Submitted:
01 February 2026
Posted:
05 February 2026
You are already at the latest version
Abstract
Reducing residual cardiovascular risk following an acute coronary syndrome (ACS) remains a major unmet clinical need. Despite substantial advances in lipid-lowering therapies, the risk of recurrent major adverse cardiovascular events (MACE) after ACS remains high, with an estimated incidence of approximately 33.4% at 5 years. Residual cardiovascular risk is driven by multiple mechanisms, including persistent inflammation, a prothrombotic status, metabolic disturbances, and the presence of atherogenic lipoproteins beyond low-density lipoprotein cholesterol (LDL-C). Lipoprotein(a) [Lp(a)] is a pro-inflammatory, pro-thrombotic and pro-atherosclerotic lipoprotein which seems to play a major role in the residual risk post ACS or atherosclerotic ischemic stroke. Elevated Lp(a) is a well-established independent and causal risk factor for atherosclerotic cardiovascular disease (ASCVD). Nevertheless, evidence regarding its prognostic value specifically after ACS remains limited, with marked heterogeneity across studies, which complicates direct comparisons and interpretation. In addition, while Lp(a) levels are predominantly genetically determined, recent studies have reported intra-individual variability, although its clinical significance remains uncertain. Finally, current therapeutic options specifically targeting Lp(a) are limited. Novel RNA-based therapies, including antisense oligonucleotides, small interfering RNAs, and emerging gene-editing approaches, have demonstrated profound and sustained reductions in circulating Lp(a) levels. Yet, whether this biological effect translates into reductions of hard clinical endpoints is under evaluation in ongoing clinical trials. This review aims to synthesize current evidence on the role Lp(a) as a major contributor to residual cardiovascular risk following ACS.
Keywords:
lipoprotein (a); acute coronary syndrome; residual cardiovascular risk
; major cardiovascular events
; emerging therapies
1. Introduction
Despite advances in treatment over the last decades, the management of residual cardiovascular risk after an acute coronary syndrome (ACS) remains a major challenge. Patients with ACS continue to face a substantial risk of recurrent major adverse cardiovascular events (MACE), with an estimated event rate of approximately 33.4% at 5 years of follow-up, and a sixfold higher risk during the first year after diagnosis [1].
Traditionally, secondary prevention strategies have been focused on multiple risk pathways, especially on low-density lipoprotein cholesterol (LDL-C) reduction. Nevertheless, evidence from randomized controlled trials (RCTs), such as the ODYSSEY Outcomes trial, which evaluated the Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) inhibitor alirocumab in patients following an ACS, has demonstrated that despite achieving intensive LDL-C control, residual risk of cardiovascular events remains substantial [2]. As a consequence, recent studies advocate the need of a more holistic approach to residual cardiovascular risk beyond traditional factors [3,4].
Lipoprotein(a) [Lp(a)] has emerged as an independent risk factor of atherosclerotic cardiovascular disease (ASCVD) and calcific aortic valvular disease (CAVD). Several mechanisms explain its pathogenic role, including proatherogenic, proinflammatory and prothrombotic effects [5,6,7]. Therefore, last European Society of Cardiology (ESC)/European Atherosclerosis Society (EAS) and American Heart Association (AHA) guidelines identify high Lp(a) levels (≥ 50 mg/dL) as a risk modifier and recommend measuring Lp(a) at least once during an individual’s lifetime [8,9].
Furthermore, growing evidence identifies Lp(a) as an important determinant of residual cardiovascular risk after an ACS. Elevated Lp(a) levels have been associated with premature and more severe ACS, with a higher risk of recurrent ischemic events after the index event, both early after the index event and during long-term follow-up [10]. Nonetheless, available data remain heterogeneous, with substantial variability in study design, patient populations, outcome definitions, and Lp(a) cut-off values, likely reflecting the linear association between Lp(a) levels and MACE rather than a clear risk threshold; consequently, robust long-term prospective evidence in ACS cohorts is still limited.
Importantly, therapeutic options specifically targeting Lp(a) are currently limited. Nevertheless, ongoing clinical trials evaluating antisense oligonucleotides (ASO) and small interfering RNA (siRNA) therapies have demonstrated substantial reductions in Lp(a) concentrations; whether these biomarker effects will translate into clinically meaningful cardiovascular benefit remains to be determined [11].
This review aims to provide a comprehensive overview of Lp(a) as a key marker of residual cardiovascular risk in ACS, highlighting its pathophysiological mechanisms, clinical implications, and emerging therapeutic options.
2. Lipoprotein(a): Structure, Metabolism and Pathophysiological Mechanisms
Lp(a) is a plasma macromolecular complex, first described in 1963 [12,13], consisting of a LDL particle containing apolipoprotein B-100 (apoB-100) covalently linked by a single disulfide bridge to apolipoprotein(a) [apo(a)] [14]. Apo(a) exhibits marked structural homology with plasminogen and other coagulation factors, as it contains multiple loop-like structures known as “kringle domains”. These include ten distinct subtypes of kringle IV (KIV) and a single kringle V domain, alongside a lysine-binding site that interferes with the receptor interactions required for plasminogen activation [15,16].
Lp(a) metabolism is largely independent of classical lipid pathways. Apo(a) is synthesized in hepatocytes and subsequently assembled with LDL particles, either at the hepatocyte surface or intracellularly, prior to secretion into the systemic circulation. The mechanisms underlying Lp(a) clearance remain incompletely understood, and the LDL-C receptor appears to play only a minor role in its catabolism [14].
From a pathophysiological perspective, Lp(a) exhibits a triad of pro-atherogenic, pro-inflammatory and pro-thrombogenic properties (Figure 1). It contributes to atherosclerosis via cholesterol deposition within the arterial intima and acts as a preferential carrier of oxidized phospholipids, which promote vascular inflammation, endothelial dysfunction, and plaque instability [17,18]. In addition, due to its structural similarity to plasminogen, Lp(a) competitively inhibits plasminogen binding and activation, leading to impaired fibrinolysis and promoting a prothrombotic state [14,19]. These synergistic effects contribute to the strong and causal association between elevated Lp(a) levels and cardiovascular disease, including coronary artery disease (CAD), stroke, and calcific aortic valve disease [20,21,22].
3. Genetic Determinants and Variability of Lipoprotein(a)
Lp(a) concentration is predominantly genetically determined, with heritability estimated at 70–90%. The LPA gene on chromosome 6 is the principal genetic determinant, and encodes apo(a).
Among apo(a) kringle domains, the KIV-2 copy number variation (CNV) accounts for a substantial proportion of interindividual variability and it inversely correlates with Lp(a) concentration: fewer repeats result in smaller apo(a) isoforms and higher Lp(a) levels. The underlying mechanism involves prolonged retention of large apo(a) isoforms in the endoplasmic reticulum, where they undergo increased proteasomal degradation, whereas smaller isoforms are more efficiently secreted.
Beyond CNVs, numerous single nucleotide polymorphisms within the LPA locus further modulate Lp(a) levels, even among individuals with identical KIV-2 repeat numbers. Notably, plasma levels differ across racial and ethnic groups, with higher concentrations observed in African and South Asian populations. These differences are largely attributed to population-specific genetic variations affecting apo(a) isoform size and LPA gene structure [5,23,24].
3.1. Intra-Individual Variability
Intra-individual variability of Lp(a) concentrations is generally low, with most individuals maintaining stable levels over time, especially those with clearly low (<30 mg/dL) or high (≥50 mg/dL) baseline values. Nonetheless, recent studies have reported significant intra-individual variability (>10 mg/dL or >25%) particularly in patients with borderline levels (30-50 mg/dL) [25,26,27,28,29,30,31,32,33,34,35,36].
As early as 1995, Nakajima et al. demonstrated an intra-individual variability of Lp(a) of 16.6%, identifying lipid metabolism–related factors, platelet-mediated prothrombotic pathways, and acute-phase reactants as key determinants of this variability [25]. In recent years, several studies have reported intra-individual Lp(a) changes exceeding 10 mg/dL or 25% in a substantial proportion of individuals, affecting approximately 15–40% of the population depending on the study. This variability may be clinically relevant for cardiovascular risk stratification, particularly within the “gray zone,” where up to 50% of individuals may be reclassified to higher or lower risk categories following repeat measurement. Nevertheless, whether intra-individual variability in Lp(a) is associated with adverse cardiovascular outcomes remains uncertain [26,27,28,29,30,31,32,33,34,35,36]. Recently, Trinder et al.an association between follow-up Lp(a) levels and incident CAD, whereas intra-individual variability was not independently associated with risk beyond follow-up Lp(a) levels [26].
Several factors have been described associated with increased Lp(a) variability; however, findings have not been consistent across all published studies (Figure 2). The factors with the strongest and most reproducible evidence include baseline Lp(a) concentration, female sex, advanced age, the presence of cardiovascular comorbidities, lipid profile and concomitant statin use [25,26,27,28,29,30,31,32,33,34,35,36].
These findings support that a second measurement may be necessary in selected cases to optimize cardiovascular risk management, especially in patients with intermediate values or clinical factors associated with higher Lp(a) variability.
4. Association Between Elevated Lipoprotein(a) Levels and Atherosclerotic Cardiovascular Disease
Across large contemporary cohorts, Lp(a) shows a continuous and largely linear association with ASCVD risk. In a large UK Biobank analysis including 460,506 participants, Lp(a) predicted incident ASCVD with a linear risk gradient [hazard ratio (HR) 1.11, 95% confidence interval (CI) 1.10–1.12, per 50 nmol/L increment], consistent relative risk across ethnic subgroups despite differences in absolute Lp(a) levels [37]. Comparable dose–response relationships have been reported in recent meta-analyses of population-based and clinical cohorts [38,39,40]. Amiri et al. demonstrated in a systematic review and dose-response meta-analysis linear dose-response analyses that Lp(a) levels were linearly associated with cardiovascular mortality in both the general population and in patients with established ASCVD, with each 50 mg/dL increase in Lp(a) corresponding to a 31% and 15% higher risk of cardiovascular mortality, respectively [38].
For clinical decision-making, multiple consensus statements and cohort analyses converge on ≥50 mg/dL (≈125 nmol/L) as a pragmatic high-risk threshold. This cut-off is adopted for risk-enhancing interpretation in both European and American guidelines. Evidence shows that cardiovascular risk increases more sharply above this level, supporting its use as a risk-enhancing factor in clinical stratification. However, randomized cardiovascular outcome trials typically use a higher threshold, usually ≥90 mg/dL, to select populations at higher absolute risk, thereby maximizing statistical power and clinical relevance [8,9,38,39,40,41].
