. 2016, 35, 359–371. [Google Scholar] [CrossRef]
- Raj, S.; Franco, V.I.; Lipshultz, S.E. Anthracycline-Induced Cardiotoxicity: A Review of Pathophysiology, Diagnosis, and Treatment. Curr. Treat. Options Cardiovasc. Med. 2014, 16, 315. [Google Scholar] [CrossRef]
- Jerusalem, G.; Lancellotti, P.; Kim, S.-B. HER2+ breast cancer treatment and cardiotoxicity: monitoring and management. Breast Cancer Res. Treat. 2019, 177, 237–250. [Google Scholar] [CrossRef]
- Benslimane, F.M.; Zakaria, Z.Z.; Shurbaji, S.; Abdelrasool, M.K.A.; Al-Badr, M.A.H.I.; Al Absi, E.S.K.; Yalcin, H.C. Cardiac function and blood flow hemodynamics assessment of zebrafish (Danio rerio) using high-speed video microscopy. Micron 2020, 136, 102876. [Google Scholar] [CrossRef]
- Santoso, F.; Farhan, A.; Castillo, A.L.; Malhotra, N.; Saputra, F.; Kurnia, K.A.; Chen, K.H.-C.; Huang, J.-C.; Chen, J.-R.; Hsiao, C.-D. An Overview of Methods for Cardiac Rhythm Detection in Zebrafish. Biomedicines 2020, 8, 329. [Google Scholar] [CrossRef]
- Ramachandran Surajambika, R.; Natarajan, R.; Nagarajan, N. Design, Synthesis, Evaluation and Toxicity Studies of Novel Acridine Derivatives in Zebra Fish Larvae. Curr. Bioact. Compd. 2024, 20. [Google Scholar] [CrossRef]
- Han, Y.; Ma, Y.; Yao, S.; Zhang, J.; Hu, C. In vivo and in silico evaluations of survival and cardiac developmental toxicity of quinolone antibiotics in zebrafish embryos (Danio rerio). Environ. Pollut. 2021, 277, 116779. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alexandria J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Ali, M.; Martinez, M.; Parekh, N. Are antioxidants a viable treatment option for male infertility? Andrologia 2021, 53. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 1–19. [Google Scholar] [CrossRef]
- Santana, C.B.L.; Sousa, D.S.; Costa, J.I. de G.; Lima, D.A.; Oliveira, E.D. Efeitos do gel complexado de naringina/β-ciclodextrina associado ao ultrassom terapêutico em biomarcadores do estresse oxidativo após lesão musculoesquelética em ratos. Res. Soc. Dev. 2024, 13, e0313645940. [Google Scholar] [CrossRef]
- Galasso, M.; Gambino, S.; Romanelli, M.G.; Donadelli, M.; Scupoli, M.T. Browsing the oldest antioxidant enzyme: catalase and its multiple regulation in cancer. Free Radic. Biol. Med. 2021, 172, 264–272. [Google Scholar] [CrossRef]
- McGhee, J.D.; von Hippel, P.H. Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974, 86, 469–489. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer US: Boston, MA, 2006. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; Gray-Goodrich, M.; Campbell, H.; Mayo, J.; Boyd, M. Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. JNCI J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Gehrcke, M.; Giuliani, L.M.; Ferreira, L.M.; Barbieri, A.V.; Sari, M.H.M.; da Silveira, E.F.; Azambuja, J.H.; Nogueira, C.W.; Braganhol, E.; Cruz, L. Enhanced photostability, radical scavenging and antitumor activity of indole-3-carbinol-loaded rose hip oil nanocapsules. Mater. Sci. Eng. C 2017, 74, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Aebi, H. Catalase in vitro; 1984; pp. 121–126. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of 2-cyano-N-phenylacrylamide Derivatives.
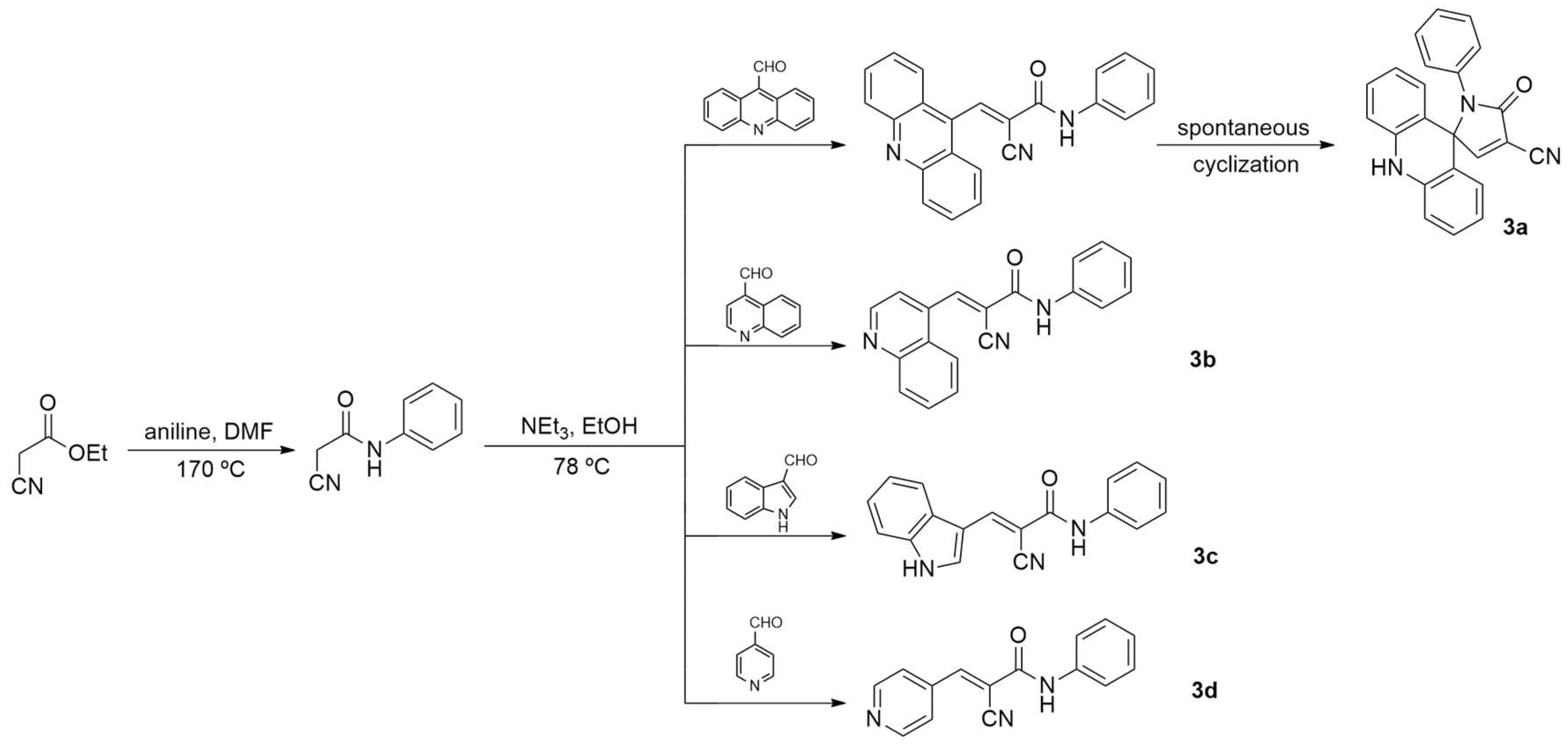
Figure 1.
