
Submitted:
23 December 2025
Posted:
24 December 2025
You are already at the latest version
Abstract
Background: Post-traumatic stress disorder (PTSD) is associated with impaired fear extinction and synaptic loss in prefrontal–limbic circuits. Converging evidence implicates glutamatergic dysfunction in these deficits, yet the genetic architecture underlying glutamatergic involvement in PTSD remains incompletely understood. Methods: We applied MAGMA gene-based and competitive gene-set analyses to the largest PTSD GWAS to date (European ancestry, effective N = 638,463; Nievergelt et al., 2024). We tested prespecified gene sets including ionotropic glutamate receptor subunits (AMPA, NMDA, kainate), downstream plasticity mediators, metabolic enzymes (e.g., CYP2D6), and negative control sets (monoaminergic and housekeeping genes). Partitioned heritability was used to assess enrichment of PTSD common-variant risk in functional categories. Results: Genome-wide significant association was observed at GRIA1 (GluA1 AMPA subunit; Z = 5.78, p = 3.77 × 10⁻⁹). Partitioned heritability revealed strong enrichment for AMPA/kainate receptors (enrichment ratio = 1.36, p ≈ 10⁻¹³), NMDA receptor subunits (ratio = 1.25, p ≈ 10⁻⁷), and metabolic enzymes including CYP2D6 (ratio = 2.08, p ≈ 10⁻⁵). A broader glutamate/plasticity gene set (n = 130) showed modest but significant enrichment (mean Z = 1.013 vs genome-wide 0.780; p = 0.024, FDR = 0.048). Negative control sets were not enriched. Conclusion: Common genetic risk for PTSD is disproportionately localized to genes encoding AMPA and NMDA receptor subunits and related metabolic enzymes. These findings provide robust genetic support for glutamatergic mechanisms in PTSD pathophysiology and highlight ionotropic glutamate receptors as promising therapeutic targets for future investigation.
Keywords:
PTSD
; AMPA
; NMDA
; plasticity
; CGR
; genomics
Introduction
Post-traumatic stress disorder (PTSD) emerges when exposure to threat collides with a brain that can no longer flexibly update fear memories. Selective serotonin re-uptake inhibitors remain first-line treatment, yet remission is uncommon—particularly in combat-related or complex presentations, where fewer than one-third of patients achieve full recovery [1]. Evidence from animal and human studies points to stress-induced loss of synapses in prefrontal–limbic networks as a primary lesion; monoaminergic drugs ease symptoms but do little to rebuild these connections [2].
Interest has therefore shifted to the “glutamate hypothesis,” which holds that rapid restoration of synaptic plasticity through the N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor systems is central to recovery. Intravenous ketamine validates this model: a brief NMDA blockade provokes an AMPA-mediated glutamate surge, releases brain-derived neurotrophic factor (BDNF), and initiates mTOR-dependent synaptogenesis. Practical use, however, is limited by infusion logistics, dissociation, and short-lived benefit.
Recently, a novel glutamatergic regimen (Cheung Glutamatergic Regimen, CGR) was developed to mimic ketamine’s plasticity cascade with inexpensive oral medicines. The protocol pairs dextromethorphan (DXM) as a low-affinity NMDA antagonist with a strong CYP2D6 inhibitor such as fluoxetine or bupropion to prolong DXM exposure, and it adds piracetam to enhance AMPA throughput [3]. Small case series report rapid, durable improvement in trauma-spectrum disorders—including somatic PTSD and trauma-linked depression—without dissociation or mania [4,5]. What remains unknown is whether the regimen targets biological processes that are genetically core to PTSD, or whether it produces benefit through more peripheral mechanisms.
The latest genome-wide association study (GWAS) of PTSD, comprising more than 1.2 million participants, identified 95 risk loci and offers the statistical power to address this question [6]. In the present work we apply partitioned heritability analysis to those summary statistics, asking whether genes that encode the CGR’s pharmacological targets—metabolic regulators (e.g., CYP2D6, SIGMAR1), glutamate-receptor subunits (GRIN and GRIA families), and downstream plasticity mediators (BDNF, mTOR)—carry a disproportionate share of PTSD genetic risk. By aligning clinical pharmacology with genomic architecture, we aim to determine whether the CGR engages the very pathways that contribute most strongly to disease liability, thereby providing a mechanistic rationale for larger controlled trials.
