Submitted:
27 January 2026
Posted:
28 January 2026
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) and post-COVID-19 syndrome share a symptom profile, including severe fatigue, cognitive dysfunction, exertional intolerance, sleep disturbances, hypervigilance, and the paradoxical state of being “wired but tired.” A well-established finding is sympathetic hyperactivity with reduced vagal tone, typically interpreted as autonomic nervous system dysfunction. Emerging evidence, however, suggests a broader disturbance across multiple neurotransmitter systems. This paper reviews current knowledge on neurotransmitter systems implicated in ME/CFS and Long COVID, focusing on potential mechanisms of dysregulation and their roles in disease pathology and symptom generation, as well as implications for treatment. In addition to abnormalities of the noradrenergic system, disturbances in serotonergic, GABAergic, and glutamatergic signaling have been reported. Contributing factors may include autoimmunity, neuroinflammation, gut dysbiosis, epigenetic influences, and stressors such as orthostatic intolerance, metabolic strain, and pain. A shift toward excitatory over inhibitory neurotransmission may cause brain overactivation, autonomic dysfunction, sensory hypersensitivity, sleep–wake disruption, and cognitive impairment. Reduced GABAergic tone combined with increased glutamatergic and noradrenergic activity may elevate skeletal muscle tone, contributing to calcium overload, mitochondrial dysfunction, exertional intolerance, and post-exertional malaise. Various pharmacological treatments may partially rebalance these neurotransmitter systems, but limited efficacy highlights the need for systematic investigation and individualized strategies.
Keywords:
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)
; Long COVID
; neurotransmitter imbalance
; autonomic dysfunction
; noradrenaline
; GABA
; glutamate
; serotonin
; hypersensitivities
Introduction
Among the key symptoms of fatigue, disturbed cognitions, and exertional intolerance with post-exertional malaise (PEM) ME/CFS patients commonly show sympathetic hyperactivity, sleep disturbances, non-restorative sleep and nocturnal hypervigilance and the feeling of overstimulation despite profound fatigue (Carruthers et al., 2011). This paradox is described by “wired but tired”. Long COVID shares many symptoms and findings with ME/ and a subset of patients fulfill the diagnostic criteria for ME/CFS (Kedor et al. 2022).
Sympathetic hyperactivity, reflected by altered heart rate variability, has been shown in numerous publications in ME/CFS (Wyller et al., 2007), (Freeman and Komaroff, 1997), (Stewart, 2000), (Van Cauwenbergh et al., 2014), (Nelson et al., 2019). It is primarily driven by increased noradrenergic activity (Ryabkova et al., 2019). Nocturnal hypervigilance, sleep disturbances and the feeling of overstimulation may result from an overactive noradrenergic system, in which the locus coeruleus plays a central role. Another consistent observation is reduced vagal activity (Sisto et al., 1995), (Beaumont et al., 2012), (Yamamoto et al., 2003), (Boneva et al., 2007), (Wyller et al., 2007), (Van Oosterwijck et al., 2021) indicating a clear imbalance between noradrenergic and cholinergic vagal signaling (Słomko et al., 2020).
In this paper, we reviewed current evidence of neurotransmitter disturbances in ME/CFS and Long COVID and compiled the findings. We provide evidence that the dysregulation of neurotransmitters may extend beyond the disturbed sympathetic–vagal balance and involve multiple neurotransmitter systems. This broader imbalance of neurotransmitters could favor excitatory over inhibitory signaling, contributing to brain overactivation and autonomic dysfunction and sensory hypersensitivities. In the second part of this paper, we try to explain how these neurotransmitter dysregulations could drive disease pathology and explain core symptoms and complaints.
The possibility should be considered that autonomic dysfunction is not the primary pathology but a secondary consequence of an underlying neurotransmitter imbalance, with additional mechanisms likely contributing, and with fully developed ME/CFS potentially aggravating this imbalance. Physiologically, the interplay of excitatory and inhibitory systems determines overall activity levels, closely tied to the sleep–wake cycle. In ME/CFS, this state-dependent balance appears disturbed both during the day and at night.
Our deeper analysis of neurotransmitter disturbances is motivated by two findings unrelated to the sympathetic nervous system and its transmitter noradrenaline. First, a recent study identified several new autoantibodies, including one against serine/arginine repetitive matrix protein 3 (SRRM3), which is linked to the GABA system (Nakano et al., 2019), (Hoheisel et al., 2025). Notably, a single nucleotide variant in SRRM3 has been associated with ME/CFS, implicating dysfunctional SRRM3, which may disrupt GABA-related splicing and impair GABAergic signaling in disease pathophysiology (Schlauch et al., 2016). Supporting this notion, a recent magnetic resonance study has reported elevated glutamate levels in ME/CFS, consistent with an imbalance of the glutamatergic-GABAergic neurotransmission (Thapaliya et al. 2025). Second, several studies have reported reduced tryptophan levels in ME/CFS and Long COVID, that could lower the availability of this precursor for the synthesis of serotonin. Together, these findings suggest that the GABAergic and serotonergic systems may also be disturbed.
Prompted by these insights we systematically reviewed the literature for further potential disturbances in major neurotransmitter systems focusing on excitatory (noradrenaline, histamine, glutamate, and dopamine) and inhibitory or modulatory neurotransmitters (GABA, serotonin, and vagal acetylcholine). We would like to emphasize that apart from sympathetic hyperactivity the evidence for disturbances of other neurotransmitters is still scarce and requires further research. Nevertheless, there are sufficient indications to seriously consider the possibility that an imbalance of the main neurotransmitter systems beyond the well-known disturbance of the sympathetic-parasympathetic axis may contribute to and potentially even play a causal role in the initiation of the disease.
Consistent with the assumptions of disturbed neurotransmitters, a range of pharmacological interventions targeting these neurotransmitters have been applied empirically in ME/CFS. Although no large-scale clinical trials have demonstrated efficacy, several drugs show beneficial effects in subsets of patients: clonidine and beta-blockers (noradrenaline modulation), vagal activation procedures and pyridostigmine (effects on vagal and sympathetic first ganglion), serotonin reuptake inhibitors (SSRI), aripiprazole (presumably dopamine receptor modulation), antihistamines, dextromethorphan or memantine (NMDA-glutamate receptor modulation), and benzodiazepines (GABAergic activation) and presumably also low-dose-naltrexone (LDN) as will be explained below. Collectively, these interventions seem to reflect a therapeutic strategy of rebalancing disturbed neurotransmitter systems, although these drugs are not administered with this explicit therapeutic concept.
The causes of these neurotransmitter disturbances may involve immune dysregulation (autoantibodies, neuroinflammation, mast cell overactivity), genetic predisposition, epigenetic regulation and gut dysbiosis which will be discussed in the following sections on the single neurotransmitters. These factors may create a vulnerability to develop ME/CFS following infection. Once ME/CFS is fully established, it seems to further augment and aggravate the neurotransmitter disturbances and autonomic dysfunction. Table 1 gives an overview of the main neurotransmitters, their localization and function. We would like to emphasize that not all neurotransmitters need to be equally affected in every patient.
While disturbances in the sympathetic–vagal axis, of GABA and serotonin signaling appear relatively pronounced, other systems might also be altered although the extent of their contribution is currently unclear. Nevertheless, together these disturbances could contribute to the overall imbalance of neurotransmitters in ME/CFS.
Disturbances of the Neurotransmitter Systems in ME/CFS
Noradrenergic System
Autoimmunity appears to play a central role in a subset of ME/CFS patients, with autoantibodies reported against several receptors. The best studied autoantibodies to date are those against the β₂AdR with higher levels correlating with symptom severity and structural alterations in the central nervous system (Sotzny et al., 2022), (Kimura et al., 2023). A recent study identified autoantibodies targeting, among others, the α1-adrenergic and α2C-adrenergic receptors (Hoheisel et al., 2025). Autoantibodies against the α1-and α2-receptors may contribute to orthostatic dysfunction, indirectly activating the noradrenergic system via orthostatic stress. In contrast, antibodies against the α2C-receptor could also directly increase noradrenergic activity by enhancing noradrenaline release from sympathetic neurons and nerve endings (Gyires et al., 2009). Indeed, antibody levels against a1- and a2-receptors were found to be significantly associated with the severity of autonomic dysfunction and fatigue (Hoheisel et al., 2025), (Freitag et al., 2021). In post-COVID-19 syndrome patients, autoantibodies against angiotensin II receptor type 1/2, against adrenoceptor beta 1/2, against muscarinic acetylcholine receptor M1/M3, and against C-X-C motif chemokine receptor 3 (CXCR3ab) were associated with altered parasympathetic and sympathetic tone (Schmitz et al., 2025).