A main consideration is that Lp(a)-associated risk is independent of, and additive to, LDL-C–mediated risk. Even among statin-treated individuals with low achieved LDL-C, elevated Lp(a) continues to confer excess ASCVD risk, supporting an additive “double-hit” model [39]. Notably, methodological analyses indicate that adjusting LDL-C for the cholesterol content of Lp(a) does not meaningfully improve CAD risk prediction, underscoring the distinct prognostic role of Lp(a) [43]. Moreover, Björnson et al. suggested that, on a per-particle basis, the proatherogenic effect of Lp(a) may be substantially greater than that of LDL-C. Notably, they reported an approximately six-fold stronger association with atherosclerotic burden for Lp(a) compared with LDL-C [44].
Mortality data further support the prognostic relevance of Lp(a) beyond nonfatal ASCVD events. In two large Danish general-population cohorts, extremely elevated Lp(a) levels (top percentiles) were associated with higher cardiovascular and all-cause mortality risk [42]. Similarly, a large systematic review and dose–response meta-analysis including 75 studies and 957,253 participants demonstrated a largely linear association between Lp(a) levels and both cardiovascular and all-cause mortality in the general population and in individuals with established ASCVD [38]. These observations align with emerging evidence linking Lp(a) to “panvascular” atherosclerotic phenotypes, plausibly contributing to cause-specific mortality beyond coronary outcomes alone [45].
Moreover, elevated Lp(a) levels were independently associated with more severe coronary disease, left main involvement, and three-vessel disease (TVD) at first myocardial infarction (MI) [46,47].
Evidence also suggests that Lp(a) may be particularly relevant in premature ACS. A recent systematic review and meta-analysis reported that elevated Lp(a) predicts both composite and individual ASCVD events in younger patients [40], supporting that high Lp(a) levels are associated with approximately a three-fold higher risk of cardiovascular events in individuals under 45 years of age [48].
Moving from association to implementation, real-world data support the incorporation of Lp(a) into ASCVD risk prediction models. In a recently developed model of ASCVD risk in individuals without prior cardiovascular disease, higher Lp(a) levels were associated with progressively increased risk, with each 25 mg/dL increment conferring a 23% higher composite ASCVD risk (adjusted HR 1.23, 95% CI 1.10–1.37) [49]. Incorporation of Lp(a) improves model discrimination and results in meaningful reclassification of individuals at borderline or intermediate-risk, highlighting potential implications for preventive interventions [49,50].
5. Lipoprotein(a) and Residual Cardiovascular Risk
Despite optimal control of traditional risk factors, particularly LDL-C. RCTs evaluating PCSK9 inhibitors, such as the ODYSSEY Outcomes trial, have demonstrated that a substantial proportion of patients continue to experience recurrent cardiovascular events even under intensive LDL-C control [2], highlighting additional pathophysiological mechanisms involved in atherosclerotic progression. Residual cardiovascular risk is thus recognized as a multifactorial phenomenon driven by uncontrolled dyslipidemia, persistent inflammation, metabolic disturbances, and a prothrombotic state [3,4,51].
5.1. Lipoprotein(a) as a Driver of Residual Cardiovascular Risk
Beyond LDL-C, lipid-related residual cardiovascular risk includes triglycerides (TG), triglyceride-rich lipoproteins, and Lp(a), all of which contribute to atherosclerosis progression and adverse cardiovascular outcomes. In this context, apoB100 and non–HDL cholesterol are widely recognized as integrative markers of the overall atherogenic lipoprotein burden [52,53]. Over recent years, Lp(a) has emerged as a major contributor to residual cardiovascular risk across both acute and chronic cardiovascular disease [10,39,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75].
In patients with established ASCVD, the most comprehensive evidence comes from participant-level meta-analysis of statin trials. Willeit et al. analyzed 29,069 patients from seven placebo-controlled statin trials and demonstrated that elevated Lp(a) independently predicted cardiovascular events in statin-treated patients. Notably, as LDL-C–attributable risk was reduced, Lp(a)-associated risk became a stronger determinant of residual risk, particularly at concentrations exceeding 50 mg/dL. In this analysis, the HR for cardiovascular events was 1.11 (95% CI, 1.00–1.22) for Lp(a) levels of 30–50 mg/dL and increased to 1.58 (95% CI, 1.08–1.58) for concentrations ≥50 mg/dL. Correspondingly, among statin-treated patients, the HR were 1.06 (95% CI, 0.94–1.21) for Lp(a) levels 30-50 mg/dL, and), and 1.43 for Lp(a) ≥50 mg/dL (95% CI, 1.15–1.76) [54].
These findings were recently confirmed by Bhatia et al. in a 2025 participant-level meta-analysis including 27,658 patients from six statin trials. Elevated Lp(a) (>50 mg/dL) was associated with increased ASCVD risk irrespective of achieved LDL-C levels. Notably, even among patients in the lowest LDL-C quartile (3.1–77.0 mg/dL), high Lp(a) conferred a 38% higher risk of cardiovascular events (HR 1.38, 95% CI 1.06–1.79). The highest risk was observed when both Lp(a) and LDL-C were elevated (HR 1.90, 95% CI 1.46–2.48), indicating that LDL-C lowering does not fully offset Lp(a)-mediated cardiovascular risk [39].
Although the available evidence in established ASCVD is extensive and robust, the prognostic impact of Lp(a) in the specific setting following an ACS remains uncertain, mainly due to the current absence of RCT using specific and potent Lp(a) inhibitors. The available evidence is still limited and inconsistent, with marked heterogeneity across study designs, populations, outcomes definitions, Lp(a) thresholds, which complicates direct comparisons across studies. This review aims to address this gap by synthesizing the current and most recent evidence supporting Lp(a) as a major contributor to residual cardiovascular risk after ACS (Table 1) [55,56,57,58,59,60,61,62,63,64,65].
Over the past decade, several studies have demonstrated a positive association between elevated Lp(a) levels and an increased risk of adverse cardiovascular outcomes following an ACS. Yang et al. demonstrated, in a multicenter study of patients with ACS, a higher incidence of a composite of recurrent cardiovascular events (all-cause death, nonfatal MI, nonfatal stroke and unplanned revascularization) as well as unplanned revascularization in patients with Lp(a) ≥30 mg/dL compared with those with Lp(a) <30 mg/dL (p < 0.05), after a median follow-up of 17 months [55]. Similar findings have been reported in single-center studies using Lp(a) thresholds of ≥32 mg/dL [56] and ≥30 mg/dL [76], after a follow-up of more than 3 years. Long-term outcomes were evaluated by Miñana et al., who demonstrated a non-linear association between Lp(a) levels and an increased risk of very long-term reinfarction from values ≥50 mg/dL (p = 0.016), with a median follow-up of 9.9 years. However, no association was observed with all-cause mortality (p = 0.934) [57].
Moreover, accumulating evidence suggests that the Lp(a) threshold at which the risk of MACE increases after an ACS may be substantially lower than the conventionally defined high-risk level (≥50 mg/dL). In a cohort of 1,758 patients with ACS who underwent emergency percutaneous coronary intervention (PCI), Takahashi et al. demonstrated that Lp(a) levels ≥15 mg/dL were associated with a significantly higher incidence of the composite endpoint of all-cause death and MI over a median follow-up of 2.2 years (HR 1.66, 95% CI 1.05–2.61, p = 0.03) [58]. Xue et al. concluded that Lp(a) levels >19.1 mg/dL were associated with higher mortality in a cohort of 1,359 patients over a median follow-up of 930 days (p < 0.001) [67]. Consistently, in a pre-specified analysis of the placebo-controlled ODYSSEY Outcomes trial, Steg et al. reported that Lp(a)-associated risk for MACE became evident at levels as low as 21.4 mg/dL [59]. Moreover, Lp(a) reduction with alirocumab independently predicted MACE reduction (HR 0.994, 95% CI 0.990–0.999, p = 0.008, per 1 mg/dL decrease) [64].
Post hoc analysis further demonstrated that, among ACS patients with elevated Lp(a), alirocumab was associated with earlier and greater reductions in both MACE and major adverse limb events (MALE) compared with patients with lower baseline Lp(a) levels. In patients with Lp(a) ≥30 or ≥50 mg/dL, the HR and 95% IC for MACE fell below 1 from 1.31 and 1.25 years onward, respectively, whereas no consistent benefit was observed in those with Lp(a) <30 mg/dL, and only a delayed effect (after 3.04 years) in those with Lp(a) <50 mg/dL. For MALE, a sustained risk reduction emerged as early as approximately 0.2 years only in patients with elevated Lp(a). This early benefit is clinically relevant, as cardiovascular risk following ACS is highest during the first year, confirming the concept that “the higher the risk, the greater the benefit” [68].
In contrast, evidence from other studies has not consistently supported an association between Lp(a) and recurrent cardiovascular events after ACS. In a post hoc nested cohort analysis of the dal-Outcomes trial, comparing dalcetrapib with placebo in patients with recent ACS, Lp(a) was not associated with recurrent ischemic cardiovascular events over a median follow-up of 29 months [60]. Similarly, Park et al. found no independent association between baseline Lp(a) levels and cardiovascular events in multivariable analyses of a Korean cohort with MI [61]. Gencer et al. reported that Lp(a) did not predict MACE after ACS, although higher Lp(a) levels were associated with failure to achieve LDL-C targets (<1.8 mmol/L) at one year [69]. Finally, Roth et al. observed no association between Lp(a) and either all-cause or cardiovascular mortality [70].
In specific subgroups, Lp(a) has also been reported as a predictor of adverse cardiovascular events after ACS. In patients aged ≥80 years, high Lp(a) levels (>30 mg/dL) have been associated with an increased risk of MACE, all-cause mortality, and cardiovascular mortality after long-term follow-up [62,71].
In patients with diabetes, the available evidence remains conflicting, mainly due to study limitations with potential biases and underpowered size. Li et al. reported that in a cohort of patients with ST-segment elevation myocardial infarction (STEMI) undergoing PCI, diabetic patients with Lp(a) levels ≥30 mg/dL had a significantly higher risk of MACE during follow-up (HR 2.08, 95% CI 1.33–3.26, p = 0.001) compared with non-diabetic patients with elevated Lp(a) [63]. Hao et al. observed that in a cohort of type 2 diabetes patients post-ACS and PCI, Lp(a) independently predicted unplanned repeat revascularization [71]. In contrast, Silveiro et al. reported that following MI, Lp(a) was independently associated with an increased risk of the composite outcome of recurrent MI and all-cause mortality in the overall population and in non-diabetic patients, but not among those with diabetes [72].
Regarding chronic kidney disease (CKD), high Lp(a) levels have been associated with an increased risk of MACE regardless of renal function, with the risk being particularly pronounced and consistent across multiple Lp(a) thresholds in patients with CKD [73].
