
Submitted:
23 December 2025
Posted:
24 December 2025
You are already at the latest version
Abstract
Protein language models (pLMs) have become indispensable tools in computational biology, driving advances in variant effect prediction, functional annotation, structure prediction, and engineering. However, their rapid expansion from millions to tens of billions of parameters introduces significant computational, accessibility, and sustainability challenges that limit practical application in environments constrained by GPU memory, hardware availability, and energy budgets. This survey presents the first comprehensive review of efficient pLMs, synthesizing recent advancements across four key dimensions. We first examine (1) dataset efficiency through meta-learning-based few-shot and scaling-law-guided data allocation; and (2) architecture efficiency via lightweight alternatives including quantized transformers, embedding compression, and convolution-based designs. Furthermore, we review (3) training efficiency through scaling-law-informed pretraining, structure-integrated multimodal approaches, and low-rank adaptations with diverse distillation strategies; and (4) inference efficiency via quantization, dense-retrieval, and structure-search methods. By providing a structured taxonomy and practical guidance, this survey enables the development of high-performance, scalable, yet sustainable next-generation pLMs.
Keywords:
protein language models
; computational biology
; efficient language models
; large language models
; artificial intelligence
; machine learning
1. Introduction
Inspired by the success of large language models (LLMs) in natural language processing (NLP) [1,2], protein language models (pLMs) have emerged as a transformative force in computational biology [3]. By treating Amino Acid (AA) sequences as a “language of life,” [4] pLMs apply self-supervised learning to vast protein databases to learn the fundamental grammar of protein structure, function, and evolution [5,6]. A comparative overview between LLMs and pLMs is shown in Figure 3, providing intuitive context for their methodological parallels and distinctions. This paradigm shift enables protein analysis at an unprecedented scale of hundreds of millions of sequences [7], offering a powerful alternative to traditional experimental methods like A foundational experimental technique used to determine the precise three-dimensional atomic structure of a protein by analyzing the diffraction pattern of X-rays passing through a protein crystal. (X-ray crystallography), Nuclear Magnetic Resonance Spectroscopy (NMR) spectroscopy, and Cryo-Electron Microscopy (Cryo-EM) which, while foundational, are often slow and costly [8,9]. The power of pLMs lies in their ability to bridge the enormous gap between the over 200 million known protein sequences and the fewer than 200 thousand experimentally determined structures [10,11], thereby accelerating drug discovery and synthetic biology [9]. A historical overview of key milestones in pLM development is summarized in Figure 2.
The remarkable capabilities of pLMs stem from their deep neural architectures, predominantly transformer-based networks [1], which are trained extensively on massive protein sequence datasets [12] to learn intrinsic biological patterns and representations. The entire pipeline of pLMs consists of dataset construction, model architecture design, training methodologies (pre-training, fine-tuning, and distillation), and inference strategies, each influencing both performance and efficiency [13,14]. Powered by large-scale computational resources, these models integrate evolutionary and structural information from massive-scale evolutionary protein corpora into parameter spaces reaching up to tens of billions [7], thereby achieving state-of-the-art accuracy on structure-prediction benchmarks such as CAMEO [15] and CASP14 [16], a level not previously reached by single-sequence models.
Despite their transformative potential, the effectiveness of large-scale pLMs comes at a substantial computational and memory cost, posing significant practical challenges. Typical large pLMs contain billions of parameters—such as the 15B parameter ESM-2 [7]—and require extensive resources; for instance, training models of this scale conventionally demands tens of thousands of GPU hours, translating to immense financial and environmental burdens [7,17,18]. This heavy reliance on compute limits accessibility for resource-constrained research groups [19]. Furthermore, deploying such extensive models in real-world scenarios, such as proteome-wide homology searches, becomes computationally formidable, severely restricting their broader applicability and practical utility [20].
Motivated by these challenges, a growing body of research has focused explicitly on improving the efficiency of pLMs across various stages of their lifecycle [21,22,23]. These efficiency-focused methods aim to achieve competitive or even superior performance while reducing resource consumption by orders of magnitude. Specifically, efficiency strategies in pLMs encompass several critical categories, the reviewed methods of which are listed in Figure 1:
- Training Efficiency: Optimizing the training procedures, including computational resource allocation guided by scaling laws [21], efficient fine-tuning techniques [22,30,31], and knowledge distillation [39] approaches that transfer knowledge from larger, teacher models to compact, efficient student models [33,34,35,36].
While recent surveys have provided broad overviews of protein language models (pLMs) and their applications [9,40], our work particularly focuses on the efficiency dimension of pLMs across dataset, architecture, training, and inference. We systematically review these efficiency-focused methodologies, highlighting their core principles, underlying mechanisms, and performance implications. While recent surveys have provided broad overviews of protein language models (pLMs) and their applications [9,40], our work particularly focuses on the efficiency dimension of pLMs across dataset, architecture, training, and inference. We systematically review these efficiency-focused methodologies, highlighting their core principles, underlying mechanisms, and performance implications. The remainder of this survey is organized as follows: Section 2 details dataset strategies, Section 3 reviews architectural innovations, Section 4 discusses training methodologies, Section 5 covers inference acceleration, and Section 6 outlines future directions.
Figure 2.
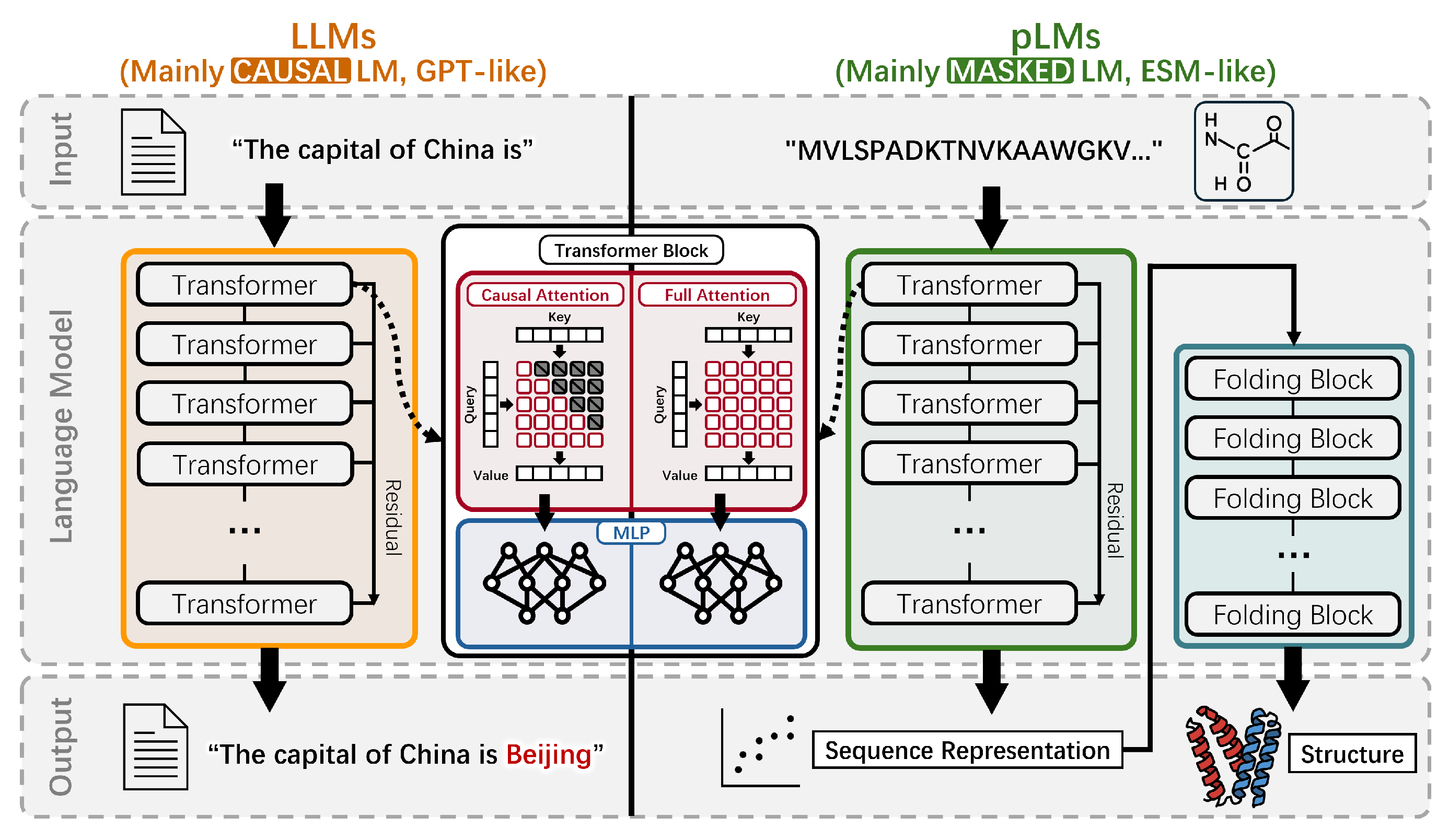
The evolution history of Protein Language Models (pLMs) from 2019 to 2025. The timeline illustrates the development and divergence of major pLM families, such as Masked pLMs and Conditional Inverse Folding pLMs, highlighting key models like the ESM series, ProtT5, and ProGen, along with their originating research labs.
Figure 2.
The evolution history of Protein Language Models (pLMs) from 2019 to 2025. The timeline illustrates the development and divergence of major pLM families, such as Masked pLMs and Conditional Inverse Folding pLMs, highlighting key models like the ESM series, ProtT5, and ProGen, along with their originating research labs.
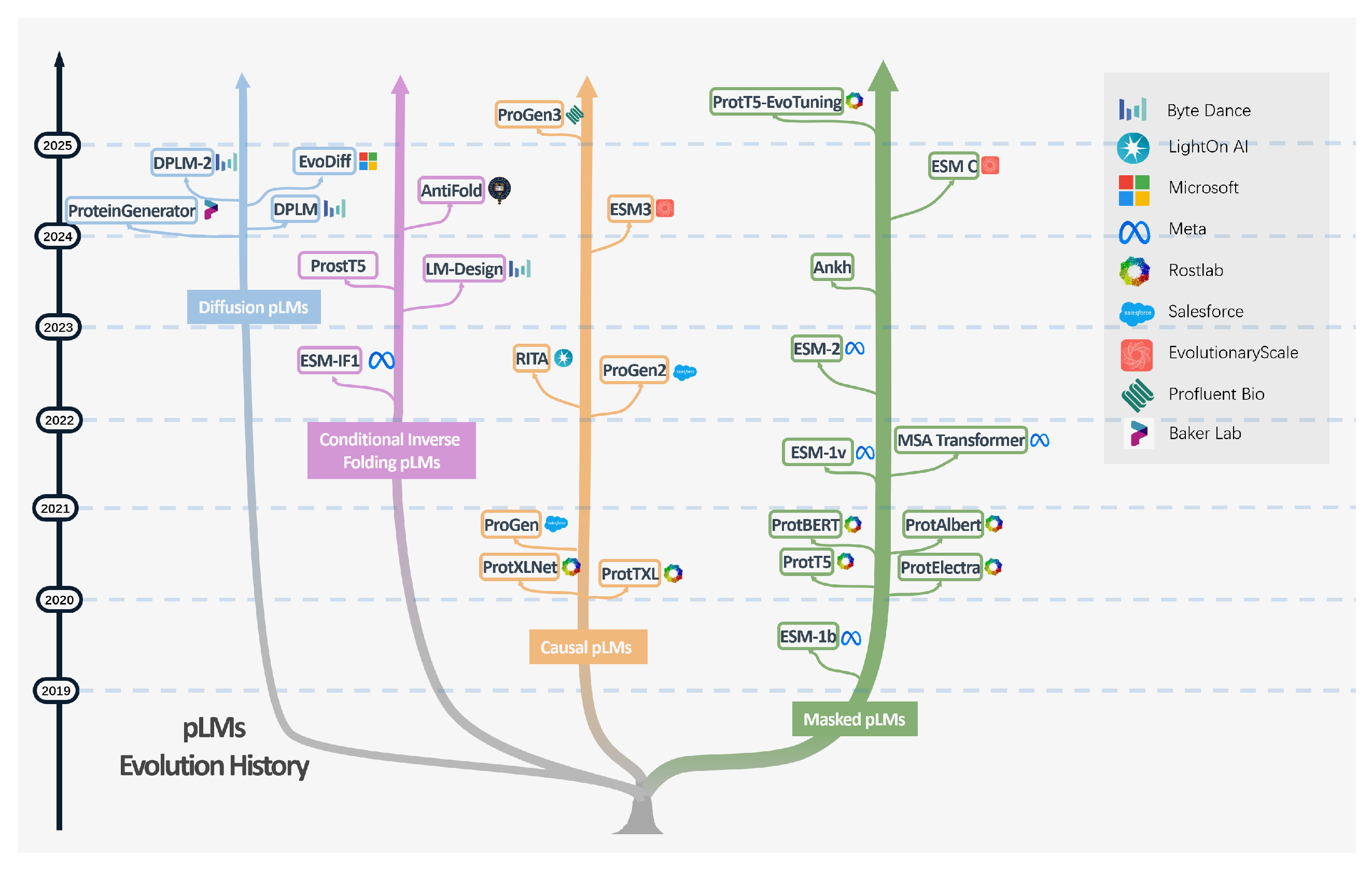
Figure 3.
A comparative overview of general Large Language Models (LLMs) and Protein Language Models (pLMs). While both are based on the Transformer architecture, LLMs (left) typically use causal attention for autoregressive text generation. In contrast, pLMs (right) often use masked language modeling (with full attention) to learn sequence representations from amino acid inputs, which can then be used for downstream tasks like structure prediction. The folding blocks denote structure-prediction components used for structure prediction, e.g., as in ESMFold [7]; their internal architectural details are omitted here for brevity.
Figure 3.
A comparative overview of general Large Language Models (LLMs) and Protein Language Models (pLMs). While both are based on the Transformer architecture, LLMs (left) typically use causal attention for autoregressive text generation. In contrast, pLMs (right) often use masked language modeling (with full attention) to learn sequence representations from amino acid inputs, which can then be used for downstream tasks like structure prediction. The folding blocks denote structure-prediction components used for structure prediction, e.g., as in ESMFold [7]; their internal architectural details are omitted here for brevity.

2. Datasets for Efficient pLMs
2.1. Background and Challenges
The training paradigm for modern language models is fundamentally data-driven, predicated on learning statistical and semantic regularities from vast corpora [42]. For Large Language Models (LLMs) like BERT [2] and the GPT series [43,44], this involves processing immense textual datasets that are rigorously deduplicated, filtered, and ultimately discretized into a finite vocabulary via tokenization [45,46]. Transposing this paradigm to computational biology, Protein Language Models (pLMs) operate on datasets of AA sequences drawn from large-scale biological repositories such as UniRef [12] and expansive Relating to the study of genetic material recovered directly from environmental samples, which often contain a vast collection of uncultured and unknown organisms. (metagenomic) collections [47]. These biological datasets undergo analogous preprocessing, including stringent sequence identity filtering and task-specific curation, to construct a suitable training corpus [21].
