
Submitted:
22 December 2025
Posted:
23 December 2025
You are already at the latest version
Abstract
The mechanisms underlying carfilzomib (CFZ)-induced cardiotoxicity remain incompletely elucidated. In this study, we used human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) to char-acterize the transcriptional impact of CFZ and to evaluate whether atorvastatin could prevent these dele-terious transcriptional changes. hiPSC-CMs were treated with 1 µM CFZ, CFZ + atorvastatin, atorvastatin, or vehicle control, followed by RNA sequencing and differential expression analysis and pathway analyses. Transcriptomic profiling revealed a marked upregulation of genes in multiple proteasome subunits, in-cluding ATPase components (PSMC1, PSMC4, PSMC5, PSMC6) and non-ATPase regulatory subunits (PSMD1, PSMD2, PSMD12), suggesting a strong compensatory activation of proteostasis and protein quality-control pathways in response to CFZ exposure. In addition, several of the most significantly altered genes were those implicated in cardiovascular disease and heart failure, such as BAG3 and FLNC and many heat-shock proteins, indicating activation of cardiac stress-response pathways relevant to CFZ-associated cardiotoxicity. Atorvastatin co-treatment partially reversed a subset of CFZ-induced transcriptional changes, particularly within cholesterol biosynthesis and lipid regulatory pathways (e.g. ACAT2 and ACTA1), but did not restore CFZ-mediated downregulation of sarcomeric genes. Together, these findings define a mul-tifactorial signature of CFZ-induced deleterious transcriptional changes and suggest that atorvastatin may provide partial metabolic, but not structural, cardioprotection.
Keywords:
carfilzomib
; cardiotoxicity
; induced pluripotent stem cells
; atorvastatin
1. Introduction
Proteasome inhibitor carfilzomib (CFZ) has been used as the backbone for first-line combination therapy for patients with relapsed or refractory multiple myeloma (MM), the second most common hematologic cancer [1,2]. However, despite its effectiveness, CFZ is associated with significant cardiovascular adverse events (CVAE), including hypertension, arrhythmias, and heart failure, with incidence estimates ranging from 8–18% [3]. A critical clinical implication of this cardiotoxicity is treatment interruption which is associated with cancer recurrence [4]. Studies have shown that early detection of and intervention for cardiotoxicity induced by other cancer therapies (i.e., anthracyclines) can improve long-term outcomes [5,6,7]. It is crucial to understand the underlying pathophysiological and inherent compensatory mechanisms of CFZ-related CVAE to develop effective strategies for early detection and management of high-risk patients [4].
Statins are a group of lipid-lowering drugs that inhibit cholesterol biosynthesis and have been approved and widely used as therapy for the prevention of coronary atherosclerotic events. Statins also have pleiotropic effects independent of their cholesterol-lowering properties [8]. Different studies, including the recently published STOP-CA randomized clinical trial, have demonstrated that statins can reduce cardiac functional impairment in patients with cancer treated with anthracyclines [9,10,11,12]. More importantly, a mouse study reported that atorvastatin prevented CFZ-mediated left ventricular dysfunction and molecular deficits in mice with cardiometabolic syndrome [13]. We hypothesize that atorvastatin may also protect against CFZ-related CVAE in humans. We used human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) model to test this hypothesis. hiPSCs can self-renew and differentiate into cell types including cardiomyocytes [14,15]. The advantages of using hiPSC-CMs are the perfect genetic homology between each line and its human counterpart, it provides a more physiologically relevant model compared to immortalized cardiac cell lines, and their demonstrated utility for high-throughput applications [16]. hiPSC-CMs exposed to other cardiotoxic medications such as doxorubicin, kinase inhibitors, and trastuzumab were able to recapitulate the phenotype of cardiac dysfunction in terms of decreased cardiomyocyte viability and contractility, impaired mitochondrial and metabolic function, as well as increased reactive oxygen species (ROS) production [16,17,18]. In this study, we aimed to investigate the underlying mechanisms of CFZ-CVAE by characterizing the transcriptional impact of CFZ treatment on hiPSC-CMs and to evaluate whether atorvastatin could prevent these deleterious transcriptional changes using RNA sequencing followed by pathway analyses.
2. Results
2.1. Validation of Pluripotent State Prior to Cardiomyocyte Differentiation
To confirm that the starting cell population had not yet undergone cardiomyocyte differentiation, we assessed the expression of key cardiac structural and mitochondrial genes in undifferentiated hiPSCs. Undifferentiated hiPSCs showed negligible expression of sarcomeric markers (MYH6, MYH7, MYL2, TNNT2) and mitochondrial regulators (ATP2A2, MFN1, MFN2), consistent with their pluripotent and non-cardiac phenotype (Supplementary Figure S1). The absence of these lineage-specific transcripts confirms that cardiomyocyte-enriched gene expression patterns observed in later experimental stages resulted from controlled differentiation rather than baseline expression in the parental hiPSC population. This validation step ensures that all downstream transcriptomic effects reflect true responses of differentiated cardiomyocytes rather than undifferentiated stem cells.
2.2. Establishing the Optimal Carfilzomib Dose for hiPSC-CMs Experiments
To determine the optimal concentration of CFZ for subsequent transcriptomic and functional analyses, we performed a dose–response experiment using 0.1 µM and 1.0 µM CFZ in hiPSC-CMs, guided by previous literature reporting CFZ cardiotoxicity within this concentration range [19]. Quantitative PCR analysis of key mitochondrial and sarcomeric genes demonstrated a clear dose-dependent effect. Mitochondrial regulators ATP2A2, MFN1, and MFN2 exhibited progressive upregulation with increasing CFZ concentrations, indicating early activation of mitochondrial stress-response programs. In contrast, core sarcomeric genes (MYH6, MYH7, and TNNT2) showed marked dose-dependent downregulation, reflecting impairment of cardiomyocyte contractile machinery. Based on these consistent transcriptional alterations and in alignment with prior studies evaluating CFZ cardiotoxicity in vitro, we selected 1.0 µM CFZ for all subsequent experiments to ensure robust and reproducible transcriptional responses while avoiding excessive cytotoxicity (Supplementary Figure S2).
2.3. Differentially Expressed Genes in CFZ vs Control
To evaluate the impact of CFZ on global gene expressions, we conducted a differential expression analysis comparing CFZ-treated hiPSC-CMs (n = 7) to controls (n = 8) using the DESeq2 framework. The dataset comprised 36,245 transcripts, of which ~11,000 had significant adjusted p-values. The differential expression was defined by a threshold of fold change (FC) ≥ 2 or ≤ 0.5 (or log2FC ≥1 or ≤ -1) and FDR-adjusted p-value < 0.05. Global gene expression profile analysis identified 6,025 differentially expressed genes (DEGs), including 3,871 downregulated genes (Supplementary Table 1) and 2,154 upregulated genes (Supplementary Table 2) in CFZ-treated hiPSC-CMs compared to controls. A volcano plot summarizing the distribution of fold changes and statistical significance is shown in Figure 1.
Among the top 100 DEGs in hiPSC-DMs treated with CFZ compared to controls, 98 were up-regulated and 2 were down-regulated. The most significantly upregulated genes were: 1). Genes in the Ubiquitin Proteasomal System, including Proteasome 26S Subunit, Non-ATPase 2 (PSMD2) (log₂FC = 2.67, padj = 2.3 × 10⁻458), Protein Tyrosine Phosphatase, Receptor Type N (PTPRN) (log₂FC = 5.84, padj = 1.14 x 10⁻340), and heat shock protein genes such as HSPA6 (log₂FC = 8.80, padj = 4.98 x 10⁻231) and HSPA1B (log₂FC = 5.20, padj = 2.13 x 10⁻222); 2). Genes in NRF2 pathway or Nuclear Receptors pathway, including Sequestosome 1 (SQSTM1) (log₂FC = 4.06, padj = 1.22 x 10⁻257), MAF basic leucine zipper transcription factor F (MAFF) (log₂FC = 4.08, padj = 1.98 x 10⁻176), heparin-binding EGF-liking growth factor (HBEGF) (log₂FC = 3.99, padj = 1.78 x 10⁻173); 3). Genes in apoptosis modulation and signaling pathway, such as BAG cochaperone 3 (BAG3) (log₂FC = 3.95, padj = 3.64 x 10⁻250) or MAPK signaling pathway such as Filamin-C (FLNC) (log₂FC = 3.92, padj = 2.19 x 10⁻238) and DUSP1 (log₂FC = 4.56, padj = 1.39 x 10⁻158) (Supplementary Table 2). Among the most significantly downregulated genes were CXXC finger protein 4 (CXXC4) (log₂FC = -2.58, padj = 1.24 x 10⁻101), Calpain 6 (CAPN6) (log₂FC = -2.89, padj = 3.60 x 10⁻84), high mobility group nucleosomal binding domain 3 (HMGN3) (log₂FC = -2.32, padj = 2.91 x 10⁻80), and fibroblast growth factor receptor 2 (FGFR2) (log₂FC = -2.34, padj = 3.51 x 10⁻76) (Supplementary Table 1).