Pharmacokinetic feasibility radar using the MPO algorithm in relation to Pfizer's drug space (space in blue).
Figure 1.
Pharmacokinetic feasibility radar using the MPO algorithm in relation to Pfizer's drug space (space in blue).
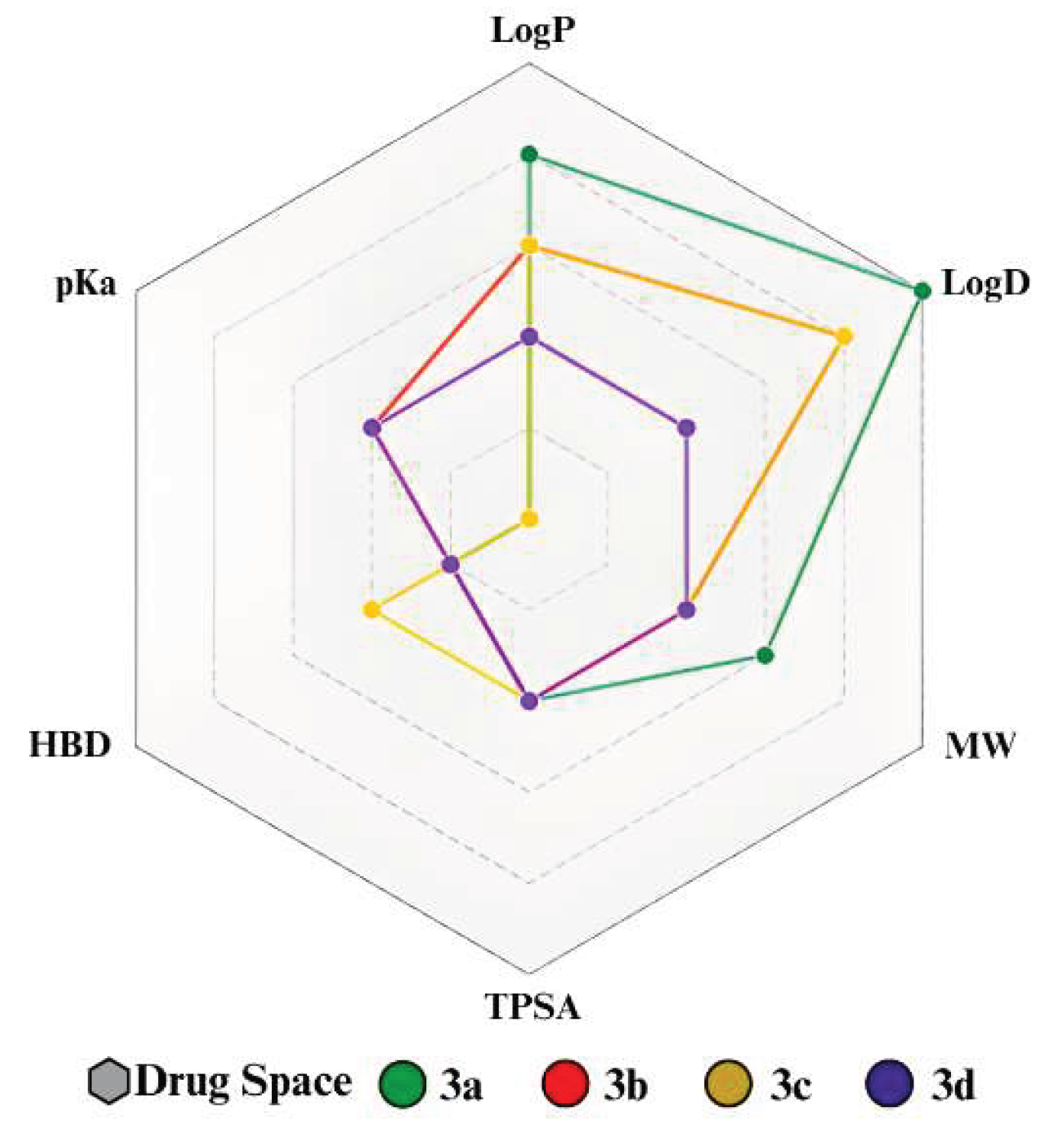
Figure 2.
Map of molecular lipophilicity potential (MLP) for compounds 3a, 3b, 3c, and 3d.
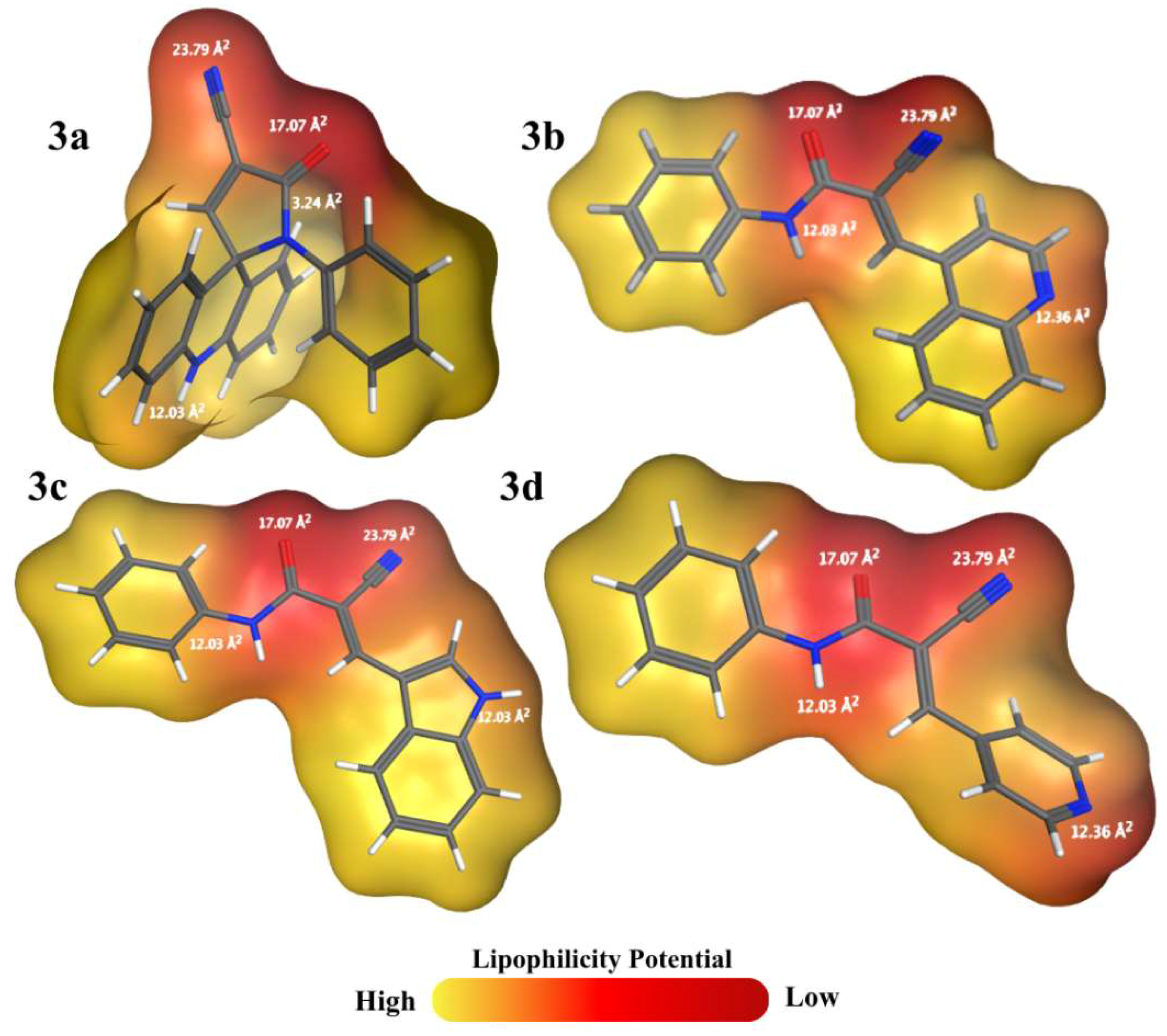
Figure 3.