Within both NLP and computational biology, an efficiency-centric approach to dataset construction seeks to maximize model utility and generalization performance under a constrained computational-budget. For LLMs, this often involves curating data for optimal scale and diversity to avoid diminishing returns on compute [48,49]. In the context of pLMs, efficiency-driven data strategies have emerged to address distinct biological challenges. Key approaches include meta-learning frameworks for few-shot adaptation, which are critical in scenarios where experimental labels are prohibitively expensive or scarce [22]. Additionally, researchers have formulated empirically-derived scaling laws that govern the co-scaling of model and data size to optimize the training trajectory within a fixed computational envelope [21]. Such methods directly confront data-related bottlenecks, aiming to enhance model performance within the practical constraints of bioinformatics research.
The development of these efficiency-focused strategies is motivated by several fundamental challenges inherent to biological data utilization:
- Paucity of Labeled Data: A critical bottleneck in many real-world applications, such as protein engineering and A field of molecular biology that aims to understand the function of genes and proteins, often at a genome-wide scale. (functional genomics), is the extreme scarcity of experimentally validated labels. Often, only a few dozen labeled protein variants are available, posing a fundamental impediment to traditional supervised fine-tuning and demanding highly data-efficient adaptation methods [22].
- Absence of Principled Scaling Guidelines: Given a finite computational-budget, determining the optimal allocation between model parameter count and training data volume remains a significant challenge. An unprincipled approach, such as aggressively scaling model size while repeatedly cycling through a small dataset, can lead to suboptimal training dynamics, including premature convergence, overfitting, and a failure to capitalize on the model’s architectural capacity [21].
To surmount these obstacles, recent work has focused on developing sophisticated meta-learning protocols and establishing a principled, theoretical foundation for balancing model and data scaling. The subsequent section reviews these methodologies in detail, with an overview provided in Table 1. See Figure 4 for an illustration of these efficient dataset strategies.
2.2. Efficient Dataset Methods
Few-Shot Learning for Protein Fitness Prediction (FSFP) [22] addresses the challenge of efficiently fine-tuning pLMs under severe data scarcity [50], enabling fitness-prediction with only tens of experimentally labeled mutants per target protein. The key innovation of FSFP lies not in increasing data volume, but in its auxiliary-task construction, evolutionary-informed pseudo-labeling, and meta-learning-driven adaptation, providing a robust solution for pLM fine-tuning in low-data scenarios.
FSFP frames fitness-prediction as a ranking task, employing a listwise-loss [51] throughout both meta-learning and fine-tuning, which better reflects practical needs in protein engineering research and applications [52]. FSFP consists of the following major components:
- Meta-Learning: These auxiliary tasks are used for meta-training via MAML [58], equipping the pLM for rapid adaptation.
- Parameter-Efficient Adaptation: To prevent overfitting under few-shot settings [59], FSFP freezes the backbone pLM and trains LoRA [41] parameters during meta-training and when transferring to target task; meta-learning yields a better initialization of the LoRA parameters, and the subsequent adaptation continues to update these parameters.
Benchmarking on 87 protein datasets [60], FSFP demonstrates superior performance compared to strong baselines. It outperforms unsupervised zero-shot inference (including ESM-1v [61], ESM-2[7], and Saprot [62]) and significantly surpasses the state-of-the-art supervised few-shot baseline, Ridge Regression [63]. Specifically, FSFP boosts the average spearman correlation by up to 0.1 and raises positive hit rates in real-world engineering by 25% [22].
Scaling laws for dataset allocation in pLM training [21] have been empirically established to maximize training efficiency under fixed compute. Similar to LLMs in NLP tasks, simply increasing model size or compute is insufficient [42,48]; the size and diversity of training data are critical. Empirical analysis shows that masked language models (MLMs) [2] and causal language models (CLMs) [44,64] follow distinct power-law relationships between model size and required data, with sublinear data scaling [65]. Repeating small datasets across many epochs yields overfitting in MLMs [66] and diminishing returns in CLMs.
To address these issues, the study introduces the UniMeta200B dataset [47,67], aggregating diverse metagenomic protein sequences with strict deduplication. This improves out-of-domain perplexity and yields more stable independent and identically distributed validation curves with reduced variance across families.
The resulting scaling laws provide explicit formulas for allocating parameters and dataset size under a compute constraint, enabling principled, data-centric decisions in pLMs development. For fitted relations and resource-allocation details, see Section 4.2. These results highlight that enhancing dataset diversity—rather than simply expanding model size or repeating data—is essential for efficient protein language modeling.

3. Efficient Architecture
3.1. Background and Challenges
Model architecture fundamentally determines the representational capability and computational complexity of language models. LLMs in NLP predominantly employ transformer architectures [1], characterized by self-attention mechanisms that effectively capture long-range-dependencies. However, the quadratic scaling of transformers in computational complexity and memory consumption with respect to input sequence length poses significant efficiency challenges [1,48]. Alternatively, convolutional neural networks (CNNs) [68,69], leveraging local receptive fields, scale linearly with sequence length and inherently incorporate positional information through sliding window operations. These distinct architectural paradigms have informed similar developments in pLMs, where transformers remain dominant due to their robust performance on capturing long-range residue interactions [5], while CNN-based methods have emerged as efficient alternative [27].
Efficiency-focused model architectures in LLMs typically include strategies such as weight quantization, embedding dimension reduction, and structural sparsity to mitigate computational overheads [70,71,72]. Analogously, recent advancements in pLM architectures have pursued quantization of weights [24], embedding compression [26], and cross-attention [1] modules conditioned on homologous sequences [25] to integrate evolutionary information efficiently [73,74]. Convolution-based models, with inherent linear scaling, further enhance computational efficiency, presenting competitive alternatives for large-scale protein modeling tasks [27].
The emergence of efficiency-focused model architectures in pLMs is largely driven by several challenges in model architecture, including:
- High Computational and Memory Cost: Standard transformer-based pLMs require quadratic time and memory with respect to sequence length, leading to prohibitive resource demands for long proteins or large models. The use of full-precision weights and high-dimensional embeddings further increases GPU and energy costs, particularly during large-scale training and deployment [1,5,24,26,27].
- Redundancy and Massive Activations in Embeddings: Protein embeddings in large models are highly compressible, indicating over-parameterization, and also exhibit massive activations in certain channels, leading to inefficient memory use [26].
- Limited Inductive Bias and Information Integration: Existing architectures may not efficiently leverage important biological information, such as evolutionary relationships or sequence homology, constraining their performance and flexibility in specialized tasks [25].
3.2. Efficient Transformer
2Bits of Protein [24] is a ternary-quantization architecture (based on [75]) which offers an efficient solution to the computational and memory bottlenecks of large pLMs. In this approach, all linear layers except the embedding and output head are quantized to ternary precision, with weights constrained to by scaling and rounding each weight matrix [76,77,78]. Specifically, for a weight matrix W, the quantized matrix is defined as:
where is the mean absolute value of W’s elements, and rounds x to the nearest integer within .
This design achieves substantial reductions in GPU memory and energy usage—prior NLP work reports up to 3.55× lower memory and 21.7× less energy consumption compared to full-precision models [76]. The ternary pLM preserves the standard transformer encoder architecture [1,7] and remains trainable from scratch with stable performance [79]. On ProteinGym [80], ternary ESM2-8M attains a mean spearman-correlation of 0.181 across 217 zero-shot tasks, competitive with efficient baselines such as ProtGPT2 [14] and UniRep [81]. Under matched compute (24 GPU-hours, ‘crammed’ training [82]), the performance gap becomes statistically insignificant, whereas under the standard training schedule, ESM2-8M is about 25% higher and statistically significant (). Thus, ternary-quantization provides a principled and highly efficient design for pLMs, substantially reducing computational cost while maintaining competitive predictive performance.
Compressed Hourglass Embedding Adaptations of Proteins (CHEAP) [26] provide an architectural solution for drastically reducing the dimensionality and memory footprint of pLMs embeddings, while retaining both sequence and structure information. CHEAP employs a transformer-based hourglass-autoencoder [83,84], which applies linear downsampling and projection to the ESMFold [7] latent embedding. Its key innovations can be divided into:
- Continuous Compression: CHEAP enables up to channel compression and sequence length compression through a learnable bottleneck, allowing backbone structure reconstruction with less than Å root-mean-squared distance [85] at channel compression, and maintaining sequence recovery above until fewer than 8 channels remain. To address abnormal massive activations in certain embedding channels, CHEAP introduces per-channel normalization [86,87], further improving compressibility.
These compressed embeddings enable efficient downstream applications—such as protein design, similarity search, and functional prediction—making advanced pLM representations accessible for resource-limited scenarios while preserving high-resolution information.
RAG-ESM [25] introduces a parameter-efficient encoder-decoder architecture that augments pretrained ESM2 [7] with a minimal number of cross-attention layers and extensive parameter-sharing. The encoder embeds a context (homologous) sequence using the original ESM2 model, while the decoder reuses pretrained ESM2 layers, inserting a few cross-attention layers to transfer information from the context embedding to the masked input sequence. Critically, self-attention and feed-forward layers in both modules share weights, so the number of new parameters is negligible compared to the backbone model. Formally, the total parameter count of RAG-ESM is
where N is the number of parameters in the pretrained ESM2 backbone, and M is the number of newly added cross-attention parameters. Conditioning on homologs during inference allows RAG-ESM to extract evolutionary information [91] without requiring MSAs [74,92,93] or significantly increasing model size.
Empirically, RAG-ESM models with 12M and 165M parameters achieve 48% and 43% lower perplexity, respectively, than their ESM2-8M and ESM2-150M baselines on masked AA prediction. Both are trained under the discrete-diffusion [94,95] using the closest homolog as context [96], with modest compute budgets (50–120 GPU-hours). Cross-attention heads naturally learn alignment [97]; several heads reach , and a logistic regression trained on all 60 heads achieves an average against Needleman–Wunsch [98] alignment matrices.
This design enables efficient and scalable homology-aware pLMs that achieve state-of-the-art performance among sequence-based models for homolog-conditioned generation and remain competitive on motif scaffolding [99], while retaining flexibility.
Cramming Protein Language Model Training in 24 GPU Hours [23] introduces an efficient transformer-based architecture specifically optimized for rapid, resource-constrained pre-training. The corresponding pre-training setup is detailed in Section 4.2.
The core approach adapts the HuggingFace ESM2 [7] backbone by removing: (1) all query, key, and value biases in attention blocks, (2) biases in intermediate linear layers, thereby reducing computational overhead while retaining model capacity to maximize per-token throughput under a stringent compute budget: model weights are initialized from scratch, and training is limited within 24 hours on a single GPU.
These architectural and implementation choices collectively allow a 67M-parameter model to achieve competitive downstream performance—on tasks such as protein fitness landscape inference [60] and protein–protein interaction classification [100]—with models trained for over 15,000× greater GPU hours. It demonstrates that by simplifying architectural details, it is feasible to obtain expressive pLMs suitable for practical research and deployment under extreme computational constraints.
3.3. Convolutional Layer Based Methods
Convolutional Autoencoding Representations of Proteins (CARP) [27] demonstrates that convolutional architectures can match or exceed transformer-based pLMs [5,7] in both predictive power and computational efficiency. CARP replaces transformer self-attention with ByteNet-style [68] dilated convolutional blocks [101,102], exponentially expanding the receptive-field while ensuring linear scaling in computation and memory with sequence length. Formally, the computational complexity per layer is:
where L is the input sequence length.
CARP-640M (640M parameters) achieves a masked language modeling loss of , close to ESM-1b’s [5], and attains higher average zero-shot mutation effect prediction (Spearman vs. ) on 41 datasets. Unlike ESM-1b, CARP processes sequences up to 4,096 residues without memory overflow and maintains stable loss and runtime for longer sequences. These results support convolutional architectures as efficient, scalable, and competitive alternatives to transformers for large protein sequence modeling.
ProtHyena [28] introduces the Hyena operator [103] as an efficient alternative to self-attention and standard convolution in pLMs. The per-layer computational complexity is:
where L is the input sequence length.
Compared to transformer models whose self-attention layers require quadratic complexity, the Hyena operator enables substantially improved scalability; compared to standard CNNs, whose long-range modeling is limited by kernel size, Hyena leverages implicit long convolutions [104] and element-wise gating [105,106] to efficiently capture both local and global dependencies. The core block applies:
where N is block depth, are long convolution filters, v is the input projection, and · denotes gating.
This enables ProtHyena to process sequences up to a million residues and achieve accuracy competitive with or better than larger transformer-based pLMs, such as ESM-1b [5], ESM-2 [7], and ProteinBERT [107], using about 10% of their parameters and up to speedup.

4. Efficient Training
4.1. Background and Challenges
Training LLMs generally follows two stages: pre-training and post-training. During pre-training, models are trained on large unlabeled corpora to learn general representations [2,43]. post-training then adapts these models to downstream objectives (including, for example, supervised fine-tuning (SFT) [49] and reinforcement learning (RL)-based tuning such as RLHF [108] or GRPO [109]). In this paper, we mainly focus on fine-tuning [110] within post-training. In parallel, distillation transfers knowledge from high-capacity teachers to compact students for efficiency [39,111,112].
Training pLMs adopt a conceptually similar pipeline to LLMs but with several domain-specific characteristics: (1) Pre-training: pLMs are trained on large corpora of unlabeled protein sequences at the amino-acid level to learn general biological representations [3,5,6,81]. (2) Post-training: pLMs are further adapted on task-specific datasets for specialized bioinformatics objectives [113], for example, function prediction [30,114]. (3) Distillation: knowledge is transferred from large teacher models to smaller students for efficient deployment, which closely mirrors the approach used in LLMs [34,36,112,115].
Given these characteristics, improving computational and data efficiency has become a key objective in pLMs training. Efficiency-focused pLMs training therefore focuses on the following bottlenecks:
-
Challenges in Pre-training:
- -
- -
- Limited Structural Integration: Most pre-training pipelines rely solely on sequence data, whereas structural information remains underutilized, which may ultimately limit both the efficiency and generalization capabilities of the models [29].
-
Challenges in Fine-tuning:
- -
- -
- -
- Low Data Efficiency: Adapting models to new tasks with limited labeled data can be inefficient, as conventional fine-tuning is prone to overfitting and resource waste [22].