To further characterize the transcriptional response to CFZ, we generated a heatmap of the top 50 differentially expressed genes between CFZ-treated and control hiPSC-CMs (Figure 2). Unsupervised hierarchical clustering clearly separated CFZ-treated samples from controls, indicating a robust and highly consistent treatment-specific signature across biological replicates. Most of the top DEGs were strongly upregulated in CFZ-treated cells (orange/red) and relatively suppressed in controls (blue), including multiple proteasome subunits.
2.4. Functional Enrichment of CFZ-Induced Gene Expression Changes
Gene Ontology (GO) enrichment analysis revealed that CFZ treatment profoundly disrupts mitochondrial function, lipid metabolism, and cardiac structural pathways in hiPSC-CMs. Mitochondrial-related GO terms showed significant enrichment, including mitochondrial genome maintenance (GO:0000002; p = 0.0043), suggesting impaired mitochondrial DNA stability and replication, as well as pathways involved in outer mitochondrial membrane organization (GO:1901028, GO:1901030; p = 0.039 and 0.053, respectively) and mitochondrial protein processing (GO:0034982; p = 0.053), indicating altered mitochondrial turnover and heightened susceptibility to apoptosis (Supplementary Figure S3). CFZ also dysregulated multiple lipid metabolic processes, with enrichment of regulation of lipoprotein lipase activity (GO:0051004; p = 0.0138), negative regulation of lipoprotein lipase activity (GO:0051005; p = 0.05), positive regulation of cholesterol storage (GO:0010886; p = 0.05), and membrane lipid catabolic process (GO:0046466; p = 0.053), highlighting disturbances in cholesterol handling and membrane lipid remodeling (Supplementary Figure S4). In addition, cardiac-related GO terms demonstrated strong enrichment, particularly negative regulation of cardiac muscle cell differentiation (GO:2000726; p = 0.052), reflecting suppression of genes required for cardiomyocyte maturation, contractility, and electrical stability. Collectively, these pathway disruptions suggest that CFZ impairs mitochondrial homeostasis, lipid metabolism, and cardiac structural development, providing mechanistic insight into the reduced contractile performance and cardiotoxicity associated with CFZ treatment (Supplementary Table 3).
2.5. Transcriptomic Clustering Reveals Metabolic Reprogramming with Limited Sarcomeric Recovery in CFZ + Atorvastatin–Treated Cardiomyocytes
Unsupervised hierarchical clustering of the top 50 differentially expressed genes between CFZ + atorvastatin and CFZ-treated hiPSC-DMs revealed two distinct expression clusters corresponding to each treatment condition, indicating a strong transcriptional shift upon atorvastatin co-treatment, as shown in Figure 3 and Supplementary Table 4. Genes associated with lipid and cholesterol metabolism, including 3-Hydroxy-3-Methylglutaryl-CoA reductase (HMGCR) (log₂FC = 0.96, padj = 2.19 x 10⁻32), 3-Hydroxy-3-Methylglutaryl-CoA synthase 1 (HMGCS1) (log₂FC = 1.21, padj = 2.21 x 10⁻17), Acetyl-CoA Acetyltransferase 2 (ACAT2) (log₂FC = 1.32, padj = 2.57 x 10⁻8) and insulin induced gene 1 (INSIG1) (log₂FC = 1.26, padj = 4.96 x 10⁻8) were upregulated in the CFZ + atorvastatin treated hiPSC-CMs compared to the cells treated with CFZ alone, consistent with reactivation of the mevalonate and cholesterol biosynthesis pathways. In contrast, sarcomeric and contractile genes such as actin alpha 1 (ACTA1), myosin heavy chain 11 (MYH11), and myosin heavy chain (MYH2) remained suppressed, suggesting that atorvastatin primarily restored metabolic pathways rather than structural cardiac programs. Additionally, modest upregulation of stress-response and signaling genes such as von Willebrand factor (VWF) (log₂FC = 1.35, padj = 3.67 x 10-5) and CYLD lysine 63 deubiquitinate (CYLD) (log₂FC = 0.67, padj = 0.0037), and partial reduction in inflammatory mediators such as interferon induced protein with tetratricopeptide repeats 2 (IFIT2) (log₂FC = -1.23, padj = 0.011), corticotropin releasing hormone binding protein (CRHBP) (log₂FC = -1.39, padj = 0.0072) were observed, reflecting a mixed protective adaptation at the transcriptional level (Supplementary Table 4).
2.6. Atorvastatin Reverses a Subset of CFZ-Induced Transcriptional Alterations
To investigate whether atorvastatin mitigates the molecular effects of CFZ treatment, we performed a comparative differential expression analysis between CFZ vs Control and CFZ + atorvastatin vs CFZ conditions, as shown in the volcano plot Figure 4. A small subset of genes (n = 12) exhibited significant changes in opposite directions across the two contrasts, indicating directional reversal of CFZ-induced transcriptional effects by atorvastatin co-treatment (Table 1). Among these, eight genes (ACAT2, PAX2, PLA2G3, VSX2, PCDHA2, DERL3, NDST4, and TMEM178B) were downregulated by CFZ but upregulated upon atorvastatin co-treatment. These genes are functionally enriched in lipid metabolic regulation (ACAT2, PLA2G3), endoplasmic reticulum (ER) stress and protein quality control (DERL3), and cellular signaling and transcriptional regulation (PAX2, VSX2, TMEM178B), suggesting that atorvastatin may restore metabolic and ER homeostasis disrupted by CFZ exposure. Conversely, four genes (ACTA1, CRHBP, IFIT2, and PTGER2) were upregulated by CFZ but suppressed by atorvastatin cotreatment. These genes are associated with sarcomeric contractile function (ACTA1), stress-response signaling (CRHBP, IFIT2), and prostaglandin receptor-mediated inflammation (PTGER2). Their downregulation following atorvastatin treatment indicates a potential attenuation of CFZ-induced cellular stress and contractile dysregulation. The gene expression levels of ACAT2 and ACTA1 are demonstrated in Figure 5 as examples. Collectively, these findings demonstrate that while most CFZ-responsive genes remain unaltered by atorvastatin, a discrete subset shows reversal of CFZ-induced expression patterns, supporting the notion that atorvastatin may exert protective transcriptional effects against CFZ-mediated cardiotoxicity, particularly through pathways involved in lipid metabolism, ER stress resolution, and cytoskeletal maintenance.
2.7. Validation of RNA-Seq Findings by qPCR
To evaluate the effects of CFZ and atorvastatin on cardiac structural and mitochondrial gene expression, we performed qPCR analysis of ATP2A2, MFN1, MFN2, MYH6, MYH7, MYL2, and TNNT2 across four experimental conditions: Control, CFZ alone, Atorvastatin alone, and CFZ + Atorvastatin. CFZ treatment markedly suppressed the expression of key sarcomeric genes (MYH6, MYH7, MYL2, TNNT2), consistent with impaired contractile gene programs observed in our RNA-seq analysis. Atorvastatin alone had a minimal impact on these cardiac markers, indicating limited transcriptional influence in atorvastatin-treated cardiomyocytes. Importantly, co-treatment with CFZ + atorvastatin did not restore sarcomeric gene expression, suggesting that atorvastatin is unable to rescue CFZ-induced transcriptional suppression of structural genes. In contrast, mitochondrial and calcium-handling genes (ATP2A2, MFN1, MFN2) showed modest increases with atorvastatin and partial normalization in the CFZ + atorvastatin condition, indicating that statins may preferentially modulate metabolic pathways rather than structural cardiac programs. Together, these qPCR findings validate our transcriptomic results, demonstrating that atorvastatin exerts a metabolic protective effect but does not reverse CFZ-induced downregulation of cardiac contractile genes Supplementary Figure S5.
3. Discussion
In this study, we investigated the transcriptomic effects of CFZ on hiPSC-CMs and assessed whether atorvastatin could mitigate CFZ-induced deleterious transcriptional alterations. We observed striking transcriptional changes induced by CFZ that are consistent with proteasome inhibitions. More importantly, our results demonstrated that CFZ profoundly disrupts cardiomyocyte homeostasis by altering pathways essential for cardiac metabolism, mitochondrial function, oxidative stress regulation, apoptosis, and sarcomeric integrity—mechanisms that closely mirror the cardiovascular adverse events observed clinically, including heart failure, arrhythmias, and cardiomyopathy. We also observed that atorvastatin co-treatment partially reversed a subset of CFZ-induced transcriptional changes, particularly within cholesterol biosynthesis and lipid regulatory pathways (e.g., ACAT2 and ACTA1), but did not restore CFZ-mediated downregulation of sarcomeric genes.
The ubiquitin proteasome pathway is the major cellular proteolytic pathways to maintain cellular proteome stability and proper cellular function and viability [20]. In patients with MM, malignant plasma cells produce excessive amounts of immunoglobulins, inducing significant proteotoxic stress, and are highly dependent on proteasome activity to survive and are therefore sensitive to proteasome inhibition [4]. By selectively and irreversibly binding the β5 subunit of the 26S proteasome and blocking its chymotrypsin-like activity that normally degrades poly-ubiquitinated proteins [21], CFZ can delay proliferation and induce cell-cycle arrest and apoptosis of malignant plasma cells in the bone marrow of patients with MM [22]. Terminally differentiated cardiomyocytes are post-mitotic cells and thus especially sensitive to proteasome inhibition [23]. In addition, the high metabolic demand of the heart increases the burden of proteostasis. Therefore, proteasome inhibition may be deleterious for the heart [4].
