
Submitted:
27 January 2026
Posted:
29 January 2026
You are already at the latest version
Abstract
Key mechanisms underlying immune thrombocytopenia (ITP) pathophysiology include impaired platelet production and macrophage-mediated platelet destruction, the latter of which is the disease driver in more than half of patients. Traditional sequential treatment approaches achieve suboptimal responses in many patients. This review summarizes ITP pathogenesis and the treatment landscape and proposes a personalized treatment approach for ITP based on underlying disease mechanisms to optimize care with immunomodulatory and bone marrow-supportive therapies (fostamatinib, rituximab, and thrombopoietin receptor agonists [TPO-RAs]). Clinical evidence of monotherapy and real-world studies of combination therapy are reviewed to support mechanism-based treatment selection, focusing on the complementary actions of fostamatinib (to target platelet destruction) and TPO-RAs (to stimulate platelet production). In prior studies, fostamatinib with or without TPO-RAs demonstrated durable platelet responses and manageable safety as second-line or later ITP treatment. The proposed treatment framework augments guidelines by recommending fostamatinib, rituximab, or TPO-RAs as second-line therapy options based on patient-specific disease characteristics and risks. Patients with inadequate response to fostamatinib or TPO-RA monotherapy may combine these therapies to address both platelet destruction and platelet production deficits. This novel mechanistic framework enables optimization of safety and efficacy based on patient-specific pathophysiology to support individualized care.
Keywords:
immune thrombocytopenia
; fostamatinib
; thrombopoietin receptor agonists
; clinical management
Introduction
Immune thrombocytopenia (ITP) is an acquired autoimmune disease characterized by a decreased platelet count (<100×109/L) that leads to increased bruising and variable bleeding.[1,2,3] ITP has an incidence of ~2 to 4 per 100,000 people per year, affecting both adults and children, and it is typically more common in adult women than men prior to age 70 years.[1,4,5] Among newly diagnosed patients, 80% have primary ITP (characterized by isolated autoimmune thrombocytopenia), and 60%-70% develop persistent or chronic ITP.[3,6] Secondary ITP is triggered by or associated with another disease or condition or by certain drugs; it may respond to treatment of the underlying disease.[6,7]
The major goals of ITP treatment are to ensure a safe platelet count, minimize significant bleeding, and optimize quality of life.[1,8,9] Although 60%-80% of patients respond to established therapies, over 50% of patients fail to maintain a sustained response, and many exhibit consistently low platelet counts in spite of treatment.[9,10] There continues to be a need for long-term effective and tolerable treatment approaches, and clinicians may benefit from expanded knowledge of treatments and how to use them most effectively in clinical practice.[9,11]
The objective of this review is to propose a unique, personalized treatment approach for ITP that focuses on underlying disease mechanisms using immunomodulatory and bone marrow-supportive therapies (fostamatinib and thrombopoietin receptor agonists [TPO-RAs]), consistent with the authors’ clinical practice; background information applicable to ITP patient management is also summarized for the purpose of aiding clinicians in optimizing care. Clarifying the role of fostamatinib in early management of ITP alongside TPO-RAs and rituximab is necessary to address unmet needs that remain when following existing treatment guidelines; the relevance of this approach in the context of personalized medicine is based on data demonstrating the efficacy and safety of fostamatinib.
Pathogenesis of Immune Thrombocytopenia
Primary ITP is characterized by complex mechanisms that involve both increased platelet destruction and impaired platelet production (Figure 1).[6,12,13]
Peripheral destruction of platelets is primarily driven by autoantibodies that target platelet surface glycoproteins such as GPIIb/IIIa and GPIb/IX.[14,15] These autoantibody-coated platelets are recognized and cleared by macrophages in the spleen and liver through Fcγ receptor-mediated phagocytosis.[15,16,17] In addition to antibody-mediated mechanisms, cytotoxic T lymphocytes can cause platelet lysis, further contributing to destruction.[15,16] Apoptosis of platelets, characterized by increased phosphatidylserine exposure and caspase activation, also plays a role in accelerating the platelet clearance observed in ITP.[14] This enhanced peripheral destruction is a key factor in patients with ITP.
Complement activation is another potential contributor to platelet destruction in ITP.[18] Autoantibodies fix complement on the surface of platelets, leading to enhanced clearance via complement receptors on macrophages and through direct complement-mediated cytolysis.[18] Studies have shown that plasma from patients with ITP exhibits increased complement activation capacity, which correlates with lower platelet counts and a reduced immature platelet fraction.[18] Complement fixation both promotes the removal of opsonized platelets and damages megakaryocytes, subsequently impacting platelet production.[18] The role of the complement system in ITP pathogenesis has therapeutic implications, as agents targeting complement components may offer promise as potential treatments.
Impaired platelet production is a significant factor in ITP pathophysiology. Autoantibodies target megakaryocytes, leading to dysfunction or apoptosis.[14] Morphological studies have demonstrated megakaryocyte damage and reduced platelet production in a majority of patients with ITP.[14] Additionally, T cell-mediated cytotoxicity against megakaryocytes and inhibitory cytokines further suppress thrombopoiesis.[15,19] The clinical relevance of impaired platelet production is supported by the efficacy of TPO-RAs, which stimulate megakaryocyte proliferation and platelet production in patients with ITP.[15,19]
Prothrombotic Characteristics of Immune Thrombocytopenia
With an incidence twice that of population-based controls, thrombotic events such as venous and arterial thromboses affect up to 8% of patients with ITP.[20,21,22,23,24,25] In these patients, ITP itself, underlying comorbidities, or ITP therapies may be the cause of thrombosis.[22] In ITP, preactivated platelets and elevated levels of thrombin, factor VIII, and von Willebrand factor combine to generate a hypercoagulable state, putting patients at risk of thrombosis.[26] In addition, microparticles composed of small surface membrane vesicles and proinflammatory cytokines contribute to the procoagulant environment.[21,23,24,27] Prothrombotic comorbidities include coronary artery disease, hypertension, diabetes, dyslipidaemia, cancer, and atrial fibrillation.[22,28] The impact of associated antiphospholipid antibodies should also be considered, as 25%-75% of patients with ITP are antiphospholipid antibody-positive; these antibodies further activate complement, causing an increased degree of platelet aggregation, thus enhancing the risk of thrombosis.[21,27,29,30,31] Some ITP treatments have been implicated in the elevated risk of thrombosis, such as TPO-RAs, corticosteroids, and splenectomy.[22,28]
Management of Immune Thrombocytopenia
Overview of Treatment Guidelines
Various guidelines and expert opinion reviews have been published that provide recommendations for the diagnosis and management of ITP, specifically, who to treat, the goals of treatment, and patient management by the International Consensus Report (ICR), the American Society of Hematology (ASH), the Spanish ITP Working Group (GEPTI), and others.[1,5,7,32,33]
Diagnosis of ITP is one of exclusion and is based on a thorough evaluation of the patient, which should include patient and family history, physical examination, laboratory investigations (complete blood count and reticulocyte count, peripheral blood film, quantitative immunoglobulin level measurement, blood group [Rh]), and viral testing (HIV, hepatitis C virus, and hepatitis B virus).[7] Asymptomatic patients who have mild (defined as a platelet count ≥100 to <150 × 109/L) or moderate (defined as a platelet count ≥50 to <100 × 109/L) thrombocytopenia should undergo observation, and patients with a higher risk of bleeding or a platelet count <30 × 109/L should receive individualized treatment; however, the platelet level necessitating treatment may vary for certain patient subpopulations, such as those who are pregnant, those who receive antiplatelet or anticoagulant drugs, or those who participate in activities with a risk of severe trauma.[7,32,34,35,36]