In patients with ACS combined with TVD, Li et al. showed that elevated Lp(a) levels remained an independent predictor of MACE (P < 0.05) and further described a non-linear association between Log10-transformed Lp(a) and MACE (p for non-linearity < 0.001), hypothesizing that very low Lp(a) levels may compromise physiological repair and anti-inflammatory processes, potentially contributing to an increased risk of adverse cardiovascular outcomes [74]. This non-linear relationship was corroborated by Wohlfahrt et al. in a cohort of 851 patients, where both individuals with Lp(a) <7 nmol/L and those with Lp(a) ≥125 nmol/L had a higher risk of the combined endpoint of recurrent ACS and cardiovascular mortality (HR 2.6, 95% CI 1.33–5.08 and HR 2.10, 95% CI 1.00–4.39, respectively) [75].
In a systematic review and meta-analysis including 16,168 patients with ACS, elevated Lp(a) levels were independently associated with an increased risk of MACE (HR 1.26, 95% CI 1.17–1.35, p < 0.01) and all-cause mortality (HR 1.36, 95% CI 1.05–1.76, p = 0.02). Across the included studies, Lp(a) thresholds for adverse outcomes varied widely (12.5–60 mg/dL), reflecting differences in populations and measurement methods. Nevertheless, the association remained consistent, suggesting that even moderate elevations may be clinically relevant [65]. Consistent findings were reported in the systematic review by Mojahedi et al., which comprehensively evaluated the prognostic role of Lp(a) in patients with ACS undergoing PCI. Among 10 studies comprising a total of 20,896 patients, elevated Lp(a) levels—using cut-off values between 23 and 50 mg/dL—were consistently associated with both in-hospital and long-term adverse cardiovascular outcomes, independent of traditional cardiovascular risk factors and even among patients receiving statin therapy with LDL-C levels within guideline-recommended ranges [10].
Overall, recent evidence underscores the importance of Lp(a) as a predictor of adverse cardiovascular events following ACS, reinforcing the need to measure Lp(a) in routine clinical practice after an acute coronary event. Nevertheless, homogeneous studies are still needed to confirm this association and to define the Lp(a) threshold at which cardiovascular risk significantly increases.
5.2. Optimal Timing for Lipoprotein(a) Assessment After Acute Coronary Syndrome
Despite Lp(a) levels are generally stable over time, the optimal timing of measurement following ACS remains uncertain. Emerging evidence proposed that Lp(a) concentrations may transiently fluctuate during the acute phase of ACS. Ziogos et al. reported that Lp(a) levels measured six months after ACS were significantly higher than those obtained within 24 hours of hospital admission, suggesting that early Lp(a) measurements may underestimate baseline cardiovascular risk [76]. On the contrary, Vavuranakis et al. observed a progressive reduction in Lp(a) concentrations one-month post-ACS compared with peri- and post-infarction values and demonstrated that this transient elevation may be mitigated by PCSK9 inhibition with evolocumab [77]. More recently, Saeki et al. described an initial decrease in Lp(a) levels within the first 12 hours of ACS, followed by a rebound to baseline values at 48 hours. Notably, the magnitude of this early decrease was independently associated with the occurrence of MACE over a three-year follow-up [78].
These studies suggest that Lp(a) measurements obtained during the acute phase may not accurately reflect baseline Lp(a)-associated cardiovascular risk. The mechanisms underlying these dynamic changes remain incompletely understood but may involve acute inflammatory responses, metabolic alterations, or lipoprotein redistribution during myocardial injury. Therefore, confirmatory Lp(a) measure 1–3 months after the coronary event is recommended to accurately evaluate Lp(a)-related cardiovascular risk [5,6,7,76,77,78].
6. Therapeutic Strategies Targeting Lipoprotein(a)
The management of elevated Lp(a) remains a significant unmet need. Because Lp(a) catabolism is largely independent of the LDL receptor, standard lipid-lowering therapies—primarily optimized for LDL-C clearance—show limited efficacy in addressing its unique atherogenic, pro-inflammatory, and pro-thrombotic burden [79].
6.1. Current Management Strategies
Statins, the cornerstone of ASCVD prevention, are ineffective for Lp(a) lowering and may paradoxically increase levels by 10–20% via LPA gene upregulation [80]. However, meta-analyses confirm this does not negate their profound cardiovascular benefit, not supporting their discontinuation [81]. Similarly, bempedoic acid effectively lowers LDL-C and hsCRP but has a negligible clinical impact on Lp(a) [82].
Although no therapies specifically targeting Lp(a) are currently available, monoclonal antibodies against PCSK9 and small interfering RNA (siRNA) agents such as inclisiran are the only approved pharmacologic classes known to reduce Lp(a) levels, demonstrating an approximate reduction of 20–30%, although the underlying mechanisms remain to be fully elucidated [83,84,85]. Emerging cholesteryl ester transfer protein inhibitors, such as obicetrapib, have also demonstrated mean Lp(a) reductions of approximately 36% when combined with ezetimibe [86]. However, the clinical implications of Lp(a) reduction with these agents for cardiovascular outcomes remain to be established.
For refractory cases with progressive disease, lipoprotein apheresis remains the most potent option available. By acutely removing 60–75% of Lp(a) per session, longitudinal registries indicate it can significantly reduce MACE rates by nearly 80% in high-risk cohorts but its use remains marginal [87].
Given the challenges in normalizing concentrations, mitigating downstream thrombotic and inflammatory risks is critical. Due to the homology between apo(a) and plasminogen, aspirin has emerged as a targeted strategy; data from the ASPREE and MESA studies suggest that aspirin significantly reduces events in primary prevention specifically in individuals with elevated Lp(a) or high genetic risk [88,89], and is already a cornerstone of secondary treatment post-ACS.
6.2. Emerging Targeted Therapies
The landscape of Lp(a) management is undergoing a paradigm shift with the advent of therapies that directly target the hepatic synthesis or assembly of the lipoprotein. These novel agents leverage advanced delivery platforms, primarily N-acetylgalactosamine conjugation, to enable selective uptake by hepatocytes via the asialoglycoprotein receptor. Comprehensive details regarding the design and current status of ongoing clinical trials for the agents discussed below are provided in Table 2 [90,91,92,93,94,95,96].
The most advanced class of therapeutics involves silencing the LPA gene transcript. Pelacarsen, an ASO, binds to LPA messenger RNA (mRNA) and triggers its degradation through the nuclear enzyme RNase H1. In pivotal clinical trials, this stoichiometric mechanism resulted in approximately an 80% reduction in Lp(a) levels, with 98% of patients achieving physiological targets (≤50 mg/dL). Notably, pelacarsen reduces oxidized phospholipids, which is expected to further lower thrombotic and inflammatory risk. Moreover, ASOs have prolonged half-lives (3–4 weeks) due to their resistance to cleavage, allowing for less frequent dosing schedules [97,98]. Results from the phase 3 HORIZON–Lp(a) trial (NCT04023552) are awaited (anticipated February 2026); this study is designed to be the first to establish whether pharmacological decrease of Lp(a) leads to a reduction in MACE in a secondary prevention setting. The HORIZON–Lp(a) trial enrolled 8,323 patients with established ASCVD, including prior MI, ischemic stroke, or symptomatic peripheral artery disease, and elevated Lp(a) concentrations (≥ 70 mg/dL). Participants were randomized, in addition to optimized lipid-lowering therapy, to receive monthly subcutaneous injections of pelacarsen 80 mg or placebo over a follow-up period of 4 to 5 years. The primary end point is a composite of cardiovascular death, nonfatal MI, nonfatal ischemic stroke, and urgent coronary revascularization requiring hospitalization. Enrollment has been completed, with final study completion anticipated in February 2026. The trial is expected to assess the long-term clinical impact of Lp(a) reduction with pelacarsen in patients with elevated Lp(a) and established ASCVD [90].
Building upon this approach, siRNA molecules offer a distinct mechanism characterized by catalytic efficiency. Once internalized, the antisense strand of the siRNA is incorporated into the RNA-Induced Silencing Complex (RISC), which binds to and degrades the target LPA mRNA. Crucially, the RISC remains active after target degradation, enabling repeated cleavage of additional mRNA transcripts. This “recycling” capacity results in potent and sustained suppression of LPA expression. Olpasiran exploits this mechanism to achieve reductions exceeding 95%, with phase 2 data demonstrating that 100% of patients achieved Lp(a) levels <50 mg/dL [99,100]. Similarly, zerlasiran has demonstrated sustained efficacy (>80% reduction) over extended dosing intervals of up to 24 weeks [101].
Newer generations of siRNAs are pushing the boundaries of durability. Lepodisiran and the investigational agent SRSD216 have exhibited promising results. In phase 2 trials, lepodisiran achieved a mean time-averaged reduction of 94%, with concentrations remaining >90% below baseline for nearly one year after a single dose, supporting the feasibility of annual dosing regimens [102,103]. Likewise, preliminary data for SRSD216 indicate similar potency and duration [96].
Distinct from gene-silencing approaches, muvalaplin is a first-in-class oral small molecule that inhibits Lp(a) formation by disrupting the non-covalent interaction between apo(a) and apoB-100. By blocking this assembly at the hepatocyte surface, it reduces circulating Lp(a) levels without affecting plasminogen. The phase 2 KRAKEN trial validated this oral approach, demonstrating that a 240 mg daily dose reduced intact Lp(a) by 85.8%, allowing 97% of high-risk patients to reach target levels, with a safety profile comparable to placebo [104].
The ultimate frontier involves the transition from chronic suppression to a potential single-course cure. CTX320 utilizes CRISPR-Cas9 technology, delivered via lipid nanoparticles, to induce permanent disruption of the LPA gene in the liver. Preclinical data have demonstrated a >95% reduction in plasma Lp(a) following a single infusion, which remained stable over long-term follow-up, suggesting the possibility of permanent correction of this genetically determined risk factor [105].
In general, these targeted future therapies demonstrate unprecedented potential, offering deep (>90%) and sustained reductions in Lp(a) levels. If this biological effect also translates in a clinical benefit through hard clinical endpoint reduction in ongoing trials, these agents may redefine the standard of care.
7. Clinical Practice Guidelines and Consensus Recommendations
The major scientific societies concur in recognizing Lp(a) as an independent determinant of cardiovascular risk and in recommending its incorporation into clinical risk assessment. The ESC and EAS, in the 2025 update of their dyslipidemia management guidelines, recommend measuring Lp(a) at least once during adulthood, with particular relevance in young individuals with premature cardiovascular disease, familial hypercholesterolemia, or a family history of early-onset cardiovascular disease. They also note that Lp(a) levels may increase after menopause, and therefore, making repeat measurements in this setting appears justified. Guidelines further recommend cascade screening of first-degree relatives when elevated concentrations are identified. In these guidelines, Lp(a) levels ≥50 mg/dL (≥105 nmol/L) are considered a cardiovascular risk modifier (Class IIa, Level of Evidence B), with the potential to reclassify risk, particularly in individuals at moderate risk or near therapeutic decision thresholds [8].
