
Submitted:
07 January 2026
Posted:
08 January 2026
You are already at the latest version
Abstract
Background. Obsessive–compulsive disorder (OCD) is highly heritable, yet the biological routes through which common variants confer risk have not been fully mapped. Glutamatergic dysregulation and faulty synaptic pruning both feature prominently in mechanistic models, but direct, large-scale genetic evaluations of these pathways are scarce.Methods. We analysed summary statistics from a recent genome-wide association meta-analysis of OCD (effective sample = 68 099). Four predefined gene sets were tested: glutamatergic signalling, synaptic pruning in shortened and expanded forms, and two negative-control sets (monoamine and housekeeping genes). Three complementary tools were applied: MAGMA for competitive gene-set enrichment, stratified LD-score regression for partitioned heritability, and S-PrediXcan for transcriptome-wide association signals across six GTEx brain tissues.Results. MAGMA detected no individual genes surpassing the genome-wide threshold. In contrast, LD-score regression showed pronounced and Bonferroni-corrected heritability enrichment for pruning-related sets (shortened set: 1.32-fold, p = 1.45 × 10⁻¹⁰³; expanded set: 1.05-fold, p = 1.13 × 10⁻¹³; pruning genes with glutamatergic overlap removed: 1.06-fold, p = 7.51 × 10⁻¹⁴). Glutamatergic sets were not enriched (all p > 0.84). S-PrediXcan produced modest but significant |Z|-score inflation in the pruning-shortened (1.19-fold, p = 0.044) and glutamatergic (1.06-fold, p = 0.032) collections. Directional TWAS signals pointed to complement and microglial drivers: C4A and the microglial marker TMEM119 both showed positive Z-scores. The negative-control sets were enriched in heritability analyses but not in TWAS, suggesting residual confounding rather than true biological relevance.Conclusions. Across convergent polygenic methods, synaptic pruning genes—independent of glutamatergic overlap—emerge as the principal enrichment signal in OCD. The results fit a model in which excessive microglia- and complement-mediated pruning disrupts cortico-striato-thalamo-cortical circuit maturation, setting the stage for compulsive behaviour. This work illustrates how integrated genomic approaches can refine mechanistic hypotheses and may inform early neurodevelopmental interventions for OCD.
Keywords:
OCD
; pruning
; genetics
; GWAS
; TWAS
; LDSC
Introduction
Obsessive–compulsive disorder (OCD) combines distressing, unwanted thoughts with repetitive acts meant to quiet the anxiety those thoughts create. The illness affects roughly 1–2 % of people worldwide and is a major source of disability, both personal and economic [1,2,3]. Although cognitive-behavioural therapy and serotonin re-uptake inhibitors help many patients, as many as 40–60 % do not achieve adequate relief, so clearer biological explanations are still needed [4].
Family, twin and population studies point to a moderate genetic influence. Heritability is estimated at about 27–47 % in adults and may reach 45–65 % in children [5,6]. Early genome-wide association studies (GWAS) were too small to detect individual risk variants, even when data were pooled across studies [7]. Larger samples have begun to change that picture: a 2024 analysis of more than 53 000 cases uncovered 30 independent loci and placed common-variant heritability near 29 % [6]. These results confirm that OCD risk is highly polygenic, but they leave open the question of which biological pathways are most relevant.
For many years glutamatergic signalling has been a prime suspect. Glutamate modulators sometimes improve symptoms, magnetic resonance spectroscopy shows elevated glutamate in cortico-striatal regions, and mouse models such as Sapap3 knock-outs display both disrupted glutamatergic synapses and compulsive grooming [4,8,9]. Even so, human genetic studies have not consistently highlighted glutamate-related genes, suggesting that other processes might be equally or more important.
One idea that is getting more and more attention is that OCD may be caused by problems with synaptic pruning during development. Research on schizophrenia indicated that excessive complement-mediated pruning, influenced by C4A variation, can reduce the thickness of cortical synapses [10]. In OCD, similar findings are coming to light: post-mortem data and PET scans show that the orbitofrontal and striatal circuits have fewer synapses [11,12]. Microglia-driven pruning in patient-derived cultures is also exaggerated and can be mitigated by pharmacological agents such as minocycline [13]. Experiments on rodents that change complement lead to repetitive, OCD-like behaviors that get better when pruning is lessened [14].
Despite these clues, systematic genetic tests that pit pruning pathways against glutamatergic pathways have been scarce. The present work tackles that gap. Using MAGMA for gene-set enrichment [15], stratified LD-score regression for heritability partitioning [16] and TWAS for predicted gene expression [17], we compared predefined sets of pruning-related genes, glutamatergic genes and negative-control sets. Our goals were threefold: first, to measure how strongly each pathway is enriched for common-variant risk; second, to see whether pruning signals stand on their own once genes shared with glutamate lists are removed; and third, to place the genetic findings in the context of existing cellular and systems-level data so that a coherent mechanistic story can emerge.
Methods
Gene-Based Testing with MAGMA
We re-analysed summary statistics from the 2025 obsessive-compulsive disorder genome-wide association study [6] that did not include 23andMe participants. After the quality checks reported by the original authors, 6,791,632 autosomal single-nucleotide polymorphisms (SNPs) with valid p values remained [15]. Using the case and control counts provided in the public file, the effective sample size was recalculated as Neff = 4 / (1/Ncases + 1/Ncontrols) = 68,099.