The goals of treatment are to manage bleeding, maintain a safe platelet count (eg, ≥20-30 × 109/L), improve health-related quality of life (HRQoL), and minimize treatment toxicity.[1,7] Treatment should be individualized to each patient, taking into consideration factors such as patient age, the degree of bleeding and comorbidities that may increase the risk of bleeding, concomitant medications, risk of adverse events, current quality of life, and patient expectations.[7,32]
Initial treatment for newly diagnosed patients typically involves corticosteroids (prednisone, dexamethasone, methylprednisolone); if a patient has active bleeding, is at high risk for bleeding, is scheduled for surgery, is contraindicated for high-dose corticosteroids, or has not responded to corticosteroids, then intravenous immunoglobin (IVIg) or IV anti-D therapy is recommended.[7] The second-line treatments that are recommended include TPO-RAs (eltrombopag, avatrombopag, or romiplostim), rituximab, and fostamatinib.[5,7,32]
Medical therapies that are suggested for subsequent treatment include immunosuppressive agents such as mycophenolate mofetil (MMF), cyclosporine A, azathioprine, danazol, and dapsone.[7] For refractory patients who have failed multiple treatments, no definitive recommendations have been made, but several options have been suggested.[5,7] Combination therapy can be considered; ideally, drugs with different mechanisms of action (MoAs) should be chosen (see Combination Therapy With Fostamatinib and Thrombopoietin Receptor Agonists).[5] Switching from one TPO-RA to another one, use of splenectomy, or hematopoietic stem cell transplantation (HSCT) in rare cases may also be considered.[7]
Treatment Landscape Overview
First-Line Treatment Options: Corticosteroids, Intravenous Immunoglobulin, and Anti-D Therapy
The standard of care for first-line treatment is a short course (6-8 weeks maximum) of corticosteroids.[7,32] Corticosteroids exert their beneficial effects by decreasing platelet clearance and increasing production of platelets; their direct effect on blood vessels may reduce bleeding as well.[7,37,38] In addition, corticosteroids broadly act on immune cells such as macrophages, B cells, and T cells to inhibit destruction of platelets.[13] Although 60%-80% of patients with ITP initially respond to the treatment, the adverse events of corticosteroids (eg, weight gain, insomnia, acne, mood changes, risk of fracture, GI symptoms [gastric irritation or ulcer formation], and glucose intolerance) make them unsuitable for long-term use.[6,32] The fact that some patients do not respond to treatment likely reflects the heterogeneity of ITP pathophysiology described above.[14] Drug-drug interactions (DDIs) with corticosteroids can occur spanning multiple therapeutic classes, which include medications such as antibiotics, HMG-CoA reductase inhibitors, anti-inflammatories, antiretrovirals, antifungals, alkylating agents, kinase inhibitors, and anticonvulsants.[39,40] The broad interaction profile reflects the complex effects on hepatic enzyme systems by corticosteroids and the influence on drug metabolism pathways.[39,40]
IVIg, another recommended first-line therapy, was approved in the U.S. in 1980 and was initially intended for treatment of primary immunodeficiency diseases.[41] IVIg reduces platelet destruction via inhibition of splenic macrophages and also enhances the clearance of antiplatelet antibodies through saturation of neonatal fragment crystallizable receptors (FcRn).[6] The majority of patients (80%) experience a transient response lasting approximately 1-4 weeks.[6] IVIg is often used as a bridging therapy to maintain a stable platelet count prior to initiation of a more long-term treatment.[42] The most common adverse events with IVIg are associated with infusion-related reactions and include fever, chills, fatigue, nausea and vomiting, muscle and joint pain, flushing, and rash; aseptic meningitis, oedema, renal failure, and thrombosis have also been reported.[5,42] DDIs may occur with concurrent administration of biologic therapies due to competitive FcRn binding, which reduces exposure to the biologic.[43]
Intravenous anti-D treatment was approved by the US Food and Drug Administration (FDA) in 1995 for treatment of ITP in Rh-positive, non-splenectomised patients as an alternative first-line treatment.[44] Like IVIg, it may also be used for patients with bleeding, those who are at high risk of bleeding, those who have surgery scheduled, or those who do not respond to corticosteroids.[7] The overall response rate is 65% and lasts a median 25 days; however, adverse events reported for this therapy are serious (multisystem organ failure, acute respiratory distress, renal failure, disseminated intravascular coagulation) and warrant caution when prescribing the treatment for patients with active autoimmune haemolysis or anaemia, as intravascular haemolysis and severe anaemia have been reported.[7,45]
Recommended Treatments for Second-Line and Later Settings
TPO-RAs, fostamatinib, and rituximab are recommended as second-line treatment options (Table 1).[1,5,7,32] The TPO-RAs consist of small molecules (avatrombopag, eltrombopag) and a peptide (romiplostim) that stimulate platelet production by activating the Janus kinase-signal transducer and activator of transcription 5 (JAK/STAT5) pathway in megakaryocytes.[46] Fostamatinib prevents phagocytosis of platelets by spleen macrophages and may also inhibit activation of antibody-generating B cells that contribute to platelet depletion.[6] Rituximab is not approved to treat ITP but is recommended for ITP in the second-line setting.[5,7] It is a monoclonal antibody that targets CD20 on B cells, triggering apoptosis of lymphocytes through two pathways: antibody-dependent cell-mediated cytotoxicity or cell lysis mediated by complement.[6] These treatments have demonstrated platelet responses in more than half of patients with ITP in second-line or later settings, and come with distinct safety considerations and drug interactions (Table 1).
Upon second-line treatment failure, recommended third-line therapies include a number of immunosuppressive agents.[5,7] Immunosuppressant drugs such as azathioprine, MMF, cyclophosphamide pulse therapy, danazol, and dapsone inhibit T and B cells, but to date, there is a lack of clinical data supporting their use for ITP treatment.[6,7,47] Response rates vary depending on the therapy used, but they have been reported to generally range from 30% to 60% (for azathioprine, MMF, danazol, and dapsone; 85% for cyclophosphamide pulse therapy), with responses achieved beginning at 1 week up to 6 months.[6,9] Adverse events of azathioprine include weakness, sweating, neutropenia, increased liver function, and increased risk of cancer; for MMF, headache, GI symptoms, fungal skin infections, and increased risk of cancer; for cyclophosphamide pulse therapy, neutropenia, infections, and deep venous thrombosis; for danazol, hirsutism, acne, amenorrhea, hepatic dysfunction; and for dapsone, GI symptoms, methemoglobinemia, rash, haemolytic anaemia in those with glucose-6-phosphate dehydrogenase deficiency.[6]
Combination therapy with Fostamatinib and Thrombopoietin Receptor Agonists
Combining fostamatinib with other therapies is a treatment strategy that has shown promising results in real-world clinical practice (Table 2). In a single-centre case series (n=15), fostamatinib was combined with either prednisone, eltrombopag, or romiplostim (depending on the treatment that was ongoing at the time of fostamatinib initiation). Eighty percent of patients (12/15) achieved a response (platelet count ≥30×109/L and at least twofold increase in the baseline count with absence of bleeding) in a median 9 days and 73% achieved a complete response (platelet count ≥100 × 109/L and absence of bleeding) after a median 13 days.[2,48] During the follow-up period (median, 119 days), 40% of patients were able to discontinue steroid or TPO-RA therapy after a median 80 days. No severe adverse events were reported, and new hypertension (in 3/15 patients) was well managed with treatment.