-
Challenges in Distillation:
- -
- -
Following sections review methods addressing challenges through efficient pre-training, fine-tuning, and distillation.
4.2. Efficient Pre-Training Methods
To orient the discussion, Figure 6 summarizes the efficient pre-training methods reviewed in this section.
Table 3.
Summary of representative training-stage efficiency methods for pLMs.
| Category | Method | Brief Description |
|---|---|---|
| Pre-training | Scaling Law [21] | Empirical study on pLMs optimal data/model allocation. |
| Structure Integration [29] | Joint sequence-structure pre-training (e.g., ProstT5). | |
| Cramming Challenge [23] | Train a performant pLM in 24 hours on a single GPU. | |
| Fine-tuning | A study of PEFT Methods [30] | LoRA for parameter-efficient tuning. |
| QLoRA [31] | 4-bit quantization + LoRA for efficient fine-tuning. | |
| SES-Adapter [32] | Structure-aware adapters for cross-modal representation. | |
| FSFP [22] | few-shot with meta-learning and LoRA. | |
| Distillation | Cross-distillation in AlphaFold3 [33] | Distillation from a single large teacher-model. |
| ProtGO [34] | Transferring functional and structural knowledge. | |
| VespaG [35] | Distillation via evolutionary expert supervision. | |
| MTDP [36] | Aggregating knowledge from multiple teacher models. |
Scaling law-guided pre-training for pLMs [21] empirically establishes optimal resource allocation between model size and data for transformer-based pLMs. On a large dataset of 939M protein sequences, over 300 models were trained to reveal that both masked (MLM) and causal (CLM) objectives follow distinct power-law scaling similar to Kaplan et al. [42] and Hestness et al. [65]. For a increase in compute, optimal MLM size increases with more data, while CLM size grows with more data. The fitted scaling relations for MLMs are:
and for CLMs are:
where C represents the total pre-training FLOPs, N is the number of forward-activated non-embedding parameters, and D represents the number of training tokens.
Repeating data leads to overfitting for MLMs and diminishing returns for CLMs [122], which is mitigated by introducing a diverse, deduplicated dataset (UniMeta200B). Transfer learning—pre-training with CLM and then fine-tuning with MLM—reduces the compute needed for optimal MLMs by up to . Large-scale validations confirm that these scaling laws yield models with better generalization and downstream performance under fixed compute budgets, particularly in practical, resource-constrained environments.
ProstT5 [29] is a T5-based model [49] that integrates structure information [123] into pLM pretraining to boost efficiency and versatility. ProstT5 unifies protein sequence (Amino Acid, AA) and structural (Three-Dimensional Interaction (token) (3Di) token) modalities within a single encoder-decoder architecture. Its key innovations in the pretraining stage can be divided into:
- Multimodal Pre-training: By encoding 3D structures from AlphaFoldDB [124] as 3Di-token sequences, and expanding the vocabulary with 3Di tokens, ProstT5 applies span-based denoising objectives to both AA and 3Di sequences, teaching the model new structural tokens while avoiding catastrophic forgetting of sequence information.
- Bi-directional Translation Objectives: ProstT5 is trained to translate between AA and 3Di representations using direction tags (<fold2AA>, <AA2fold>), enabling both “folding” (AA→3Di) and “inverse folding” (3Di→AA), thus robustly linking sequence and structure information.
This bilingual setup allows ProstT5-predicted 3Di strings to be used for structure-level similarity search (e.g., with Foldseek [125]), achieving The task of identifying distant evolutionary relationships between proteins, typically when their sequence similarity is very low but their 3D structures are still related. (remote homology detection) [126] sensitivity nearly matching experimental structures, but with a speedup of over three orders of magnitude (e.g., 43 seconds vs. 48 hours for full The complete set of proteins expressed by an organism, cell, or tissue at a given time. (proteome) annotation) [57,125]. Embedding-based annotation transfer further shows improved CATH fold classification [127] accuracy over ProtT5 [6] and ESM-1b [5], and The task of predicting the local secondary structures of a protein (e.g., alpha-helices, beta-sheets) from its primary amino acid sequence. (secondary structure prediction) with ProstT5 matches or exceeds prior state-of-the-art. In inverse folding [63,99], ProstT5 generates diverse sequences with predicted structures closely matching targets, demonstrating both enhanced efficiency and versatility.
Overall, integrating structure into pLM pretraining via bilingual modeling delivers substantial gains in inference speed and structural generalization, advancing protein design and large-scale annotation capabilities.
Cramming Protein Language Model Training in 24 GPU Hours [23] proposes a pre-training paradigm specifically tailored for rapid and resource-constrained scenarios. In this framework, a transformer-based pLM is trained from scratch under a strict 24 GPU-hour budget, with no use of pre-trained models at any stage. The pre-training pipeline consists of several key components:
- Random initialization: All model weights are initialized randomly.
- On-the-fly masked language modeling: Masked language modeling is performed directly on UniRef50 [12] splits, with all data processing (e.g., tokenization, filtering, sorting) occurring on-the-fly during training, and no offline pre-processing.
- Large effective batch size: Training employs a batch size of 128, sequence length of 512, and gradients are accumulated over 16 steps for an effective batch of 2048 sequences.
- Critical hyperparameter tuning: The optimizer is AdamW [128] with carefully tuned parameters. The learning rate schedule is central: the optimal regime employs a fast warmup [129,130] to a peak learning rate of over 1,000 steps, followed by a slow linear decay [131] to near zero over the remaining steps, with training capped at 50,000 updates.
These pre-training choices allow the resulting 67M-parameter model to match or approach the downstream performance of much larger models such as ESM2-3B [7]—trained for over 15,000× more GPU hours—on tasks including protein fitness landscape inference and PPI classification. The architectural design supporting these results is detailed in Section 3.2. This work demonstrates that, through systematic pre-training design and hyperparameter tuning [132,133], performant pLMs can be obtained at a fraction of the conventional computational cost, enabling broader accessibility for rapid biological modeling.

4.3. Efficient Fine-Tuning Methods
Fine-tuning in pLMs involves adapting a pre-trained model to specific downstream biological tasks using relatively small amounts of labeled protein data [30]. This step is inherently efficiency-focused, enabling rapid model adaptation without extensive retraining. Advanced PEFT techniques, such as LoRA [134] and structure-aware adapters [32], further minimize computational and memory demands while maintaining high performance. Subsequent subsections review these PEFT methods and associated efficiency gains.
To summarize the discussion, Figure 7 provides an overview of the key techniques covered for efficient fine-tuning.
A study of Parameter-efficient fine-tuning (PEFT) methods systematically compared full-model fine-tuning with several PEFT approaches—including LoRA [41], DoRA [135], IA3 [136], and prefix tuning [137]—across diverse pLMs and protein prediction tasks [30]. Its main findings include:
- Efficiency and accuracy: By freezing most model weights and only updating a small fraction (e.g., 0.25% for LoRA), PEFT methods, such as LoRA, achieved nearly equivalent accuracy to full fine-tuning.
- Training speedup and memory savings: For larger models, LoRA offered up to a 4.5× training speedup with comparable GPU memory requirements, and required saving only adapter weights, further improving efficiency.
- Compatibility and resource efficiency: LoRA, DoRA, and similar methods are compatible with mixed precision, gradient accumulation, and CPU-offloading, enabling fine-tuning even on 8GB or 16GB GPUs.
- Method comparison: Among PEFT methods, LoRA was generally most compute-efficient for large pLMs, with DoRA sometimes up to 30% slower, and all PEFT methods yielded average prediction gains of 61.3% compared to using static embeddings.
PEFT—especially LoRA—provides a practical solution for adapting large pLMs in resource-constrained environments, with all tested PEFT methods demonstrating substantial improvements over static, pre-trained embeddings.
Quantization with LoRA (QLoRA) [31] combines 4-bit quantization with LoRA to enable highly efficient fine-tuning of pLMs while maintaining strong performance [138]. By quantizing most model weights to 4 bits and updating only a small set of adapter parameters, QLoRA substantially reduces memory and computation costs.
Clancy et al. [31] systematically evaluated QLoRA across multiple pLMs, including ESM-2 [7], ESM C [139], ProtBERT [107], ProtT5-half [6], and Ankh-base [140], and diverse tasks, including Green Fluorescent Protein (GFP) fluorescence [141], protease stability [142], and protein secondary structure prediction [16,143,144]. Experiments show that QLoRA achieves an average GPU memory reduction of 46.7%, with generative models such as ProtGPT2 and ProLLaMA reaching up to 76.4% reduction.
Performance remains largely preserved: for ESM C 600M, fluorescence prediction SpearmanR is 0.850 for QLoRA versus 0.863 for full precision, and secondary structure accuracy remains identical at 0.870. In generative tasks, 4-bit quantization yields negligible differences in predicted protein quality, with metrics such as Foldseek pass rate, local distance difference test, and predicted local distance difference test showing no significant decline.
Overall, QLoRA lowers hardware barriers for fine-tuning and inference of large pLMs, making efficient protein modeling feasible on modest computing infrastructure.
Few-Shot Learning for Protein Fitness Prediction (FSFP) [22] builds on the dataset construction and meta-learning strategies described in Section 2.2, and focuses on PEFT for robust fitness prediction with minimal labeled data.
After meta-training on auxiliary tasks constructed from related proteins and MSA-based pseudo-labels, FSFP fine-tunes only a small subset of parameters (e.g., via LoRA with rank ) to mitigate overfitting. Fitness prediction is formalized as a ranking problem, with ListMLE loss [145] used to optimize the ordering of variants. On 87 deep mutational scanning datasets, FSFP outperforms both zero-shot and regression-based baselines, improves spearman-correlation by up to 0.1 with just 20 labeled mutants, and demonstrates strong extrapolation to unseen mutations. Wet-lab validation further highlights its efficiency and real-world utility.
SES-Adapter [32] is a structure-aware, PEFT framework for pLMs, integrating sequence and structural information via cross-modal multi-head attention. In SES-Adapter, the pLM backbone is frozen, and only the adapter module is trained. Structural features representations are serialized via, such as, FoldSeek [125] or DSSP [146], into embeddings and serve as queries (Q), while pLM sequence embeddings serve as keys and values (K, V). Uniquely, both Q and K are first embedded with rotary position embedding (RoPE). For an embedding vector at position p, RoPE performs a rotation on each coordinate pair :
where is a position-dependent rotation angle varying with p and the dimension index i.
This fusion enables SES-Adapter to consistently outperform vanilla pLMs and prior tuning strategies: on nine diverse benchmarks, it yields up to 11% (avg. 3%) accuracy improvement, convergence acceleration up to 1034% (avg. 362%), and maintains robustness to structural noise (performance variation <0.6% across different structure predictors). SES-Adapter thus offers an efficient, scalable, and model-agnostic solution for fine-tuning pLMs in protein research.

4.4. Efficient Training via Distillation
knowledge distillation [39] in pLMs is a widely adopted model compression technique that transfers knowledge from larger, high-capacity teacher models to smaller, efficient student models. It typically involves guiding student models to mimic teacher outputs, intermediate representations, or other informative signals[147]. Distillation inherently emphasizes efficiency, enabling deployment of lightweight models that substantially reduce computational and memory requirements [148,149] while retaining predictive capabilities comparable to larger models [150,151]. Recent literature highlights diverse distillation strategies—including single-teacher [33], expert-guided [35], cross-modal [34], and multi-teacher adaptive methods [36]—consistently demonstrating significant efficiency improvements.
Cross-distillation of AlphaFold 3 [33] is a strategy for mitigating hallucinations and improving data efficiency in diffusion-based protein structure prediction [152]. Its core methodological steps are:
- Joint supervision with ground-truth and teacher outputs: AlphaFold 3 is explicitly optimized to align its outputs not only with experimental ground-truth structures, but also with reliable teacher-generated structures. This enables the model to learn more physically realistic disorder regions, as teacher-generated structures tend to represent unstructured segments as extended loops, reducing hallucinated compact order in intrinsically disordered regions.
On the CAID 2 benchmark [16], cross-distillation led to better alignment between predicted confidence (predicted local distance difference test) and actual disorder, effectively suppressing generative artifacts. By increasing both the diversity and quantity of usable training data, intra-chain metrics reach 97% of their maximum within the first 20,000 training steps, reflecting rapid and efficient learning. Overall, cross-distillation exemplifies how leveraging both ground-truth and high-quality model-generated supervision can simultaneously reduce hallucinations and accelerate robust protein structure learning in next-generation diffusion-based models.
ProtGO [34] is a multimodal protein representation learning framework that distills functional knowledge from a teacher to a student network via distribution alignment in latent space. The teacher leverages sequence, structure, and Gene Ontology (GO) (GO) [157,158] function annotations, while the student uses only sequence and structure, making it practical for annotation-scarce scenarios. Instead of aligning individual embeddings, ProtGO minimizes the KL-divergence [159] between the batch-level Gaussian-distributed graph embeddings of teacher and student:
where and are graph-level embeddings of the teacher and student models, and , denote their estimated batch-wise distributions (Gaussian with mean and variance computed over each batch). The overall loss for training the student is:
where is the cross entropy loss for downstream classification and is a trade-off hyperparameter.
This strategy enables the student to inherit rich functional information, achieving state-of-the-art results: e.g., 60.5% accuracy for fold classification (3.8 points higher than prior methods), 89.4% for enzyme reaction prediction, and top values on GO term and A numerical classification code for enzymes, based on the chemical reactions they catalyze. (Enzyme Commission (EC) number) prediction [160]. Ablation studies confirm the distillation module is critical for performance (fold accuracy drops from 60.5% to 57.8% when removed). ProtGO thus demonstrates the efficiency and effectiveness of distribution-based multimodal distillation for robust protein representation learning.
Expert-guided distillation in VespaG [35] offers an efficient strategy for protein The task of predicting the functional impact (e.g., on fitness, stability) of a mutation or a set of mutations on a protein. (variant effect prediction) by training a lightweight neural network (student) to imitate an evolutionary expert model (teacher). Here, the teacher is GEMME [55]—a MSA (MSA)-based method that generates mutational effect scores—while the student is VespaG, a feed-forward neural network with a single hidden layer. VespaG takes embeddings from a pretrained pLM (ESM-2 [7]) as input and learns to predict GEMME’s variant effect scores for each possible mutation. The student is trained by minimizing the mean squared error between its predictions and the GEMME scores :
This distillation approach removes the need for log-odds calculations and MSAs at inference time, enabling VespaG to rapidly predict mutational landscapes across entire proteomes on standard hardware. On the ProteinGym [80] benchmark (over 3 million variants), VespaG achieves a mean spearman-correlation of , matching state-of-the-art methods and sometimes surpassing its expert teacher in certain protein families. Importantly, VespaG is over three orders of magnitude faster than MSA-based methods. This demonstrates that expert-guided distillation with pLM embeddings yields robust, scalable, and practical variant effect predictors.