Noradrenergic activity may also rise if catecholamine metabolism is impaired. Some studies showed that ME/CFS is associated with genetic variations in catechol-O-methyltransferase (COMT), the main enzymes degrading catecholamines (Sommerfeldt et al., 2011), (Hall et al., 2016), but could not be confirmed by others (Polli et al., 2022), (Löbel et al., 2015). An epigenetic investigation, however, reported increased MB-COMT DNA methylation, and this was considered an independent factor contributing to the symptoms and pathophysiology of ME/CFS with concomitant fibromyalgia (Polli et al., 2022). Decreased expression of COMT would enhance the activity of both noradrenaline and dopamine (discussed below) and thus contribute to the neurotransmitter imbalance. These factors may represent risk conditions that, under stress, disturb the balance between excitatory and inhibitory neurotransmitter systems. An open question is whether DNA methylation of COMT is a primary or secondary event.
Stress is a major pathogenic factor in ME/CFS. Most patients suffer from orthostatic intolerance and reduced cerebral blood flow (van Campen), and in Long COVID this has been shown to be prevalent already early in the disease course (van Campen et al., 2021), (Campen and Visser, 2022). Orthostatic dysfunction causes orthostatic stress, further activating the sympathetic system. Additional stressors—metabolic strain from poor skeletal muscle energetic situation, respiratory stress, pain, and psychosocial stress—likely stimulate the system. The expected sympathetic hyperactivity, brain overactivation and nocturnal hypervigilance are consistent with enhanced excitatory noradrenergic influences. However, a surprising finding requires a deeper consideration: noradrenaline was decreased in the liquor of patients (Walitt et al., 2024) while sympathetic tone was found raised in the same patients as in many other studies. There is no easy answer to these paradoxical findings. A conclusion is that the noradrenergic system must be dysregulated. But how? High sympathetic tone with tachycardia as in ME/CFS cannot be reconciled with cerebral catecholamine depletion. Experimental catecholamine depletion by reserpine causes among other effects, bradycardia, a decrease in blood pressure, hypothermia and sedation (Shore and Giachetti, 1978). ME/CFS patients cannot sleep, show tachycardia, often have subfebrile temperature, are usually not depressive, and are “wired”. These contradictory findings can be reconciled by the assumption that Locus Coeruleus is permanently overstimulated by stressors. Such permanent stimulation requires much energy, however supply is impaired due to malperfusion and disturbed neurovascular coupling (van Campen et al., 2020) (Shan et al., 2018), (Shan et al., 2020), (Tanaka et al., 2006). As a result, the Locus Coeruleus may not be able to recover and fail to generate an adequate sympathetic response during exercise and orthostatic stress. Orthostatic dysfunction and the poor energetic situation in skeletal muscle (see below) may even require a higher-than-normal level of sympathetic activation for everyday activities in ME/CFS. Assuming that sympathetic tone is chronically elevated but cannot be further raised, adrenergic receptors may not be sufficiently activated during exercise and for proper orthostatic regulation. In patients with connective tissue abnormalities and dilated veins due to weakness of the vascular connective tissue, the noradrenergic stimulation required to maintain adequate cardiac preload when standing and to further increase it during exercise is likely higher than normal. Contractile stimulation, however, may even be lower due to a disturbed noradrenergic system (too high at rest – too low on demand). The observation that ME/CFS patients show normal hemodynamics at rest but substantially reduced cardiac output during exercise in an invasive CPET study supports this interpretation (Jothi et al., 2025). Still, the potential contribution of hypovolemia to the diminished cardiac output should also be considered.
Apart from impaired contraction of capacitance vessels by insufficient α -adrenergic stimulation to enhance cardiac preload for proper orthostatic regulation and for exercise the following mechanisms may be involved related to ß-adrenergic receptors: ß2-adrengeric receptors (ß2AdR) are the most sensitive adrenergic receptors towards desensitization as known in heart failure and in experimental medicine (Lohse et al., 2003), (Cotecchia et al., 2012). In ME/CFS these could be desensitized by a permanently high sympathetic tone, and they could be dysfunctional due to autoantibodies. ß2AdRs raise blood flow to skeletal muscle, brain and heart and dilate bronchi enabling exercise. Thus, desensitization of ß2AdR, the inability to sufficiently stimulate them during exercise and autoantibodies could strongly interact to reduce ß2AdR receptor function, thereby impairing exercise capacity and thus contribute to exercise intolerance.
Proper function of ß2AdR is not only important with respect to adequate perfusion and respiration but also for skeletal muscle function. In skeletal muscle not only perfusion, but, probably even more important, stimulation of the Na+/K+-ATPase may be impaired by a disturbed ß2AdR function. This is because during exercise the stimuli of this ion transporter are ß2AdR and calcitonin-gene-related peptide (CGRP) released from sensory nerve fibers. CGRP, physiologically released from small sensory nerve fibers, is likely diminished as soon as small fiber neuropathy has developed. The functional consequences of a disturbed overall function of ß2AdR for skeletal muscle pathophysiology and exercise intolerance will be highlighted below in a separate section.
ß2AdRs should be more disturbed than ß1AdRs due to their high sensitivity to desensitization, but an impaired maximal stimulatory capacity of noradrenergic output as argued above during exercise would also affect and impair the ß1AdR-component. What are the effects of these disturbances on the heart? Chronotropic incompetence during exercise has been reported which can be fully explained by deficient ß-adrenergic stimulation as both ß-adrenergic receptor subtypes exert chronotropic effects (Davenport et al., 2019). Implicating deficient ß-adrenergic stimulation for causing chronotropic incompetence automatically leads to the assumption of an impaired inotropy and lusitropy (impaired diastolic relaxation for adequate cardiac filling) since these actions are also mediated via ßAdR-mediated rise in cAMP. A paper reported shortening of QTc interval in ME/CFS patients (Naschitz et al., 2006) which can only be attributed to dysfunction of ß2AdR because the cardiac action potential is physiologically shortened by ß1AdR-activation but prolonged by ß2AdR-activation (Wang et al., 2012), (Novodvorsky et al., 2018). This means both ß-adrenergic receptor subtypes have opposing effects on the action potential duration (reflected by the QT-time in the ECG). Shortening of the action potential also has a mild negative inotropic effect forcing down the calcium current and thereby calcium influx in the heart beyond the direct effect of diminished cAMP on the different mechanisms involved in cardiac calcium dynamics.
Pyridostigmine, which has been shown to improve orthostatic dysregulation and muscular fatigue in ME/CFS (Joseph et al., 2022), (Schlömer et al., 2025), can stimulate and amplify sympathetic tone in ME/CFS: The first ganglia of both the sympathetic and the parasympathetic system are cholinergic (nicotinic). Raising acetylcholine by inhibition of degradation by pyridostigmine theoretically can raise the activity of both arms of the autonomic nervous system. Since inhibition of cholinesterase is an amplifying mechanism, it will particularly raise the activity of the system that is already centrally activated. It means that, when sympathetic drive starts for exercise or for orthostatic regulation, pyridostigmine preferentially amplifies sympathetic tone. By contrast, when central vagal tone is predominant at rest, vagus nerve becomes more stimulated. Thus, pyridostigmine can theoretically improve the situation during both exercise and recovery. Pyridostigmine, which does not penetrate the blood brain barrier, may have other favorable effect via enhanced acetylcholine concentrations at the vagal nerve terminals and as a neuromuscular transmitter that also activates the Na+/K+-ATPase at rest (not during exercise) (Pirkmajer and Chibalin, 2016). Additionally, effects of acetylcholine via the α7-nicotinic (α7-nAChR)- and M3-cholinergic receptors subtype on endothelia cells are considered beneficial (Wang et al., 2001).
Stress-induced sympathetic activation typically reduces vagal tone. Further impairment may arise from gut dysbiosis, which disturbs the gut–brain axis (Iqbal et al., 2025), (Simadibrata et al., 2023), (Zhang et al., 2023), (Oh et al., 2025). This axis largely depends on serotonin release from enterochromaffin cells, linking vagal activity to serotonergic signaling (Barton et al., 2023), (Kaelberer et al., 2018). Thus, disturbed gut microbiome might therefore weaken vagal activity. Whether efferent vagal output e.g. to the heart and cardiovascular system is influenced by reduced serotonergic afferent vagal input from the gut remains to be demonstrated, however. Therapeutic interventions that stimulate the vagus nerve or enhance acetylcholine release may help improve the vagal tone.
Serotonin
Serotonin, a modulatory neurotransmitter, is likely to be of relevance in ME/CFS. Reduced tryptophan availability—caused by gut dysbiosis (Iqbal et al., 2025), (Simadibrata et al., 2023), (Zhang et al., 2023), (Oh et al., 2025) and diversion of tryptophan into the kynurenine pathway via increased IDO2 activity —is assumed to cause decreased serotonin synthesis (Guo et al., 2023), (Rus, 2025). Several studies show that the amount of tryptophan in blood is decreased in post-COVID-19-syndrome patients (Cysique et al., 2022), (Cron, 2023), (Chilosi et al., 2022). Another possible mechanism lowering the level of the serotonin precursor tryptophan is an increased activity by tryptophan-catabolizing enzyme indoleamine 2,3-dioxygenase (IDO)1, which is typically induced by interferon in viral infections (Guo et al., 2023).