Another case series of patients with ITP (n=5) who transitioned from TPO-RAs (which included romiplostim, eltrombopag, and avatrombopag) to fostamatinib therapy demonstrated that the combinations were effective and well tolerated, as all patients achieved platelet counts of 100 × 109/L or more with no adverse events reported.[49] In an international, multicentre, retrospective study, the combination of avatrombopag with fostamatinib was effective: a response (defined as a platelet count >30 × 109/L) was achieved in 7/18 patients, and 8/18 achieved a complete response (defined as a platelet count >100 × 109/L), with a median time to best response of 15 days. Adverse events occurred in 33% of patients (6/18) and included headache and grade 2 liver toxicity determined to be related to avatrombopag, and grade 1 diarrhoea and grade 4 neutropenia determined to be related to fostamatinib; these events were in line with known safety data.[50]
TPO-RAs have also been used in combination with immunosuppressive agents or steroids. In a retrospective, observational study that included patients with multirefractory ITP, eltrombopag or romiplostim was taken with MMF, azathioprine, cyclophosphamide, cyclosporin, or everolimus.[51] Thirty out of 39 patients (77%) achieved a response, with 24 patients achieving a complete response within a median 30 days (median duration of response, 15 months); however, severe treatment-related adverse events were observed in 31%.[51] Another retrospective, observational study of patients with multirefractory ITP examined combination therapy of a TPO-RA (eltrombopag, romiplostim, or avatrombopag) with an immunosuppressive or immunomodulatory treatment (corticosteroids, purine synthesis inhibitors, or fostamatinib).[52] Seventy-five out of 97 patients (77.3%) achieved platelet counts of ≥30 × 109/L with at least doubling of baseline counts, with a median time to response of 14 days.[52] The most common safety event reported was infection requiring treatment in 15 patients (15.5%), 14 of whom required hospitalization.[52] A case series in patients with multirefractory ITP who were unresponsive to TPO-RAs showed that the addition of prednisone to their treatment regimen improved outcomes by increasing the platelet count to acceptable levels (consistently above 30 × 109/L).[53] Adverse effects were observed in 60% of patients which included headaches, hypertension, elevated liver enzymes on eltrombopag, avascular necrosis of the hip, hyperglycaemia, mood changes, and weight gain.[53] In addition, an open-label study assessing eltrombopag in combination with dexamethasone showed that the combination was effective, with all patients (n=12) achieving a response by day 33 and 50% having a platelet count ≥100 × 109/L after 6 months.[54]
Splenectomy
Splenectomy is generally only recommended for surgical candidates (few comorbidities, up-to-date immunizations) following failure of pharmacological therapies and after 1 to 2 years after ITP diagnosis.[1,7,32,55] The response to splenectomy has been reported to be sustained for a decade in 78% of patients, with relapse-free survival rates ranging from 63% to 94%; despite these encouraging statistics, almost 19% of patients have no formal response to the procedure.[56,57] The risk for postoperative complications must also be considered and includes infections, thrombosis, and haemorrhage; some events may be fatal.[58]
New and Emerging Treatments
ITP treatment is undergoing rapid advancement, with emerging therapies designed to target specific disease mechanisms and help address the unmet needs of many patients.[59] Rilzabrutinib was recently FDA-approved for ITP, and a variety of other agents that target novel immune pathways (such as blockade of antibody reuptake mechanisms, inhibition of immune cells, and the complement cascade) are currently undergoing investigation (belimumab, ianalumab, bortezomib, sutimlimab, efgartigimod, iptacopan, daratumumab, and mezagitamab).[59] Although evidence collected to date suggests that these latter medications may offer promise as effective therapeutic options in the future, more analysis is needed to evaluate their efficacy and safety.[59]
Newer Management Approaches Contrasted with Traditional Guidelines
Newer management approaches for patients with ITP suggest incorporating TPO-RAs into the treatment paradigm earlier, as first-line options, which contrasts with traditional guidelines that have prioritized corticosteroids despite high relapse rates and metabolic complications.[7,60,61] Additionally, while splenectomy remains a second-line option in older protocols, more recent strategies include use of targeted biologics such as neonatal Fc receptor antagonists and complement pathway blockers, which address specific immune mechanisms rather than providing general immune suppression.[61] The 2019 ASH guidelines suggest IVIg or IV anti-D as acute interventions, but emerging therapies such as efgartigimod may offer alternatives to reduce pathogenic antibody recycling.[7,62] The traditional sequencing of therapy is currently undergoing reevaluation, challenged by promising combination therapies that target deficient platelet production and increased platelet destruction simultaneously (such as in trials that have combined TPO-RAs with immunomodulators).[48] Recent shifts in the treatment landscape also emphasize the importance of taking into consideration HRQoL, cumulative treatment toxicity, and the cost of treatment, moving beyond just platelet count thresholds to guide treatment choices and resource utilization.[60,61]
Personalized Approach for Optimizing Immune Thrombocytopenia Treatment by Targeting Underlying ITP Mechanism with Fostamatinib and TPO-RAs
Published guidelines from the ICR recommend TPO-RAs, rituximab and fostamatinib as second-line treatment options, while the ASH guidelines published that same year did not include fostamatinib among recommendations for second-line therapy.[7,32] This discrepancy was due to the timing of evidence collected for appraisal (the ASH panel evaluated data published only up until May 2017, while the ICR group included articles published by July 2018).[7,32] Analyses of fostamatinib in the second-line setting have since reported platelet responses ≥50 × 109/L among 78% (25/32) of evaluated patients, with 64% (16/25) of responders demonstrating a consistent platelet response ≥50 × 109/L.[63] The proposed treatment approach outlined below incorporates fostamatinib earlier in the sequencing of therapeutic agents based on real-world and clinical data demonstrating its effectiveness and manageable safety profile and is designed to offer healthcare providers a novel framework with clinical value.
Since ITP is a heterogeneous disease that may manifest differently in each patient, selection of the most appropriate second-line treatment option may depend on determining whether platelet destruction by macrophages or inadequate platelet production is responsible for an insufficient platelet count at the time of patient evaluation (Figure 2).[60] A more nuanced understanding of the mechanisms underlying the patient-specific disease pathophysiology offers individual-level insight to providers and patients and can guide selection of the next appropriate therapy. This approach is designed to take advantage of the complementary mechanisms of action of fostamatinib, which inhibits platelet destruction, and TPO-RAs, which stimulate platelet production.
On the basis of clinical and real-world evidence in a small number of patients, and while taking into account the underlying drug mechanism of action, treatment with fostamatinib monotherapy as the first-choice second-line therapy may be beneficial in patients who have an inadequate response or are experiencing intolerance to corticosteroids, particularly in those who are also at high risk of cardiovascular or thromboembolic events, or who are receiving secondary prophylaxis with antiplatelet or anticoagulant drugs.[5,48,50,63,64,65,66,67,68,69,70] Fostamatinib could be added to corticosteroids that are then tapered with the intention of transitioning to fostamatinib monotherapy, which can be initiated to assess platelet destruction by macrophages. If the response to fostamatinib is inadequate, a TPO-RA can be added to overcome the potential platelet production issue (Table 2). If the combination is intolerable, then reduction in fostamatinib dose can be considered, or the TPO-RA can be used alone. Patients with ITP who are intolerant of or who have an inadequate response to fostamatinib could be switched to a TPO-RA to overcome impaired platelet production. This proposed treatment sequencing requires additional evidence and should be further investigated in clinical trials and real-word practice.
Use of a TPO-RA as a single second-line agent may not be informative in determining the underlying disease mechanism leading to low platelet counts. In a patient who initiates a TPO-RA after corticosteroid therapy and experiences an inadequate response, we propose adding fostamatinib to the current TPO-RA to assess whether the increased number of platelets can be spared from destruction by macrophages.[49] If the combination is intolerable, an alternative TPO-RA monotherapy could be considered. In the case of intolerance to the initial TPO-RA, the therapy may be replaced with fostamatinib monotherapy. In order to determine if fostamatinib is efficacious, the production of platelets must increase on its own after treatment with the TPO-RA is terminated, indicating that platelet destruction has been inhibited.
Rituximab might be considered in a patient with underlying rheumatologic or autoimmune conditions. Similarly to fostamatinib and TPO-RAs, rituximab could be added to corticosteroids that are then tapered to transition to rituximab monotherapy. If a patient experiences intolerability or inadequate response to rituximab, fostamatinib or TPO-RA monotherapy could be considered as a third-line therapy. As in the second-line setting described above, fostamatinib may be beneficial in patients with known platelet destruction pathophysiology, in those with cardiovascular or thrombotic risk factors, or in those who are receiving antiplatelet or anticoagulant drugs. Subsequent treatment should follow a similar approach as in the case of second-line fostamatinib or TPO-RA monotherapy.
If these approaches fail to improve platelet count, other therapies, such as splenectomy, azathioprine, cyclosporin A, cyclophosphamide, danazol, dapsone, MMF, sirolimus/everolimus, or participation in clinical trials may be initiated.
Considerations
When first-line treatments cannot be used due to tolerability issues, there are factors that should be considered when selecting the next therapy. The benefit-to-risk ratio should be carefully examined when weighing potential efficacy against toxicity in the selection of the subsequent agent or procedure. The patient-specific platelet target should be determined, especially in those patients with thrombotic risk, as some treatments may increase this risk (such as TPO-RAs, with an incidence of 2.6%-8.9% in thromboembolic events in studies lasting 2-8 years; corticosteroids, a hazard ratio of 3.3 (95% CI, 1.0-11.0) for venous thromboembolism and arterial thromboembolism with prednisone use; and splenectomy, with a prevalence of venous thromboembolism ranging from 1.4% to 16%).[55,64,71,72] Measurement of the immature platelet fraction can provide insight into a patient’s relative risk of bleeding or thrombosis due to the reactivity of immature platelets.[71] A lower fraction of immature platelets is associated with an increased risk of bleeding and a reduced likelihood of thrombosis, whereas a higher fraction is correlated with the opposite.[71] In addition, care should be taken to ensure the use of minimally effective doses and to taper concomitant medications when indicated in order to avoid or minimize potential adverse events.