Multi-teacher distillation in MTDP [36] enables efficient and high-fidelity embedding by aggregating knowledge from multiple large pre-trained models, specifically ESM2-33 and ProtT5-XL-UniRef50.MTDP student, a T5-based model with ∼20M parameters, is trained via an adaptive teacher selection mechanism that dynamically assigns the most suitable guidance to each sample using RL strategy [161]. The training objective jointly minimizes a masked language modeling loss () and a distillation loss () defined by the KL divergence between student and teacher output distributions:
where,
where N is sequence length, V is vocabulary size, / are the true/predicted amino acids at position i, is the indicator function, / are the teacher/student output probabilities, and denotes KL divergence. At each step, selected teacher T for each sample is chosen via a RL-based scheduling mechanism [161].
Compared to its teachers (650M and 120M parameters), the MTDP student is dramatically more efficient, achieving ±1.5% accuracy of large models on tasks like function and The specific location or compartment within a cell where a protein resides and performs its function (e.g., nucleus, mitochondria, cytoplasm). (subcellular localization) prediction, and requiring ∼70% less encoding time for 10,000 protein sequences. Ablation studies confirm multi-teacher distillation outperforms single-teacher setups. MTDP can be deployed on modest GPU hardware, providing a scalable, resource-efficient solution for large-scale, real-time protein informatics applications.

5. Efficient Inference
5.1. Background and Challenges
Inference in language models refers to the process of generating outputs, such as predictions or embeddings, based on trained models given new input data. Autoregressive models (e.g. GPT series [44]) generate text by predicting subsequent tokens, while masked language models (e.g. BERT [2]) infer masked tokens or produce contextual embeddings for various tasks. This inference typically involves passing inputs through multiple transformer layers to capture contextual relationships and generate meaningful, task-relevant representations.
Analogously, pLMs infer structural, functional, or evolutionary properties from protein sequences. Given a protein sequence, pLM inference generates embeddings that encode rich biological information, facilitating downstream tasks like The computational task of identifying proteins that share a common evolutionary ancestor (homologs), which is often challenging when sequence similarity is low (remote homologs). (homolog detection), structure prediction, and functional annotation [5].
Efficiency-focused inference methods in LLMs encompass techniques such as quantization [162,163] and embedding-based dense retrieval, aiming to reduce computational costs and memory usage while preserving model performance [164,165]. Similarly, efficient inference in pLMs integrates analogous strategies, including PTQ [38], embedding-based retrieval via dual-encoder architectures [20], and structure-informed similarity scoring [37]. An overview of these efficient inference strategies is provided in Figure 8.
The emergence of efficiency-focused inference methods in pLMs is largely motivated by several challenges related to inference process, including:
- Resource Constraints: The inference process of large pLMs require substantial computation and memory, hindering deployment in resource-limited settings [38].
- Inference Latency: Traditional alignment-based and MSA methods are inherently slow and cannot efficiently scale to large databases, which is critical for many practical applications, such as proteome-wide A bioinformatics method used to identify sequences with a shared evolutionary ancestry, typically by comparing a query sequence against a database. (homology search) [20,37].
The following section reviews methods that address these issues, with an overview provided in Table 4.
5.2. Efficient Inference
PTQ4Protein [38] enables efficient inference of large pLMs such as ESMFold [7] by applying PTQ [79] to both weights and activations. Baseline experiments show that standard 8-bit uniform quantization [162] substantially reduces memory usage () but leads to significant accuracy loss—mainly due to the asymmetric and wide activation distributions before LayerNorm layers. To address this, PTQ4Protein employs piecewise linear quantization for activations, splitting the value range at breakpoints and , and quantizing each region separately:
where denotes the full activation range, and are the central breakpoints, b is the total bit width, and denotes uniform quantization within each segment. Each represents the activation value in the central and two tail regions, respectively.
Experiments demonstrate that PTQ4Protein achieves nearly lossless accuracy: for 8-bit quantization, TM-score [166] drops by only 0.36% (CASP14) [16] and 0.08% (CAMEO) [167], with memory usage reduced to 25% of baseline. Even at 6 bits, accuracy loss remains below 0.5%. Ablation studies show ESM-2 [7] is particularly robust, and the visual fidelity of predicted structures is preserved. Overall, PTQ4Protein provides a practical solution for deploying large pLMs on resource-limited hardware with minimal accuracy degradation.
PLMSearch [37] is an efficient inference framework for large-scale remote homology search that leverages pLM embeddings and structure-informed similarity prediction. Its pipeline consists of:
- PfamClan-based pre-filtering: PfamClan [168] assigns proteins to evolutionarily related superfamilies (clans); this stage retains only protein pairs sharing the same clan, ensuring high recall while greatly reducing the candidate search space.
- SS-predictor: A bilinear neural network is trained to predict structural similarity (TM-score [169]) between protein pairs based on pLM embeddings; it integrates predicted TM-scores with cosine similarity, capturing global and local sequence features for remote homolog detection.
- PLMAlign: For top-scoring pairs, per-residue alignment is performed using pLM embeddings to refine hit precision and enhance biological relevance.
A key efficiency innovation is the precomputation and caching of all target protein embeddings, enabling scalable, million-level searches via a single forward pass through the SS-predictor. By restricting downstream alignment to high-confidence pre-filtered pairs, PLMSearch dramatically reduces computation while maintaining high sensitivity. On standard benchmarks, PLMSearch achieves orders-of-magnitude faster search and higher sensitivity than conventional structure or sequence-based methods. These results establish PLMSearch as a scalable, structure-aware solution for accurate and ultra-fast remote protein homology inference.
Dense Homolog Retriever (DHR) [20] is an structure-aware inference framework for large-scale protein homolog detection, integrating pLMs with deep dense retrieval. Its workflow consists of:
- Dual-encoder architecture: Both query and candidate sequences are embedded into dense vectors using a pair of ESM-initialized encoders, allowing alignment-free similarity calculation.
- Contrastive learning: The dual-encoders are trained to embed homologous pairs close together and nonhomologous pairs far apart, enabling effective discrimination via dot-product scoring.
As an MSA prefilter, DHR accelerates MSA (with JackHMMER [172]) by up to 93-fold, increases MSA diversity (log Meff [173]), and improves AlphaFold2 [174] structure prediction—boosting TM-score [169] by up to 0.03 and lowering root-mean-squared distance [85] by 0.15 Å on hard targets. DHR also maintains robust performance on massive datasets such as BFD [175] or [47] MGnify (over 500 million sequences) [57], demonstrating its practicality for structure-aware, scalable, and accurate protein inference at unprecedented speed.

6. Future Directions
Quantum Algorithms for Protein Structure Prediction
A central challenge in classical, physics-driven approaches to protein folding (like A computational simulation method that models the physical (molecular dynamics (MD))) is their immense computational complexity. These methods simulate the intricate A multidimensional surface representing the potential energy (energy landscape) of a protein, a task that scales poorly on classical hardware. This has motivated the exploration of quantum algorithms, which are naturally suited for simulating complex physical systems. An overview of quantum–classical learning pipeline is provided in Figure 9.
The core physical principle of protein folding is that a protein chain will configure itself into its most stable, lowest-energy state. In physics, the total energy of a system is described by a mathematical operator known as a Hamiltonian. Therefore, a protein’s most stable 3D structure can be reframed as finding the lowest possible energy value of this The energy operator of a system, representing the total energy (Hamiltonian). This lowest-energy solution is known as the ground state.
Quantum Protein Structure Prediction (QPSP) attempts to solve this problem by first simplifying it [176,177,178,179]. The vast, continuous space of all possible protein folds is too complex to model directly. Instead, the protein is “discretized” by placing its residues onto the points of a 3D grid, or lattice. Common choices include cubic lattice [180,181,182] and tetrahedral lattice, which build upon earlier 2D models [183,184,185].
With the problem now on a lattice, the Hamiltonian is assembled from terms that encode the The potential energy associated with a given molecular or (structural energy): terms that capture the interactions between nearby An individual amino acid unit within a protein chain; in lattice (residue) and terms that act as constraints to enforce a structurally valid chain. Finding the ground state of this lattice-based Hamiltonian becomes an optimization task for a quantum computer. Research in this emerging field is focused on improving the realism of these models—progressing from 2D to 3D lattices—and developing more efficient encoding (e.g., turn-based rather than coordinate-based) to reduce the number of qubits required.
A fundamental limitation remains: native protein structures are not confined to a discrete lattice. This mismatch limits the fidelity of such models. Consequently, the accuracy of any lattice-based prediction is ultimately constrained by the resolution of the lattice, thereby necessitating models with higher degrees of freedom to yield more realistic fits [186].
Hybrid quantum–classical Transformers as a path toward quantum PLMs.
A more immediate and pragmatic future direction lies in creating hybrid quantum-classical models. Instead of attempting to replace the entire classical pipeline, this approach seeks to identify computationally expensive sub-components of existing models (like PLMs) and "enhance" them with a quantum subroutine.
The self-attention mechanism is a prime target. At its core, the attention score is a similarity measure—typically a simple dot product—between a Query (Q) vector and a Key (K) vector. This score determines how much influence one token has on another. Recent work on hybrid transformers [187] proposes the replacement of this classical dot product.
In this hybrid model, the Q and K vectors are used to prepare quantum states. A quantum circuit then measures the fidelity (or overlap) between these two states, and this measurement becomes the new attention score. This is not just a faster way to compute a dot product; it is a fundamentally different, quantum-native similarity metric.
This pathway is promising for several reasons. As a new similarity metric, this quantum-derived similarity is a specialized nonlinear function that may be able to capture complex relationships between token embeddings that a simple dot product misses, potentially improving model generalization in the low-data regimes common to protein engineering. It also offers theoretical efficiency: while the primary benefit may be representative power, this quantum subroutine also offers a theoretical cost reduction for the scoring portion, from to , where n is the sequence length and d the embedding dimension. Furthermore, it has An acronym for Noisy Intermediate-Scale Quantum, referring (NISQ)-era (Noisy Intermediate-Scale Quantum) feasibility, as the model remains "mostly classical," retaining standard tokenization, embedding, and feed-forward blocks. Crucially, the quantum subroutine only requires a The fundamental unit of quantum information, represented by (qubit) count that scales as , making its parameter-efficiency a viable candidate for NISQ hardware. Finally, it represents a practical prototyping path that can be prototyped today. Using frameworks like CUDA-Q [188,189], the quantum circuit can be simulated on classical CPU/GPU hardware and integrated directly into a standard model’s training loop, which allows for rapid, end-to-end validation of the concept.
Positive results from such simulations would position hybrid attention as a credible and practical first step toward future, quantum-enhanced PLMs for protein modeling.
7. Conclusions
In this survey, we systematically reviewed advancements in efficient pLMs across four dimensions: dataset optimization, model architecture, training strategies, and inference. We highlighted a spectrum of techniques—from meta-learning and scaling law-guided design to PEFT, knowledge distillation, and quantization—that collectively address the formidable computational demands of modern pLMs. These approaches enable substantial cost reductions, democratize accessibility, and lay the groundwork for scalable, practical deployment in computational biology.
Despite this substantial progress, significant challenges remain. While efficient pLMs achieve competitive results on standard benchmarks, a performance gap often emerges in complex tasks such as de novo structure prediction or out-of-distribution generalization. The optimal strategies for downstream adaptation, such as fine-tuning protocols and representation pooling in extreme resource-limited settings, also remain underexplored. Future research must not only extend these efficiency paradigms to more diverse biological tasks but also systematically benchmark adaptation strategies and develop even more scalable solutions through advances in optimization, quantization, and architectural innovation.
We anticipate that continued innovation in these areas, coupled with broader community engagement, will be essential for unlocking the full potential of efficient pLMs and accelerating biological discovery.