Gene analysis was carried out with MAGMA version 1.10 [15]. SNPs were mapped to protein-coding genes from the NCBI37.3 (hg19) build, adding a 35-kb margin upstream of the transcription start site and 10 kb downstream of the stop site (command option "--annotate window=35,10"). Linkage disequilibrium (LD) was estimated with the European reference panel from 1000 Genomes Phase 3 ("--bfile g1000_eur"). The analysis used the mean χ² model, which combines SNP p values while weighting them by LD, and took the effective sample size (68,099) into account. In total 18,279 genes carried at least one annotated SNP and were tested. Bonferroni correction for these 18,279 tests set genome-wide significance at p < 2.74 × 10⁻⁶.
Six predefined gene collections were evaluated with a competitive test that compares the average Z-score of genes in a set against the genome-wide average. Gene Z-scores were produced by transforming each MAGMA gene p value as Z = Φ⁻¹(1 – p). A one-sided one-sample t test assessed whether the mean Z within a set exceeded the global mean. The six collections were:
- Focused Glutamatergic (23 genes) – candidate glutamatergic targets collated from earlier pharmacological and genetic studies.
- Expanded_Glutamatergic (130 genes) – a broader glutamatergic pathway panel spanning 14 functional categories.
- Pruning_Shortened (38 genes) – a core list of synaptic pruning genes covering 10 biological themes.
- Pruning_Expanded (262 genes) – an extensive pruning pathway panel covering 25 themes, including complement, autophagy and immune modulators.
- Negative_Control_Monoamine (101 genes) – monoaminergic genes (dopaminergic, serotonergic, noradrenergic and histaminergic) used as a negative control.
- Negative_Control_Housekeeping (182 genes) – highly expressed housekeeping genes involved in basic cellular functions, also serving as a negative control.
For each set, significance was adjusted for testing six collections. Bonferroni correction required p < 0.0083; false-discovery-rate (FDR) q values were also reported.
Partitioned Heritability Analysis
To determine whether common-variant risk was concentrated in specific biological themes, stratified linkage disequilibrium score regression (LDSC) was applied following the framework of Finucane and colleagues [16]. European ancestry LD scores from phase 3 of the 1000 Genomes Project served as the reference. Genic coordinates were taken from GRCh37/hg19 and were extended 10 kb upstream and downstream so that proximal regulatory elements were retained. For every gene set a binary annotation was created, restricted to the autosomes. Single-nucleotide polymorphisms (SNPs) were divided into two mutually exclusive groups—annotated and non-annotated—and LDSC was run once for each annotation. Enrichment was expressed as the ratio of the mean χ² statistic in annotated SNPs to the corresponding mean in non-annotated SNPs; a second ratio in which the χ² values were normalised by their mean LD score provided an LD-adjusted estimate. Seven annotations were analysed: the six collections assessed in the earlier MAGMA work and an additional "Subtracted_Pruning" panel obtained by removing the 37 genes shared by the expanded pruning and expanded glutamatergic lists, leaving 225 unique pruning genes. Significance was evaluated with a one-tailed Mann–Whitney U test that compared the χ² distributions for annotated versus background SNPs. A Bonferroni threshold of p < 0.0071 (0.05/7) was applied.
Transcriptome-Wide Association
Genetically regulated expression was interrogated with S-PrediXcan, the summary-data implementation of PrediXcan [17]. Prediction weights derived with the MASHR procedure from GTEx v8 were downloaded for six brain regions that have been implicated in obsessive-compulsive circuitry: frontal cortex (BA9), anterior cingulate cortex (BA24), hippocampus, amygdala, nucleus accumbens, and caudate. GWAS summary statistics were aligned to the GTEx models; 6.79 million autosomal variants overlapped and were retained.
For each gene-tissue pair, the S-PrediXcan Z-score was calculated by multiplying the vector of SNP weights by the vector of GWAS Z-scores and scaling by the LD-adjusted variance of the weighted SNP set. In total, about 62,800 gene-tissue tests were produced. Within every tissue model, false-discovery rate (FDR) control was applied, and an additional Bonferroni threshold was used for gene-level correction.
Enrichment analyses were carried out for the seven hypothesis-driven gene sets described earlier. For each set, the mean absolute Z-score was compared with that of all other genes; significance was assessed with one-tailed Mann–Whitney tests.