Clinical Implications
When considering the sequencing of second-line therapies for patients with ITP, particularly for those with high thrombotic risk and those with comorbidities or contraindications to other medical treatments, it may be beneficial to position fostamatinib earlier in therapy.[1,5,73,74,75]
The literature suggests that managing patients with refractory ITP requires a multimodal approach integrating novel targeted therapies with strategic treatment sequencing and combination regimens. Combination therapies targeting multiple pathogenic mechanisms concurrently show improved efficacy compared to single agents, with TPO-RA-based combinations achieving response rates around 70% in heavily pretreated patients who had failed multiple prior therapies including splenectomy and rituximab.[76,77,78,79]
Data on combination treatment approaches utilizing fostamatinib beyond TPO-RAs are limited but emerging. Some case reports and expert reviews suggest that fostamatinib could potentially be used in combination with immunosuppressants (such as mycophenolate mofetil or cyclosporin), IVIg, corticosteroids, and even rituximab, particularly in refractory patients.[49,66,77,80] However, similar to combinations of fostamatinib with TPO-RAs, the evidence remains largely limited to case reports and retrospective analyses, with most peer-reviewed studies emphasizing the need for prospective controlled trials to establish the safety and efficacy of fostamatinib combinations before they can be routinely recommended.[49,80,81]
Future Directions
Topics and considerations for future investigation and clinical research should include an analysis of biomarkers predicting treatment response. In order to further elucidate and confirm the effectiveness of fostamatinib in the treatment of patients with ITP, the development of head-to-head trials comparing fostamatinib with TPO-RAs and rituximab should be initiated. Finally, a critical evaluation of data gaps regarding treatment discontinuation strategies should be undertaken to clarify therapeutic challenges related to transitioning patients to more effective and tolerable therapies.
Conclusions
The goals of ITP treatment are to increase platelet count and reduce the risk of bleeding. Despite significant advances, the optimal management of ITP remains a clinical challenge. Fostamatinib alone or in combination with TPO-RAs has demonstrated both efficacy and safety in various ITP patient populations.[48,63,65,70] This review provides an overview of the current ITP treatment landscape and offers guidance to clinicians for developing a personalized treatment approach that optimizes clinical use of fostamatinib and TPO-RAs. This proposed treatment approach may help elucidate individual ITP pathophysiology, potentially leading to improved patient outcomes by illustrating a clear pathway for treatment adjustments based on patient response.
Acknowledgments & Funding
The authors thank Nicole L. Day, PhD, Ted Stanek, PhD, Scott Bergfeld, PhD, and Stephen Bublitz, ELS, of the Spark Division of Woven Health Collective, LLC, for medical writing and editorial assistance, which were funded by Rigel Pharmaceuticals, Inc.
Disclosures
EBC: Speakers Bureau: Alnylam, Rigel; MPL: Membership on advisory board: 22qSociety, Argenx, CdLS Foundation, Dova, Octapharma, PDSA, Principia, and Rigel; Consultancy: Argenx, Dova, Janssen, Novartis, Principia, Rigel, Sanofi, Sobi; Research funding: Argenx, Dova, FWGBD, NIH, Novartis, Octapharma, PDSA, Principia, Sanofi, Sysmex; MEMC: Grants from Amgen, Novartis, and Sobi, and served as an advisor or speaker for Amgen, Grifols, Novartis, Novo Nordisk, Sanofi, and Takeda.
Permissions
Figure 1 is used with permission from Audia et al., (2021); permission conveyed through Copyright Clearance Center, Inc.
Author Contributions
MEMC: Reviewed and revised the paper; MPL: Reviewed and revised the paper; EBC: Reviewed and revised the paper.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
References
- Bussel, J; Cooper, N; Boccia, R; Zaja, F; Newland, A. Immune thrombocytopenia. Expert Rev Hematol. 2021, 14(11), 1013–1025. [Google Scholar] [CrossRef]
- Rodeghiero, F; Stasi, R; Gernsheimer, T; et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009, 113(11), 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Liu, XG; Hou, Y; Hou, M. How we treat primary immune thrombocytopenia in adults. J Hematol Oncol. 2023, 16(1), 4. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, PE; Hall, SA; Feudjo-Tepie, M; Mitrani-Gold, FS; Logie, J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol. 2009, 83(2), 83–89. [Google Scholar] [CrossRef]
- Mingot-Castellano, ME; Canaro Hirnyk, M; Sánchez-González, B; et al. Recommendations for the clinical approach to immune thrombocytopenia: Spanish ITP Working Group (GEPTI). J Clin Med. 2023, 12(20), 6422. [Google Scholar] [CrossRef]
- Mititelu, A; Onisâi, MC; Roșca, A; Vlădăreanu, AM. Current understanding of immune thrombocytopenia: a review of pathogenesis and treatment options. Int J Mol Sci. 2024, 25(4), 2163. [Google Scholar] [CrossRef]
- Provan, D; Arnold, DM; Bussel, JB; et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019, 3(22), 3780–3817. [Google Scholar] [CrossRef] [PubMed]
- McMillan, R; Bussel, JB; George, JN; Lalla, D; Nichol, JL. Self-reported health-related quality of life in adults with chronic immune thrombocytopenic purpura. Am J Hematol. 2008, 83(2), 150–154. [Google Scholar] [CrossRef]
- Mingot-Castellano, ME; Bastida, JM; Caballero-Navarro, G; et al. Novel therapies to address unmet needs in ITP. Pharmaceuticals (Basel) 2022, 15(7), 779. [Google Scholar] [CrossRef]
- Podolanczuk, A; Lazarus, AH; Crow, AR; Grossbard, E; Bussel, JB. Of mice and men: an open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood 2009, 113(14), 3154–3160. [Google Scholar] [CrossRef]
- Al-Samkari, H. 2025 update on clinical trials in immune thrombocytopenia. Am J Hematol. 2024, 99(11), 2178–2190. [Google Scholar] [CrossRef] [PubMed]
- Cines, DB; Bussel, JB; Liebman, HA; Luning Prak, ET. The ITP syndrome: pathogenic and clinical diversity. Blood 2009, 113(26), 6511–6521. [Google Scholar] [CrossRef]
- Audia, S; Mahévas, M; Nivet, M; Ouandji, S; Ciudad, M; Bonnotte, B. Immune thrombocytopenia: recent advances in pathogenesis and treatments. Hemasphere 2021, 5(6), e574. [Google Scholar] [CrossRef]
- Lev, PR; Goette, NP; Marta, RF. Pathophysiological mechanisms leading to low platelet count in immune thrombocytopenia. J Immunol Sci. 2020, 4(2), 1–7. [Google Scholar] [CrossRef]
- Nugent, D; McMillan, R; Nichol, JL; Slichter, SJ. Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. Br J Haematol. 2009, 146(6), 585–596. [Google Scholar] [CrossRef]
- Zufferey, A; Kapur, R; Semple, JW. Pathogenesis and therapeutic mechanisms in immune thrombocytopenia (ITP). J Clin Med. 2017, 6(2), 16. [Google Scholar] [CrossRef]
- Provan, D; Semple, JW. Recent advances in the mechanisms and treatment of immune thrombocytopenia. EBioMedicine 2022, 76, 103820. [Google Scholar] [CrossRef]
- Peerschke, EI; Andemariam, B; Yin, W; Bussel, JB. Complement activation on platelets correlates with a decrease in circulating immature platelets in patients with immune thrombocytopenic purpura. Br J Haematol. 2010, 148(4), 638–645. [Google Scholar] [CrossRef]
