Submitted:
22 December 2025
Posted:
23 December 2025
You are already at the latest version
Abstract
NK cells are key components of the innate immune system, capable of recognizing and eliminating tumor or virus-infected cells and able to modulate both innate and adaptive immune responses. This makes NK cells attractive candidates for cancer immunotherapy, through passive approaches such as adoptive NK-cell transfer, or active approaches aimed at enhancing endogenous NK-cell activity in vivo. Promising results have emerged from preclinical studies and early-phase clinical trials. Nevertheless, the therapeutic efficacy of NK cell–based approaches is often limited by several factors, such as the poor NK cell persistence in vivo, the inefficient tumor infiltration, and the immunosuppressive milieu typical of the tumor microenvironment. The preclinical development of NK cell–based therapies relies largely on animal models. Humanized mouse models have evolved from early immunodeficient strains to more advanced systems incorporating human cytokines, which more effectively support NK cell development, maturation, and function. These models have substantially improved our understanding of human NK cell biology and enabled the evaluation of novel therapeutic strategies. However, further optimization is still required to better recapitulate the tissue-specific heterogeneity of human NK cells and their conditioning by the tumor microenvironment. In this review, we provide an overview of recent advances in the generation of humanized mouse models for NK cell–based cancer immunotherapy, discussing their advantages and limitations and highlighting how emerging technologies may contribute to the development of more predictive preclinical platforms.
Keywords:
natural killer (NK) cells
; NK cell-based immunotherapy
; humanized mouse models
; cancer immunotherapy
; CAR-NK cells
; NK cell engagers (NKCEs)
; tumor microenvironment
1. Introduction
Natural killer (NK) cells are cytotoxic members of the innate lymphoid cell (ILC) family which play a central role in early defense against cancer and viral infections [1,2]. Upon recognition through a variety of activating receptors, NK cells eliminate transformed cells by releasing perforin- and granzyme-containing granules and through the activation of death receptor pathways, such as Fas/FasL, on transformed/infected cells. In addition to their cytolytic capacity, NK cells produce chemokines and cytokines, including IFN-γ, that shape and amplify both innate and adaptive immune responses [2]. NK cells originate from hematopoietic stem cells (HSCs) in the bone marrow (BM) and continue their maturation in secondary lymphoid tissues, following both linear and, as recently described, branched developmental pathways [3]. Moreover, NK cells include tissue-resident populations with distinct phenotypic and functional features, underlining the complexity of NK cell biology [4]. NK cell activity depends on a fine balance between inhibitory and activating receptors, which sense the presence of specific ligands on target cells [5]. Inhibitory receptors, such as Killer Ig-like Receptors (KIRs) and CD94/NKG2A, bind HLA class I molecules to ensure self-tolerance, driving NK cell “education”, and enabling competent effector functions [2,6,7,8]. Additional inhibitory pathways, including immune checkpoint receptors such as TIGIT, TIM-3, and PD-1, negatively regulate NK cell responses and are often exploited within the tumor microenvironment (TME) [8,9]. Conversely, activating receptors enable NK cells to recognize stress signals on transformed cells. Key activating receptors include natural cytotoxicity receptors (NCRs) (NKp30, NKp44, NKp46), NKG2D, activating KIRs, CD94/NKG2C, DNAM-1, and CD16A (FcγRIIIa), the latter mediating antibody-dependent cellular cytotoxicity (ADCC) [5,8,10]. The integration of these signals determines whether NK cells initiate cytotoxicity or cytokine production.
Human peripheral blood (PB) NK cells are classically divided into two major subsets: CD56bright cells, which are potent cytokine producers with limited basal cytotoxicity, and the more mature and highly cytotoxic CD56dim population, which expresses CD16A and higher perforin levels [11]. A third subset of tissue-resident NK cells is found in organs such as the BM, lymph nodes, spleen and liver [12,13] and is characterized by CD69 expression, and phenotypic and functional traits distinct from circulating NK cells [12,13,14]. Another specialized population of adaptive or memory-like NK cells emerges following chronic stimulation, such as by CMV infection, and displays enhanced responsiveness upon reactivation [8,15]. Recent single-cell multi-omics approaches have further expanded the understanding of NK cell heterogeneity, revealing multiple NK cell states with distinct transcriptional programs, metabolic profiles, and developmental trajectories, challenging traditional classifications based solely on surface markers. These studies also show that NK cell composition differs markedly across tissues and tumors, often being quite different from that observed in PB [3].
NK cell ability to eliminate malignant cells, combined with a favorable safety profile and minimal risk of graft-versus-host disease (GvHD) compared with T cell-based therapies [16,17,18] makes them strong candidates for both adoptive transfer and in vivo engagement strategies. Despite significant progress, the clinical translation of NK cell-based therapies, especially for solid tumors, still requires substantial optimization. A key obstacle is the difficulty of accurately modeling NK cell behavior in preclinical settings, including their persistence and trafficking in vivo, and interactions with human immune and stromal compartments. NK cell-based immunotherapies include a broad range of strategies, from adoptive passive approaches to active engagement modalities, and their fast validation depends on the availability of robust preclinical platforms. In this context, humanized mouse models have gained increasing attention. This review outlines the development, strengths, and limitations of current humanized mouse platforms and discusses how their refinement could improve their ability to anticipate clinical outcomes.
2. NK Cell-Based Immunotherapies in Oncology: Approaches, Mechanisms, and Challenges
NK cell ability to combine natural cytotoxicity with cytokine-mediated modulation of innate and adaptive immunity has made NK cells highly attractive tools in cancer immunotherapy [19,20,21,22]. Over the past decade, NK cell-based therapies have progressed from fundamental experimental approaches to clinically actionable platforms and can be broadly categorized into passive and active approaches [23]. Passive strategies involve the adoptive transfer into patients of in vitro cytokine-expanded and/or genetically engineered NK cells. These therapies use NK cells from healthy donors (HDs) or from cancer patients providing recipient patients with a population of highly cytotoxic and viable NK cells [24,25]. Active strategies, on the other hand, are mainly aimed at harnessing and potentiating the patient’s own endogenous NK cells, and can include the administration of molecular engagers such as monoclonal antibodies (mAbs) [26], bispecific (BiKEs) or tri-specific (TriKEs) NK cell engagers [27,28,29], and the recently described tri- or tetra-specific Antibody-based NK cell Engager Therapeutics (ANKETs) [30,31,32]. All these tools are designed to be administered to patients, facilitating in vivo NK cell activation and tumor recognition.
The distinction between passive and active strategies reflects key differences in mechanistic approach, scalability, and translational challenges. Passive therapies allow to precisely define the NK cell product in terms of cell number, phenotype, grade of activation and genetic modifications, supporting the creation of tailored products. Some examples of genetic manipulation are Chimeric Antigen Receptor (CAR)-NK [33,34] or induced pluripotent stem cell (iPSC)-derived NK cells [35]. On the contrary, active strategies exploit existing NK pools, avoiding the complexities of cell manufacturing and potentially promoting more durable in vivo responses. Despite differences, both approaches share similar hurdles, such as the NK cell trafficking to tumor sites and the immunosuppression occurring in the tumor TME [36]. Understanding the strengths and limitations of passive and active strategies is therefore critical for designing efficient patient treatments, and innovative preclinical models are necessary to analyze the efficacy and compare different therapeutic strategies.
2.1. Passive Strategies: Adoptive NK Cell Transfer
For passive strategies, the main challenge is certainly to obtain an adequate NK cell product in terms of number, functionality, and in vivo persistence [37,38,39]. Thus, technological advances, including optimized cytokine-based expansion protocols [40,41], feeder-cell co-culture systems, and gene engineering [42], are being used to transform NK cells into a viable therapeutic platform (Figure 1). Multiple sources of NK cells are now employed: the most common source is represented by the PB of patients or HDs, but additional sources are umbilical cord blood (UBC), BM, and human embryonic stem cells (hESCs) or iPSC [43,44,45]. Additionally, the NK cell product can be represented by immortalized NK cell lines such as NK-92 [46]. iPSC-derived NK and NK cell lines in particular offer unlimited scalability, standardized phenotypes, and the possibility of complex genetic modifications, thus representing a promising tool for generating “off-the-shelf” therapies that can be consistently manufactured with defined functional properties [47]. Ex vivo cytokine stimulation remains central in adoptive NK cell therapy. The cytokines used span from the seminal interleukin-2 (IL-2) [48] to IL-15 or IL-21, which play key roles in NK cell differentiation, proliferation, survival, and activation [49,50,51]. Special attention was paid to IL-15 due to reduced toxicity as compared to IL-2 and its ability to support NK persistence without expanding regulatory T cells. Along this side, IL-15 super-agonists (for example, N-803/ALT-803) have been developed [53]. Interestingly, combinations of IL-12 and IL-18 can generate cytokine-induced memory-like NK cells, which exhibit enhanced cytotoxicity and prolonged activity in vivo [53,54]. Nonetheless, in clinical and preclinical studies, in vivo long-term persistence beyond two to three weeks remains uncommon [55,56], and trafficking into solid tumor sites is frequently inefficient [57], highlighting an important weakness in current passive NK cell-based therapies.
Genetic engineering has enabled a new generation of adoptive NK cell-based therapies, most notably the infusion of CAR-NK cells. These products combine the innate antigen-specific recognition and cytotoxic potential of NK cells with an additive mechanism represented by the expression of an engineered tumor-targeting chimeric receptor [17,58]. For instance, adult PBMCs and UBC-derived CD19 CAR-NK cells have demonstrated promising clinical efficacy in hematologic malignancies, achieving durable responses without severe cytokine release syndrome or neurotoxicity [59,60]. Further engineering strategies aim to enhance CAR-NK cell persistence, improve tumor homing, and overcome inhibitory pathways. In this context, it was shown that co-expression of IL-15 [61] or IL-21 promotes CAR-NK expansion and survival [62].
Beyond CAR-based approaches, additional genetic engineering strategies are being explored to enhance NK cell efficacy. For example, the introduction of a gene coding for chemokine receptors (e.g., CXCR4, CCR7, or CCL21) facilitates trafficking to tumor sites [63,64,65]. Moreover, the CRISPR/Cas9-mediated knockout of inhibitory receptors such as NKG2A or TIGIT is also being explored [66,67,68].
Finally, iPSC-derived NK cells represent an especially versatile platform, allowing for a scalable and standardized “off-the-shelf” product [35,69]. Unlike primary NK cells, which show a huge donor-related heterogeneity and limited expansion capacity, homogeneous iPSC-NK cell products can be obtained, addressing the reproducibility of the results and enabling complex genetic engineering. In recent years, several studies have highlighted the potential of these cells in advanced preclinical models. For example, CRISPR/Cas9-mediated deletion of the CISH protein generated iPSC-NK cells with enhanced JAK-STAT signaling, improved proliferation under low cytokine conditions, in vivo persistence, metabolic fitness, and resistance to functional exhaustion [70]. Similarly, overexpression of CD226 enhanced cytotoxicity against acute myeloid leukemia (AML) cells, demonstrating how targeted activation receptor modifications can improve the antitumor efficacy [71]. Another innovative approach involved engineering strategies to obtain a high-affinity Fc receptor, such as that derived from the CD64/CD16A fusion. The expression of this construct, integrated with IL-15 coding modules, allowed iPSC-NK cells to be preloaded with tumor antigen-specific mAbs for flexible targeting of multiple tumors, including lymphoma. Importantly, iPSC-derived NK cells expressing the CD64/16 FcγR showed cytotoxic activity preserved after cryopreservation [72].