In our study, among the most prominent transcriptomic changes induced by CFZ treatment include the significant upregulation of multiple proteasome subunits—including ATPase components (PSMC1, PSMC4, PSMC5, PSMC6), non-ATPase regulatory subunits (PSMD1, PSMD2, PSMD12)—indicates a robust compensatory response to proteasome inhibition. These genes are essential for substrate recognition, unfolding, and degradation, suggesting that CFZ-treated cardiomyocytes activate proteostasis and protein quality-control pathways to counteract the accumulation of misfolded proteins [24]. Among other notable upregulated genes were SQSTM1, BAG3, FLNC, HSPA6 and HSPA1B. SQSTM1 acts as a ubiquitin-binding scaffold that redirects the overload of poly-ubiquitinated proteins toward autophagy. BAG3, together with the stress-inducible Hsp70 isoforms (HSPA6 and HSPA1B) form a chaperone-assisted selective autophagy complex. Our results confirmed that CFZ’s blockage of the proteasome elicits a multilayered adaptive network to rebuild proteasome capacity or reroute the surplus of ubiquitinated proteins into autophagy. Importantly, several of these genes have been directly implicated in cardiovascular disease and heart failure and have been identified in a previous hiPSC study [19], including BAG3 [25,26,27], multiple heat-shock proteins (HSPA1A/B/L, HSPH1, HSPB8) [28]. These genes mediate essential processes such as sarcomere maintenance, mitochondrial respiration, oxidative stress defense, apoptosis, and proteostasis—all of which are disrupted in CFZ-induced cardiotoxicity. Their consistent upregulation in CFZ-treated samples further confirms activation of cardiac stress-response pathways known to contribute to heart failure. Importantly, genetic variants in BAG3 and FLNC are also associated with dilated myopathy in genome-wide association studies [29,30,31].
CFZ-treated hiPSC-CMs also exhibited extensive downregulation of genes critical for cardiac structure and contractility, including MYH6, MYH7, MYL2, and TNNT2 [32]. Suppression of these sarcomeric genes is strongly associated with impaired contraction, reduced force generation, and cardiomyocyte differentiation—all features reported in patients who develop CFZ-associated systolic dysfunction and heart failure [3,33]. These transcriptomic findings align with clinical and preclinical studies demonstrating impaired cardiac output, altered calcium handling, and structural remodeling after CFZ exposure [34,35].
In addition to structural gene suppression, CFZ significantly altered mitochondrial pathways. Enrichment of mitochondrial genome maintenance, outer membrane organization, and mitochondrial protein processing pathways suggests that CFZ disrupts mitochondrial homeostasis [19,36,37]. Mitochondrial instability is directly linked to reduced ATP production [38], bioenergetic failure [39,40,41], and increased ROS, which are established drivers of heart failure pathogenesis [42]. Oxidative stress emerged as a key feature of the CFZ transcriptomic signature. Genes involved in oxidative phosphorylation, oxidative RNA/DNA demethylation, and oxidative stress–induced apoptosis were significantly enriched [43,44]. These pathways reflect mitochondrial-derived ROS accumulation and redox imbalance—mechanisms known to contribute to cardiomyocyte apoptosis and left ventricular dysfunction [42]. These findings are consistent with prior studies showing that CFZ increases oxidative stress markers, reduces mitochondrial membrane potential, and triggers cardiomyocyte injury [42,45]. Apoptotic pathways were also robustly represented among CFZ-regulated genes. Enrichment of intrinsic apoptotic signaling, caspase activation, and execution-phase apoptosis supports the idea that CFZ induces programmed cell death in cardiomyocytes [46,47]. Apoptosis is a hallmark of many forms of chemotherapy-induced cardiotoxicity and correlates strongly with clinical cardiac dysfunction [42,48,49].
In our study, while CFZ significantly altered cardiomyocyte structural and mitochondrial pathways, atorvastatin’s protective effects were primarily confined to metabolic regulation. Atorvastatin co-treatment consistently upregulated genes involved in cholesterol biosynthesis and lipid homeostasis, including HMGCR [50,51], HMGCS1 [52], INSIG1 [53], and notably ACAT2 [54], which was markedly suppressed by CFZ and restored by atorvastatin. However, atorvastatin did not rescue CFZ-induced downregulation of sarcomeric genes, such as MYH6, MYH7, MYL2, and TNNT2. This indicates that while atorvastatin may correct metabolic disturbances, it does not reverse the structural deterioration or contractile gene suppression that underlies CFZ-induced systolic dysfunction. This difference underscores the complexity of CFZ cardiotoxicity and suggests that atorvastatin therapy alone may offer partial metabolic protection but not full preservation of cardiac function.
Collectively, our findings support a multifactorial model of CFZ-induced cardiotoxicity in which several interconnected mechanisms contribute to cardiomyocyte injury. CFZ exposure disrupts mitochondrial function, leading to impaired energy production and increased vulnerability to metabolic stress. This mitochondrial instability is accompanied by excessive oxidative stress and accumulation of ROS, which further damages cellular components and exacerbates redox imbalance. In parallel, CFZ activates intrinsic apoptotic pathways, promoting programmed cell death and loss of viable cardiomyocytes. These molecular disturbances are compounded by the marked suppression of key structural and contractile genes essential for maintaining sarcomere integrity and cardiac mechanical function. Additionally, CFZ alters lipid and cholesterol metabolism, a process fundamental for membrane stability, mitochondrial health, and cellular signaling. Together, these molecular signatures closely mirror the clinical phenotypes observed in CFZ-treated patients, including reduced left ventricular ejection fraction, diminished contractile reserve, arrhythmias, and heart failure. Notably, the partial restoration of metabolic pathways by atorvastatin—particularly those related to cholesterol regulation, ER stress mitigation, and mitochondrial stabilization—suggests a promising, though incomplete, cardioprotective potential that warrants further investigation.
It is important to acknowledge some limitations. This study used a single hiPSC line and focused on acute (24 h) drug exposure, which may not fully capture chronic or cumulative toxicity. Additional models incorporating electrophysiology, contractility, calcium imaging, and mitochondrial respiration are needed to confirm the functional consequences of the transcriptomic changes observed here. Further studies should evaluate dose- and timing-dependent atorvastatin effects, compare multiple statins, and investigate combination therapies targeting both metabolic and structural injury mechanisms.
4. Materials and Methods
4.1. Human-Induced Pluripotent Stem Cell-Derived cardiomyocyte Differentiation
hiPSCs (line SCTi003-A from Stem Cell Technologies, USA) were differentiated toward cardiomyocytes using small molecules or growth factors based on the manufacturer’s protocol. Briefly, hiPSCs were cultured on Matrigel®-coated plates in mTeSR™ Plus. For dissociation, cells were washed with D-PBS, treated with Gentle Cell Dissociation Reagent for 8–10 minutes at 37 °C, and resuspended in mTeSR™ media with 10 μM Y-27632. Following centrifugation (300 x g, 5 min), cells were seeded at a density of 3.58*105 cells/well onto Matrigel®-coated plates. After 24 hours of incubation, the media were refreshed, and cells were cultured to >95% confluency before differentiation. Cardiomyocyte differentiation was initiated once hiPSCs reached >95% confluency. On Day 0, the culture media was replaced with the STEMdiff™ Ventricular Cardiomyocyte Differentiation Medium A, supplemented with Matrigel® (1:100). Media changes were performed every two days, using Medium B on Day 2, Medium C on Days 4 and 6, and STEMdiff™ Cardiomyocyte Maintenance Medium from Day 8 onward. Beating cardiomyocytes were observed by Day 8, and cells were ready for assay by Day 15. For long-term maintenance, the media were changed every 2 days using Cardiomyocyte Maintenance Medium.
4.2. Drug Preparation and Treatment
The stock solution of CFZ (Selleck Chemicals, USA) was at the concentration of 10 mmol/L. Based on the pharmacokinetic profile of CFZ, the peak plasma concentration is 5.88 µmol/L. Following the initial testing at 1, 2, 10, and 20 µmol/L, a dose range of 0.01 to 10 µmol/L was established [19]. A concentration of 1 µmol/L for CFZ was determined for all the following experiments. On the day of each experiment, CFZ was diluted in STEMdiff™ Cardiomyocyte Maintenance Medium to achieve a 2× final concentration and kept on ice in the dark. Dimethyl sulfoxide (DMSO) at 0.2% (v/v), equivalent to the highest drug concentration, was used as a vehicle control. Atorvastatin (Selleck Chemicals, USA) at 10 µmol/L was evaluated as a potential cardioprotective agent against CFZ-induced deleterious transcriptional changes, with its stock solution (10 mmol/L) prepared similarly to CFZ.
hiPSC-CMs were treated on Day 15 of differentiation with the following conditions: (1) CFZ alone (1 µM), (2) atorvastatin alone (1 µM), (3) CFZ + atorvastatin (same concentration), and (4) untreated controls. All treatments were adjusted to contain equivalent concentrations of DMSO (0.2% v/v). Cells were incubated with drugs for 24 hours, after which total RNA was harvested on Day 16 using the RNeasy Fibrous Tissue Mini Kit (Qiagen, USA) according to the manufacturer’s instructions.