Levels of serotonin or its metabolite 5-methoxytryptamin in blood are found decreased or unchanged (Wong et al., 2023), (Raij and Raij, 2024), (Nagy-Szakal et al., 2018), (Mathé et al., 2025). In another ME/CFS-study, plasma serotonin was not altered in women but significantly reduced in men. The ratio of tryptophan to serotonin was elevated in patients suggesting lower serotonin levels according to the authors (Abujrais et al., 2024). Therefore, serum serotonin does not seem to be a reliable biomarker. Furthermore, serotonin does not cross the blood–brain barrier to act as a neurotransmitter. In the brain, serotonin is generated from tryptophan so that tryptophan levels in blood, which are diminished, may be more relevant as a proxy. Serotonin’s role in stimulating afferent vagal activity in the gut via chromaffin cells through gut–brain interactions may also affect vagal function in Long COVID and ME/CFS (Barton et al., 2023), (Kaelberer et al., 2018).
Clinical observations suggest that serotonin reuptake inhibitors (SSRIs) can provide some symptomatic relief, supporting the hypothesis of serotoninergic dysregulation (Rus et al., 2023). SSRIs were shown to improve fatigue, cognitive impairment and hypersensitivity in ME/CFS. Raising serotonin is probably not beneficial in all respects: Post-COVID-19-syndrome patients are more sensitive to side effects of SSRIs than other patients (Rus et al., 2023). Serotonin particularly in the spinal cord can raise motor neuron excitability and output via the 5-HT2 receptor subtype (Giorgi et al., 2025), (Ghosh and Pearse, 2015), (Goodlich et al., 2023). In severe ME/CFS skeletal muscle membrane is most likely depolarized and therefore hyperexcitable (Wirth and Steinacker, 2025; discussion below) and serotonin may have the potential to worsen this aspect of muscle symptomatology.
GABA
Several findings suggest an impairment of the GABAergic system in ME/CFS which is the most important inhibitory system in the brain. This impairment may result from TRPM3 dysfunction as well as from genetic or autoimmune disturbances of SRRM3, a protein involved in GABA signaling. Both a SNP and autoantibodies against SRRM3 have been found and linked to ME/CFS, raising suspicion that GABAergic signaling is disturbed (Hoheisel et al., 2025), (Schlauch et al., 2016). A pilot study also reported reduced urinary GABA levels (Taenzer et al., 2023). TRPM3 dysfunction has been documented in natural killer cells, and low-dose naltrexone (LDN) has been shown to restore ion channel function function (Cabanas et al., 2019), (Cabanas et al., 2018), (Eaton-Fitch et al., 2022). Since TRPM3 is also expressed in sensory Aδ- and C-fibers and in neurons of the CNS where it is involved in the secretion of GABA (Seljeset et al., 2023), LDN may indirectly improve GABAergic signaling in the brain by improving TRPM3 function (Löhn and Wirth, 2024). This also fits into the concept of neurotransmitter rebalancing.
GABA plays a critical role not only in sleep regulation but also in skeletal muscle tone, where it exerts a relaxing effect (Delgado-Ramírez et al., 2023), (Thorstensen et al., 2024). Conversely, noradrenergic and glutamatergic activity increases muscle tone. Thus, an imbalance between noradrenaline, glutamate and GABA at the muscular level may contribute to ME/CFS muscle pathology (Wirth and Steinacker, 2025) . Additionally, GABA normally counterbalances excitatory glutamate, suggesting that impaired GABA function could explain observed increases in glutamate (see below in the section glutamate). The pathophysiological consequences of an elevated skeletal muscle tone as a consequence of an imbalance of these neurotransmitters involved in setting the muscle tone will be discussed below. Benzodiazepines (e.g. lorazepam), which enhance GABA receptor activity, are one of the most effective treatments for fatigue and sensory hypersensitivity in ME/CFS, however, their clinical use is limited by the risk of dependency.
Glutamate
A recent MRI study reports elevated brain level of glutamate and N-acetyl-aspartate in the brains of Long COVID and ME/CFS patients (Thapaliya et al., 2025). Glutamate is the most important excitatory neurotransmitter in the brain. Since GABAergic signaling normally inhibits glutamatergic activity, reduced GABAergic function could contribute to this increase (see section on GABA). An increase density of AMPA-type glutamate receptors on the post-synaptic neural cell surface was found to be associated with cognitive impairment in Long COVID in a recent study using [11 C]K-2 AMPAR PET imaging (Fujimoto et al., 2025), demonstrating a possible altered glutamatergic receptor signaling, rather than altered transmitter levels itself. The data therefore suggests disturbance of the neurotransmitter systems in a broad sense like in the case of alpha2-C-adrenergic receptors.
There is increasing evidence suggesting a role of neuroinflammation in ME/CFS (VanElzakker et al., 2018), (Tate et al., 2022a), (Glassford, 2017), (Lee et al., 2024). Low grade neuroinflammation indicated by cytokines found in cerebrospinal fluid may act on astrocytes and influence the handling of the neurotransmitters glutamate and GABA (Bastos et al., 2025). Astrocytes take up 80% of the neuronally released glutamate but only 20% of the released GABA (Garaschuk and Verkhratsky, 2019). Both neurotransmitters are recycled via the astrocytes and returned to the neurons as glutamine, which, in turn, release glutamate and GABA. Cytokines, generated in the process of neuroinflammation, are supposed to reduce both glutamate and GABA uptake in astrocytes. Since astrocytic glutamate uptake is much higher than that of GABA, the cytokine-induced inhibition of neurotransmitter uptake in astrocytes should result in an imbalance in favor of glutamate over GABA. This would increase neuronal excitatory effects of glutamate in the brain (Hu et al., 2000).
Memantine and Dextromethorphan, which act as NMDA (glutamate) receptor antagonists, have been discussed as potential treatment, although data on their efficacy in ME/CFS are not available. GABA has crucial inhibitory effects in the brain directly counteracting the excitatory effects of glutamate. The efficacy of benzodiazepines activating the GABA system indirectly supports the notion that dysregulation of the glutamate receptor plays an important role in disturbing the neurotransmitter balance.
Glycine
In the central nervous system glycine acts as an inhibitory neurotransmitter in certain regions of the CNS (e.g. spinal cord, brainstem). In addition, as a co-agonist at the NMDA receptor, it is involved in glutamatergic signal transmission (excitatory or modulatory). Glycine can be transported via the blood-brain-barrier by amino acid transporters (ASC transporter), but quantitatively its generation as a local neurotransmitter in the CNS from serine via the one-carbon metabolism seems to be by far more important (Shank et al., 1973). Serine and its derivatives sarcosine were found elevated in the liquor of ME/CFS patients with a decrease in 5-methyltetrahydrofolate (5MTHF) suggesting general dysfunction of folate and one-carbon metabolism in ME/CFS (Baraniuk, 2025) . Speculatively, this could reduce the availability of glycine as a local neurotransmitter in the CNS.
Dopamine
Dopaminergic activity may also be enhanced due to genetic or more likely epigenetic changes of COMT, such as altered DNA methylation, as explained above (Polli et al., 2022), reducing dopamine breakdown and raising its levels but direct evidence is missing. Clinically, low-dose aripiprazole, a dopamine receptor modulator, has shown some benefit. Its use at low doses is consistent with a broader therapeutic concept of neurotransmitter rebalancing.
Histamine
Many Long COVID and ME/CFS patients show increased mast cell activity. Peripherally, histamine likely contributes to pathophysiology through vascular effects: (i) inadequate arterial dilation causing blood diversion (“steal phenomenon”), and (ii) venous dilation via H2-receptors reducing venous contractility and preload (venous pooling) (Wirth and Löhn, 2022). Hypovolemia due to vascular leakage may further aggravate these effects. As histamine is unable to cross an intact blood–brain barrier, mast cells as resident immune cells in the brain must be considered a potential pathological source of histamine within the central nervous system. Centrally, histamine functions as an excitatory neurotransmitter, especially in the tuberomammillary nucleus (TMN) of the hypothalamus (Blandina et al., 2012), suggesting a potential role in hyperexcitability. Whether histamine acting as a central excitatory transmitter contributes to the disease remains to be investigated (Shank et al., 1973). An open question is whether a dysfunctional blood brain barrier could allow histamine to enter the brain. A disturbance of the blood brain barrier has been proposed in ME/CFS and Long COVID (Bested et al., 2001), (Tate et al., 2022b).
Among the antihistamines, brain penetrating drugs include diphenhydramine as a H1-histamine receptor antagonist and cimetidine blocking H2-receptors.
Summary of the neurotransmitter disturbances: We have compiled the available information on potential disturbances of neurotransmitters in ME/CFS beyond the well-known dysregulation of the sympathetic and vagal system. There are preliminary data supporting the notion that additional neurotransmitter systems may also be affected, including glutamate, serotonin and GABA, whereas for histamine, glycine and dopamine such indications remain hypothetical.