More recently, engineering strategies have combined CCL19, CCR2B, NKG2D, high-affinity CD16A, and IL-15 into a single iPSC-NK line, resulting in cells with improved tumor infiltration and enhanced antitumor activity in solid tumor models [73]. These studies underscore how the iPSC-NK platform represents a rational, flexible engineering strategy, paving the way for advanced pre-manufactured NK cell therapies in cancer immunotherapy. However, limitations for the use in vivo of iPSC-NK are present, including the incomplete terminal maturation with reduced cytotoxicity and cytokine production, still poorly characterized differences among different iPSC clones, and their potential dangerousness due to genomic instability with the risk to acquire chromosomal abnormalities or mutations during reprogramming and in vitro expansion [47,69]. Moreover, iPSC-based strategies require complex manufacturing and high costs.
Concerning the NK cell differentiation, it is worth noting that in vivo, the physiologic process requires key cell-to-cell signals provided by stromal cells. In their absence, the NK cell maturation trajectory and/or their functional programming may be distorted. Future refinement of iPSC differentiation protocols, incorporation of human stromal co-culture systems, and engineering of more physiologic cytokine niches within humanized mice will be fundamental to improving the translational efficacy of iPSC-NK platforms.
2.2. Active Strategies: NK Cell Engagers (NKCEs)
While passive therapies deliver an in vitro optimized NK cell product, active strategies aim to potentiate/expand endogenous NK populations in vivo through targeted molecular engagement. These approaches include chimeric or humanized mAbs that block inhibitory axes such as immune checkpoints (IgG4 mAbs) or trigger ADCC (IgG1 mAbs), as well as engineered tools such as BiKEs, TriKEs, and ANKETs, which engage multiple NK receptors and tumor antigens (Figure 1). Active strategies are very appealing because they do not require specialized infrastructure for cell manipulation. Together with their scalability and their potential to engage NK cells across multiple tissue compartments, they are among the therapeutic strategies with the broadest applicability to date.
mAbs such as Rituximab (anti-CD20), Trastuzumab (anti-HER2), and Cetuximab (anti-EGFR) induce ADCC by engaging NK cells expressing CD16A, which bind the Fc region of IgG antibodies, thereby triggering degranulation and tumor cell lysis [74,75]. Next-generation Fc-engineered antibodies, such as Margetuximab (anti-HER2), [76] or afucosylated antibodies, such as Obinutuzumab (anti-CD20) [77], have been shown to have good clinical efficacy by enhancing NK cell activation in both hematological and solid tumors [78,79,80]. Moreover, the association of these ADCC-optimized antibodies with pro-inflammatory cytokines (e.g., IL-15 superagonists) [52,81,82] or checkpoint blockade (e.g., anti-NKG2A [83]) further boosts their efficacy. However, it must be considered that CD16A expression is not a phenotypic trait proper to all human NK cells. In fact, while approximately 90% of blood NK cells in the adult HDs expressed CD16A, in other tissues, its expression is confined to a variable percentage of cells, which in some cases could be very low [3]. This prompted researchers to develop bi/tri/tetra-specific NK cell engagers able to overcome this limitation.
With these premises, several groups have started investigating NKCEs in solid cancers, using their modular structure to combine the engagement of NK activating receptors with the recognition of tumor-associated antigens (TAAs) in epithelial malignancies [31]. Although still early, these studies underscore the versatility of NKCEs and their potential to extend NK-cell-based immunotherapy beyond onco-hematological disease.
These multi-specific molecules are engineered tools combining different single-chain variable fragment (scFv) portions of mAbs specific for different NK activating receptors (such as CD16A, NKp30, NKp46, NKG2D) and TAAs, forming a functional effector/target bridge that activates NK cell cytotoxicity [31,32,84,85,86]. Preclinical studies of CD16A×CD33 [28,87] or CD16A×CD123 BiKEs [88] demonstrated effective activation of NK cells against primary leukemic and myelodysplastic stem and progenitor cells, highlighting their potential as immunotherapeutic strategies for AML, myelodysplastic syndrome and acute lymphoblastic leukemia (ALL). In a separate study, dual targeting of glioblastoma cells using bispecific killer cell engagers directed at EGFR and HER2 enabled efficient killing by NKG2D-CAR-engineered NK cells [89]. Recent work has expanded the specificity of NKCEs, providing proof of concept that multi-specific molecules can be tailored toward diverse TAAs and employed across different oncological settings. TriKEs frequently incorporate an IL-15 moiety, sustaining NK proliferation and cytotoxic function during engagement [90,91]. TriKEs have been shown to induce higher IFN-γ production than BiKEs, indicating stronger NK cell activation and tumor control in preclinical mouse models. Importantly, the degree of cytokine release remains within a favorable safety profile. In pediatric B-cell precursor-ALL, NKCEs triggering either NKp46 or NKp30, in combination with CD16A, and targeting CD19 or CD20 on tumor cells, demonstrated potent enhancement of NK cell degranulation and IFN-γ production [92]. More recently, Gauthier et al. reported a trifunctional NKCE targeting CD123 in AML and co-engaging NKp46 and CD16A on NK cells [86]. This NKCE was shown to overcome CD64-mediated resistance: the high-affinity IgG receptor CD64 sequesters conventional anti-CD123 IgG1 mAbs, rendering them effective only against CD64⁻ target cells. In contrast, the CD123-NKCE retained its activity regardless of CD64 expression. Moreover, it induced NK cell activation only in the presence of AML cells and maintained a safe inflammatory and cytokine-release profile. In fact, in xenogeneic and non-human primate studies, it provided enhanced disease control, prolonged depletion of CD123⁺ cells, and no detectable toxicity, supporting its progression toward clinical development [86].
Novel anti-CD20 tetrameric NK cell engagers (ANKETs) have been recently validated in preclinical models. These molecules are designed to selectively co-engage on NK cells NKp46, CD16A, through an Fc portion, and the IL-2Rβγ chain through an IL-2 variant (IL-2v), while simultaneously targeting CD20, to enable directed cytotoxicity. IL-2v carrying a point-mutation does not bind to the IL-2Rα (CD25) chain, constitutively expressed by regulatory T cells (Tregs) and by other activated cells, including endothelial cells, thus limiting Treg expansion and other adverse effects such as pulmonary edema [93,94]. aCD20/aNKp46/Fc/IL2v tetra-specific ANKETs activate and expand NK cells in vitro and promote their accumulation within tumors in vivo, resulting in superior tumor control in aggressive lymphoma mouse models compared with conventional antibodies such as the anti-CD20 Obinutuzumab. Importantly, in non-human primates, the molecule induced B-cell depletion with only minimal and transient cytokine release, indicating a favorable safety profile [32]. Based on that, Demaria et al. focused on the detailed characterization of IPH6501, a clinical-grade aCD20/aNKp46/Fc/IL2v ANKET developed for the treatment of B-cell non-Hodgkin lymphoma (B-NHL) [95]. This study reinforced the earlier results by demonstrating superior cytotoxic activity in vitro compared with both therapeutic anti-CD20 IgG1 antibodies (Rituximab and Obinutuzumab) and CD20-specific T-cell engagers (TCEs). Moreover, at comparable functional doses, IPH6501 induced lower and transient levels of pro-inflammatory cytokines (TNF-α and IL-6) than CD20-TCEs, suggesting a reduced risk of cytokine release syndrome (CRS). In addition, single-cell RNA-seq (scRNA-seq) demonstrated that the treatment activates all major NK cell subsets, favoring the emergence of Cytokine-Induced Memory-Like (CIML) NK cells, known for potent antitumor activity and persistence. Finally, IPH6501 induced an antigen-independent antitumor activity by upregulating the expression of key activating receptors such as NKG2D. This effect may be beneficial, as it could exploit the natural NK cytotoxicity, helping to counteract the survival and expansion of CD20-negative tumor variants that commonly emerge in patients following CD20-targeted immunotherapies [95].
Collectively, these results highlight ANKETs as versatile next generation immunotherapeutic agents capable of harnessing NK cells for potent yet safe antitumor responses. NKCEs show a favorable safety profile also because NK cells produce fewer pro-inflammatory cytokines than T cells and are mainly activated in the presence of target tumor cells. Despite these advantages, both passive and active NK cell-based strategies face key limitations: limited NK cell persistence and trafficking in vivo, manufacturing complexity and immunogenicity risks for passive approaches, and reduced responsiveness of patient NK cells or their suppression in the TME for active ones. The limited clinical evidence, particularly in solid tumors, calls for caution and underscores the need for preclinical models that better recapitulate human NK cells and the TME [96,97,98,99]. The following chapters provide an overview of efforts addressing these challenges.
3. Preclinical Humanized Mouse Models for Assessing NK Cell-Based Strategies
3.1. Early Immunodeficient Models
The seminal approaches for the preclinical evaluation of NK cell therapy through animal models comprised immunodeficient strains such as Non Obese Diabetic/Severe Combined Immunodeficient (NOD/SCID) mouse [100], NOD/SCID/γcnull (NSG) mouse [101], and NOG mouse (NOD/Shi-scid/IL2Rγnull) [102] which lack functional T, B and NK cell compartments (by virtue of IL2Rγ knockout) (Figure 2). These mice allowed the xenotransplantation of human tumor cells, including those patient-derived (PDX models), and intravenous injection of human immune effectors, including NK cells from HDs or engineered NK cell lines, enabling the first evaluation of human tumor engraftment and the NK cell-mediated anti-tumor activity [103,104,105,106,107].
Nevertheless, these models exhibited major translational gaps. One key limitation is the poor interspecies cross-reactivity, the lack of homologous cytokines, such as IL-15, IL-21, able to efficiently sustain human NK cell differentiation and activation. Consequently, human NK cells engrafted in such hosts are short-lived and exhibit limited expansion or functional maturation [108,109]. In addition to that, the human stromal and vascular microenvironment is progressively replaced by host-derived murine stroma, which differ from human counterparts in extracellular matrix (ECM) composition and signaling networks. These differences result in non-identical chemokine gradients and signaling networks compared with human tumors, potentially influencing immune cell trafficking cues, including those relevant for NK cell recruitment and function [110,111,112,113,114,115]. Furthermore, in vitro documented crosstalk between human NK cells and other immune lineages, such as dendritic cells, macrophages, and T cells, cannot be modelled in these animal models.
3.2. Progress Towards the First Humanized Immunodeficient Models to Study Human NK Cells
To address some of the above-mentioned issues, the field has advanced to generate humanized mouse models in which human hematopoietic stem cells and progenitor cells (HSPCs, CD34⁺) are engrafted into immunodeficient recipients, thereby generating a full human immune system within the murine host. These HSPC-engrafted humanized systems are more suitable for investigating not only the NK cell anti-tumor activity but also NK cell ontogeny, differentiation, trafficking, and infiltration into human xenografted tumors.
The most widely used humanized mouse models are NSG-SGM3 [116], MISTRG [109], SRG-15 [117], and NSG-IL15 models [118]. Specifically, NSG-SGM3 mice are based on the highly immunodeficient NSG background and are transgenic for human SCF, GM-CSF, and IL-3; these cytokines enhance the development of human myeloid lineages, which in turn support NK cell maturation and function, although the endogenous levels of human IL-15 are still scarce [116]. MISTRG mice (M-CSFh/h IL-3/GM-CSFh/h SIRPαh/h TPOh/h RAG2-/- IL2Rγ-/-) incorporate knock-ins of human M-CSF, IL-3/GM-CSF, TPO (thrombopoietin), and SIRPα in mice deficient for T/B and NK cells, enabling efficient development not only of human myeloid cells but also of NK cells that display maturation, cytotoxicity, and tissue distribution more closely resembling that in humans [109]. In this model, the human engraftment is favored by the presence of transgenic human SIRPα, which provides a “don’t eat me signal” reducing the phagocytosis of human stem cells and developing an immune compartment.
In SRG-15 mice having a RAG2⁻/⁻ IL2Rγ⁻/⁻ background, the gene coding for the murine IL-15 is replaced with the human counterpart, along with a knock-in of the human SIRPα. This results in a physiological expression of IL-15 in a tissue- and cell-specific manner, which results in robust populations of human NK cells trafficking, infiltrating xenografted tumors and interacting with human T cells, dendritic cells and macrophages [117]. In particular, in SRG-15 mice, de novo-derived human NK cells have been shown to infiltrate lymphoma xenografts and mediate ADCC following Rituximab treatment.