Methods
We analysed summary statistics from the most recent post-traumatic stress disorder (PTSD) genome-wide association study of European ancestry participants (effective N = 638,463) [6]. After excluding non-autosomal variants, 7,004,486 SNPs with valid p values were retained. Gene-based and gene-set tests were run with MAGMA v1.10 [7]. Each single-nucleotide polymorphism was assigned to a gene if it lay within 35 kb upstream or 10 kb downstream of the transcriptional boundaries defined in NCBI build 37.3. Linkage disequilibrium was modelled with the 1000 Genomes European reference panel (g1000_eur).
Four prespecified gene groups were evaluated. (i) The “CGR target” set comprised 23 loci that correspond to the pharmacological components of the novel glutamatergic regimen, including NMDA and AMPA receptor subunits, plasticity mediators and metabolic enzymes. (ii) An “expanded glutamate/plasticity” panel (130 genes) added all known ionotropic glutamate receptor genes, transporters, key metabolic enzymes and canonical downstream effectors such as BDNF–TrkB, mTOR and CaMKII. (iii) One hundred and twenty-one monoaminergic genes served as a negative control for pathway specificity. (iv) One hundred and thirty-four broadly expressed housekeeping and brain-enriched genes provided an additional control for baseline polygenicity.
Competitive gene-set analysis in MAGMA compared each set’s mean Z statistic with the genome-wide distribution. Significance was judged at three levels: nominal (p < 0.05), within-set Bonferroni (correcting for the number of genes per set) and genome-wide Bonferroni (18,012 tested genes). One-sided t tests contrasted mean Z values of each set with the genome-wide mean. Gene-set level p-values were further corrected for multiple testing across the four gene sets using Bonferroni and Benjamini-Hochberg FDR procedures. All post-processing and visualisation were carried out in Python 3.11 with custom scripts.
To test whether common PTSD risk is preferentially localised to the CGR target regions, a stripped-down partitioned heritability approach was applied. GWAS summary statistics were reformatted to include SNP ID, effective N, z-score, and allele information. A binary annotation file marking SNPs inside the extended CGR intervals was generated from the 1000 Genomes .bim index. Mean χ2 statistics were calculated separately for annotated and unannotated SNPs. Differences were assessed with both Welch’s t-test and the Mann–Whitney U test. Standard errors were estimated by delete-one-block jack-knife (200 equal-sized blocks). Enrichment was defined as the ratio of the mean χ2 value for annotated SNPs to that for the remainder of the genome.
Results
MAGMA produced gene-based statistics for 18,012 loci, capturing 54.4% of input SNPs. After genome-wide Bonferroni correction (p < 2.78 × 10−6) 235 genes were significant, 972 were suggestive at p < 0.001 and 3,959 reached nominal significance.
Of the 23 CGR targets, 21 were present in the output. Six were nominally significant (Table 1). GRIA1 (encoding the GluA1 AMPA subunit) showed the strongest signal (Z = 5.78, p = 3.77 × 10−9; genome-wide corrected p = 6.9 × 10−5). CYP2D6 (Z = 3.92, p = 7.49 × 10−5) also surpassed the set-level Bonferroni threshold. The remaining nominal genes were GRIN2A, ARC, NTRK2 and PTEN. The mean Z statistic for the entire CGR list (1.006) exceeded the genome-wide average (0.780) but did not reach statistical enrichment (one-sided t = 0.64, p = 0.26; set-level Bonferroni-corrected p = 0.78).

Table 1.
Detailed Target Gene Results.