References
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser; Polosukhin, I. Attention is all you need. Advances in neural information processing systems 2017, 30. [Google Scholar]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. Proceedings of the Proceedings of the 2019 conference of the North American chapter of the association for computational linguistics: human language technologies 2019, volume 1 (long and short papers), 4171–4186. [Google Scholar]
- Bepler, T.; Berger, B. Learning protein sequence embeddings using information from structure. arXiv 2019, arXiv:1902.08661. [Google Scholar] [CrossRef]
- Asgari, E.; Mofrad, M.R. Continuous distributed representation of biological sequences for deep proteomics and genomics. PloS one 2015, 10, e0141287. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE transactions on pattern analysis and machine intelligence 2021, 44, 7112–7127. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Peng, C.X.; Liang, F.; Xia, Y.H.; Zhao, K.L.; Hou, M.H.; Zhang, G.J. Recent advances and challenges in protein structure prediction. Journal of Chemical Information and Modeling 2023, 64, 76–95. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhao, W.; Zhang, J.; Jin, Y.; Zhang, H.; Ren, Z.; Sun, R.; Wang, H.; Wan, G.; Lu, P.; et al. Protein large language models: A comprehensive survey. arXiv arXiv:2502.17504. [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic acids research 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: the universal protein knowledgebase in 2021. Nucleic acids research 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; Consortium, U. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Ofer, D.; Brandes, N.; Linial, M. The language of proteins: NLP, machine learning & protein sequences. Computational and Structural Biotechnology Journal 2021, 19, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nature communications 2022, 13, 4348. [Google Scholar] [CrossRef]
- Haas, J.; Roth, S.; Arnold, K.; Kiefer, F.; Schmidt, T.; Bordoli, L.; Schwede, T. The Protein Model Portal—a comprehensive resource for protein structure and model information. Database 2013, 2013, bat031. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)—Round XIV. Proteins: Structure, Function, and Bioinformatics 2021, 89, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Strubell, E.; Ganesh, A.; McCallum, A. Energy and Policy Considerations for Deep Learning in NLP. arXiv 2019, arXiv:1906.02243. [Google Scholar] [CrossRef]
- Strubell, E.; Ganesh, A.; McCallum, A. Energy and policy considerations for modern deep learning research. Proceedings of the Proceedings of the AAAI conference on artificial intelligence 2020, Vol. 34, 13693–13696. [Google Scholar] [CrossRef]
- Kaplan, J.; McCandlish, S.; Henighan, T.; Brown, T.B.; Chess, B.; Child, R.; Gray, S.; Radford, A.; Wu, J.; Amodei, D. Scaling laws for neural language models. arXiv 2020, arXiv:2001.08361. [Google Scholar] [CrossRef]
- Hong, L.; Hu, Z.; Sun, S.; Tang, X.; Wang, J.; Tan, Q.; Zheng, L.; Wang, S.; Xu, S.; King, I.; et al. Fast, sensitive detection of protein homologs using deep dense retrieval. Nature Biotechnology 2024, 1–13. [Google Scholar] [CrossRef]
- Cheng, X.; Chen, B.; Li, P.; Gong, J.; Tang, J.; Song, L. Training compute-optimal protein language models. Advances in Neural Information Processing Systems 2024, 37, 69386–69418. [Google Scholar]
- Zhou, Z.; Zhang, L.; Yu, Y.; Wu, B.; Li, M.; Hong, L.; Tan, P. Enhancing efficiency of protein language models with minimal wet-lab data through few-shot learning. Nature Communications 2024, 15, 5566. [Google Scholar] [CrossRef]
- Frey, N.C.; Joren, T.; Ismail, A.A.; Goodman, A.; Bonneau, R.; Cho, K.; Gligorijević, V. Cramming Protein Language Model Training in 24 GPU Hours. bioRxiv 2024, 2024–05. [Google Scholar] [CrossRef]
- Turnbull, O.M.; Baioumy, M.; Deane, C. 2Bits of Protein: Efficient Protein Language Models at the Scale of 2-bits. In Proceedings of the ICML 2024 Workshop on Efficient and Accessible Foundation Models for Biological Discovery, 2024. [Google Scholar]
- Sgarbossa, D.; Bitbol, A.F. RAG-ESM: Improving pretrained protein language models via sequence retrieval. bioRxiv 2025, 2025–04. [Google Scholar] [CrossRef]
- Lu, A.X.; Yan, W.; Yang, K.K.; Gligorijevic, V.; Cho, K.; Abbeel, P.; Bonneau, R.; Frey, N.C. Tokenized and continuous embedding compressions of protein sequence and structure. Patterns 2025, 6. [Google Scholar] [CrossRef]
- Yang, K.K.; Fusi, N.; Lu, A.X. Convolutions are competitive with transformers for protein sequence pretraining. Cell Systems 2024, 15, 286–294. [Google Scholar] [CrossRef]
- Zhang, Y.; Okumura, M. Prothyena: A fast and efficient foundation protein language model at single amino acid resolution. bioRxiv 2024, 2024–01. [Google Scholar] [CrossRef]
- Heinzinger, M.; Weissenow, K.; Sanchez, J.G.; Henkel, A.; Mirdita, M.; Steinegger, M.; Rost, B. Bilingual language model for protein sequence and structure. NAR Genomics and Bioinformatics 2024, 6, lqae150. [Google Scholar] [CrossRef] [PubMed]
- Schmirler, R.; Heinzinger, M.; Rost, B. Fine-tuning protein language models boosts predictions across diverse tasks. Nature Communications 2024, 15, 7407. [Google Scholar] [CrossRef]
- Clancy, S.; Zeisler, I.Y.; Bayat, P.; Xie, M.; White, V.; Perkins, S.; Bayat, S.; Pardee, K. Assessing Quantization and Efficient Fine-Tuning for Protein Language Models. In Proceedings of the ICLR 2025 Workshop on Generative and Experimental Perspectives for Biomolecular Design, 2025. [Google Scholar]
- Tan, Y.; Li, M.; Zhou, B.; Zhong, B.; Zheng, L.; Tan, P.; Zhou, Z.; Yu, H.; Fan, G.; Hong, L. Simple, efficient, and scalable structure-aware adapter boosts protein language models. Journal of Chemical Information and Modeling 2024, 64, 6338–6349. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Tan, C.; Xu, Y.; Gao, Z.; Xia, J.; Wu, L.; Li, S.Z. Protgo: Function-guided protein modeling for unified representation learning. Advances in Neural Information Processing Systems 2024, 37, 88581–88604. [Google Scholar]
- Marquet, C.; Schlensok, J.; Abakarova, M.; Rost, B.; Laine, E. Expert-guided protein language models enable accurate and blazingly fast fitness prediction. Bioinformatics 2024, 40, btae621. [Google Scholar] [CrossRef]
- Shang, J.; Peng, C.; Ji, Y.; Guan, J.; Cai, D.; Tang, X.; Sun, Y. Accurate and efficient protein embedding using multi-teacher distillation learning. Bioinformatics 2024, 40, btae567. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Z.; You, R.; Xie, C.; Wei, H.; Xiong, Y.; Yang, J.; Zhu, S. PLMSearch: Protein language model powers accurate and fast sequence search for remote homology. Nature communications 2024, 15, 2775. [Google Scholar] [CrossRef]
- Peng, S.; Yang, F.; Sun, N.; Chen, S.; Jiang, Y.; Pan, A. Exploring Post-Training Quantization of Protein Language Models. In Proceedings of the 2023 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 2023; IEEE; pp. 602–608. [Google Scholar]
- Hinton, G.; Vinyals, O.; Dean, J. Distilling the knowledge in a neural network. arXiv 2015, arXiv:1503.02531. [Google Scholar] [CrossRef]
- Wu, L.; Huang, Y.; Lin, H.; Li, S.Z. A survey on protein representation learning: Retrospect and prospect. arXiv 2022, arXiv:2301.00813. [Google Scholar] [CrossRef]
- Hu, E.J.; Shen, Y.; Wallis, P.; Allen-Zhu, Z.; Li, Y.; Wang, S.; Wang, L.; Chen, W.; et al. Lora: Low-rank adaptation of large language models. ICLR 2022, 1, 3. [Google Scholar]
- Kaplan, J.; McCandlish, S.; Henighan, T.; Brown, T.B.; Chess, B.; Child, R.; Gray, S.; Radford, A.; Wu, J.; Amodei, D. Scaling laws for neural language models. arXiv 2020, arXiv:2001.08361. [Google Scholar] [CrossRef]
- Radford, A.; Narasimhan, K.; Salimans, T.; Sutskever, I.; et al. Improving language understanding by generative pre-training. Advances in neural information processing systems; 2018. [Google Scholar]
- Brown, T.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.D.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language models are few-shot learners. Advances in neural information processing systems 2020, 33, 1877–1901. [Google Scholar]
- Sennrich, R.; Haddow, B.; Birch, A. Neural machine translation of rare words with subword units. arXiv 2015, arXiv:1508.07909. [Google Scholar]
- Kudo, T.; Richardson, J. SentencePiece: A simple and language independent subword tokenizer and detokenizer for neural text processing. arXiv 2018, arXiv:1808.06226. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Almeida, A.; Beracochea, M.; Boland, M.; Burgin, J.; Cochrane, G.; Crusoe, M.R.; Kale, V.; Potter, S.C.; Richardson, L.J.; et al. MGnify: the microbiome analysis resource in 2020. Nucleic acids research 2020, 48, D570–D578. [Google Scholar] [CrossRef]
- Hoffmann, J.; Borgeaud, S.; Mensch, A.; Buchatskaya, E.; Cai, T.; Rutherford, E.; Casas, D.d.L.; Hendricks, L.A.; Welbl, J.; Clark, A.; et al. Training compute-optimal large language models. arXiv 2022, arXiv:2203.15556. [Google Scholar] [CrossRef]
- Raffel, C.; Shazeer, N.; Roberts, A.; Lee, K.; Narang, S.; Matena, M.; Zhou, Y.; Li, W.; Liu, P.J. Exploring the limits of transfer learning with a unified text-to-text transformer. Journal of machine learning research 2020, 21, 1–67. [Google Scholar]
- Romero, P.A.; Krause, A.; Arnold, F.H. Navigating the protein fitness landscape with Gaussian processes. Proceedings of the National Academy of Sciences 2013, 110, E193–E201. [Google Scholar] [CrossRef]
- Cao, Z.; Qin, T.; Liu, T.Y.; Tsai, M.F.; Li, H. Learning to rank: from pairwise approach to listwise approach. In Proceedings of the Proceedings of the 24th international conference on Machine learning, 2007; pp. 129–136. [Google Scholar]
- Buller, A.R.; Van Roye, P.; Cahn, J.K.; Scheele, R.A.; Herger, M.; Arnold, F.H. Directed evolution mimics allosteric activation by stepwise tuning of the conformational ensemble. Journal of the American Chemical Society 2018, 140, 7256–7266. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Fields, S. Deep mutational scanning: a new style of protein science. Nature methods 2014, 11, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Melamed, D.; Young, D.L.; Gamble, C.E.; Miller, C.R.; Fields, S. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly (A)-binding protein. Rna 2013, 19, 1537–1551. [Google Scholar] [CrossRef] [PubMed]
- Laine, E.; Karami, Y.; Carbone, A. GEMME: a simple and fast global epistatic model predicting mutational effects. Molecular biology and evolution 2019, 36, 2604–2619. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic acids research 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: making protein folding accessible to all. Nature methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Finn, C.; Abbeel, P.; Levine, S. Model-agnostic meta-learning for fast adaptation of deep networks. In Proceedings of the International conference on machine learning. PMLR, 2017; pp. 1126–1135. [Google Scholar]
- Snell, J.; Swersky, K.; Zemel, R. Prototypical networks for few-shot learning. Advances in neural information processing systems 2017, 30. [Google Scholar]
- Notin, P.; Kollasch, A.; Ritter, D.; Van Niekerk, L.; Paul, S.; Spinner, H.; Rollins, N.; Shaw, A.; Orenbuch, R.; Weitzman, R.; et al. Proteingym: Large-scale benchmarks for protein fitness prediction and design. Advances in Neural Information Processing Systems 2023, 36, 64331–64379. [Google Scholar]
- Meier, J.; Rao, R.; Verkuil, R.; Liu, J.; Sercu, T.; Rives, A. Language models enable zero-shot prediction of the effects of mutations on protein function. Advances in neural information processing systems 2021, 34, 29287–29303. [Google Scholar]
- Su, J.; Han, C.; Zhou, Y.; Shan, J.; Zhou, X.; Yuan, F. Saprot: Protein language modeling with structure-aware vocabulary. BioRxiv 2023, 2023–10. [Google Scholar]
- Hsu, C.; Verkuil, R.; Liu, J.; Lin, Z.; Hie, B.; Sercu, T.; Lerer, A.; Rives, A. Learning inverse folding from millions of predicted structures. In Proceedings of the International conference on machine learning. PMLR, 2022; pp. 8946–8970. [Google Scholar]
- Radford, A.; Wu, J.; Child, R.; Luan, D.; Amodei, D.; Sutskever, I.; et al. Language models are unsupervised multitask learners. OpenAI blog 2019, 1, 9. [Google Scholar]
- Hestness, J.; Narang, S.; Ardalani, N.; Diamos, G.; Jun, H.; Kianinejad, H.; Patwary, M.M.A.; Yang, Y.; Zhou, Y. Deep learning scaling is predictable, empirically. arXiv 2017, arXiv:1712.00409. [Google Scholar] [CrossRef]
- Roelofs, R.; Shankar, V.; Recht, B.; Fridovich-Keil, S.; Hardt, M.; Miller, J.; Schmidt, L. A meta-analysis of overfitting in machine learning. Advances in neural information processing systems 2019, 32. [Google Scholar]
- Nayfach, S.; Roux, S.; Seshadri, R.; Udwary, D.; Varghese, N.; Schulz, F.; Wu, D.; Paez-Espino, D.; Chen, I.M.; Huntemann, M.; et al. A genomic catalog of Earth’s microbiomes. Nature biotechnology 2021, 39, 499–509. [Google Scholar] [CrossRef]
- Kalchbrenner, N.; Espeholt, L.; Simonyan, K.; Oord, A.; Graves, A.; Kavukcuoglu, K. Neural machine translation in linear time. arXiv arXiv:1610.10099. [CrossRef]
- Gehring, J.; Auli, M.; Grangier, D.; Yarats, D.; Dauphin, Y.N. Convolutional sequence to sequence learning. In Proceedings of the International conference on machine learning. PMLR, 2017; pp. 1243–1252. [Google Scholar]
- Tay, Y.; Dehghani, M.; Bahri, D.; Metzler, D. Efficient Transformers: A Survey. arXiv 2022, arXiv:cs. [Google Scholar] [CrossRef]
- Frantar, E.; Ashkboos, S.; Hoefler, T.; Alistarh, D. Gptq: Accurate post-training quantization for generative pre-trained transformers. arXiv 2022, arXiv:2210.17323. [Google Scholar]
- Frankle, J.; Carbin, M. The lottery ticket hypothesis: Finding sparse, trainable neural networks. arXiv 2018, arXiv:1803.03635. [Google Scholar]
- Hopf, T.A.; Ingraham, J.B.; Poelwijk, F.J.; Schärfe, C.P.; Springer, M.; Sander, C.; Marks, D.S. Mutation effects predicted from sequence co-variation. Nature biotechnology 2017, 35, 128–135. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Courbariaux, M.; Hubara, I.; Soudry, D.; El-Yaniv, R.; Bengio, Y. Binarized neural networks: Training deep neural networks with weights and activations constrained to+ 1 or-1. arXiv 2016, arXiv:1602.02830. [Google Scholar]
- Ma, S.; Wang, H.; Ma, L.; Wang, L.; Wang, W.; Huang, S.; Dong, L.; Wang, R.; Xue, J.; Wei, F. The era of 1-bit llms: All large language models are in 1.58 bits. arXiv 2024, arXiv:2402.177641. [Google Scholar] [CrossRef]
- Li, F.; Liu, B.; Wang, X.; Zhang, B.; Yan, J. Ternary weight networks. arXiv arXiv:1605.04711.
