Submitted:
02 February 2026
Posted:
05 February 2026
You are already at the latest version
Abstract
Core-binding factor acute myeloid leukemia (CBF-AML), defined by the chromosomal rearrangements t(8;21)(q22;q22) and inv(16)(p13q22)/t(16;16), has traditionally been classified as a favorable-risk subtype of AML. Nevertheless, relapse occurs in approximately 30–40% of patients, highlighting substantial biological and clinical heterogeneity within this entity. In this review, we synthesize emerging evidence delineating the distinct molecular architecture and prognostic divergence between RUNX1::RUNX1T1 and CBFB::MYH11 AML.
We focus on the clinical implications of measurable residual disease (MRD) dynamics and recurrent cooperating lesions, including KIT, FLT3, RAS pathway genes, cohesin-complex alterations, and ZBTB7A, and critically evaluate their impact on relapse risk and therapeutic decision-making. Current treatment paradigms, centered on intensive chemotherapy and high-dose cytarabine consolidation, are reviewed alongside evolving targeted, epigenetic, and immunotherapeutic strategies, with emphasis on distinguishing practice-changing interventions from investigational approaches.
Collectively, the available data challenge a uniform “favorable-risk” designation for CBF-AML and support a shift toward fusion-specific, MRD-guided, and molecularly integrated management strategies. We propose a contemporary framework in which dynamic MRD assessment and biological risk modifiers inform treatment intensification, including selective use of targeted agents and allogeneic transplantation, with the goal of achieving durable remissions and reducing relapse in this heterogeneous leukemia subtype.
Keywords:
CBF-AML
; RUNX1::RUNX1T1 CBFB::MYH11
; MRD
1. Introduction
Acute myeloid leukemia (AML) is a diverse cancer caused by the rapid growth of immature myeloid precursor cells in the bone marrow [1]. In recent years, there has been remarkable change regarding AML classifications. Modern classifications combine cytogenetic data with the new knowledge from molecular genetics [2]. These updates have made it easier to define AML subtypes, estimate prognosis, and improve treatment decisions. Among these different subtypes, core binding factor AML (CBF-AML) defined by particular genetic rearrangements in CBF complex including RUNX1::RUNX1T1 or CBFB::MYH11. Although CBF-AML is considered a favorable-risk AML subtype, recent data show significant genetic and clinical differences among patients. This favorable cytogenetics yet non-negligible relapse risk, suggests that baseline classification alone is insufficient to guide contemporary clinical decision-making. About 30% of CBF-AML patients experience a relapse, which underscores the need for continued research into prognostic factors and new treatment methods [3]. In this narrative review, we critically synthesize current evidence on the molecular landscape, prognostic modifiers, measurable residual disease (MRD), and therapeutic strategies in CBF-AML, with an emphasis on clinically actionable factors. Rather than providing an exhaustive catalog of genetic alterations, this review aims to contextualize emerging data within a practical, risk-adapted framework to inform treatment decisions in adult patients with CBF-AML.
Recent studies report that CBF-AML is one of the more common cytogenetic subtypes of AML, accounting for about 12–15% of adult AML cases [4]. CBF-AML is a unique cytogenetic classification characterized by chromosomal alterations that impact the RUNX1 and CBFB genes. These genes are essential for the formation of the CBF transcription complex, highlighting the distinct nature of this subtype among the various forms of AML [4]. The detected alterations, t(8;21)(q22;q22) RUNX1-RUNX1T1 and inv(16)(p13.1q22) or t(16;16)(p13;q22) CBFB-MYH11, are associated with impaired hematopoietic differentiation and leukemogenesis [5,6,7].
Prognostic variability and clinical challenges in CBF-AML are evident between its two main cytogenetic subgroups: inv(16) and t(8;21). The overall survival (OS) and relapse risk differ between these subtypes. Studies report that the 3-year OS rate for patients with inv(16) is approximately 57.3%, while it is lower, around 35.5%, for those with t(8;21) [4]. Long-term follow-up demonstrates that relapse occurs in about 30% to 40% of CBF-AML patients overall, but inv(16) patients tend to have a better prognosis with lower relapse rates compared to the t(8;21) subgroup [3,8,9]. These subtype-specific differences demonstrate that CBF-AML is not a homogeneous favorable-risk entity and underscore the need for subtype-directed risk stratification and individualized therapeutic strategies to reduce relapse and improve long-term survival.
Methods
This narrative review was conducted to synthesize and critically evaluate current evidence on the molecular heterogeneity, measurable residual disease (MRD), and therapeutic decision-making in core binding factor acute myeloid leukemia (CBF-AML). A comprehensive literature search was performed using PubMed/MEDLINE, Web of Science, and Scopus, covering publications from January 2000 through June 2025.
Search terms included combinations of: “core binding factor AML,” “CBF-AML,” “RUNX1::RUNX1T1,” “CBFB::MYH11,” “measurable residual disease,” “MRD,” “KIT mutation,” “FLT3,” “RAS,” “cohesin,” “gemtuzumab ozogamicin,” “epigenetic therapy,” and “allogeneic stem cell transplantation.” Reference lists of relevant articles were manually screened to identify additional key studies.
Eligible studies included peer-reviewed original research articles, prospective and retrospective clinical studies, cooperative group trials, meta-analyses, and authoritative reviews focusing on adult CBF-AML. Case reports, conference abstracts without full peer-reviewed publication, non–peer-reviewed sources, and studies lacking clinical or molecular relevance were excluded. When conflicting data were identified, greater weight was given to larger cohorts, prospective studies, randomized trials, and meta-analyses.
Evidence was synthesized qualitatively with emphasis on clinical relevance, reproducibility of findings, and implications for risk stratification and therapeutic decision-making. Given the heterogeneity of study designs and outcomes, no formal meta-analysis was performed.
2. Clinical and Therapeutic Challenges in CBF-AML
A subset of CBF-AML cases arises as therapy-related AML (t-CBF-AML), typically following prior cytotoxic therapy such as alkylating agents, topoisomerase II inhibitors or radiation [10]. Recent analyses suggest that approximately 15–20% of CBF-AML cases are therapy-related rather than de novo [11]. These patients are often older, with history of antecedent malignancies or prior cytotoxic exposure, and frequently exhibit reduced organ reserve and increased comorbidities [11]. Although CBF-AML in general retains high sensitivity to intensive cytarabine-based therapy, multiple studies now report inferior OS and higher relapse rates in t-CBF-AML compared to de novo CBF-AML [11,12,13]. Importantly, emerging data suggest that the inferior outcomes in t-CBF-AML are driven predominantly by host-related factors (such as older age, prior treatment-related organ toxicity and reduced biologic reserve), rather than by fundamentally distinct leukemia biology [11]. For example, in a recent single-center retrospective cohort of CBF-AML patients, the t-CBF-AML group had a median age of 64 years compared to 48 years for de novo cases (p = 0.001); 5-year OS for t-inv(16) was significantly lower than that of de novo inv(16) (58.3% vs. 83.2%, p = 0.033) [11]. Consensus guidelines now emphasize that treatment decisions including choice of induction intensity, number of consolidation cycles, maintenance strategies and consideration of allogeneic stem-cell transplant in first remission should not rest solely on the therapy-related status, but rather be guided by a comprehensive risk stratification incorporating MRD kinetics, additional adverse features and patient fitness [10,14,15].
Limitations of Current Standard Therapies (7+3 Regimen, Consolidation)
The backbone of frontline therapy in fit CBF-AML patients remains intensive induction chemotherapy (7+3: an anthracycline administered for 3 days plus 7 days of cytarabine) followed by repeated cycles of intensive cytarabine as consolidation therapy [16]. Although this approach induces CR in approximately 90% of patients, long-term outcomes remain suboptimal [17]. Pooled data indicate that even with optimal therapy, 3-year OS is ~65–70% and 5-year OS only ~58–65%, reflecting persistent relapse [17]. Thus, standard 7+3 plus consolidation cures only about half of CBF-AML patients, underscoring a plateau of benefit from existing regimens. In an effort to overcome this limitation, multiple trials have investigated the addition of targeted agents to the 7+3 backbone. Results have been heterogeneous: for example, the CALGB 10801 phase II study of dasatinib plus induction and consolidation reported a favorable 3-year disease-free survival (DFS) of ~75% and OS of ~77% [18], whereas a subsequent phase III randomized trial (NCT02013648) did not demonstrate a significant improvement in event-free survival (EFS) or OS with dasatinib (4-year EFS ≈41% vs 44%; 4-year OS ≈76% vs 78%) [19]. This discrepancy underscores the challenges of translating early-phase signals into consistent survival benefit in unselected CBF-AML populations.
By contrast, more robust evidence supports the integration of gemtuzumab ozogamicin (GO) into frontline therapy. An individual-patient meta-analysis of five randomized controlled trials (n=3,325) demonstrated that adding gemtuzumab ozogamicin (GO) to induction chemotherapy significantly reduced relapse and improved OS at 5–6 years, but this benefit was confined mainly to patients with favorable or intermediate cytogenetic risk rather than those with adverse cytogenetics [20]. These findings underscore that, while the 7+3 backbone remains foundational, future improvements in CBF-AML outcomes will likely come from integrating molecular profiling with clinical risk factors to guide personalized therapeutic strategies beyond cytogenetic risk alone.
3. Molecular Basis of Heterogeneity in CBF-AML
The defining leukemogenic event in CBF-AML is disruption of the CBF transcriptional complex through the RUNX1::RUNX1T1 or CBFB::MYH11 fusion proteins, establishing a distinct cytogenetic subtype of AML. Despite this shared initiating lesion, CBF-AML exhibits marked biological and clinical heterogeneity that cannot be explained by cytogenetics alone.
Emerging evidence indicates that the two canonical CBF fusion proteins differ in their downstream transcriptional programs, epigenetic interactions, and dependence on cooperating mutations, leading to divergent patterns of disease biology and treatment response. These differences provide a mechanistic basis for the observed variability in relapse risk, sensitivity to chemotherapy, and response to adjunctive therapies among patients with CBF-AML. Consequently, CBF fusions should be viewed not as uniform prognostic markers, but as initiating events whose clinical impact is shaped by additional molecular and context-dependent factors.
3.1. The t(8;21)(q22;q22) Translocation and RUNX1-RUNX1T1 Fusion Gene
The t(8;21)(q22;q22) chromosomal translocation, present in approximately 5–10% of adult AML cases, results in fusion of the RUNX1 gene on chromosome 21 with RUNX1T1 on chromosome 8 and defines a major subset of CBF-AML [21]. The breakpoint typically occurs between exons 5 and 6 of RUNX1, juxtaposing its RUNT DNA-binding domain with the NHR1–4 repression domains of RUNX1T1, thereby generating the oncogenic RUNX1::RUNX1T1 fusion transcript [22].
RUNX1-RUNX1T1 can be detected in the peripheral blood and bone marrow of t(8;21) AML patients and is a sensitive marker for MRD monitoring [23]. It is also thought of as an early leukemogenic event, being detectable in the neonatal blood spots of patients destined to develop AML, and persisting in clones at relapse, emphasizing its importance in the initiation and maintenance of disease [24]. However, the presence of RUNX1::RUNX1T1 alone is insufficient to drive overt leukemia, as evidenced by long latency periods and the requirement for cooperating genetic and epigenetic alterations. This distinction has important clinical implications, as it explains both the relative chemosensitivity of t(8;21) AML and the substantial heterogeneity in relapse risk.
Structural complexity further contributes to heterogeneity within t(8;21) AML. Cryptic or variant translocations may escape detection by conventional cytogenetics and require fluorescence in situ hybridization (FISH) or next-generation sequencing (NGS) for identification. Moreover, alternative RUNX1::RUNX1T1 splice variants have been described and may influence transcriptional output and clinical behavior, although their independent prognostic value remains incompletely defined and is not yet actionable in routine practice [25,26].
inv(16)(p13q22)/t(16;16)(p13;q22) and the CBFB::MYH11 Fusion
The chromosomal inversion inv(16)(p13q22) and its less frequent variant t(16;16)(p13;q22) define a distinct molecular subset of CBF-AML, historically associated with the FAB M4Eo phenotype but more accurately characterized by its unique biological and clinical features. These rearrangements result in fusion of the CBFB gene at 16q22 with MYH11 at 16p13, generating the CBFB::MYH11 oncofusion [22,27].