Considering these disturbances in a context leads to the conclusion that excitatory neurotransmitter activity may predominate over inhibitory, potentially explaining sympathetic hyperactivity, brain overactivation and hypersensitivities and skeletal muscle pathology as illustrated in Figure 1.
Role of Neurotransmitter Imbalance in the Pathophysiology of ME/CFS and Their Potential Contribution to Typical Symptoms
After presenting the results of our search suggesting a predominance of excitatory over inhibitory transmitters in ME/CFS, we aim to explain how this neurotransmitter imbalance could contribute to the typical symptomatology of ME/CFS. These considerations raise the possibility that ME/CFS symptoms are also influenced to a relevant extent by disturbed neurotransmitter regulation. As outlined earlier, this imbalance could be driven by autoantibodies, mast cell overactivity, low level neuroinflammation, tryptophan deficiency, genetic predisposition or epigenetic changes, and exposure to various stressors, with orthostatic stress likely playing a particularly important role.
Autoantibodies against peripheral neurotransmitter receptors and other structures can certainly play an important role in the disease pathology in ME/CFS such as antibodies against β₂AdR, alpha1-adrenergic receptors, M3-cholinergic receptors and the potassium-dependent sodium-calcium-exchanger NCKX3/ SLC24A3 to name a few (Hoheisel et al., 2025), but the disturbance of centrally acting neurotransmitter may be particularly relevant for the severity of the neurological symptoms, sleep disturbances and hypersensitivities and may also influence skeletal muscle pathophysiology.
Sleep Disturbances, Cognitive Impairment, and Sensory Hypersensitivities
A predominance of excitatory over inhibitory neurotransmitter systems will lead to sleep disturbances, nocturnal hypervigilance and brain overstimulation. This obviously hyper-alert state is associated with overactivation of brain regions that could also cause sensory and perceptual disturbances and hypersensitivities. As explained in a recent publication (Sandoval et al., 2025) these disturbances may be due to an overactivation of the anterior insula within the salience network, leading to excessive sensitivities. The authors further explain that this is likely related to the low excitability threshold of the anterior insula, with excessive stimuli leading to overmobilization of responses in non-threatening situations (Wortinger et al., 2016), (Schimmelpfennig et al., 2023). In support of these notions, increased activation of the salience network was found in patients with ME/CFS (Manca et al., 2021). Therefore, we consider the possibility that the symptoms involving sensitivity to lights, noises, and smells could be the result of an overactivation of the anterior insula by excitatory neurotransmitters while inhibitory influences are impaired. Thus, brain and particularly insula overactivation could lead to enhanced stimulus uptake in all sensory organs, while stimulus processing may be impaired because of a reduced global and local cerebral blood flow (van Campen et al., 2020) (Shan et al., 2018), (Shan et al., 2020), (Tanaka et al., 2006). The strongly enhanced incoming sensory information would not be appropriately processed to finally judge them realistically as non-threatening which could further enhance the hyper-alert state. Elevated neuronal activity increases energy demand, yet cerebral energy supply is impaired due to both reduced large-artery blood flow and disturbed neurovascular coupling (van Campen et al., 2020) (Shan et al., 2018), (Shan et al., 2020), (Tanaka et al., 2006). Broad brain overactivation also diminishes the ability to concentrate and focus on a single demanding cognitive task. Combined with reduced cerebral perfusion and severely disturbed sleep, these disturbances could account for the cognitive impairments commonly observed in patients as explained previously (Wirth et al., 2021). Reduced ATP-to-phosphocreatine (PCr) ratio in neuropsychiatric post-COVID patient was observed via 31P magnetic resonance spectroscopy in the cingulate cortex indicating impaired brain energy metabolism and, lower ATP/PCr ratios specifically correlated with poorer cognitive performance (Weber-Fahr et al., 2026).
Additional factors could worsen these disturbances: blood–brain barrier permeability appears to be increased, and intracranial pressure may be slightly elevated (Bragée et al., 2020), (Hasan-Olive et al., 2019).
Skeletal Muscle Pathophysiology
Enhanced noradrenergic and particularly glutamatergic activity, combined with reduced GABAergic signaling, increases skeletal muscle tone (Burgess et al., 2008). As we have described in a recent publication, skeletal muscle membrane—particularly in severely affected patients—appears to be in a state of depolarization, likely reflecting disturbed electrophysiology due to insufficient activity of the Na+/K+-ATPase required to maintain the negative resting membrane potential (Wirth and Steinacker, 2025). As the resting membrane potential then gets closer to the activation threshold causing hyperexcitability, increased central muscle tone due to a predominance of excitatory over inhibitory neurotransmitters involved in skeletal muscle tone regulation could therefore easily trigger abnormal excitations. These inappropriate excitations may manifest clinically as fasciculations and cramps and lead to sodium influx and potassium efflux, thereby further increasing the workload of the already impaired Na⁺/K⁺-ATPase. As a result, Na⁺/K⁺-ATPase can no longer restore the physiological membrane potential; depolarization worsens, calcium overload ensues, and mitochondria can be damaged. Mechanistically this involves reversal of the Na⁺/Ca²⁺ exchanger (NCX), which begins importing calcium instead of exporting it in the setting of elevated intracellular sodium and a more positive membrane potential.
High stress levels, finally mediated by the excitatory neurotransmitters, additionally promote vasoconstriction and desensitize β₂-adrenergic receptors. These receptors, together with calcitonin gene-related peptide (CGRP) during exercise are the only hormonal activators of the Na⁺/K⁺-ATPase. Its insufficient stimulation contributes to intracellular sodium accumulation and calcium overload and subsequent mitochondrial and myocyte injury.
Why Current Treatments Show Limited Efficacy in ME/CFS
At present, no treatment for ME/CFS provides satisfactory improvement, let alone disease remission. Several factors may explain the limited efficacy of currently available therapeutic approaches. Once ME/CFS is established, the dominant pathophysiological mechanisms appear to shift from early neurotransmitter imbalances toward self-sustaining pathomechanisms, including mitochondrial dysfunction, persistent skeletal muscle depolarization, oxidative stress, and impaired tissue perfusion. These processes mutually reinforce one another through complex feedback loops involving sympathetic overactivity, hypovolemia, inflammation, and chronic stress responses. As a result, cardinal clinical symptoms such as exercise intolerance and post-exertional malaise (PEM) become largely independent of the original disease trigger.
The effectiveness of pharmacological interventions targeting neurotransmitter systems is further constrained by substantial interindividual variability in the specific neurotransmitters affected, as well as by the fact that multiple physiological systems can be disturbed simultaneously. This limitation is particularly evident in patients with clear autoimmune signatures, with autoantibodies directed against multiple targets and receptors, often extending beyond classical neurotransmitter-related structures to include peripheral and non-neuronal structures. In such cases, interventions aimed at only one or two neurotransmitter systems may be insufficient or poorly aligned with the patient’s dominant pathophysiological pathomechanisms. For these patients, therapeutic strategies targeting autoimmune mechanisms may hold greater promise.
More effective rebalancing approaches may require carefully titrated combinations of drugs that address multiple neurotransmitter systems as well as central regulation of muscle tone, potentially including antihistamines. However, poor drug tolerability and marked interindividual variability pose major challenges to combination therapies. These factors necessitate very low starting doses, slow and cautious titration, and a high degree of clinical patience. Additionally, many patients appear to rely on elevated sympathetic activation as a compensatory mechanism to counteract metabolic fatigue and orthostatic dysfunction. While this persistent sympathetic overactivation may offer short-term functional compensation, it likely contributes to disease perpetuation and worsening over time. Consequently, in some patients, reducing sympathetic overshoot with sympatholytic agents may be beneficial, whereas in others it may aggravate symptoms and functional impairment.
Conclusions
Growing evidence indicates that, in addition to the noradrenergic system, several other neurotransmitter systems—particularly glutamate, serotonin and GABA—are dysregulated in ME/CFS. This imbalance, characterized by excessive excitatory relative to inhibitory signaling, may drive neural overactivation and autonomic dysfunction. These disturbances may cause key neurological symptoms and contribute to skeletal-muscle dysfunction that manifests as exercise intolerance, post-exertional malaise (PEM), fasciculations and cramps. The aim of future research should be to systematically investigate which neurotransmitter systems are affected across the ME/CFS patient population and to determine whether interindividual differences exist. Assuming substantial heterogeneity exists, a long-term objective should be to identify each patient’s specific neurotransmitter abnormalities to enable optimally individualized, targeted rebalancing therapies.
Author Contributions
KW: Writing – original draft, writing – review and editing. JCS: Writing – original draft, writing – review and editing.
Funding
This research received no external funding.