4.3. Quantitative Reverse Transcription– Polymerase Chain Reaction
Total RNA was extracted using the Zymo total RNA mini kit (Zymo, USA) according to the manufacturer’s instructions. One microgram of total RNA was used for complementary DNA synthesis using the High-Capacity RNA-to-cDNA (Applied Biosystems, USA), and the reaction mixture was incubated using a VeritiPro thermal cycler (Applied Biosystems) as follows: 37 °C for 1 hour and 95 °C for 5 minutes. The reaction mixture was further diluted, and 10 ng complementary DNA as the template was subjected to quantitative reverse transcription–polymerase chain reaction (RT-PCR), which was performed in triplicate for each gene using a Taqman Fast Advanced Master Mix (Applied Biosystems). Real-time polymerase chain reaction (PCR) conditions included the initial denaturation step at 95 °C for 20 seconds, 40 cycles of 2-step with 1 second of denaturation at 95 °C, followed by 20 seconds of annealing at 60 °C using QuantStudio™ 12K Flex Real–Time PCR System (Applied Biosystems). The messenger RNA levels of the genes examined were normalized to GAPDH. The primers used for qPCR are listed in Supplementary Table 5.
4.4. RNA Library and RNA Sequencing
Total RNA was extracted from hiPSC-CMs exposed to different treatment conditions: control (n = 8), CFZ (n = 7), atorvastatin (n = 5), and CFZ + atorvastatin (n = 7) using RNeasy Fibrous Tissue Mini Kit (Qiagen, USA). RNA integrity and quality were confirmed prior to library preparation and sequencing. Sample quality was assessed using Agilent TapeStation 4200 (Agilent Technologies, Inc.). One hundred ng of total RNA was used for library construction using the NEB Ultra II Directional RNA-seq library preparation kit for Illumina according to the manufacturer’s protocol. First, mRNA was isolated from 100 ng total RNA using the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs, catalog # E7490). This was then followed by RNA library construction with the NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs, catalog #E7760) according to the manufacturer’s user guide. Briefly, RNA was fragmented in NEBNext First Strand Synthesis Buffer via incubation at 94 °C for the desired time. This step was followed by first-strand cDNA synthesis using reverse transcriptase and random hexamer primer. Synthesis of ds-cDNA was performed using the 2nd strand master mix provided in the kit, followed by end-repair and adaptor ligation. At this point, Illumina adaptors were ligated to the sample. Finally, each library (uniquely barcoded) was enriched by 12 cycles of amplification and purified with Agencourt AMPure beads (Beckman Coulter, catalog # A63881). 27 barcoded libraries were sized on the TapeStation 4200 and quantified with the Qubit® 2.0 Fluorometer. Finally, these 27 individual libraries were pooled in equimolar concentration and sequenced on NovaSeq X 10B folowcell with 2x150 cycles run. One lane generated 1.25 billion pair-end reads with an average Q30% ≥ 92.5% and Cluster PF= 85.4%. FastQ files were generated using the BCL2fastQ function in the Illumina BaseSpace portal. One NovaSeq X, 2x150 cycles lane resulted in an average of 50 million demultiplexed, paired-end reads when sequencing a pool of 27 samples. RNASeq libraries were constructed at the University of Florida Interdisciplinary Center for Biotechnology Research (ICBR) Gene Expression Core (https://biotech.ufl.edu/gene-expression-genotyping/, RRID:SCR_019145), and sequenced at the UF ICBR NextGen Sequencing Core using NovaSeq X Sequencer. Principal Component Analysis (PCA) was performed on variance-stabilized transformed (VST) expression data to visualize sample clustering. The PCA plot revealed that samples segregated along the second principal component based on sequencing batch, independent of treatment conditions. We performed these experiments in two different batches and our analysis confirmed the presence of a batch effect. To mitigate its influence on downstream differential expression analysis, batch information was incorporated as a covariate in the DESeq2 design formula (batch + condition), a widely accepted strategy to adjust for unwanted variation [20,21].
4.5. RNA-Seq Analysis and Differential Expression Analysis
Transcriptomic data was processed through a standardized RNA-seq analysis pipeline. Raw sequencing reads underwent quality control using FastQC, and adapter trimming with Trim Galore. Transcript-level quantification was performed using Salmon [22], a lightweight and accurate tool for transcript abundance estimation. Quantified transcripts were aggregated to gene-level counts using tximport [23] in R, with reference to the GENCODE human gene annotation database. Differential expression analysis was conducted using DESeq2 [21], incorporating batch effects and treatment conditions in the design formula. Pairwise comparisons were performed between treatment groups to identify differentially expressed genes (DEGs), with significance defined as an adjusted p-value < 0.05 (Benjamini–Hochberg false discovery (FDR) correction) and an absolute log2 fold change (FC) > 1. Visualization of results included volcano plots and MA plots generated with ggplot2 [24], and heatmaps of the top DEGs created using pheatmap. Gene identifiers were mapped to gene symbols using biomaRt [25]. Functional enrichment analyses for biological processes such as apoptosis, oxidative stress, cardiotoxicity, lipid metabolism, and mitochondrial pathways were performed using the enrichR package [26], leveraging curated pathway databases.
5. Conclusions
In this hiPSC-CM study, we found that CFZ induces extensive transcriptomic remodeling consistent with clinically observed cardiovascular toxicity, driven by mitochondrial dysfunction, oxidative stress, apoptosis, and repression of contractile genes. Atorvastatin partially mitigates CFZ-induced metabolic disturbances—especially cholesterol-related pathways—but does not reverse structural gene downregulation. These findings underscore the need for multi-target cardioprotective strategies and support further evaluation of statins as a potential cardioprotective agent in reducing CFZ-associated cardiac risk.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, M.T, W.D, and Y.G.; methodology, M.T.; validation, M.T; formal analysis, M.T.; resources, F.Y., Y.Z., and K.A.B; data curation, F.Y. and Y.Z..; writing—original draft preparation, M.T. and Y.G.; writing—review and editing, W.D. Y.Z. M.G., K.A.B. and Y.G .; visualization, M.T.; supervision, Y.G; funding acquisition, M.T, W.D, Y.G. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported in part by the University of Florida College of Pharmacy Recurring Opportunity Seed Program for Education and Research (PROSPER) award. The funder had no role in the design and conduct of the study, collection, management, analysis, and interpretation of the data, preparation, review, or approval of the manuscript, and decision to submit the manuscript for publication.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CFZ | Carfilzomib |
| CVAE hiPSC-CMs |
Cardiovascular adverse event human induced pluripotent stem cell–derived cardiomyocytes |
| ROS MM |
Reactive Oxygen Species multiple myeloma |
References
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer Statistics, 2025. CA Cancer J Clin 2025, 75, 10–45. [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: 2024 Update on Diagnosis, Risk-Stratification, and Management. Am J Hematol 2024, 99, 1802–1824. [CrossRef]
- Waxman, A.J.; Clasen, S.; Hwang, W.-T.; Garfall, A.; Vogl, D.T.; Carver, J.; O’Quinn, R.; Cohen, A.D.; Stadtmauer, E.A.; Ky, B.; et al. Carfilzomib-Associated Cardiovascular Adverse Events. JAMA Oncol 2018, 4, e174519. [CrossRef]
- Georgiopoulos, G.; Makris, N.; Laina, A.; Theodorakakou, F.; Briasoulis, A.; Trougakos, I.P.; Dimopoulos, M.-A.; Kastritis, E.; Stamatelopoulos, K. Cardiovascular Toxicity of Proteasome Inhibitors: Underlying Mechanisms and Management Strategies: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol 2023, 5, 1–21. [CrossRef]
- Asselin, B.L.; Devidas, M.; Chen, L.; Franco, V.I.; Pullen, J.; Borowitz, M.J.; Hutchison, R.E.; Ravindranath, Y.; Armenian, S.H.; Camitta, B.M.; et al. Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children’s Oncology Group Randomized Trial Pediatric Oncology Grou. Journal of Clinical Oncology 2016, 34, 854–862. [CrossRef]
- Koutsoukis, A.; Ntalianis, A.; Repasos, E.; Kastritis, E.; Dimopoulos, M.A.; Paraskevaidis, I. Cardio-Oncology: A Focus on Cardiotoxicity. European Cardiology Review 2018, 13, 64–69. [CrossRef]
- Henry, M.L.; Niu, J.; Zhang, N.; Giordano, S.H.; Chavez-MacGregor, M. Cardiotoxicity and Cardiac Monitoring Among Chemotherapy-Treated Breast Cancer Patients. JACC Cardiovasc Imaging 2018, 11, 1084–1093. [CrossRef]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the Evidence for the Efficacy and Safety of Statin Therapy. The Lancet 2016, 388, 2532–2561. [CrossRef]