Results
MAGMA Analysis
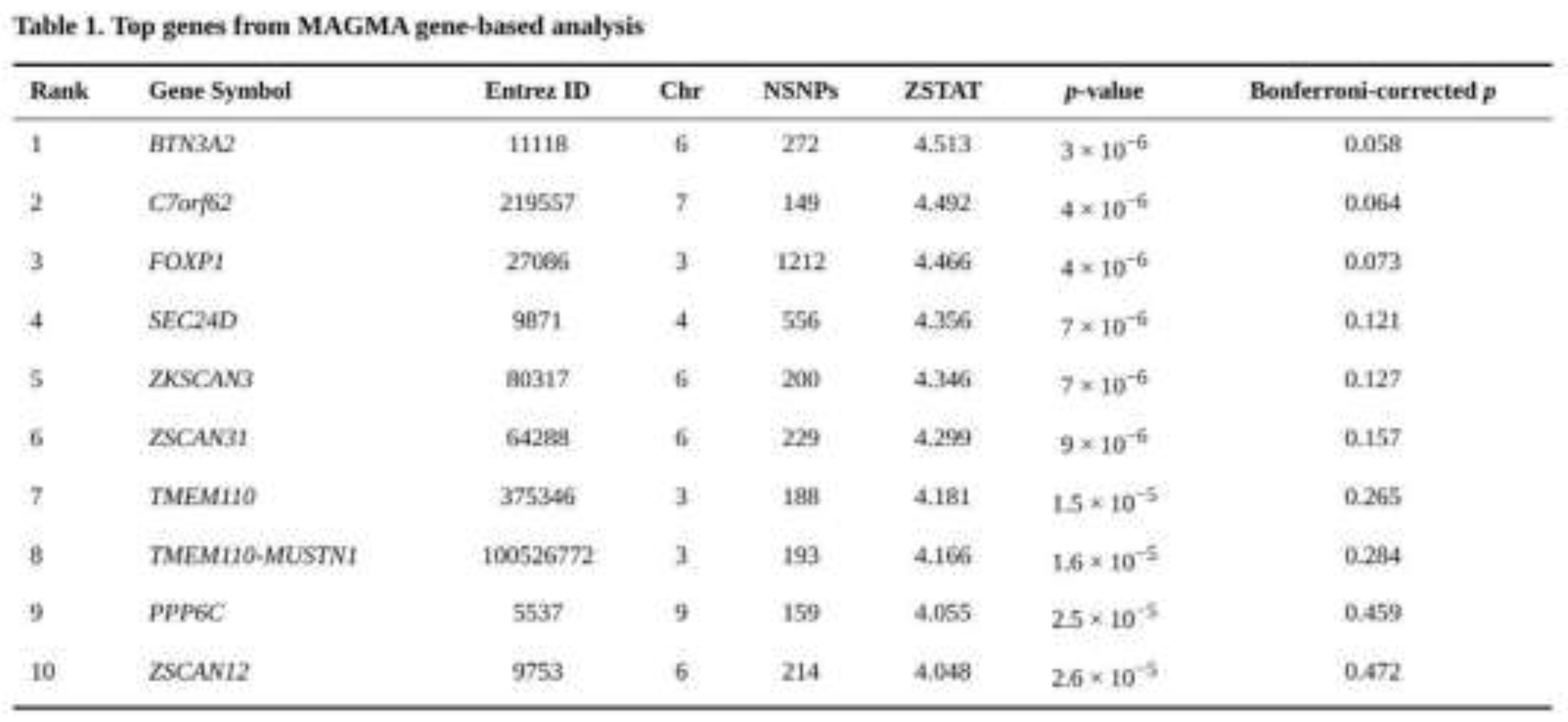
None of the 18,279 genes reached the Bonferroni threshold. Two hundred genes were suggestive at p < 0.001 and 2,070 showed nominal evidence at p < 0.05. The smallest nominal p value occurred at BTN3A2 (p = 3 × 10⁻⁶; Bonferroni-adjusted p = 0.058). Other prominent signals were C7orf62 and FOXP1 (both p = 4 × 10⁻⁶). Several of the leading associations clustered in the histone region on chromosome 6, including HIST1H4C, HIST1H3C and HIST1H3B. The 20 strongest [associations are summarised in Table 1.
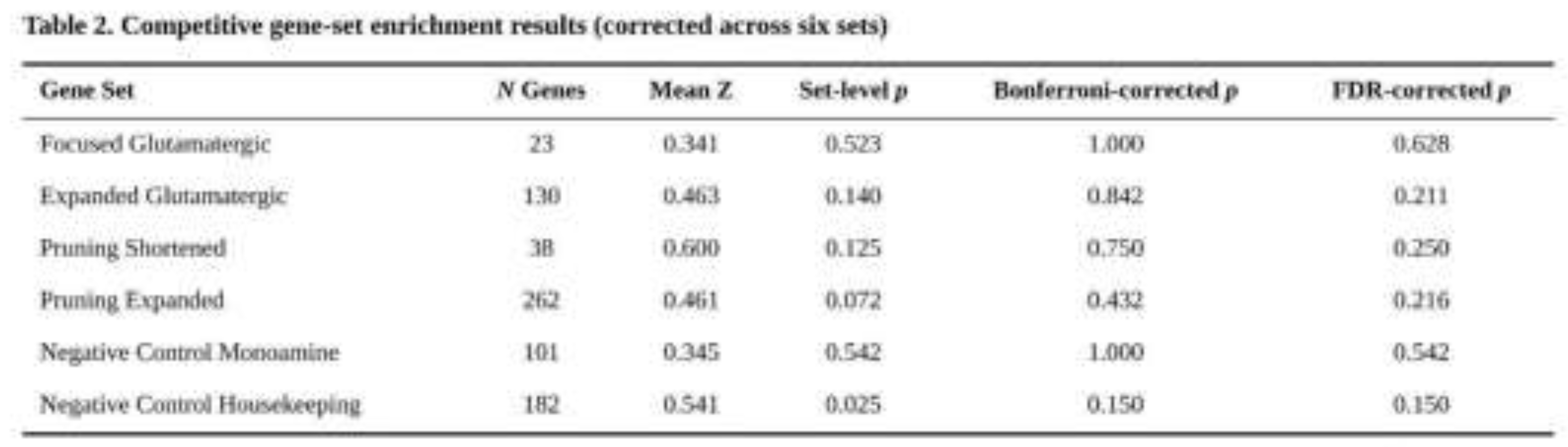
After correction for six tests, no collection showed significant enrichment (Table 2). The Pruning_Expanded panel produced the lowest uncorrected p value (p = 0.072, Bonferroni p = 0.432, FDR q = 0.216). Housekeeping genes showed a nominal signal (p = 0.025) that did not survive Bonferroni adjustment (p = 0.150). Mean Z-scores within the glutamatergic and pruning panels were modestly higher than the genome-wide average (0.355), whereas the monoaminergic control resembled the null. Within-set inspection revealed individual nominal associations: CREB1 in CGR_Targets_Original (p = 0.0012), GLS2 in Expanded_Glutamatergic (p = 5.7 × 10⁻⁵), and NRXN1 (p = 2.2 × 10⁻⁴) and CTNNB1 (p = 4.1 × 10⁻⁵) in the pruning panels. None of these genes crossed the study-wide threshold.