Under physiological conditions, CBFβ, encoded by CBFB, heterodimerizes with RUNX1 to stabilize DNA binding and facilitate transcriptional activation of genes essential for myeloid differentiation. In contrast, the CBFB::MYH11 fusion protein disrupts this process by sequestering RUNX1 into cytoplasmic aggregates, thereby preventing formation of functional CBF complexes and suppressing RUNX1-dependent transcriptional programs [23,28]. Structurally, the fusion retains the N-terminal domain of CBFβ fused to oligomerization-capable regions of MYH11, which contribute to aberrant intracellular localization and dominant-negative activity [29]. Functionally, CBFB-MYH11 blocks myeloid differentiation, leading to an accumulation of immature progenitors, and acts as a dominant-negative inhibitor of RUNX1-mediated differentiation programs [30]. Similar to RUNX1::RUNX1T1, CBFB::MYH11 acts as an initiating lesion that is insufficient on its own to induce overt leukemia. Cooperative genetic events, most commonly involving signaling pathways (e.g., KIT, RAS), epigenetic regulators (e.g., TET2, ASXL2), RNA splicing machinery, or cohesin complex components, are typically required to complete leukemic transformation and modulate disease behavior [31,32]. Clinically, the CBFB::MYH11 fusion transcript serves as a highly specific molecular marker for diagnosis and measurable residual disease (MRD) monitoring in inv(16)/t(16;16) AML. Although patients with this rearrangement generally demonstrate favorable initial responses to intensive chemotherapy, relapse remains a significant clinical challenge, particularly in the presence of high-risk secondary mutations. These observations underscore that, despite shared classification as favorable-risk AML, inv(16)/t(16;16) cases exhibit clinically relevant heterogeneity that must be integrated into risk-adapted treatment strategies [33]. (Figure 1)
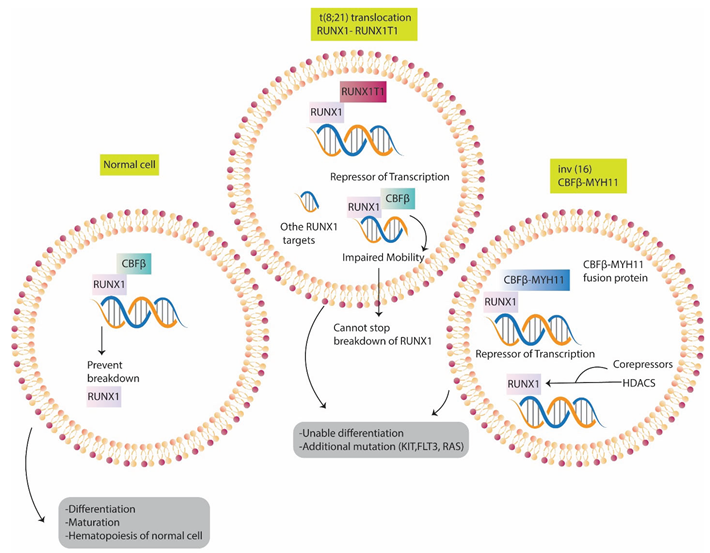
3.2. Clinical Phenotype and Treatment Sensitivity: Divergent Implications of CBF
Fusion Subtypes
Although both RUNX1::RUNX1T1 and CBFB::MYH11 fusion proteins disrupt normal CBF function through altered DNA binding and transcriptional repression, they differ in mechanistic nuance and in their patterns of cooperating genetic events. These biological differences translate into distinct clinical phenotypes and differential sensitivity to therapy. For example, recent work demonstrated that in pediatric CBF-AML, exon 17 mutations of the proto-oncogene KIT (notably D816 and N822) were strongly associated with adverse outcomes only in RUNX1::RUNX1T1 cases, whereas no similar prognostic impact was observed in CBFB::MYH11 patients [34]. While extrapolation to adult populations should be approached with caution, these findings highlight fusion-specific interactions between signaling mutations and disease biology. More direct evidence of differential treatment sensitivity comes from large retrospective adult cohorts. A large retrospective study (n=536) found that when using standard-dose versus intermediate-dose cytarabine induction, CBFB::MYH11 patients had no significant difference in outcome, but RUNX1::RUNX1T1 patients derived a statistically significant survival benefit from intermediate dosing (5-year OS 77.7% vs 60.3%, P<0.001) [35]. These data suggest that fusion subtype influences responsiveness to cytarabine-based regimens and that uniform treatment strategies may obscure clinically relevant differences.
Despite these documented differences, major clinical guidelines (such as those from the European Leukemia Net and other international consortia) continue to categorize both RUNX1::RUNX1T1 and CBFB::MYH11 AML under the single favorable risk group but stop short of recommending distinct therapeutic pathways for each fusion subtype. For example, MRD monitoring is recommended in CBF-AML generally, but there is no current guideline-endorsed, fusion-specific algorithm (differing induction dose, consolidation strategy, oหr HSCT indication) for RUNX1::RUNX1T1 versus CBFB::MYH11 [36].
Taken together, the evidence indicates that key differences in treatment response and prognosis exist between RUNX1::RUNX1T1 and CBFB::MYH11 AML. According to these results, treating both subtypes with the same approach is probably not the best course of action. Rather, they encourage the creation of molecularly informed, subtype-specific management algorithms that take into account fusion type, cooperating mutations, MRD clearance kinetics, and possibly fusion-specific maintenance or intensification of therapy. This molecular stratification should form the scientific basis for future prospective clinical trials and eventual guideline refinement.
3.3. Cooperation with Secondary Mutations and Therapeutic Challenges
Although the primary fusion events in CBF-AML (such as RUNX1::RUNX1T1 and CBFB::MYH11) clearly initiate the leukemic process, they are insufficient alone to drive full malignant transformation. Instead, secondary mutations in genes involving signal transduction, chromatin regulation, and the cohesin complex are frequently required and contribute to the observed clinical heterogeneity and therapeutic resistance [37]
Among these cooperating events, mutations affecting signal transduction pathways—particularly KIT and RAS family genes—are the most consistently observed and have the clearest clinical relevance. Alterations in epigenetic regulators (such as TET2, ASXL2, and DNMT3A), cohesin complex components, and RNA splicing factors occur less uniformly and appear to modulate disease biology in a context-dependent manner rather than acting as universal prognostic determinants.
Importantly, the distribution and impact of these secondary mutations differ between RUNX1::RUNX1T1 and CBFB::MYH11 AML, providing a molecular basis for divergent treatment responses and relapse patterns. From a therapeutic perspective, this layered mutational architecture limits the effectiveness of uniform treatment strategies and underscores the need for risk-adapted approaches that integrate fusion subtype with cooperating genetic lesions (Table 1).
3.4. FLT3 Mutations in CBF-AML: Infrequent but Clinically Relevant
Mutations in the FLT3 gene, including internal tandem duplications (FLT3-ITD) and tyrosine kinase domain (FLT3-TKD) alterations, are detected in a minority of patients with CBF-AML and occur less frequently than in AML with normal cytogenetics [49]. These mutations cause ligand-independent activation of the FLT3 signaling pathway. As a result, they enhance cell proliferation and block normal differentiation, contributing to leukemogenesis [50]. FLT3 mutations particularly FLT3-ITD have been associated with adverse clinical outcomes in selected CBF-AML cohorts. In an international collaborative study of 97 CBF-AML patients, FLT3-ITD was shown to be an independent adverse prognostic factor [51]. From a therapeutic perspective, while FLT3 inhibitors (such as quizartinib or other next-generation agents) offer hope, their effectiveness in CBF-AML is still under investigation and the frequent emergence of resistance mechanisms remains a significant challenge for long-term disease control [52,53]. Moreover, several FLT3 inhibitors such as Midostaurin and Gilteritinib have become an integral part of therapy in FLT3-mutated AML, but their specific effectiveness in CBF-AML remains insufficiently proven [54]. Ongoing research is therefore focused on optimizing the use of FLT3 inhibition in CBF-AML through next-generation agents and rational combination strategies, such as pairing FLT3 inhibitors with epigenetic therapies or BCL2 inhibition, to overcome resistance and improve long-term outcomes [55].
3.5. KIT Mutations in CBF-AML: Subtype-Specific Risk Modifiers with Unresolved Therapeutic Implications
Mutations of the KIT gene are among the most common cooperating events in CBF-AML, especially in the t(8;21) subgroup [38]. In CBF-AML, KIT mutations are detectable in roughly 20 % to 45 % of all cases [56]. In t(8;21) cases, high allelic ratios of KIT mutations have been linked to higher white blood cell counts, shorter event-free survival (EFS) and increased relapse risk, whereas this adverse effect appears less consistent in the inv(16) subgroup [38]. These data suggest that accurate mutation detection is crucial for effective management, particularly because outcomes differ by subtype, highlighting subtype-specific differences in clinical outcomes.
Despite the biological rationale for targeting KIT in CBF-AML, clinical evidence remains limited. For instance, the phase II CALGB 10801 study combined Dasatinib plus standard chemotherapy in CBF-AML and reported a 90% complete remission rate and 3-year OS of ≈77%, but outcomes were similar in patients with and without KIT mutations, suggesting the inhibitor may not fully overcome the adverse impact of KIT lesions [57]. Consistently, earlier studies evaluating single-agent dasatinib maintenance in high-risk CBF-AML, including KIT-mutated cases, failed to meaningfully reduce relapse rates [58]. These findings underscore key therapeutic gaps: the lack of randomized trials specifically in CBF-AML, unclear patient-selection criteria (e.g., high allelic ratio of KIT mutations), and the need to define whether KIT inhibition adds true long-term benefit beyond optimized chemotherapy.
3.6. RAS Pathway Mutations in CBF-AML: Frequent but Largely Prognostically Neutral
RAS pathway gene mutations, including NRAS and KRAS, are frequently observed in CBF-AML, with a higher prevalence in inv(16) CBF-AML compared to t(8;21) CBF-AML. High mutant allelic ratios of NRAS and KRAS mutations are associated with the absence of receptor tyrosine kinase mutations, such as KIT and FLT3, and are linked to a favorable prognosis, suggesting that RAS mutations may define a distinct subgroup of CBF-AML with unique clinical outcomes [59,60].
However, based on our knowledge, no studies have demonstrated a significantly different prognostic impact of RAS mutations between the inv(16) and t(8;21) subtypes, indicating that their role in disease progression may vary depending on the specific genetic context. Additionally, RAS mutations are commonly found in AML and myelodysplastic syndromes (MDS) and are associated with disease progression and poor prognosis [61]. Notably, RAS mutations often co-occur with cohesin mutations as AML progresses from MDS to secondary AML (sAML), suggesting a cooperative role in leukemogenesis [62].
From a therapeutic perspective, targeting RAS directly has been historically difficult, so current efforts have shifted to inhibiting downstream effectors. For example, in a phase II trial of the MEK inhibitor Binimetinib (MEK162) in relapsed/refractory AML with RAS-mutations, only 1 out of 13 evaluable patients (≈8%) achieved a complete response, indicating modest activity at best [44]. Another phase II study combining the MEK-inhibitor Trametinib with azacitidine and venetoclax in RAS-pathway-mutated AML showed a 25% response rate in 16 heavily pre-treated patients, but the median OS was only ~2.4 months and toxicity was substantial [63]. These data imply that while RAS-pathway targeting is biologically plausible in AML, its translation into effective therapy in CBF-AML is still far from optimal.
3.7. ASXL1 and ASXL2 Mutations in CBF-AML: Frequency Does Not Equal Prognostic Impact
Mutations in ASXL family genes occur in CBF-AML with distinct distributions and differing clinical implications. ASXL2 mutations are relatively frequent in RUNX1::RUNX1T1 (t(8;21)) AML, reported in approximately 22–23% of cases, whereas ASXL1 mutations are less common and are largely mutually exclusive with ASXL2 alterations [45]. From a prognostic perspective, ASXL1 mutations are well established as adverse risk factors across myeloid malignancies, including AML more broadly [46]. However, their independent prognostic impact within CBF-AML appears less consistent and may be modulated by fusion subtype and co-occurring genetic events. In contrast, despite their relatively high prevalence, ASXL2 mutations in CBF-AML have not demonstrated a reproducible, independent association with inferior survival when compared with ASXL2–wild-type cases [45]. At present, the therapeutic implications of ASXL1 and ASXL2 mutations in CBF-AML remain limited. Although epigenetic dysregulation provides a biological rationale for exploring hypomethylating agents or histone deacetylase inhibition, there is currently no clinical trial evidence supporting a mutation-specific benefit of these approaches in CBF-AML [64]. Consequently, ASXL1 and ASXL2 mutations should be regarded as markers of biological heterogeneity rather than actionable drivers of treatment decisions in current clinical practice.