- Dehnavi, S.; Kiani, A.; Sadeghi, M.; Biregani, A.F.; Banach, M.; Atkin, S.L.; Jamialahmadi, T.; Sahebkar, A. Targeting AMPK by Statins: A Potential Therapeutic Approach. Drugs 2021, 81, 923–933. [CrossRef]
- Acar, Z.; Kale, A.; Turgut, M.; Demircan, S.; Durna, K.; Demir, S.; Meriç, M.; Ağaç, M.T. Efficiency of Atorvastatin in the Protection of Anthracycline-Induced Cardiomyopathy. J Am Coll Cardiol 2011, 58, 988–989. [CrossRef]
- Neilan, T.G.; Quinaglia, T.; Onoue, T.; Mahmood, S.S.; Drobni, Z.D.; Gilman, H.K.; Smith, A.; Heemelaar, J.C.; Brahmbhatt, P.; Ho, J.S.; et al. Atorvastatin for Anthracycline-Associated Cardiac Dysfunction. JAMA 2023, 330, 528. [CrossRef]
- Hundley, W.G.; D’Agostino, R.; Crotts, T.; Craver, K.; Hackney, M.H.; Jordan, J.H.; Ky, B.; Wagner, L.I.; Herrington, D.M.; Yeboah, J.; et al. Statins and Left Ventricular Ejection Fraction Following Doxorubicin Treatment. NEJM Evidence 2022, 1. [CrossRef]
- Efentakis, P.; Varela, A.; Lamprou, S.; Papanagnou, E.-D.; Chatzistefanou, M.; Christodoulou, A.; Davos, C.H.; Gavriatopoulou, M.; Trougakos, I.; Dimopoulos, M.A.; et al. Implications and Hidden Toxicity of Cardiometabolic Syndrome and Early-Stage Heart Failure in Carfilzomib-Induced Cardiotoxicity. Br J Pharmacol 2024, 181, 2964–2990. [CrossRef]
- Burridge, P.W.; Holmström, A.; Wu, J.C. Chemically Defined Culture and Cardiomyocyte Differentiation of Human Pluripotent Stem Cells. Curr Protoc Hum Genet 2015, 87. [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [CrossRef]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.-G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Recapitulate the Predilection of Breast Cancer Patients to Doxorubicin-Induced Cardiotoxicity. Nat Med 2016, 22, 547–556. [CrossRef]
- Kitani, T.; Ong, S.-G.; Lam, C.K.; Rhee, J.-W.; Zhang, J.Z.; Oikonomopoulos, A.; Ma, N.; Tian, L.; Lee, J.; Telli, M.L.; et al. Human-Induced Pluripotent Stem Cell Model of Trastuzumab-Induced Cardiac Dysfunction in Patients With Breast Cancer. Circulation 2019, 139, 2451–2465. [CrossRef]
- Sharma, A.; Burridge, P.W.; McKeithan, W.L.; Serrano, R.; Shukla, P.; Sayed, N.; Churko, J.M.; Kitani, T.; Wu, H.; Holmström, A.; et al. High-Throughput Screening of Tyrosine Kinase Inhibitor Cardiotoxicity with Human Induced Pluripotent Stem Cells. Sci Transl Med 2017, 9. [CrossRef]
- Forghani, P.; Rashid, A.; Sun, F.; Liu, R.; Li, D.; Lee, M.R.; Hwang, H.; Maxwell, J.T.; Mandawat, A.; Wu, R.; et al. Carfilzomib Treatment Causes Molecular and Functional Alterations of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. J Am Heart Assoc 2021, 10, e022247. [CrossRef]
- Pohl, C.; Dikic, I. Cellular Quality Control by the Ubiquitin-Proteasome System and Autophagy. Science 2019, 366, 818–822. [CrossRef]
- Jayaweera, S.P.E.; Wanigasinghe Kanakanamge, S.P.; Rajalingam, D.; Silva, G.N. Carfilzomib: A Promising Proteasome Inhibitor for the Treatment of Relapsed and Refractory Multiple Myeloma. Front Oncol 2021, 11, 740796. [CrossRef]
- Yee, A.J. The Role of Carfilzomib in Relapsed/Refractory Multiple Myeloma. Ther Adv Hematol 2021, 12, 20406207211019612. [CrossRef]
- Willis, M.S.; Townley-Tilson, W.H.D.; Kang, E.Y.; Homeister, J.W.; Patterson, C. Sent to Destroy: The Ubiquitin Proteasome System Regulates Cell Signaling and Protein Quality Control in Cardiovascular Development and Disease. Circ Res 2010, 106, 463–478. [CrossRef]
- Haertle, L.; Buenache, N.; Cuesta Hernández, H.N.; Simicek, M.; Snaurova, R.; Rapado, I.; Martinez, N.; López-Muñoz, N.; Sánchez-Pina, J.M.; Munawar, U.; et al. Genetic Alterations in Members of the Proteasome 26S Subunit, AAA-ATPase (PSMC) Gene Family in the Light of Proteasome Inhibitor Resistance in Multiple Myeloma. Cancers (Basel) 2023, 15. [CrossRef]
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J.; Stroud, M.J.; Zhang, Z.; Ma, X.; Mu, Y.; et al. Loss-of-Function Mutations in Co-Chaperone BAG3 Destabilize Small HSPs and Cause Cardiomyopathy. Journal of Clinical Investigation 2017, 127, 3189–3200. [CrossRef]
- Knezevic, T.; Myers, V.D.; Gordon, J.; Tilley, D.G.; Sharp, T.E.; Wang, J.; Khalili, K.; Cheung, J.Y.; Feldman, A.M. BAG3: A New Player in the Heart Failure Paradigm. Heart Fail Rev 2015, 20, 423–434. [CrossRef]
- Lakda, S.; Davies, R.H.; Hingorani, A.D.; Paliwal, N. A Systematic Review of GWAS on CMR Imaging Traits: Genetic Insights into Cardiovascular Structure, Function, and Diseases. EBioMedicine 2025, 121, 105992. [CrossRef]
- Patnaik, S.; Nathan, S.; Kar, B.; Gregoric, I.D.; Li, Y.-P. The Role of Extracellular Heat Shock Proteins in Cardiovascular Diseases. Biomedicines 2023, 11. [CrossRef]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol 2016, 68, 2440–2451. [CrossRef]
- Franaszczyk, M.; Bilinska, Z.T.; Sobieszczańska-Małek, M.; Michalak, E.; Sleszycka, J.; Sioma, A.; Małek, Ł.A.; Kaczmarska, D.; Walczak, E.; Włodarski, P.; et al. The BAG3 Gene Variants in Polish Patients with Dilated Cardiomyopathy: Four Novel Mutations and a Genotype-Phenotype Correlation. J Transl Med 2014, 12, 192. [CrossRef]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Züchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-Wide Studies of Copy Number Variation and Exome Sequencing Identify Rare Variants in BAG3 as a Cause of Dilated Cardiomyopathy. Am J Hum Genet 2011, 88, 273–282. [CrossRef]
- Oberwallner, B.; Brodarac, A.; Anić, P.; Šarić, T.; Wassilew, K.; Neef, K.; Choi, Y.-H.; Stamm, C. Human Cardiac Extracellular Matrix Supports Myocardial Lineage Commitment of Pluripotent Stem Cells. Eur J Cardiothorac Surg 2015, 47, 416–425; discussion 425. [CrossRef]
- Danhof, S.; Schreder, M.; Rasche, L.; Strifler, S.; Einsele, H.; Knop, S. ‘Real-Life’ Experience of Preapproval Carfilzomib-Based Therapy in Myeloma - Analysis of Cardiac Toxicity and Predisposing Factors. Eur J Haematol 2016, 97, 25–32. [CrossRef]
- Holmberg, S.R.; Williams, A.J. Patterns of Interaction between Anthraquinone Drugs and the Calcium-Release Channel from Cardiac Sarcoplasmic Reticulum. Circ Res 1990, 67, 272–283. [CrossRef]
- Chang, Y.-F.; Su, W.-C.; Su, C.-C.; Chung, M.-W.; Chang, J.; Li, Y.-Y.; Kao, Y.-J.; Chen, W.-P.; Daniels, M.J. High-Throughput Optical Controlling and Recording Calcium Signal in IPSC-Derived Cardiomyocytes for Toxicity Testing and Phenotypic Drug Screening. J Vis Exp 2022. [CrossRef]
- Jannuzzi, A.T.; Arslan, S.; Yilmaz, A.M.; Sari, G.; Beklen, H.; Méndez, L.; Fedorova, M.; Arga, K.Y.; Karademir Yilmaz, B.; Alpertunga, B. Higher Proteotoxic Stress Rather than Mitochondrial Damage Is Involved in Higher Neurotoxicity of Bortezomib Compared to Carfilzomib. Redox Biol 2020, 32, 101502. [CrossRef]
- Jannuzzi, A.T.; Korkmaz, N.S.; Gunaydin Akyildiz, A.; Arslan Eseryel, S.; Karademir Yilmaz, B.; Alpertunga, B. Molecular Cardiotoxic Effects of Proteasome Inhibitors Carfilzomib and Ixazomib and Their Combination with Dexamethasone Involve Mitochondrial Dysregulation. Cardiovasc Toxicol 2023, 23, 121–131. [CrossRef]
- Fiore, C.; Trézéguet, V.; Le Saux, A.; Roux, P.; Schwimmer, C.; Dianoux, A.C.; Noel, F.; Lauquin, G.J.-M.; Brandolin, G.; Vignais, P.V. The Mitochondrial ADP/ATP Carrier: Structural, Physiological and Pathological Aspects. Biochimie 1998, 80, 137–150. [CrossRef]
- Zhuang, L.; Jia, K.; Chen, C.; Li, Z.; Zhao, J.; Hu, J.; Zhang, H.; Fan, Q.; Huang, C.; Xie, H.; et al. DYRK1B-STAT3 Drives Cardiac Hypertrophy and Heart Failure by Impairing Mitochondrial Bioenergetics. Circulation 2022, 145, 829–846. [CrossRef]
- Da Dalt, L.; Cabodevilla, A.G.; Goldberg, I.J.; Norata, G.D. Cardiac Lipid Metabolism, Mitochondrial Function, and Heart Failure. Cardiovasc Res 2023, 119, 1905–1914. [CrossRef]
- Hinton, A.; Claypool, S.M.; Neikirk, K.; Senoo, N.; Wanjalla, C.N.; Kirabo, A.; Williams, C.R. Mitochondrial Structure and Function in Human Heart Failure. Circ Res 2024, 135, 372–396. [CrossRef]
- Chen, Q.; Zheng, A.; Xu, X.; Shi, Z.; Yang, M.; Sun, S.; Wang, L.; Wang, Y.; Zhao, H.; Xiao, Q.; et al. Nrf3-Mediated Mitochondrial Superoxide Promotes Cardiomyocyte Apoptosis and Impairs Cardiac Functions by Suppressing Pitx2. Circulation 2025, 151, 1024–1046. [CrossRef]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-Induced Oxidative Stress and Toxicity. J Toxicol 2012, 2012, 1–13. [CrossRef]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid Med Cell Longev 2015, 2015, 13. [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury through Mitochondrial ROS. Nature 2014, 515, 431–435. [CrossRef]
- Rezvani, M. Apoptosis-Related Genes Expressed in Cardiovascular Development and Disease: An EST Approach. Cardiovasc Res 2000, 45, 621–629. [CrossRef]
- Zhu, Q.; Luo, Y.; Wen, Y.; Wang, D.; Li, J.; Fan, Z. Semaglutide Inhibits Ischemia/Reperfusion-Induced Cardiomyocyte Apoptosis through Activating PKG/PKCε/ERK1/2 Pathway. Biochem Biophys Res Commun 2023, 647, 1–8. [CrossRef]
- Makin, G.; Hickman, J.A. Apoptosis and Cancer Chemotherapy. Cell Tissue Res 2000, 301, 143–152.