Similarly, the 2018 guidelines from the AHA and the American College of Cardiology recognize Lp(a) as a risk-enhancing factor. They recommend its measurement in individuals with a personal or family history of premature cardiovascular disease not explained by traditional risk factors and consider levels ≥50 mg/dL (≥125 nmol/L) to be clinically relevant, potentially influencing therapeutic decision-making, especially in patients at intermediate cardiovascular risk [9].
In contrast, while current ESC and AHA ACS guidelines strongly emphasize aggressive secondary prevention strategies—such as intensive LDL-C lowering and comprehensive risk factor control—they do not yet provide specific recommendations on Lp(a) measurement or management, referring instead to general cardiovascular prevention and dyslipidemia guidelines [106].
8. Conclusions
Residual cardiovascular risk after an ACS remains a substantial unmet clinical challenge, driven not only by LDL-C but also by other atherogenic lipoproteins, persistent inflammation, metabolic dysregulation, and prothrombotic states. Among these factors, Lp(a) has emerged as a major and independent contributor to residual cardiovascular risk.
Accumulating evidence indicates that Lp(a)-associated risk after ACS may become clinically relevant at concentrations below the conventional high-risk threshold (≥50 mg/dL), with several studies reporting increased MACE risk at levels as low as 15–30 mg/dL.
Transient fluctuations may occur during the acute phase of ACS, therefore, Lp(a) measurement may optimally be reperformed 1–3 months after the index event to accurately assess baseline Lp(a)-related cardiovascular risk.
While currently available therapies have limited effects on Lp(a), novel targeted approaches—including ASO, siRNAs, oral inhibitors, and emerging gene-editing strategies—have demonstrated unprecedented profound and sustained reductions in Lp(a) concentrations. Ongoing outcome trials will clarify whether Lp(a) lowering translates into meaningful clinical benefit, Lp(a) may evolve from a risk marker to a modifiable therapeutic target.
Author Contributions
Conceptualization, N.G.-A. and F.C.; methodology, N.G.-A. F.C.; investigation, N.G.-A, R.F.-H., J.I.L.-V., F.P.-R., A.R.-M., J.M.G.-P., M.J.-S., A.P.-L., F.J.P.-M., A.P.-C., P.S. and F.C.; writing-original draft preparation, N.G.-A, R.F.-H., J.I.L.-V., F.P.-R. and F.C.; writing—review and editing, N.G.-A, R.F.-H., J.I.L.-V., F.P.-R., A.R.-M., J.M.G.-P., M.J.-S., A.P.-L., F.J.P.-M., A.P.-C., P.S. and F.C.; visualization: N.G.-A, R.F.-H., J.I.L.-V., F.P.-R., A.R.-M., J.M.G.-P., M.J.-S., A.P.-L., F.J.P.-M., A.P.-C., P.S. and F.C.; supervision, F.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACS | Acute Coronary Syndrome |
| MACE | Major Adverse Cardiovascular Events |
| LDL-C | Low-Density Lipoprotein Cholesterol |
| Lp(a) | Lipoprotein(a) |
| ASCVD | Atherosclerotic Cardiovascular Disease |
| RCT | Randomized Clinical Trial |
| PCSK9 | Proprotein Convertase Subtilisin/Kexin Type 9 |
| CAVD | Calcific Aortic Valvular Disease |
| ESC | European Society of Cardiology |
| EAS | European Atherosclerosis Society |
| AHA | American Heart Association |
| ASO | Antisense Oligonucleotide |
| siRNA | Small Interfering RNA |
| ApoB-100 | Apolipoprotein B-100 |
| Apo(a) | Apolipoprotein(a) |
| KIV | Kringle IV |
| CAD | Coronary Artery Disease |
| CNV | Copy Number Variation |
| HDL-C | High-Density Lipoprotein Cholesterol |
| HsCRP | High-Sensitivity C-Reactive Protein |
| HR | Hazard Ratio |
| CI | Confidence Interval |
| TVD | Three-Vessel Disease |
| MI | Myocardial Infarction |
| TG | Triglycerides |
| PCI | Percutaneous Coronary Intervention |
| MALE | Major Adverse Limb Events |
| STEMI | ST-Segment Elevation Myocardial Infarction |
| CKD | Chronic Kidney Disease |
| mRNA | Messenger RNA |
| PAD | Peripheral Artery Disease |
| RISC | RNA-Induced Silencing Complex |
References
- Steen, D.L.; Khan, I.; Andrade, K.; Koumas, A.; Giugliano, R.P. Event rates and risk factors for recurrent cardiovascular events and mortality in a contemporary post-acute coronary syndrome population representing 239,234 patients during 2005 to 2018 in the United States. J. Am. Heart Assoc. 2022, 11, e024200. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Leiter, L.A.; Kim, J.H.; Kuder, J.F.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Lawler, P.R.; Bhatt, D.L.; Godoy, L.C.; Lüscher, T.F.; Bonow, R.O.; Verma, S.; Ridker, P.M. Targeting cardiovascular inflammation: next steps in clinical translation. Eur. Heart J. 2021, 42, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Sabouret, P.; Angoulvant, D.; Pathak, A.; Fysekidis, M.; Laterra, G.; Costa, F.; Angelini, F.; Bocchino, P.P.; Montalescot, G.; Biondi-Zoccai, G. How to fill the GAPS-I in secondary prevention: Application of a strategy based on GLP1 analogues, antithrombotic agents, PCSK9 inhibitors, SGLT2 inhibitors and immunomodulators. Panminerva Med. 2022, 64, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein(a) and cardiovascular disease. Lancet 2024, 404, 1255–1264. [Google Scholar] [CrossRef]
- Tsioulos, G.; Kounatidis, D.; Vallianou, N.G.; Poulaki, A.; Kotsi, E.; Christodoulatos, G.S.; Tsilingiris, D.; Karampela, I.; Skourtis, A.; Dalamaga, M. Lipoprotein(a) and Atherosclerotic Cardiovascular Disease: Where Do We Stand? Int. J. Mol. Sci. 2024, 25, 3537. [Google Scholar] [CrossRef]
- Duarte Lau, F.; Giugliano, R.P. Lipoprotein(a) and its significance in cardiovascular disease: a review. JAMA Cardiol. 2022, 7, 760–769. [Google Scholar] [CrossRef]
- Mach, F.; Koskinas, K.C.; Roeters van Lennep, J.E.; Tokgözoğlu, L.; Badimon, L.; Baigent, C.; Binder, C.J.; Catapano, A.L.; De Backer, G.; Delgado, V.; et al. 2025 focused update of the 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur. Heart J. 2025, 46, 4359–4378. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC Guideline on the management of blood cholesterol. Circulation 2019, 139, e1082–e1143. [Google Scholar] [CrossRef]
- Mojahedi, A.; Chen, O.; Skopicki, H.A.; Rahman, T.; Sadeghian, A. Lipoprotein(a) as a predictor of cardiovascular risk in acute coronary syndrome patients undergoing percutaneous coronary intervention: a systematic review. Rev. Cardiovasc. Med. 2025, 26, 42784. [Google Scholar] [CrossRef]
- Mansoor, T.; Ismayl, M.; Parikh, S.; Nambi, V.; Virani, S.S.; Mehta, A.; Jia, X.; Minhas, A.M.K. Emerging pharmacological strategies in lipoprotein(a) reduction. Proc. (Baylor Univ. Med. Cent.) 2025, 38, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Berg, K. A new serum type system in man—the LP system. Acta Pathol. Microbiol. Scand. 1963, 59, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.; Mohr, J. Genetics of the LP system. Acta Genet. Stat. Med. 1963, 13, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K. Structure, function, and metabolism of lipoprotein(a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef]
- Kratzin, H.; Armstrong, V.W.; Niehaus, M. Structural relationship of an apolipoprotein(a) phenotype (570 kDa) to plasminogen: homologous kringle domains are linked by carbohydrate-rich regions. Biol. Chem. Hoppe Seyler 1987, 368, 1533–1544. [Google Scholar] [CrossRef]
- Boffa, M.B. Beyond fibrinolysis: the confounding role of Lp(a) in thrombosis. Atherosclerosis 2022, 349, 72–81. [Google Scholar] [CrossRef]
- Tsimikas, S.; Hall, J.L. Lipoprotein(a) as a Potential Causal Genetic Risk Factor of Cardiovascular Disease: A Rationale for Increased Efforts to Understand its Pathophysiology and Develop Targeted Therapies. J. Am. Coll. Cardiol. 2012, 60, 716–721. [Google Scholar] [CrossRef]
- Koutsogianni, A.D.; Liberopoulos, E.; Tellis, K.; Tselepis, A.D. Oxidized phospholipids and lipoprotein(a): An update. Eur. J. Clin. Invest. 2022, 52, e13710. [Google Scholar] [CrossRef]
- Boffa, M.B.; Koschinsky, M.L. Lipoprotein(a): truly a direct prothrombotic factor in cardiovascular disease? J. Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef]
- Tasdighi, E.; Adhikari, R.; Almaadawy, O.; Leucker, T.M.; Blaha, M.J. Lp(a): Structure, Genetics, Associated Cardiovascular Risk, and Emerging Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2024, 64, 135–157. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Borén, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur. Heart J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Elevated Lipoprotein(a) and Risk of Aortic Valve Stenosis in the General Population. J. Am. Coll. Cardiol. 2014, 63, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Volgman, A.S.; Koschinsky, M.L.; Mehta, A.; Rosenson, R.S. Genetics and Pathophysiological Mechanisms of Lipoprotein(a)-Associated Cardiovascular Risk. J. Am. Heart Assoc. 2024, 13, e033654. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Soffer, G.; Ginsberg, H.N.; Berglund, L.; Duell, P.B.; Heffron, S.P.; Kamstrup, P.R.; Lloyd-Jones, D.M.; Marcovina, S.M.; Yeang, C.; Koschinsky, M.L. Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e48–e60. [Google Scholar] [CrossRef]
- Nakajima, K.; Hata, Y. Intraindividual Variations in Lipoprotein(a) Levels and Factors Related to These Changes. J. Atheroscler. Thromb. 1996, 2, 96–106. [Google Scholar] [CrossRef]