Finally, in NSG-IL15 mice, human IL-15 is constitutively expressed (via transgenic or knock-in design), and provide particularly robust human NK cell reconstitution: high frequencies of mature and functional NK cells across multiple tissues, support for long-term survival without inducing GvHD, and enable physiologically relevant NK-mediated control of both tumor and infections [118,119,120]. Experiments on patient-derived melanoma in NSG-IL15 mice showed slower tumor growth compared to NSG mice, indicating that the enhanced NK-cell compartment confers a significantly improved tumor-control effect [119].
Thus, growing evidence pointed out the superior reliability of humanized mouse models for studying human NK cell behavior in cancer, as compared with first-generation standard models such as NOD/SCID or NSG mice.
4. Burning Challenges and Limitations of Current Humanized Mouse Models for Studying NK Cell-Based Therapies
Despite the described progress in the establishment of preclinical humanized mouse models, significant limitations remain, primarily due to inter-species differences in immune system composition, cytokine signaling, tissue microenvironments, and NK cell biology. These discrepancies substantially affect the interpretation and the translation of preclinical findings, limiting the accurate prediction of the immunotherapies’ benefits [121,122,123]. In healthy humans, PB-NK cells constitute approximately 5–15% of circulating lymphocytes, with a major proportion (~90%) of highly cytotoxic CD56dim cells. A minor fraction (~10%) is represented by poor cytotoxic and cytokine-secreting CD56bright cells [124,125]. As previously mentioned, CD56dim NK cells mediate direct cytotoxicity via perforin and granzymes, whereas CD56bright NK cells mainly orchestrate innate and adaptive immune responses through cytokine and chemokine secretion, influencing dendritic cell (DC), macrophage [126] maturation and T-cell priming [2,127].
In most humanized mouse models, the peripheral NK compartment not only is consistently under-represented but also does not mirror the human distribution between the two main subsets [107,119,128]. For example, NSG mice engrafted with human CD34⁺ HSCs typically show a predominance of CD56bright NK cells in the peripheral blood, whereas the CD56dim/CD16⁺ subset is virtually absent. On the contrary, the more complex NSG-Tg(Hu-IL-15) model [119] supports the development of both CD56bright and CD56dim NK cell compartments, resulting in a significantly improved representation of circulating human NK cells. Although this model does not fully recapitulate the human NK cell scenario, it more closely resembles the relative proportions of the major circulating NK cell subsets observed in humans. Moreover, NSG-Tg(Hu-IL-15) mice showed higher levels of functional granzyme A/B+, perforin+ NK cells in both PB and spleen compared to NSG model. Even more advanced models such as the previously described MISTRG model, despite representing a significant improvement with respect to first-generation humanized models in supporting human NK cell development, NK cell maturation, tissue distribution and functional diversity, remain incomplete and highly dependent on additional human cytokine support. Thus, nowadays we are far to have a mouse model that fully recapitulates the human NK cell compartment [108,117,129].
In fact, human NK cells exhibit a huge tissue heterogeneity, with specific subsets residing in secondary lymphoid organs and peripheral tissues [3,5,130,131,132]. For instance, liver-resident CD56brightCD69+CXCR6+ NK cells display low cytotoxicity but high cytokine output, contributing to immune surveillance and tolerance [12,131,133,134]. Lung NK cells acquire adaptive-like, memory features following viral exposure [135,136], whereas uterine NK cells play critical roles in vascular remodeling during pregnancy [137,138]. Lymphoid tissue NK cells regulate DC maturation and T cell priming, demonstrating the interplay between NK cells and adaptive immunity [139,140,141].
Conventional humanized mouse models fail to faithfully reproduce this tissue-specific heterogeneity because the signals required for tissue-resident NK differentiation, such as local cytokines, adhesion molecules, and stromal cues, are largely absent due to the lack of interspecies cross-reaction [110,112,113,114]. In fact, in humanized mice, multiple species-specific mismatches exist. Stromal and tissue-resident immune compartments remain predominantly murine. For instance, murine IL-15 poorly bind human receptors, leading to incomplete NK maturation, reduced survival, and impaired cytotoxicity [119,142]. Consequently, humanized mice harbor PB-like CD56dim NK cells in circulation but virtually lack tissue-resident NK phenotypes. These data indicate that conventional humanized mice inadequately model organ-specific NK cell properties, severely limiting studies on solid tumors where tissue-resident NK cells are critical for controlling tumor growth and metastasis.
Cytokine-humanized mouse models improved human NK cell development. However, the various strategies used to introduce human cytokines can generate expression patterns that deviate from physiological conditions, sometimes resulting in temporarily abnormally higher systemic cytokine levels. This can lead to altered hematopoiesis and over-activation of NK cells, which also present defects in maturation and licensing [114,143,144]. Thus, cytokine-engineered models only partially correct NK developmental defects, and the cytokine environments they establish may influence NK homeostasis, potentially obscuring processes such as exhaustion or the switch on of negative regulatory programs.
As previously noted, cross-species differences in the chemokine compartment represent another limiting factor. Mice and humans differ in the presence and regulation of chemokines (e.g. absence of CXCL8 in mice, differential CXCL9/10/11 and CCL5 signaling), leading to a non-physiological NK recruitment, retention, and organ-specific trafficking [112,113]. Importantly, the human NK cell “education” or “licensing”, a process that leads to a progressive acquisition of the cytolytic machinery, relies on interactions of NK cells with autologous HLA class I+ DCs and macrophages. This process is poorly recapitulated in the presence of murine myeloid cells, leading to incomplete licensing, altered receptor expression, and reduced secretion of IFN-γ and cytotoxic granules [128,143,145,146]. In many humanized tumor models, the human stromal compartment is progressively replaced by murine stromal cells (fibroblasts, ECM, vasculature, other stromal elements), thereby altering the cellular and molecular composition of the TME [147,148,149]. Given species-specific differences in stroma-derived signals (e.g., cytokines, growth factors), this replacement may compromise the fidelity of cancer–stroma and immune–stroma interactions present in the human TME. Even if direct experimental data comparing oxygen, nutrient, or metabolite gradients between human tissues and murine-stromal xenografts remain unexplored, it can be hypothesized that such stromal replacement could produce altered metabolic landscapes with potential consequences for the metabolic adaptation, survival, trafficking, and effector function of human NK cells engrafted in humanized mouse models. Therefore, when interpreting results from NK cell-based immunotherapy studies in in vivo models, the impact of stromal replacement should be carefully considered. Most xenografted humanized mouse systems lack human antigen expression on normal murine tissues [123,150]. Tissue-humanized mouse models, such as humanized liver, have been developed; however, they remain technically challenging and often preserve murine tissue/metabolism, limiting their utility for informative organ-specific toxicity studies. Simultaneous humanization of hematopoietic and non-hematopoietic tissues remains largely incomplete [151]. The limited reconstitution and the poor tumor infiltration of functional human NK cells in conventional mouse models [152], together with the progressive replacement of human stromal components by murine cells in humanized models [114], have historically hampered the right evaluation of the negative impact of the TME-mediated immunosuppression in patients. In fact, phenotypic and functional profiling of tumor-resident NK cells in humans has clearly demonstrated the presence of exhausted cells, expressing high levels of inhibitory immune checkpoint receptors (e.g., PD-1, TIGIT, TIM3), downregulation of activating (e.g., NKp30, NKG2D, and DNAM-1), and enriched in the CD16A- population [153,154,155,156,157]. Thus, ideally, only the reproduction of these NK cell properties in the preclinical mouse model could allow a reasonable evaluation of therapies’ efficacy.
In this context, MISTRG-6-15 mice with knocked-in human IL-6 and IL-15 provide robust support for the expansion of IL-15-armored or CAR-engineered NK cells and have been used to study NK cell exhaustion in the settings of chronic infections such as HIV. This study showed that NK cells significantly contribute to anti-HIV-1 responses in vivo, but this activity was reduced in lymphoid organs, suggesting a tissue-specific responsiveness of NK cells to certain therapies [158]. So far, published studies specifically evaluating NK cell exhaustion in tumor-bearing MISTRG-6-15 mice are still absent.
Recent evidence indicates that sustained CAR-NK activation, driven by continuous CAR signaling through the CD3ζ domain or by elevated levels of IL-15, upregulates the transcription factor CREM, which functions as a negative regulator. CREM induction is primarily mediated by the PKA–CREB axis, activated downstream of ITAM-dependent CAR signaling as well as IL-15 stimulation. CREM drives epigenetic and transcriptional reprogramming of CAR-NK cells, repressing key effector genes such as GZMB and IFNG while promoting exhaustion- and stress-associated programs. Together with that, engineered CAR-NK cells co-expressing soluble IL-15 have been linked to severe toxicity in animal models, as systemic accumulation of IL-15 can trigger uncontrolled CAR-NK expansion. Safer strategies such as membrane-bound IL-15 provide spatially restricted and more physiologic cytokine signaling: it enhances the efficacy and persistence of NK cells while maintaining a favorable safety profile, avoiding uncontrolled proliferation, acute toxicity, and organ damage associated with soluble IL-15 [159,160,161].
In summary, current humanized mouse models, although providing significant help for the study of NK cell-based immunotherapies, still demonstrate critical limitations in mirroring human NK biology. This underscores the need for next-generation approaches, including multiple knock-in strategies, organoid integration, iPSC-derived NK cells, and CRISPR-based tissue humanization, to enhance NK maturation, preserve tissue heterogeneity, enable functional plasticity, and improve the translational relevance of preclinical studies.
5. Next-Generation Humanized Mouse Models and Innovative Technologies Unraveling Human NK Cell Dynamics in Tumor Immunity
As mentioned above, NSG mice expressing transgenic human IL-15 exhibit improved development, survival, and in vivo function of NK cells [118,119,120]. Nevertheless, it was demonstrated that a constitutive IL-15 expression could drive supra-physiological NK-cell expansion, which can obscure emerging exhaustion and checkpoint regulation, and has also been shown to promote functional NK-cell dysfunction [162].
To address these potential drawbacks and recognizing that insufficient human cytokine support was a major bottleneck for human NK cell development in vivo, more recent cytokine-enhanced humanized strains have incorporated additional human cytokines to further improve NK-cell robustness. Beyond the strains already described in Section 2.2, expanded cytokine knock-in models such as NSG-hIL7-hIL15 [163] and MISTRG6 (expressing human M-CSF, IL-3, GM-CSF, SIRPα, TPO, and IL-6) [164] have demonstrated even greater improvements in human NK cell development and survival. Matsuda et al. showed that NSG-hIL7-hIL15 mice engrafted with human CD34⁺ HSPCs generate extremely high NK-cell frequencies, approximately 43% of human CD45⁺ cells in PB, far exceeding earlier models [163]. A particularly compelling study used MISTRG6 mice engrafted with patient-derived BM HSPCs, followed by implantation of a matched PDX tumor. This strategy created a fully autologous, genetically matched humanized model for the prospective study of tumor-immune interactions in patients with solid tumors. Humanization of the IL-6 locus in MISTRG6 mice enhanced HSPC engraftment, enabling autologous modeling of tumor-immune dynamics directly from a patient’s BM aspirate. The autologous tumors recreated key elements of the human TME, including activation of both innate and adaptive immunity, thereby providing a powerful platform for preclinical drug testing [165]. Nevertheless, detailed phenotypic maturation of these NK cells (e.g. expression of KIR, CD57) was not deeply characterized in that context.