- Evangelidis, P; Tragiannidis, K; Gavriilaki, E; Tragiannidis, A. Impact of thrombopoietin receptor agonists on pathophysiology of pediatric immune thrombocytopenia. Curr Issues Mol Biol. 2025, 47(1), 65. [Google Scholar] [CrossRef] [PubMed]
- Rodeghiero, F. Is ITP a thrombophilic disorder? Am J Hematol. 2016, 91(1), 39–45. [Google Scholar] [CrossRef]
- Swan, D; Newland, A; Rodeghiero, F; Thachil, J. Thrombosis in immune thrombocytopenia - current status and future perspectives. Br J Haematol. 2021, 194(5), 822–834. [Google Scholar] [CrossRef]
- Zhou, F; Zhang, S; Li, R; et al. A retrospective study of risk factors for thrombosis in patients with ITP. Hematology 2025, 30(1), 2472461. [Google Scholar] [CrossRef]
- Lambert, C; Maitland, H; Ghanima, W. Risk-based and individualised management of bleeding and thrombotic events in adults with primary immune thrombocytopenia (ITP). Eur J Haematol. 2024, 112(4), 504–515. [Google Scholar] [CrossRef]
- Takagi, S; Suzuki, I; Watanabe, S. Risk of thromboembolism in patients with immune thrombocytopenia. J Hematol Thrombo Dis. 2015, 3, 1. [Google Scholar]
- Langeberg, WJ; Schoonen, WM; Eisen, M; Gamelin, L; Stryker, S. Thromboembolism in patients with immune thrombocytopenia (ITP): a meta-analysis of observational studies. Int J Hematol. 2016, 103(6), 655–664. [Google Scholar] [CrossRef]
- van Dijk, WEM; Poolen, GC; Huisman, A; et al. Evaluation of the procoagulant state in chronic immune thrombocytopenia before and after eltrombopag treatment-a prospective cohort study. J Thromb Haemost. 2023, 21(4), 1020–1031. [Google Scholar] [CrossRef]
- Doobaree, IU; Nandigam, R; Bennett, D; Newland, A; Provan, D. Thromboembolism in adults with primary immune thrombocytopenia: a systematic literature review and meta-analysis. Eur J Haematol. 2016, 97(4), 321–330. [Google Scholar] [CrossRef]
- Goncalves, I; Lewis, C; Grainger, B; et al. Thrombosis in patients with immune thrombocytopenia: incidence, risk, and clinical outcomes. Res Pract Thromb Haemost. 2024, 8(1), 102342. [Google Scholar] [CrossRef] [PubMed]
- Liang, H; Duan, L; Long, M; et al. Analysis of risk factors and the establishment of a predictive model for thrombosis in patients with immune thrombocytopenia. Clin Appl Thromb Hemost. 2025, 31, 10760296241301398. [Google Scholar] [CrossRef]
- Moulis, G; Audemard-Verger, A; Arnaud, L; et al. Risk of thrombosis in patients with primary immune thrombocytopenia and antiphospholipid antibodies: a systematic review and meta-analysis. Autoimmun Rev. 2016, 15(3), 203–209. [Google Scholar] [CrossRef] [PubMed]
- Uthman, I; Godeau, B; Taher, A; Khamashta, M. The hematologic manifestations of the antiphospholipid syndrome. Blood Rev. 2008, 22(4), 187–194. [Google Scholar] [CrossRef]
- Neunert, C; Terrell, DR; Arnold, DM; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3(23), 3829–3866. [Google Scholar] [CrossRef]
- Neunert, CE; Arnold, DM; Grace, RF; Kuhne, T; McCrae, KR; Terrell, DR. The 2022 review of the 2019 American Society of Hematology guidelines on immune thrombocytopenia. Blood Adv. 2024, 8(13), 3578–3582. [Google Scholar] [CrossRef] [PubMed]
- Erkurta, MA; Kayaa, E; Berbera, I; Koroglua, M; Kuku, I. Thrombocytopenia in adults: review article. J Hematol. 2012, 1(2-23), 44–53. [Google Scholar] [CrossRef]
- Jia, M; Wang, Z; Hu, F. Causal relationship between physical activity and platelet traits: a Mendelian randomization study. Front Physiol. 2024, 15, 1371638. [Google Scholar] [CrossRef]
- Ruggeri, M; Fortuna, S; Rodeghiero, F. Heterogeneity of terminology and clinical definitions in adult idiopathic thrombocytopenic purpura: a critical appraisal from a systematic review of the literature. Haematologica 2008, 93(1), 98–103. [Google Scholar] [CrossRef] [PubMed]
- Gernsheimer, T; Stratton, J; Ballem, PJ; Slichter, SJ. Mechanisms of response to treatment in autoimmune thrombocytopenic purpura. N Engl J Med. 1989, 320(15), 974–980. [Google Scholar] [CrossRef]
- Kitchens, CS. Amelioration of endothelial abnormalities by prednisone in experimental thrombocytopenia in the rabbit. J Clin Invest. 1977, 60(5), 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Feldweg, AM; Leddy, JP. Drug interactions affecting the efficacy of corticosteroid therapy a brief review with an illustrative case. J Clin Rheumatol. 1999, 5(3), 143–150. [Google Scholar] [CrossRef]
- Jacobs, TG; Marzolini, C; Back, DJ; Burger, DM. Dexamethasone is a dose-dependent perpetrator of drug-drug interactions: implications for use in people living with HIV. J Antimicrob Chemother. 2022, 77(3), 568–573. [Google Scholar] [CrossRef]
- Lünemann, JD; Quast, I; Dalakas, MC. Efficacy of intravenous immunoglobulin in neurological diseases. Neurotherapeutics 2016, 13(1), 34–46. [Google Scholar] [CrossRef]
- Onisâi, M; Vlădăreanu, AM; Spînu, A; Găman, M; Bumbea, H. Idiopathic thrombocytopenic purpura (ITP) - new era for an old disease. Rom J Intern Med. 2019, 57(4), 273–283. [Google Scholar] [CrossRef]
- Salerno, SN; Deng, R; Kakkar, T. Physiologically-based pharmacokinetic modeling of immunoglobulin and antibody coadministration in patients with primary human immunodeficiency. CPT Pharmacometrics Syst Pharmacol. 2022, 11(10), 1316–1327. [Google Scholar] [CrossRef]
- Despotovic, JM; Lambert, MP; Herman, JH; et al. RhIG for the treatment of immune thrombocytopenia: consensus and controversy (CME). Transfusion 2012, 52(5), 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Naithani, R; Kumar, R; Mahapatra, M; Tyagi, S; Saxena, R. Efficacy and safety of anti-D for treatment of adults with immune thrombocytopenia. Platelets 2009, 20(7), 525–527. [Google Scholar] [CrossRef] [PubMed]
- Ghanima, W; Cooper, N; Rodeghiero, F; Godeau, B; Bussel, JB. Thrombopoietin receptor agonists: ten years later. Haematologica 2019, 104(6), 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Kochhar, M; Neunert, C. Immune thrombocytopenia: a review of upfront treatment strategies. Blood Rev. 2021, 49, 100822. [Google Scholar] [CrossRef]
- Passucci, M; Laganà, A; Donzelli, L; et al. Fostamatinib combined with TPO-RAs or steroids as a bridge to monotherapy or as time-limited continuous treatment in relapsed chronic ITP: a single-centre case series. Br J Haematol. 2024, 204(5), 2129–2132. [Google Scholar] [CrossRef]
- Hughes, DM; Toste, C; Nelson, C; Escalon, J; Blevins, F; Shah, B. Transitioning from thrombopoietin agonists to the novel SYK inhibitor fostamatinib: a multicenter, real-world case series. J Adv Pract Oncol. 2021, 12(5), 508–517. [Google Scholar]
- Mingot-Castellano, ME; Bastida, JM; Ghanima, W; et al. Avatrombopag plus fostamatinib combination as treatment in patients with multirefractory immune thrombocytopenia. Br J Haematol. 2024, 205(4), 1551–1555. [Google Scholar] [CrossRef]
- Crickx, E; Ebbo, M; Rivière, E; et al. Combining thrombopoietin receptor agonists with immunosuppressive drugs in adult patients with multirefractory immune thrombocytopenia, an update on the French experience. Br J Haematol. 2023, 202(4), 883–889. [Google Scholar] [CrossRef]