- Yaomura, T.; Tsuboi, N.; Urahama, Y.; Hobo, A.; Sugimoto, K.; Miyoshi, J.; Matsuguchi, T.; Reiji, K.; Matsuo, S.; Yuzawa, Y. Serine/Threonine Kinase, Cot/Tpl2, Regulates Renal Cell Apoptosis in Ischaemia/Reperfusion Injury. Nephrology (Carlton) 2008, 13, 397–404. [CrossRef]
- Mohassel, P.; Mammen, A.L. Anti-HMGCR Myopathy. J Neuromuscul Dis 2018, 5, 11–20. [CrossRef]
- Selva-O’Callaghan, A.; Alvarado-Cardenas, M.; Pinal-Fernández, I.; Trallero-Araguás, E.; Milisenda, J.C.; Martínez, M.Á.; Marín, A.; Labrador-Horrillo, M.; Juárez, C.; Grau-Junyent, J.M. Statin-Induced Myalgia and Myositis: An Update on Pathogenesis and Clinical Recommendations. Expert Rev Clin Immunol 2018, 14, 215–224. [CrossRef]
- Yi, S.A.; Sun, L.; Rao, Y.; Ordureau, A.; Lewis, J.S.; An, H. Activity-Based Probes and Chemical Proteomics Uncover the Biological Impact of Targeting HMG-CoA Synthase 1 in the Mevalonate Pathway. Journal of Biological Chemistry 2025, 301, 110660. [CrossRef]
- Theusch, E.; Kim, K.; Stevens, K.; Smith, J.D.; Chen, Y.-D.I.; Rotter, J.I.; Nickerson, D.A.; Medina, M.W. Statin-Induced Expression Change of INSIG1 in Lymphoblastoid Cell Lines Correlates with Plasma Triglyceride Statin Response in a Sex-Specific Manner. Pharmacogenomics J 2017, 17, 222–229. [CrossRef]
- Pramfalk, C.; Angelin, B.; Eriksson, M.; Parini, P. Cholesterol Regulates ACAT2 Gene Expression and Enzyme Activity in Human Hepatoma Cells. Biochem Biophys Res Commun 2007, 364, 402–409. [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.-Y.; Dillies, M.-A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS One 2016, 11, e0157022. [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol 2014, 15, 550. [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat Methods 2017, 14, 417–419. [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Res 2015, 4, 1521. [CrossRef]
- Wickham, Hadley. Ggplot2 : Elegant Graphics for Data Analysis; Springer, 2016; ISBN 9783319242774.
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package BiomaRt. Nat Protoc 2009, 4, 1184–1191. [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinformatics 2013, 14, 128. [CrossRef]
Figure 1.
Volcano plot of differentially expressed genes in carfilzomib-treated hiPSC-CMs compared with controls. Volcano plot illustrating transcriptome-wide differential gene expression following 24-hour treatment of hiPSC-CMs with 1 µM carfilzomib (CFZ). Each point represents a gene, plotted by log₂ fold change (x-axis) and –log₁₀(adjusted p-value) (y-axis). Significantly upregulated genes (log₂FC ≥ 1 and FDR < 0.05) are shown in red, while significantly downregulated genes (log₂FC ≤ –1 and FDR < 0.05) are shown in blue. Non-significant genes are visualized in grey. CFZ treatment induced substantial transcriptional remodeling, characterized by a large cluster of downregulated structural and sarcomeric genes, alongside marked upregulation of stress-response, proteostasis, and lipid-regulatory pathways. Threshold lines indicate log₂FC = ±1 and FDR = 0.05. Differential expression analysis was performed using DESeq2.
Figure 1.
Volcano plot of differentially expressed genes in carfilzomib-treated hiPSC-CMs compared with controls. Volcano plot illustrating transcriptome-wide differential gene expression following 24-hour treatment of hiPSC-CMs with 1 µM carfilzomib (CFZ). Each point represents a gene, plotted by log₂ fold change (x-axis) and –log₁₀(adjusted p-value) (y-axis). Significantly upregulated genes (log₂FC ≥ 1 and FDR < 0.05) are shown in red, while significantly downregulated genes (log₂FC ≤ –1 and FDR < 0.05) are shown in blue. Non-significant genes are visualized in grey. CFZ treatment induced substantial transcriptional remodeling, characterized by a large cluster of downregulated structural and sarcomeric genes, alongside marked upregulation of stress-response, proteostasis, and lipid-regulatory pathways. Threshold lines indicate log₂FC = ±1 and FDR = 0.05. Differential expression analysis was performed using DESeq2.
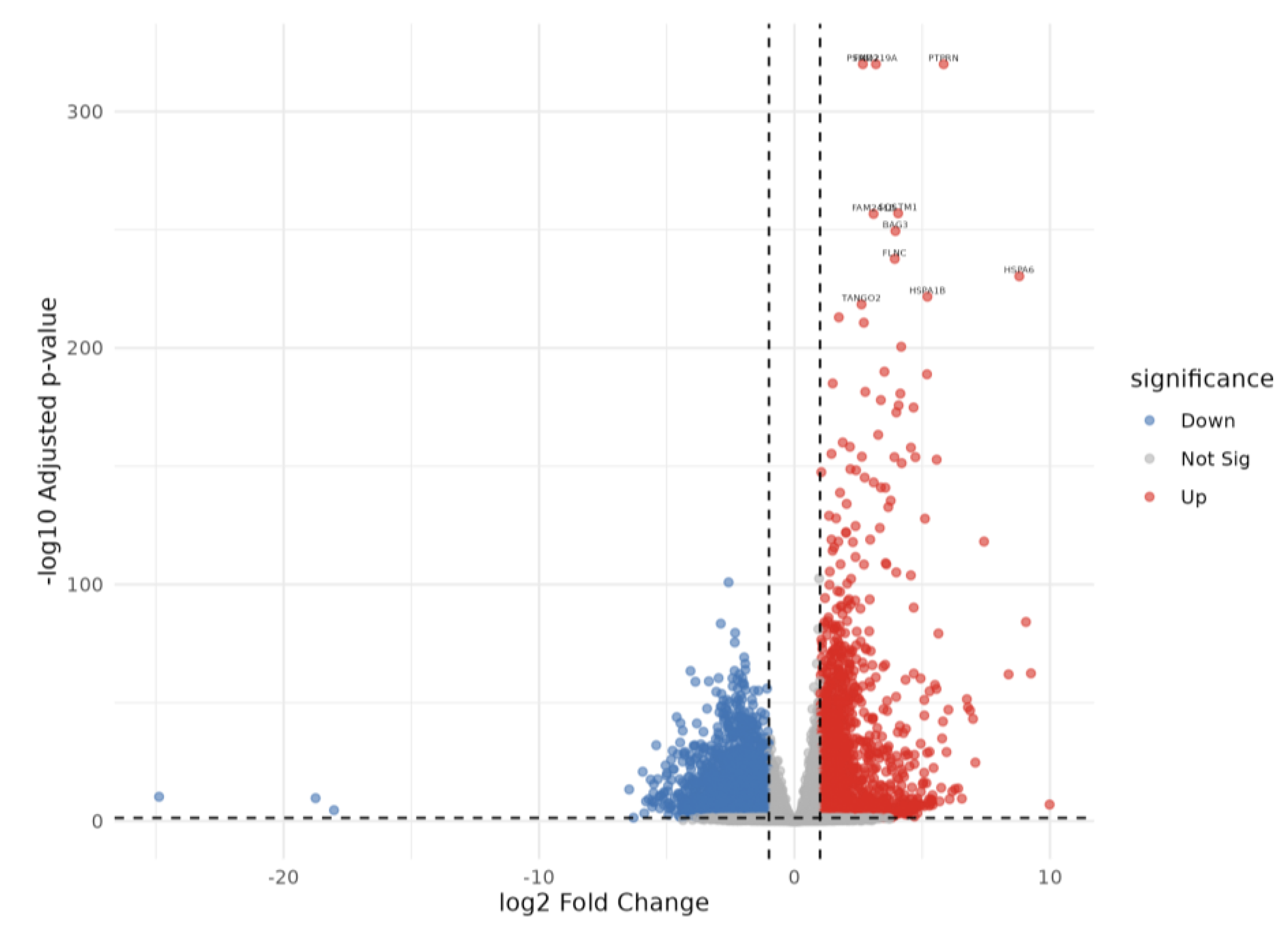
Figure 2.