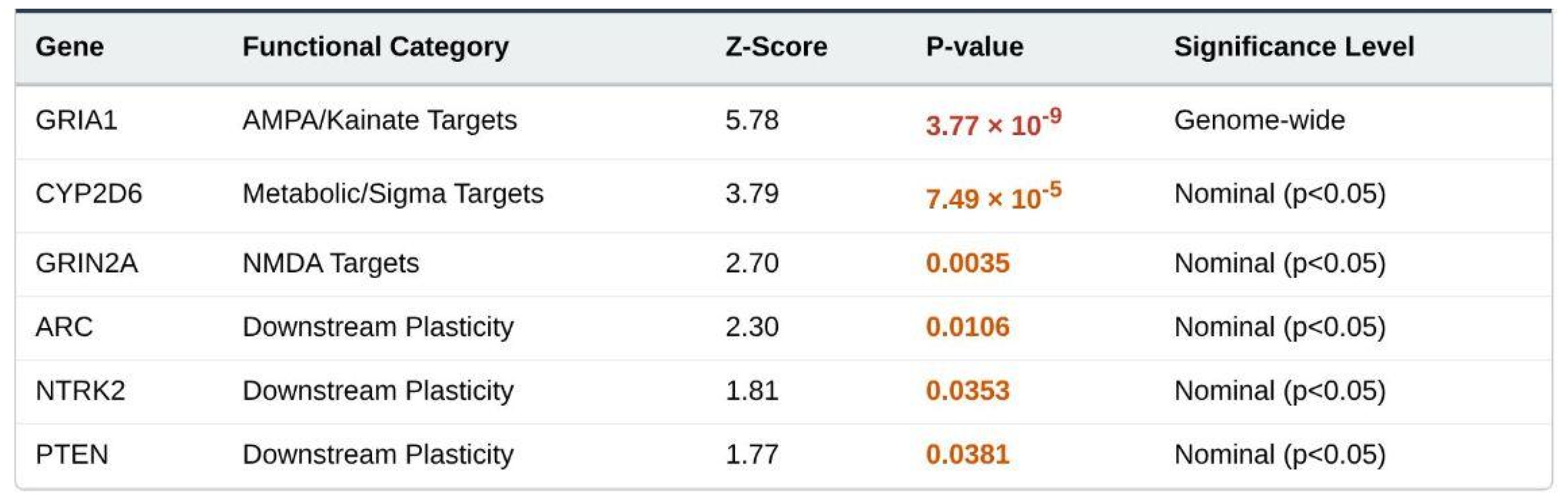
Table 2.
Detailed Target Gene Results (Expanded Panel).
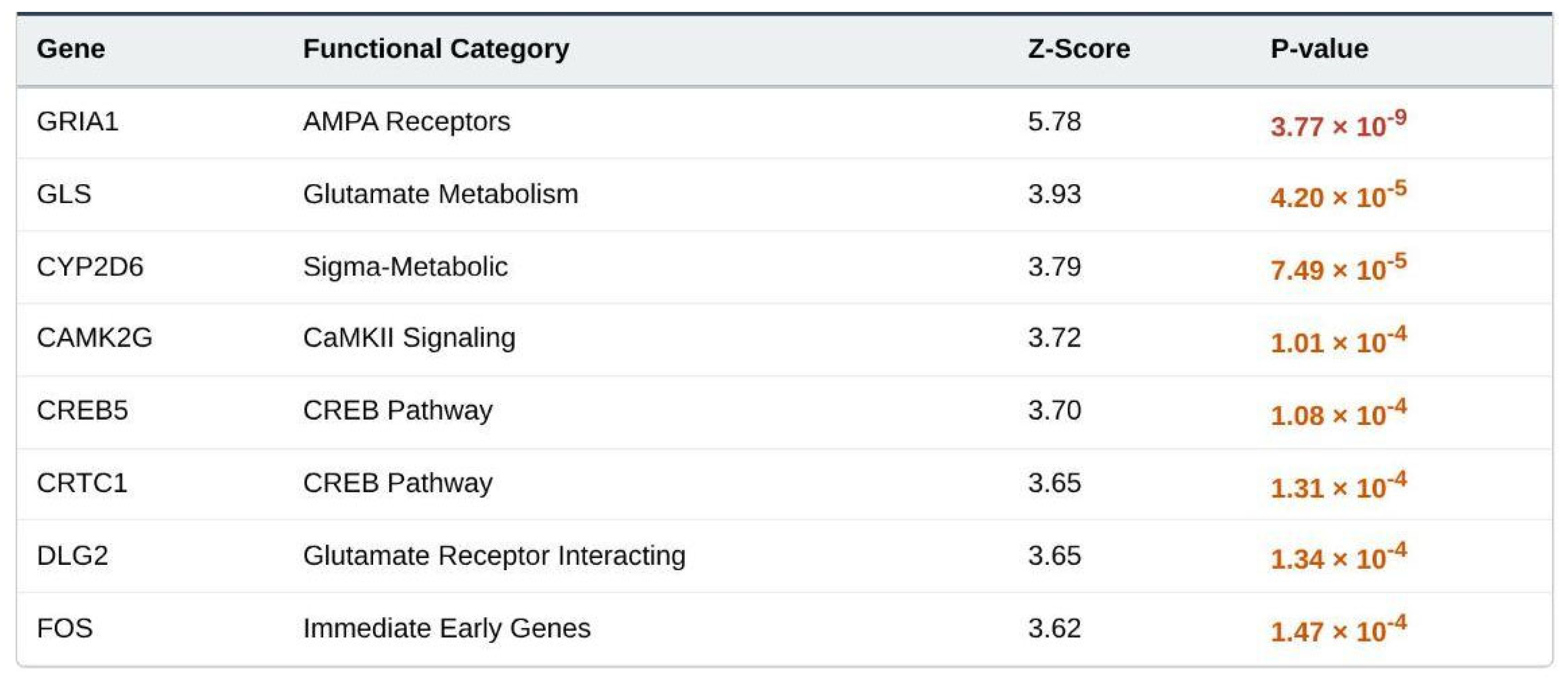
The expanded glutamate/plasticity panel contained 126 evaluable genes. Thirty were nominally associated, eight survived the set-level Bonferroni correction and GRIA1 again ranked first. The group’s mean Z (1.013) was significantly higher than background (one-sided t = 1.98, p = 0.024; set-level Bonferroni-corrected p = 0.096; FDR = 0.048), indicating modest but consistent enrichment of PTSD risk within broader glutamatergic and plasticity pathways.
Negative control sets showed weaker or variable patterns. Of 121 monoaminergic genes, nine were nominally significant and three endured set-level Bonferroni adjustment; DRD2 produced the top signal (Z = 