NK cell development and function have been much better studied in MISTRG-6-15 (knock-in of human IL-6 and IL-15) humanized mice. In this model, Sungur et al. showed rapid NK cell expansion and, upon HIV-1 infection, strong degranulation, cytotoxicity (against the K562 cell line), and pro-inflammatory cytokine production in non-lymphoid tissues [129]. In these mice, NK cells in lymphoid organs showed more immature phenotypes (lower CD16A) and reduced function, mirroring some features of human tissue-resident NK cells. Importantly, depletion of NK cells (via anti-NKp46) in vivo led to significantly higher HIV-1 replication, demonstrating that these NK cells exert functional antiviral control in vivo [129].
Importantly, “double-humanized” platforms, such as human-BM-Liver-Thymus (hu-BLT) mice, models combining HSPCs engraftment with tissue-specific human grafts, enable the study of NK cell trafficking and infiltration into organ-specific niches. Experimental studies in hu-BLT systems show that human NK cells (transferred or endogenously reconstituted) can reach xenografted tumors, produce IFN-γ, mediate cytotoxicity, and in some cases restore or potentiate endogenous NK cell function. An additional hu-BLT study has reported NK-cell tumor infiltration and functional activation (IFN-γ, cytotoxicity) [166,167]. However, these platforms also do not yet fully recapitulate human vasculature, chemokine gradients, and stromal architecture, which should be considered when interpreting tissue-level immune behaviors [167]. Taken together, the available evidence demonstrates that cytokine-enhanced humanized mouse strains substantially overcome the key limitations of first-generation platforms. By providing sustained human cytokine support, these models achieve markedly higher levels of human NK cell reconstitution, promote the emergence of more mature and functionally competent NK subsets (including CD56dim/CD16bright, KIR⁺, perforin⁺/granzyme⁺ populations), and support robust cytotoxic and cytokine-producing activity in vivo. Importantly, they enable physiologically relevant NK cell trafficking, higher tumor infiltration, and therapeutic responsiveness within human tumor xenograft settings, features largely absent in classical models. Nevertheless, even in cytokine-optimized platforms, the licensing and education of NK cells are constrained by incomplete humanization of the MHC class I landscape. Mouse models expressing KIR2DL2 receptors together with their HLA-Cw3 ligand have demonstrated that HLA class I influences KIR repertoire and education [168]. In humanized NSG mice, for example, KIR-mediated NK education critically depends on the presence of matching HLA ligands in trans, and mixed-HLA reconstitution can abrogate this education [169]. Although emerging transgenic and knock-in strategies offer promising routes, full recreation of a physiological KIR-HLA class I axis -including classical (HLA-A, -B, -C) and nonclassical (HLA-E/F) class I molecules remains largely unexplored.
6. Conclusions and Future Perspectives
Collectively, the studies described and discussed in this review position cytokine-enhanced humanized mice as powerful and increasingly indispensable systems for dissecting human NK cell biology and evaluating NK-mediated anti-cancer immunotherapies. Yet, their continued refinement, particularly with respect to HLA class I humanization, tissue-specific immune architecture, and preservation of NK-cell education, will be essential to fully unlock their predictive and translational potential.
In addition to that, the combination of multi-omics analytic platforms will speed up the optimization of the models. Historically, in vivo studies of human NK cells were largely limited to measurements of frequency, surface phenotype, cytotoxicity, and cytokine production, providing little insight into their temporal dynamics or tissue-specific behavior. The integration of longitudinal monitoring with single-cell multi-omics, intravital imaging, and in vivo tracking (PET/SPECT) has transformed this landscape, offering a far more detailed view of NK cell biology in both systemic circulation and TME. For example, in a humanized mouse model with IL-15 knock-in, researchers combined scRNA-seq with repeated functional assays to track NK cells over time in hepatocellular carcinoma xenografts [152]. This approach identified transient subpopulations exhibiting activation markers early after infusion and, later, upregulation of exhaustion-associated genes such as PDCD1 and TIGIT, which would have been missed by conventional analyses. Functional readouts correlated with transcriptional states, revealing progressive loss of cytotoxicity in specific tissue compartments and highlighting the dynamic interplay between NK activation and suppression in vivo. Similarly, longitudinal studies in MISTRG-6-15 humanized mice demonstrated that NK cells initially expand and display high cytotoxic and metabolic activity, but over weeks, some subsets undergo transcriptional and metabolic reprogramming toward reduced oxidative phosphorylation and glycolytic capacity [170]. By integrating single-cell transcriptomics with functional assays, these studies revealed how metabolic exhaustion develops in vivo, insights inaccessible in static models. Regarding the possibility of tracking NK cells, recent studies highlighted the importance of integrating PET scanning in preclinical investigation. PET imaging using 89Zr-oxine–labeled human NK cells has enabled whole-body tracking of adoptively transferred NK cells over time: in a model of HER2-positive breast cancer, 89Zr-labeled NK cells were visualized by PET/CT for up to seven days, revealing their accumulation in liver and spleen, and enhanced tumor infiltration when co-administered with Trastuzumab [171]. Similarly, in non-human primates, serial PET/CT of 89Zr-oxine–labeled NK cells showed initial localization in the lungs followed by migration to the liver and spleen, demonstrating the feasibility of quantitatively tracking NK cell biodistribution in vivo [172]. Together, these technologies transcend traditional endpoint measurements by providing time-resolved, spatially resolved, and molecularly detailed insights into NK cell trafficking, activation, dysfunction, and therapeutic responsiveness, insights that were largely inaccessible with earlier in vitro or in vivo assays.
To conclude, encouraging recent innovations have already delivered major advances in humanized models, imaging technologies, and integrative analytical pipelines, greatly enhancing their resolution and physiological relevance. Building on this strong base, continued refinement of these platforms will further expand our ability to capture the full spectrum of human NK cell behavior and more reliably predict therapeutic responses, ultimately empowering the rational prioritization of immunotherapeutic strategies and helping minimize the risk of clinical setbacks.
Author Contributions
Vitale C. conceptualized and wrote the manuscript; Ruiba A. conceptualized and wrote the manuscript. Dondero A. contributed to manuscript writing; Serra M. contributed to manuscript writing. Tassistro A. contributed to manuscript writing; Bottino C. conceptualized and edited the manuscript; Castriconi R. conceptualized and edited the manuscript.
Acknowledgments
To Fondazione NB-OPEN Odv and Ministry of Health (Ricerca Corrente 2025) for supporting our research activity. To Fondazione NB-OPEN Odv for the scholarship awarded to Dr. Alessia Ruiba.
Conflicts of Interests
Nothing to disclose.
Abbreviations
The following abbreviations are used in this manuscript:
| ADCC | Antibody-Dependent Cellular Cytotoxicity |
| ALL | Acute Lymphoblastic Leukemia |
| AML | Acute Myeloid Leukemia |
| ANKETs | Antibody-based NK cell Engager Therapeutics |
| B-NHL | B cell Non-Hodgkin Lymphoma |
| BiKEs | Bispecific NK cell Engagers |
| BM | Bone Marrow |
| CAR-NK | Chimeric Antigen Receptor-Natura Killer |
| CIML | Cytokine Induced Memory Like |
| CRS | Cytokine Release Syndrome |
| DC | Dendritic Cell |
| GvHD | Graft versus Host Disease |
| HD | Healthy donor |
| hESCs | Human Embryonic Stem Cells |
| HSC | Hematopoietic Stem Cell |
| HSPCs | Hematopoietic Stem and Progenitor Cells |
| hu-BLT | Human BM Liver Thymus |
| IL-2 | Interleukin-2 |
| IL-2v | IL-2 variant |
| ILC | Innate Lymphoid Cell |
| iPSC | Induced Pluripotent Stem Cell |
| KIR | Killer Ig-like Receptor |
| mAbs | monoclonal Antibodies |
| MISTRG | M-CSFh/h IL-3/GM-CSFh/h SIRPαh/h TPOh/h RAG2-/- IL2Rg-/- |
| NCR | Natural cytotoxicity receptors |
| NK | Natural Killer |
| NKCEs | NK Cell Engagers |
| NOD/SCID | Non Obese Diabetic/Severe Combined Immunodeficient |
| NOG | NOD/Shi-scid/IL2Rγnull |
| NSG | NOD/SCID/γcnull |
| NSG-IL15 | transgenic for human IL-15 |
| NSG-SGM3 | NOD-scid IL2rγ⁻/⁻ transgenic for human SCF, GM-CSF, and IL-3 |
| PB | Peripheral Blood |
| PDX | Patient Derived Xenograft |
| sc RNA-seq | single-cell RNA-sequencing |
| scFv | single-chain variable Fragment |
| SRG-15 | RAG2⁻/⁻ IL2Rg⁻/⁻ transgenic for human IL-15 |
| TAAs | Tumor Associated Antigens |
| TCEs | T Cell Engagers |
| TME | Tumor Microenvironment |
| Tregs | Regulatory T cells |
| TriKEs | Trispecific NK cell Engagers |
| UBC | Umbilical Cord Blood |
References
- L. Chiossone, P. Y. Dumas, M. Vienne, and E. Vivier, “Author Correction: Natural killer cells and other innate lymphoid cells in cancer,” Nat Rev Immunol, vol. 18, no. 11, p. 726, Nov. 2018. [CrossRef]
- F. Bellora et al., “Human NK cells and NK receptors,” Immunol Lett, vol. 161, no. 2, pp. 168–173, 2014. [CrossRef]
- L. Rebuffet et al., “High-dimensional single-cell analysis of human natural killer cell heterogeneity,” Nat Immunol, vol. 25, no. 8, pp. 1474–1488, Aug. 2024. [CrossRef]