Heatmap showing the top 50 differentially expressed genes between carfilzomib–treated and control hiPSC-derived cardiomyocytes. Rows represent genes and columns represent samples, with expression Z-score–scaled across samples. carfilzomib-treated cells cluster distinctly from controls and show marked upregulation of proteasome- and stress-response genes (e.g., PSMC5, PSMD1, HSPA1A) and downregulation of metabolic and regulatory genes. Batch annotations indicate that clustering is driven primarily by treatment conditions.
Figure 2.
Heatmap showing the top 50 differentially expressed genes between carfilzomib–treated and control hiPSC-derived cardiomyocytes. Rows represent genes and columns represent samples, with expression Z-score–scaled across samples. carfilzomib-treated cells cluster distinctly from controls and show marked upregulation of proteasome- and stress-response genes (e.g., PSMC5, PSMD1, HSPA1A) and downregulation of metabolic and regulatory genes. Batch annotations indicate that clustering is driven primarily by treatment conditions.
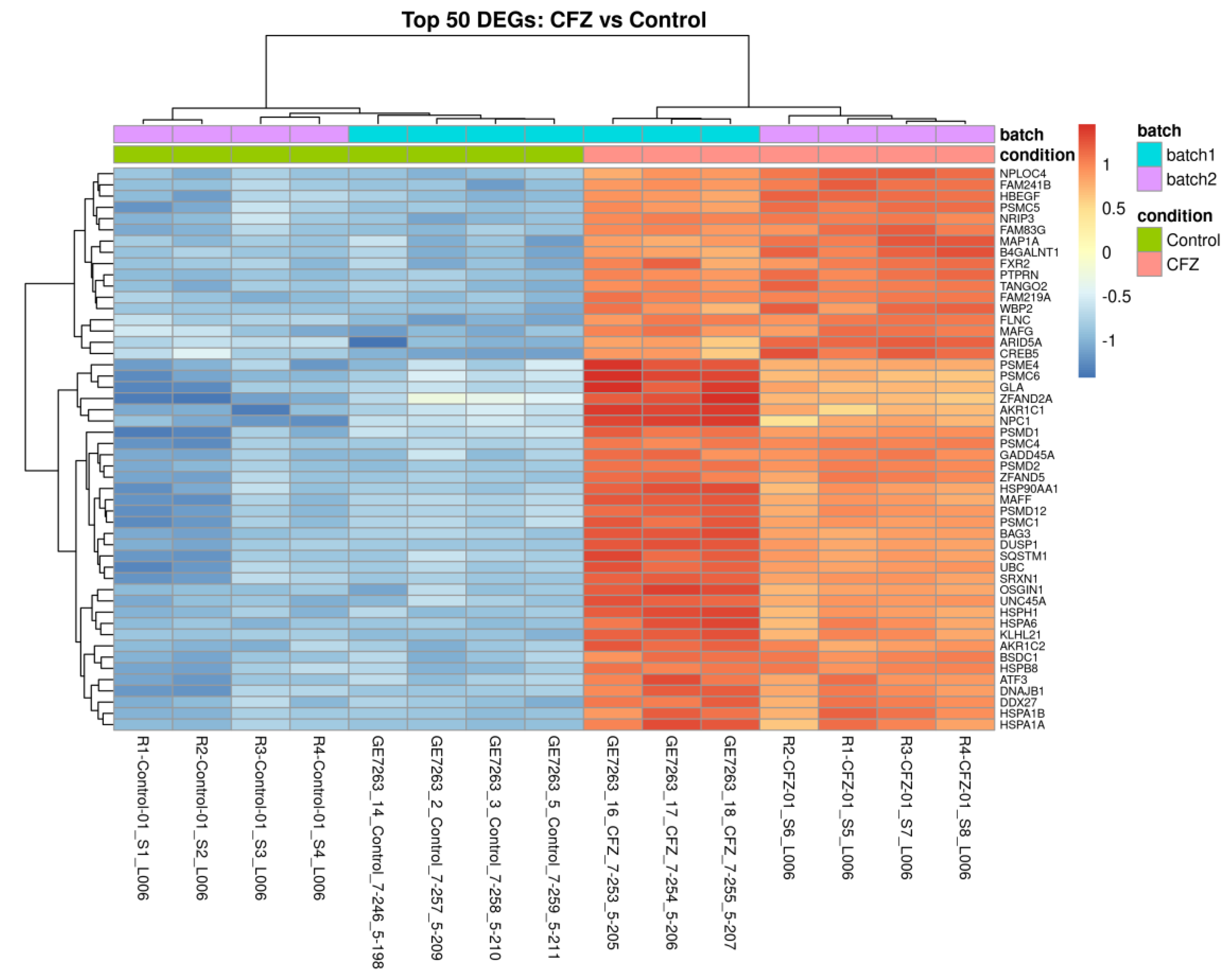
Figure 3.
Heatmap of the top 50 differentially expressed genes comparing carfilzomib + Atorvastatin–treated hiPSC-CMs to carfilzomib alone. Samples cluster distinctly by treatment, indicating a clear transcriptional shift with atorvastatin co-treatment. Key lipid-regulatory genes (HMGCR, HMGCS1, INSIG1, ACAT2) show marked upregulation with atorvastatin, reflecting activation of cholesterol and metabolic pathways, whereas sarcomeric and contractile genes (ACTA1, MYH11, MYLPF) remain suppressed. This pattern suggests that atorvastatin primarily restores metabolic processes without fully rescuing carfilzomib-induced structural gene downregulation.
Figure 3.
Heatmap of the top 50 differentially expressed genes comparing carfilzomib + Atorvastatin–treated hiPSC-CMs to carfilzomib alone. Samples cluster distinctly by treatment, indicating a clear transcriptional shift with atorvastatin co-treatment. Key lipid-regulatory genes (HMGCR, HMGCS1, INSIG1, ACAT2) show marked upregulation with atorvastatin, reflecting activation of cholesterol and metabolic pathways, whereas sarcomeric and contractile genes (ACTA1, MYH11, MYLPF) remain suppressed. This pattern suggests that atorvastatin primarily restores metabolic processes without fully rescuing carfilzomib-induced structural gene downregulation.

Figure 4.
Volcano plot highlighting differentially expressed genes between carfilzomib + atorvastatin vs carfilzomib-treated hiPSC-CMs compared with carfilzomib vs. controls. A subset of genes (e.g., ACAT2, DERL3, NDST4, PLA2G3, PAX2, TMEM178B) show opposite directional changes compared to carfilzomib alone, indicating partial reversal of carfilzomib-induced transcriptional effects by atorvastatin co-treatment, particularly in lipid metabolism and ER-stress–related pathways.
Figure 4.
Volcano plot highlighting differentially expressed genes between carfilzomib + atorvastatin vs carfilzomib-treated hiPSC-CMs compared with carfilzomib vs. controls. A subset of genes (e.g., ACAT2, DERL3, NDST4, PLA2G3, PAX2, TMEM178B) show opposite directional changes compared to carfilzomib alone, indicating partial reversal of carfilzomib-induced transcriptional effects by atorvastatin co-treatment, particularly in lipid metabolism and ER-stress–related pathways.
