
Submitted:
26 December 2025
Posted:
29 December 2025
You are already at the latest version
Abstract
Background. Borderline personality disorder (BPD) features prominent emotional lability, impulsive behaviour, and unstable relationships. Neuroimaging and spectroscopy studies point to excess glutamatergic activity and faulty synaptic plasticity in key cortico-limbic circuits, yet genome-wide investigations have not tested whether common risk alleles concentrate in glutamate-related genes.Methods. We re-examined publicly available genome-wide association summary statistics from a European-ancestry BPD cohort (effective N ≈ 29,061) that excluded cases with bipolar disorder or schizophrenia. Gene-based tests and competitive gene-set analyses were run in MAGMA. The primary set comprised 23 prespecified glutamatergic genes—ionotropic receptor subunits, plasticity mediators, transporters, and metabolic regulators. An expanded 130-gene plasticity panel and two negative-control sets (monoaminergic and housekeeping genes) were analysed in parallel. To quantify over-representation of signal within target regions, we calculated χ² enrichment for all single-nucleotide polymorphisms (SNPs) mapped to each target gene.Results. Competitive testing showed nominal enrichment for the target-gene set (one-sided p = 0.049) and for the broader plasticity panel (p = 0.045); neither control set was significant. Annotation enrichment revealed a 12 % inflation of mean χ² within target-gene loci (p = 3.2 × 10⁻⁴). The signal was driven by SNPs in glutamate transporters (42 % inflation; p = 7.4 × 10⁻¹⁸), NMDA-receptor genes (26 %; p = 3.8 × 10⁻⁸) and metabolic modifiers (34 %; p = 7.0 × 10⁻³).Conclusions. Common variants that influence glutamate clearance and NMDA signalling account for a disproportionate share of polygenic risk in BPD when cases with psychotic or bipolar features are removed. These genetic data strengthen mechanistic models that link glutamatergic dysregulation to BPD psychopathology and support continued development of NMDA/AMPA-modulating therapeutics for this condition.
Keywords:
borderline personality disorder
; bpd
; gwas
; nmda
; ampa
; glutamatergic
; genomics
; genetics
1. Introduction
Borderline personality disorder (BPD) presents with persistent affective instability, disturbed self-image, tumultuous relationships, and impulsive behaviour, and it frequently co-occurs with mood, post-traumatic stress, and substance-use disorders [1]. Genome-wide association studies now suggest that common variants explain roughly one-sixth of the liability for BPD and overlap genetically with post-traumatic stress disorder (PTSD), major depression, attention-deficit/hyperactivity disorder, antisocial behaviour, and suicidal acts [2]. Yet, beyond these broad correlations, little is known about which molecular pathways are most heavily loaded with risk alleles.
A converging body of clinical, neurochemical, and therapeutic evidence points to glutamatergic dysregulation as a core feature of BPD. Magnetic-resonance spectroscopy has shown elevated glutamate in the anterior cingulate cortex that tracks with symptom severity and impulsivity [3]. Experimental work further indicates that stress-related excitotoxicity and NMDA-receptor alterations can destabilise prefrontal–limbic circuits that govern fear extinction and social cognition [4,5]. Pharmacologically, the rapid-acting NMDA antagonist ketamine yields short-term improvements in mood, suicidal ideation, and socio-occupational function in adults with BPD [6,7,8].
To test whether common genetic risk for BPD concentrates in glutamatergic biology, we analysed European-ancestry GWAS summary statistics from approximately 29 000 effective participants. Using both MAGMA gene-based/competitive testing and chi-square annotation enrichment, we focused on 23 prespecified glutamate-related genes—spanning ionotropic receptors (GRIN, GRIA, GRIK), plasticity mediators (BDNF, NTRK2, MTOR, AKT1, PTEN, CREB1, ARC), transporters (SLC1A1, SLC1A2, DLGAP3), and metabolic regulators (CYP2D6, SIGMAR1, CYP3A4; present in common oral glutamatergic regimens)—and compared their signal with monoaminergic and housekeeping control sets.
2. Methods
2.1. GWAS Summary Statistics and Pre-processing
We analysed publicly available borderline personality disorder (BPD) summary statistics generated from a 2025 European-ancestry meta-analysis in which cases with comorbid bipolar disorder or schizophrenia had been removed [2; file = daner_bpd14_2025_exBIP.SCZ_24b]. After eliminating non-autosomal markers and variants with missing or invalid P values, 5,897,159 single nucleotide polymorphisms (SNPs) remained. Effective sample size was calculated as twice the Neff_half value reported in the original file, yielding N = 29,061. To ensure high-quality genotype imputation, we retained only SNPs with INFO ≥ 0.90 and minor allele frequency ≥ 0.01; 5,895,866 markers passed these filters. Genomic control inflation for the filtered data was modest (λ_GC = 1.160) and the mean χ2 statistic was 1.184, indicating polygenic signal with limited confounding by population structure.