6.06, p = 6.76 × 10−10). The monoamine mean Z (1.078) did not differ from background after correction (p = 0.11; set-level Bonferroni p = 0.44). The housekeeping list (134 genes) yielded 12 nominal signals, one persisting after within-set correction, and an average Z of 0.906 (p = 0.23; set-level Bonferroni p = 0.92).
Table 3.
Primary Gene Set Enrichment Results.
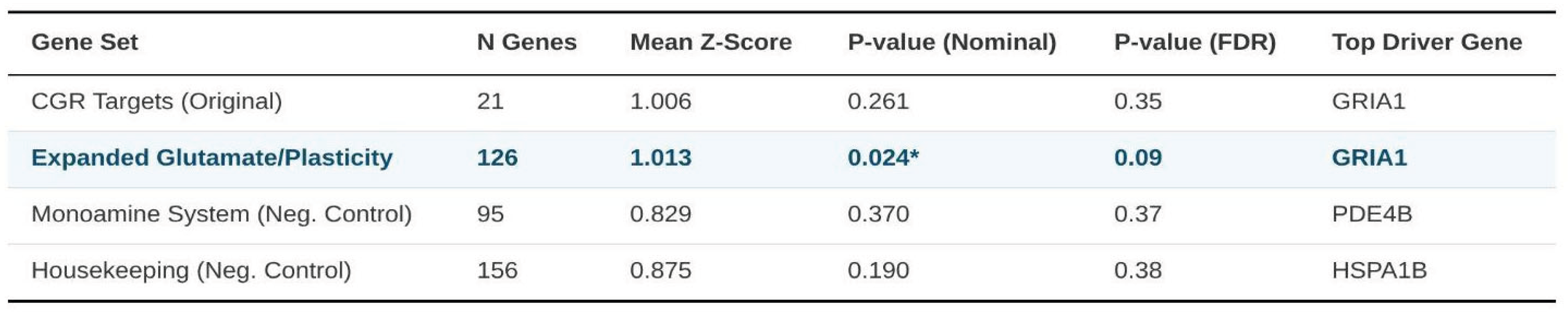
Across all four groups, a Kruskal–Wallis test detected no global difference in Z-score distributions (χ2 = 0.39, df = 3, p = 0.91). Even so, the expanded glutamate/plasticity set presented the most coherent over-representation of genome-wide association signals, while enrichment within the smaller CGR list centred on a subset of receptor and metabolic genes—most notably GRIA1 and CYP2D6.