- S. D. Scoville, A. G. Freud, and M. A. Caligiuri, “Modeling Human Natural Killer Cell Development in the Era of Innate Lymphoid Cells,” Front Immunol, vol. 8, no. MAR, Mar. 2017. [CrossRef]
- L. Quatrini, M. Della Chiesa, S. Sivori, M. C. Mingari, D. Pende, and L. Moretta, “Human NK cells, their receptors and function,” Eur J Immunol, vol. 51, no. 7, pp. 1566–1579, Jul. 2021. [CrossRef]
- F. Borrego, M. J. Robertson, J. Ritz, J. Peña, and R. Solana, “CD69 is a stimulatory receptor for natural killer cell and its cytotoxic effect is blocked by CD94 inhibitory receptor,” Immunology, vol. 97, no. 1, pp. 159–165, 1999. [CrossRef]
- N. Anfossi et al., “Human NK cell education by inhibitory receptors for MHC class I,” Immunity, vol. 25, no. 2, pp. 331–342, Aug. 2006. [CrossRef]
- S. Sivori et al., “NK cells and ILCs in tumor immunotherapy,” Mol Aspects Med, vol. 80, Aug. 2021. [CrossRef]
- L. Ni and C. Dong, “New B7 Family Checkpoints in Human Cancers,” Mol Cancer Ther, vol. 16, no. 7, pp. 1203–1211, Jul. 2017. [CrossRef]
- A. Moretta et al., “Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis,” Annu Rev Immunol, vol. 19, pp. 197–223, 2001. [CrossRef]
- A.G. Freud, B. L. Mundy-Bosse, J. Yu, and M. A. Caligiuri, “The broad spectrum of human natural killer cell diversity,” Immunity, vol. 47, no. 5, p. 820, Nov. 2017. [CrossRef]
- H. Peng and R. Sun, “Liver-resident NK cells and their potential functions,” Cell Mol Immunol, vol. 14, no. 11, p. 890, Nov. 2017. [CrossRef]
- G. Lugthart et al., “Human Lymphoid Tissues Harbor a Distinct CD69+CXCR6+ NK Cell Population,” J Immunol, vol. 197, no. 1, pp. 78–84, Jul. 2016. [CrossRef]
- J. E. Melsen et al., “Human Bone Marrow-Resident Natural Killer Cells Have a Unique Transcriptional Profile and Resemble Resident Memory CD8+ T Cells,” Front Immunol, vol. 9, Aug. 2018. [CrossRef]
- M. López-Botet, A. De Maria, A. Muntasell, M. Della Chiesa, and C. Vilches, “Adaptive NK cell response to human cytomegalovirus: Facts and open issues,” Semin Immunol, vol. 65, Jan. 2023. [CrossRef]
- L. Peng, G. Sferruzza, L. Yang, L. Zhou, and S. Chen, “CAR-T and CAR-NK as cellular cancer immunotherapy for solid tumors,” Cell Mol Immunol, vol. 21, no. 10, pp. 1089–1108, Oct. 2024. [CrossRef]
- R. Basar, M. Daher, and K. Rezvani, “Next-generation cell therapies: the emerging role of CAR-NK cells,” Blood Adv, vol. 4, no. 22, pp. 5868–5876, Nov. 2020. [CrossRef]
- T. J. Laskowski, A. Biederstädt, and K. Rezvani, “Natural killer cells in antitumour adoptive cell immunotherapy,” Nature Reviews Cancer 2022 22:10, vol. 22, no. 10, pp. 557–575, Jul. 2022. [CrossRef]
- S. Liu, V. Galat, Y. Galat4, Y. K. A. Lee, D. Wainwright, and J. Wu, “NK cell-based cancer immunotherapy: from basic biology to clinical development,” J Hematol Oncol, vol. 14, no. 1, Dec. 2021. [CrossRef]
- N. Shimasaki, A. Jain, and D. Campana, “NK cells for cancer immunotherapy,” Nat Rev Drug Discov, vol. 19, no. 3, pp. 200–218, Mar. 2020. [CrossRef]
- S. Ma, J. Yu, and M. A. Caligiuri, “Natural killer cell-based immunotherapy for cancer,” J Immunol, vol. 214, no. 7, pp. 1444–1456, Jul. 2025. [CrossRef]
- M. D. Blunt and S. I. Khakoo, “Harnessing natural killer cell effector function against cancer,” Immunotherapy advances, vol. 4, no. 1, 2023. [CrossRef]
- J. A. Myers and J. S. Miller, “Exploring the NK cell platform for cancer immunotherapy,” Nat Rev Clin Oncol, vol. 18, no. 2, pp. 85–100, Feb. 2021. [CrossRef]
- A. Biederstädt and K. Rezvani, “Engineered natural killer cells for cancer therapy,” Cancer Cell, vol. 43, no. 11, pp. 1987–2013, Nov. 2025. [CrossRef]
- J. S. Miller et al., “Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer,” Blood, vol. 105, no. 8, pp. 3051–3057, Apr. 2005. [CrossRef]
- W. Wang, A. K. Erbe, J. A. Hank, Z. S. Morris, and P. M. Sondel, “NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy,” Front Immunol, vol. 6, no. JUL, 2015. [CrossRef]
- A. Wiernik et al., “Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition,” Clin Cancer Res, vol. 19, no. 14, pp. 3844–3855, Jul. 2013. [CrossRef]
- M. K. Gleason et al., “CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets,” Blood, vol. 123, no. 19, p. 3016, May 2014. [CrossRef]
- F. Oh, M. Felices, B. Kodal, J. S. Miller, and D. A. Vallera, “Immunotherapeutic Development of a Tri-Specific NK Cell Engager Recognizing BCMA,” Immuno, vol. 3, no. 2, pp. 237–249, Jun. 2023. [CrossRef]
- A. Fenis, O. Demaria, L. Gauthier, E. Vivier, and E. Narni-Mancinelli, “New immune cell engagers for cancer immunotherapy,” Nat Rev Immunol, vol. 24, no. 7, pp. 471–486, Jul. 2024. [CrossRef]
- A. Zhu, Y. Bai, Y. Nan, and D. Ju, “Natural killer cell engagers: From bi-specific to tri-specific and tetra-specific engagers for enhanced cancer immunotherapy,” Clin Transl Med, vol. 14, no. 11, p. e70046, Nov. 2024. [CrossRef]
- O. Demaria et al., “Antitumor immunity induced by antibody-based natural killer cell engager therapeutics armed with not-alpha IL-2 variant,” Cell Rep Med, vol. 3, no. 10, p. 100783, Oct. 2022. [CrossRef]
- T. Li, M. Niu, W. Zhang, S. Qin, J. Zhou, and M. Yi, “CAR-NK cells for cancer immunotherapy: recent advances and future directions,” Front Immunol, vol. 15, p. 1361194, Feb. 2024. [CrossRef]
- E. Liu et al., “Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors,” N Engl J Med, vol. 382, no. 6, pp. 545–553, Feb. 2020. [CrossRef]
- P. Karagiannis and S. Il Kim, “iPSC-Derived Natural Killer Cells for Cancer Immunotherapy,” Mol Cells, vol. 44, no. 8, pp. 541–548, Aug. 2021. [CrossRef]
- M. Chen et al., “Recent advances in tumor immunotherapy based on NK cells,” Front Immunol, vol. 16, p. 1595533, Aug. 2025. [CrossRef]
- G. D. Lizana-Vasquez, M. Torres-Lugo, R. B. Dixon, J. D. Powderly, and R. F. Warin, “The application of autologous cancer immunotherapies in the age of memory-NK cells,” Front Immunol, vol. 14, p. 1167666, May 2023. [CrossRef]
- N. Sakamoto et al., “Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer,” J Transl Med, vol. 13, no. 1, pp. 277-, Aug. 2015. [CrossRef]
- M. Tarannum, R. Romee, and R. M. Shapiro, “Innovative Strategies to Improve the Clinical Application of NK Cell-Based Immunotherapy,” Front Immunol, vol. 13, p. 859177, Mar. 2022. [CrossRef]
- S. Kundu, L. Durkan, M. O’Dwyer, and E. Szegezdi, “Protocol for isolation and expansion of natural killer cells from human peripheral blood scalable for clinical applications,” Biol Methods Protoc, vol. 10, no. 1, Jan. 2025. [CrossRef]
- S. Khanal and N. Bhattarai, “A Scalable Protocol for Ex Vivo Production of CAR-Engineered Human NK Cells,” Methods Protoc, vol. 8, no. 5, p. 102, Sep. 2025. [CrossRef]
- E. M. McErlean and H. O. McCarthy, “Non-viral approaches in CAR-NK cell engineering: connecting natural killer cell biology and gene delivery,” J Nanobiotechnology, vol. 22, no. 1, Dec. 2024. [CrossRef]
- M. G. Lamb, H. G. Rangarajan, B. P. Tullius, and D. A. Lee, “Natural killer cell therapy for hematologic malignancies: successes, challenges, and the future,” Stem Cell Research & Therapy 2021 12:1, vol. 12, no. 1, pp. 211-, Mar. 2021. [CrossRef]
- Y. Harada et al., “Clinical Applications of Natural Killer Cells,” Natural Killer Cells, Dec. 2017. [CrossRef]
- O. Lim, M. Y. Jung, Y. K. Hwang, and E. C. Shin, “Present and Future of Allogeneic Natural Killer Cell Therapy,” Front Immunol, vol. 6, no. JUN, p. 286, 2015. [CrossRef]
- H. Klingemann, “The NK-92 cell line-30 years later: its impact on natural killer cell research and treatment of cancer,” Cytotherapy, vol. 25, no. 5, pp. 451–457, May 2023. [CrossRef]
- J. Sun, M. Elliott, and F. Souza-Fonseca-Guimaraes, “Engineered iPSC-derived natural killer cells: recent innovations in translational innate anti-cancer immunotherapy,” Clin Transl Immunology, vol. 14, no. 7, p. e70045, Jan. 2025. [CrossRef]
- E. A. Grimm, A. Mazumder, H. Z. Zhang, and S. A. Rosenberg, “Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes,” J Exp Med, vol. 155, no. 6, pp. 1823–1841, 1982. [CrossRef]
- A.K. Ali, N. Nandagopal, and S. H. Lee, “IL-15–PI3K–AKT–mTOR: A Critical Pathway in the Life Journey of Natural Killer Cells,” Front Immunol, vol. 6, no. JUN, p. 355, 2015. [CrossRef]
- A.H. Pillet, J. Thèze, and T. Rose, “Interleukin (IL)-2 and IL-15 have different effects on human natural killer lymphocytes,” Hum Immunol, vol. 72, no. 11, pp. 1013–1017, Nov. 2011. [CrossRef]
- Y. Yang and A. Lundqvist, “Immunomodulatory Effects of IL-2 and IL-15; Implications for Cancer Immunotherapy,” Cancers (Basel), vol. 12, no. 12, p. 3586, Dec. 2020. [CrossRef]
- P. R. Rhode et al., “Comparison of the superagonist complex, ALT-803, to IL15 as cancer immunotherapeutics in animal models,” Cancer Immunol Res, vol. 4, no. 1, pp. 49–60, Jan. 2016. [CrossRef]
- I. Terrén, A. Orrantia, G. Astarloa-Pando, A. Amarilla-Irusta, O. Zenarruzabeitia, and F. Borrego, “Cytokine-Induced Memory-Like NK Cells: From the Basics to Clinical Applications,” Front Immunol, vol. 13, p. 884648, May 2022. [CrossRef]
- J.W. Leong et al., “Preactivation with IL-12, IL-15, and IL-18 Induces CD25 and a Functional High-Affinity IL-2 Receptor on Human Cytokine-Induced Memory-like Natural Killer Cells,” Biology of Blood and Marrow Transplantation, vol. 20, no. 4, pp. 463–473, Apr. 2014. [CrossRef]
- P. R. Kennedy, M. Felices, and J. S. Miller, “Challenges to the broad application of allogeneic natural killer cell immunotherapy of cancer,” Stem Cell Research & Therapy 2022 13:1, vol. 13, no. 1, pp. 165-, Apr. 2022. [CrossRef]
- F. Fang et al., “Advances in NK cell production,” Cell Mol Immunol, vol. 19, no. 4, p. 460, Apr. 2022. [CrossRef]
- V. K. Singh, “Natural killer cell-based Immunotherapy for Solid tumors: A Comprehensive Review,” 2025, Accessed: Nov. 13, 2025. [Online]. Available: https://ijbc.ir.