(a) Metabolization points by glucuronidation (b) Metabolization points by stable oxidation for the compounds 3a, 3b, 3c, and 3d.
Figure 3.
(a) Metabolization points by glucuronidation (b) Metabolization points by stable oxidation for the compounds 3a, 3b, 3c, and 3d.
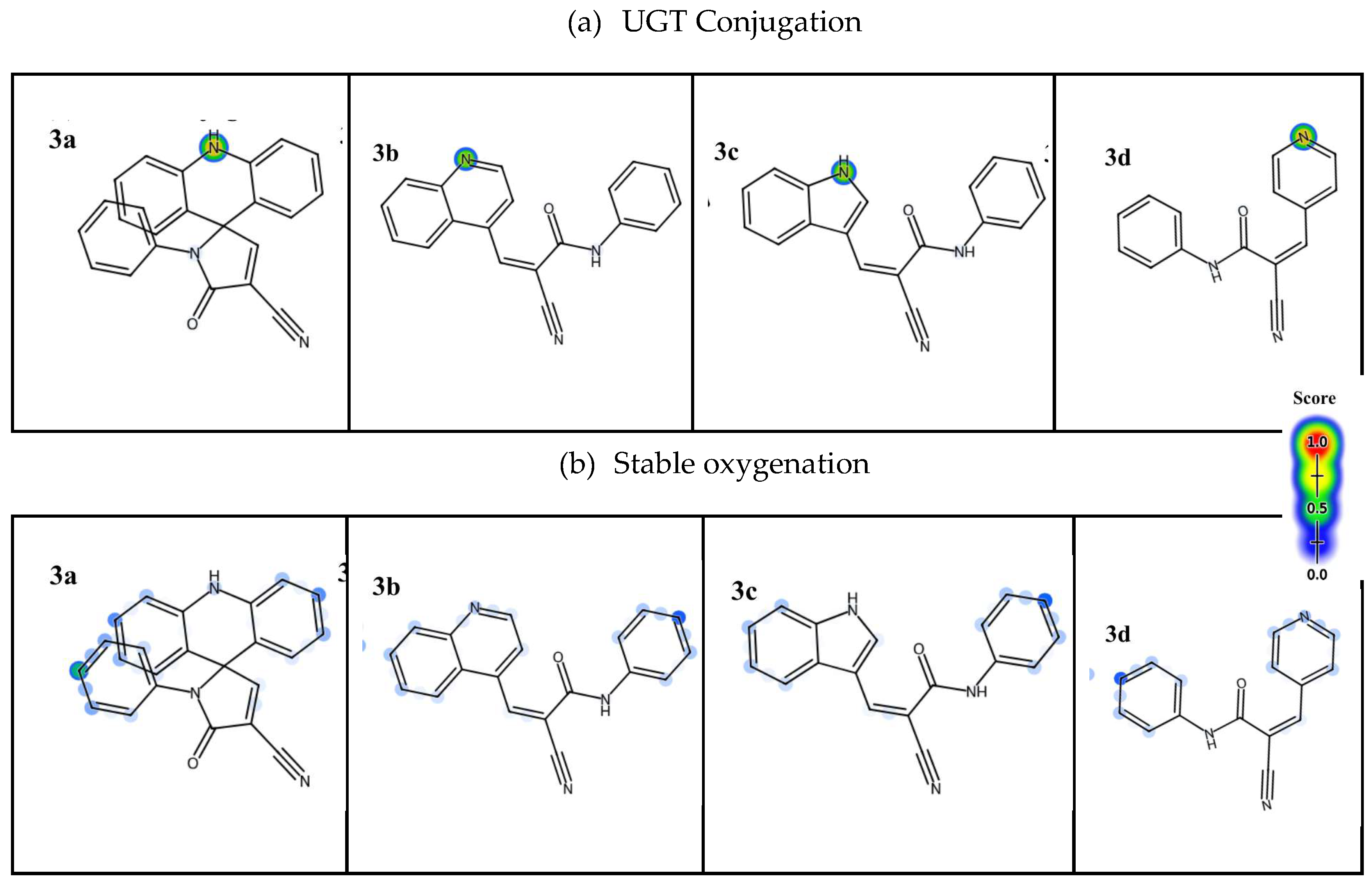
Figure 4.
Toxicity points generated by STopTox for the compounds 3a, 3b, 3c, and 3d.
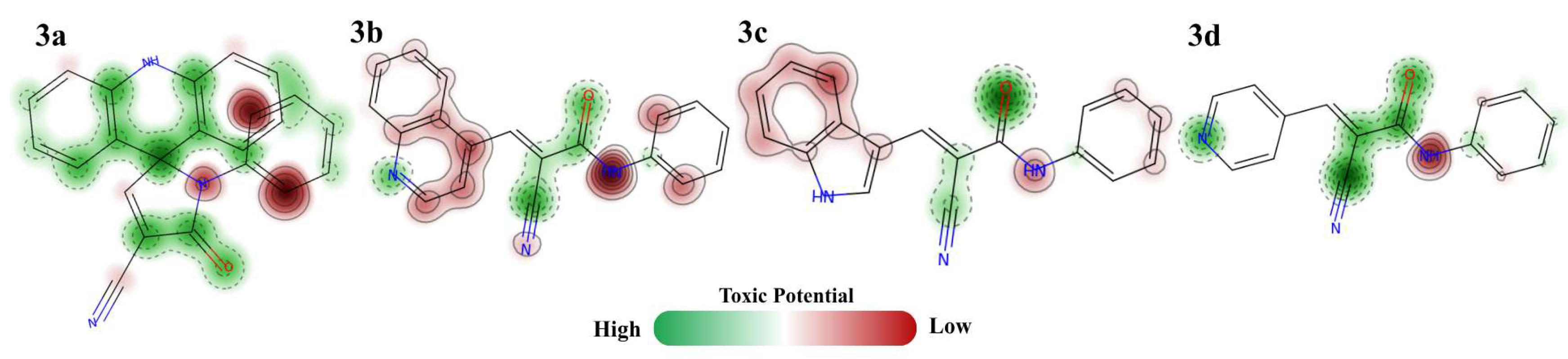
Figure 5.