- Zhu, C.; Han, S.; Mao, H.; Dally, W.J. Trained ternary quantization. arXiv 2016, arXiv:1612.01064. [Google Scholar]
- Banner, R.; Nahshan, Y.; Soudry, D. Post training 4-bit quantization of convolutional networks for rapid-deployment. Advances in neural information processing systems 2019, 32. [Google Scholar]
- Notin, P.; Dias, M.; Frazer, J.; Marchena-Hurtado, J.; Gomez, A.N.; Marks, D.; Gal, Y. Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval. In Proceedings of the International Conference on Machine Learning. PMLR, 2022; pp. 16990–17017. [Google Scholar]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified rational protein engineering with sequence-based deep representation learning. Nature methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.C.; Joren, T.; Ismail, A.A.; Goodman, A.; Bonneau, R.; Cho, K.; Gligorijević, V. Cramming protein language model training in 24 gpu hours. bioRxiv 2024, 2024–05. [Google Scholar] [CrossRef]
- Newell, A.; Yang, K.; Deng, J. Stacked hourglass networks for human pose estimation. In Proceedings of the European conference on computer vision, 2016; Springer; pp. 483–499. [Google Scholar]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114. [Google Scholar]
- Kabsch, W. A solution for the best rotation to relate two sets of vectors. Foundations of Crystallography 1976, 32, 922–923. [Google Scholar] [CrossRef]
- Ba, J.L.; Kiros, J.R.; Hinton, G.E. Layer normalization. arXiv 2016, arXiv:1607.06450. [Google Scholar] [CrossRef]
- Ioffe, S.; Szegedy, C. Batch normalization: Accelerating deep network training by reducing internal covariate shift. In Proceedings of the International conference on machine learning. pmlr, 2015; pp. 448–456. [Google Scholar]
- Mentzer, F.; Minnen, D.; Agustsson, E.; Tschannen, M. Finite scalar quantization: Vq-vae made simple. arXiv 2023, arXiv:2309.15505. [Google Scholar] [CrossRef]
- Van Gerven, M.; Bohte, S. Artificial neural networks as models of neural information processing. 2017. [Google Scholar] [CrossRef]
- Van Den Oord, A.; Vinyals, O.; et al. Neural discrete representation learning. Advances in neural information processing systems 2017, 30. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. Journal of molecular biology 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nature methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.M.; Liu, J.; Verkuil, R.; Meier, J.; Canny, J.; Abbeel, P.; Sercu, T.; Rives, A. MSA transformer. In Proceedings of the International conference on machine learning. PMLR, 2021; pp. 8844–8856. [Google Scholar]
- Austin, J.; Johnson, D.D.; Ho, J.; Tarlow, D.; Van Den Berg, R. Structured denoising diffusion models in discrete state-spaces. Advances in neural information processing systems 2021, 34, 17981–17993. [Google Scholar]
- Hoogeboom, E.; Nielsen, D.; Jaini, P.; Forré, P.; Welling, M. Argmax flows and multinomial diffusion: Learning categorical distributions. Advances in neural information processing systems 2021, 34, 12454–12465. [Google Scholar]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature biotechnology 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Vig, J.; Belinkov, Y. Analyzing the structure of attention in a transformer language model. arXiv 2019, arXiv:1906.04284. [Google Scholar] [CrossRef]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. Journal of molecular biology 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Dauparas, J.; Anishchenko, I.; Bennett, N.; Bai, H.; Ragotte, R.J.; Milles, L.F.; Wicky, B.I.; Courbet, A.; de Haas, R.J.; Bethel, N.; et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science 2022, 378, 49–56. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic acids research 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Yu, F.; Koltun, V. Multi-scale context aggregation by dilated convolutions. arXiv 2015, arXiv:1511.07122. [Google Scholar]
- Chen, L.C.; Papandreou, G.; Kokkinos, I.; Murphy, K.; Yuille, A.L. Deeplab: Semantic image segmentation with deep convolutional nets, atrous convolution, and fully connected crfs. IEEE transactions on pattern analysis and machine intelligence 2017, 40, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.; Massaroli, S.; Nguyen, E.; Fu, D.Y.; Dao, T.; Baccus, S.; Bengio, Y.; Ermon, S.; Ré, C. Hyena hierarchy: Towards larger convolutional language models. In Proceedings of the International Conference on Machine Learning. PMLR, 2023; pp. 28043–28078. [Google Scholar]
- Gu, A.; Goel, K.; Ré, C. Efficiently modeling long sequences with structured state spaces. arXiv 2021, arXiv:2111.00396. [Google Scholar]
- Dauphin, Y.N.; Fan, A.; Auli, M.; Grangier, D. Language modeling with gated convolutional networks. In Proceedings of the International conference on machine learning. PMLR, 2017; pp. 933–941. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural computation 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef]
- Ouyang, L.; Wu, J.; Jiang, X.; Almeida, D.; Wainwright, C.; Mishkin, P.; Zhang, C.; Agarwal, S.; Slama, K.; Ray, A.; et al. Training language models to follow instructions with human feedback. Advances in neural information processing systems 2022, 35, 27730–27744. [Google Scholar]
- Shao, Z.; Wang, P.; Zhu, Q.; Xu, R.; Song, J.; Bi, X.; Zhang, H.; Zhang, M.; Li, Y.; Wu, Y.; et al. Deepseekmath: Pushing the limits of mathematical reasoning in open language models. arXiv 2024, arXiv:2402.03300. [Google Scholar]
- Howard, J.; Ruder, S. Universal language model fine-tuning for text classification. arXiv 2018, arXiv:1801.06146. [Google Scholar] [CrossRef]
- Gou, J.; Yu, B.; Maybank, S.J.; Tao, D. Knowledge distillation: A survey. International journal of computer vision 2021, 129, 1789–1819. [Google Scholar] [CrossRef]
- Sanh, V.; Debut, L.; Chaumond, J.; Wolf, T. DistilBERT, a distilled version of BERT: smaller, faster, cheaper and lighter. arXiv 2019, arXiv:1910.01108. [Google Scholar]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Briefings in bioinformatics 2017, 18, 851–869. [Google Scholar] [CrossRef]
- Gligorijević, V.; Renfrew, P.D.; Kosciolek, T.; Leman, J.K.; Berenberg, D.; Vatanen, T.; Chandler, C.; Taylor, B.C.; Fisk, I.M.; Vlamakis, H.; et al. Structure-based protein function prediction using graph convolutional networks. Nature communications 2021, 12, 3168. [Google Scholar] [CrossRef]
- Sun, Z.; Yu, H.; Song, X.; Liu, R.; Yang, Y.; Zhou, D. Mobilebert: a compact task-agnostic bert for resource-limited devices. arXiv arXiv:2004.02984.
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: a simple way to prevent neural networks from overfitting. The journal of machine learning research 2014, 15, 1929–1958. [Google Scholar]
- Ding, N.; Qin, Y.; Yang, G.; Wei, F.; Yang, Z.; Su, Y.; Hu, S.; Chen, Y.; Chan, C.M.; Chen, W.; et al. Delta tuning: A comprehensive study of parameter efficient methods for pre-trained language models. arXiv 2022, arXiv:2203.06904. [Google Scholar] [CrossRef]
- Houlsby, N.; Giurgiu, A.; Jastrzebski, S.; Morrone, B.; De Laroussilhe, Q.; Gesmundo, A.; Attariyan, M.; Gelly, S. Parameter-efficient transfer learning for NLP. In Proceedings of the International conference on machine learning. PMLR, 2019; pp. 2790–2799. [Google Scholar]
- Lester, B.; Al-Rfou, R.; Constant, N. The power of scale for parameter-efficient prompt tuning. arXiv 2021, arXiv:2104.08691. [Google Scholar] [CrossRef]
- Li, X.L.; Liang, P. Prefix-tuning: Optimizing continuous prompts for generation. arXiv 2021, arXiv:2101.00190. [Google Scholar] [CrossRef]
- Mirzadeh, S.I.; Farajtabar, M.; Li, A.; Levine, N.; Matsukawa, A.; Ghasemzadeh, H. Improved knowledge distillation via teacher assistant. Proceedings of the Proceedings of the AAAI conference on artificial intelligence 2020, Vol. 34, 5191–5198. [Google Scholar] [CrossRef]
- Hernandez, D.; Brown, T.; Conerly, T.; DasSarma, N.; Drain, D.; El-Showk, S.; Elhage, N.; Hatfield-Dodds, Z.; Henighan, T.; Hume, T.; et al. Scaling laws and interpretability of learning from repeated data. arXiv 2022, arXiv:2205.10487. [Google Scholar] [CrossRef]
- Jing, B.; Eismann, S.; Suriana, P.; Townshend, R.J.; Dror, R. Learning from protein structure with geometric vector perceptrons. arXiv arXiv:2009.01411. [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic acids research 2024, 52, D368–D375. [Google Scholar] [CrossRef]
- Van Kempen, M.; Kim, S.S.; Tumescheit, C.; Mirdita, M.; Lee, J.; Gilchrist, C.L.; Söding, J.; Steinegger, M. Fast and accurate protein structure search with Foldseek. Nature biotechnology 2024, 42, 243–246. [Google Scholar] [CrossRef]
- Rost, B. Twilight zone of protein sequence alignments. Protein engineering 1999, 12, 85–94. [Google Scholar] [CrossRef]
- Sillitoe, I.; Bordin, N.; Dawson, N.; Waman, V.P.; Ashford, P.; Scholes, H.M.; Pang, C.S.; Woodridge, L.; Rauer, C.; Sen, N.; et al. CATH: increased structural coverage of functional space. Nucleic acids research 2021, 49, D266–D273. [Google Scholar] [CrossRef]
- Loshchilov, I.; Hutter, F. Decoupled weight decay regularization. arXiv 2017, arXiv:1711.05101. [Google Scholar]
- Goyal, P.; Dollár, P.; Girshick, R.; Noordhuis, P.; Wesolowski, L.; Kyrola, A.; Tulloch, A.; Jia, Y.; He, K. Accurate, large minibatch sgd: Training imagenet in 1 hour. arXiv 2017, arXiv:1706.02677. [Google Scholar]
- Liu, L.; Liu, X.; Gao, J.; Chen, W.; Han, J. Understanding the difficulty of training transformers. arXiv arXiv:2004.08249. [CrossRef]
- Loshchilov, I.; Hutter, F. Sgdr: Stochastic gradient descent with warm restarts. arXiv 2016, arXiv:1608.03983. [Google Scholar]
- Bergstra, J.; Bengio, Y. Random search for hyper-parameter optimization. The journal of machine learning research 2012, 13, 281–305. [Google Scholar]
- Snoek, J.; Larochelle, H.; Adams, R.P. Practical bayesian optimization of machine learning algorithms. Advances in neural information processing systems 2012, 25. [Google Scholar]
- Hu, E.J.; Shen, Y.; Wallis, P.; Allen-Zhu, Z.; Li, Y.; Wang, S.; Wang, L.; Chen, W.; et al. Lora: Low-rank adaptation of large language models. ICLR 2022, 1, 3. [Google Scholar]
- Liu, S.Y.; Wang, C.Y.; Yin, H.; Molchanov, P.; Wang, Y.C.F.; Cheng, K.T.; Chen, M.H. Dora: Weight-decomposed low-rank adaptation. In Proceedings of the Forty-first International Conference on Machine Learning, 2024. [Google Scholar]
- Liu, H.; Tam, D.; Muqeeth, M.; Mohta, J.; Huang, T.; Bansal, M.; Raffel, C.A. Few-shot parameter-efficient fine-tuning is better and cheaper than in-context learning. Advances in Neural Information Processing Systems 2022, 35, 1950–1965. [Google Scholar]
- Li, X.L.; Liang, P. Prefix-tuning: Optimizing continuous prompts for generation. arXiv 2021, arXiv:2101.00190. [Google Scholar] [CrossRef]
- Dettmers, T.; Pagnoni, A.; Holtzman, A.; Zettlemoyer, L. Qlora: Efficient finetuning of quantized llms. Advances in neural information processing systems 2023, 36, 10088–10115. [Google Scholar]
- Team, E. “ESM Cambrian: Revealing the mysteries of proteins with unsupervised learning”. 2024. Available online: https://www.evolutionaryscale.ai/blog/esm-cambrian.
- Elnaggar, A.; Essam, H.; Salah-Eldin, W.; Moustafa, W.; Elkerdawy, M.; Rochereau, C.; Rost, B. Ankh: Optimized protein language model unlocks general-purpose modelling. arXiv 2023, arXiv:2301.06568. [Google Scholar]
- Sarkisyan, K.S.; Bolotin, D.A.; Meer, M.V.; Usmanova, D.R.; Mishin, A.S.; Sharonov, G.V.; Ivankov, D.N.; Bozhanova, N.G.; Baranov, M.S.; Soylemez, O.; et al. Local fitness landscape of the green fluorescent protein. Nature 2016, 533, 397–401. [Google Scholar] [CrossRef]
- Rocklin, G.J.; Chidyausiku, T.M.; Goreshnik, I.; Ford, A.; Houliston, S.; Lemak, A.; Carter, L.; Ravichandran, R.; Mulligan, V.K.; Chevalier, A.; et al. Global analysis of protein folding using massively parallel design, synthesis, and testing. Science 2017, 357, 168–175. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic acids research 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Soenderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins: Structure, Function, and Bioinformatics 2019, 87, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Liu, T.Y.; Wang, J.; Zhang, W.; Li, H. Listwise approach to learning to rank: theory and algorithm. In Proceedings of the Proceedings of the 25th international conference on Machine learning, 2008; pp. 1192–1199. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers: Original Research on Biomolecules 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Zagoruyko, S.; Komodakis, N. Paying more attention to attention: Improving the performance of convolutional neural networks via attention transfer. arXiv 2016, arXiv:1612.03928. [Google Scholar]
- Cheng, Y.; Wang, D.; Zhou, P.; Zhang, T. A survey of model compression and acceleration for deep neural networks. arXiv 2017, arXiv:1710.09282. [Google Scholar]
- Han, S.; Mao, H.; Dally, W.J. Deep compression: Compressing deep neural networks with pruning, trained quantization and huffman coding. arXiv 2015, arXiv:1510.00149. [Google Scholar]
- Wang, L.; Yoon, K.J. Knowledge distillation and student-teacher learning for visual intelligence: A review and new outlooks. IEEE transactions on pattern analysis and machine intelligence 2021, 44, 3048–3068. [Google Scholar] [CrossRef] [PubMed]
- Geffen, Y.; Ofran, Y.; Unger, R. DistilProtBert: a distilled protein language model used to distinguish between real proteins and their randomly shuffled counterparts. Bioinformatics 2022, 38, ii95–ii98. [Google Scholar] [CrossRef]
- Watson, J.L.; Juergens, D.; Bennett, N.R.; Trippe, B.L.; Yim, J.; Eisenach, H.E.; Ahern, W.; Borst, A.J.; Ragotte, R.J.; Milles, L.F.; et al. De novo design of protein structure and function with RFdiffusion. Nature 2023, 620, 1089–1100. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. biorxiv 2021, 2021–10. [Google Scholar]
- Židek, A. AlphaFold v.2.3.0 Technical Note. 2022. Available online: https://github.com/google-deepmind/alphafold/blob/main/docs/technical_note_v2.3.0.md.