- Mingot-Castellano, M-E; Alcalde-Mellado, P; Entrena-Ureña, L; et al. Combining an immunomodulatory drug with a TPO-RA to treat multirefractory ITP patients: the Spanish ITP group experience. In Br J Haematol.; in press; p. 52. [Google Scholar]
- Poston, JN; Gernsheimer, TB. Glucocorticoids promote response to thrombopoietin-receptor agonists in refractory ITP: a case series. Int J Hematol. 2019, 110(2), 255–259. [Google Scholar] [CrossRef]
- Gómez-Almaguer, D; Herrera-Rojas, MA; Jaime-Pérez, JC; et al. Eltrombopag and high-dose dexamethasone as frontline treatment of newly diagnosed immune thrombocytopenia in adults. Blood 2014, 123(25), 3906–3908. [Google Scholar] [CrossRef]
- Chaturvedi, S; Arnold, DM; McCrae, KR. Splenectomy for immune thrombocytopenia: down but not out. Blood 2018, 131(11), 1172–1182. [Google Scholar] [CrossRef]
- Chater, C; Terriou, L; Duhamel, A; et al. Reemergence of splenectomy for ITP second-line treatment? Ann Surg. 2016, 264(5), 772–777. [Google Scholar] [CrossRef] [PubMed]
- Tada, K; Ohta, M; Saga, K; et al. Long-term outcomes of laparoscopic versus open splenectomy for immune thrombocytopenia. Surg Today 2018, 48(2), 180–185. [Google Scholar] [CrossRef] [PubMed]
- Vianelli, N; Palandri, F; Polverelli, N; et al. Splenectomy as a curative treatment for immune thrombocytopenia: a retrospective analysis of 233 patients with a minimum follow up of 10 years. Haematologica 2013, 98(6), 875–880. [Google Scholar] [CrossRef] [PubMed]
- Lambert, MP. On the horizon: upcoming new agents for the management of ITP. Hematology Am Soc Hematol Educ Program. 2024, 2024(1), 692–699. [Google Scholar] [CrossRef]
- Madkhali, MA. Recent advances in the management of immune thrombocytopenic purpura (ITP): a comprehensive review. Medicine (Baltimore) 2024, 103(3), e36936. [Google Scholar] [CrossRef]
- Jiang, D; Al-Samkari, H; Panch, SR. Changing paradigms in ITP management: newer tools for an old disease. Transfus Med Rev. 2022, 36(4), 188–194. [Google Scholar] [CrossRef]
- Newland, AC; Sánchez-González, B; Rejtő, L; et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am J Hematol. 2020, 95(2), 178–187. [Google Scholar] [CrossRef] [PubMed]
- Boccia, R; Cooper, N; Ghanima, W; et al. Fostamatinib is an effective second-line therapy in patients with immune thrombocytopenia. British Journal of Haematology 2020, 190(6), 933–938. [Google Scholar] [CrossRef]
- Cooper, N; Altomare, I; Thomas, MR; et al. Assessment of thrombotic risk during long-term treatment of immune thrombocytopenia with fostamatinib. Ther Adv Hematol. 2021, 12, 20406207211010875. [Google Scholar] [CrossRef]
- Bussel, JB; Arnold, DM; Boxer, MA; et al. Long-term fostamatinib treatment of adults with immune thrombocytopenia during the phase 3 clinical trial program. Am J Hematol. 2019, 94(5), 546–553. [Google Scholar] [CrossRef] [PubMed]
- Lee, EJ; Izak, M; Bussel, JB. Long-term sustained response to fostamatinib in two patients with chronic refractory immune thrombocytopenia (ITP). Br J Haematol. 2020, 189(2), 379–382. [Google Scholar] [CrossRef] [PubMed]
- Liu, J; Hsia, CC. The efficacy and safety of fostamatinib in elderly patients with immune thrombocytopenia: a single-center, real-world case series. Adv Hematol. 2022, 2022, 8119270. [Google Scholar] [CrossRef]
- Auteri, G; Biondo, M; Mazzoni, C; et al. Sustained response off therapy after fostamatinib: a chronic refractory ITP case report. Heliyon 2023, 9(2), e13462. [Google Scholar] [CrossRef]
- Moore, DC; Elmes, JB; Arnall, JR; Pineda-Roman, M. Real-world clinical outcomes with fostamatinib for the treatment of refractory chronic immune thrombocytopenia: a single-center experience. Blood Coagul Fibrinolysis 2024, 35(6), 316–320. [Google Scholar]
- Bussel, J; Arnold, DM; Grossbard, E; et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: results of two phase 3, randomized, placebo-controlled trials. Am J Hematol. 2018, 93(7), 921–930. [Google Scholar] [CrossRef]
- Provan, D; Thachil, J; Álvarez Román, MT. Addressing thrombosis concerns in immune thrombocytopenia: the role of fostamatinib in immune thrombocytopenia management. Expert Rev Hematol. 2024, 17(1-3), 55–66. [Google Scholar]
- Ruggeri, M; Tosetto, A; Palandri, F; et al. Thrombotic risk in patients with primary immune thrombocytopenia is only mildly increased and explained by personal and treatment-related risk factors. J Thromb Haemost. 2014, 12(8), 1266–1273. [Google Scholar] [CrossRef]
- Lucchesi, A; Fattizzo, B; De Stefano, V; et al. Use and positioning of fostamatinib in the management of primary chronic immune thrombocytopenia: an Italian expert opinion. Ther Adv Hematol. 2023, 14, 20406207221147777. [Google Scholar] [PubMed]
- González-López, TJ; Bermejo-Vega, N; Cardesa-Cabrera, R; et al. Fostamatinib effectiveness and safety for immune thrombocytopenia in clinical practice. Blood 2024, 144(6), 646–656. [Google Scholar] [CrossRef]
- Ghanima, W; Cuker, A; Michel, M. Insights on treatment of adult ITP: algorithm for management and role of multimodal therapy. Hematology Am Soc Hematol Educ Program. 2024, 2024(1), 678–684. [Google Scholar] [CrossRef]
- Vianelli, N; Auteri, G; Buccisano, F; et al. Refractory primary immune thrombocytopenia (ITP): current clinical challenges and therapeutic perspectives. Ann Hematol. 2022, 101(5), 963–978. [Google Scholar]
- Gudbrandsdottir, S; Leven, E; Imahiyerobo, A; Lee, CS; Bussel, J. Combination of thrombopoietin receptor agonists, immunosuppressants and intravenous immunoglobulin as treatment of severe refractory immune thrombocytopenia in adults and children. Br J Haematol. 2020, 189(2), e37–e40. [Google Scholar] [CrossRef] [PubMed]
- Mahévas, M; Gerfaud-Valentin, M; Moulis, G; et al. Characteristics, outcome, and response to therapy of multirefractory chronic immune thrombocytopenia. Blood 2016, 128(12), 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y; Shi, H; Liu, H; Zhou, L. Current therapeutic strategies and perspectives in refractory ITP: what have we learned recently? Front Immunol. 2022, 13, 953716. [Google Scholar] [CrossRef]
- Ghanima, W; Lucas Boronat, FJ; Carrai, V; Rackwitz, S. Sustained response off treatment after fostamatinib in refractory immune thrombocytopenia: a series of four case reports. Hematology 2025, 30(1), 2456687. [Google Scholar] [CrossRef]
- Kou, R; Zhao, L; Tham, D; et al. Fostamatinib for immune thrombocytopenic purpura in adult patients: a systematic review and meta-analysis. EJHaem 2024, 5(4), 651–660. [Google Scholar] [CrossRef]
- PROMACTA® (eltrombopag) tablets, for oral use. PROMACTA® (eltrombopag) for oral suspension [prescribing information]; Novartis Pharmaceuticals Corporation: East Hanover, NJ, June 2025.
- NPLATE® (romiplostim) for injection, for subcutaneous use [prescribing information]; Amgen Inc.: Thousand Oaks, California, February 2025.
- DOPTELET® (avatrombopag) tablets, for oral use; DOPTELET® SPRINKLE (avatrombopag) oral granules [prescribing information]; AkaRx, Inc.: Morrisville, North Carolina, July 2025.
- TAVALISSE® (fostamatinib) tablets, for oral use [prescribing information]; Rigel Pharmaceuticals, Inc.: South San Francisco, CA, December 2024.
- RITUXAN® (rituximab) injection, for intravenous use [prescribing information]; Biogen: Cambridge, MA, April 2021.