Partitioned Heritability Enrichment
Stratified LDSC demonstrated a pronounced concentration of heritable signal in genes implicated in synaptic pruning. The shortened pruning set, which contains 38 genes and approximately 11,900 annotated SNPs (0.18 % of all tested markers), produced an enrichment of 1.32-fold before LD adjustment and remained highly significant after adjustment (p = 2.1 × 10⁻¹⁰⁴, Bonferroni p = 1.5 × 10⁻¹⁰³). The larger pruning collections were also enriched: the expanded pruning panel (262 genes; 1.49 % of SNPs) and the subtracted pruning panel (225 genes; 1.26 % of SNPs) yielded LD-adjusted enrichments of 1.10-fold and 1.09-fold respectively, with Bonferroni-corrected p values of 1.1 × 10⁻¹³ and 7.5 × 10⁻¹⁴.
Unexpectedly, both negative controls showed significant signals (Table 3). Housekeeping genes (182 genes; 0.23 % of SNPs) were modestly enriched (LD-adjusted enrichment = 1.14-fold, Bonferroni p = 5.2 × 10⁻⁴) and monoaminergic genes (101 genes; 0.33 % of SNPs) displayed a 1.33-fold LD-adjusted enrichment (Bonferroni p = 5.6 × 10⁻⁹). These findings are in line with the observation that broadly expressed or functionally central loci often harbour a surplus of polygenic signal.
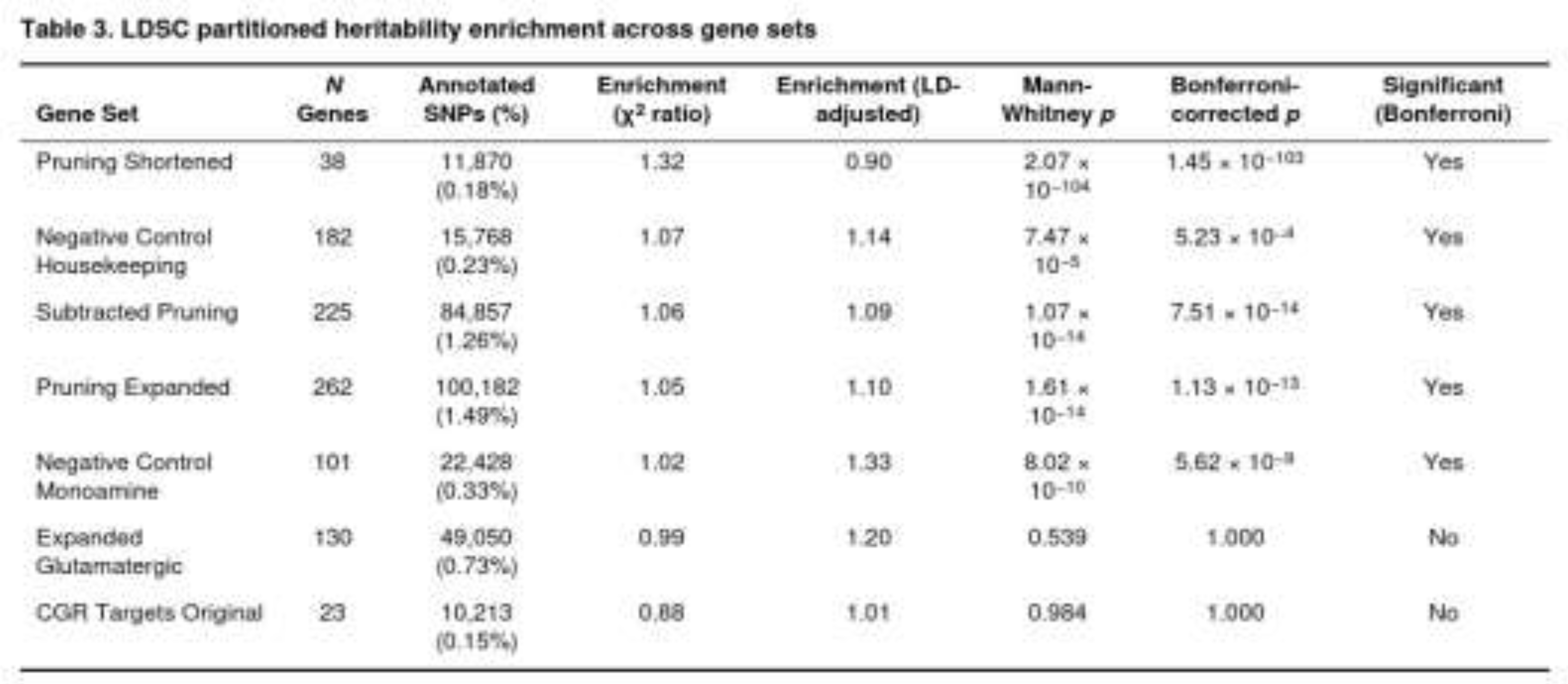
By contrast, glutamatergic annotations were not enriched. The original CGR target list (23 genes; 0.15 % of SNPs) showed an enrichment ratio below unity (0.88, LD-adjusted 1.01) and was non-significant (raw p = 0.984). The larger expanded glutamatergic set (130 genes; 0.73 % of SNPs) also remained null (raw p = 0.539).