- Lee, D.H.; et al. Pseudo-label: The simple and efficient semi-supervised learning method for deep neural networks. In Proceedings of the Workshop on challenges in representation learning, ICML. Atlanta, 2013; Vol. 3, p. 896. [Google Scholar]
- Xie, Q.; Luong, M.T.; Hovy, E.; Le, Q.V. Self-training with noisy student improves imagenet classification. In Proceedings of the Proceedings of the IEEE/CVF conference on computer vision and pattern recognition, 2020; pp. 10687–10698. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: tool for the unification of biology. Nature genetics 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The gene ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef] [PubMed]
- Kullback, S.; Leibler, R.A. On information and sufficiency. The annals of mathematical statistics 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Bairoch, A. The ENZYME database in 2000. Nucleic acids research 2000, 28, 304–305. [Google Scholar] [CrossRef]
- Yuan, F.; Shou, L.; Pei, J.; Lin, W.; Gong, M.; Fu, Y.; Jiang, D. Reinforced multi-teacher selection for knowledge distillation. Proceedings of the Proceedings of the AAAI conference on artificial intelligence 2021, Vol. 35, 14284–14291. [Google Scholar] [CrossRef]
- Jacob, B.; Kligys, S.; Chen, B.; Zhu, M.; Tang, M.; Howard, A.; Adam, H.; Kalenichenko, D. Quantization and training of neural networks for efficient integer-arithmetic-only inference. In Proceedings of the Proceedings of the IEEE conference on computer vision and pattern recognition, 2018; pp. 2704–2713. [Google Scholar]
- Nagel, M.; Fournarakis, M.; Amjad, R.A.; Bondarenko, Y.; Van Baalen, M.; Blankevoort, T. A white paper on neural network quantization. arXiv 2021, arXiv:2106.08295. [Google Scholar] [CrossRef]
- Bondarenko, Y.; Nagel, M.; Blankevoort, T. Understanding and overcoming the challenges of efficient transformer quantization. arXiv 2021, arXiv:2109.12948. [Google Scholar] [CrossRef]
- Lewis, P.; Perez, E.; Piktus, A.; Petroni, F.; Karpukhin, V.; Goyal, N.; Küttler, H.; Lewis, M.; Yih, W.t.; Rocktäschel, T.; et al. Retrieval-augmented generation for knowledge-intensive nlp tasks. Advances in neural information processing systems 2020, 33, 9459–9474. [Google Scholar]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins: Structure, Function, and Bioinformatics 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Haas, J.; Barbato, A.; Behringer, D.; Studer, G.; Roth, S.; Bertoni, M.; Mostaguir, K.; Gumienny, R.; Schwede, T. Continuous Automated Model EvaluatiOn (CAMEO) complementing the critical assessment of structure prediction in CASP12. Proteins: Structure, Function, and Bioinformatics 2018, 86, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic acids research 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic acids research 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Douze, M.; Jégou, H. Billion-scale similarity search with GPUs. IEEE Transactions on Big Data 2019, 7, 535–547. [Google Scholar] [CrossRef]
- Malkov, Y.A.; Yashunin, D.A. Efficient and robust approximate nearest neighbor search using hierarchical navigable small world graphs. IEEE transactions on pattern analysis and machine intelligence 2018, 42, 824–836. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS computational biology 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Morcos, F.; Pagnani, A.; Lunt, B.; Bertolino, A.; Marks, D.S.; Sander, C.; Zecchina, R.; Onuchic, J.N.; Hwa, T.; Weigt, M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proceedings of the National Academy of Sciences 2011, 108, E1293–E1301. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. Clustering huge protein sequence sets in linear time. Nature communications 2018, 9, 2542. [Google Scholar] [CrossRef]
- Robert, A.; Barkoutsos, P.K.; Woerner, S.; Tavernelli, I. Resource-efficient quantum algorithm for protein folding. npj Quantum Information 2021, 7. [Google Scholar] [CrossRef]
- Chandarana, P.; Hegade, N.N.; Montalban, I.; Solano, E.; Chen, X. Digitized Counterdiabatic Quantum Algorithm for Protein Folding. Physical Review Applied 2023, 20. [Google Scholar] [CrossRef]
- Doga, H.; Raubenolt, B.; Cumbo, F.; Joshi, J.; DiFilippo, F.P.; Qin, J.; Blankenberg, D.; Shehab, O. A Perspective on Protein Structure Prediction Using Quantum Computers. Journal of Chemical Theory and Computation 2024, 20, 3359–3378. [Google Scholar] [CrossRef]
- Li, R.H.; Doga, H.; Raubenolt, B.; Mostame, S.; DiSanto, N.; Cumbo, F.; Joshi, J.; Linn, H.; Gaffney, M.; Holden, A.; et al. Quantum Algorithm for Protein Structure Prediction Using the Face-Centered Cubic Lattice. arXiv 2025, arXiv:2507.08955. [Google Scholar] [CrossRef]
- Wong, R.; Chang, W.L. Quantum speedup for protein structure prediction. IEEE Transactions on NanoBioscience 2021, 20, 323–330. [Google Scholar] [CrossRef]
- Babej, T.; Ing, C.; Fingerhuth, M. Coarse-grained lattice protein folding on a quantum annealer. arXiv 2018, arXiv:1811.00713. [Google Scholar] [CrossRef]
- Fingerhuth, M.; Babej, T.; Ing, C. A quantum alternating operator ansatz with hard and soft constraints for lattice protein folding. arXiv 2018, arXiv:1810.13411. [Google Scholar] [CrossRef]
- Perdomo, A.; Truncik, C.; Tubert-Brohman, I.; Rose, G.; Áspuru-Guzik, A. Construction of model hamiltonians for adiabatic quantum computation and its application to finding low-energy conformations of lattice protein models. Physical Review A 2008, 78, 012320. [Google Scholar] [CrossRef]
- Perdomo-Ortiz, A.; Dickson, N.; Drew-Brook, M.; Rose, G.; Áspuru-Guzik, A. Finding low-energy conformations of lattice protein models by quantum annealing. Scientific Reports 2012, 2, 571. [Google Scholar] [CrossRef]
- Babbush, R.; Perdomo-Ortiz, A.; O’Gorman, B.; Macready, W.; Áspuru-Guzik, A. Construction of energy functions for lattice heteropolymer models: Efficient encodings for constraint satisfaction programming and quantum annealing. In Advances in Chemical Physics; John Wiley & Sons, Inc., 2014; Vol. 155, pp. 201–244. [Google Scholar] [CrossRef]
- Godzik, A.; Kolinski, A.; Skolnick, J. Lattice representations of globular proteins: How good are they? Journal of Computational Chemistry 1993, 14, 1194–1202. [Google Scholar] [CrossRef]
- Smaldone, A.M.; Shee, Y.; Kyro, G.W.; Farag, M.H.; Chandani, Z.; Kyoseva, E.; Batista, V.S. A Hybrid Transformer Architecture with a Quantized Self-Attention Mechanism Applied to Molecular Generation. arXiv 2025, arXiv:quant. [Google Scholar] [CrossRef] [PubMed]
- NVIDIA Corporation. CUDA-Q: A Platform for Hybrid Quantum–Classical Computing Version 0.12.0. 2025. Available online: https://github.com/NVIDIA/cuda-quantum.
- NVIDIA Corporation. CUDA-Q Documentation (latest). 2025. Available online: https://nvidia.github.io/cuda-quantum/latest/.
Figure 1.
An overview and taxonomy of the efficient Protein Language Model methodologies discussed in this survey. We structure our review into four primary pillars—Dataset, Architecture, Training, and Inference—and highlight the key approaches and representative works within each category. ([21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38]).
Figure 1.
An overview and taxonomy of the efficient Protein Language Model methodologies discussed in this survey. We structure our review into four primary pillars—Dataset, Architecture, Training, and Inference—and highlight the key approaches and representative works within each category. ([21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38]).
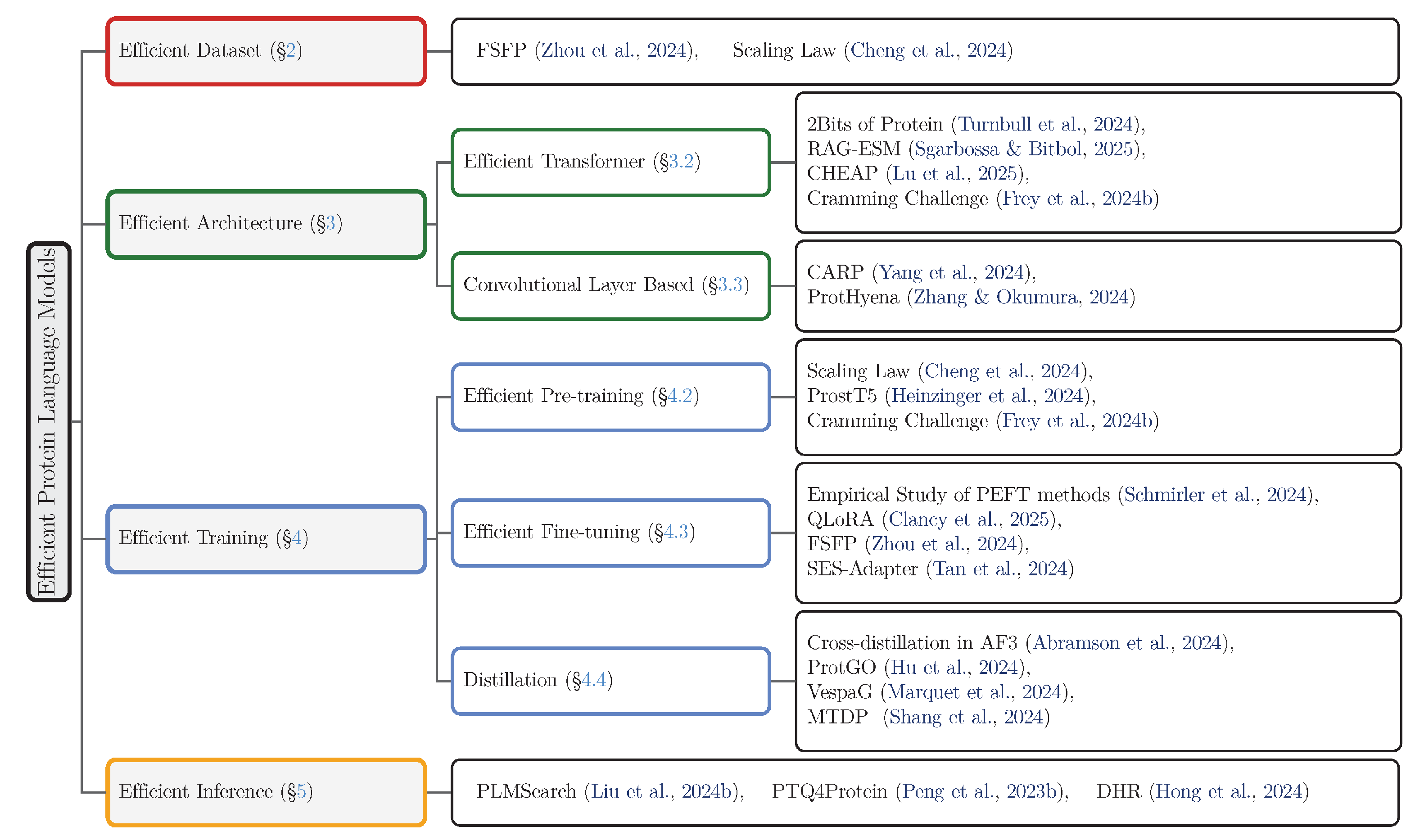
Figure 4.
Conceptual illustration of efficient dataset strategies. The left subfigure, ’Improve Utilization,’ represents using methods like scaling laws for better data selection and allocation from large-scale corpora. The right subfigure, ’Adapt to Severely Scarce Data,’ represents few-shot learning approaches, for example using auxiliary tasks to extend limited datasets.
Figure 4.
Conceptual illustration of efficient dataset strategies. The left subfigure, ’Improve Utilization,’ represents using methods like scaling laws for better data selection and allocation from large-scale corpora. The right subfigure, ’Adapt to Severely Scarce Data,’ represents few-shot learning approaches, for example using auxiliary tasks to extend limited datasets.
Figure 5.
Overview of efficient architecture methodologies. These strategies diverge from the standard Transformer (left), branching into ’Efficient Transformer’ approaches (top right) such as low-bit quantization, embedding compression, and parameter reduction/reuse, and ’CNN Alternatives’ (bottom right) which replace attention blocks with convolutional layers to achieve linear scaling complexity.
Figure 5.
Overview of efficient architecture methodologies. These strategies diverge from the standard Transformer (left), branching into ’Efficient Transformer’ approaches (top right) such as low-bit quantization, embedding compression, and parameter reduction/reuse, and ’CNN Alternatives’ (bottom right) which replace attention blocks with convolutional layers to achieve linear scaling complexity.
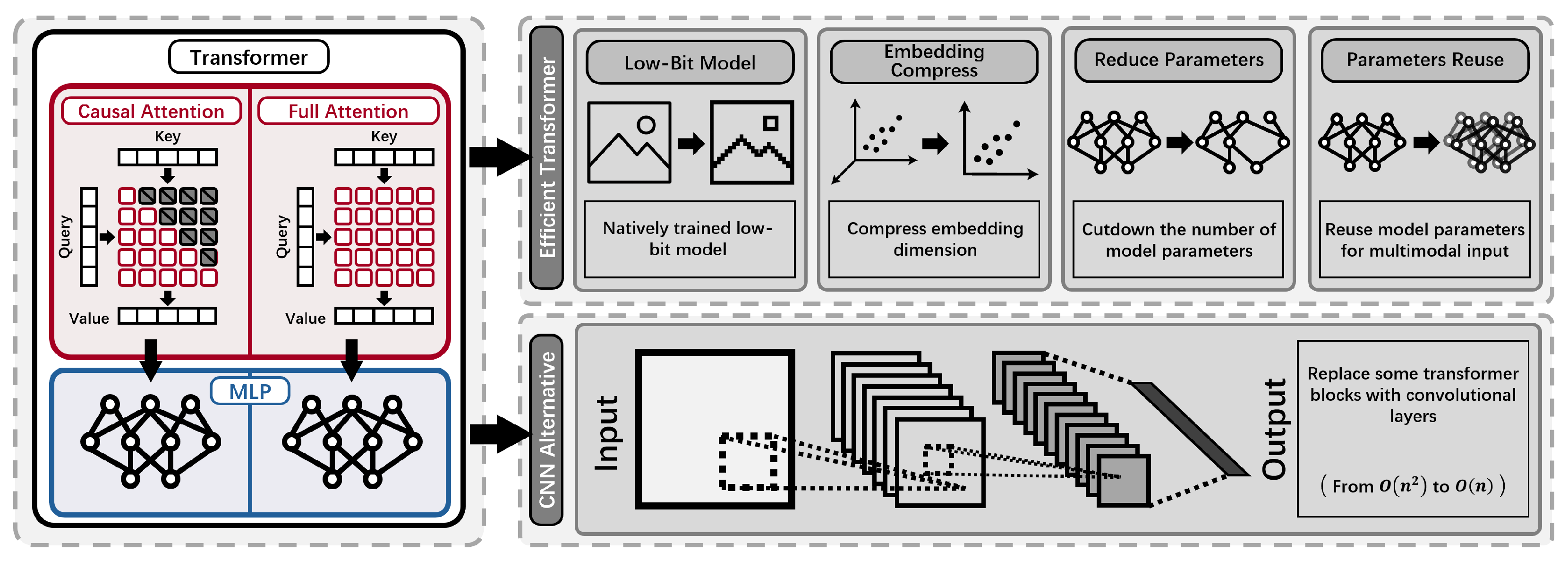
Figure 6.