3.8. Cohesin Complex Alterations in CBF-AML: Markers of Clonal Evolution Rather than Immediate Therapeutic Targets
Alterations affecting components of the cohesin complex, including STAG2, RAD21, SMC1A, and SMC3, have been increasingly recognized across myeloid malignancies and are also observed in a subset of patients with CBF-AML [65]. These mutations alter chromatin looping, transcriptional regulation, and contribute to genomic instability; in the context of CBF-AML they often co-occur with RAS-pathway lesions and may herald disease progression or resistance to therapy [66]. Current research aims to map the vulnerabilities specific to cohesin-mutant leukemic clones and to test therapeutic strategies targeting chromatin-architecture dependencies. In terms of treatment, preclinical research such as PARP-inhibitor sensitivity in STAG2-mutant cells indicates that cohesin-mutated AML may have synthetic-lethality potential, which may be significant in CBF-AML if such lesions exist [67].
3.9. ZBTB7A Mutations in CBF-AML: Recurrent in t(8;21) but Clinically Unresolved
Recent research has identified recurrent mutations in the transcription factor gene ZBTB7A among patients with CBF-AML, particularly within the t(8;21) subtype. One study reported ZBTB7A mutations in 23 % of t(8;21) cases (13 of 56) and demonstrated that these lesions often consisted of missense or truncating changes affecting the zinc-finger C-terminal domain [47]. According to functional studies, ZBTB7A suppression impairs its function as a transcriptional repressor, biases progenitor differentiation toward monocytic lineage, and increases glycolysis, indicating a joint carcinogenic role with the RUNX1::RUNX1T1 fusion [68]. A Japanese cohort study likewise found ZBTB7A mutations exclusively in t(8;21) patients (9.8 %) but observed no statistically significant impact on OS or event-free survival [48]. Altogether, these findings implicate ZBTB7A as a potential tumor-suppressor gene cooperating in t(8;21)-driven leukemogenesis; however, the clinical utility of its mutation status such as for risk stratification or therapeutic targeting has not yet been established and warrants further investigation.
3.10. WT1 Alterations in CBF-AML: Adverse Marker or Surrogate of High-Risk Biology?
Mutations of the tumor-suppressor gene WT1 appear in a subset of CBF-AML cases, often co-existing with high-risk lesions such as FLT3-ITD or KIT [69]. Clinical data show that WT1-mutated AML patients tend to have worse outcomes and higher relapse risk; meanwhile, WT1-directed immunotherapies (such as WT1 vaccines and T-cell therapies) are under investigation, although their use in front-line CBF-AML remains experimental [69,70]. Available evidence suggests that WT1 alterations in CBF-AML may primarily reflect an underlying high-risk molecular background rather than act as standalone drivers of adverse prognosis. Consequently, while WT1 mutation status may contribute to risk assessment when integrated with other molecular features, it does not currently mandate distinct therapeutic intervention in CBF-AML.
4. Molecular Risk Stratification and MRD-Guided Prognostic Modeling
Molecular risk stratification and MRD-guided prognostic modeling have become essential tools in the management of CBF-AML [71]. Molecular risk stratification refers to the classification of patients based on specific genetic and epigenetic abnormalities that influence disease behavior and relapse risk [72]. Integration of these molecular markers improves the precision of traditional cytogenetic classification by identifying patients at higher risk of relapse despite favorable baseline features [71].
Although patients may have cytogenetically favorable features like t(8;21) or inv(16) certain mutations such as KIT and FLT3, along with epigenetic changes play a key role in disease severity and treatment resistance [73,74]. Thus, integrating these molecular abnormalities into risk stratification models allows clinicians to identify patients at hidden high risk and tailor more precise, risk-adapted therapies [59]. This advances beyond the traditional cytogenetic classification by capturing the underlying biological heterogeneity that impacts prognosis and treatment response.
MRD refers to the small quantity of leukemic cells that remain in a patient after treatment, which are below the detection limit of standard microscopic methods [75]. MRD plays a crucial role in predicting relapse risk and informs therapeutic decisions, such as whether treatment intensification or allogeneic stem cell transplantation is warranted [36]. At present, qPCR is the most established and commonly applied method of assessing MRD in CBF-AML. This method enables the highly sensitive detection of leukemia-specific fusion transcripts such as RUNX1-RUNX1T1 in t(8;21) and CBFB-MYH11 in inv(16)/t(16;16) AML with a sensitivity of 10⁻⁴ to 10⁻⁶ [71]. Several studies have showed that the prognosis of MRD quantification via qPCR has very strong prognostic utility for CBF-AML. For example, a finding of ≥ 2-log reduction in transcript levels following the second consolidation cycle is associated with an increased relapse rate [76]. Consecutive MRD determinations allow for dynamic risk assessment and can incentivize pre-emptive actions such as intensifying consolidation therapy, and considering allogeneic hematopoietic stem cell transplantation (allo-HSCT) in MRD positive patients [77,78]. Limitation to qPCR is that it is only applicable to patients with a known and trackable fusion transcript, and is not applicable for other subtypes of AML.
NGS has become a new major player in the field of MRD detection since it is a method with high genomic coverage that enables assessment of a broad spectrum of somatic mutations associated with AML [79]. In contrast to qPCR that can only address fusion transcripts, NGS can monitor mutation clearance or clonal evolution of several genes involved in leukaemogenesis [80] NGS has clear advantages over qPCR in situations where qPCR does not work due to the absence of fusion transcripts, or when tracking multiple co-occurring mutations is required [81]. Further, NGS also permits the detection of subclonal populations and preleukemic clones which may persist after therapy and contribute to relapse.
The MRD in CBF-AML presents several distinct challenges despite its recognized prognostic value. First, although RT-qPCR or flow cytometric methods (FCM) can detect low levels of residual disease, a significant majority of relapses occur too quickly after molecular detection for intervention to change outcome. One study found that ~74 % of relapses progressed to morphologic disease in under 100 days, offering inadequate lead time for pre-emptive treatment [82]. Second, there is no universally accepted standard for MRD thresholds (e.g., log-reduction magnitude), optimal timing of sampling (bone marrow vs. peripheral blood, every 3 months vs. more frequent), or which assay (qPCR vs. NGS vs. FCM) should be used for CBF-AML specifically (71,82,83). Overcoming these obstacles will require harmonized methods, more frequent or real-time monitoring, and prospective trials that link MRD-guided decisions with improved outcomes. (Table 2)
5. Emerging Therapeutic Strategies Addressing Heterogeneity
5.1. Targeted Therapy in CBF-AML
Targeted therapies in AML recent approaches to the biological delineation of AML by molecular profiling, identifying recurrent genetic and epigenetic alterations in AML, have promoted the development of targeted therapies directed toward specific leukemogenic pathways [86].
5.2. KIT Inhibition
KIT activating mutations are observed in 20-40% of CBF-AML cases, with a higher rate of relapse and OS [39]. These mutations lead to constitutive signaling of the KIT receptor tyrosine kinase (RTK), leading to leukemia [43]. There has been preclinical activity with TKIs dasatinib and midostaurin in models of KIT-mutated AML, and both agents are in clinical trials [87]. Dasatinib plus chemotherapy works well with reducing MRD in patients with CBF-AML and KIT mutations [88].
5.3. Exploratory Strategies: Menin Inhibition and Novel Combinations
Menin inhibitors disrupt oncogenic transcriptional programs driven by KMT2A rearrangements and aberrant HOXA/MEIS1 activation. While these agents have shown compelling activity in KMT2A-rearranged and NPM1-mutated AML, their relevance to CBF-AML remains exploratory [89]. While these agents have shown compelling activity in KMT2A-rearranged and NPM1-mutated AML, their relevance to CBF-AML remains exploratory. Preclinical studies suggest that a subset of CBF-AML may exhibit sensitivity to menin inhibition, but clinical validation is currently lacking, and this strategy should be considered hypothesis-generating at present [90].
5.4. Practice-Changing Targeted Therapy: Gemtuzumab Ozogamicin
Gemtuzumab ozogamicin (GO) is an anti-CD33-directed antibody-drug conjugate that delivers a chemotherapeutic agent (calicheamicin) directly to leukemic cells expressing CD33, which is expressed on the majority of AML blasts [91]. GO was initially approved in 2000, and was withdrawn in 2010 due to toxicity and lack of efficacy, but was re-approved in 2017 after new dosing and favorable results in clinical trials [92]. CBF-AML shows a marked sensitivity to GO. These subtypes have often been observed to show strong CD33 expression and low multidrug resistance gene expression, which make them particularly suitable for treatment with GO [93,94]. A study demonstrated that adding GO to intensive chemotherapy (IC) significantly benefits patients with CBF-AML. In a retrospective multi-center analysis including 265 patients, those treated with GO showed a higher 2-year OS rate (90% vs. 80%) and improved 2-year event-free survival (51% vs. 36%) compared to those who did not receive GO [93]. Moreover, the ALFA-0701 study and other meta-analyses reported that, in patients with favorable-risk cytogenetics, including CBF-AML, the addition of GO to standard chemotherapy significantly increased event-free survival and reduced relapse rates [95,96]. These findings have resulted in the incorporation of GO into frontline therapy for CBF-AML in many clinical practice recommendations. While effective, GO use must be accompanied by frequent monitoring due to the risks of hepatotoxicity and veno-occlusive disease risks, especially in patients who will go onto hematopoietic stem cell transplantation in the future. Choosing the right patient and dose of GO (i.e., low-dose fractionated) is critical to maximize benefit and limit toxicity [97].
5.5. Promising but Unproven Approaches: FLT3 Inhibition
Currently, several FLT3 inhibitors are being studied for their efficacy in treating CBF-AML, particularly in patients with FLT3 mutations. Key FLT3 inhibitors under investigation include: Midostaurin: The first FLT3 inhibitor approved by the FDA, used in combination with chemotherapy in newly diagnosed FLT3-mutated AML. It shows clinical benefit by improving survival when added to standard induction regimens [98].
Gilteritinib: A potent and selective FLT3 inhibitor active against both FLT3-ITD and TKD mutations [99]. Studies demonstrate good tolerability and efficacy, including ongoing studies combining gilteritinib with chemotherapy in newly diagnosed and relapsed AML [54,100].
Crenolanib: A second-generation FLT3 inhibitor targeting both ITD and TKD mutations, including [101]. It is being evaluated in phase I/II trials both as monotherapy and in combination with chemotherapy or ICT [102].
Sorafenib: Sorafenib, a multi-kinase inhibitor targeting FLT3-ITD mutations, has demonstrated efficacy in multiple clinical settings. In a randomized controlled trial, sorafenib maintenance stem cell transplantation significantly improved relapse-free survival in FLT3-ITD AML patients [103]. Despite its therapeutic benefits, sorafenib’s use is tempered by notable toxicity concerns. High rates of toxicity-related treatment interruption have been reported, indicating the need for careful management and dose adjustments in clinical practice [104]. Overall, while effective, sorafenib’s tolerability profile necessitates vigilant monitoring to maintain its clinical benefits.
5.6. Epigenetic Therapies in CBF-AML: Context-Dependent and Largely Investigational
Epigenetic dysregulation, particularly aberrant DNA methylation and altered histone acetylation, is a central pathogenic mechanism in AML [107]. As a result, inhibitors targeting DNA methyltransferases (DNMT) and histone deacetylases (HDAC) are actively under investigation as potential therapeutic agents, with several compounds being evaluated in clinical trials for their ability to restore normal gene expression patterns and suppress leukemic cell growth [107,108,109]. DNA methyltransferase inhibitors (DNMTi) such as azacitidine and decitabine can restore normal gene expression and indicate that leukemic cells are sensitivized to chemotherapy [110].