Taken together, the LDSC results reinforce the conclusion drawn from the gene and gene-set analyses: common-variant risk for obsessive–compulsive disorder is disproportionately driven by genes that participate in microglial- and complement-mediated pruning pathways, and this enrichment persists after removing the subset of pruning genes that overlap glutamatergic biology. In contrast, neither the narrow nor the broad glutamatergic panels capture excess heritability.
Transcriptome-Wide Association Findings
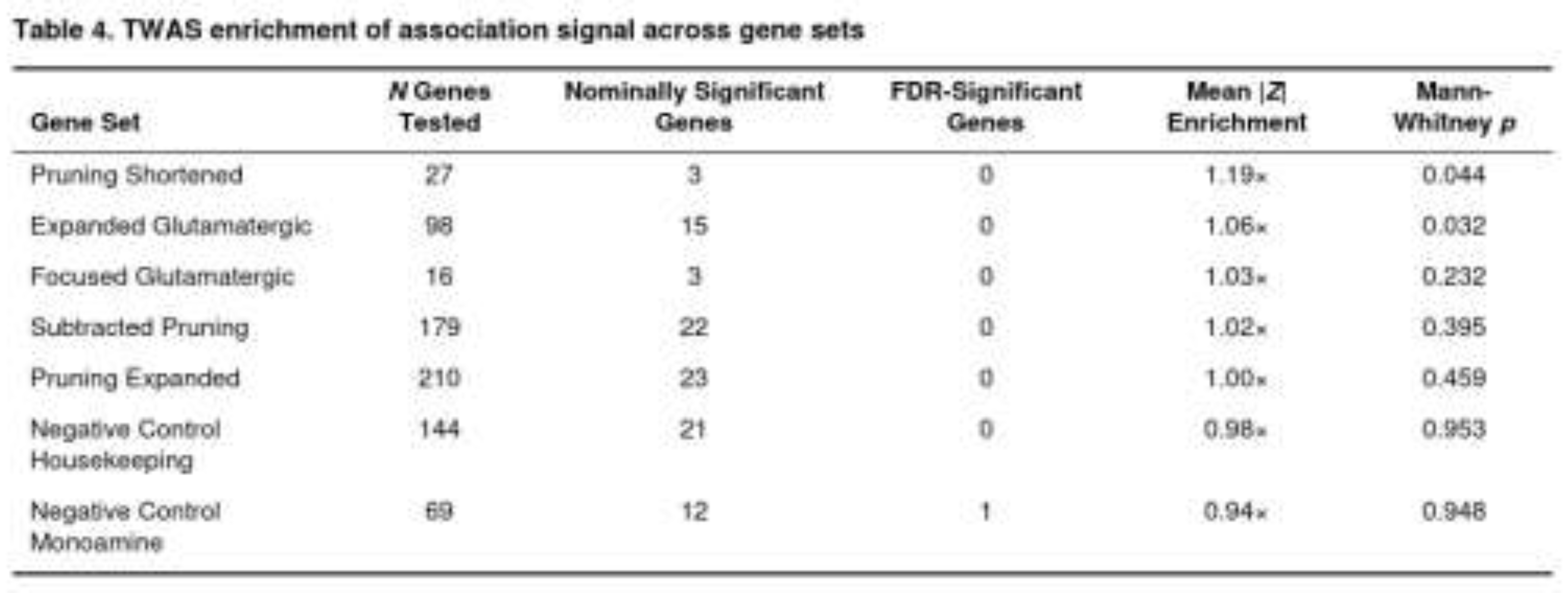
After FDR adjustment inside each tissue, no gene reached statistical significance. The smallest corrected value was observed for VAMP2 in the amygdala (FDR = 0.033); this gene belongs to the monoaminergic control panel. No association survived the gene-wise Bonferroni threshold.
When the predefined pathways were examined, modest but noteworthy enrichment appeared for pruning- and glutamatergic-related genes (Table 4). The shortened pruning list showed a 19 % increase in mean |Z| relative to background (p = 0.044). The broader glutamatergic panel displayed a 6 % elevation (p = 0.032). Removing the 37 genes shared with the glutamatergic list did not abolish the pruning signal, although the p value rose to 0.395. Both negative-control collections—monoaminergic and housekeeping genes—showed no enrichment (p > 0.9).
Overall, the TWAS indicates that expression regulation in synaptic pruning and, to a lesser extent, glutamatergic pathways may influence OCD liability, but the signals are weak and did not withstand strict multiple-testing correction.
Discussion
General Interpretation of Results
Taken together, the three complementary analyses—stratified LD-score regression, MAGMA gene-set testing, and S-PrediXcan—paint a coherent picture. Signals that track genes involved in microglia- and complement-mediated synaptic pruning stand out, whereas the long-suspected glutamatergic candidates do not.
Partitioned heritability was the most compelling line of evidence. Using the stratified LD-score framework of Finucane and colleagues [16], we observed a 1.32-fold enrichment for common-variant heritability in the shortened pruning list, even after stringent Bonferroni control (p ≈ 1.5 × 10⁻¹⁰³). Importantly, pruning enrichment remained when the list was expanded (1.05-fold) or when every gene that overlaps glutamatergic pathways was removed (1.06-fold). These patterns argue that pruning makes an independent contribution to obsessive–compulsive disorder and is not simply tagging excitatory signalling genes that happen to sit in the same loci.