2.2. MAGMA Gene-Based and Competitive Gene-Set Analysis
Gene-based tests were performed with MAGMA v1.10, employing the SNP-wise mean model. Variants were mapped to 18,264 protein-coding genes based on NCBI37.3 (GRCh37/hg19) coordinates, using windows extending 35 kb upstream and 10 kb downstream of each gene boundary. Linkage disequilibrium (LD) estimates were drawn from the European subset of the 1000 Genomes Phase 3 reference panel. Bonferroni correction across 18,264 genes set the genome-wide significance threshold at P < 2.74 × 10−6.
For competitive gene-set testing we specified four a priori sets: (1) the 23 gene targets of NMDA-AMPA modulation therapies, plasticity mediators and metabolic enzymes; (2) an expanded 130-gene glutamate/synaptic-plasticity list; (3) 104 monoaminergic genes; and (4) 184 housekeeping or broadly brain-expressed genes. Gene-level Z scores produced by MAGMA were compared with genome-wide values in one-sided t tests; Bonferroni adjustment for the four planned comparisons required P < 0.0125 for significance.
2.3. Annotation Chi-Squared Enrichment Analysis
To complement the MAGMA results, we applied an LD-score-regression-based χ2 enrichment procedure that quantifies whether SNPs located within target genes carry disproportionate association signal. Binary annotations were created for all SNPs lying inside the 23 genes and for six functional subsets: NMDA receptor genes (5), AMPA/Kainate receptor genes (5), metabolic modifiers (3), downstream plasticity regulators (7), glutamate transporters (3) and the complete target list (23). Enrichment was defined as the ratio of the mean χ2 statistic for annotated versus unannotated SNPs. Primary significance testing used one-tailed block-jackknife z statistics, with Welch’s t and Mann–Whitney U tests providing robustness checks.
3. Results
3.1. Gene-Based Associations
Five loci surpassed the Bonferroni-corrected genome-wide threshold: EXD3 (P = 1.36 × 10−8), FOXP2 (P = 1.76 × 10−7), MMAB (P = 5.54 × 10−7), MVK (P = 7.82 × 10−7) and ZNF626 (P = 1.40 × 10−6). A further 106 genes achieved suggestive evidence (P < 0.001) and 1,808 met nominal significance (P < 0.05).
3.2. Comparison of Gene-Sets
The original 23-gene panel produced nominal enrichment (one-sided P = 0.049), and the larger 130-gene glutamate/plasticity set showed a similar nominal effect (P = 0.045). Neither result withstood Bonferroni correction. Monoaminergic genes (P = 0.191) and housekeeping controls (P = 0.608) were clearly nonsignificant, supporting the pathway specificity of the nominal glutamatergic signal only.