Azacitidine is primarily indicated for the treatment of AML patients who are not candidates for intensive chemotherapy, including some with CBF-AML in specific settings. Clinical studies have shown that azacitidine, either alone or in combination with other agents, provides clinical benefit by improving OS and delaying progression, especially in older or unfit patients [111]. A recent multicenter retrospective study demonstrated moderate clinical activity and good tolerability of hypomethylating agents like azacitidine in relapsed/refractory CBF-AML patients and those unfit for intensive chemotherapy [112]. However, there are studies reporting differences in response to azacitidine between inv(16) and t(8;21) subtypes of CBF-AML. For instance, a recent multicenter retrospective analysis showed that azacitidine combined with venetoclax (VEN-HMA) resulted in higher complete remission rates and better 2-year OS trends in patients with inv(16)/t(16;16) AML compared to those with t(8;21) AML, where responses were suboptimal [113]. The poorer response in t(8;21) AML may be related to distinct molecular features and mutations such as KIT D816, which was frequently associated with treatment failure in this subgroup [113]. These findings suggest that although both subtypes are grouped as CBF-AML, they behave differently biologically and respond differently to hypomethylating agent-based therapies like azacitidine. Moreover, genomic studies highlight that mutations in chromatin modifiers and cohesins are more common in t(8;21) AML, contributing to its distinct clinical behavior and possibly its differential response to epigenetic therapies [110].
Decitabine has shown clinical utility in CBF-AML primarily as a maintenance therapy and in patients unfit for intensive chemotherapy [114]. A multicenter retrospective French study reported moderate efficacy of hypomethylating agents including decitabine in relapsed/refractory CBF-AML, with an overall response rate of about 49% and an improved survival in responders [112]. Maintenance therapy with decitabine after achieving first remission has been evaluated in clinical trials, including patients with CBF-AML, showing safety and potential to prolong disease-free survival, especially in patients with measurable residual disease after remission [115]. However, larger randomized controlled trials specifically assessing decitabine’s impact exclusively in CBF-AML remain limited, and further studies are warranted to fully define its role. There is clinical evidence comparing decitabine response in inv(16) versus t(8;21) subtypes of CBF-AML. A phase 2 study of decitabine maintenance therapy in 31 CBF-AML patients (14 with t(8;21), 17 with inv(16)) found that both groups benefitted from decitabine, with many patients achieving molecular remission [116]. A study by He et al. investigated why patients with the t(8;21) chromosomal translocation in AML respond better to decitabine-based treatments compared to other AML subtypes. The study analyzed DNA methylation patterns in bone marrow samples from patients with t(8;21) and non-t(8;21) AML. It identified 1,377 regions with differential methylation associated specifically with t(8;21) AML that responded to decitabine treatment. Analysis revealed specific regions of aberrant methylation linked to the t(8;21) subtype, with LIN7A found to be hypermethylated and transcriptionally repressed in these patients. Notably, reduced expression of LIN7A correlated with poorer clinical outcomes. Functional experiments demonstrated that treatment with decitabine led to demethylation and reactivation of LIN7A, which in turn enhanced the effectiveness of concurrent cytarabine therapy by promoting leukemic cell apoptosis. Conversely, lowering LIN7A levels impaired this therapeutic response, indicating that LIN7A is a crucial mediator and potential biomarker for predicting decitabine sensitivity in t(8;21) AML [117]. Therefore, decitabine represents a promising epigenetic therapeutic option in CBF-AML, especially as maintenance therapy for patients who achieve remission. While evidence suggests efficacy in both inv(16) and t(8;21) subtypes, with molecular markers such as LIN7A influencing sensitivity, further large-scale, randomized trials are necessary to better define its optimal clinical use and personalize treatment strategies.
Histone deacetylase inhibitors (HDACis) have been studied as epigenetic therapies in CBF-AML [108]. Among the HDACis explored, panobinostat and vorinostat have shown potential to enhance efficacy [118]. Natural compounds such as baicalein, which inhibit HDAC1 and HDAC8, have demonstrated growth suppression and apoptosis induction specifically in CBF-AML cell lines, including those with AML1-ETO and CBFβ-MYH11 fusions [109]. These inhibitors may promote degradation of fusion proteins and restore acetylation of important regulatory proteins, contributing to their anti-leukemic effects [64,109]. However, HDACis as monotherapy have shown limited clinical efficacy in AML, and their greatest promise lies in combination regimens with chemotherapy or other epigenetic agents like DNMT inhibitors [64]. Currently, there are no extensive clinical studies explicitly comparing the response to panobinostat among different mutations within CBF-AML. Most available data on panobinostat focus on its efficacy in t(8;21) AML models, showing promising preclinical results and synergy with other agents like arsenic trioxide, but mutation-specific differential responses within CBF-AML have not been clearly delineated [119]. Additionally, preclinical data using models of t(8;21) AML have demonstrated a robust antileukemic response to panobinostat, independent of p53 status or classical apoptotic pathways, suggesting subtype-specific molecular mechanisms of sensitivity [120]. While these findings imply possible differential responses in CBF subtypes, comprehensive comparative clinical trials assessing panobinostat’s efficacy across CBF-AML cytogenetic subgroups are lacking. More research is needed to clarify whether distinct molecular features translate to varied therapeutic outcomes with panobinostat between the inv(16) and t(8;21) subtypes. Studies investigating vorinostat in CBF-AML are relatively scarce and have produced inconsistent findings. For example, one clinical trial reported that administering higher doses of cytarabine in younger AML patients, with or without vorinostat, did not improve treatment outcomes [121]. In contrast, preclinical research has demonstrated that vorinostat induces apoptosis and differentiation, suggesting that epigenetic modulation by vorinostat might offer potential therapeutic benefits in CBF-AML [122]. This dichotomy highlights the need for further focused studies to clarify vorinostat's clinical value in this AML subtype.
Overall, epigenetic therapies in CBF-AML should be viewed as adjunctive or investigational approaches rather than standard frontline options. Their optimal use will depend on careful patient selection, disease context (e.g., unfit, relapsed, or MRD-positive settings), and integration with molecular and MRD-guided risk stratification. Prospective, subtype-focused clinical trials are required to clarify their role in personalized CBF-AML management. (Table 3)
5.7. Role of Hematopoietic Stem Cell Transplantation in High-Risk and Relapsed Patients
Most patients with CBF-AML achieve durable remissions with intensive chemotherapy alone, particularly when high-dose cytarabine is incorporated into consolidation regimens. Consequently, allogeneic hematopoietic stem cell transplantation (allo-HSCT) is not routinely recommended in first complete remission (CR1) for patients with standard-risk CBF-AML [126]. Allo-HSCT can be seen in limited circumstances when there is a high relapse risk even with favorable cytogenetics [127]. The allo-HSCT is primarily recommended for patients with CBF-AML who exhibit MRD after initial treatments or harbor KIT gene mutations [40]. These mutations, particularly frequent in cases with the t(8;21) translocation, are linked to a poorer prognosis and an increased chance of relapse [128]. In addition, other molecular abnormalities with adverse prognostic implications may also justify consideration for allo-HSCT. Patients with standard-risk CBF-AML typically receive consolidation chemotherapy without routine allo-HSCT in first remission, due to generally favorable outcomes. However, those with high-risk features such as MRD positivity, KIT mutations (especially exon 17 mutations), and certain co-occurring genomic alterations are candidates for allo-HSCT as a more intensive therapeutic approach to improve long-term survival [40,128,129]. Studies emphasize the heterogeneity within CBF-AML subtypes and the need for tailored treatment strategies based on molecular risk profiling [130,131]. This approach aligns with evidence from recent clinical studies and consensus guidelines, underscoring the importance of molecular and residual disease evaluation in transplant decision-making for CBF-AML.
Timing of allo-HSCT is important. Transplantation in patients in CR2 after relapse is also associated with a better survival benefit than non-transplant but inferior to upfront transplant in patients with molecular high-risk [71,127]. There is a delicate balance replacing the risk of relapse against the risk of morbidity/mortality from transplant. Individualized assessments based on age, donor availability, comorbidities, and dynamic molecular assessments must also be incorporated into the decision to undergo allo-HSCT in CR1(132). Although autologous transplantation has been investigated in CBF-AML, it has no reported benefit over high-dose chemotherapy and lacks a graft-versus-leukemia effect. In CBF-AML, the choice between autologous stem cell transplantation (ASCT) and allo-HSCT remains complex. Data from the EBMT registry (2024) analyzing 1901 patients transplanted in CR1 showed that allo-HSCT was associated with higher non-relapse mortality compared to ASCT [133]. Outcomes of matched sibling donor allo-HSCT were comparable to ASCT, suggesting that allo-HSCT may not confer a universal survival advantage for standard-risk patients in CR1 and is often reserved for those with higher risk or relapse [133]. Therefore, personalized treatment decisions based on molecular risk, MRD, and patient condition are essential in determining the optimal transplantation approach for CBF-AML.
6. Conclusions
CBF-AML constitutes a biologically distinct yet clinically heterogeneous subgroup of AML defined by RUNX1::RUNX1T1 and CBFB::MYH11 fusions. Although high remission rates are routinely achieved with intensive chemotherapy, relapse remains a persistent challenge driven by fusion-specific biology, cooperating molecular lesions, and dynamic MRD behavior. Accumulating evidence demonstrates that favorable cytogenetics alone are insufficient to capture relapse risk or guide contemporary treatment decisions.
This review highlights that molecular heterogeneity within CBF-AML has direct clinical implications. Secondary mutations, most notably KIT in a fusion-dependent context, modify relapse risk, while MRD kinetics consistently emerge as the most powerful determinant of outcome and therapeutic escalation. Accordingly, MRD-guided strategies, rather than uniform cytogenetic classification, should form the backbone of risk-adapted management in CBF-AML. Among targeted approaches, gemtuzumab ozogamicin represents the most clearly validated adjunct to frontline therapy, whereas kinase inhibitors, epigenetic therapies, and immunotherapeutic strategies currently remain context-dependent or investigational.
Looking forward, the clinical paradigm in CBF-AML must evolve from a binary favorable-risk label toward an integrated, biology-driven framework that incorporates fusion subtype, cooperating mutations, and MRD dynamics. Prospective clinical trials specifically designed to test fusion- and MRD-adapted therapeutic algorithms are essential to refine patient selection for treatment intensification, including allogeneic transplantation. Ultimately, durable remission in CBF-AML will depend on personalized strategies that translate molecular insight into rational, risk-adapted clinical decision-making.
References
- Hasserjian RP. Acute myeloid leukemia: advances in diagnosis and classification. Int J Lab Hematol [Internet]. 2013 Jun [cited 2025 Oct 30];35(3):358–66. Available from: https://pubmed.ncbi.nlm.nih.gov/23590662/.
- Appelbaum FR. WHO, what, when, where, and why: New classification systems for acute myeloid leukemia and their impact on clinical practice. Best Pract Res Clin Haematol [Internet]. 2023 Dec 1 [cited 2025 Oct 30];36(4). Available from: https://pubmed.ncbi.nlm.nih.gov/38092471/.
- Orvain C, Bertoli S, Peterlin P, Desbrosses Y, Dumas PY, Iat A, et al. Molecular relapse after first-line intensive therapy in patients with CBF or NPM1-mutated acute myeloid leukemia – a FILO study. Leuk 2024 389 [Internet]. 2024 Jul 17 [cited 2025 Oct 30];38(9):1949–57. Available from: https://www.nature.com/articles/s41375-024-02335-2.
- Borthakur G, Kantarjian H. Core binding factor acute myelogenous leukemia-2021 treatment algorithm. Blood Cancer J 2021 116 [Internet]. 2021 Jun 16 [cited 2025 Oct 30];11(6):1–5. Available from: https://www.nature.com/articles/s41408-021-00503-6.
- Kundu M, Chen A, Anderson S, Kirby M, Xu LP, Castilla LH, et al. Role of Cbfb in hematopoiesis and perturbations resulting from expression of the leukemogenic fusion gene Cbfb-MYH11. Blood [Internet]. 2002 Oct 1 [cited 2025 Oct 30];100(7):2449–56. Available from: https://pubmed.ncbi.nlm.nih.gov/12239155/.
- Link KA, Chou FS, Mulloy JC. Core binding factor at the crossroads: Determining the fate of the HSC. J Cell Physiol. 2010 Jan;222(1):50–6.
- De Bruijn MFTR, Speck NA. Core-binding factors in hematopoiesis and immune function. Oncogene [Internet]. 2004 May 24 [cited 2025 Oct 30];23(24):4238–48. Available from: https://pubmed.ncbi.nlm.nih.gov/15156179/.