- Trinder, M.; Paruchuri, K.; Haidermota, S.; Bernardo, R.; Zekavat, S.M.; Gilliland, T.; Januzzi, J.L., Jr.; Natarajan, P. Repeat Measures of Lipoprotein(a) Molar Concentration and Cardiovascular Risk. J. Am. Coll. Cardiol. 2022, 79, 617–628. [Google Scholar] [CrossRef]
- Sung, D.E.; Lee, M.Y.; Kwon, M.J.; Sung, K.C. Longitudinal changes and borderline reclassification of Lipoprotein(a) compared with conventional lipids in over 230,000 adults. Atherosclerosis. 2025, 410, 120524. [Google Scholar] [CrossRef]
- Awad, K.; Mahmoud, A.K.; Abbas, M.T.; Alsidawi, S.; Ayoub, C.; Arsanjani, R.; Farina, J.M. Intra-individual variability in lipoprotein(a) levels: findings from a large academic health system population. Eur. J. Prev. Cardiol. 2025, 32, 716–721. [Google Scholar] [CrossRef]
- Harb, T.; Ziogos, E.; Blumenthal, R.S.; Gerstenblith, G.; Leucker, T.M. Intra-individual variability in lipoprotein(a): the value of a repeat measure for reclassifying individuals at intermediate risk. Eur. Heart J. Open. 2024, 4, oeae064. [Google Scholar] [CrossRef]
- Joo, H.J.; Yun, S.G.; Park, J.H.; Hong, S.J.; Yu, C.W.; Shin, S.Y.; Kim, E.J. Predictors of lipoprotein(a) variability in clinical practice and their impact on cardiovascular risk. Lipids Health Dis. 2025, 24, 250. [Google Scholar] [CrossRef]
- Deshotels, M.R.; Sun, C.; Nambi, V.; Virani, S.S.; Matsushita, K.; Yu, B.; Ballantyne, C.M.; Hoogeveen, R.C. Temporal Trends in Lipoprotein(a) Concentrations: The Atherosclerosis Risk in Communities Study. J. Am. Heart Assoc. 2022, 11, e026762. [Google Scholar] [CrossRef]
- Zhang, S.S.; Hu, W.Y.; Li, Y.J.; Yu, J.; Sang, S.; Alsalman, Z.M.; Xie, D.Q. Lipoprotein(a) variability is associated with mean follow-up C-reactive protein in patients with coronary artery disease following percutaneous coronary intervention. World J. Clin. Cases. 2022, 10, 12909–12919. [Google Scholar] [CrossRef] [PubMed]
- Gołąb, A.M.; Konieczyńska, M.; Ząbczyk, M.; Undas, A.; Natorska, J. Intraindividual variability of lipoprotein(a) in the middle-aged population: the impact of smoking. Pol. Arch. Intern. Med. 2024, 134, 16889. [Google Scholar] [CrossRef] [PubMed]
- Matta, M.G.; Schreier, L.; Lavalle-Cobo, A.; Garcia-Zamora, S.; Ferraresi, A.; Madsen, A.; Bellini, S.; Ramos, G.; Roubicek, P.; Corral, P. Temporal variability of Lp(a) in clinically stable patients: implications for cardiovascular risk assessment. Med. Clin. (Barc). 2024, 163, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Marcovina, S.M.; Viney, N.J.; Hughes, S.G.; Xia, S.; Witztum, J.L.; Tsimikas, S. Temporal variability in lipoprotein(a) levels in patients enrolled in the placebo arms of IONIS-APO(a)Rx and IONIS-APO(a)-LRx antisense oligonucleotide clinical trials. J. Clin. Lipidol. 2018, 12, 122–129.e2. [Google Scholar] [CrossRef]
- de Boer, L.M.; Hof, M.H.; Wiegman, A.; Stroobants, A.K.; Kastelein, J.J.P.; Hutten, B.A. Lipoprotein(a) levels from childhood to adulthood: data in nearly 3,000 children who visited a pediatric lipid clinic. Atherosclerosis 2022, 349, 227–232. [Google Scholar] [CrossRef]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lp(a) (Lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease: New insights from a large national biobank. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 465–474. [Google Scholar] [CrossRef]
- Amiri, M.; Raeisi-Dehkordi, H.; Verkaar, A.J.C.F.; Wu, Y.; van Westing, A.C.; Berk, K.A.; Bramer, W.M.; Aune, D.; Voortman, T. Circulating lipoprotein(a) and all-cause and cause-specific mortality: A systematic review and dose–response meta-analysis. Eur. J. Epidemiol. 2023, 38, 485–499. [Google Scholar] [CrossRef]
- Bhatia, H.S.; Wandel, S.; Willeit, P.; Lesogor, A.; Bailey, K.; Ridker, P.M.; Nestel, P.; Simes, J.; Tonkin, A.; Schwartz, G.G.; et al. Independence of lipoprotein(a) and low-density lipoprotein cholesterol–mediated cardiovascular risk: A participant-level meta-analysis. Circulation 2025, 151, 312–321. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, N.; Tse, G.; Li, G.; Sun, Y.; Liu, T. Association between lipoprotein(a) and premature atherosclerotic cardiovascular disease: A systematic review and meta-analysis. Eur. Heart J. Open. 2024, 4, oeae031. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. High lipoprotein(a) and high risk of mortality. Eur. Heart J. 2019, 40, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Arnold, N.; Blaum, C.; Goßling, A.; Brunner, F.J.; Bay, B.; Zeller, T.; Ferrario, M.M.; Brambilla, P.; Cesana, G.; Leoni, V.; et al. Impact of lipoprotein(a) level on low-density lipoprotein cholesterol– or apolipoprotein B–related risk of coronary heart disease. J. Am. Coll. Cardiol. 2024, 84, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Björnson, E.; Adiels, M.; Taskinen, M.R.; Burgess, S.; Chapman, M.J.; Packard, C.J.; Borén, J. Lipoprotein(a) is markedly more atherogenic than LDL: an apolipoprotein B-based genetic analysis. J. Am. Coll. Cardiol. 2024, 83, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, Z.; Dong, J.; Yu, M.; Zhou, Y. Lipoprotein(a) and panvascular disease. Lipids Health Dis. 2025, 24, 186. [Google Scholar] [CrossRef]
- Wang, Z.; Xiao, S.; Liu, N. Association of lipoprotein(a) with coronary severity in patients with new-onset acute myocardial infarction: A large cross-sectional study. Clin. Chim. Acta. 2023, 540, 117220. [Google Scholar] [CrossRef]
- Kozieł-Siołkowska, M.; Mitręga, K.; Podolecki, T.; Olma, A.; Kalarus, Z.; Streb, W. Lipoprotein(a) as an independent predictor of elevated SYNTAX score. J. Clin. Med. 2024, 13, 7109. [Google Scholar] [CrossRef]
- Rallidis, L.S.; Pavlakis, G.; Foscolou, A.; Kotakos, C.; Katsimardos, A.; Drosatos, A.; Zolindaki, M.; Panagiotakos, D.B. High levels of lipoprotein(a) and premature acute coronary syndrome. Atherosclerosis. 2018, 269, 29–34. [Google Scholar] [CrossRef]
- Fan, W.; Wu, C.; Wong, N.D. Lipoprotein(a) atherosclerotic cardiovascular disease risk score development and prediction in primary prevention from real-world data. Circ. Genom. Precis. Med. 2025, 18, e004631. [Google Scholar] [CrossRef]
- Cordero, A.; Salinas, J.M.; Quintanilla, M.A.; López-Ayala, J.M.; Blasco, Á.; Flores, E. Reclassification of low or intermediate cardiovascular risk by determining lipoprotein(a) levels. Biomedicines. 2025, 13, 2648. [Google Scholar] [CrossRef]
- Patel, K.V.; Pandey, A.; de Lemos, J.A. Conceptual framework for addressing residual atherosclerotic cardiovascular disease risk in the era of precision medicine. Circulation. 2018, 137, 2551–2553. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Ballantyne, C.M. Residual cardiovascular risk at low LDL: Remnants, lipoprotein(a), and inflammation. Clin. Chem. 2021, 67, 143–153. [Google Scholar] [CrossRef]
- Katsi, V.; Argyriou, N.; Fragoulis, C.; Tsioufis, K. The Role of Non-HDL Cholesterol and Apolipoprotein B in Cardiovascular Disease: A Comprehensive Review. J. Cardiovasc. Dev. Dis. 2025, 12, 256. [Google Scholar] [CrossRef] [PubMed]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet. 2018, 392, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, H.; Yu, X.; Tong, L.; Chen, X.; Qi, L.; Cui, C.; Cheng, L.; Cai, L.; et al. Elevated lipoprotein(a) levels as an independent predictor of long-term recurrent events in patients with acute coronary syndrome: an observational, retrospective cohort study. Coron. Artery Dis. 2022, 33, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Dai, K.; Shiode, N.; Yoshii, K.; Kimura, Y.; Matsuo, K.; Jyuri, Y.; Tomomori, S.; Higaki, T.; Oi, K.; Kawase, T.; et al. Impact of Lipoprotein(a) on Long-Term Outcomes in Patients With Acute Myocardial Infarction. Circ. J. 2023, 87, 1356–1361. [Google Scholar] [CrossRef]
- Miñana, G.; Cordero, A.; Fácila, L.; Company, M.; Fernández-Cisnal, A.; Valero, E.; Carratalá, A.; Navarro, J.; Llergo, J.T.; Fernández-Olmo, R.; et al. Lipoprotein(a) and long-term recurrent infarction after an acute myocardial infarction. Am. J. Cardiol. 2024, 211, 9–16. [Google Scholar] [CrossRef]
- Takahashi, D.; Wada, H.; Ogita, M.; Yasuda, K.; Nishio, R.; Takeuchi, M.; Shitara, J.; Tsuboi, S.; Dohi, T.; Suwa, S.; et al. Impact of Lipoprotein(a) as a Residual Risk Factor in Long-Term Cardiovascular Outcomes in Patients With Acute Coronary Syndrome Treated With Statins. Am. J. Cardiol. 2022, 168, 11–16. [Google Scholar] [CrossRef]
- Steg, P.G.; Szarek, M.; Jukema, J.W.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Fazio, S.; Garon, G.; Goodman, S.G.; Harrington, R.A.; et al. Relation of Low-Density Lipoprotein Cholesterol, High-Sensitivity C-Reactive Protein, and Lipoprotein(a) Each to Future Cardiovascular Events and Death After Acute Coronary Syndrome on High-Intensity Statin Therapy: An Analysis of the Placebo Arm of ODYSSEY OUTCOMES. Circulation 2025, 151, 1047–1050. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Ballantyne, C.M.; Barter, P.J.; Davidson, M.H.; Kallend, D.; Leiter, L.A.; Sisk, C.; Tershakovec, A.M.; Wright, R.S.; et al. Association of Lipoprotein(a) With Risk of Recurrent Ischemic Events Following Acute Coronary Syndrome: Analysis of the dal-Outcomes Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 164–168. [Google Scholar] [CrossRef]