Heritability was next partitioned by functional category. Significant enrichment was confined to three groups: AMPA/kainate receptors (enrichment ratio = 1.36, p = 1.05 × 10−13), metabolic targets such as CYP2D6 (ratio = 2.08, p = 4.08 × 10−5) and NMDA receptors (ratio = 1.25, p = 6.49 × 10−7). When all 23 genes were pooled, the enrichment estimate was modest and failed to reach significance (ratio = 1.16, p = 0.414). No excess heritability was observed for genes classified under plasticity signalling or glutamate transport.
Collectively, these results indicate that common variation in loci encoding AMPA and NMDA receptor subunits, together with key metabolic enzymes, accounts for a disproportionate share of the PTSD polygenic signal relative to genome-wide expectations.
Discussion
Principal Findings
The present MAGMA-based interrogation of the largest PTSD GWAS currently available (effective N = 638 463) indicates that common variation influencing glutamatergic transmission and activity–dependent synaptic remodeling carries a disproportionate share of disorder liability. Among the 23 a priori Cheung Glutamatergic Regimen (CGR) targets, six reached nominal significance, with GRIA1 exhibiting genome-wide evidence of association (Z = 5.78, p = 3.77 × 10−9) and CYP2D6 ranking second (p = 7.49 × 10−5). Partitioned heritability highlighted significant enrichment for AMPA/kainate receptor genes (enrichment = 1.36), NMDA receptor subunits (1.25) and glutamate metabolic enzymes (2.08). Although the pooled CGR list showed only modest enrichment (1.16, p = .41), a broader panel of 130 glutamate–plasticity genes displayed a significant shift in test statistics (mean Z = 1.013 vs. genome-wide mean = 0.780; p = .024; set-level FDR = 0.048). Collectively, these outcomes suggest that PTSD polygenic risk clusters at discrete receptor and metabolic nodes rather than being diffusely distributed across the glutamatergic pathway.