Emission spectra of free and ssDNA-bound EB, in the absence and presence of 3a derivative.
Figure 5.
Emission spectra of free and ssDNA-bound EB, in the absence and presence of 3a derivative.
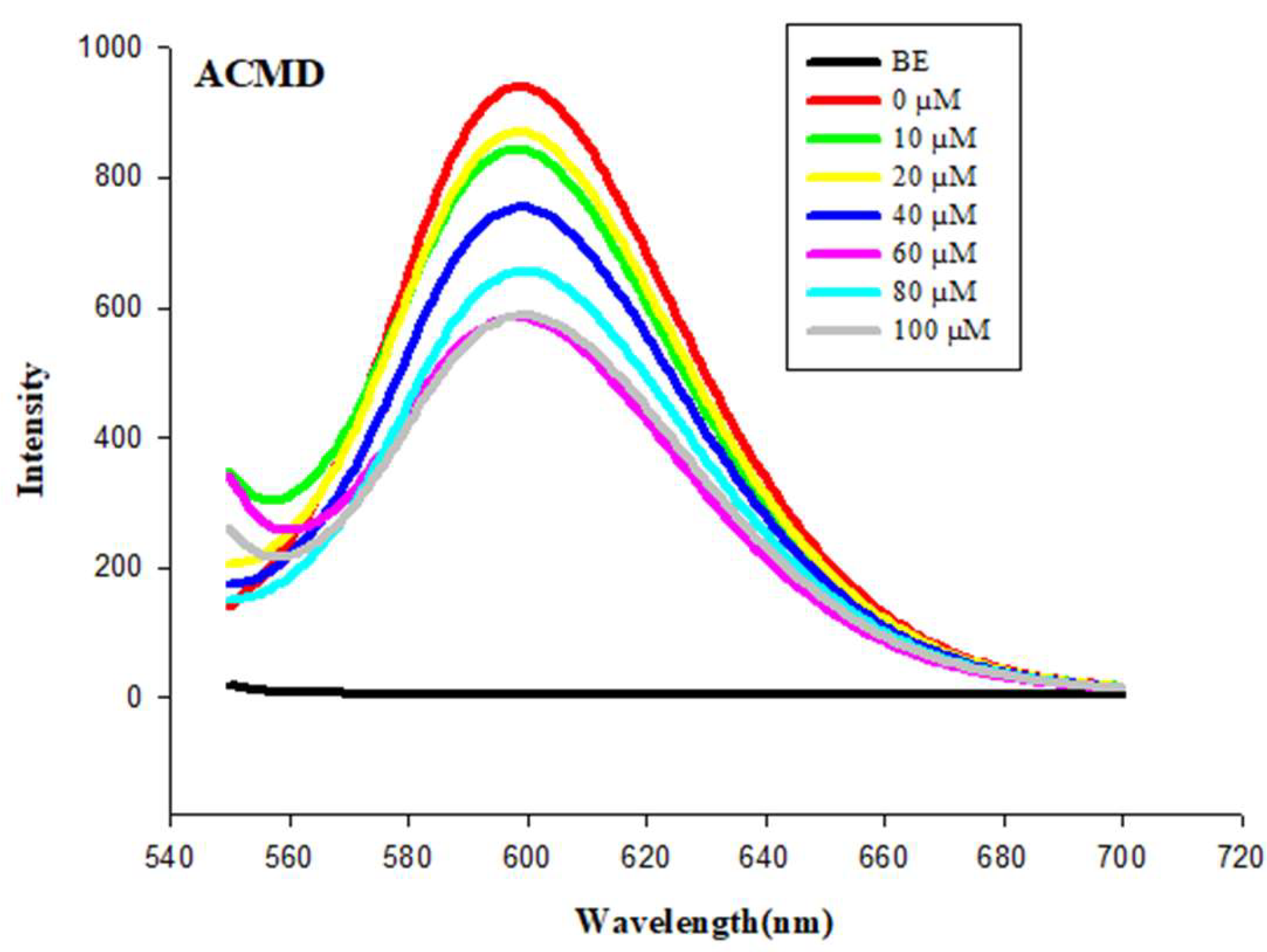
Figure 6.
Effect of m-AMSA, 3a, 3b, 3c, and 3d at 100 μM in the Topo IIα Enzymatic Inhibition Assay.
Figure 6.
Effect of m-AMSA, 3a, 3b, 3c, and 3d at 100 μM in the Topo IIα Enzymatic Inhibition Assay.
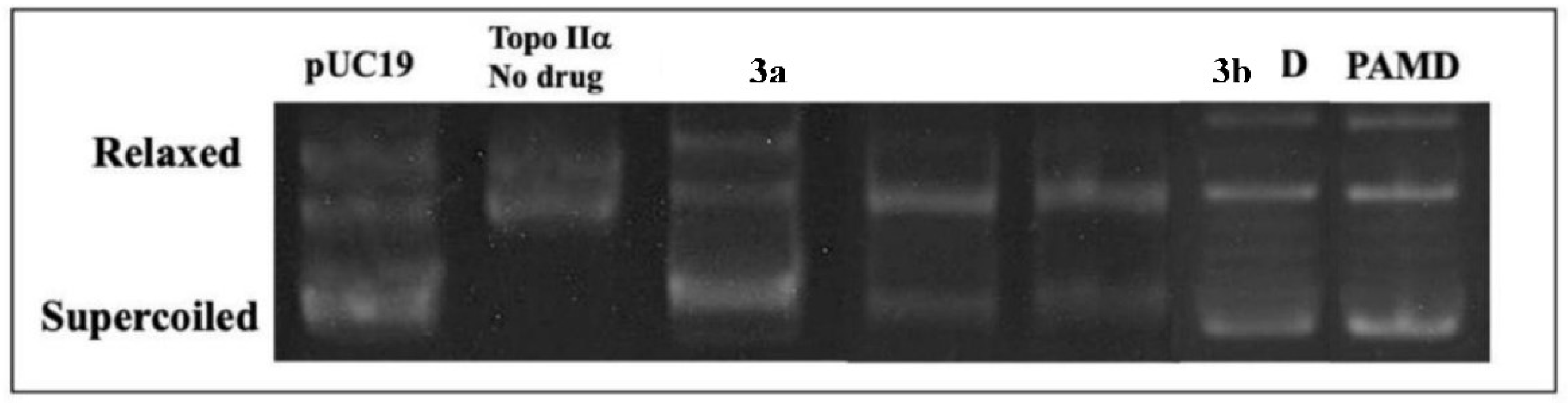
Figure 7.