- L.V. Jørgensen, E. B. Christensen, M. B. Barnkob, and T. Barington, “The clinical landscape of CAR NK cells,” Experimental Hematology & Oncology 2025 14:1, vol. 14, no. 1, pp. 46-, Mar. 2025. [CrossRef]
- R. Basar, M. Daher, and K. Rezvani, “Next-generation cell therapies: the emerging role of CAR-NK cells,” Blood Adv, vol. 4, no. 22, pp. 5868–5876, Nov. 2020. [CrossRef]
- S. Müller et al., “High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia,” Front Immunol, vol. 10, Jan. 2020. [CrossRef]
- L. Herrera et al., “Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells,” Sci Rep, vol. 9, no. 1, Dec. 2019. [CrossRef]
- E. Liu et al., “Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity,” Leukemia, vol. 32, no. 2, pp. 520–531, Feb. 2018. [CrossRef]
- Y. Zhang, C. Zhang, M. He, W. Xing, R. Hou, and H. Zhang, “Co-expression of IL-21-Enhanced NKG2D CAR-NK cell therapy for lung cancer,” BMC Cancer, vol. 24, no. 1, Dec. 2024. [CrossRef]
- X. Wang et al., “Co-expression of IL-15 and CCL21 strengthens CAR-NK cells to eliminate tumors in concert with T cells and equips them with PI3K/AKT/mTOR signal signature,” J Immunother Cancer, vol. 13, no. 6, Jun. 2025. [CrossRef]
- M. Carlsten et al., “Efficient mRNA-Based Genetic Engineering of Human NK Cells with High-Affinity CD16 and CCR7 Augments Rituximab-Induced ADCC against Lymphoma and Targets NK Cell Migration toward the Lymph Node-Associated Chemokine CCL19,” Front Immunol, vol. 7, no. MAR, Mar. 2016. [CrossRef]
- F. F. Feigl et al., “Efficient Redirection of NK Cells by Genetic Modification with Chemokine Receptors CCR4 and CCR2B,” Int J Mol Sci, vol. 24, no. 4, Feb. 2023. [CrossRef]
- M.F. Hasan et al., “Knockout of the inhibitory receptor TIGIT enhances the antitumor response of ex vivo expanded NK cells and prevents fratricide with therapeutic Fc-active TIGIT antibodies,” J Immunother Cancer, vol. 11, no. 12, p. e007502, Dec. 2023. [CrossRef]
- T. Bexte et al., “CRISPR-Cas9 based gene editing of the immune checkpoint NKG2A enhances NK cell mediated cytotoxicity against multiple myeloma,” Oncoimmunology, vol. 11, no. 1, p. 2081415, 2022. [CrossRef]
- T. Bexte et al., “CRISPR/Cas9 editing of NKG2A improves the efficacy of primary CD33-directed chimeric antigen receptor natural killer cells,” Nature Communications 2024 15:1, vol. 15, no. 1, pp. 8439-, Sep. 2024. [CrossRef]
- W. Qiao, P. Dong, H. Chen, and J. Zhang, “Advances in Induced Pluripotent Stem Cell-Derived Natural Killer Cell Therapy,” Cells 2024, Vol. 13, Page 1976, vol. 13, no. 23, p. 1976, Nov. 2024. [CrossRef]
- H. Zhu et al., “Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity,” Cell Stem Cell, vol. 27, no. 2, pp. 224-237.e6, Aug. 2020. [CrossRef]
- R. Cai, B. Lu, X. Zhao, S. Zhou, and Y. Li, “iPSC-derived NK cells engineered with CD226 effectively control acute myeloid leukemia,” Exp Hematol Oncol, vol. 14, no. 1, pp. 93-, Dec. 2025. [CrossRef]
- K. M. Snyder et al., “iPSC-derived natural killer cells expressing the FcγR fusion CD64/16A can be armed with antibodies for multitumor antigen targeting,” J Immunother Cancer, vol. 11, no. 12, p. e007280, Dec. 2023. [CrossRef]
- Y. Fukutani et al., “Human iPSC-derived NK cells armed with CCL19, CCR2B, high-affinity CD16, IL-15, and NKG2D complex enhance anti-solid tumor activity,” Stem Cell Research and Therapy , vol. 16, no. 1, pp. 373-, Dec. 2025. [CrossRef]
- M. Gauthier, C. Laroye, D. Bensoussan, C. Boura, and V. Decot Nat, “Natural killer cells and monoclonal antibodies: Two partners for successful antibody dependent cytotoxicity against tumor cells,” Crit Rev Oncol Hematol, vol. 160, p. 103261, 2021. [CrossRef]
- R. Vincken, U. Armendáriz-Martínez, and A. Ruiz-Sáenz, “ADCC: the rock band led by therapeutic antibodies, tumor and immune cells,” Front Immunol, vol. 16, p. 1548292, Apr. 2025. [CrossRef]
- H. S. Rugo et al., “Efficacy of Margetuximab vs Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial,” JAMA Oncol, vol. 7, no. 4, pp. 573–584, Apr. 2021. [CrossRef]
- D. N. Vo et al., “NK cell activation and recovery of NK cell subsets in lymphoma patients after obinutuzumab and lenalidomide treatment,” Oncoimmunology, vol. 7, no. 4, p. e1409322, Apr. 2017. [CrossRef]
- H. J. van der Horst, I. S. Nijhof, T. Mutis, and M. E. D. Chamuleau, “Fc-Engineered Antibodies with Enhanced Fc-Effector Function for the Treatment of B-Cell Malignancies,” Cancers (Basel), vol. 12, no. 10, p. 3041, Oct. 2020. [CrossRef]
- D. T. Abdeldaim and K. Schindowski, “Fc-Engineered Therapeutic Antibodies: Recent Advances and Future Directions,” Pharmaceutics, vol. 15, no. 10, p. 2402, Oct. 2023. [CrossRef]
- K. J. Dixon, J. Wu, and B. Walcheck, “Engineering Anti-Tumor Monoclonal Antibodies and Fc Receptors to Enhance ADCC by Human NK Cells,” Cancers 2021, Vol. 13, Page 312, vol. 13, no. 2, p. 312, Jan. 2021. [CrossRef]
- Y. Chu et al., “Combinatorial immunotherapy of N-803 (IL-15 superagonist) and dinutuximab with ex vivo expanded natural killer cells significantly enhances in vitro cytotoxicity against GD2+ pediatric solid tumors and in vivo survival of xenografted immunodeficient NSG mice,” J Immunother Cancer, vol. 9, no. 7, p. 2267, Jul. 2021. [CrossRef]
- Z. Antosova et al., “SOT101 induces NK cell cytotoxicity and potentiates antibody-dependent cell cytotoxicity and anti-tumor activity,” Front Immunol, vol. 13, p. 989895, Oct. 2022. [CrossRef]
- S. P. Patel et al., “Phase 1/2 study of monalizumab plus durvalumab in patients with advanced solid tumors,” J Immunother Cancer, vol. 12, no. 2, Feb. 2024. [CrossRef]
- M. Felices et al., “CD16-IL15-CD33 Trispecific Killer Engager (TriKE) induces NK cell expansion, persistence, and myeloid blast antigen specific killing.,” The Journal of Immunology, vol. 196, no. 1_Supplement, pp. 75.8-75.8, May 2016. [CrossRef]
- S. K. Phung, J. S. Miller, and M. Felices, “Bi-specific and Tri-specific NK Cell Engagers: The New Avenue of Targeted NK Cell Immunotherapy,” Mol Diagn Ther, vol. 25, no. 5, pp. 577–592, Sep. 2021. [CrossRef]
- L. Gauthier et al., “Control of acute myeloid leukemia by a trifunctional NKp46-CD16a-NK cell engager targeting CD123,” Nat Biotechnol, vol. 41, no. 9, pp. 1296–1306, Sep. 2023. [CrossRef]
- S. B. Reusing et al., “CD16xCD33 Bispecific Killer Cell Engager (BiKE) as potential immunotherapeutic in pediatric patients with AML and biphenotypic ALL,” Cancer Immunology, Immunotherapy, vol. 70, no. 12, pp. 3701–3708, Dec. 2021. [CrossRef]
- N. Schmitt et al., “The bispecific innate cell engager AFM28 eliminates CD123+ leukemic stem and progenitor cells in AML and MDS,” Nat Commun, vol. 16, no. 1, Dec. 2025. [CrossRef]
- A. Kiefer et al., “Dual Targeting of Glioblastoma Cells with Bispecific Killer Cell Engagers Directed to EGFR and ErbB2 (HER2) Facilitates Effective Elimination by NKG2D-CAR-Engineered NK Cells,” Cells, vol. 13, no. 3, Feb. 2024. [CrossRef]
- M. Felices et al., “Potent Cytolytic Activity and Specific IL15 Delivery in a Second-Generation Trispecific Killer Engager,” Cancer Immunol Res, vol. 8, no. 9, pp. 1139–1149, Sep. 2020. [CrossRef]
- P. Winidmanokul, A. Panya, and S. Okada, “Tri-specific killer engager: unleashing multi-synergic power against cancer,” Open Exploration 2019 5:2, vol. 5, no. 2, pp. 432–448, Apr. 2024. [CrossRef]
- N. Colomar-Carando et al., “Exploiting Natural Killer Cell Engagers to Control Pediatric B-cell Precursor Acute Lymphoblastic Leukemia,” Cancer Immunol Res, vol. 10, no. 3, pp. 291–302, Mar. 2022. [CrossRef]
- A.N. Shouse, K. M. LaPorte, and T. R. Malek, “Interleukin-2 signaling in the regulation of T cell biology in autoimmunity and cancer,” Immunity, vol. 57, no. 3, pp. 414–428, Mar. 2024. [CrossRef]
- E. F. Conant, K. R. Fox, and W. T. Miller, “Pulmonary edema as a complication of interleukin-2 therapy,” AJR Am J Roentgenol, vol. 152, no. 4, pp. 749–752, 1989. [CrossRef]