- Patel, VL; Mahévas, M; Lee, SY; et al. Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood 2012, 119(25), 5989–5995. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pathogenesis of immune thrombocytopenia. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; AMR, Ashwell-Morell receptor; BAFF, B-cell activating factor; CD, cluster of differentiation; CDC, complement-dependent cytotoxicity; CMC, cytotoxic T lymphocyte-mediated cytotoxicity; cMpl, thrombopoietin receptor; CR, complement receptor; CTL, cytotoxic T cell; FcγR, Fc gamma receptor; GP, glycoprotein; ICOS, inducible T-cell costimulator; ICOS-L, ICOS ligand; IL, interleukin; ITP, immune thrombocytopenia; JAK, Janus kinase; MAC, membrane attack complex; MEK, megakaryocyte; MHC, major histocompatibility complex; mRNA, messenger RNA; PD-1, programmed cell death protein 1 (CD279); PD-L1, programmed cell death ligand 1 (CD274); STAT, signal transducer and activator of transcription; Tc, cytotoxic T cell; TCR, T-cell receptor; TFH, T follicular helper cell; Th, helper T cell; TPO, thrombopoietin; Treg, regulatory T cell. Used with permission from Audia et al.[13]; permission conveyed through Copyright Clearance Center, Inc.
Figure 1.
Pathogenesis of immune thrombocytopenia. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; AMR, Ashwell-Morell receptor; BAFF, B-cell activating factor; CD, cluster of differentiation; CDC, complement-dependent cytotoxicity; CMC, cytotoxic T lymphocyte-mediated cytotoxicity; cMpl, thrombopoietin receptor; CR, complement receptor; CTL, cytotoxic T cell; FcγR, Fc gamma receptor; GP, glycoprotein; ICOS, inducible T-cell costimulator; ICOS-L, ICOS ligand; IL, interleukin; ITP, immune thrombocytopenia; JAK, Janus kinase; MAC, membrane attack complex; MEK, megakaryocyte; MHC, major histocompatibility complex; mRNA, messenger RNA; PD-1, programmed cell death protein 1 (CD279); PD-L1, programmed cell death ligand 1 (CD274); STAT, signal transducer and activator of transcription; Tc, cytotoxic T cell; TCR, T-cell receptor; TFH, T follicular helper cell; Th, helper T cell; TPO, thrombopoietin; Treg, regulatory T cell. Used with permission from Audia et al.[13]; permission conveyed through Copyright Clearance Center, Inc.
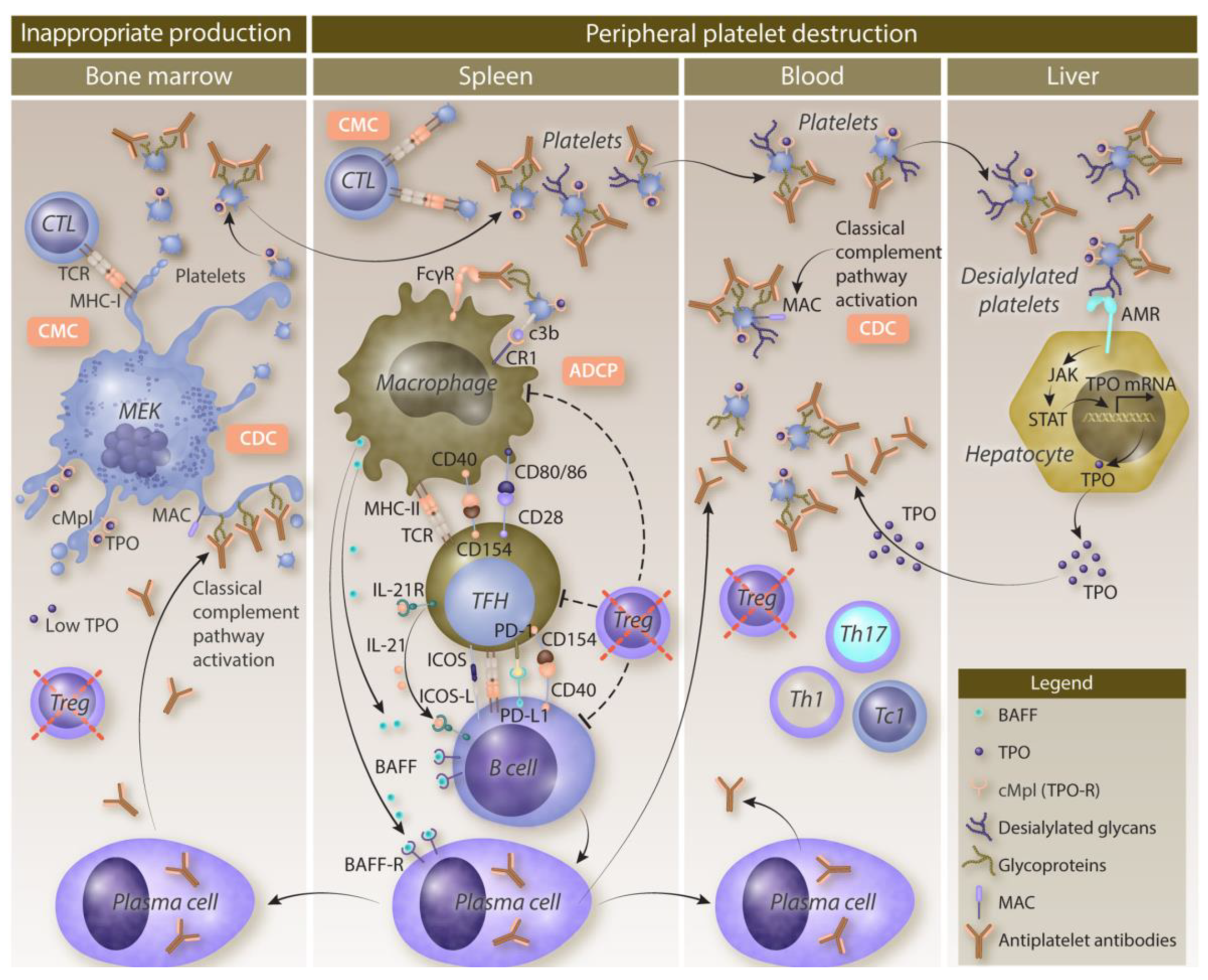
Figure 2.
Proposed personalized treatment approach based on underlying ITP mechanism. AE, adverse event; CS, corticosteroids; CV, cardiovascular; FOS, fostamatinib; TPO-RA, thrombopoietin receptor agonist. Immunoglobulins are reserved for emergency situations requiring rapid response. aIn cases of thrombosis during TPO-RA therapy, the TPO-RA dose is reduced to maintain a safe platelet count for anticoagulation, with a gradual transition to fostamatinib over 3-4 weeks once the acute event has stabilized. bIf an adverse event occurs at the current dose of fostamatinib, the dose of fostamatinib may be reduced prior to considering a switch in therapy. cFostamatinib is preferred in patients with known platelet destruction pathophysiology, in those with CV or thromboembolic risk, or in those who are receiving antiplatelet or anticoagulant drugs.
Figure 2.
Proposed personalized treatment approach based on underlying ITP mechanism. AE, adverse event; CS, corticosteroids; CV, cardiovascular; FOS, fostamatinib; TPO-RA, thrombopoietin receptor agonist. Immunoglobulins are reserved for emergency situations requiring rapid response. aIn cases of thrombosis during TPO-RA therapy, the TPO-RA dose is reduced to maintain a safe platelet count for anticoagulation, with a gradual transition to fostamatinib over 3-4 weeks once the acute event has stabilized. bIf an adverse event occurs at the current dose of fostamatinib, the dose of fostamatinib may be reduced prior to considering a switch in therapy. cFostamatinib is preferred in patients with known platelet destruction pathophysiology, in those with CV or thromboembolic risk, or in those who are receiving antiplatelet or anticoagulant drugs.