Docking poses of the 3b derivative with their respective interaction modes with DNA and amino acid residues of the topoisomerase active site, including hydrogen bonding interactions and π-π stacking interactions of the derivative's aromatic ring with nitrogenous bases A12-D, C8-E, G13-D.
Figure 7.
Docking poses of the 3b derivative with their respective interaction modes with DNA and amino acid residues of the topoisomerase active site, including hydrogen bonding interactions and π-π stacking interactions of the derivative's aromatic ring with nitrogenous bases A12-D, C8-E, G13-D.
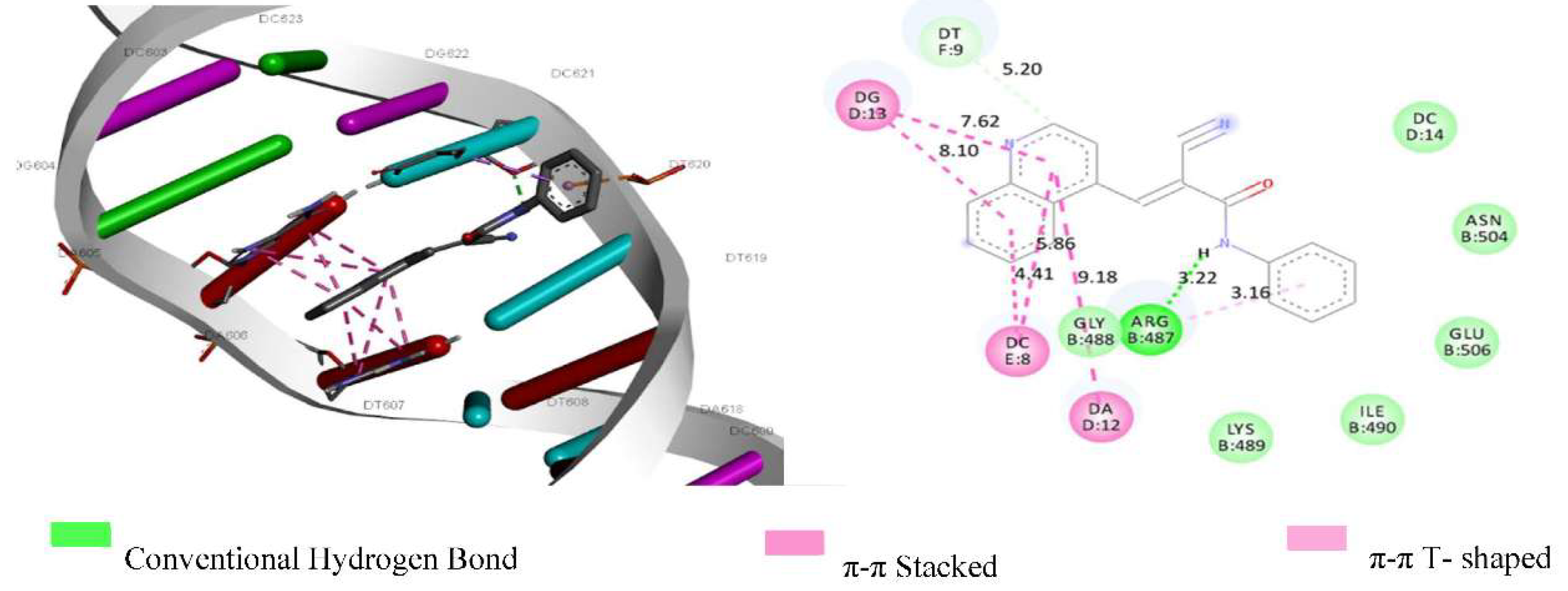
Figure 8.
Embryonic development of zebrafish after 72 hpf in contact with the derivatives: 3a, 3b, 3c, and 3d.
Figure 8.
Embryonic development of zebrafish after 72 hpf in contact with the derivatives: 3a, 3b, 3c, and 3d.
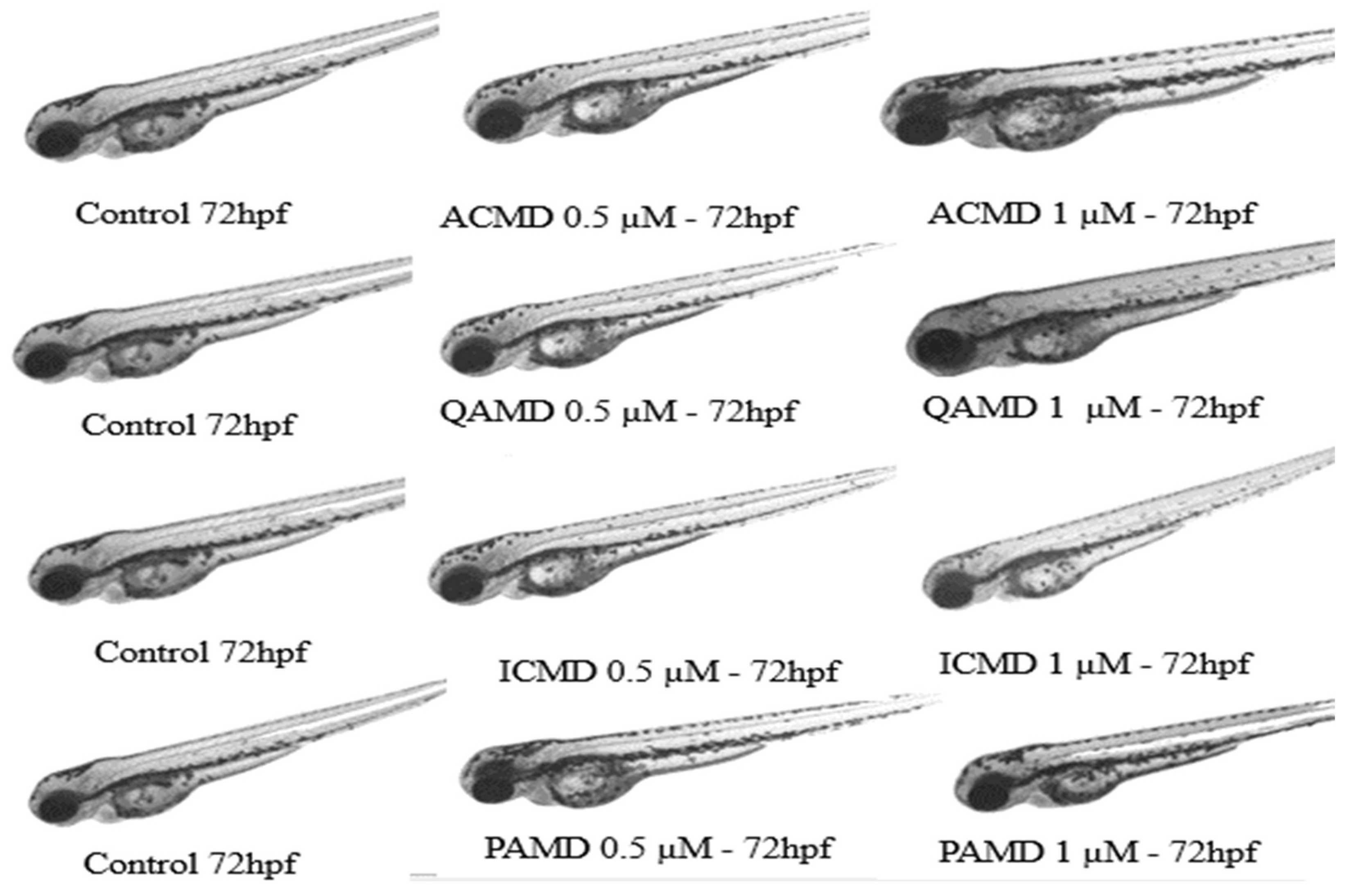
Figure 9.