- Talami A, Bettelli F, Pioli V, Giusti D, Gilioli A, Colasante C, et al. How to Improve Prognostication in Acute Myeloid Leukemia with CBFB-MYH11 Fusion Transcript: Focus on the Role of Molecular Measurable Residual Disease (MRD) Monitoring. Biomed 2021, Vol 9, Page 953 [Internet]. 2021 Aug 3 [cited 2025 Oct 30];9(8):953. Available from: https://www.mdpi.com/2227-9059/9/8/953/htm.
- Khan M, Cortes J, Qiao W, Alzubaidi MA, Pierce SA, Ravandi F, et al. Outcomes of patients with relapsed core binding factor-positive acute myeloid leukemia. Clin Lymphoma Myeloma Leuk [Internet]. 2017 Jan 1 [cited 2025 Oct 30];18(1):e19. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC5861376/.
- George B, Yohannan B, Mohlere V, Gonzalez A. Therapy-related core binding factor acute myeloid leukemia. Int J Hematol Oncol [Internet]. 2023 Mar [cited 2025 Oct 31];12(1):IJH43. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC9979104/.
- Chiu M, Schimmer AD, Schuh AC, Bankar A, Richard-Carpentier G, Sibai H, et al. Genomic profiles and outcomes in de novo versus therapy-related core binding factor AML. Blood Cancer J 2024 141 [Internet]. 2024 Oct 31 [cited 2025 Oct 31];14(1):1–5. Available from: https://www.nature.com/articles/s41408-024-01166-9.
- Borthakur G, Kantarjian H. Core binding factor acute myelogenous leukemia-2021 treatment algorithm. Blood Cancer J 2021 116 [Internet]. 2021 Jun 16 [cited 2025 Oct 31];11(6):1–5. Available from: https://www.nature.com/articles/s41408-021-00503-6.
- Rogers HJ, Wang X, Xie Y, Davis AR, Thakral B, Wang SA, et al. Comparison of therapy-related and de novo core binding factor acute myeloid leukemia: A bone marrow pathology group study. Am J Hematol [Internet]. 2020 Jul 1 [cited 2025 Oct 31];95(7):799–808. Available from: /doi/pdf/10.1002/ajh.25814.
- Fareed S, Soliman DS, Al-Mashdali AF, Gameil A, Mulikandathil Y, Alshurafa A, et al. Acute Myeloid Leukemia with Core Binding Factor Rearrangements: A 10-Year Cancer Center Experience. Oncol [Internet]. 2025 [cited 2025 Oct 31]; Available from: https://dx.doi.org/10.1159/000544049.
- Shimony S, Stahl M, Stone RM. Acute Myeloid Leukemia: 2025 Update on Diagnosis, Risk-Stratification, and Management. Am J Hematol [Internet]. 2025 May 1 [cited 2025 Oct 31];100(5):860. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC11966364/.
- Begna KH, Xu X, Gangat N, Alkhateeb H, Patnaik MM, Al-Kali A, et al. Core-binding factor acute myeloid leukemia: long-term outcome of 70 patients uniformly treated with “7+3.” Blood Cancer J 2022 124 [Internet]. 2022 Apr 7 [cited 2025 Oct 31];12(4):1–4. Available from: https://www.nature.com/articles/s41408-022-00654-0.
- Ronnacker J, Muller PJ, Mikesch JH, Zukunft S, Weinbergerová B, Šrámek J, et al. Gemtuzumab ozogamicin in first-line treatment of CBF-AML: insights from a retrospective multi-center analysis. Leuk 2025 399 [Internet]. 2025 Jul 21 [cited 2025 Oct 31];39(9):2174–80. Available from: https://www.nature.com/articles/s41375-025-02700-9.
- Marcucci G, Geyer S, Laumann K, Zhao W, Bucci D, Uy GL, et al. Combination of dasatinib with chemotherapy in previously untreated core binding factor acute myeloid leukemia: CALGB 10801. Blood Adv [Internet]. 2020 Feb 25 [cited 2025 Oct 31];4(4):696–705. Available from: https://pubmed.ncbi.nlm.nih.gov/32092139/.
- Dasatinib Misses Survival End Points in Core-Binding Factor AML | CancerNetwork [Internet]. [cited 2025 Oct 31]. Available from: https://www.cancernetwork.com/view/dasatinib-misses-survival-end-points-in-core-binding-factor-aml?utm_source=chatgpt.com.
- Hills RK, Castaigne S, Appelbaum FR, Delaunay J, Petersdorf S, Othus M, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol [Internet]. 2014 Aug 1 [cited 2025 Oct 31];15(9):986–96. Available from: https://www.thelancet.com/action/showFullText?pii=S1470204514702815.
- Bruserud O, Reikvam H, Hatfield KJ, Kittang AO, Hovland R. Acute Myeloid Leukemia with the t(8;21) Translocation: Clinical Consequences and Biological Implications. J Biomed Biotechnol [Internet]. 2011 [cited 2025 Nov 8];2011:104631. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC3100545/.
- Al-Harbi S, Aljurf M, Mohty M, Almohareb F, Ahmed SOA. An update on the molecular pathogenesis and potential therapeutic targeting of AML with t(8;21)(q22;q22.1);RUNX1-RUNX1T1. Blood Adv [Internet]. 2020 Jan 14 [cited 2025 Oct 31];4(1):229–38. Available from: https://pubmed.ncbi.nlm.nih.gov/31935293/.
- Day RB, Hickman JA, Xu Z, Katerndahl CDS, Ferraro F, Ramakrishnan SM, et al. Proteogenomic analysis reveals cytoplasmic sequestration of RUNX1 by the acute myeloid leukemia–initiating CBFB::MYH11 oncofusion protein. J Clin Invest [Internet]. 2024 Feb 15 [cited 2025 Oct 31];134(4). Available from: https://www.researchgate.net/publication/376312045_Proteogenomic_analysis_reveals_cytoplasmic_sequestration_of_RUNX1_by_the_acute_myeloid_leukemia-initiating_CBFBMYH11_oncofusion_protein.
- Grinev V V., Barneh F, Ilyushonak IM, Nakjang S, Smink J, van Oort A, et al. RUNX1/RUNX1T1 mediates alternative splicing and reorganises the transcriptional landscape in leukemia. Nat Commun 2021 121 [Internet]. 2021 Jan 22 [cited 2025 Oct 31];12(1):1–16. Available from: https://www.nature.com/articles/s41467-020-20848-z.
- Grinev V V., Migas AA, Kirsanava AD, Mishkova OA, Siomava N, Ramanouskaya T V., et al. Decoding of exon splicing patterns in the human RUNX1-RUNX1T1 fusion gene. Int J Biochem Cell Biol [Internet]. 2015 Nov 9 [cited 2025 Oct 31];68:48–58. Available from: https://pubmed.ncbi.nlm.nih.gov/26320575/.
- Li X, Liu G. Acute Myeloid Leukemias with Variant RUNX1::RUNX1T1: Report of Three Cases. Hematopathology [Internet]. 2023 May 1 [cited 2025 Oct 31];8(1). Available from: http://journals.librarypublishing.arizona.edu/hemepath/article/id/5668/.
- Detection of inv(16) and t(16;16) by fluorescence in situ hybridization in acute myeloid leukemia M4Eo | Haematologica [Internet]. [cited 2025 Oct 31]. Available from: https://haematologica.org/article/view/1679.
- Mandoli A, Singh AA, Jansen PWTC, Wierenga ATJ, Riahi H, Franci G, et al. CBFB-MYH11/RUNX1 together with a compendium of hematopoietic regulators, chromatin modifiers and basal transcription factors occupies self-renewal genes in inv(16) acute myeloid leukemia. Leukemia [Internet]. 2014 [cited 2025 Oct 31];28(4):770–8. Available from: https://pubmed.ncbi.nlm.nih.gov/24002588/.
- Pulikkan JA, Castilla LH. Preleukemia and Leukemia-initiating cell activity in inv(16) acute myeloid Leukemia. Front Oncol [Internet]. 2018 Apr 26 [cited 2025 Oct 31];8(APR):324072. Available from: www.frontiersin.org.
- Hyde RK, Liu PP. RUNX1 repression-independent mechanisms of leukemogenesis by fusion genes CBFB-MYH11 and AML1-ETO (RUNX1-RUNX1T1). J Cell Biochem [Internet]. 2010 Aug 1 [cited 2025 Oct 31];110(5):1039–45. Available from: https://pubmed.ncbi.nlm.nih.gov/20589720/.
- Sinha C, Cunningham LC, Liu PP. Core Binding Factor Acute Myeloid Leukemia: New Prognostic Categories and Therapeutic Opportunities. Semin Hematol [Internet]. 2015 Jul 1 [cited 2025 Oct 31];52(3):215–22. Available from: https://pubmed.ncbi.nlm.nih.gov/26111469/.
- Döhner K, Döhner H. Molecular characterization of acute myeloid leukemia. Haematologica [Internet]. 2008 Jul 1 [cited 2025 Oct 31];93(7):976–82. Available from: https://haematologica.org/article/view/4939.
- Zhang W, Wang H, Zhang P, Li H, Ma X, Liu H. Rare type I CBFβ/MYH11 fusion transcript in primary acute myeloid leukemia with inv(16)(p13.1q22): a case report. Brazilian J Med Biol Res = Rev Bras Pesqui medicas e Biol [Internet]. 2021 [cited 2025 Oct 31];54(12). Available from: https://pubmed.ncbi.nlm.nih.gov/34730684/.
- Srinivasan S, Dhamne C, Patkar N, Chatterjee G, Moulik NR, Chichra A, et al. KIT exon 17 mutations are predictive of inferior outcome in pediatric acute myeloid leukemia with RUNX1::RUNX1T1. Pediatr Blood Cancer [Internet]. 2024 Feb 1 [cited 2025 Oct 31];71(2). Available from: https://pubmed.ncbi.nlm.nih.gov/38014874/.
- Yang M, Wang W, Zhang G, Qiu S, Liu B, Mi Y, et al. Clinical characteristics and therapeutic determinants of RUNX1::RUNX1T1 differ from those of CBFB::MYH11 acute myeloid leukemia. Haematologica [Internet]. 2025 Apr 30 [cited 2025 Oct 31];110(10):2281–92. Available from: https://haematologica.org/article/view/12048.
- Schuurhuis GJ, Heuser M, Freeman S, Buccisano F, Cloos J, Grimwade D, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. [cited 2025 Oct 31]; Available from: www.bloodjournal.org.
- Yu S, Yang S, Hu L, Duan W, Zhao T, Qin Y, et al. Genetic abnormalities predict outcomes in patients with core binding factor acute myeloid leukemia. Ann Hematol [Internet]. 2025 Feb 1 [cited 2025 Oct 31];104(2):997–1006. Available from: https://pubmed.ncbi.nlm.nih.gov/39966122/.
- Ayatollahi H, Shajiei A, Sadeghian MH, Sheikhi M, Yazdandoust E, Ghazanfarpour M, et al. Prognostic Importance of C-KIT Mutations in Core Binding Factor Acute Myeloid Leukemia: A Systematic Review. Hematol Oncol Stem Cell Ther [Internet]. 2017 Mar 1 [cited 2025 Nov 4];10(1):1–7. Available from: https://pubmed.ncbi.nlm.nih.gov/27613372/.
- Paschka P, Marcucci G, Ruppert AS, Mrózek K, Chen H, Kittles RA, et al. Adverse Prognostic Significance of KIT Mutations in Adult Acute Myeloid Leukemia With inv(16) and t(8;21): A Cancer and Leukemia Group B Study. J Clin Oncol [Internet]. 2006 Aug 20 [cited 2025 Nov 5];24(24):3904–11. Available from: https://ascopubs.org/doi/10.1200/JCO.2006.06.9500.
- Qin YZ, Jiang Q, Wang Y, Jiang H, Xu LP, Zhao XS, et al. The impact of the combination of KIT mutation and minimal residual disease on outcome in t(8;21) acute myeloid leukemia. Blood Cancer J [Internet]. 2021 Apr 1 [cited 2025 Nov 7];11(4):67. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC8016839/.
- Quan X, Deng J. Core binding factor acute myeloid leukemia: Advances in the heterogeneity of KIT, FLT3, and RAS mutations (Review). Mol Clin Oncol [Internet]. 2020 [cited 2025 Nov 13];13(2):95. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC7366242/.