Data availability statement
The original contributions presented in the study are included in the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Abujrais, S.; Vallianatou, T.; Bergquist, J. Untargeted Metabolomics and Quantitative Analysis of Tryptophan Metabolites in Myalgic Encephalomyelitis Patients and Healthy Volunteers: A Comparative Study Using High-Resolution Mass Spectrometry. ACS Chemical Neuroscience 2024, 15, 3525–3534. [Google Scholar] [CrossRef]
- Baraniuk, J.N. Exertional Exhaustion (Post-Exertional Malaise, PEM) Evaluated by the Effects of Exercise on Cerebrospinal Fluid Metabolomics–Lipidomics and Serine Pathway in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. International Journal of Molecular Sciences 2025, 26, 1282. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.R.; Londregan, A.K.; Alexander, T.D.; Entezari, A.A.; Covarrubias, M.; Waldman, S.A. Enteroendocrine cell regulation of the gut-brain axis. Frontiers in Neuroscience 2023, 17, 1272955. [Google Scholar] [CrossRef] [PubMed]
- Bastos, V.C.; Greene, K.A.; Tabachnikova, A.; Bhattacharjee, B.; Sjögren, P.; Bertilson, B.; Reifert, J.; Zhang, M.; Kamath, K.; Shon, J. Cerebrospinal fluid immune phenotyping reveals distinct immunotypes of myalgic encephalomyelitis/chronic fatigue syndrome. The Journal of Immunology 2025, vkaf087. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, A.; Burton, A.R.; Lemon, J.; Bennett, B.K.; Lloyd, A.; Vollmer-Conna, U. Reduced cardiac vagal modulation impacts on cognitive performance in chronic fatigue syndrome. PloS one 2012, 7, e49518. [Google Scholar] [CrossRef]
- Bested, A.; Saunders, P.; Logan, A. Chronic fatigue syndrome: neurological findings may be related to blood–brain barrier permeability. Medical hypotheses 2001, 57, 231–237. [Google Scholar] [CrossRef]
- Blandina, P.; Munari, L.; Provensi, G.; Passani, M.B. Histamine neurons in the tuberomamillary nucleus: a whole center or distinct subpopulations? Frontiers in systems neuroscience 2012, 6, 33. [Google Scholar] [CrossRef]
- Boneva, R.S.; Decker, M.J.; Maloney, E.M.; Lin, J.-M.; Jones, J.F.; Helgason, H.G.; Heim, C.M.; Rye, D.B.; Reeves, W.C. Higher heart rate and reduced heart rate variability persist during sleep in chronic fatigue syndrome: a population-based study. Autonomic Neuroscience 2007, 137, 94–101. [Google Scholar] [CrossRef]
- Bragée, B.; Michos, A.; Drum, B.; Fahlgren, M.; Szulkin, R.; Bertilson, B.C. Signs of Intracranial Hypertension, Hypermobility, and Craniocervical Obstructions in Patients With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Frontiers in Neurology 2020, 11. [Google Scholar] [CrossRef]
- Burgess, C.; Lai, D.; Siegel, J.; Peever, J. An endogenous glutamatergic drive onto somatic motoneurons contributes to the stereotypical pattern of muscle tone across the sleep–wake cycle. Journal of Neuroscience 2008, 28, 4649–4660. [Google Scholar] [CrossRef]
- Cabanas, H.; Muraki, K.; Balinas, C.; Eaton-Fitch, N.; Staines, D.; Marshall-Gradisnik, S. Validation of impaired Transient Receptor Potential Melastatin 3 ion channel activity in natural killer cells from Chronic Fatigue Syndrome/ Myalgic Encephalomyelitis patients. Molecular Medicine 2019, 25, 14. [Google Scholar] [CrossRef]
- Cabanas, H.; Muraki, K.; Eaton, N.; Balinas, C.; Staines, D.; Marshall-Gradisnik, S. Loss of Transient Receptor Potential Melastatin 3 ion channel function in natural killer cells from Chronic Fatigue Syndrome/Myalgic Encephalomyelitis patients. Molecular Medicine 2018, 24, 44. [Google Scholar] [CrossRef]
- van Campen, C.M.; Visser, F.C. Long-Haul COVID patients: prevalence of POTS are reduced but cerebral blood flow abnormalities remain abnormal with longer disease duration. Presented at the Healthcare, MDPI; 2022; p. 2105. [Google Scholar]
- Chilosi, M.; Doglioni, C.; Ravaglia, C.; Martignoni, G.; Salvagno, G.L.; Pizzolo, G.; Bronte, V.; Poletti, V. Unbalanced IDO1/IDO2 endothelial expression and skewed keynurenine pathway in the pathogenesis of COVID-19 and post-COVID-19 pneumonia. Biomedicines 2022, 10, 1332. [Google Scholar] [CrossRef]
- Cotecchia, S.; Stanasila, L.; Diviani, D. Protein-protein interactions at the adrenergic receptors. Current drug targets 2012, 13, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Cron, R.Q. Immunologic prediction of long COVID. Nature Immunology 2023, 24, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Cysique, L.A.; Jakabek, D.; Bracken, S.G.; Allen-Davidian, Y.; Heng, B.; Chow, S.; Dehhaghi, M.; Pires, A.S.; Darley, D.R.; Byrne, A. Post-acute COVID-19 cognitive impairment and decline uniquely associate with kynurenine pathway activation: a longitudinal observational study; Medrxiv, 2022-06. [Google Scholar]
- Davenport, T.E.; Lehnen, M.; Stevens, S.R.; VanNess, J.M.; Stevens, J.; Snell, C.R. Chronotropic Intolerance: An Overlooked Determinant of Symptoms and Activity Limitation in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome? Frontiers in Pediatrics 2019, 7, 82. [Google Scholar] [CrossRef]
- Delgado-Ramírez, X.; Alvarado-Cervantes, N.S.; Jiménez-Barrios, N.; Raya-Tafolla, G.; Felix, R.; Martínez-Rojas, V.A.; Delgado-Lezama, R. GABAB Receptors Tonically Inhibit Motoneurons and Neurotransmitter Release from Descending and Primary Afferent Fibers. Life 2023, 13, 1776. [Google Scholar] [CrossRef]
- Eaton-Fitch, N.; Du Preez, S.; Cabanas, H.; Muraki, K.; Staines, D.; Marshall-Gradisnik, S. Impaired TRPM3-dependent calcium influx and restoration using Naltrexone in natural killer cells of myalgic encephalomyelitis/chronic fatigue syndrome patients. Journal of Translational Medicine 2022, 20, 94. [Google Scholar] [CrossRef]
- Freeman, R.; Komaroff, A.L. Does the chronic fatigue syndrome involve the autonomic nervous system? The American journal of medicine 1997, 102, 357–364. [Google Scholar] [CrossRef]
- Freitag, H.; Szklarski, M.; Lorenz, S.; Sotzny, F.; Bauer, S.; Philippe, A.; Kedor, C.; Grabowski, P.; Lange, T.; Riemekasten, G.; Heidecke, H.; Scheibenbogen, C. Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J Clin Med 2021, 10, 3675. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Abe, H.; Eiro, T.; Tsugawa, S.; Tanaka, M.; Hatano, M.; Nakajima, W.; Ichijo, S.; Arisawa, T.; Takada, Y. Systemic increase of AMPA receptors associated with cognitive impairment of long COVID. Brain Communications 2025, 7, fcaf337. [Google Scholar] [CrossRef]
- Garaschuk, O.; Verkhratsky, A. GABAergic astrocytes in Alzheimer’s disease. Aging (Albany NY) 2019, 11, 1602. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Pearse, D.D. The role of the serotonergic system in locomotor recovery after spinal cord injury. Frontiers in neural circuits 2015, 8, 151. [Google Scholar] [CrossRef]
- Giorgi, A.; Heckman, C.; Perreault, M.-C. Spinally Projecting Serotonergic Neurons in Motor Network Modulation. Journal of Neurophysiology, 2025.