Heart rate of zebrafish embryos. Heartbeats were quantified at 72 hpf, and the experiment was performed in quadruplicate by two researchers. ↑ represents a percentage increase in heart rate and ↓ a decrease compared to the control. Values are presented as means ± SE (n = 20).
Figure 9.
Heart rate of zebrafish embryos. Heartbeats were quantified at 72 hpf, and the experiment was performed in quadruplicate by two researchers. ↑ represents a percentage increase in heart rate and ↓ a decrease compared to the control. Values are presented as means ± SE (n = 20).
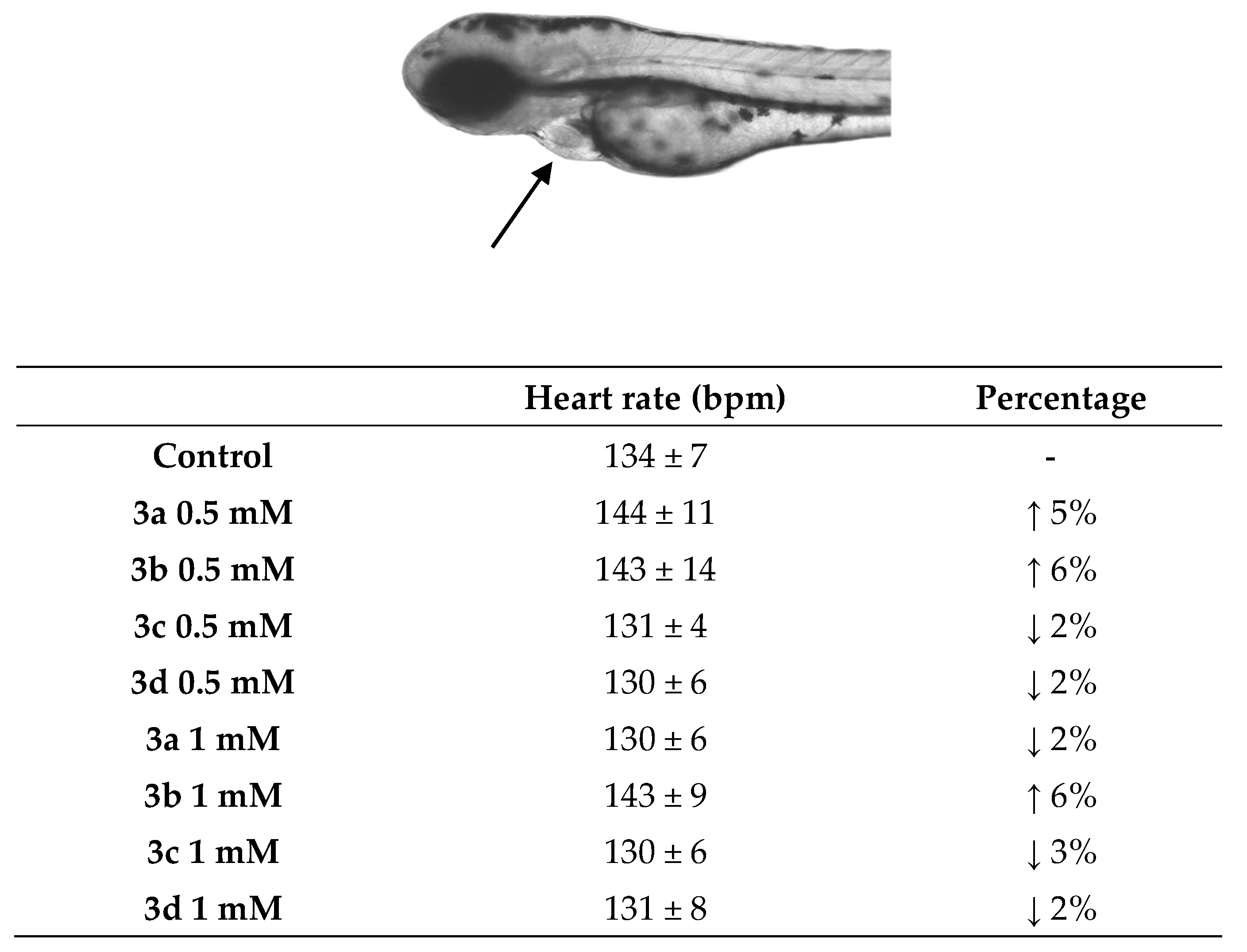
Figure 10.
The results represent enzymatic activity (U/μg of protein). The asterisk (*) represents a value of p = 0.001.A -catalase activity, ' * represents a value of p < 0.03 and 'B' represents superoxide dismutase activity * represents value of p < 0.01. Values are presented as means ± standard error (n = 20).
Figure 10.
The results represent enzymatic activity (U/μg of protein). The asterisk (*) represents a value of p = 0.001.A -catalase activity, ' * represents a value of p < 0.03 and 'B' represents superoxide dismutase activity * represents value of p < 0.01. Values are presented as means ± standard error (n = 20).