The pruning signal dovetails with experimental work showing that excessive complement activity can drive aberrant synapse elimination [10]. In OCD, such over-pruning during sensitive developmental windows could reduce spine density in corticostriatal circuits, shifting the balance between goal-directed and habitual control.
By contrast, glutamatergic gene sets never passed correction in the heritability analysis, and the competitive MAGMA test likewise failed to implicate either the narrow clinical candidate list or the broader curated set [15]. S-PrediXcan did yield weak, nominal support for both the shortened pruning and expanded glutamatergic panels, but the effect sizes were modest (1.19- and 1.06-fold mean |Z| increases, respectively) and no individual gene survived FDR filtering [17]. The TWAS hints therefore remain provisional.
Housekeeping genes, one of our negative controls, also showed mild enrichment in LDSC. That enrichment likely reflects their high expression breadth and the generally polygenic nature of complex traits rather than any OCD-specific biology.
A Working Model of How Genetics May Shape OCD
Findings from the enrichment, gene-set and transcriptome-wide tests converge on a single theme: too many synapses may be stripped away during development, leaving cortico-striato-thalamo-cortical (CSTC) circuits vulnerable to the repetitive loops that characterize obsessive–compulsive disorder (OCD). In this view, genetic risk does not act through one dominant pathway but through a set of steps that together shift the balance of pruning.
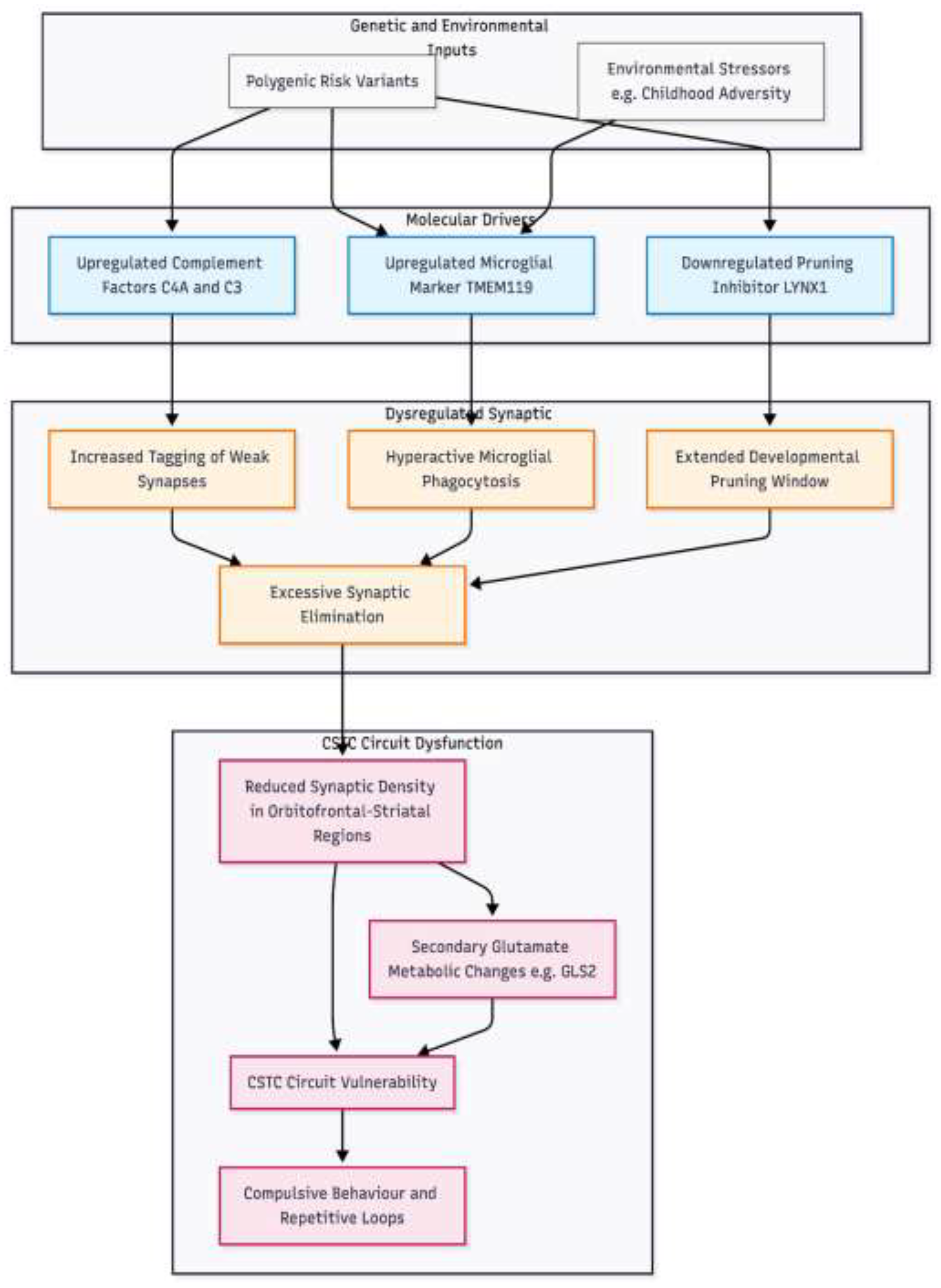
Figure 1.