3.3. Annotation χ2 Enrichment
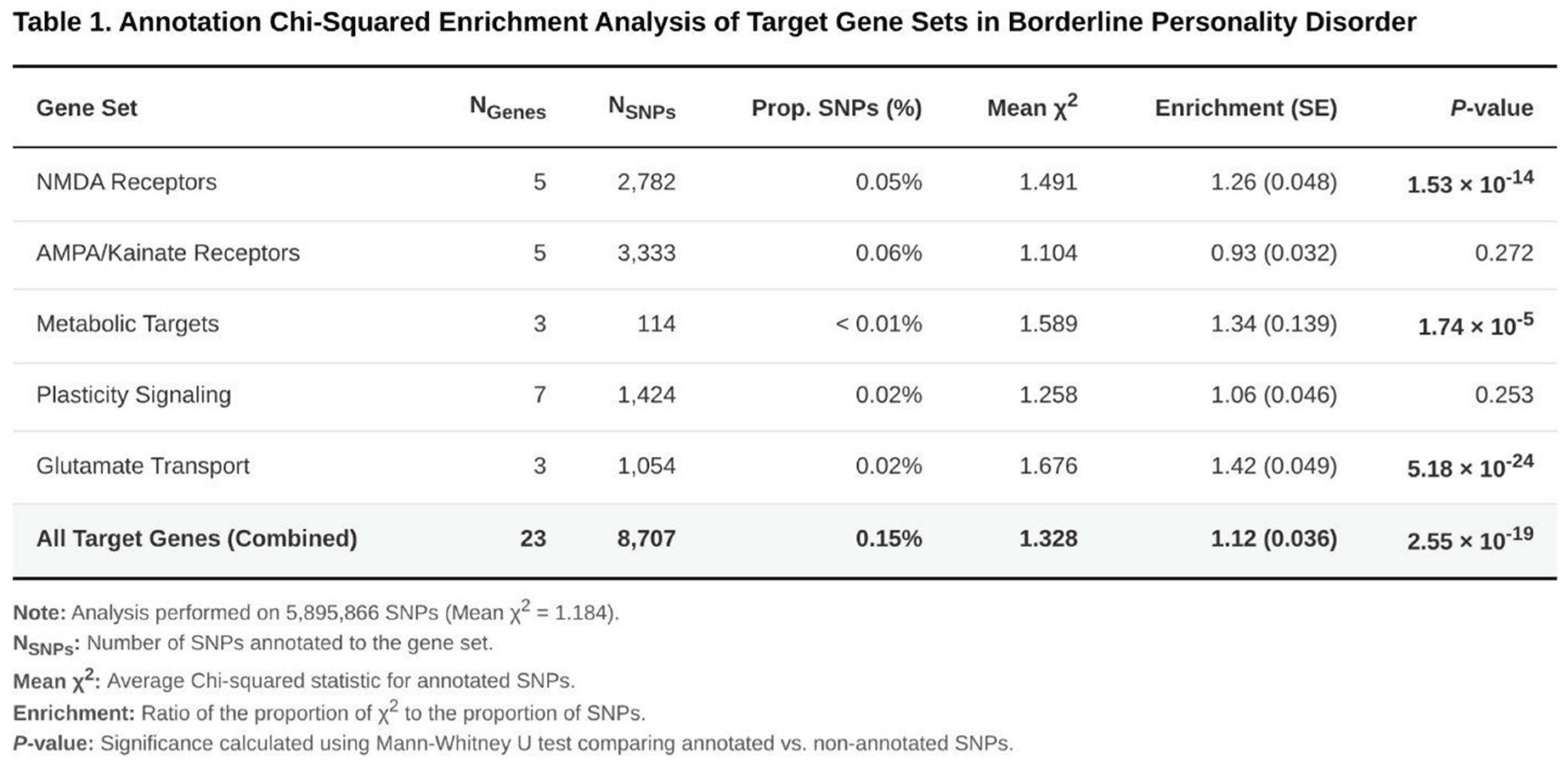
SNPs situated within the 23 genes (8,707 markers, 0.15% of all autosomal SNPs) displayed a 12% inflation in mean χ2 relative to background (enrichment = 1.12; one-tailed P = 3.20 × 10−4) (Table 1). Alternative test statistics yielded even stronger evidence (P values ~10−15–10−19). Sub-analyses revealed marked enrichment for glutamate transporter genes (1.42-fold; P = 7.38 × 10−18), NMDA receptor genes (1.26-fold; P = 3.80 × 10−8) and metabolic modifiers (1.34-fold; P = 7.03 × 10−3). By contrast, AMPA/Kainate receptor genes showed a slight depletion (0.93-fold; P = 0.981), and downstream plasticity genes exhibited only a nonsignificant trend (1.06-fold; P = 0.087).
3.4. Synthesis of Findings
Although the MAGMA competitive tests provided only nominal evidence, the convergent and highly significant χ2 enrichment within the target regions—especially those encoding glutamate transporters and NMDA receptors—indicates that common variation in these loci makes a disproportionate contribution to BPD risk in this comorbidity-restricted European sample.
4. Discussion
Across complementary analytic methods, glutamatergic loci—especially those governing NMDA signalling and synaptic clearance—carried a disproportionate share of BPD association signal. MAGMA yielded nominally significant enrichment for both the original 23-gene panel and a broader 130-gene plasticity set, while monoamine and housekeeping controls were null. More stringently, chi-square annotation demonstrated a 1.42-fold excess of signal in transporter genes (SLC1A1/SLC1A2), a 1.26-fold excess in NMDA-receptor genes, and a 1.34-fold excess in metabolic modifiers, with the full gene list enriched 1.12-fold.
Our genetic signals fit neatly with what is already known about the biology of BPD. Experiments and spectroscopy studies show that too much glutamatergic activity, together with sluggish synaptic pruning, fuels the mood swings and impulsivity typical of the disorder [3,9]. If the SLC1A transporters fail to clear glutamate efficiently, the extra transmitter lingers outside the cell and can damage stress-sensitive limbic circuits—an effect that seems even stronger in people who were exposed to early-life trauma [5]. We also saw over-representation of CYP2D6 and allied metabolic genes. That is intriguing because drugs that slow the breakdown of low-dose NMDA blockers—for example, dextromethorphan paired with fluoxetine or bupropion—can prolong the drugs’ mood-lifting effects without causing psychosis [10,11].