Relation to Experimental and Clinical Literature
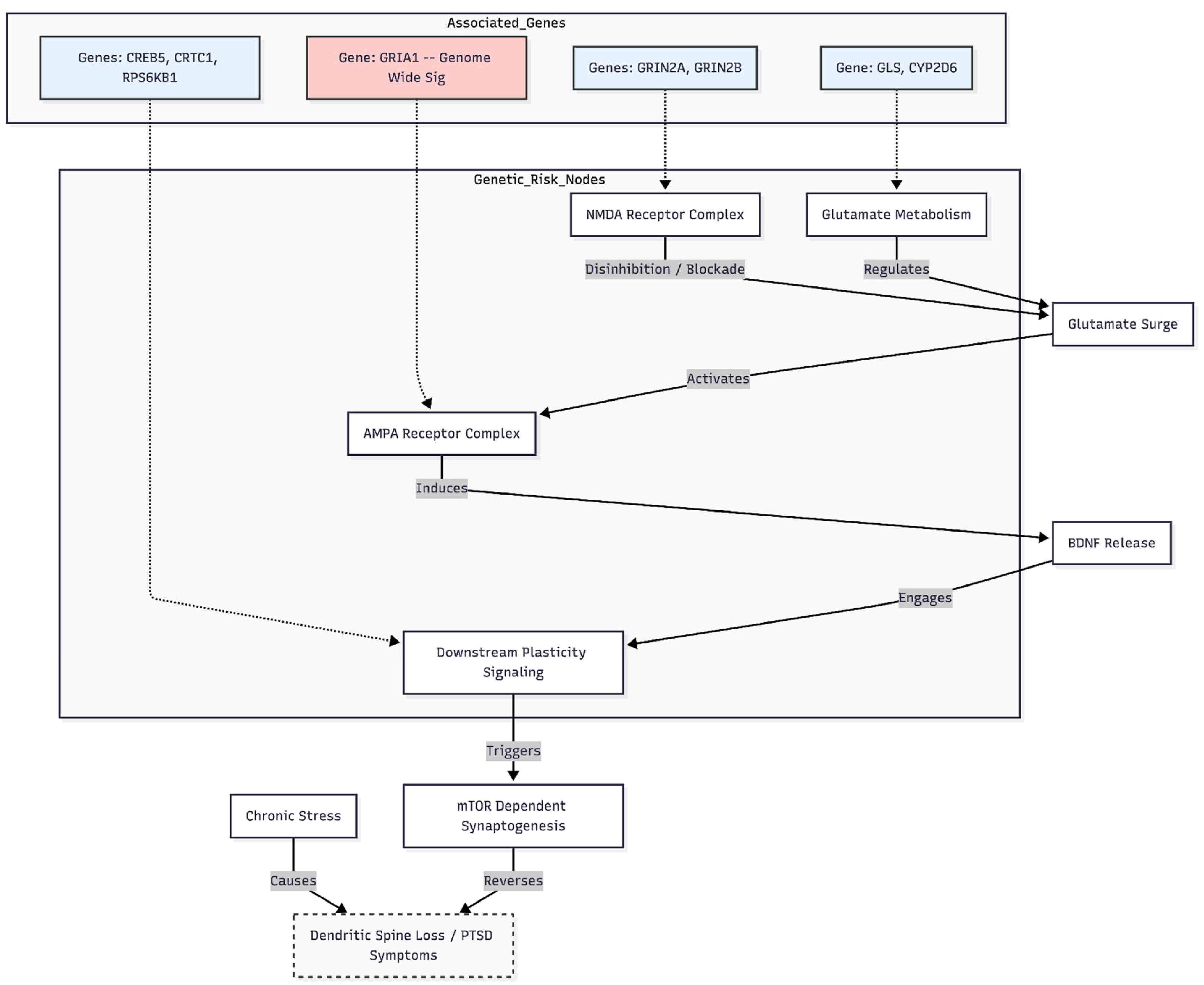
The robust GRIA1 signal dovetails with pre-clinical work demonstrating that AMPA throughput is required for ketamine’s fast-acting antidepressant and anti-PTSD properties [8]. Acute NMDA blockade triggers a glutamate surge, activates postsynaptic AMPA receptors, releases BDNF and engages mTOR-dependent synaptogenesis—events that reverse stress-induced spine loss in medial prefrontal and hippocampal circuits [2,9]. Enrichment at GRIN2A and other NMDA genes supports this cascade, consistent with evidence that NMDA disinhibition initiates downstream plasticity [10] (Figure 1).
The link to CYP2D6 is interesting. In addition to breaking down drugs in the liver, CNS CYP2D6 also affects the production of dopamine and the turnover of neurosteroids [11]. Fast metabolic variants could make people more vulnerable by changing the levels of catecholamines or neurosteroids, while slow variants could make dextromethorphan (DXM) stay in the brain longer when combined with CYP2D6 inhibitors. This is exactly what the CGR is based on [3,12].
Methodological Considerations
Several caveats merit emphasis. First, summary statistics were restricted to European ancestry; transferability to other populations, where allele frequencies and LD differ, is uncertain [6]. Second, the hypothesis-driven CGR list was small and may not capture all relevant glutamatergic elements. Third, MAGMA’s window-based annotation and reliance on common SNPs can miss distal regulatory variants or rare alleles of large effect. Fourth, although receptor and enzyme clusters are enriched relative to size, the absolute heritability explained remains small, a hallmark of PTSD’s polygenicity. Finally, gene-set level multiple testing correction across only four sets is conservative; larger hypothesis-driven panels may warrant FDR-based approaches in future work.