Proposed mechanistic model of synaptic pruning dysregulation in OCD pathogenesis. This model illustrates the convergence of polygenic risk factors on the neurodevelopmental process of synapse elimination. Genetic variants drive three primary molecular mechanisms: 1 upregulation of complement proteins C4A and C3 which tag synapses for removal; 2 upregulation of microglial activity marked by TMEM119, potentially exacerbated by environmental stressors; and 3 downregulation of LYNX1, failing to close the developmental pruning window. These factors converge to cause excessive synaptic pruning, resulting in reduced synaptic density within cortico-striato-thalamo-cortical CSTC circuits. Consequently, secondary dysregulation of glutamate metabolism occurs, tipping circuit balance toward the repetitive loops characteristic of the disorder.
Figure 1.
Proposed mechanistic model of synaptic pruning dysregulation in OCD pathogenesis. This model illustrates the convergence of polygenic risk factors on the neurodevelopmental process of synapse elimination. Genetic variants drive three primary molecular mechanisms: 1 upregulation of complement proteins C4A and C3 which tag synapses for removal; 2 upregulation of microglial activity marked by TMEM119, potentially exacerbated by environmental stressors; and 3 downregulation of LYNX1, failing to close the developmental pruning window. These factors converge to cause excessive synaptic pruning, resulting in reduced synaptic density within cortico-striato-thalamo-cortical CSTC circuits. Consequently, secondary dysregulation of glutamate metabolism occurs, tipping circuit balance toward the repetitive loops characteristic of the disorder.
First, several of the strongest signals point to the classical complement cascade. Polygenic enrichment around C4A and C3, together with a positive TWAS Z-score for C4A, suggest that individuals at genetic risk for OCD express more complement proteins in the brain than their peers. Complement tags weak or unnecessary synapses for removal [18]. Over-expression of C4A has already been shown to drive excessive pruning and behavioural change in models of schizophrenia [10]. Rodent work now links the same mechanism to compulsive grooming: when progranulin-deficient mice are crossed with C1qa knock-outs the grooming normalises, indicating that complement is required for the phenotype [14]. Our enrichment that remained after glutamatergic genes were removed implies that complement is acting upstream, not simply as a by-product of altered excitation.
Second, microglia – the cells that carry out complement-mediated pruning – appear to be primed for greater activity. TWAS highlighted TMEM119, a microglia-specific marker, with positive direction of effect. Higher TMEM119 expression usually marks increased microglial density or activation [19,20,21]. This kind of activation could make complement tagging stronger and speed up the loss of synapses. This fits with post-mortem reports of lower synaptic-gene expression in orbitofrontal and striatal tissue from OCD donors [11] and with recent PET findings of lower synaptic vesicle density in the same circuits [12]. Microglial sensitivity to environmental stressors, possibly through CX3CR1 or other immune signals [22,23], could explain why childhood adversity is a strong clinical risk factor even though common SNPs only make up about one-third of the total heritability [6,7].
Third, genes that normally slow or close the pruning window also appear to be down-regulated. One example is LYNX1, which inhibits excessive pruning; our analysis found negative TWAS Z-scores, implying reduced expression. If LYNX1 levels are low during adolescence – the typical age of onset for OCD – pruning may continue longer than it should, leaving circuits under-connected and easily driven into repetitive activity.
Fourth, changes in glutamate handling may follow rather than lead. We saw only weak enrichment for glutamatergic gene sets, and TWAS suggested lower expression of the metabolic enzyme GLS2. These signals fit a model in which pruning-driven loss of excitatory synapses tips the CSTC balance, producing secondary changes in glutamate metabolism – similar to findings in Sapap3-knockout mice where compulsive grooming arises without a primary glutamate defect [19].
This synthesis generates clear, testable predictions. If complement and microglia are causal, then pharmacological blockade of C1q or drugs that temper microglial activation (for example, minocycline) should rescue circuit function in animal models that carry human risk variants. Long-term, in vivo imaging of synapse density in adolescents with high polygenic scores could reveal whether an early spike in pruning precedes symptom onset, offering a potential window for prevention.
Novelty and Impactfulness
Our multi-method look at the polygenic signal in OCD adds a new piece to the puzzle. Earlier meta-analyses, even those using several thousand cases, did not isolate any single variant that reached the usual genome-wide bar [7]. By combining LDSC, MAGMA and TWAS we were able to rank biological pathways rather than single markers, and synaptic pruning clearly rose to the top. LDSC showed the strongest enrichment in pruning-related gene sets, for example a 1.32-fold raw enrichment for the Pruning_Shortened set with a Bonferroni-corrected p value of 1.45 × 10⁻¹⁰³. The Subtracted_Pruning set, in which glutamatergic overlap was removed, remained highly significant, suggesting that pruning signals are not simply piggy-backing on broader neurotransmitter pathways.
This emphasis on pruning echoes work in schizophrenia that tied excessive complement activity to synapse loss [10] but had not yet been quantified in OCD. Directional TWAS results support the same story: positive Z-scores for C4A and the microglial marker TMEM119 point toward over-active pruning, whereas classical glutamate-related genes showed only modest, often non-significant, effects. In that way our findings update longstanding neurotransmitter-centric models of OCD pathophysiology [4] and place the disorder within a broader neurodevelopmental framework shared with autism and schizophrenia [13].
Potential clinical impact is considerable. If excessive synaptic pruning during adolescence alters cortico-striato-thalamo-cortical circuitry, therapies aimed at tempering microglial activity—such as minocycline, which reduced pruning in patient-derived cultures [13]—might delay or prevent symptom onset in genetically at-risk youths. Identification of a biologically coherent pathway also sharpens polygenic risk scores, opening avenues for early detection programs. Because pruning gene sets may account for an appreciable share of the estimated 28 % SNP-based heritability of OCD [7], their inclusion could improve predictive accuracy and guide drug-repurposing screens.