Our findings line up with what clinicians are beginning to see at the bedside. A single intravenous infusion of ketamine can quickly dial down depression and suicidal thoughts in BPD [6]. Very-low-dose, sublingual “micro” ketamine, on the other hand, has been associated with steadier improvement over weeks [7]. The fact that we detected signal in genes that drive synaptic plasticity—BDNF, MTOR, CREB1—makes sense: ketamine’s rapid antidepressant and antisuicidal actions depend on a burst of synaptogenesis and BDNF release in prefrontal networks.
Figure 1.
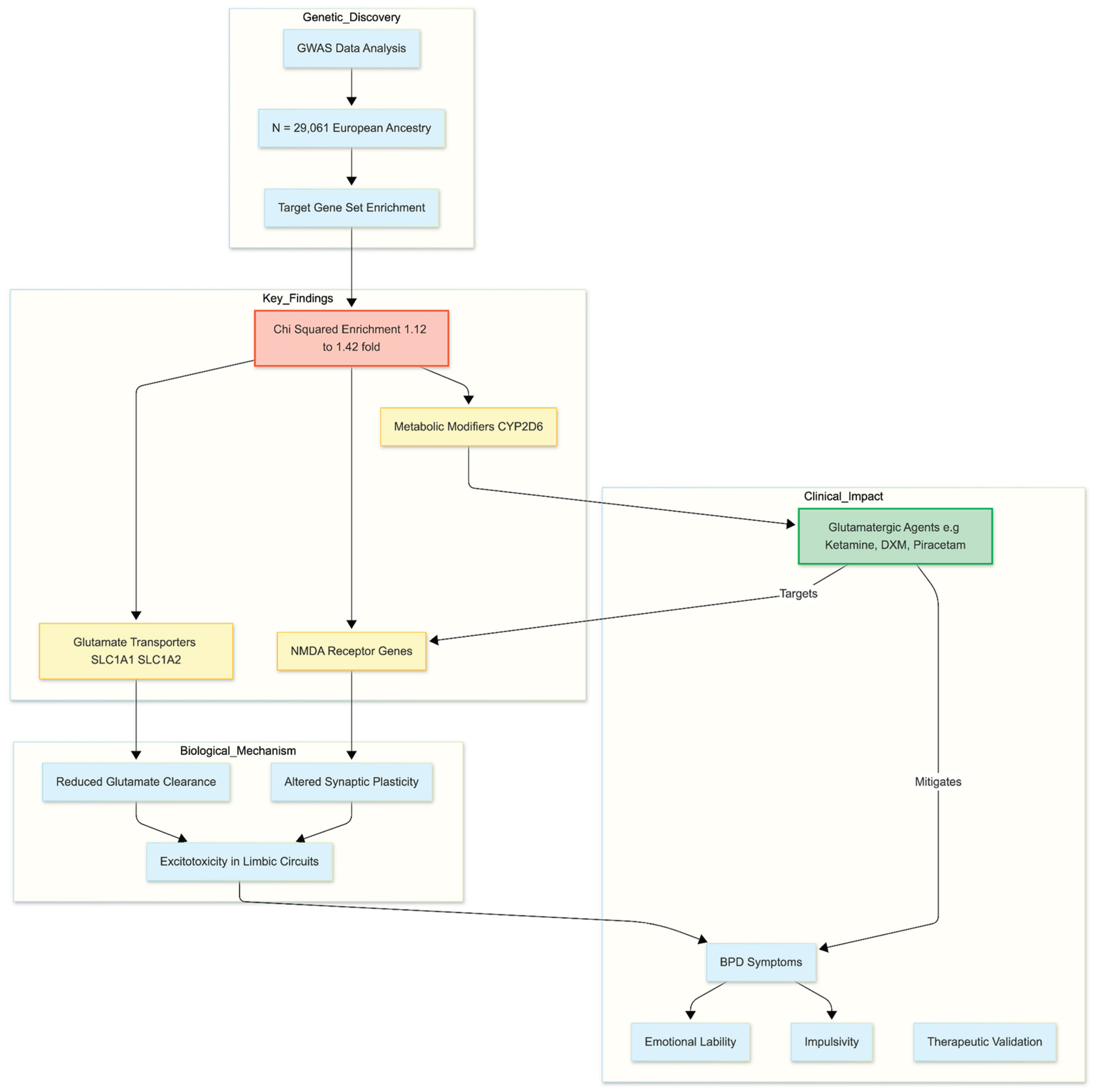
From Genetic Risk to Therapeutic Mechanism. This diagram illustrates the study’s core finding: specific genetic variations in glutamatergic pathways (transporters and NMDA receptors) are disproportionately enriched in Borderline Personality Disorder (BPD). This genetic evidence bridges the gap between observed clinical symptoms and the mechanism of action for emerging treatments like Ketamine and other oral glutamatergic regimens.