- O. Demaria et al., “Preclinical assessment of IPH6501, a first-in-class IL2v-armed tetraspecific NK cell engager directed against CD20 for R/R B-NHL, in comparison with a CD20-targeting T cell engager.,” Journal of Clinical Oncology, vol. 42, no. 16_suppl, pp. 7030–7030, Jun. 2024. [CrossRef]
- J. Shang, S. Hu, and X. Wang, “Targeting natural killer cells: from basic biology to clinical application in hematologic malignancies,” Exp Hematol Oncol, vol. 13, no. 1, Dec. 2024. [CrossRef]
- M. H. Shin, E. Oh, and D. Minn, “Current Developments in NK Cell Engagers for Cancer Immunotherapy: Focus on CD16A and NKp46,” Immune Netw, vol. 24, no. 5, p. e34, Oct. 2024. [CrossRef]
- F. Zhang, H. Soleimani Samarkhazan, Z. Pooraskari, and A. Bayani, “Beyond CAR-T: Engineered NK cell therapies (CAR-NK, NKCEs) in next-generation cancer immunotherapy,” Crit Rev Oncol Hematol, vol. 214, p. 104912, Oct. 2025. [CrossRef]
- T. Huan, B. Guan, H. Li, X. Tu, C. Zhang, and B. Tang, “Principles and current clinical landscape of NK cell engaging bispecific antibody against cancer,” Hum Vaccin Immunother, vol. 19, no. 2, p. 2256904, 2023. [CrossRef]
- J. C. M. Van der Loo, H. Hanenberg, R. J. Cooper, F. Y. Luo, E. N. Lazaridis, and D. A. Williams, “Nonobese Diabetic/Severe Combined Immunodeficiency (NOD/SCID) Mouse as a Model System to Study the Engraftment and Mobilization of Human Peripheral Blood Stem Cells,” Blood, vol. 92, no. 7, pp. 2556–2570, Oct. 1998. [CrossRef]
- M. Ito et al., “NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells,” Blood, vol. 100, no. 9, pp. 3175–3182, Nov. 2002. [CrossRef]
- M. Ito, K. Kobayashi, and T. Nakahata, “NOD/Shi-scid IL2rgamma(null) (NOG) mice more appropriate for humanized mouse models,” Curr Top Microbiol Immunol, vol. 324, pp. 53–76, 2008. [CrossRef]
- Castriconi et al., “Human NK cell infusions prolong survival of metastatic human neuroblastoma-bearing NOD/scid mice,” Cancer Immunol Immunother, vol. 56, no. 11, pp. 1733–1742, Nov. 2007. [CrossRef]
- J. S. Hoogstad-van Evert et al., “Umbilical cord blood CD34+ progenitor-derived NK cells efficiently kill ovarian cancer spheroids and intraperitoneal tumors in NOD/SCID/IL2Rgnull mice,” Oncoimmunology, vol. 6, no. 8, Aug. 2017. [CrossRef]
- U. Siegler, C. P. Kalberer, P. Nowbakht, S. Sendelov, S. Meyer-Monard, and A. Wodnar-Filipowicz, “Activated natural killer cells from patients with acute myeloid leukemia are cytotoxic against autologous leukemic blasts in NOD/SCID mice,” Leukemia, vol. 19, no. 12, pp. 2215–2222, 2005. [CrossRef]
- M. Shiokawa et al., “In vivo assay of human NK-dependent ADCC using NOD/SCID/gammac(null) (NOG) mice,” Biochem Biophys Res Commun, vol. 399, no. 4, pp. 733–737, Sep. 2010. [CrossRef]
- M. Parodi et al., “Murine models to study human NK cells in human solid tumors,” Front Immunol, vol. 14, 2023. [CrossRef]
- J. T. Kim, G. Bresson-Tan, and J. A. Zack, “Current Advances in Humanized Mouse Models for Studying NK Cells and HIV Infection,” Microorganisms, vol. 11, no. 8, Aug. 2023. [CrossRef]
- R. A et al., “Corrigendum: Development and function of human innate immune cells in a humanized mouse model,” Nat Biotechnol, vol. 35, no. 12, p. 1211, 2017. [CrossRef]
- M. De Jong and T. Maina, “Of Mice and Humans: Are They the Same?—Implications in Cancer Translational Research,” Journal of Nuclear Medicine, vol. 51, no. 4, pp. 501–504, Apr. 2010. [CrossRef]
- S. Mun, H. J. Lee, and P. Kim, “Rebuilding the microenvironment of primary tumors in humans: a focus on stroma,” Experimental & Molecular Medicine 2024 56:3, vol. 56, no. 3, pp. 527–548, Mar. 2024. [CrossRef]
- H. Xu et al., “New genetic and epigenetic insights into the chemokine system: the latest discoveries aiding progression toward precision medicine,” Cell Mol Immunol, vol. 20, no. 7, pp. 739–776, Jul. 2023. [CrossRef]
- G. Chamberlain, K. Wright, A. Rot, B. Ashton, and J. Middleton, “Murine mesenchymal stem cells exhibit a restricted repertoire of functional chemokine receptors: comparison with human,” PLoS One, vol. 3, no. 8, Aug. 2008. [CrossRef]
- N. B. Horowitz, I. Mohammad, U. Y. Moreno-Nieves, I. Koliesnik, Q. Tran, and J. B. Sunwoo, “Humanized Mouse Models for the Advancement of Innate Lymphoid Cell-Based Cancer Immunotherapies,” Front Immunol, vol. 12, Apr. 2021. [CrossRef]
- A. Zlotnik, O. Yoshie, and H. Nomiyama, “The chemokine and chemokine receptor superfamilies and their molecular evolution,” Genome Biol, vol. 7, no. 12, Dec. 2006. [CrossRef]
- M. Wunderlich, F.-S. Chou, B. Mizukawa, K. A. Link, and J. C. Mulloy, “A New Immunodeficient Mouse Strain, NOD/SCID IL2Rγ−/− SGM3, Promotes Enhanced Human Hematopoietic Cell Xenografts with a Robust T Cell Component.,” Blood, vol. 114, no. 22, pp. 3524–3524, Nov. 2009. [CrossRef]
- D. Herndler-Brandstetter et al., “Humanized mouse model supports development, function, and tissue residency of human natural killer cells,” Proc Natl Acad Sci U S A, vol. 114, no. 45, pp. E9626–E9634, Nov. 2017. [CrossRef]
- M. A. Brehm, K.-E. Aryee, L. Bruzenksi, D. L. Greiner, L. D. Shultz, and J. Keck, “Transgenic expression of human IL15 in NOD-scid IL2rgnull (NSG) mice enhances the development and survival of functional human NK cells,” The Journal of Immunology, vol. 200, no. Supplement_1, pp. 103.20-103.20, May 2018. [CrossRef]
- K. E. Aryee et al., “Enhanced development of functional human NK cells in NOD-scid-IL2rgnull mice expressing human IL15,” FASEB J, vol. 36, no. 9, Sep. 2022. [CrossRef]
- S.A. Abeynaike et al., “Human Hematopoietic Stem Cell Engrafted IL-15 Transgenic NSG Mice Support Robust NK Cell Responses and Sustained HIV-1 Infection,” Viruses, vol. 15, no. 2, p. 365, Feb. 2023. [CrossRef]
- J. Chuprin et al., “Humanized mouse models for immuno-oncology research,” Nat Rev Clin Oncol, vol. 20, no. 3, p. 192, Mar. 2023. [CrossRef]
- T.M. Allen et al., “Humanized immune system mouse models: progress, challenges and opportunities,” Nature Immunology 2019 20:7, vol. 20, no. 7, pp. 770–774, Jun. 2019. [CrossRef]
- J. Chen et al., “The development and improvement of immunodeficient mice and humanized immune system mouse models,” Front Immunol, vol. 13, p. 1007579, Oct. 2022. [CrossRef]
- M. A. Cooper et al., “Human natural killer cells: a unique innate immunoregulatory role for the CD56bright subset,” Blood, vol. 97, no. 10, pp. 3146–3151, May 2001. [CrossRef]
- A. Poli, T. Michel, M. Thérésine, E. Andrès, F. Hentges, and J. Zimmer, “CD56bright natural killer (NK) cells: an important NK cell subset,” Immunology, vol. 126, no. 4, p. 458, 2009. [CrossRef]
- F. Bellora et al., “The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes,” Proc Natl Acad Sci U S A, vol. 107, no. 50, pp. 21659–21664, Dec. 2010. [CrossRef]
- E. Vivier, E. Tomasello, M. Baratin, T. Walzer, and S. Ugolini, “Functions of natural killer cells,” Nat Immunol, vol. 9, no. 5, pp. 503–510, May 2008. [CrossRef]
- L. Yin, X.-J. Wang, D.-X. Chen, X.-N. Liu, and X.-J. Wang, “Humanized mouse model: a review on preclinical applications for cancer immunotherapy,” Am J Cancer Res, vol. 10, no. 12, p. 4568, 2020, Accessed: Dec. 05, 2025. [Online]. Available.
- C. M. Sungur et al., “Human NK cells confer protection against HIV-1 infection in humanized mice,” J Clin Invest, vol. 132, no. 24, Dec. 2022. [CrossRef]
- M. Shen et al., “Unraveling NK cell heterogeneity through single-cell sequencing: insights from physiological and tumor contexts for clinical applications,” Front Immunol, vol. 16, p. 1612352, Jul. 2025. [CrossRef]
- E. Hashemi and S. Malarkannan, “Tissue-Resident NK Cells: Development, Maturation, and Clinical Relevance,” Cancers (Basel), vol. 12, no. 6, pp. 1–23, Jun. 2020. [CrossRef]
- T. Le, R. K. Reeves, and L. R. McKinnon, “The functional diversity of tissue-resident natural killer cells against infection,” Immunology, vol. 167, no. 1, pp. 28–39, Sep. 2022. [CrossRef]
- Z. Pan, Y. S. Ye, C. Liu, and W. Li, “Role of liver-resident NK cells in liver immunity,” Hepatol Int, vol. 19, no. 2, pp. 315–324, Apr. 2025. [CrossRef]