- Park, J.S.; Cho, K.H.; Hong, Y.J.; Kim, M.C.; Sim, D.S.; Kim, J.H.; Ahn, Y.; Jeong, M.H.; et al. Baseline Lipoprotein(a) Levels and Long-Term Cardiovascular Outcomes After Acute Myocardial Infarction. J. Korean Med. Sci. 2023, 38, e102. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, H.H.; Jin, J.L.; Yan, X.N.; Dong, Q.; Li, J.J.; Wang, Y.; Chen, L.; Zhao, H.; Li, X.; et al. Lipoprotein(a) and cardiovascular death in oldest-old (≥80 years) patients with acute myocardial infarction: A prospective cohort study. Atherosclerosis 2020, 312, 54–59. [Google Scholar] [CrossRef]
- Li, N.; Zhou, J.; Chen, R.; Zhao, X.; Li, J.; Zhou, P.; Liu, C.; Chen, Y.; Wang, Y.; Song, L.; et al. Prognostic impacts of diabetes status and lipoprotein(a) levels in patients with ST-segment elevation myocardial infarction: a prospective cohort study. Cardiovasc. Diabetol. 2023, 22, 151. [Google Scholar] [CrossRef]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xia, M.; Liang, C.; Pu, F.; Liu, S.; Jia, D.; Zhang, Y.; Li, H.; Chen, X.; Zhao, Q.; et al. Prognostic value of elevated lipoprotein(a) in patients with acute coronary syndromes: a systematic review and meta-analysis. Front. Cardiovasc. Med. 2024, 11, 1362893. [Google Scholar] [CrossRef] [PubMed]
- Rigattieri, S.; Cristiano, E.; Tempestini, F.; Lo Monaco, M.; Cava, F.; Bongiovanni, M.; Tifi, P.; Berni, A.; Volpe, M.; Rossi, L.; et al. Lipoprotein(a) and the risk of recurrent events in patients with acute myocardial infarction treated by percutaneous coronary intervention. Minerva Cardiol. Angiol. 2023, 71, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Jian, S.; Zhou, W.; Zhou, Q.; Xiang, J.; Zhu, Y.; Xiang, Z.; Yang, H.; Liu, G.; Luo, S.; et al. Associations of Lipoprotein(a) With Coronary Atherosclerotic Burden and All-Cause Mortality in Patients With ST-Segment Elevation Myocardial Infarction Treated With Primary Percutaneous Coronary Intervention. Front. Cardiovasc. Med. 2021, 8, 638679. [Google Scholar] [CrossRef]
- Ray, K.K.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Fazio, S.; Goodman, S.G.; Harrington, R.A.; Jukema, J.W.; White, H.D.; Steg, P.G.; et al. Lipoprotein(a) Levels and Time to Cardiovascular Benefit With Alirocumab in Patients With Recent Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2025, 86, 2071–2076. [Google Scholar] [CrossRef]
- Gencer, B.; Rigamonti, F.; Nanchen, D.; Vuilleumier, N.; Kern, I.; Aghlmandi, S.; Klingenberg, R.; Räber, L.; Auer, R.; Carballo, D.; et al. Prognostic value of elevated lipoprotein(a) in patients with acute coronary syndromes. Eur. J. Clin. Invest. 2019, 49, e13117. [Google Scholar] [CrossRef]
- Roth, C.; Krychtiuk, K.A.; Gangl, C.; Schrutka, L.; Distelmaier, K.; Wojta, J.; Hengstenberg, C.; Berger, R.; Speidl, W.S.; Müller, P.; et al. Lipoprotein(a) plasma levels are not associated with survival after acute coronary syndromes: An observational cohort study. PLoS One 2020, 15, e0227054. [Google Scholar] [CrossRef]
- Hao, Y.; Yang, Y.; Wang, Y.; Li, J. Relationship between lipoprotein(a) and revascularization after percutaneous coronary intervention in type 2 diabetes mellitus patients with acute coronary syndrome. Curr. Med. Res. Opin. 2022, 38, 1663–1672. [Google Scholar] [CrossRef]
- Silverio, A.; Cancro, F.P.; Di Maio, M.; Bellino, M.; Esposito, L.; Centore, M.; Carrizzo, A.; Di Pietro, P.; Borrelli, A.; De Luca, G.; et al. Lipoprotein(a) levels and risk of adverse events after myocardial infarction in patients with and without diabetes. J. Thromb. Thrombolysis 2022, 54, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, Y.; Yu, L.; Zhu, L.; Wang, Z.; Jiao, S.; Wu, C.; Tu, Y.; Wu, Y.; Guo, Z.; et al. The relationship between lipoprotein(a) and cardiovascular events in acute coronary syndrome patients with and without chronic kidney disease. Atherosclerosis 2022, 349, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xu, S.; Shen, J.; Sun, F. The nonlinear association between lipoprotein(a) and major adverse cardiovascular events in acute coronary syndrome patients with three-vessel disease. Sci. Rep. 2025, 15, 1720. [Google Scholar] [CrossRef] [PubMed]
- Wohlfahrt, P.; Jenča, D.; Melenovský, V.; Franeková, J.; Jabor, A.; Šramko, M.; Staněk, V.; Želízko, M.; Poledne, R.; Piťha, J.; et al. Very low lipoprotein(a) and increased mortality risk after myocardial infarction. Eur. J. Intern. Med. 2021, 91, 33–39. [Google Scholar] [CrossRef]
- Ziogos, E.; Vavuranakis, M.A.; Harb, T.; Foran, P.L.; Blaha, M.J.; Jones, S.R.; Lai, S.; Gerstenblith, G.; Leucker, T.M. Lipoprotein(a) concentrations in acute myocardial infarction patients are not indicative of levels at six month follow-up. Eur. Heart J. Open 2023, 3, oead035. [Google Scholar] [CrossRef]
- Vavuranakis, M.A.; Jones, S.R.; Ziogos, E.; Blaha, M.J.; Williams, M.S.; Foran, P.; Schindler, T.H.; Lai, S.; Schulman, S.P.; Gerstenblith, G.; et al. The trajectory of lipoprotein(a) during the peri- and early postinfarction period and the impact of proprotein convertase subtilisin/kexin type 9 inhibition. Am. J. Cardiol. 2022, 171, 1–6. [Google Scholar] [CrossRef]
- Saeki, Y.; Sawaguchi, J.; Akita, S.; Takamura, T.A.; Fujibayashi, K.; Wakasa, M.; Akao, H.; Kitayama, M.; Kawai, Y.; Kajinami, K. Initial decrease in the lipoprotein(a) level is a novel prognostic biomarker in patients with acute coronary syndrome. World J. Cardiol. 2024, 16, 329–338. [Google Scholar] [CrossRef]
- Greco, A.; Finocchiaro, S.; Spagnolo, M.; Faro, D.C.; Mauro, M.S.; Raffo, C.; Sangiorgio, G.; Imbesi, A.; Laudani, C.; Mazzone, P.M.; et al. Lipoprotein(a) as a Pharmacological Target: Premises, Promises, and Prospects. Circulation 2025, 151, 400–415. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- de Boer, L.M.; Oorthuys, A.O.J.; Wiegman, A.; Langendam, M.W.; Kroon, J.; Spijker, R.; et al. Statin therapy and lipoprotein(a) levels: a systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2022, 29, 779–792. [Google Scholar] [CrossRef]
- Ridker, P.M.; Lei, L.; Ray, K.K.; Ballantyne, C.M.; Bradwin, G.; Rifai, N. Effects of bempedoic acid on CRP, IL-6, fibrinogen and lipoprotein(a) in patients with residual inflammatory risk: a secondary analysis of the CLEAR harmony trial. J. Clin. Lipidol. 2023, 17, 167–175. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk: insights from the FOURIER trial. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Watts, G.F.; Robinson, J.G.; Minini, P.; Sasiela, W.J.; Edelberg, J.; Louie, M.J.; Raal, F.J. Effect of Alirocumab on Lipoprotein(a) Over ≥1.5 Years (from the Phase 3 ODYSSEY Program). Am. J. Cardiol. 2017, 119, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Khalil, I.; Rahman, M.T.; Hossain, I. The Impact of Inclisiran on Lipid Profiles in Adults with Hyperlipidemia: A Meta-Analysis and Meta-Regression of Randomized Controlled Trials. Am. J. Cardiol. 2025, 250, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, M.I.; Safiullah, M.; Farhan, K.; Ali, A.; Abdullah, S.; Ali, A.; Ahmed, R. Obicetrapib and its impact on lipid parameters: a comprehensive meta-analysis of the latest evidence. J. Clin. Lipidol. 2025, 19, 1536–1549. [Google Scholar] [CrossRef]
- Roeseler, E.; Julius, U.; Heigl, F.; Spitthoever, R.; Heutling, D.; Breitenberger, P.; Leebmann, J.; Klingel, R.; Hafner, G.; Pöss, J.; et al. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2019–2027. [Google Scholar] [CrossRef]
- Lacaze, P.; Bakshi, A.; Riaz, M.; Polekhina, G.; Owen, A.; Bhatia, H.S.; Natarajan, P.; Wolfe, R.; Beilin, L.; Nicholls, S.J.; et al. Aspirin for primary prevention of cardiovascular events in relation to lipoprotein(a) genotypes. J. Am. Coll. Cardiol. 2022, 80, 1287–1298. [Google Scholar] [CrossRef]
- Bhatia, H.S.; Trainor, P.; Carlisle, S.; Tsai, M.Y.; Criqui, M.H.; DeFilippis, A.; Tsimikas, S. Aspirin and cardiovascular risk in individuals with elevated lipoprotein(a): the Multi-Ethnic Study of Atherosclerosis. J. Am. Heart Assoc. 2024, 13, e033562. [Google Scholar] [CrossRef]
- Cho, L.; Nicholls, S.J.; Nordestgaard, B.G.; Landmesser, U.; Tsimikas, S.; Blaha, M.J.; Leitersdorf, E.; Lincoff, A.M.; Lesogor, A.; Manning, B.; et al. Design and rationale of Lp(a)HORIZON trial: assessing the effect of lipoprotein(a) lowering with pelacarsen on major cardiovascular events in patients with CVD and elevated Lp(a). Am. Heart J. 2025, 287, 1–9. [Google Scholar] [CrossRef]
- ACCLAIM-Lp(a) Investigators. A Study to Investigate the Effect of Lepodisiran on the Reduction of Major Adverse Cardiovascular Events in Adults With Elevated Lipoprotein(a) (ACCLAIM-Lp(a)). ClinicalTrials.gov 2025, NCT06292013. Available online: https://clinicaltrials.gov/ct2/show/NCT06292013 (accessed on 7 January 2026).
- MOVE-Lp(a) Investigators. Assessing the Impact of Muvalaplin on Major Cardiovascular Events in Adults With Elevated Lipoprotein(a) (MOVE-Lp(a)). ClinicalTrials.gov 2025, NCT07157774. Available online: https://clinicaltrials.gov/ct2/show/NCT07157774 (accessed on 7 January 2026).
- OCEAN(a) Investigators. Olpasiran Trials of Cardiovascular Events and Lipoprotein(a) Reduction (OCEAN(a)) - Outcomes Trial. ClinicalTrials.gov 2025, NCT05581303. Sponsor: Amgen. Available online: https://clinicaltrials.gov/ct2/show/NCT05581303 (accessed on 7 January 2026).
- OCEAN(a)-PreEvent Investigators. OCEAN(a)-PreEvent: Olpasiran Trials of Cardiovascular Events And LipoproteiN(a) Reduction to Prevent First Major Cardiovascular Events. ClinicalTrials.gov 2025, NCT07136012. Available online: https://clinicaltrials.gov/ct2/show/NCT07136012 (accessed on 7 January 2026).