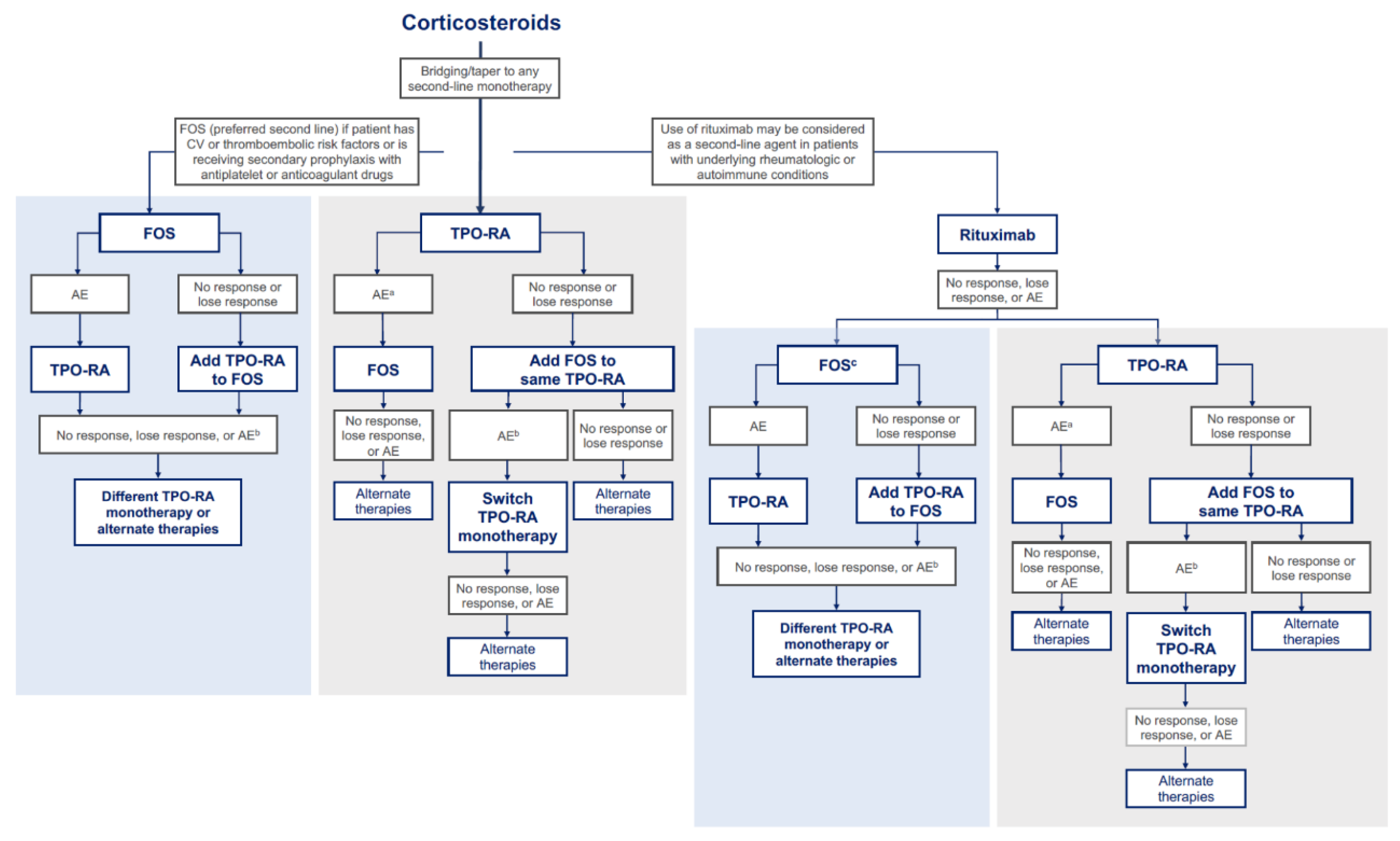

Table 1.
Pharmacological therapies recommended by treatment guidelines for second-line treatment of immune thrombocytopenia.
Table 1.
Pharmacological therapies recommended by treatment guidelines for second-line treatment of immune thrombocytopenia.
| Treatment (First FDA Approval for ITP) | Mechanism of Action | Indicated Patient Population | Efficacy in Second-Line or Later Settings | Safety | Drug-Drug Interactions |
|---|---|---|---|---|---|
| Eltrombopag[82] (2008) | TPO-RA that stimulates platelet production | Adults and children ≥1 year old with chronic ITP | 59% (43/73) and 70% (19/27) of adult patients experienced a platelet response ≥50 × 109/L in 2 studies 60% of 134 adult patients had a sustained platelet responsea |
Common AEs include anaemia, nausea, pyrexia, ALT increased, cough, fatigue, headache, and diarrhoea | Requires coordination of dose with foods or supplements that contain polyvalent cations (eg, iron, calcium, magnesium, zinc) Use caution with coadministration of substrates of OATP1B1 or BCRP |
| Romiplostim[83] (2008) | TPO-RA that stimulates platelet production | Adults with ITP and insufficient response to corticosteroids Children ≥1 year old with ITP for ≥6 months and insufficient response to corticosteroids |
88% (36/41) of adult patients experienced a platelet response ≥50 × 109/L 61% (25/41) adult patients had a durable platelet responsea |
Common AEs in adults include arthralgia, dizziness, insomnia, myalgia, pain in extremity, abdominal pain, shoulder pain, dyspepsia, and paraesthesia Common AEs in paediatric patients include contusion, upper respiratory tract infection, and oropharyngeal pain |
None reported |
| Avatrombopag[84] (2019) | TPO-RA that stimulates platelet production | Adults with chronic ITP and insufficient response to prior treatment | 66% (21/32) patients had a platelet response ≥50 × 109/L at day 8 of treatment 32 patients had a median of 12.4 weeks with a platelet response ≥50 × 109/L without rescue therapy |
Common AEs include headache, fatigue, contusion, epistaxis, upper respiratory tract infection, arthralgia, gingival bleeding, petechiae, nasopharyngitis | Dose adjustments are necessary with concomitant CYP3A4 or CYP2C9 inhibitors or inducers |
| Fostamatinib[65,85] (2018) | SYK inhibitor that prevents phagocytosis of platelets by spleen macrophages | Adults with chronic ITP and insufficient response to ≥1 prior treatment | 44% (64/146) patients had a platelet response ≥50 × 109/L 18% (27/146) responders had a stable platelet responseb |
Common AEs include diarrhoea, hypertension, nausea, respiratory infection, dizziness, ALT/AST increase, rash, abdominal pain, fatigue, chest pain, and neutropenia | CYP3A4 inhibitors may require dose modification Avoid concomitant CYP3A4 inducers |
| Rituximab[6,86,87] (Not FDA-approved for ITP) | Monoclonal antibody that targets CD20 on B cells to trigger apoptosis | Studies have been performed in adults and children ≥2 years old with ITP | 57% (215/376) adult patients with ITP had a platelet response ≥50 × 109/L 43% of 60 adult patients had a 1-year platelet response ≥50 × 109/L |
Common infusion-related AEs include chills, bronchospasm, neutropenia, serum sickness, increased infection risk, PML, hypogammaglobulinemia | None reported |
aA sustained or durable treatment response was defined as a platelet response ≥50 × 109/L for 6 of the last 8 weeks of treatment without the use of rescue medication. bA stable platelet response was defined as a platelet response ≥50 × 109/L for ≥4 of 6 bi-weekly visits during weeks 14 to 24 of treatment without the use of rescue medication. AE, adverse event; BCRP, breast cancer resistance protein; DVT, deep vein thrombosis; ITP, immune thrombocytopenia; OATP1B1, organic anion transporting polypeptide 1B1; PML, progressive multifocal leukoencephalopathy; SYK, spleen tyrosine kinase; TPO-RA, thrombopoietin receptor agonist.
Table 2.
Literature on fostamatinib and TPO-RA combination treatment.
| Citation | Patient Population | Efficacy | Safety | Follow-up Period |
|---|---|---|---|---|
| Hughes et al.[49] (2021) | Patients transitioning from thrombopoietin agonists |
|
|
|
| Passucci et al.[48] (2024) | Relapsed or refractory chronic ITP patients who received fostamatinib with prednisone or TPO-RAs as a bridge to fostamatinib monotherapy or time-limited continuous treatment (N=15) |
|
|
|
| Mingot-Castellano et al.[50] (2024) | Patients with multirefractory ITP (N=18) |
|
|
|
AE, adverse event; DVT, deep vein thrombosis; ITP, immune thrombocytopenia; TPO-RA, thrombopoietin receptor agonist.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