Clinical Implications
These genetic signals reinforce emerging clinical observations that oral NMDA–AMPA modulation can relieve trauma-related symptoms rapidly [3,4,5]. Enrichment at GRIA1, GRIN2A and CYP2D6 implies that patients with higher polygenic load at these loci might benefit most from regimens such as DXM plus CYP2D6 inhibition ± AMPA potentiation. Prospective trials should incorporate genotyping to test whether these variants predict response, advancing precision glutamatergic therapy for PTSD.
Future Directions
Two priorities follow. First, larger multi-ancestry GWAS with fine-mapping are needed to validate receptor-centric and metabolic signals. Second, mechanistic studies should explore how naturally occurring variants in CYP2D6, GRIA1 and related genes alter stress circuitry and drug response in cellular and animal models.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
None declared.
References
- Krystal, J.H.; Abdallah, C.G.; Averill, L.A.; et al. Synaptic Loss and the Pathophysiology of PTSD: Implications for Ketamine as a Prototype Novel Therapeutic. Current psychiatry reports 2017, 19(10), 74. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nature medicine 2016, 22(3), 238–249. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N. Oral Glutamatergic Augmentation for Trauma-Related Disorders with Fluoxetine- / Bupropion- Potentiated Dextromethorphan ± Piracetam: A Four-Patient Case Series. Preprints 2025. [Google Scholar] [CrossRef]
- Cheung, N. Rapid Stabilization of Trauma-Associated Depression and Suicidality via a Novel Oral Pharmacological Strategy: A Retrospective Case Series; Zenodo, 2025. [Google Scholar] [CrossRef]
- Cheung, N. Targeting the NMDA–AMPA Axis to Restore Synaptic Plasticity in Trauma-Related Disorders: Mechanistic Principles and Clinical Observations. Preprints 2025. [Google Scholar] [CrossRef]
- Nievergelt, C.M.; Maihofer, A.X.; Atkinson, E.G.; et al. Genome-wide association analyses identify 95 risk loci and provide insights into the neurobiology of post-traumatic stress disorder. Nature genetics 2024, 56(5), 792–808. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS computational biology 2015, 11(4), e1004219. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Iijima, M.; Chaki, S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behavioural brain research 2011, 224(1), 107–111. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lee, B.; Liu, R.J.; et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329(5994), 959–964. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Moaddel, R.; Morris, P.J.; et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016, 533(7604), 481–486. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhen, Y.; Miksys, S.; et al. Potential role of CYP2D6 in the central nervous system. Xenobiotica 2013, 43(11), 973–984. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, B.; Bunn, H.; Santalucia, M.; et al. Dextromethorphan-bupropion (Auvelity) for the Treatment of Major Depressive Disorder. Clinical psychopharmacology and neuroscience 2023, 21(4), 609–616. [Google Scholar] [CrossRef] [PubMed]
- Feder, A.; Costi, S.; Rutter, S.B.; et al. A Randomized Controlled Trial of Repeated Ketamine Administration for Chronic Posttraumatic Stress Disorder. The American journal of psychiatry 2021, 178(2), 193–202. [Google Scholar] [CrossRef] [PubMed]
- Duek, O.; Korem, N.; Li, Y.; et al. Long term structural and functional neural changes following a single infusion of Ketamine in PTSD. Neuropsychopharmacology 2023, 48(11), 1648–1658. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mechanistic Pathway of PTSD Risk Variants.
Note. This diagram illustrates how the significant genetic hits (GRIA1, GRIN2A, RPS6KB1) map onto the glutamate-plasticity cascade described in the discussion.
Figure 1.
Mechanistic Pathway of PTSD Risk Variants.
Note. This diagram illustrates how the significant genetic hits (GRIA1, GRIN2A, RPS6KB1) map onto the glutamate-plasticity cascade described in the discussion.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