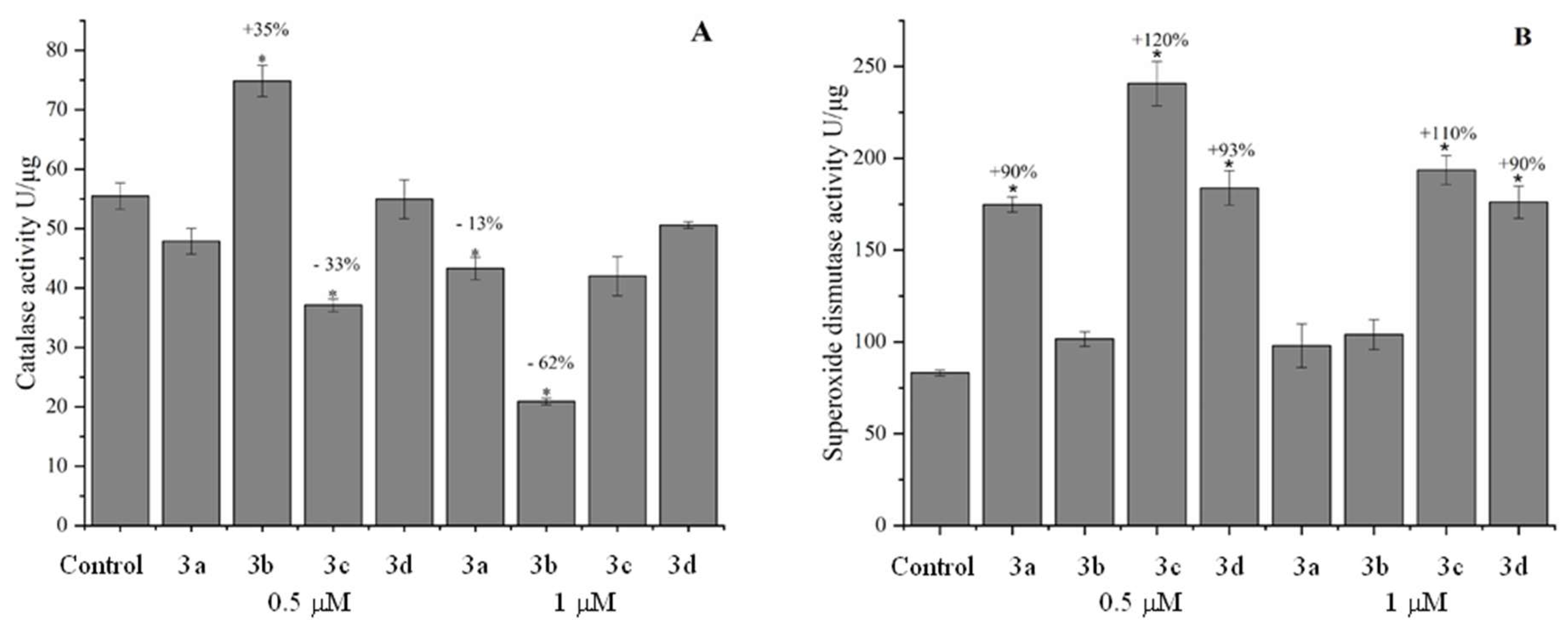

Table 1.
General physicochemical properties of compounds, characteristics of Lipinski, Ghose, Veber, Egan, and Muegge druglikeness rules according to SwissADME and MPO of 3a, 3b, 3c, and 3d compounds.
Table 1.
General physicochemical properties of compounds, characteristics of Lipinski, Ghose, Veber, Egan, and Muegge druglikeness rules according to SwissADME and MPO of 3a, 3b, 3c, and 3d compounds.
| Properties | 3a | 3b | 3c | 3d |
| LogP | 4.03 | 3.54 | 3.48 | 2.16 |
| LogD | 4.03 | 3.54 | 3.48 | 2.16 |
| MW | 348.49 g/mol | 299.33 g/mol | 287.32 g/mol | 249.27 g/mol |
| TPSA | 56.13 Å2 | 64.98 Å2 | 65.25 Å2 | 65.25 Å2 |
| pKa | -1.22 | 4.59 | -4.30 | 4.72 |
| HBD | 1.00 | 1.00 | 2.00 | 1.00 |
| HBA | 3.0 | 3.0 | 2.0 | 3.0 |
| RB | 1.0 | 4.0 | 4.0 | 4.0 |
| MR | 110.26 | 90.22 | 86.78 | 72.72 |
| CF | C23H15N3O | C19H13N3O | C18H13N3O | C15H11N3O |
| Criteria | ||||
| Lipinski | Yes | Yes | Yes | Yes |
| Ghose | Yes | Yes | Yes | Yes |
| Veber | Yes | Yes | Yes | Yes |
| Egan | Yes | Yes | Yes | Yes |
| Muegge | Yes | Yes | Yes | Yes |
| MPO | 4.24 | 4.71 | 4.52 | 4.67 |
Table 2.
General pharmacokinetic parameters obtained for the compounds 3a, 3b, 3c, and 3d.
| Parameters | 3a | 3b | 3c | 3d | Etoposide |
| Permeability Caco-2 | 6.60x10-5 cm/s | 1.11x10-5 cm/s | 1.15x10-5 cm/s | 8.57x10-6 cm/s | 9.31x10-7 cm/s |
| Permeability MDCK | 2.23x10-5 cm/s | 1.48x10-5 cm/s | 1.45x10-5 cm/s | 1.72x10-5 cm/s | 4.66x10-6 cm/s |
| Plasma protein binding | 98.25% | 98.55% | 98.65% | 95.46% | 95.03% |
| Distribution volume | 1.18 L/Kg |
0.59 L/Kg |
0.50 L/Kg |
0.56 L/Kg |
0.23 L/Kg |
| Permeability to the blood-brain barrier | 0% | 0.1% | 0.2% | 0.7% | 0.8% |
| Plasma clearance | 6.041 L/h/Kg |
0.2772 L/h/Kg | 0.30972 L/h/Kg | 0.30222 L/h/Kg | 0.14934 L/h/Kg |
| Half-life | 3h 23min | 1h 29min | 1h 15min | 1h 09min | 1h 04min |
Table 3.
Probability results of compounds being enzyme targets for CYP1A2, CYP2C19, and CYP3A4.
| Target | Probability | |||
| 3a | 3b | 3c | 3d | |
| CYP1A2 | 82.1% | 3.4% | 63.2% | 97.9% |
| CYP2C19 | 100% | 0% | 0% | 0.4% |
| CYP3A4 | 100% | 7% | 24.3% | 59.5 |
Table 4.
Prediction and probability of toxicity for the compounds 3a, 3b, 3c, and 3d.
| Target | Prediction and probability | ||||
| 3a | 3b | 3c | 3d | Etoposide | |
| Hepatotoxicity | Active (57%) |
Active (57%) |
Active (51%) |
Active (57%) |
Inactive (84%) |
| Nephrotoxicity | Inactive (76%) |
Inactive (71%) |
Inactive (64%) |
Inactive (71%) |
Active (66%) |
| Cardiotoxicity | Active (92%) |
Inactive (83%) |
Inactive (84%) |
Inactive (81%) |
Active (70%) |
| Carcinogenicity | Inactive (56%) |
Inactive (62%) |
Inactive (52%) |
Inactive (65%) |
Inactive (64%) |
| Mutagenicity | Inactive (52%) |
Inactive (64%) |
Inactive (56%) |
Inactive (61%) |
Inactive (85%) |
| Cytotoxicity | Inactive (80%) |
Inactive (80%) |
Inactive (71%) |
Inactive (80%) |
Inactive (90%) |
Table 5.
UV-vis absortion data of derivatives in the presence and absence of ssDNA.
| Compound | Ring | λmax free (nm) a | λmax bond (nm) | Δλ (nm) | Hyper. (%)b |
Hypochr. (%)c |
Kb d M−1 |
| 3a | Acridine | 363 | 404 | 71 | - | 58.67 | 2.23 x 105 |
| 3b | Quinoline | 326 | 404 | 78 | - | 91.63 | 1.41 x 105 |
| 3c | Indole | 390 | 400 | 10 | 54.81 | - | 5.26 x 104 |
| 3d | Pyridine | 272 | 269 | 3 | - | 37.12 | 6.46 x 104 |
Table 6.
Fluorescent emission data of EB-ssDNA complexes in the absence and the presence of compounds.
Table 6.