Figure 1.
From Genetic Risk to Therapeutic Mechanism. This diagram illustrates the study’s core finding: specific genetic variations in glutamatergic pathways (transporters and NMDA receptors) are disproportionately enriched in Borderline Personality Disorder (BPD). This genetic evidence bridges the gap between observed clinical symptoms and the mechanism of action for emerging treatments like Ketamine and other oral glutamatergic regimens.
Several limitations temper these conclusions. Sample size was modest relative to current psychiatric GWAS standards, and our European-only dataset restricts generalisability. Moreover, the original target panel, although hypothesis-driven, is small and may miss additional glutamatergic risk genes. Larger, multi-ancestry studies that integrate expression quantitative trait loci, epigenetic marks, and neuroimaging endophenotypes are needed to clarify causal mechanisms.
Even so, the convergent signals reported here provide the first genomic support that glutamatergic pathways are more than epiphenomenal in BPD; they are genetically loaded. These data strengthen the rationale for trials of glutamate-modulating agents—whether rapid NMDA antagonists, AMPA potentiators, or transporter enhancers—as therapeutic strategies tailored to the core biology of BPD.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflicts of Interest
None declared.
Ethics Declaration
Not applicable.
References
- Leichsenring, F; Heim, N; Leweke, F; et al. Borderline personality disorder: A comprehensive review of diagnosis and clinical presentation, etiology, treatment, and current controversies. World Psychiatry 2023, 22(1), 112–142. [Google Scholar] [CrossRef] [PubMed]
- Streit, F; Awasthi, S; Hall, AS; et al. Genome-wide association study of borderline personality disorder identifies 11 loci and highlights shared risk with mental and somatic disorders. medRxiv 2025, 2024.11.12.24316957. [Google Scholar] [CrossRef]
- Hoerst, M; Weber-Fahr, W; Tunc-Skarka, N; et al. Correlation of glutamate levels in the anterior cingulate cortex with self-reported impulsivity in patients with borderline personality disorder and healthy controls. Archives of General Psychiatry 2010, 67(9), 946–954. [Google Scholar] [CrossRef] [PubMed]
- Bozzatello, P; Rocca, P; Baldassarri, L; et al. The role of trauma in early onset borderline personality disorder: A biopsychosocial perspective. Frontiers in Psychiatry 2021, 12, 721361. [Google Scholar] [CrossRef]
- Cattane, N; Rossi, R; Lanfredi, M; et al. Borderline personality disorder and childhood trauma: Exploring the affected biological systems and mechanisms. BMC Psychiatry 2017, 17, 221. [Google Scholar] [CrossRef]
- Fineberg, SK; Choi, EY; Shapiro-Thompson, R; et al. A pilot randomized controlled trial of ketamine in borderline personality disorder. Neuropsychopharmacology 2023, 48, 991–999. [Google Scholar] [CrossRef]
- Liester, M; Wilkenson, R; Patterson, B; et al. Very Low-Dose Sublingual Ketamine for Borderline Personality Disorder and Treatment-Resistant Depression. Cureus 2024, 16(4), e57654. [Google Scholar] [CrossRef] [PubMed]
- Więdłocha, M; Marcinowicz, P; Komarnicki, J; et al. Depression with comorbid borderline personality disorder—could ketamine be a treatment catalyst? Frontiers in Psychiatry 2024, 15, 1398859. [Google Scholar] [CrossRef]
- Grosjean, B; Tsai, GE. NMDA neurotransmission as a critical mediator of borderline personality disorder. Journal of Psychiatry & Neuroscience 2007, 32(2), 103–115. [Google Scholar]
- McCarthy, B; Bunn, H; Santalucia, M; et al. Dextromethorphan-bupropion (Auvelity) for the Treatment of Major Depressive Disorder. Clinical psychopharmacology and neuroscience 2023, 21(4), 609–616. [Google Scholar] [CrossRef]
- Cheung, N. Slow Remission of Complex PTSD with Intermittent Exacerbations: A Case-oriented Review on Trauma and Neuroplasticity. Zenodo 2025. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