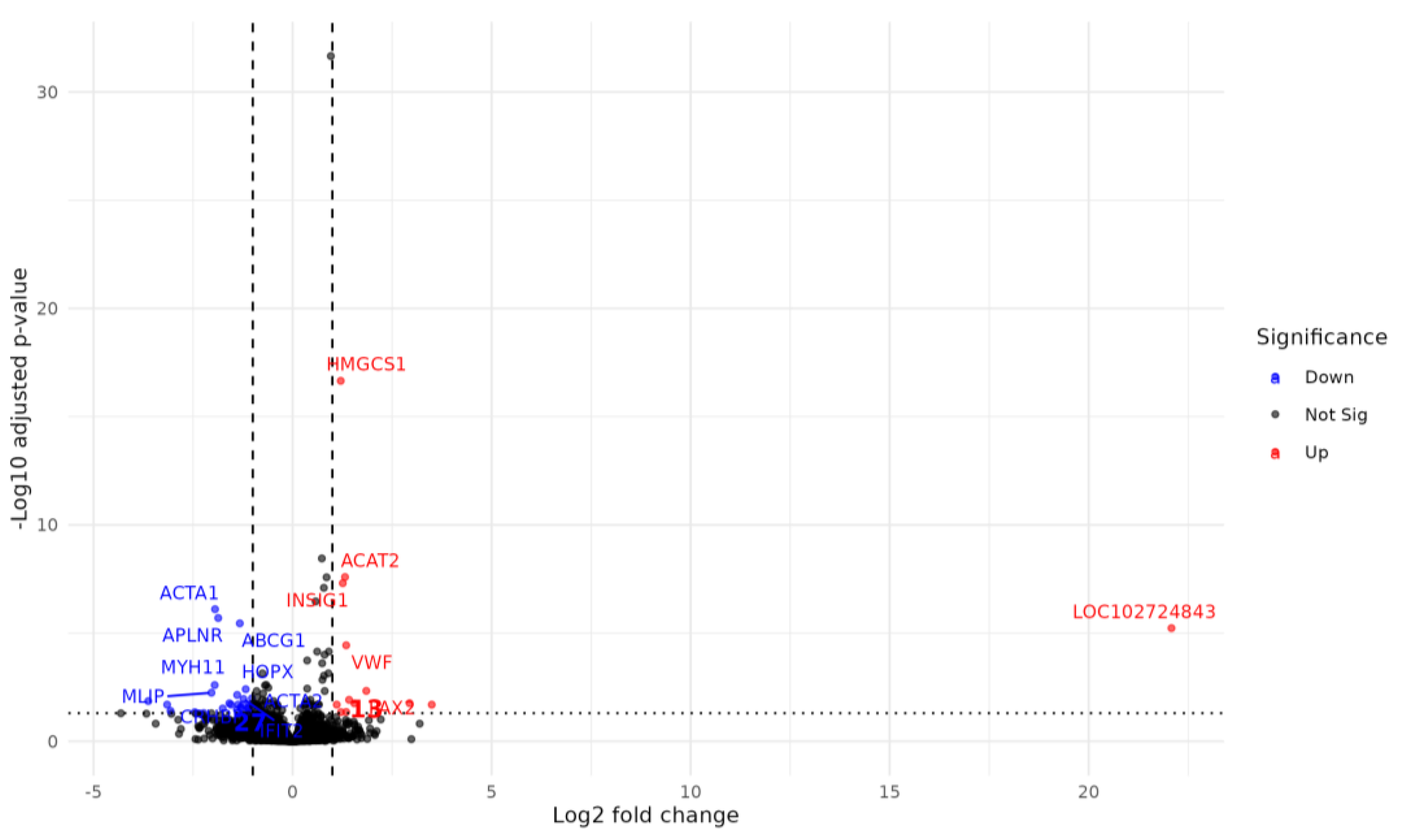
Figure 5.
Gene expression of top genes treated with control, carfilzomib, or carfilzomib + atorvastatin. a). ACAT2 gene; c). ACTA1 gene. CFZ: carfilzomib.
Figure 5.
Gene expression of top genes treated with control, carfilzomib, or carfilzomib + atorvastatin. a). ACAT2 gene; c). ACTA1 gene. CFZ: carfilzomib.
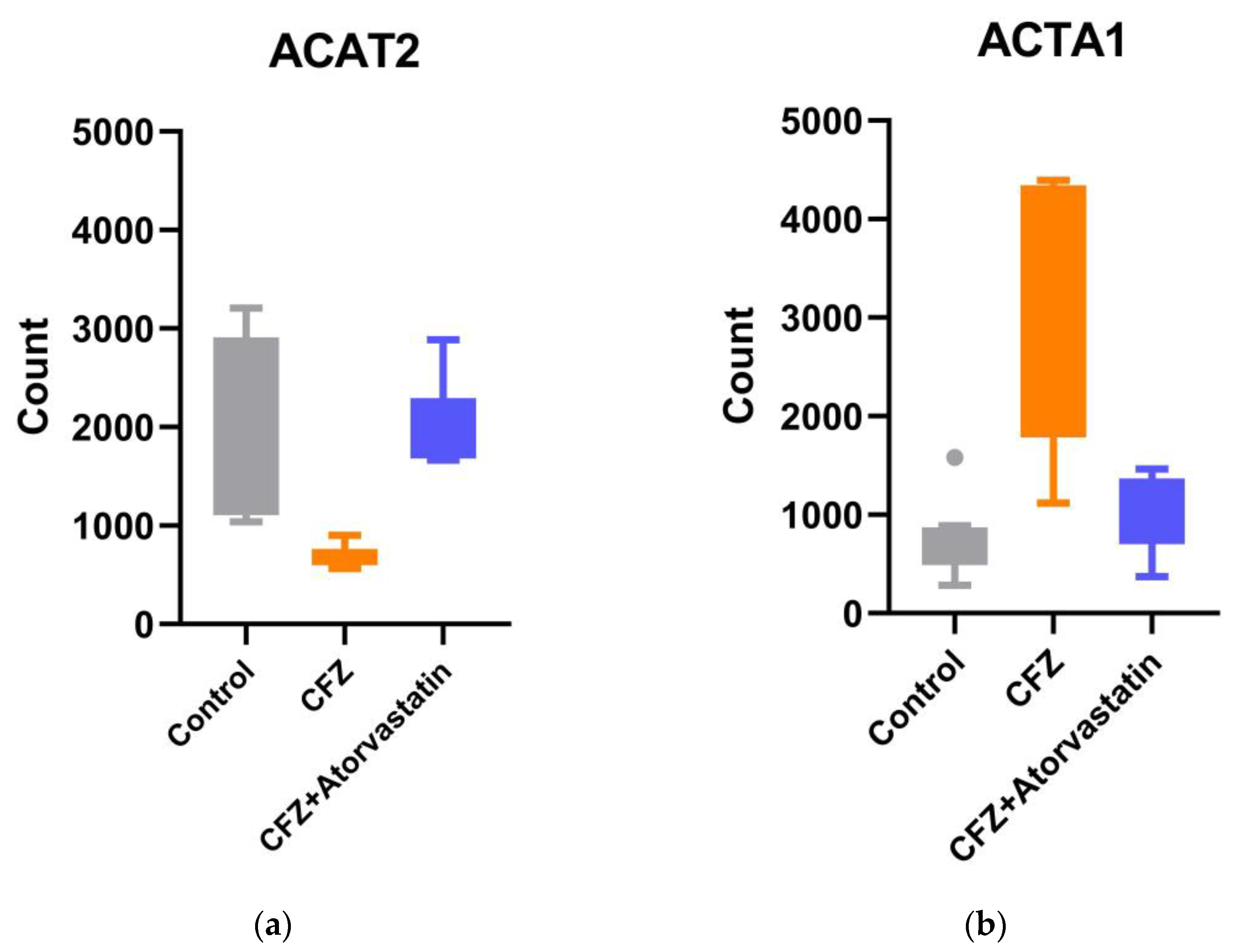

Table 1.
Differentially expressed genes reversed by atorvastatin in CFZ-treated hiPSC-CM.
| Gene Symbol | GENE Name | GENE ID | CFZ vs. Controls | CFZ + atorvastatin vs. CFZ | ||
| Log2 Fold Change | Padj | Log2 Fold Change | Padj | |||
| ACAT2 | Acetyl-CoA acetyltransferase 2 | ENSG00000120437 | -1.20 | 7.53E-10 | 1.32 | 2.57E-08 |
| ACTA1 | actin alpha 1, skeletal muscle | ENSG00000143632 | 2.31 | 1.68E-13 | -1.95 | 7.89E-07 |
| PAX2 | paired box 2 | ENSG00000075891 | -4.57 | 5.45E-29 | 1.85 | 0.0047 |
| CRHBP | corticotropin releasing hormone binding protein | ENSG00000145708 | 1.71 | 1.79E-07 | -1.39 | 0.0072 |
| IFIT2 | interferon induced protein with tetratricopeptide repeats 2 | ENSG00000119922 | 1.20 | 7.15E-05 | -1.23 | 0.011 |
| PLA2G3 | phospholipase A2 group III | ENSG00000100078 | -1.97 | 1.06E-08 | 1.42 | 0.012 |
| VSX2 | visual system homeobox 2 | ENSG00000119614 | -4.69 | 1.23E-10 | 2.94 | 0.017 |
| PCDHA2 | protocadherin alpha 2 | ENSG00000204969 | -1.20 | 0.0036 | 1.55 | 0.018 |
| PTGER2 | prostaglandin E receptor 2 (EP2) | ENSG00000125384 | 1.02 | 0.0148 | -1.59 | 0.018 |
| DERL3 | derlin 3 | ENSG00000099958 | -2.82 | 3.01E-25 | 1.11 | 0.020 |
| NDST4 | N-deacetylase/N-sulfotransferase 4 | ENSG00000138653 | -6.47 | 4.64E-14 | 3.50 | 0.020 |
| TMEM178B | transmembrane protein 178B | ENSG00000261115 | -2.28 | 4.35E-12 | 1.22 | 0.045 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