- ADD-VANTAGE Investigators. Lp(a) Lowering Study of Pelacarsen (TQJ230) With Background Inclisiran in Participants With Elevated Lp(a) and Established ASCVD (ADD-VANTAGE). ClinicalTrials.gov 2025, NCT06813911. Available online: https://clinicaltrials.gov/ct2/show/NCT06813911 (accessed on 20 January 2026).
- SRSD216 Investigators. A Study of SRSD216 in Patients With Elevated Lipoprotein(a). ClinicalTrials.gov. 2025, NCT07172646. Available online: https://clinicaltrials.gov/study/NCT07172646 (accessed on 7 January 2026).
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.G.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Niman, S.; Goldfaden, R.F.; Ashchi, M.; Bisharat, M.; Huston, J.; Hartmann, H.; Choksi, R. A Review of the Clinical Pharmacology of Pelacarsen: A Lipoprotein(a)-Lowering Agent. Am. J. Cardiovasc. Drugs 2022, 22, 47–54. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small interfering RNA to reduce lipoprotein(a) in cardiovascular disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Rosenson, R.S.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; Murphy, S.A.; et al. The off-treatment effects of olpasiran on lipoprotein(a) lowering: OCEAN(a)-DOSE extension period results. J. Am. Coll. Cardiol. 2024, 84, 790–797. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Watts, G.F.; Koren, M.J.; Fok, H.; Nicholls, S.J.; Rider, D.A.; Cho, L.; Romano, S.; Melgaard, C.; et al. Zerlasiran, a short interfering RNA targeting lipoprotein(a): a phase 2 randomized clinical trial. JAMA 2024, 331, 1215–1224. [Google Scholar] [CrossRef]
- Nissen, S.E.; Linnebjerg, H.; Shen, X.; Wolski, K.; Ma, X.; Lim, S.; Michael, L.F.; Ruotolo, G.; Gribble, G.; Navar, A.M.; et al. Lepodisiran, an extended-duration short interfering RNA targeting lipoprotein(a): a randomized dose-escalation clinical trial. JAMA 2023, 330, 2075–2083. [Google Scholar] [CrossRef]
- Faisal, A.; Basit, A.; Iftikhar, A.; Saifullah, M.; Rehmaan, M.K.U.; Basil, A.M. Lepodisiran: from genetic targeting to cardiovascular promise: a detailed narrative review of the literature. World J. Cardiol. 2025, 17, 109657. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ni, W.; Rhodes, G.M.; Nissen, S.E.; Navar, A.M.; Michael, L.F.; Haupt, A.; Krege, J.H. Oral muvalaplin for lowering of lipoprotein(a): a randomized clinical trial. JAMA. 2025, 222–231. [Google Scholar] [CrossRef]
- Gurevitz, C.; Bajaj, A.; Khera, A.V.; Do, R.; Schunkert, H.; Musunuru, K.; Rosenson, R.S. Gene therapy and genome editing for lipoprotein disorders. Eur. Heart J. 2025, 46, 3420–3433. [Google Scholar] [CrossRef]
- Rao, S.V.; O’Donoghue, M.L.; Ruel, M.; Rab, T.; TamisHolland, J.E.; Alexander, J.H.; Baber, U.; Baker, H.; Cohen, M.G.; CruzRuiz, M.; et al. 2025 ACC/AHA/ACEP/NAEMSP/SCAI guideline for the management of patients with acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2025, 85, 2135–2237. [Google Scholar] [CrossRef]
Figure 1.
Pathophysiological Mechanisms of Lipoprotein(a).
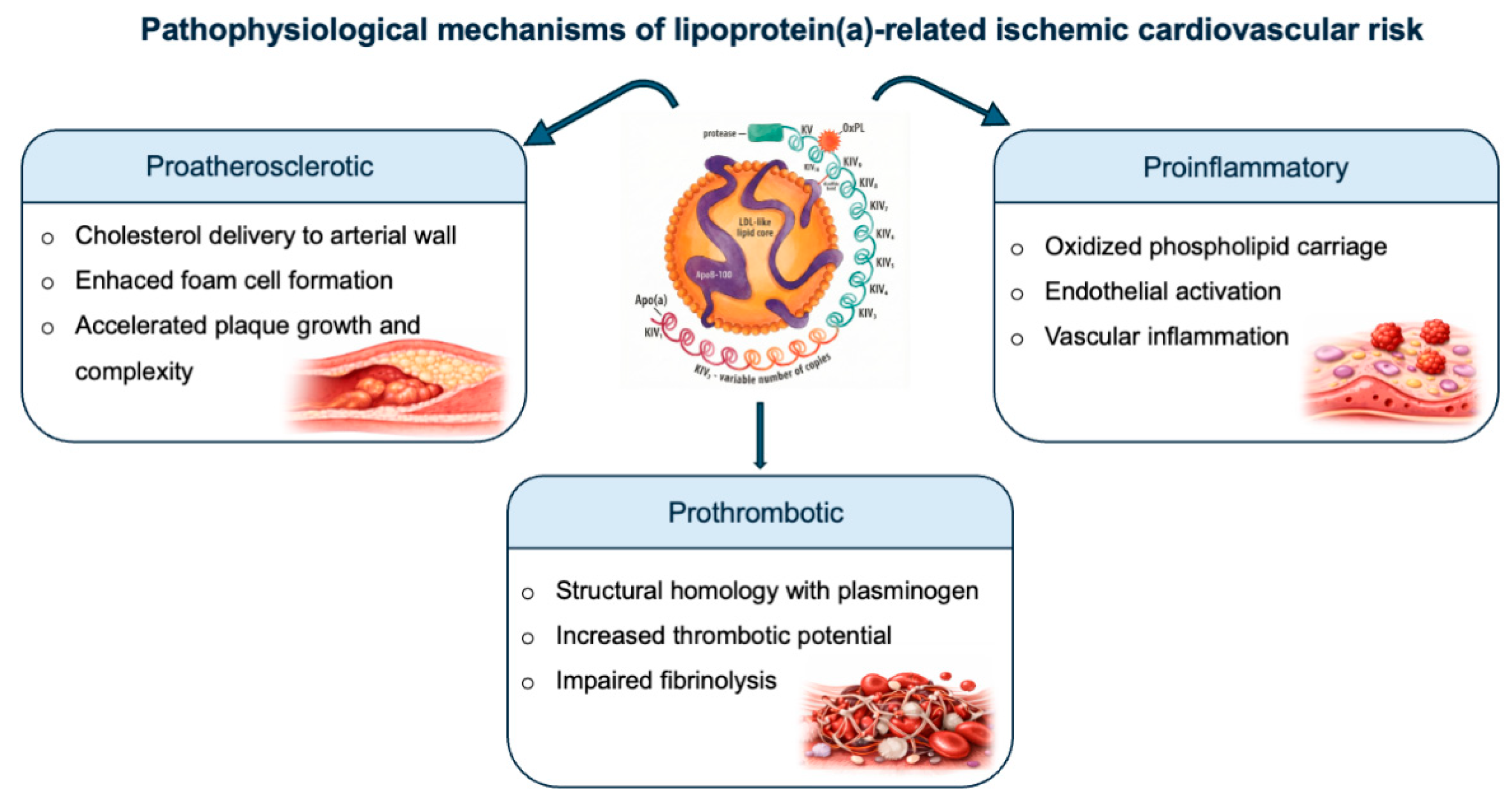
Figure 2.
Comprehensive summary of clinical and laboratory predictors for lipoprotein(a) variability evaluated across studies. Footer: Abbreviations: Lp(a): lipoprotein (a); Comorb: comorbidities [ASCVD, Hypertension, Diabetes, Smoking, BMI (Body Mass Index), SCORE2 (Systematic Coronary Risk Estimation 2)]; CBC: Complete Blood Count (Platelets, Hemoglobin, White Blood Cells); Lipid: lipid profile [Total Cholesterol, LDL-C, HDL-C (High-Density Lipoprotein Cholesterol), Triglycerides, Apolipoproteins, Phospholipids]; Inflam: Inflammatory markers [(Erythrocyte Sedimentation Rate, High-Sensitivity C-Reactive Protein (hsCRP)].
Figure 2.
Comprehensive summary of clinical and laboratory predictors for lipoprotein(a) variability evaluated across studies. Footer: Abbreviations: Lp(a): lipoprotein (a); Comorb: comorbidities [ASCVD, Hypertension, Diabetes, Smoking, BMI (Body Mass Index), SCORE2 (Systematic Coronary Risk Estimation 2)]; CBC: Complete Blood Count (Platelets, Hemoglobin, White Blood Cells); Lipid: lipid profile [Total Cholesterol, LDL-C, HDL-C (High-Density Lipoprotein Cholesterol), Triglycerides, Apolipoproteins, Phospholipids]; Inflam: Inflammatory markers [(Erythrocyte Sedimentation Rate, High-Sensitivity C-Reactive Protein (hsCRP)].