- Glassford, J.A. The neuroinflammatory etiopathology of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Frontiers in physiology 2017, 8, 88. [Google Scholar] [CrossRef]
- Goodlich, B.I.; Del Vecchio, A.; Horan, S.A.; Kavanagh, J.J. Blockade of 5-HT2 receptors suppresses motor unit firing and estimates of persistent inward currents during voluntary muscle contraction in humans. The Journal of Physiology 2023, 601, 1121–1138. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Appelman, B.; Mooij-Kalverda, K.; Houtkooper, R.H.; van Weeghel, M.; Vaz, F.M.; Dijkhuis, A.; Dekker, T.; Smids, B.S.; Duitman, J.W. Prolonged indoleamine 2, 3-dioxygenase-2 activity and associated cellular stress in post-acute sequelae of SARS-CoV-2 infection. EBioMedicine 2023, 94. [Google Scholar] [CrossRef] [PubMed]
- Gyires, K.; Zádori, Z.S.; Török, T.; Mátyus, P. α2-Adrenoceptor subtypes-mediated physiological, pharmacological actions. Neurochemistry international 2009, 55, 447–453. [Google Scholar] [CrossRef]
- Hall, K.T.; Kossowsky, J.; Oberlander, T.F.; Kaptchuk, T.J.; Saul, J.P.; Wyller, V.B.; Fagermoen, E.; Sulheim, D.; Gjerstad, J.; Winger, A. Genetic variation in catechol-O-methyltransferase modifies effects of clonidine treatment in chronic fatigue syndrome. The pharmacogenomics journal 2016, 16, 454–460. [Google Scholar] [CrossRef]
- Hasan-Olive, M.M.; Hansson, H.-A.; Enger, R.; Nagelhus, E.A.; Eide, P.K. Blood-Brain Barrier Dysfunction in Idiopathic Intracranial Hypertension. Journal of Neuropathology & Experimental Neurology 2019, 78, 808–818. [Google Scholar] [CrossRef]
- Hoheisel, F.; Fleischer, K.M.; Rubarth, K.; Sepúlveda, N.; Bauer, S.M.; Konietschke, F.; Kedor, C.; Stein, A.E.; Wittke, K.; Seifert, M. Exploratory Study on Autoantibodies to Arginine-rich Human Peptides Mimicking Epstein-Barr Virus in Women with Post-COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Frontiers in Immunology 2025, 16, 1650948. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Ehrlich, L.C.; Peterson, P.K.; Chao, C.C. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 2000, 7, 153–159. [Google Scholar] [CrossRef]
- Iqbal, N.T.; Khan, H.; Khalid, A.; Mahmood, S.F.; Nasir, N.; Khanum, I.; de Siqueira, I.; Van Voorhis, W. Chronic inflammation in post-acute sequelae of COVID-19 modulates gut microbiome: a review of literature on COVID-19 sequelae and gut dysbiosis. Molecular Medicine 2025, 31, 22. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Pari, R.; Miller, S.; Warren, A.; Stovall, M.C.; Squires, J.; Chang, C.-J.; Xiao, W.; Waxman, A.B.; Systrom, D.M. Neurovascular Dysregulation and Acute Exercise Intolerance in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Randomized, Placebo-Controlled Trial of Pyridostigmine. CHEST 2022, 162, 1116–1126. [Google Scholar] [CrossRef]
- Jothi, S.; Insel, M.; Claessen, G.; Kubba, S.; Howden, E.J.; Ruiz-Carmona, S.; Levine, T.; Rischard, F.P. Long COVID and chronic fatigue syndrome/myalgic encephalitis share similar pathophysiologic mechanisms of exercise limitation. Physiological Reports 2025, 13, e70535. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361, eaat5236. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Sato, W.; Maikusa, N.; Ota, M.; Shigemoto, Y.; Chiba, E.; Arizono, E.; Maki, H.; Shin, I.; Amano, K.; Matsuda, H.; Yamamura, T.; Sato, N. Free-water-corrected diffusion and adrenergic/muscarinic antibodies in myalgic encephalomyelitis/chronic fatigue syndrome. Journal of Neuroimaging 2023, 33, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Sato, W.; Son, C.-G. Brain-regional characteristics and neuroinflammation in ME/CFS patients from neuroimaging: A systematic review and meta-analysis. Autoimmunity Reviews 2024, 23, 103484. [Google Scholar] [CrossRef]
- Löbel, M.; Mooslechner, A.A.; Bauer, S.; Günther, S.; Letsch, A.; Hanitsch, L.G.; Grabowski, P.; Meisel, C.; Volk, H.-D.; Scheibenbogen, C. Polymorphism in COMT is associated with IgG3 subclass level and susceptibility to infection in patients with chronic fatigue syndrome. Journal of Translational Medicine 2015, 13, 264. [Google Scholar] [CrossRef]
- Löhn, M.; Wirth, K.J. Potential pathophysiological role of the ion channel TRPM3 in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and the therapeutic effect of low-dose naltrexone. J Transl Med 2024, 22, 630. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What is the role of β-adrenergic signaling in heart failure? Circulation Research 2003, 93, 896–906. [Google Scholar] [CrossRef]
- Manca, R.; Khan, K.; Mitolo, M.; De Marco, M.; Grieveson, L.; Varley, R.; Wilkinson, I.D.; Venneri, A. Modulatory effects of cognitive exertion on regional functional connectivity of the salience network in women with ME/CFS: A pilot study. Journal of the Neurological Sciences 2021, 422, 117326. [Google Scholar] [CrossRef]
- Mathé, P.; Götz, V.; Stete, K.; Walzer, D.; Hilger, H.; Pfau, S.; Hofmann, M.; Rieg, S.; Kern, W.V. No reduced serum serotonin levels in patients with post-acute sequelae of COVID-19. Infection 2025, 53, 463–466. [Google Scholar] [CrossRef]
- Nagy-Szakal, D.; Barupal, D.K.; Lee, B.; Che, X.; Williams, B.L.; Kahn, E.J.R.; Ukaigwe, J.E.; Bateman, L.; Klimas, N.G.; Komaroff, A.L.; Levine, S.; Montoya, J.G.; Peterson, D.L.; Levin, B.; Hornig, M.; Fiehn, O.; Lipkin, W.I. Insights into myalgic encephalomyelitis/chronic fatigue syndrome phenotypes through comprehensive metabolomics. Scientific Reports 2018, 8, 10056. [Google Scholar] [CrossRef]
- Naschitz, J.E.; Mussafia-Priselac, R.; Kovalev, Y.; Zaigraykin, N.; Elias, N.; Rosner, I.; Slobodin, G. Patterns of Hypocapnia on Tilt in Patients with Fibromyalgia, Chronic Fatigue Syndrome, Nonspecific Dizziness, and Neurally Mediated Syncope. The American Journal of the Medical Sciences 2006, 331, 295–303. [Google Scholar] [CrossRef]
- Nelson, M.J.; Bahl, J.S.; Buckley, J.D.; Thomson, R.L.; Davison, K. Evidence of altered cardiac autonomic regulation in myalgic encephalomyelitis/chronic fatigue syndrome: A systematic review and meta-analysis. Medicine 2019, 98, e17600. [Google Scholar] [CrossRef]
- Novodvorsky, P.; Bernjak, A.; Robinson, E.J.; Iqbal, A.; Macdonald, I.A.; Jacques, R.M.; Marques, J.L.; Sheridan, P.J.; Heller, S.R. Salbutamol-induced electrophysiological changes show no correlation with electrophysiological changes during hyperinsulinaemic–hypoglycaemic clamp in young people with Type 1 diabetes. Diabetic Medicine 2018, 35, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; An, S.; Park, K.; Lee, S.; Han, Y.M.; Koh, S.-J.; Lee, J.; Gim, H.; Kim, D.; Seo, H. Gut Microbial Signatures in Long COVID: Potential Biomarkers and Therapeutic Targets; Infectious Diseases and Therapy, 2025; pp. 1–15. [Google Scholar]
- Pirkmajer, S.; Chibalin, A.V. Na,K-ATPase regulation in skeletal muscle. Am J Physiol Endocrinol Metab 2016, 311, E1–E31. [Google Scholar] [CrossRef] [PubMed]
- Polli, A.; Hendrix, J.; Ickmans, K.; Bakusic, J.; Ghosh, M.; Monteyne, D.; Velkeniers, B.; Bekaert, B.; Nijs, J.; Godderis, L. Genetic and epigenetic regulation of Catechol-O-methyltransferase in relation to inflammation in chronic fatigue syndrome and Fibromyalgia. Journal of Translational Medicine 2022, 20, 487. [Google Scholar] [CrossRef]
- Raij, T.; Raij, K. Association between fatigue, peripheral serotonin, and L-carnitine in hypothyroidism and in chronic fatigue syndrome. Frontiers in Endocrinology 2024, 15, 1358404. [Google Scholar] [CrossRef] [PubMed]
- Rus, C.P. Disruptions in serotonin-and kynurenine pathway metabolism in post-COVID: biomarkers and treatment. Frontiers in Neurology 2025, 16, 1532383. [Google Scholar] [CrossRef] [PubMed]
- Rus, C.P.; de Vries, B.E.; de Vries, I.E.; Nutma, I.; Kooij, J.S. Treatment of 95 post-Covid patients with SSRIs. Scientific Reports 2023, 13, 18599. [Google Scholar] [CrossRef]
- Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Neuroimmunology: what role for autoimmunity, neuroinflammation, and small fiber neuropathy in fibromyalgia, chronic fatigue syndrome, and adverse events after human papillomavirus vaccination? International journal of molecular sciences 2019, 20, 5164. [Google Scholar] [CrossRef]
- Sandoval, A.E.; Li, M.; Jason, L.A. Two Neurocognitive Domains Identified for Patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Post-Acute Sequelae of COVID-19. Frontiers in Neurology 2025, 16, 1612548. [Google Scholar] [CrossRef]
- Schimmelpfennig, J.; Topczewski, J.; Zajkowski, W.; Jankowiak-Siuda, K. The role of the salience network in cognitive and affective deficits. Frontiers in human neuroscience 2023, 17, 1133367. [Google Scholar] [CrossRef] [PubMed]
- Schlauch, K.A.; Khaiboullina, S.F.; De Meirleir, K.L.; Rawat, S.; Petereit, J.; Rizvanov, A.A.; Blatt, N.; Mijatovic, T.; Kulick, D.; Palotás, A. Genome-wide association analysis identifies genetic variations in subjects with myalgic encephalomyelitis/chronic fatigue syndrome. Translational psychiatry 2016, 6, e730–e730. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, B.; Garbsch, R.; Schäfer, H.; Bär, C.; Chatterjee, S.; Riemekasten, G.; Schulze-Forster, K.; Heidecke, H.; Schultheiß, C.; Binder, M. Autonomic dysfunction and vasoregulation in long COVID-19 are linked to anti-GPCR autoantibodies. Journal of Allergy and Clinical Immunology, 2025.