- Ramirez RMC, Ramirez RMC, Pachas MT de JC, Dongo RJZ. Prevalence and Prognosis of Secondary Genetic Aberrations Among Patients With Core Binding Factor Acute Myeloid Leukemia: A Mitelman Database Analysis. World J Oncol [Internet]. 2023 Nov 17 [cited 2025 Nov 13];14(6):488–98. Available from: https://www.wjon.org/index.php/wjon/article/view/1661.
- Goemans BF, Zwaan CM, Miller M, Zimmermann M, Harlow A, Meshinchi S, et al. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia [Internet]. 2005 [cited 2025 Nov 5];19(9):1536–42. Available from: https://pubmed.ncbi.nlm.nih.gov/16015387/.
- Maiti A, Naqvi K, Kadia TM, Borthakur G, Takahashi K, Bose P, et al. Phase II Trial of MEK Inhibitor Binimetinib (MEK162) in RAS-mutant Acute Myeloid Leukemia. Clin Lymphoma, Myeloma Leuk [Internet]. 2019 Mar 1 [cited 2025 Nov 4];19(3):142-148.e1. Available from: https://pubmed.ncbi.nlm.nih.gov/30635233/.
- Jahn N, Agrawal M, Bullinger L, Weber D, Corbacioglu A, Gaidzik VI, et al. Incidence and Prognostic Relevance of ASXL2 Mutations in Adult CBF-AML with t(8;21)(q22;q22): A Study of the German-Austrian AML Study Group (AMLSG). Blood. 2015 Dec 3;126(23):3818–3818.
- Medina EA, Delma CR, Yang FC. ASXL1/2 mutations and myeloid malignancies. J Hematol Oncol [Internet]. 2022;15(1):1–18. Available from: . [CrossRef]
- Hartmann L, Dutta S, Opatz S, Vosberg S, Reiter K, Leubolt G, et al. ZBTB7A mutations in acute myeloid leukaemia with t(8;21) translocation. Nat Commun [Internet]. 2016 Jun 2 [cited 2025 Nov 8];7:11733. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC4895769/.
- Kawashima N, Akashi A, Nagata Y, Kihara R, Ishikawa Y, Asou N, et al. Clinical significance of ASXL2 and ZBTB7A mutations and C-terminally truncated RUNX1-RUNX1T1 expression in AML patients with t(8;21) enrolled in the JALSG AML201 study. Ann Hematol [Internet]. 2019 Jan 30 [cited 2025 Nov 8];98(1):83–91. Available from: https://pubmed.ncbi.nlm.nih.gov/30251205/.
- Li S, Li N, Chen Y, Zheng Z, Guo Y. FLT3-TKD in the prognosis of patients with acute myeloid leukemia: A meta-analysis. Front Oncol. 2023 Feb 17;13:1086846.
- Guan W, Zhou L, Li Y, Yang E, Liu Y, Lv N, et al. Profiling of somatic mutations and fusion genes in acute myeloid leukemia patients with FLT3-ITD or FLT3-TKD mutation at diagnosis reveals distinct evolutionary patterns. Exp Hematol Oncol [Internet]. 2021 Dec 1 [cited 2025 Nov 2];10(1). Available from: https://pubmed.ncbi.nlm.nih.gov/33836835/.
- Kayser S, Kramer M, Martínez-Cuadrón D, Grenet J, Metzeler KH, Sustkova Z, et al. Characteristics and outcome of patients with core-binding factor acute myeloid leukemia and FLT3-ITD: results from an international collaborative study. Haematologica [Internet]. 2022 Apr 1 [cited 2025 Nov 2];107(4):836–43. Available from: https://haematologica.org/article/view/haematol.2021.278645.
- Cortes JE, Khaled S, Martinelli G, Perl AE, Ganguly S, Russell N, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol [Internet]. 2019 Jul 1 [cited 2025 Nov 2];20(7):984–97. Available from: https://pubmed.ncbi.nlm.nih.gov/31175001/.
- Bazinet A, Bataller A, Kadia T, Daver N, Short NJ, Yilmaz M, et al. A retrospective study of outcomes across time and treatment regimens in newly diagnosed, FMS-like tyrosine kinase 3 (FLT3)-mutated acute myeloid leukemia. Cancer [Internet]. 2025 Mar 15 [cited 2025 Nov 2];131(6):e35813. Available from: /doi/pdf/10.1002/cncr.35813.
- Travaglini S, Gurnari C, Ottone T, Voso MT. Advances in the pathogenesis of FLT3 -mutated acute myeloid leukemia and targeted treatments. Curr Opin Oncol [Internet]. 2024 Nov 1 [cited 2025 Nov 6];36(6):569–76. Available from: https://journals.lww.com/co-oncology/fulltext/2024/11000/advances_in_the_pathogenesis_of_flt3_mutated_acute.15.aspx.
- Pommert L, Tarlock K. The evolution of targeted therapy in pediatric AML: gemtuzumab ozogamicin, FLT3/IDH/BCL2 inhibitors, and other therapies. Hematology [Internet]. 2022 Dec 9 [cited 2025 Nov 9];2022(1):603–10. Available from: https://dx.doi.org/10.1182/hematology.2022000358.
- Mrózek K, Bloomfield CD. Clinical Significance of the Most Common Chromosome Translocations in Adult Acute Myeloid Leukemia. J Natl Cancer Inst Monogr [Internet]. 2008 [cited 2025 Nov 4];(39). Available from: https://academic.oup.com/jncimono/article/2008/39/52/953332.
- Marcucci G, Geyer S, Laumann K, Zhao W, Bucci D, Uy GL, et al. Combination of dasatinib with chemotherapy in previously untreated core binding factor acute myeloid leukemia: CALGB 10801. Blood Adv [Internet]. 2020 Feb 25 [cited 2025 Nov 4];4(4):696. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC7042984/.
- Boissel N, Renneville A, Leguay T, Lefebvre PC, Recher C, Lecerf T, et al. Dasatinib in high-risk core binding factor acute myeloid leukemia in first complete remission: a French Acute Myeloid Leukemia Intergroup trial. Haematologica [Internet]. 2015 [cited 2025 Nov 4];100(6):780–5. Available from: https://pubmed.ncbi.nlm.nih.gov/25715404/.
- Hu Z, Tang X, Chen F, Li T, Liu Y, Zhou G, et al. Molecular genetics profiling of core-binding factor acute myeloid leukemia in pediatrics. Ther Adv Hematol [Internet]. 2025 Jan 1 [cited 2025 Nov 4];16. Available from: https://pubmed.ncbi.nlm.nih.gov/40290757/.
- Kantarjian HM, DiNardo CD, Kadia TM, Daver NG, Altman JK, Stein EM, et al. Acute myeloid leukemia management and research in 2025. CA Cancer J Clin [Internet]. 2025 Jan 1 [cited 2025 Nov 4];75(1):46–67. Available from: /doi/pdf/10.3322/caac.21873.
- (PDF) Rapid mutational analysis of N-ras proto-oncogene in hematologic malignancies: A study of 77 Greek patients [Internet]. [cited 2025 Nov 4]. Available from: https://www.researchgate.net/publication/11812910_Rapid_mutational_analysis_of_N-ras_proto-oncogene_in_hematologic_malignancies_A_study_of_77_Greek_patients.
- Alawieh D, Cysique-Foinlan L, Willekens C, Renneville A. RAS mutations in myeloid malignancies: revisiting old questions with novel insights and therapeutic perspectives. Blood Cancer J [Internet]. 2024 Dec 1 [cited 2025 Nov 4];14(1). Available from: https://pubmed.ncbi.nlm.nih.gov/38658558/.
- Desikan SP, Ravandi F, Pemmaraju N, Konopleva M, Loghavi S, Jabbour EJ, et al. A Phase II Study of Azacitidine, Venetoclax, and Trametinib in Relapsed or Refractory Acute Myeloid Leukemia Harboring RAS Pathway-Activating Mutations. Acta Haematol [Internet]. 2022 Sep 1 [cited 2025 Nov 4];145(5):529–36. Available from: https://pubmed.ncbi.nlm.nih.gov/35717939/.
- José-Enériz ES, Gimenez-Camino N, Agirre X, Prosper F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers (Basel) [Internet]. 2019 Nov 1 [cited 2025 Nov 6];11(11):1794. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC6896008/.
- Conneely SE, Quezada A, Kurtz KJ, Zhang N, Fuente JD La, Mercer N, et al. Cohesin haploinsufficiency is tolerated in Cbfb::MYH11-driven murine acute myeloid leukemia. Exp Hematol [Internet]. 2025 Oct [cited 2025 Nov 8];105287. Available from: https://pubmed.ncbi.nlm.nih.gov/41173205/.
- Martín-Izquierdo M, Abáigar M, Hernández-Sánchez JM, Tamborero D, López-Cadenas F, Ramos F, et al. Co-occurrence of cohesin complex and Ras signaling mutations during progression from myelodysplastic syndromes to secondary acute myeloid leukemia. Haematologica [Internet]. 2021 Aug 1 [cited 2025 Nov 8];106(8):2215–23. Available from: https://haematologica.org/article/view/9805.
- Tothova Z, Valton AL, Gorelov RA, Vallurupalli M, Krill-Burger JM, Holmes A, et al. Cohesin mutations alter DNA damage repair and chromatin structure and create therapeutic vulnerabilities in MDS/AML. JCI Insight [Internet]. 2021 Feb 8 [cited 2025 Nov 8];6(3). Available from: . [CrossRef]
- Redondo Monte E, Wilding A, Leubolt G, Kerbs P, Bagnoli JW, Hartmann L, et al. ZBTB7A prevents RUNX1-RUNX1T1-dependent clonal expansion of human hematopoietic stem and progenitor cells. Oncogene [Internet]. 2020 Apr 9 [cited 2025 Nov 8];39(15):3195. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC7142018/.
- Park SH, Lee HJ, Kim IS, Kang JE, Lee EY, Kim HJ, et al. Incidences and Prognostic Impact of c-KIT, WT1, CEBPA, and CBL Mutations, and Mutations Associated With Epigenetic Modification in Core Binding Factor Acute Myeloid Leukemia: A Multicenter Study in a Korean Population. Ann Lab Med [Internet]. 2015 May 1 [cited 2025 Nov 8];35(3):288–97. Available from: https://pubmed.ncbi.nlm.nih.gov/25932436/.
- Kreutmair S, Pfeifer D, Waterhouse M, Takács F, Graessel L, Döhner K, et al. First-in-human study of WT1 recombinant protein vaccination in elderly patients with AML in remission: a single-center experience. Cancer Immunol Immunother [Internet]. 2022 Dec 1 [cited 2025 Nov 8];71(12):2913–28. Available from: https://pubmed.ncbi.nlm.nih.gov/35476127/.
- Liu Yin JA, O’Brien MA, Hills RK, Daly SB, Wheatley K, Burnett AK. Minimal residual disease monitoring by quantitative RT-PCR in core binding factor AML allows risk stratification and predicts relapse: results of the United Kingdom MRC AML-15 trial. Blood [Internet]. 2012 Oct 4 [cited 2025 Nov 5];120(14):2826–35. Available from: https://dx.doi.org/10.1182/blood-2012-06-435669.
- Yoon JH, Kim H, Shin SH, Jeon YW, Kim JH, Lee SE, et al. Molecular and Cytogenetic Risk Stratification For Core-Binding Factor-Positive Adult AML With Analysis Of Post-Remission Treatment Outcomes Including Transplantation. Blood. 2013 Nov 15;122(21):1301–1301.
- Tarlock K, Gerbing RB, Ries RE, Smith JL, Leonti A, Huang BJ, et al. Prognostic impact of cooccurring mutations in FLT3-ITD pediatric acute myeloid leukemia. Blood Adv [Internet]. 2024 May 14 [cited 2025 Nov 7];8(9):2094–103. Available from: https://dx.doi.org/10.1182/bloodadvances.2023011980.
- Fan J, Gao L, Chen J, Hu S. Influence of KIT mutations on prognosis of pediatric patients with core-binding factor acute myeloid leukemia: a systematic review and meta-analysis. Transl Pediatr [Internet]. 2020 Dec 1 [cited 2025 Nov 4];9(6):72633–733. Available from: https://tp.amegroups.org/article/view/56363/html.