- I. Filipovic et al., “29-Color Flow Cytometry: Unraveling Human Liver NK Cell Repertoire Diversity,” Front Immunol, vol. 10, p. 494855, Nov. 2019. [CrossRef]
- S. Soleimanian and R. Yaghobi, “Harnessing Memory NK Cell to Protect Against COVID-19,” Front Pharmacol, vol. 11, p. 560516, Aug. 2020. [CrossRef]
- D. Brownlie et al., “Expansions of adaptive-like NK cells with a tissue-resident phenotype in human lung and blood,” Proc Natl Acad Sci U S A, vol. 118, no. 11, p. e2016580118, Mar. 2021. [CrossRef]
- X. W. Wei, Y. C. Zhang, F. Wu, F. J. Tian, and Y. Lin, “The role of extravillous trophoblasts and uterine NK cells in vascular remodeling during pregnancy,” Front Immunol, vol. 13, p. 951482, Jul. 2022. [CrossRef]
- M. M. Faas and P. de Vos, “Uterine NK cells and macrophages in pregnancy,” Placenta, vol. 56, pp. 44–52, Aug. 2017. [CrossRef]
- G. Ferlazzo et al., “Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs,” Proc Natl Acad Sci U S A, vol. 101, no. 47, pp. 16606–16611, Nov. 2004. [CrossRef]
- R. Thomas and X. Yang, “NK-DC Crosstalk in Immunity to Microbial Infection,” J Immunol Res, vol. 2016, 2016. [CrossRef]
- O. Chijioke and C. Münz, “Dendritic cell derived cytokines in human natural killer cell differentiation and activation,” Front Immunol, vol. 4, no. NOV, 2013. [CrossRef]
- N. D. Huntington et al., “IL-15 trans-presentation promotes human NK cell development and differentiation in vivo,” J Exp Med, vol. 206, no. 1, pp. 25–34, Jan. 2009. [CrossRef]
- C. Flahou, T. Morishima, H. Takizawa, and N. Sugimoto, “Fit-For-All iPSC-Derived Cell Therapies and Their Evaluation in Humanized Mice With NK Cell Immunity,” Front Immunol, vol. 12, p. 662360, Apr. 2021. [CrossRef]
- S. A. Mian, F. Anjos-Afonso, and D. Bonnet, “Advances in Human Immune System Mouse Models for Studying Human Hematopoiesis and Cancer Immunotherapy,” Front Immunol, vol. 11, Feb. 2021. [CrossRef]
- C. Zalfa and S. Paust, “Natural Killer Cell Interactions With Myeloid Derived Suppressor Cells in the Tumor Microenvironment and Implications for Cancer Immunotherapy,” Front Immunol, vol. 12, May 2021. [CrossRef]
- S. Guil-Luna, C. Sedlik, and E. Piaggio, “Humanized Mouse Models to Evaluate Cancer Immunotherapeutics,” Annu Rev Cancer Biol, vol. 5, no. Volume 5, 2021, pp. 119–136, Mar. 2020. [CrossRef]
- A. Gu, J. Li, M. Y. Li, and Y. Liu, “Patient-derived xenograft model in cancer: establishment and applications,” MedComm (Beijing), vol. 6, no. 2, Feb. 2025. [CrossRef]
- J. Jin, K. Yoshimura, M. Sewastjanow-Silva, S. Song, and J. A. Ajani, “Challenges and Prospects of Patient-Derived Xenografts for Cancer Research,” Cancers (Basel), vol. 15, no. 17, Sep. 2023. [CrossRef]
- S. J. Jackson and G. J. Thomas, “Human tissue models in cancer research: looking beyond the mouse,” Dis Model Mech, vol. 10, no. 8, p. 939, Aug. 2017. [CrossRef]
- B. Olson, Y. Li, Y. Lin, E. T. Liu, and A. Patnaik, “Mouse Models for Cancer Immunotherapy Research,” Cancer Discov, vol. 8, no. 11, pp. 1358–1365, Nov. 2018. [CrossRef]
- R. Stripecke et al., “Innovations, challenges, and minimal information for standardization of humanized mice,” EMBO Mol Med, vol. 12, no. 7, p. e8662, Jul. 2020. [CrossRef]
- W. N. Liu et al., “Single-cell RNA sequencing reveals anti-tumor potency of CD56+ NK cells and CD8+ T cells in humanized mice via PD-1 and TIGIT co-targeting,” Mol Ther, vol. 32, no. 11, pp. 3895–3914, Nov. 2024. [CrossRef]
- P. Morcillo-Martín-Romo et al., “The Role of NK Cells in Cancer Immunotherapy: Mechanisms, Evasion Strategies, and Therapeutic Advances,” Biomedicines 2025, Vol. 13, Page 857, vol. 13, no. 4, p. 857, Apr. 2025. [CrossRef]
- R. Paolini and R. Molfetta, “Dysregulation of DNAM-1-Mediated NK Cell Anti-Cancer Responses in the Tumor Microenvironment,” Cancers 2023, Vol. 15, Page 4616, vol. 15, no. 18, p. 4616, Sep. 2023. [CrossRef]
- P. Carrega et al., “Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells,” Cancer, vol. 112, no. 4, pp. 863–875, Feb. 2008. [CrossRef]
- H. Jia, H. Yang, H. Xiong, and K. Q. Luo, “NK cell exhaustion in the tumor microenvironment,” Front Immunol, vol. 14, p. 1303605, Nov. 2023. [CrossRef]
- J. Bi and Z. Tian, “NK cell exhaustion,” Front Immunol, vol. 8, no. JUN, p. 278004, Jun. 2017. [CrossRef]
- J. T. Kim and J. A. Zack, “A humanized mouse model to study NK cell biology during HIV infection,” J Clin Invest, vol. 132, no. 24, Dec. 2022. [CrossRef]
- H. Rafei et al., “CREM is a regulatory checkpoint of CAR and IL-15 signalling in NK cells,” Nature 2025 643:8073, vol. 643, no. 8073, pp. 1076–1086, Jun. 2025. [CrossRef]
- X. Xu et al., “Signaling intact membrane-bound IL-15 enables potent anti-tumor activity and safety of CAR-NK cells,” Front Immunol, vol. 16, p. 1658580, Sep. 2025. [CrossRef]
- D. Feng et al., “Expression of membrane-bound Interleukin-15 sustains the growth and survival of CAR-NK cells,” Int Immunopharmacol, vol. 166, p. 115577, Dec. 2025. [CrossRef]
- M. Felices et al., “Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect,” JCI Insight, vol. 3, no. 3, Feb. 2018. [CrossRef]
- M. Matsuda et al., “Human NK cell development in hIL-7 and hIL-15 knockin NOD/SCID/IL2rgKO mice,” Life Sci Alliance, vol. 2, no. 2, p. e201800195, 2019. [CrossRef]
- A. Chen et al., “Comparison of NSG-Quad and MISTRG-6 humanized mice for modeling circulating and tumor-infiltrating human myeloid cells,” Mol Ther Methods Clin Dev, vol. 33, no. 2, p. 101487, Jun. 2025. [CrossRef]
- M. Chiorazzi et al., “Autologous humanized PDX modeling for immuno-oncology recapitulates features of the human tumor microenvironment,” J Immunother Cancer, vol. 11, no. 7, Jul. 2023. [CrossRef]
- K. Kaur, P. Topchyan, and A. Jewett, “Supercharged Natural Killer (sNK) Cells Inhibit Melanoma Tumor Progression and Restore Endogenous NK Cell Function in Humanized BLT Mice,” Cancers (Basel), vol. 17, no. 15, p. 2430, Aug. 2025. [CrossRef]
- K. Kaur and A. Jewett, “Differences in Tumor Growth and Differentiation in NSG and Humanized-BLT Mice; Analysis of Human vs. Humanized-BLT-Derived NK Expansion and Functions,” Cancers (Basel), vol. 15, no. 1, p. 112, Jan. 2023. [CrossRef]
- F. Koning Christelle Retière, D. Salvatori, D. H. Raulet, J. Jeroen van Bergen, A. Thompson, and M. van Pel, “Humanized Mouse Model Expression Level and Frequency in a HLA Reduces Killer Cell Ig-like Receptor Downloaded from,” J Immunol The Journal of Immunology at, vol. 190, pp. 2880–2885, 2013. [CrossRef]
- V. Landtwing et al., “Cognate HLA absence in trans diminishes human NK cell education,” J Clin Invest, vol. 126, no. 10, pp. 3772–3782, Oct. 2016. [CrossRef]
- L. Li et al., “Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering,” Sci Adv, vol. 9, no. 30, p. eadd6997, Jul. 2023. [CrossRef]
- T. T. Pham et al., “In Vivo PET Imaging of 89Zr-Labeled Natural Killer Cells and the Modulating Effects of a Therapeutic Antibody,” J Nucl Med, vol. 65, no. 7, pp. 1035–1042, Jul. 2024. [CrossRef]
- N. Sato et al., “In-vivo tracking of adoptively transferred natural killer-cells in rhesus macaques using 89Zirconium-oxine cell labeling and PET imaging,” Clin Cancer Res, vol. 26, no. 11, p. 2573, Jun. 2020. [CrossRef]
Figure 1.
Summary of the main passive and active NK cell-based immunotherapeutic strategies. Created in https://BioRender.com.
Figure 1.
Summary of the main passive and active NK cell-based immunotherapeutic strategies. Created in https://BioRender.com.
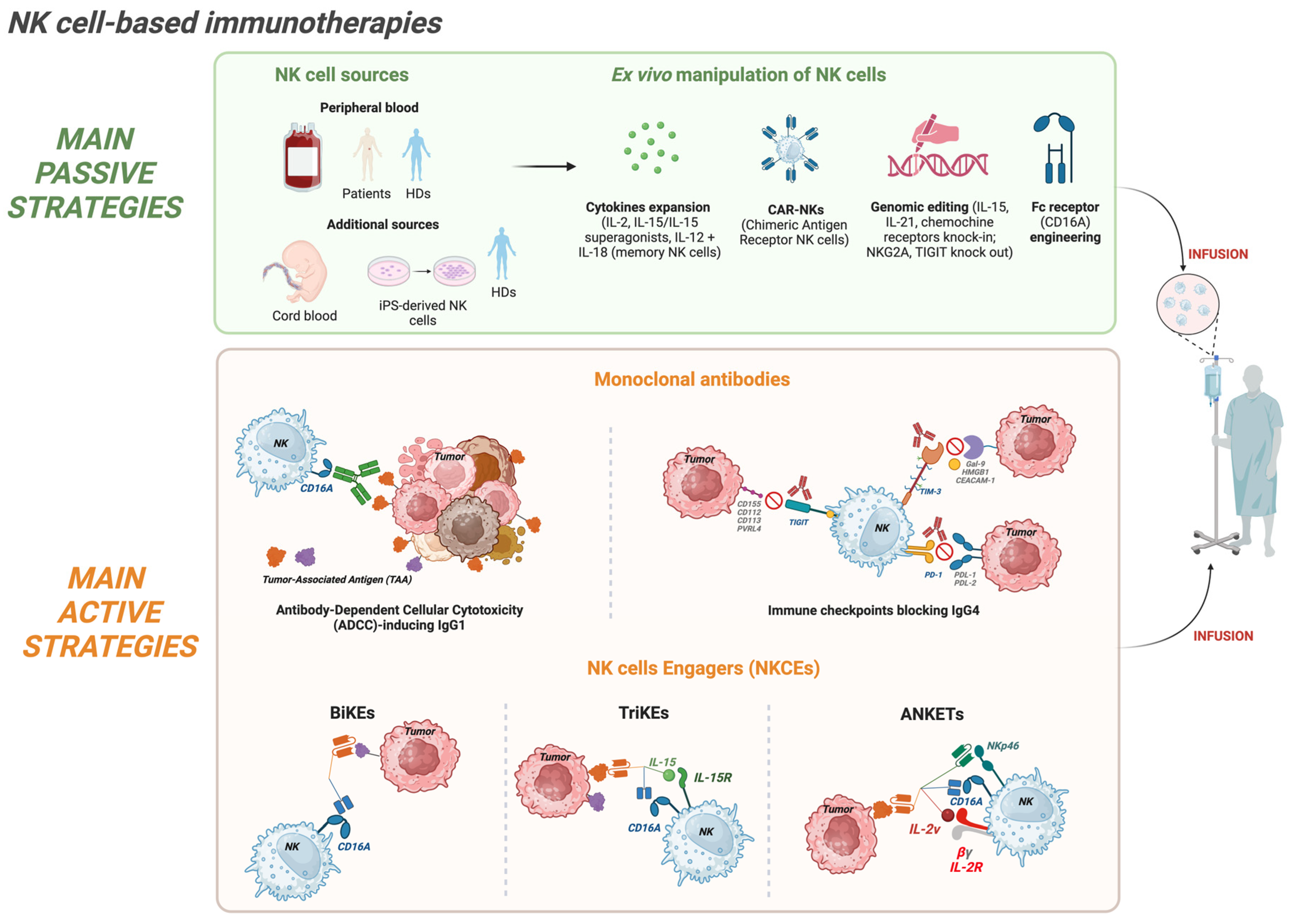
Figure 2.
Progressive evolution of humanized mouse models for studying human NK cell biology and anti-tumor functions. Created in https://BioRender.com.
Figure 2.
Progressive evolution of humanized mouse models for studying human NK cell biology and anti-tumor functions. Created in https://BioRender.com.
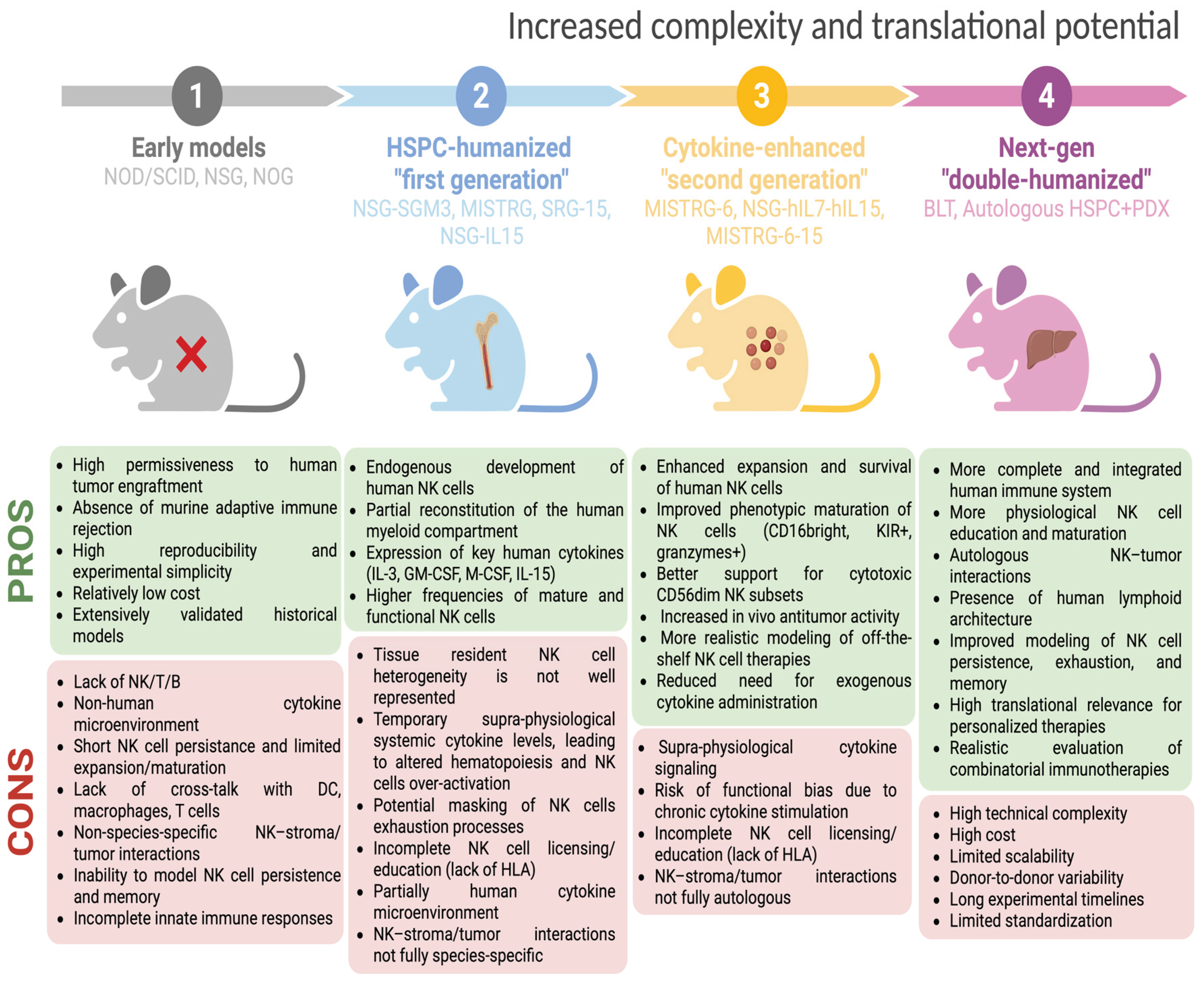
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