Fluorescent emission data of EB-ssDNA complexes in the absence and the presence of compounds.
| Compound/ Ring | EB-ssDNA | |||
| λmáx free (nm)a |
λmáx bound (nm)a |
Hypochr. (%)b | Ksv (M-1) c | |
| 3a/ Acridine | 599 | 599 | 59.64 | 0.67 x 103 |
| 3b/ Quinoline | 599 | 600 | 73.14 | 0.63 x 103 |
| 3c/ Indole | 599 | 600 | 53.29 | 0.53 x 103 |
| 3d/ Pyridine | 599 | 600 | 69.72 | 0.58 x 103 |
a Data regarding the emissive profile of the EB-ssDNA complex in the presence of different concentrations of the test compounds (10-100 µM). b Hipochromism resulting from the formation of the interaction between derivatives with the EB-ssDNAc Stern – Volmer suppression constant (Ksv) obtained based on fluorescence data with ssDNA.
Table 7.
Data obtained for the derivatives with the targets 1G3X - Intercalation of a 9Acridine-peptide drug in a DNA dodecamer, 1BNA - Structure of a B-DNA dodecamer. Conformation and dynamics, 5GWK - Human Topoisomerase IIα in complex with DNA and etoposide by molecular docking.
Table 7.
Data obtained for the derivatives with the targets 1G3X - Intercalation of a 9Acridine-peptide drug in a DNA dodecamer, 1BNA - Structure of a B-DNA dodecamer. Conformation and dynamics, 5GWK - Human Topoisomerase IIα in complex with DNA and etoposide by molecular docking.
| Compound | Binding energy (kcal/mol) Alvo: 1G3X |
Binding energy (kcal/mol) Alvo: 1BNA- |
Binding energy (kcal/mol) Alvo: 5GWK |
Type of bond | Residues |
| Ligand 9Acridine | -6.99 | _ | - | - | - |
| Ligand Etoposide | - | - | -11.2 | Hydrogen boding; π -Alkyl; stacking π- π; Van der Waals; | MET763, PRO803, ASP463, ARG487, G13-D, G10-F C8-E, GLY488; GLY462 |
| 3a | -8.07 | -6.94 | -8.52 | Hydrogen boding; π -Alkyl; stacking π- π; π-Cation; π-Anion | ALA614; ASP108; LYS 190-466; ARG787; HIS146 |
| 3b | -9.23 | -10.37 | -8.84 | Hydrogen boding; stacking π- π | ARG197; TYR148; CYS200-246; LEU250; ALA151; G13-D, C8-E, A12-D, ARG 487 |
| 3c | -8.75 | -10.16 | -8.76 | Hydrogen boding; π-Cation; π-Sigma; π-Sulfur | ALA26-151; LYS106; GLN29; ARG487; PHE149; TYR148; CYS245-200-246; LEU250; T9-F; A12D |
| 3d | -7.35 | -8.65 | -7.2 | Hydrogen boding π-Cation; π-Sigma; π-Sulfur; π -Alkyl | ALA151-26; LYS106; ARG487; TYR150; CYS200-246; LEU250; GLY248 |
A12; C8; Gx; T9 = Nitrogenous bases Adenine, Cytosine, Guanine, and Thymine. D; E = DNA strands segments. ARG; ASP; TYR; SER; GLU, LYS; GLY, MET, PRO = Amino acid residues - Topoisomerase IIα enzyme and DNA.
Table 8.
GI50 values in µM for the compounds 3a, 3b, 3c, and 3d tested in the breast cancer cell lines MCF-7, T47-D, and the non-tumoral cell line HaCaT. Percentage of hemolysis of the derivatives in human erythrocytes.
Table 8.
GI50 values in µM for the compounds 3a, 3b, 3c, and 3d tested in the breast cancer cell lines MCF-7, T47-D, and the non-tumoral cell line HaCaT. Percentage of hemolysis of the derivatives in human erythrocytes.
| Derivative | Cell linesa GI50 (µM) | |||
| MCF-7 | T47-D | HaCaT | % hemolysis in human erythrocytesd | |
| m-AMSAb | 3.27 | 0.14 | 1.44 | 19% |
| 3a | 34.41 | 3.42 | >100 | 6.66% |
| 3b | 33.62 | 8.07 | 2.98 | 3.08% |
| 3c | 9.44 | n.a | 4.29 | 4.14% |
| 3d | 33.27 | n.a | 2.96 | 2.05% |
aMCF-7 (breast adenocarcinoma); T47-D and HaCaT (immortalized human keratinocyte). Cell lines were exposed to the compounds in DMSO/RPMI/FBS 5% at 37 °C, 5% CO2, for 48 h. bm -AMSA positive controls. Triton X-100 positive controls (100%) and m-AMSAd , n.a= Inactive at the highest concentration.
Table 9.
Zebrafish Embryo Acute Test. The acute test was performed following the Fish Embryo Toxicity (FET) Test of OECD [60] with modifications. Zebrafish embryos are exposed before 90 minutes post fertilization. The E3 medium was used as negative control and 4 mg.L-1 of 3,4- dichloroaniline was used as positive control. The experiment is performed in duplicate (n=20).
Table 9.
Zebrafish Embryo Acute Test. The acute test was performed following the Fish Embryo Toxicity (FET) Test of OECD [60] with modifications. Zebrafish embryos are exposed before 90 minutes post fertilization. The E3 medium was used as negative control and 4 mg.L-1 of 3,4- dichloroaniline was used as positive control. The experiment is performed in duplicate (n=20).
| Derivative | Lethality parameters [60] | Exposure time (h) / Concentration / Percentage (%) of embryos within lethality parameters [60] | ||
| 24 h | 48 h | 72h | ||
| Negative Control | Embryo coagulation(%) | 10% | 0% | 0% |
| Lack of somite formation (%) | 0% | 0% | 0% | |
| No tail detachment (%) | 0% | 0% | 0% | |
| Absence of heartbeat (%) | 0% | 0% | 0% | |
| 3,4-dichloroaniline | Embryo coagulation (%) | 100% | - | - |
| 24h/ 0.5 and 1 µM | 48h/ 0.5 and 1 µM | 72h/ 0.5 and 1 µM | ||
| 3a |
Embryo coagulation (%) | 10% and 0% | 0% and % | 0% and 0 % |
| Lack of somite formation (%) | 0% | 0% | 0% | |
| No tail detachment (%) | 0% | 0% | 0% | |
| Absence of heartbeat (%) | 0% | 0% | 0% | |
| 3b | Embryo coagulation (%) | 0% and 2.5% | 0% | 0% |
| Lack of somite formation (%) | 0% | 0% | 0% | |
| No tail detachment (%) | 0% | 0% | 0% | |
| Absence of heartbeat (%) | 0% | 0% | 0% | |
| 3c | Embryo coagulation (%) | 0% and 5% | 0% | 0% |
| Lack of somite formation (%) | 0% | 0% | 0% | |
| No tail detachment (%) | 0% | 0% | 0% | |
| Absence of heartbeat (%) | 0% | 0% | 0% | |
| 3d | Embryo coagulation (%) | 0% | 0% | 0% |
| Lack of somite formation (%) | 0% | 0% | 0% | |
| No tail detachment (%) | 0% | 0% | 0% | |
| Absence of heartbeat (%) | 0% | 0% | 0% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