- Seljeset, S.; Liebowitz, S.; Bright, D.P.; Smart, T.G. Pre- and postsynaptic modulation of hippocampal inhibitory synaptic transmission by pregnenolone sulphate. Neuropharmacology 2023, 233, 109530. [Google Scholar] [CrossRef]
- Shan, Z.Y.; Barnden, L.R.; Kwiatek, R.A.; Bhuta, S.; Hermens, D.F.; Lagopoulos, J. Neuroimaging characteristics of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): a systematic review. Journal of Translational Medicine 2020, 18, 335. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.Y.; Finegan, K.; Bhuta, S.; Ireland, T.; Staines, D.R.; Marshall-Gradisnik, S.M.; Barnden, L.R. Brain function characteristics of chronic fatigue syndrome: A task fMRI study. NeuroImage: Clinical 2018, 19, 279–286. [Google Scholar] [CrossRef]
- Shank, R.; Aprison, M.; Baxter, C. Precursors of glycine in the nervous system: comparison of specific activities in glycine and other amino acids after administration of [U-14C] glucose,[3, 4-14C] glucose,[1-14C] glucose,[U-14C] serine or [1, 5-14C] citrate to the rat. Brain Research 1973, 52, 301–308. [Google Scholar] [CrossRef]
- Shore, P.A.; Giachetti, A. Reserpine: basic and clinical pharmacology. In Handbook of Psychopharmacology; Neuroleptics and Schizophrenia. Springer, 1978; Volume 10, pp. 197–219. [Google Scholar]
- Simadibrata, D.M.; Lesmana, E.; Gunawan, J.; Quigley, E.M.; Simadibrata, M. A systematic review of gut microbiota profile in COVID-19 patients and among those who have recovered from COVID-19. Journal of Digestive Diseases 2023, 24, 244–261. [Google Scholar] [CrossRef]
- Sisto, S.A.; Tapp, W.; Drastal, S.; Bergen, M.; DeMasi, I.; Cordero, D.; Natelson, B. Vagal tone is reduced during paced breathing in patients with the chronic fatigue syndrome. Clinical Autonomic Research 1995, 5, 139–143. [Google Scholar] [CrossRef]
- Słomko, J.; Estévez-López, F.; Kujawski, S.; Zawadka-Kunikowska, M.; Tafil-Klawe, M.; Klawe, J.J.; Morten, K.J.; Szrajda, J.; Murovska, M.; Newton, J.L. Autonomic phenotypes in chronic fatigue syndrome (CFS) are associated with illness severity: a cluster analysis. Journal of Clinical Medicine 2020, 9, 2531. [Google Scholar] [CrossRef]
- Sommerfeldt, L.; Portilla, H.; Jacobsen, L.; Gjerstad, J.; Wyller, V.B. Polymorphisms of adrenergic cardiovascular control genes are associated with adolescent chronic fatigue syndrome. Acta Paediatr 2011, 100, 293–8. [Google Scholar] [CrossRef]
- Sotzny, F.; Filgueiras, I.S.; Kedor, C.; Freitag, H.; Wittke, K.; Bauer, S.; Sepúlveda, N.; Mathias da Fonseca, D.L.; Baiocchi, G.C.; Marques, A.H.C.; Kim, M.; Lange, T.; Plaça, D.R.; Luebber, F.; Paulus, F.M.; De Vito, R.; Jurisica, I.; Schulze-Forster, K.; Paul, F.; Bellmann-Strobl, J.; Rust, R.; Hoppmann, U.; Shoenfeld, Y.; Riemekasten, G.; Heidecke, H.; Cabral-Marques, O.; Scheibenbogen, C. Dysregulated autoantibodies targeting vaso- and immunoregulatory receptors in Post COVID Syndrome correlate with symptom severity. Frontiers in immunology 13, 2022.
- Stewart, J.M. Autonomic Nervous System Dysfunction in Adolescents with Postural Orthostatic Tachycardia Syndrome and Chronic Fatigue Syndrome Is Characterized by Attenuated Vagal Baroreflex and Potentiated Sympathetic Vasomotion. Pediatric Research 2000, 48, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sadato, N.; Okada, T.; Mizuno, K.; Sasabe, T.; Tanabe, H.C.; Saito, D.N.; Onoe, H.; Kuratsune, H.; Watanabe, Y. Reduced responsiveness is an essential feature of chronic fatigue syndrome: A fMRI study. BMC Neurology 2006, 6, 9. [Google Scholar] [CrossRef]
- Tate, W.; Walker, M.; Sweetman, E.; Helliwell, A.; Peppercorn, K.; Edgar, C.; Blair, A.; Chatterjee, A. Molecular mechanisms of neuroinflammation in ME/CFS and long COVID to sustain disease and promote relapses. Frontiers in neurology 2022a, 13, 877772. [Google Scholar] [CrossRef] [PubMed]
- Tate, W.; Walker, M.; Sweetman, E.; Helliwell, A.; Peppercorn, K.; Edgar, C.; Blair, A.; Chatterjee, A. Molecular mechanisms of neuroinflammation in ME/CFS and long COVID to sustain disease and promote relapses. Frontiers in neurology 2022b, 13, 877772. [Google Scholar] [CrossRef] [PubMed]
- Thapaliya, K.; Marshall-Gradisnik, S.; Eaton-Fitch, N.; Eftekhari, Z.; Inderyas, M.; Barnden, L. Imbalanced brain neurochemicals in long COVID and ME/CFS: a preliminary study using MRI. The American journal of medicine 2025, 138, 567–574. [Google Scholar] [CrossRef]
- Thorstensen, J.R.; Henderson, T.T.; Kavanagh, J.J. Serotonergic and noradrenergic contributions to motor cortical and spinal motoneuronal excitability in humans. Neuropharmacology 2024, 242, 109761. [Google Scholar] [CrossRef]
- van Campen, C.; Rowe, P.C.; Visser, F.C. Cerebral Blood Flow Is Reduced in Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients During Mild Orthostatic Stress Testing: An Exploratory Study at 20 Degrees of Head-Up Tilt Testing. Healthcare (Basel) 2020, 8. [Google Scholar] [CrossRef]
- van van Campen, C.M.C.; Rowe, P.C.; Visser, F.C. Orthostatic symptoms and reductions in cerebral blood flow in Long-Haul COVID-19 patients: similarities with myalgic encephalomyelitis/chronic fatigue syndrome. Medicina 2021, 58, 28. [Google Scholar] [CrossRef]
- Van Cauwenbergh, D.; Nijs, J.; Kos, D.; Van Weijnen, L.; Struyf, F.; Meeus, M. Malfunctioning of the autonomic nervous system in patients with chronic fatigue syndrome: a systematic literature review. European journal of clinical investigation 2014, 44, 516–526. [Google Scholar] [CrossRef]
- Van Oosterwijck, J.; Marusic, U.; De Wandele, I.; Meeus, M.; Paul, L.; Lambrecht, L.; Moorkens, G.; Danneels, L.; Nijs, J. Reduced parasympathetic reactivation during recovery from exercise in myalgic encephalomyelitis/chronic fatigue syndrome. Journal of clinical medicine 2021, 10, 4527. [Google Scholar] [CrossRef]
- VanElzakker, M.B.; Brumfield, S.A.; Lara Mejia, P.S. Neuroinflammation and Cytokines in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A Critical Review of Research Methods. Front Neurol 2018, 9, 1033. [Google Scholar] [CrossRef]
- Walitt, B.; Singh, K.; LaMunion, S.R.; Hallett, M.; Jacobson, S.; Chen, K.; Enose-Akahata, Y.; Apps, R.; Barb, J.J.; Bedard, P. Deep phenotyping of post-infectious myalgic encephalomyelitis/chronic fatigue syndrome. Nature communications 2024, 15, 907. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Zhu, H.; Wang, S.; Zhang, X.; Xu, D.; Cao, K.; Zou, J. Increased response to β2-adrenoreceptor stimulation augments inhibition of IKr in heart failure ventricular myocytes, 2012.
- Wang, Y.; Pereira, E.; Maus, A.; Ostlie, N.; Navaneetham, D.; Lei, S.; Albuquerque, E.; Conti-Fine, B. Human bronchial epithelial and endothelial cells express α7 nicotinic acetylcholine receptors. Molecular pharmacology 2001, 60, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Weber-Fahr, W.; Dommke, S.; Sack, M.; Alzein, N.; Becker, R.; Demirakca, T.; Ende, G.; Schilling, C. Reduced ATP-to-phosphocreatine ratios in neuropsychiatric post-COVID condition: Evidence from 31P magnetic resonance spectroscopy. Biological Psychiatry, 2026.