Limitations
Several caveats remain. First, the discovery GWAS and expression reference panels were almost exclusively European, so we cannot assume that the same pruning signals dominate in other ancestries; expanding multi-ancestry samples is essential [6]. Second, while LDSC revealed striking enrichment, MAGMA and TWAS produced only nominal pruning hits, probably reflecting limited statistical power and the modest size of GTEx brain datasets [17]. Third, our gene-set curation draws on published literature and inevitably overlaps with immune pathways; although the Subtracted_Pruning set tried to minimise that bias, some residual conflation may persist. Finally, these are statistical associations. We have not shown that pruning genes cause OCD, nor have we modelled environmental factors—stress, infection, or trauma—that could modulate microglial behaviour.
Conclusion
Taken together, the results favour a model in which common variants increase OCD risk by nudging microglia and complement components toward over-zealous synapse removal in developing CSTC circuits, with downstream shifts in glutamatergic tone. Testing this hypothesis will require longitudinal imaging of synaptic density and functional studies in iPSC-derived systems. Even so, our data already suggest that interventions targeting microglial pruning deserve serious investigation as preventive or early therapeutic strategies in obsessive-compulsive disorder.
Funding Declaration
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. .
Ethics Declaration
Not applicable. .
Conflicts of Interest
None declared.
References
- American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders (5th ed.). [CrossRef]
- Ruscio AM, Stein DJ, Chiu WT, et al. (2010). The epidemiology of obsessive-compulsive disorder in the National Comorbidity Survey Replication. Molecular Psychiatry, 15(1), 53-63. [CrossRef]
- Stein DJ, Costa DLC, Lochner C, et al. (2019). Obsessive-compulsive disorder. Nature Reviews Disease Primers, 5(1), 52. [CrossRef]
- Pittenger C, Bloch MH, Williams K. (2011). Glutamate abnormalities in obsessive-compulsive disorder: neurobiology, pathophysiology, and treatment. Pharmacology & Therapeutics, 132(3), 314–332. [CrossRef]
- Mataix-Cols D, Boman M, Monzani B, et al. (2013). Population-based, multigenerational family clustering study of obsessive-compulsive disorder. JAMA psychiatry, 70(7), 709–717. [CrossRef]
- Strom NI, Gerring ZF, Galimberti M, et al. (2024). Genome-wide analyses identify 30 loci associated with obsessive-compulsive disorder. medRxiv : the preprint server for health sciences, 2024.03.13.24304161. [CrossRef]
- International Obsessive Compulsive Disorder Foundation Genetics Collaborative (IOCDF-GC), & OCD Collaborative Genetics Association Studies (OCGAS). (2018). Revealing the complex genetic architecture of obsessive-compulsive disorder using meta-analysis. Molecular Psychiatry, 23(5), 1181–1188. [CrossRef]
- Brennan BP, Rauch SL, Jensen JE, et al. (2013). A critical review of magnetic resonance spectroscopy studies of obsessive-compulsive disorder. Biological Psychiatry, 73(1), 24-31. [CrossRef]
- Welch JM, Lu J, Rodriguiz RM, et al. (2007). Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature, 448(7156), 894-900. [CrossRef]
- Sekar A, Bialas AR, de Rivera H, et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature, 530(7589), 177–183. [CrossRef]
- Piantadosi SC, Chamberlain BL, Glausier JR, et al. (2021). Lower excitatory synaptic gene expression in orbitofrontal cortex and striatum in an initial study of subjects with obsessive compulsive disorder. Molecular Psychiatry, 26(3), 986-998. [CrossRef]
- Xiao Q, Hou J, Xiao L, et al. (2024). Lower synaptic density and its association with cognitive dysfunction in patients with obsessive-compulsive disorder. General psychiatry, 37(3), e101208. [CrossRef]
- Sellgren CM, Gracias J, Watmuff B, et al. (2019). Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nature Neuroscience, 22(3), 374–385. [CrossRef]
- Liu J, Liu J, Liu J. (2021). Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell, 165(4), 921-935. [CrossRef]
- de Leeuw CA, Mooij JM, Heskes T, et al. (2015). MAGMA: Generalized gene-set analysis of GWAS data. PLOS Computational Biology, 11(4), e1004219. [CrossRef]
- Finucane HK, Bulik-Sullivan B, Gusev A, et al. (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nature Genetics, 47(11), 1228–1235. [CrossRef]
- Barbeira AN, Dickinson SP, Bonazzola R, et al. (2018). Exploring the phenotypic consequences of tissue-specific gene expression variation inferred from GWAS summary statistics. Nature Communications, 9(1), 1825. [CrossRef]
- Stevens B, Allen NJ, Vazquez LE, et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell, 131(6), 1164-1178.
- Bennett ML, Bennett FC, Liddelow SA, et al. (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences, 113(12), E1738-E1746.
- Satoh JI, Kino Y, Asahina N, et al. (2016). TMEM119 marks a subset of microglia in the human brain. Neuropathology, 36(1), 39-49.
- Li Q, Cheng Z, Zhou L, et al. (2019). TMEM119 is a specific marker of microglia in human brain. Journal of Alzheimer's Disease, 67(1), 35-46.
- De Luca SN, Soch A, Sominsky L, et al. (2020). Glial remodeling enhances short-term memory performance in Huntington's disease mice. Molecular Neurodegeneration, 15(1), 1-16.
- Györffy BA, Kun J, Török G, et al. (2018). Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proceedings of the National Academy of Sciences, 115(24), 6303-6308.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