- Voso MT, Ottone T, Lavorgna S, Venditti A, Maurillo L, Lo-Coco F, et al. MRD in AML: The Role of New Techniques. Front Oncol [Internet]. 2019 Jul 23 [cited 2025 Nov 7];9:655. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC6664148/.
- Cilloni D, Renneville A, Hermitte F, Hills RK, Daly S, Jovanovic J V., et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: A European LeukemiaNet Study. J Clin Oncol [Internet]. 2009 Nov 1 [cited 2025 Nov 7];27(31):5195–201. Available from: https://ascopubs.org/doi/10.1200/JCO.2009.22.4865.
- Ivey A, Hills RK, Simpson MA, Jovanovic J V., Gilkes A, Grech A, et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N Engl J Med [Internet]. 2016 Feb 4 [cited 2025 Nov 7];374(5):422–33. Available from: https://www.nejm.org/doi/full/10.1056/NEJMoa1507471.
- Schlenk RF, Kayser S, Bullinger L, Kobbe G, Casper J, Ringhoffer M, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood [Internet]. 2014 Sep 30 [cited 2025 Nov 7];124(23):3441–9. Available from: https://pubmed.ncbi.nlm.nih.gov/25270908/.
- Li Y, Solis-Ruiz J, Yang F, Long N, Tong CH, Lacbawan FL, et al. NGS-defined measurable residual disease (MRD) after initial chemotherapy as a prognostic biomarker for acute myeloid leukemia. Blood Cancer J 2023 131 [Internet]. 2023 Apr 24 [cited 2025 Nov 7];13(1):1–9. Available from: https://www.nature.com/articles/s41408-023-00833-7.
- Svaton M, Skotnicova A, Reznickova L, Rennerova A, Valova T, Kotrova M, et al. NGS-Based MRD Quantitation: An Alternative to qPCR Validated on a Large Consecutive Cohort of Children with ALL. Blood. 2021 Nov 23;138(Supplement 1):1314.
- Yao Q, Bai Y, Kumar S, Au E, Orfao A, Chim CS. Minimal Residual Disease Detection by Next-Generation Sequencing in Multiple Myeloma: A Comparison With Real-Time Quantitative PCR. Front Oncol. 2021 Jan 29;10:611021.
- Puckrin R, Atenafu EG, Claudio JO, Chan S, Gupta V, Maze D, et al. Measurable residual disease monitoring provides insufficient lead-time to prevent morphological relapse in the majority of patients with core-binding factor acute myeloid leukemia. Haematologica [Internet]. 2020 Jan 1 [cited 2025 Nov 7];106(1):56. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC7776265/.
- Puckrin R, Atenafu EG, Claudio JO, Chan S, Gupta V, Maze D, et al. Measurable residual disease monitoring provides insufficient lead-time to prevent morphologic relapse in the majority of patients with core-binding factor acute myeloid leukemia. Haematologica [Internet]. 2021 Jan 1 [cited 2025 Nov 7];106(1):56–63. Available from: https://haematologica.org/article/view/9590.
- Heuser M, Freeman SD, Ossenkoppele GJ, Buccisano F, Hourigan CS, Ngai LL, et al. 2021 Update on MRD in acute myeloid leukemia: a consensus document from the European LeukemiaNet MRD Working Party. Blood [Internet]. 2021 Dec 30 [cited 2025 Nov 13];138(26):2753–67. Available from: https://dx.doi.org/10.1182/blood.2021013626.
- Onecha E, Linares M, Rapado I, Ruiz-Heredia Y, Martinez-Sanchez P, Cedena T, et al. A novel deep targeted sequencing method for minimal residual disease monitoring in acute myeloid leukemia. Haematologica [Internet]. 2018 Jan 31 [cited 2025 Nov 13];104(2):288. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC6355493/.
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med [Internet]. 2016 Jun 9 [cited 2025 Nov 5];374(23):2209–21. Available from: https://www.nejm.org/doi/full/10.1056/NEJMoa1516192.
- Cioccio J, Claxton D. Therapy of acute myeloid leukemia: therapeutic targeting of tyrosine kinases. Expert Opin Investig Drugs [Internet]. 2019 Apr 3 [cited 2025 Nov 5];28(4):337–49. Available from: https://pubmed.ncbi.nlm.nih.gov/30775933/.
- Arock M, Wedeh G, Hoermann G, Bibi S, Akin C, Peter B, et al. Preclinical human models and emerging therapeutics for advanced systemic mastocytosis. Haematologica [Internet]. 2018 Oct 31 [cited 2025 Nov 5];103(11):1760. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC6278969/.
- Yokoyama A, Somervaille TCP, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell [Internet]. 2005 Oct 21 [cited 2025 Nov 5];123(2):207–18. Available from: https://pubmed.ncbi.nlm.nih.gov/16239140/.
- Krivtsov A V., Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell [Internet]. 2019 Dec 9 [cited 2025 Nov 5];36(6):660-673.e11. Available from: https://pubmed.ncbi.nlm.nih.gov/31821784/.
- Jurcic JG. What happened to anti-CD33 therapy for acute myeloid Leukemia? Curr Hematol Malig Rep [Internet]. 2012 Mar 23 [cited 2025 Nov 5];7(1):65–73. Available from: https://link.springer.com/article/10.1007/s11899-011-0103-0.
- Swaminathan M, Cortes JE. Update on the role of gemtuzumab-ozogamicin in the treatment of acute myeloid leukemia. Ther Adv Hematol [Internet]. 2023 Jan 1 [cited 2025 Nov 5];14. Available from: https://journals.sagepub.com/doi/full/10.1177/20406207231154708.
- Ronnacker J, Muller PJ, Mikesch JH, Zukunft S, Weinbergerová B, Šrámek J, et al. Gemtuzumab ozogamicin in first-line treatment of CBF-AML: insights from a retrospective multi-center analysis. Leuk 2025 399 [Internet]. 2025 Jul 21 [cited 2025 Nov 5];39(9):2174–80. Available from: https://www.nature.com/articles/s41375-025-02700-9.
- Khan N, Hills RK, Virgo P, Couzens S, Clark N, Gilkes A, et al. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leuk 2017 315 [Internet]. 2016 Oct 31 [cited 2025 Nov 5];31(5):1059–68. Available from: https://www.nature.com/articles/leu2016309.
- Hills RK, Castaigne S, Appelbaum FR, Delaunay J, Petersdorf S, Othus M, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol [Internet]. 2014 Aug 1 [cited 2025 Nov 5];15(9):986–96. Available from: https://www.thelancet.com/action/showFullText?pii=S1470204514702815.
- Castaigne S, Pautas C, Terré C, Raffoux E, Bordessoule D, Bastie JN, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet [Internet]. 2012 Apr 21 [cited 2025 Nov 5];379(9825):1508–16. Available from: https://www.thelancet.com/action/showFullText?pii=S0140673612604851.
- Amadori S, Venditti A, Voso MT, Annino L, De Fabritiis P, Alimena G, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: Results of the randomized phase III EORTC-GIMEMA AML-19 Trial. J Clin Oncol [Internet]. 2016 Mar 20 [cited 2025 Nov 5];34(9):972–9. Available from: https://ascopubs.org/doi/10.1200/JCO.2015.64.0060.
- Kiyoi H. FLT3 INHIBITORS: RECENT ADVANCES AND PROBLEMS FOR CLINICAL APPLICATION. Nagoya J Med Sci [Internet]. 2015 [cited 2025 Nov 5];77(1–2):7. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC4361503/.
- Shah V, Shah A, Jain P, Panchal V, Fernandes BCA, Gangasani S, et al. AML-1203: Efficacy of Gilteritinib Versus Salvage Chemotherapy in Relapsed or Refractory FLT3-Mutated Acute Myeloid Leukemia: A Meta-Analysis. Clin Lymphoma Myeloma Leuk. 2025 Sep 1;25:S492–3.
- Pratz KW, Cherry M, Altman JK, Cooper BW, Podoltsev NA, Cruz JC, et al. Gilteritinib in Combination with Induction and Consolidation Chemotherapy and as Maintenance Therapy: A Phase IB Study in Patients with Newly Diagnosed AML. J Clin Oncol [Internet]. 2023 Sep 10 [cited 2025 Nov 6];41(26):4236–46. Available from: https://ascopubs.org/doi/10.1200/JCO.22.02721.
- Wang ES, Goldberg AD, Tallman M, Walter RB, Karanes C, Sandhu K, et al. Crenolanib and Intensive Chemotherapy in Adults With Newly Diagnosed FLT3-Mutated AML. J Clin Oncol [Internet]. 2024 May 20 [cited 2025 Nov 6];42(15):1776–87. Available from: https://ascopubs.org/doi/pdf/10.1200/JCO.23.01061.
- Garciaz S, Hospital MA. FMS-Like Tyrosine Kinase 3 Inhibitors in the Treatment of Acute Myeloid Leukemia: An Update on the Emerging Evidence and Safety Profile. Onco Targets Ther [Internet]. 2023 [cited 2025 Nov 6];16:31. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC9869913/.
- Bazarbachi A, Labopin M, Battipaglia G, Djabali A, Passweg J, Socié G, et al. Sorafenib improves survival of FLT3-mutated acute myeloid leukemia in relapse after allogeneic stem cell transplantation: a report of the EBMT Acute Leukemia Working Party. Haematologica [Internet]. 2019 Sep 1 [cited 2025 Nov 6];104(9):e398–401. Available from: https://haematologica.org/article/view/9059.
- Morin S, Giannotti F, Mamez AC, Pradier A, Masouridi-Levrat S, Simonetta F, et al. Real-world experience of sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for FLT3-ITD AML reveals high rates of toxicity-related treatment interruption. Front Oncol [Internet]. 2023 [cited 2025 Nov 6];13:1095870. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC10050716/.
- Yamaura T, Nakatani T, Uda K, Ogura H, Shin W, Kurokawa N, et al. A novel irreversible FLT3 inhibitor, FF-10101, shows excellent efficacy against AML cells with FLT3 mutations. Blood [Internet]. 2018 Jan 25 [cited 2025 Nov 6];131(4):426–38. Available from: https://dx.doi.org/10.1182/blood-2017-05-786657.
- Ferng TT, Terada D, Ando M, Tarver TC, Chaudhary F, Lin KC, et al. The irreversible FLT3 inhibitor FF-10101 is active against a diversity of FLT3 inhibitor resistance mechanisms. Mol Cancer Ther [Internet]. 2022 May 1 [cited 2025 Nov 6];21(5):844. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC9081245/.
- Beghini A, Beghini A. Core Binding Factor Leukemia: Chromatin Remodeling Moves Towards Oncogenic Transcription. Cancers 2019, Vol 11, [Internet]. 2019 Dec 7 [cited 2025 Nov 6];11(12). Available from: https://www.mdpi.com/2072-6694/11/12/1973.
- Zhang J, Gao X, Yu L. Roles of Histone Deacetylases in Acute Myeloid Leukemia With Fusion Proteins. Front Oncol [Internet]. 2021 Sep 1 [cited 2025 Nov 6];11:741746. Available from: www.frontiersin.org.
- Yu X, Li H, Hu P, Qing Y, Wang X, Zhu M, et al. Natural HDAC-1/8 inhibitor baicalein exerts therapeutic effect in CBF-AML. Clin Transl Med [Internet]. 2020 Aug 1 [cited 2025 Nov 6];10(4):e154. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/ctm2.154.
- Kuykendall A, Duployez N, Boissel N, Lancet JE, Welch JS. Acute Myeloid Leukemia: The Good, the Bad, and the Ugly. Am Soc Clin Oncol Educ B [Internet]. 2018 May [cited 2025 Nov 6];(38):555–73. Available from: https://ascopubs.org/doi/10.1200/EDBK_199519.
- Vanegas YM, Badar T. Clinical Utility of Azacitidine in the Management of Acute Myeloid Leukemia: Update on Patient Selection and Reported Outcomes. Cancer Manag Res [Internet]. 2022 Dec 1 [cited 2025 Nov 6];14:3527. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC9793740/.
- Gabellier L, Peterlin P, Thepot S, Hicheri Y, Paul F, Gallego-Hernanz MP, et al. Hypomethylating agent monotherapy in core binding factor acute myeloid leukemia: a French multicentric retrospective study. Ann Hematol [Internet]. 2024 Mar 1 [cited 2025 Nov 6];103(3):759. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC10867066/.