Conceptual illustration of efficient pretraining strategies: Improve Utilization — optimizing resource allocation such as model size and data scaling; Refined Pipeline — developing more streamlined and effective training pipeline designs; Diverse Datasets — incorporating more varied datasets, including multimodal data, to enhance generalization and robustness.
Figure 6.
Conceptual illustration of efficient pretraining strategies: Improve Utilization — optimizing resource allocation such as model size and data scaling; Refined Pipeline — developing more streamlined and effective training pipeline designs; Diverse Datasets — incorporating more varied datasets, including multimodal data, to enhance generalization and robustness.
Figure 7.
A comparison of Full Fine-Tuning (left) with several mainstream parameter Parameter-Efficient Fine-Tuning (PEFT) methods (right) for pLMs. In full fine-tuning, all model parameters are trainable. PEFT methods keep the backbone parameters frozen (blue) and introduce a small number of new trainable parameters (pink). Examples shown include LoRA, which adds low-rank adaptation matrices; Q-LoRA, which combines LoRA with quantization; IA3, which trains scaling vectors; and Prefix Tuning, which adds trainable prefixes to key and value vectors.
Figure 7.
A comparison of Full Fine-Tuning (left) with several mainstream parameter Parameter-Efficient Fine-Tuning (PEFT) methods (right) for pLMs. In full fine-tuning, all model parameters are trainable. PEFT methods keep the backbone parameters frozen (blue) and introduce a small number of new trainable parameters (pink). Examples shown include LoRA, which adds low-rank adaptation matrices; Q-LoRA, which combines LoRA with quantization; IA3, which trains scaling vectors; and Prefix Tuning, which adds trainable prefixes to key and value vectors.
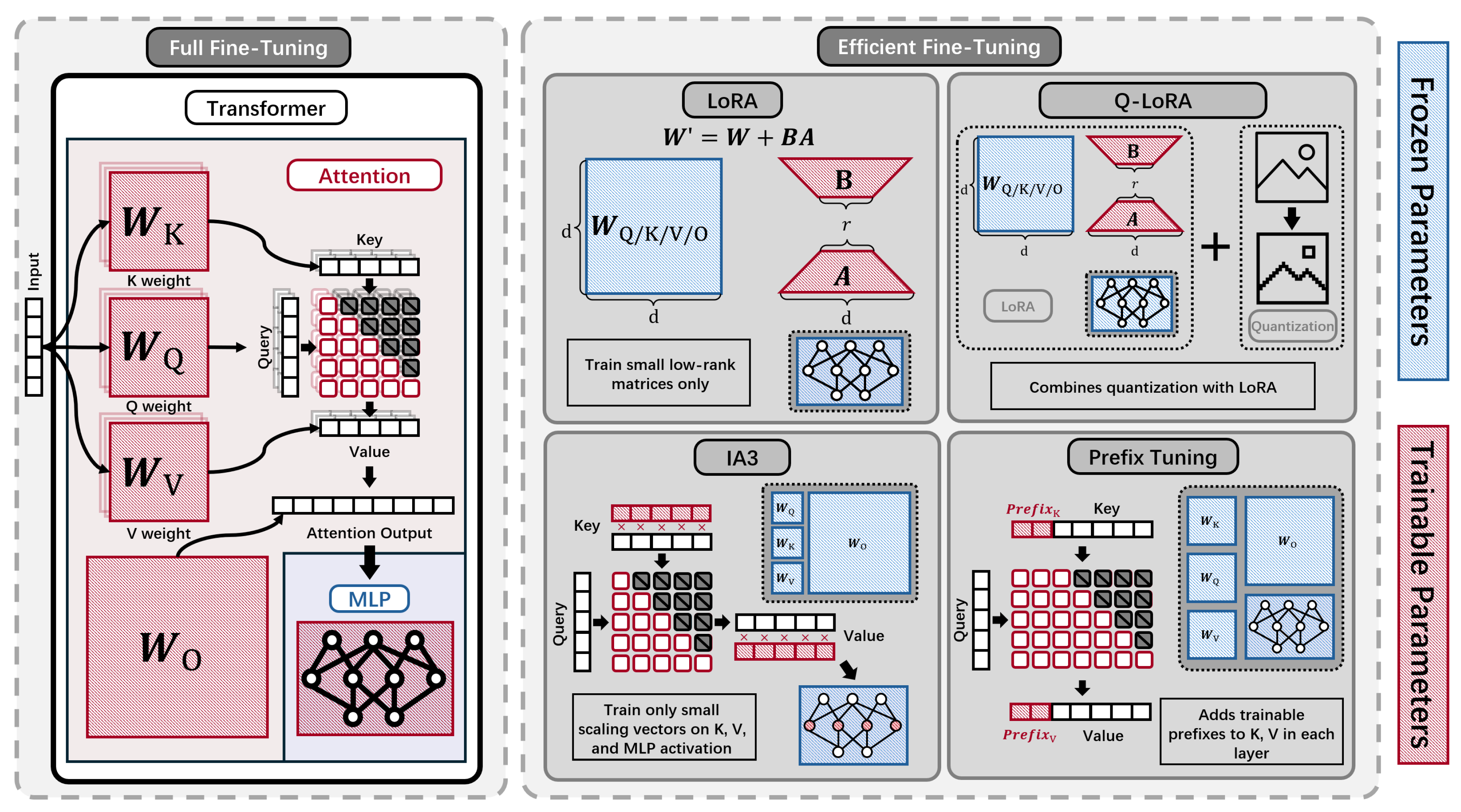
Figure 8.
Strategies for improving inference efficiency: Post-Training Quantization (PTQ) — reduces numerical precision to compress model size and accelerate inference; Improve Downstream Performance — applies pLMs to enhance downstream task performance and practical applicability.
Figure 8.
Strategies for improving inference efficiency: Post-Training Quantization (PTQ) — reduces numerical precision to compress model size and accelerate inference; Improve Downstream Performance — applies pLMs to enhance downstream task performance and practical applicability.
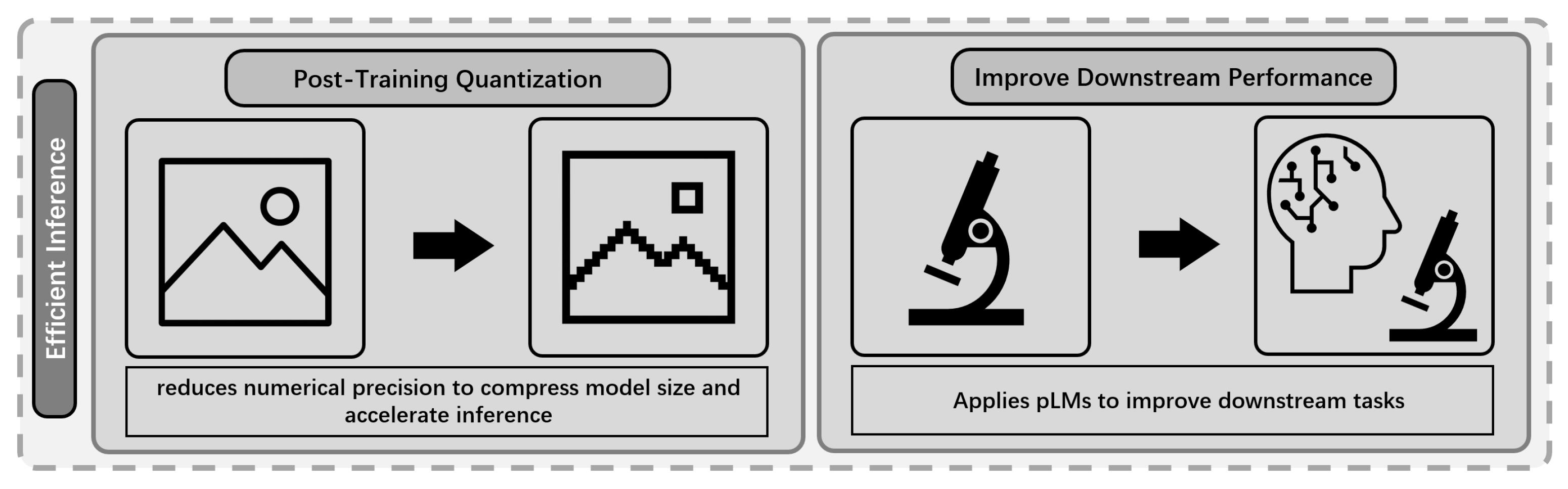
Figure 9.
Conceptual illustration of a hybrid quantum–classical learning framework. Input data X are first processed through classical preprocessing steps such as tokenization and embedding, and subsequently encoded into a parameterized quantum circuit via a data-dependent unitary . The quantum circuit consists of trainable single-qubit rotation gates and entangling operations, and is executed on a quantum processing unit (QPU), where measurements produce output probability distributions. These measurement outcomes are used to evaluate a classical objective function , whose feedback is computed on classical hardware (CPU/GPU) to iteratively update the circuit parameters . This variational optimization loop illustrates a potential direction for integrating quantum computing with machine learning pipelines in future work.
Figure 9.
Conceptual illustration of a hybrid quantum–classical learning framework. Input data X are first processed through classical preprocessing steps such as tokenization and embedding, and subsequently encoded into a parameterized quantum circuit via a data-dependent unitary . The quantum circuit consists of trainable single-qubit rotation gates and entangling operations, and is executed on a quantum processing unit (QPU), where measurements produce output probability distributions. These measurement outcomes are used to evaluate a classical objective function , whose feedback is computed on classical hardware (CPU/GPU) to iteratively update the circuit parameters . This variational optimization loop illustrates a potential direction for integrating quantum computing with machine learning pipelines in future work.
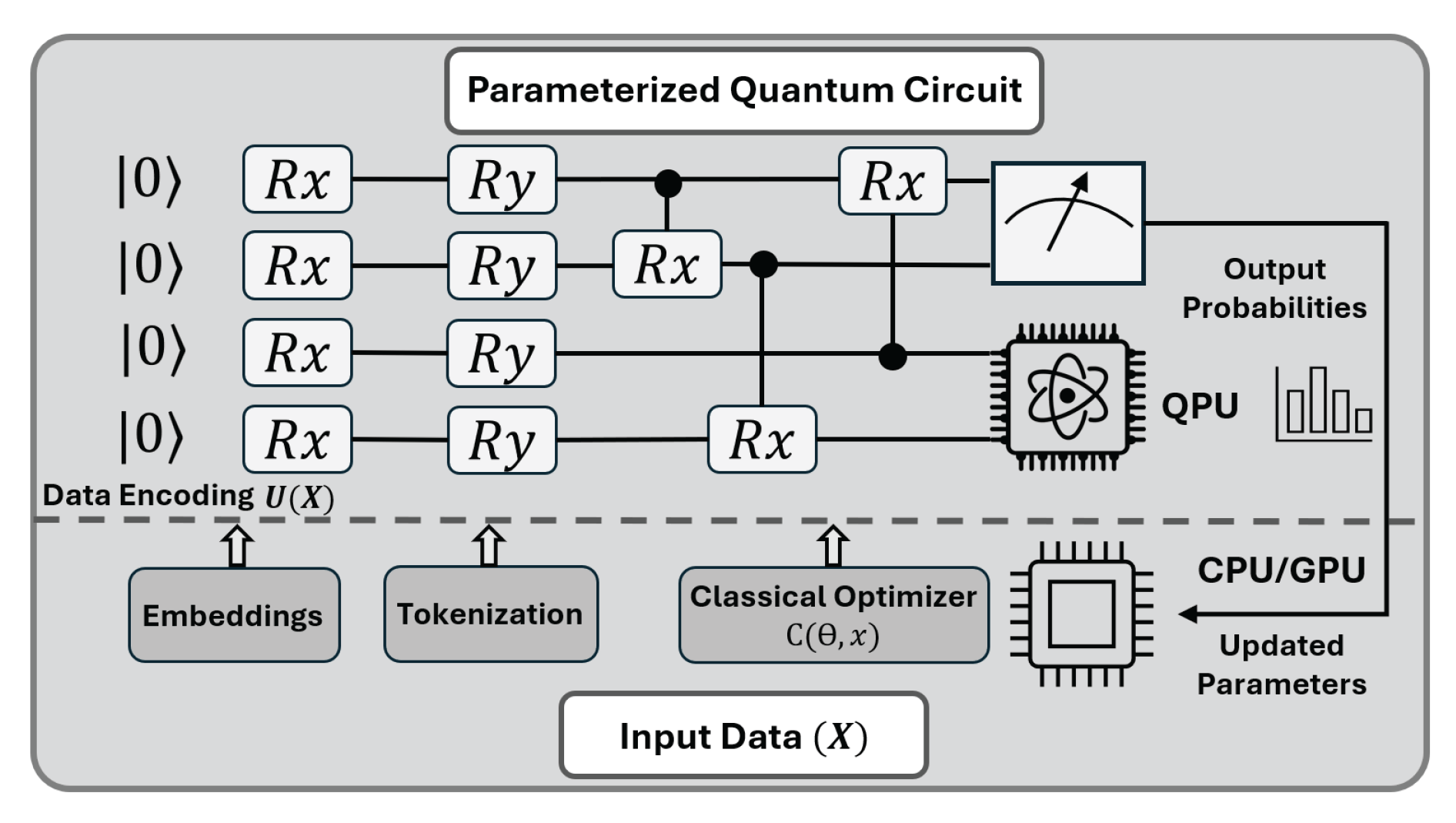
Table 2.
Summary of representative efficient model architectures for pLMs.
| Category | Method | Brief Description |
|---|---|---|
| Transformer-based | 2Bits of Protein [24] | Training with ternary weights for memory and compute efficiency. |
| RAG-ESM [25] | Lightweight retrieval-augmented model with parameter-sharing. | |
| CHEAP [26] | Compressing high-dimensional embeddings while preserving structure. | |
| Cramming Challenge [23] | Train a performant pLM in 24 hours on a single GPU. | |
| CNN-based | CARP [27] | Using dilated-convolutions (ByteNet-style) to replace attention layers. |
| Hyena operator [28] | Implicit long convolutions for subquadratic scaling on long sequences. |
Table 4.
Summary of representative inference-stage efficiency methods for pLMs.
| Category | Method | Brief Description |
|---|---|---|
| Inference | PLMSearch [37] | Efficient remote homology search using learned embeddings. |
| PTQ4Protein [38] | PTQ for reducing inference memory and compute. | |
| DHR [20] | Transforms protein homology detection from pairwise alignment into vector retrieval. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