- Wirth, K.; Steinacker, J.M. The Potential Causes Of Myasthenia And Fasciculations In The Severely Ill Me/Cfs-Patient: Role Of Disturbed Electrophysiology, 2025.
- Wirth, K.J.; Löhn, M. Orthostatic Intolerance after COVID-19 Infection: Is Disturbed Microcirculation of the Vasa Vasorum of Capacitance Vessels the Primary Defect? Medicina 2022, 58, 1807. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.J.; Scheibenbogen, C.; Paul, F. An attempt to explain the neurological symptoms of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Journal of Translational Medicine 2021, 19, 471. [Google Scholar] [CrossRef]
- Wong, A.C.; Devason, A.S.; Umana, I.C.; Cox, T.O.; Dohnalová, L.; Litichevskiy, L.; Perla, J.; Lundgren, P.; Izzo, L.T.; Kim, J. Serotonin reduction in post-acute sequelae of viral infection. Cell 2023, 186, 4851–4867. [Google Scholar] [CrossRef] [PubMed]
- Wortinger, L.A.; Endestad, T.; Melinder, A.M.D.; Øie, M.G.; Sevenius, A.; Bruun Wyller, V. Aberrant resting-state functional connectivity in the salience network of adolescent chronic fatigue syndrome. PLoS One 2016, 11, e0159351. [Google Scholar] [CrossRef]
- Wyller, V.B.; Saul, J.; Amlie, J.P.; Thaulow, E. Sympathetic predominance of cardiovascular regulation during mild orthostatic stress in adolescents with chronic fatigue. Clinical physiology and functional imaging 2007, 27, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; LaManca, J.J.; Natelson, B.H. A measure of heart rate variability is sensitive to orthostatic challenge in women with chronic fatigue syndrome. Experimental biology and medicine 2003, 228, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhou, Y.; Ma, Y.; Chen, P.; Tang, J.; Yang, B.; Li, H.; Liang, M.; Xue, Y.; Liu, Y. Gut microbiota dysbiosis correlates with long COVID-19 at one-year after discharge. Journal of Korean medical science 2023, 38. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Neurotransmitter balance may be disturbed by autoantibodies, genetic predisposition, epigenetic modifications, neuroinflammation, and physiological stressors. A predominance of excitatory over inhibitory neurotransmission can lead to brain overactivation and autonomic dysfunction, resulting in downstream disturbances. These processes contribute to neurological symptoms and skeletal muscle dysfunction and may exacerbate exercise intolerance and PEM.
Figure 1.
Neurotransmitter balance may be disturbed by autoantibodies, genetic predisposition, epigenetic modifications, neuroinflammation, and physiological stressors. A predominance of excitatory over inhibitory neurotransmission can lead to brain overactivation and autonomic dysfunction, resulting in downstream disturbances. These processes contribute to neurological symptoms and skeletal muscle dysfunction and may exacerbate exercise intolerance and PEM.
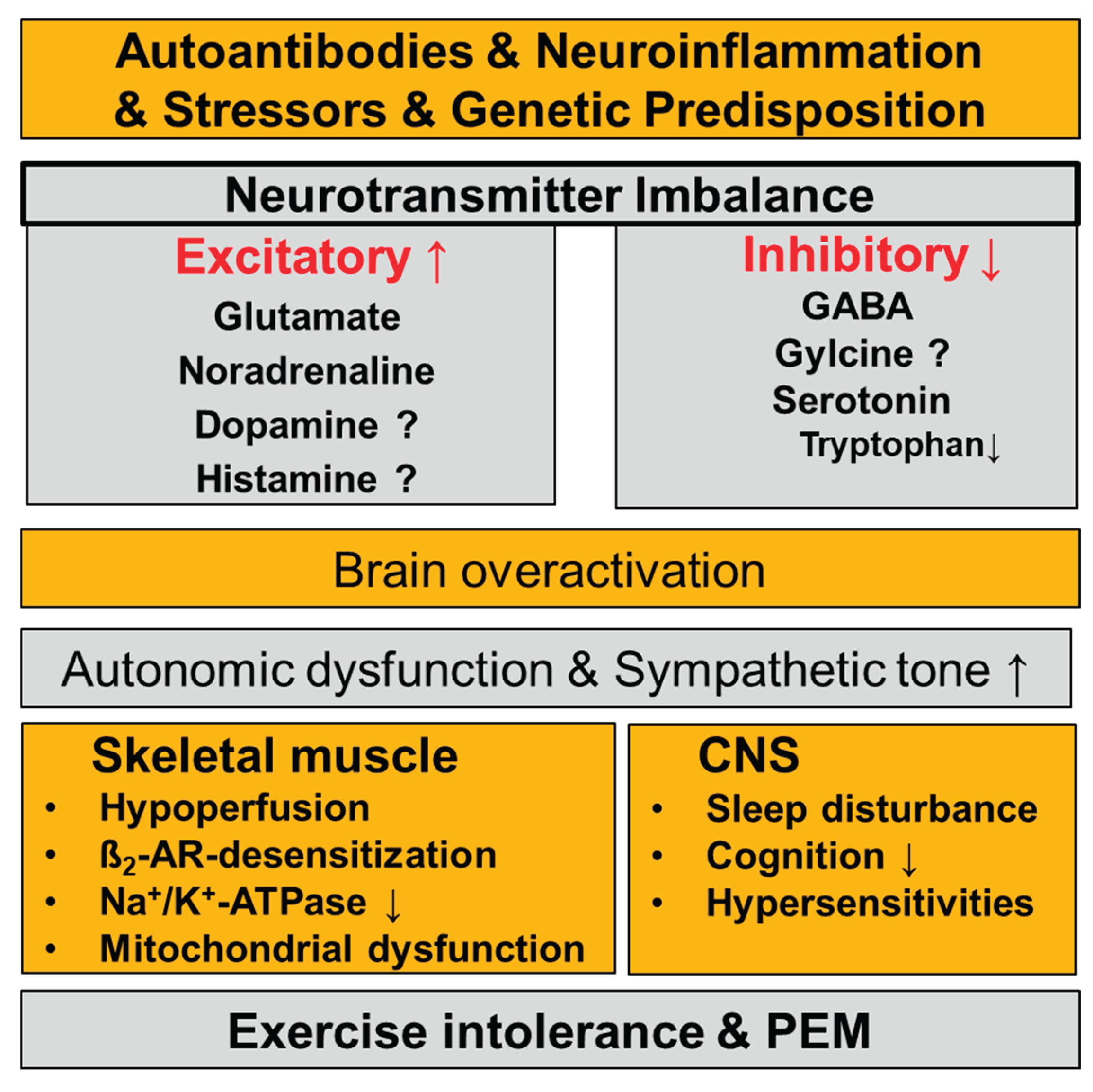

Table 1.
Overview of neurotransmitters, their receptors, main locations and key functions.
| Neurotransmitter | Effect | Receptors | Main locations | Key functions |
|---|---|---|---|---|
| Glutamate | Excitatory (main CNS excitatory transmitter) | Ionotropic: NMDA, AMPA, Kainate Metabotropic: mGluR (I–III) | Widely distributed in CNS (cortex, hippocampus, cerebellum) | Synaptic plasticity, learning & memory, motor control, sensory processing |
| Noradrenaline | Mainly excitatory | α₁, α₂, β₁, β₂, β3-adrenergic | Locus coeruleus, widespread CNS projections; peripheral sympathetic nerves | Arousal, attention, stress response, mood, blood pressure regulation |
| Acetylcholine (ACh) | Excitatory or inhibitory (receptor-dependent) | Nicotinic (ionotropic). Muscarinic (M1–M5) (metabotropic) |
Neuromuscular junction, autonomic nervous system, basal forebrain | Muscle contraction, memory & learning, autonomic control |
| Histamine | Excitatory/ modulatory |
H₁, H₂, H₃, H₄ | Hypothalamus (tuberomammillary nucleus), peripheral | Wakefulness, attention, appetite control, immune responses |
|
GABA (γ-aminobutyric acid) |
Inhibitory | GABA-A (ionotropic, Cl⁻), GABA-B (metabotropic), GABA-C | Brain and spinal cord | Inhibitory control, anxiety regulation, sleep, muscle tone |
| Glycine | Inhibitory | Glycine receptor (ionotropic, Cl⁻), Co-agonist at NMDA receptors | Spinal cord, brainstem | Motor and sensory inhibition, reflex control, NMDA receptor modulation |
| Serotonin (5-HT) | Mostly inhibitory/ modulatory |
5-HT₁–₇ (mostly metabotropic; 5-HT₃ ionotropic) | Raphe nuclei, widespread CNS | Mood, sleep, appetite, pain modulation, emotional regulation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