- Zhang K, Zhang X, Xu Y, Xue S, Qiu H, Tang X, et al. Efficacy of venetoclax combined with hypomethylating agents in young, and unfit patients with newly diagnosed core binding factor acute myeloid leukemia. Blood Cancer J 2023 131 [Internet]. 2023 Oct 11 [cited 2025 Nov 6];13(1):1–4. Available from: https://www.nature.com/articles/s41408-023-00928-1.
- Santini V, Lübbert M, Wierzbowska A, Ossenkoppele GJ. The Clinical Value of Decitabine Monotherapy in Patients with Acute Myeloid Leukemia. Adv Ther [Internet]. 2021 Apr 1 [cited 2025 Nov 6];39(4):1474. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC8989816/.
- Bhattacharya S, Banerjee A, Dolai TK, Sadhukhan SK, Med IJ, Oncol P. Role of Maintenance Therapy with Decitabine in Acute Myeloid Leukemia in First Remission: A Prospective Interventional Study from a Tertiary Care Center in Eastern India. 2025 [cited 2025 Nov 6]; Available from: .
- Arslan S, Zhang J, Dhakal P, Moran JA, Naidoo N, Lombardi Story J, et al. Outcomes of Therapy with Venetoclax Combined with Hypomethylating Agents in Favorable-Risk Acute Myeloid Leukemia (AML). Blood [Internet]. 2020 Nov 5 [cited 2025 Nov 6];136(Supplement 1):41–2. Available from: https://dx.doi.org/10.1182/blood-2020-142780.
- He S, Li Y, Shi X, Wang L, Cai D, Zhou J, et al. DNA methylation landscape reveals LIN7A as a decitabine-responsive marker in patients with t(8;21) acute myeloid leukemia. Clin Epigenetics [Internet]. 2023 Dec 1 [cited 2025 Nov 6];15(1):37. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC9983225/.
- Imai Y, Maru Y, Tanaka J. Action mechanisms of histone deacetylase inhibitors in the treatment of hematological malignancies. Cancer Sci [Internet]. 2016 Nov 1 [cited 2025 Nov 6];107(11):1543. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC5132279/.
- Salmon JM, Bots M, Vidacs E, Stanley KL, Atadja P, Zuber J, et al. Combining the differentiating effect of panobinostat with the apoptotic effect of arsenic trioxide leads to significant survival benefit in a model of t(8;21) acute myeloid leukemia. Clin Epigenetics [Internet]. 2015 Jan 22 [cited 2025 Nov 7];7(1):2. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC4308003/.
- Bots M, Verbrugge I, Martin BP, Salmon JM, Ghisi M, Baker A, et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood [Internet]. 2014 Feb 27 [cited 2025 Nov 7];123(9):1341. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC3938147/.
- Garcia-Manero G, Podoltsev NA, Othus M, Pagel JM, Radich JP, Fang M, et al. Standard versus high-dose cytarabine with or without vorinostat for AML. Leukemia [Internet]. 2023 [cited 2025 Nov 7];38(1):58. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC11729399/.
- Silva G, Cardoso BA, Belo H, Almeida AM. Vorinostat Induces Apoptosis and Differentiation in Myeloid Malignancies: Genetic and Molecular Mechanisms. PLoS One [Internet]. 2013 Jan 8 [cited 2025 Nov 7];8(1):e53766. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC3540071/.
- Collados-Ros A, Muro M, Legaz I. Gemtuzumab Ozogamicin in Acute Myeloid Leukemia: Efficacy, Toxicity, and Resistance Mechanisms—A Systematic Review. Biomedicines [Internet]. 2024 Jan 1 [cited 2025 Nov 13];12(1):208. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC10813452/.
- Travaglini S, Gurnari C, Ottone T, Voso MT. Advances in the pathogenesis of FLT3 -mutated acute myeloid leukemia and targeted treatments. Curr Opin Oncol [Internet]. 2024 Nov 1 [cited 2025 Nov 13];36(6):569–76. Available from: https://journals.lww.com/co-oncology/fulltext/2024/11000/advances_in_the_pathogenesis_of_flt3_mutated_acute.15.aspx.
- José-Enériz ES, Gimenez-Camino N, Agirre X, Prosper F, José-Enériz ES, Gimenez-Camino N, et al. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, Vol 11, [Internet]. 2019 Nov 14 [cited 2025 Nov 13];11(11). Available from: https://www.mdpi.com/2072-6694/11/11/1794.
- Wang T, Chen S, Chen J, Liu T, Zhang T, Qiu H, et al. Allogeneic Hematopoietic Stem Cell Transplantation Improved Survival for Adult Core Binding Factor Acute Myelogenous Leukemia Patients with Intermediate- and Adverse-Risk Genetics in the 2017 European LeukemiaNet. Transplant Cell Ther [Internet]. 2021 Feb 1 [cited 2025 Nov 7];27(2):173.e1-173.e9. Available from: https://pubmed.ncbi.nlm.nih.gov/33830030/.
- Halaburda K, Labopin M, Mailhol A, Socié G, Craddock C, Aljurf M, et al. Allogeneic stem cell transplantation in second complete remission for core binding factor acute myeloid leukemia: a study from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica [Internet]. 2020 Jun 1 [cited 2025 Nov 7];105(6):1723–30. Available from: https://haematologica.org/article/view/9449.
- Kawashima N, Ishikawa Y, Atsuta Y, Sugiura I, Sawa M, Dobashi N, et al. Prospective Evaluation of Prognostic Relevance of KIT Mutations in Core-Binding Factor Acute Myeloid Leukemia: Results from the JALSG CBF-AML209-KIT Study. Blood [Internet]. 2018 Nov 29 [cited 2025 Nov 7];132(Supplement 1):438–438. Available from: https://dx.doi.org/10.1182/blood-2018-99-113931.
- Duan W, Liu X, JIA J, Wang J, Gong L, Jiang Q, et al. Both the Subtypes of Kit Mutation and Minimal Residual Disease Are Associated with Prognosis in Core Banding Factor Acute Myeloid Leukemia. Blood [Internet]. 2020 Nov 5 [cited 2025 Nov 7];136(Supplement 1):4–5. Available from: https://dx.doi.org/10.1182/blood-2020-141905.
- Jahn N, Terzer T, Sträng E, Dolnik A, Cocciardi S, Panina E, et al. Genomic heterogeneity in core-binding factor acute myeloid leukemia and its clinical implication. Blood Adv [Internet]. 2020 Dec 22 [cited 2025 Nov 2];4(24):6342. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC7757000/.
- Quan X, Deng J. Core binding factor acute myeloid leukemia: Advances in the heterogeneity of KIT, FLT3, and RAS mutations (Review). Mol Clin Oncol [Internet]. 2020 [cited 2025 Nov 2];13(2):95. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC7366242/.
- Cornelissen JJ, Blaise D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood [Internet]. 2016 Jan 7 [cited 2025 Nov 7];127(1):62–70. Available from: https://pubmed.ncbi.nlm.nih.gov/26660427/.
- Al Hamed R, Labopin M, Wu D, Gedde-Dahl T, Aljurf M, Forcade E, et al. Allogeneic stem cell transplantation in de novo core-binding factor acute myeloid leukemia in first complete remission: data from the EBMT. Bone Marrow Transplant [Internet]. 2024 Oct 1 [cited 2025 Nov 7];59(10):1458–65. Available from: https://pubmed.ncbi.nlm.nih.gov/39095548/.

Table 1.
Key Secondary Mutations and Prognostic Implications in Core Binding Factor AML.
| Gene/Mutation Class | Prevalence (Overall CBF-AML) | Prognostic Impact in t(8;21) (RUNX1::RUNX1T1) | Prognostic Impact in inv(16) (CBFB::MYH11) | Key References |
| KIT (Exon 17 D816/N822) | 20%–45% (Higher in t(8;21)) | Highly Adverse (Increased relapse risk, shorter EFS/OS). Guide for Allo-HSCT in CR1. | Less Consistent/Less Significant Adverse Impact | [34,38,39,40] |
| FLT3-ITD/TKD | Detected in a subset; 4%-11% (Less common than NK-AML) | Adverse (Independent factor for poorer survival/relapse) | Adverse (Predictive of short progression-free survival (PFS)) | [41] |
| RAS (NRAS/KRAS) | Frequent (Overall 15-20%); Highest in inv(16) (up to 36%) | Prognosis varies; potentially favorable when isolated (no RTK mutations) | Higher prevalence; outcome influenced by co-mutations/allelic ratio | [42,43,44] |
| ASXL2 | Frequent in t(8;21) (22%-23%) | No statistically significant independent adverse prognosis demonstrated to date | Less Common; Role not defined | [45,46] |
| ZBTB7A | Primarily in t(8;21) (up to 23%) | Implicated in leukemogenesis; clinical utility/prognosis not established | Rare/Absent | [47,48] |
Table 2.
Comparison of Molecular Methods for Minimal Residual Disease (MRD) Monitoring in CBF-AML.
| Methodology | Primary Target(s) | Typical Sensitivity (LOD) | Key Advantages | Limitations in CBF-AML | References |
| RT-qPCR (Real-Time Quantitative PCR) | Fusion transcripts (RUNX1::RUNX1T1, CBFB::MYH11) | High (10-4 to 10-6) | Highly specific, standardized, and cost-effective; established prognostic marker. | Limited to tracking only the fusion transcript; fails to capture clonal evolution of secondary mutations. | [71,84] |
| NGS (Next-Generation Sequencing) | Co-occurring somatic mutations (e.g., KIT, FLT3, RAS, cohesin) & Fusion transcripts (DNA-based) | Moderate to High (10-4 to 10-5 for SNVs/Indels) | Comprehensive monitoring of multiple mutations and clonal evolution; detects preleukemic persistence. | Lower sensitivity than optimized qPCR for fusion transcripts; lack of standardized thresholds; complex bioinformatics. | [85] |
| Flow Cytometry (MFC) (Multi-parameter Flow Cytometry) | Aberrant immunophenotypes (e.g., CD19 co-expression in t(8;21)) | Moderate 10-3 to 10-4) | Rapid, can be applied to many AML subtypes, widely available. | Requires sufficient aberrant markers; interpretation can be challenging in monocytic subtypes (inv(16) M4Eo). | [85] |
Table 3.
Summary of Key Targeted and Epigenetic Therapeutic Strategies for Core Binding Factor AML.
| Agent Class (Example Agent) | Mechanism of Action | Relevance/Role in CBF-AML | Clinical Status/Key Findings | Key References |
| Anti-CD33 ADC (Gemtuzumab Ozogamicin - GO) | Monoclonal antibody-drug conjugate; targeted delivery of calicheamicin toxin to CD33+ cells upon internalization. | Frontline therapy; high CD33 expression and low Multi-Drug Resistance (MDR) in CBF-AML. | Significantly improves OS/EFS, reducing relapse; established standard of care in favorable-risk AML. | [123] |
| FLT3 Inhibitors (Midostaurin, Gilteritinib) | Blocks ligand-independent activation of FLT3 Receptor Tyrosine Kinase (RTK). | Targets adverse FLT3-ITD/TKD mutations associated with poor outcomes. | Used widely in FLT3-mutated AML; specific benefit in CBF-AML requires focused investigation due to mixed results. | [51,98,100,124] |
| KIT Inhibitors (Dasatinib) | Blocks KIT RTK constitutive signaling. | Targets adverse KIT mutations, particularly in the t(8;21) subtype. | Phase III studies failed to show clear EFS/OS benefit; therapeutic challenge remains despite prognostic significance. | [19,57,87] |
| DNMT Inhibitors (Azacitidine, Decitabine) | Restores gene expression by inhibiting DNA Methyltransferases (e.g., LIN7A reactivation). | Used in unfit/older patients and maintenance therapy. Potential for synergy (e.g., with Venetoclax). | AZA/VEN response superior in inv(16) vs. t(8;21); Decitabine promising for maintenance, guided by molecular markers. | [113,114,117] |
| HDAC Inhibitors (Panobinostat, Vorinostat) | Targets Histone Deacetylases, restoring acetylation and gene expression; promotes fusion protein degradation. | Potential for combination synergy with IC or DNMTi; robust preclinical activity in t(8;21) models. | Limited clinical monotherapy efficacy; require randomized trials in combination regimens tailored to specific CBF subtypes. | [118,120,122,125] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