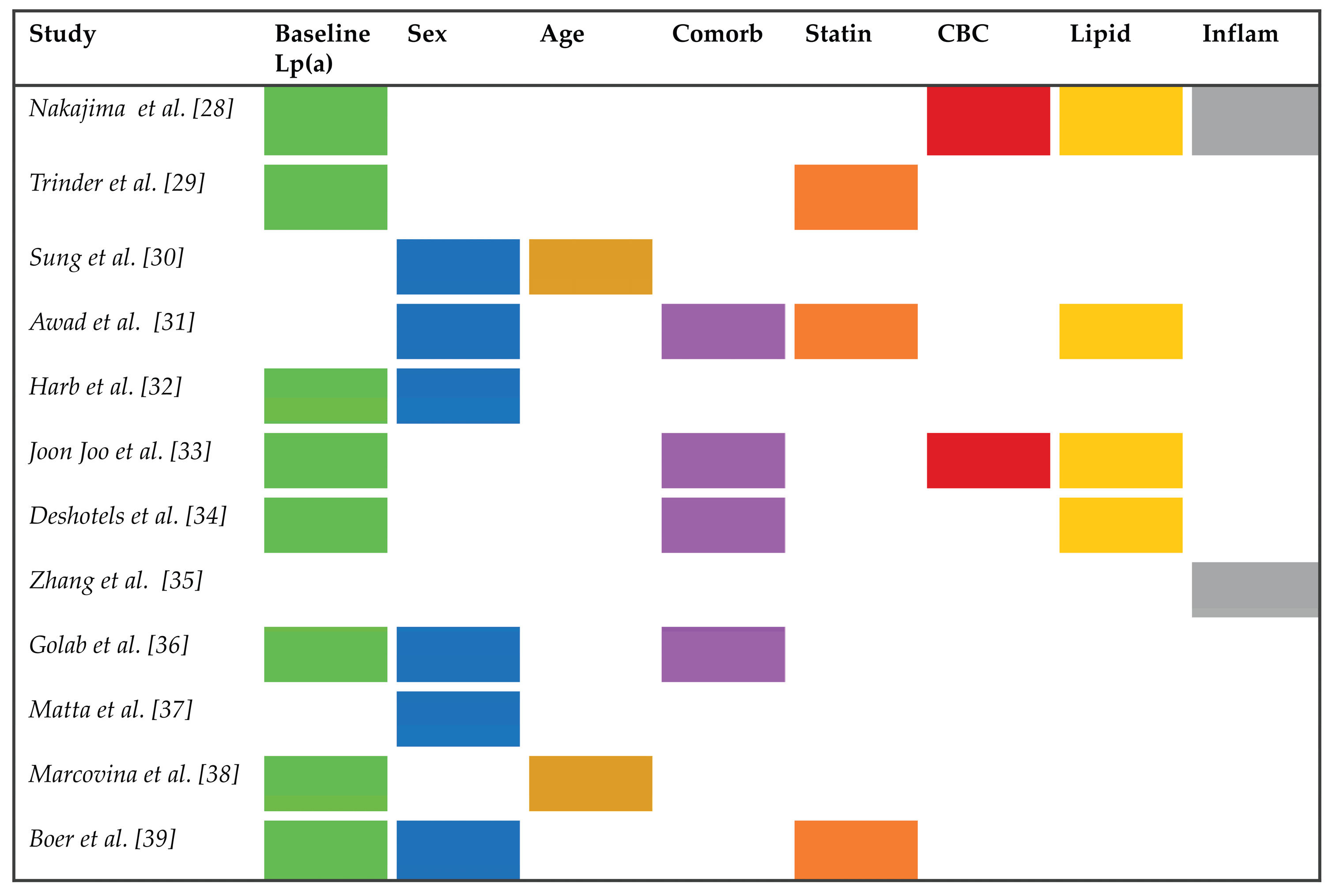

Table 1.
Summary of studies evaluating the impact of lipoprotein(a) after acute coronary syndrome.
| Study (year) |
Study design |
Study population |
STEMI | Mean of Lp(a) (mg/dL) |
Lp(a) threshold (mg/dL) |
Median follow-up | Outcome | Conclusion | |
|
Yang et al. (2022) [55] |
Retrospective multicenter cohort | 765 | 431 (56.3%) | 13.41 | ≥30 | 17 months | Composite of all-cause death, nonfatal MI, nonfatal stroke and unplanned revascularization | Each 1-SD increase in Lp(a) predicted MACE (HR 1.29, 95% CI 1.11–1.48, p=0.001) and revascularization (HR 1.59, 95% CI 1.31-1.93, p=0.001). | |
|
Dai et al. (2023) [56] |
Prospective cohort | 262 | NR | 21 | ≥32 | 4.6 years | Composite of CV death, nonfatal MI, and readmission for heart failure | Lp(a) ≥32 mg/dL independently predicted MACE (HR 2.84, 95% CI l 1.25–6.60, p=0.01). | |
|
Miñana et al. (2024) [57] |
Retrospective cohort | 1,223 | 417 (34.1%) | 28.8 | Q1 ≤8.9, Q2 9-21.6, Q3 21.6-40, Q4 40-74.6, Q5 74.6-305 | 9.9 years | MI | Lp(a) ≥50 mg/dL nonlinearly associated with reinfarction (p=0.016); not associated with long-term all-cause mortality. | |
|
Takahashi et al. (2022) [58] |
Prospective cohort | 1,758 | 872 (77.1%) | 15 | ≥15 | 2.2 years | Composite of all-cause death and MI | Risk of MACE was higher in high Lp(a) group (≥15mg/dL). | |
|
Steg et al. (2025) [59] |
Post-hoc placebo-group RCT (ODYSSEY-OUTCOMES) | 9,149 | 3,235 (35.4%) | 21.4 | NR | 2.8 years | Composite of CAD death, nonfatal MI, fatal or nonfatal stroke, or hospitalization for UA | Lp(a) below 21.4mg/dL robust predicted MACE. | |
|
Schwartz et al. (2018) [60] |
Post-hoc nested cohort analysis (dal-OUTCOMES) | 4,139 | NR | 12.3 | NR | 29 months | Composite of CAD death, a major nonfatal MI, hospitalization for UA, or resuscitated cardiac arrest, or a fatal or nonfatal stroke | Lp(a) no associated with ischemic cardiovascular events. | |
|
Park et al. (2023) [61] |
Prospective cohort | 1,908 | 695 (36.4%) | 17 | I<30, II 30–49, III ≥50 | 3 years | Composite of nonfatal MI, nonfatal stroke, and CV death |
Lp(a) no associated with MACE. |
|
|
Zhang et al. (2020) [62] |
Prospective cohort | 1,008 (≥80 years) | NR | 13 | ≤10, 10-30, >30 | 32.26 months | CV death | Lp(a) >30 mg/dL linked to CV death (HR 1.52, 95% CI 1.08–2.13, p=0.016) and lower event-free survival. | |
|
Li et al. (2023) [63] |
Prospective cohort | 1,543 (677 DM, 866 non-DM) | 100% | 16.9 DM, 17.3 non-DM | ≥30 | 3.96 years | Composite of all-cause death, recurrence of MI, and stroke | MACE risk increased linearly above Lp(a) 16.9 mg/dL in DM; no effect in non-DM. | |
|
Bittner et al. (2020) [64] |
Pre-specified analysis of RCT (ODYSSEY Outcomes) |
18,924 | 6,536 (34.5%) | 21.2 | Q1 <6.7, Q2 6.7-21.2, Q3 21.2-59.6, Q4 >59.6 | 2.8 years | Composite of CV death, nonfatal MI, stroke, or hospitalization for UA | Alirocumab lowered Lp(a) by 5 mg/dL and reduced MACE risk (HR 0.85, 95% CI 0.78–0.93, p<0.001) | |
|
Wang et al. (2024) [65] |
Meta-analysis | 18,168 | NR | NR | From 12.5 to 60 | From 2.93 to 66 months | Composite of all-cause death, stroke, non-fatal MI, and coronary revascularization | High Lp(a) associated with increased MACE risk (HR 1.26, 95% CI 1.17–1.35, p< 0.001) and all-cause mortality (HR 1.36, 95% CI: 1.05–1.76, p = 0.02). |
Footer: Abbreviations: STEMI: ST-segment elevation myocardial infarction; Lp(a): lipoprotein(a); MI: myocardial infarction; SD: standard deviation; MACE: major adverse cardiovascular events; HR: hazard ratio; CI: confidence interval; NR: not reported; CV: cardiovascular; Q: quartile; RCT: randomized controlled trial; CAD: coronary artery disease; UA: unstable angina; DM: diabetes mellitus.
Table 2.
Ongoing and upcoming clinical trials investigating emerging therapies targeting lipoprotein(a).
Table 2.
Ongoing and upcoming clinical trials investigating emerging therapies targeting lipoprotein(a).
| Molecule | NCT Number / Trial Name | Design | Inclusion Criteria | Primary Outcomes | Study Period | ||
| Pelacarsen [90] |
NCT04023552 Lp(a)HORIZON |
Phase 3 Double-blind Placebo-controlled |
Lp(a) ≥175 nmol/L + MI ≥3m and ≤10y/ischemic stroke ≥3m and ≤10y/PAD | Time to first MACE with Lp(a)≥70 mg/dL or ≥90 mg/dL | Dic 2019 – Feb 2026 | ||
| Lepodisiran [91] |
NCT06292013 ACCLAIM-Lp(a) |
Phase 3 Double-blind Placebo-controlled |
Lp(a) ≥175 nmol/L + Age ≥18y with ASCVD or ≥55y with high CV risk | Time to MACE-4 (CV death, nonfatal MI, nonfatal ischemic stroke, and urgent coronary revascularization) | Mar 2024 – Mar 2029 | ||
| Muvalaplin [92] |
NCT07157774 MOVE-Lp(a) |
Phase 3 Double-blind Placebo-controlled |
Lp(a) ≥175 nmol/L + ASCVD or high CV risk |
Time to MACE-4 (CV death, nonfatal MI, nonfatal ischemic stroke, and urgent coronary revascularization) | Sep 2025 – Mar 2031 |
||
| Olpasiran [93] |
NCT05581303 OCEAN(a)-Outcomes |
Phase 3 Double-blind Placebo-controlled |
Age 18-85y + Lp(a) ≥200 nmol/L + ASCVD | Time to first CHD death, MI, or urgent coronary revascularization |
Dec 2022 – Dec 2026 | ||
| Olpasiran [94] |
NCT07136012 OCEAN(a)-PreEvent |
Phase 3 Double-blind Placebo-controlled |
Age ≥50y + Lp(a) ≥200 nmol/L + multiple CV risk factors |
Time to CHD death, MI, or urgent revascularization | Aug 2025 – Oct 2031 | ||
| Pelacarsen + Inclisiran vs Placebo + Inclisiran [95] |
NCT06813911 ADD-VANTAGE |
Phase 3 Double-blind Placebo-controlled |
Age 18-80y + ASCVD + Lp(a) ≥175 nmol/L + LDL-C >70 mg/dL despite statins + Run-in period of inclisiran | Change from baseline Lp(a) and LDL-C | Apr 2025 – Dec 2027 | ||
| SRSD216 [96] |
NCT07172646 SRSD216 Study |
Phase I/II Double-blind Placebo-controlled |
BMI 18-40 kg/m2 |
Safety, tolerability, and adverse events | Apr 2025 – Apr 2027 |
Footer: Abbreviations: NCT: National Clinical Trial; Inc: Inclusion; Lp(a): Lipoprotein(a); Y: years; ASCVD: Atherosclerotic Cardiovascular Disease; CV: Cardiovascular; Exc: Exclusion; NYHA: New York Heart Association; HF: Heart Failure; D: Days; HTA: Hypertension; UACR: Urinary Albumin-to-Creatinine Ratio; MACE-4: Major Adverse Cardiovascular Events (CV death, nonfatal MI, nonfatal ischemic stroke, urgent coronary revascularization); MI: Myocardial Infarction; AST: Aspartate Aminotransferase; ALT: Alanine Aminotransferase; ULN: Upper Limit of Normal; BT: Total Bilirubin; CHD: Coronary Heart Disease; LDL-C: Low-Density Lipoprotein Cholesterol; PAD: Peripheral Artery Disease; PCSK9i: Proprotein Convertase Subtilisin/Kexin Type 9 inhibitor; TG: Triglycerides; BMI: Body Mass Index; M: Months. Data regarding study design, inclusion criteria, and estimated completion dates were retrieved from the U.S. National Library of Medicine (ClinicalTrials.gov) database, accessed January 2026.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



