Submitted:
18 December 2025
Posted:
19 December 2025
You are already at the latest version
Abstract
The consequences of stacking multiple insect-resistance and herbicide-tolerance genes, particularly across the entire plant life cycle, remain inadequately understood. This study investigated the impact of stacked-trait transgenic soybean (expressing cry1Ac, vip3Aa19, mOsPPO2, and pat genes) on rhizosphere microbial communities across five growth stages (pre-sowing, V3, R3, R5, R8). Using 16S rRNA and ITS sequencing, we compared the rhizosphere microbiome of the transgenic modified soybean (GMO) with its non-transgenic control check (CK). Results showed transient but significant shifts in soil properties (e.g., available nitrogen) and microbial beta-diversity during the V3 stages. However, plant developmental stage was the predominant factor shaping microbial succession, with its effect outweighing that of the transgene. No persistent changes in microbial alpha-diversity were observed. We conclude that the influence of this stacked-trait soybean on the rhizosphere is growth-stage-specific and represents a minor, recoverable perturbation rather than a sustained ecological impact. These findings contribute to the ecological safety assessment of multi-gene transgenic crops.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Genetically Modified Soybean
2.2. Determination of Physical and Chemical Properties of Rhizosphere Soil
2.3. DNA Extraction and Amplicon Sequencing
2.4. Bioinformatic Analysis of Amplicon Sequencing Data
3. Results
3.1. The Influence of GMO on Physical and Chemical Properties of Soil
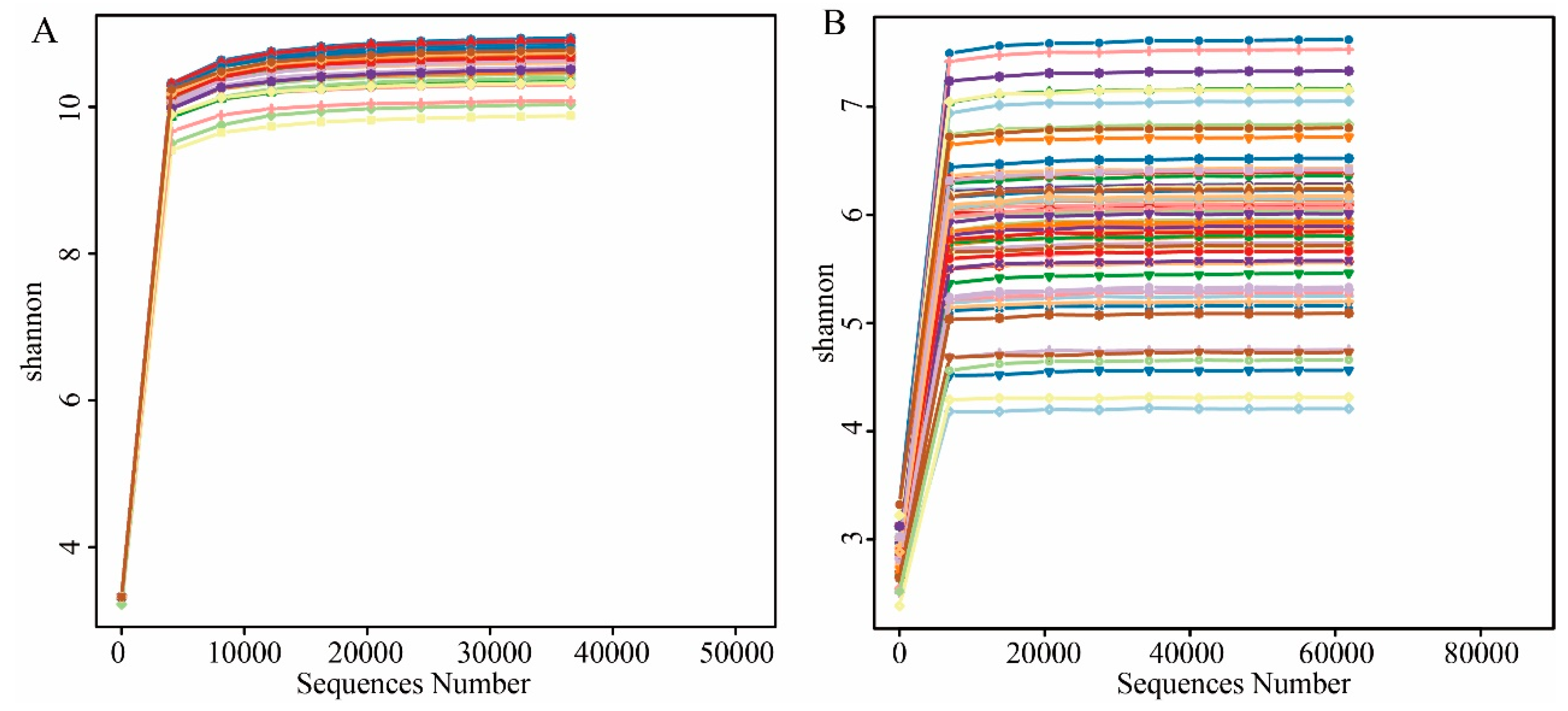
3.2. The Basic Information of Amplicon Sequencing Data
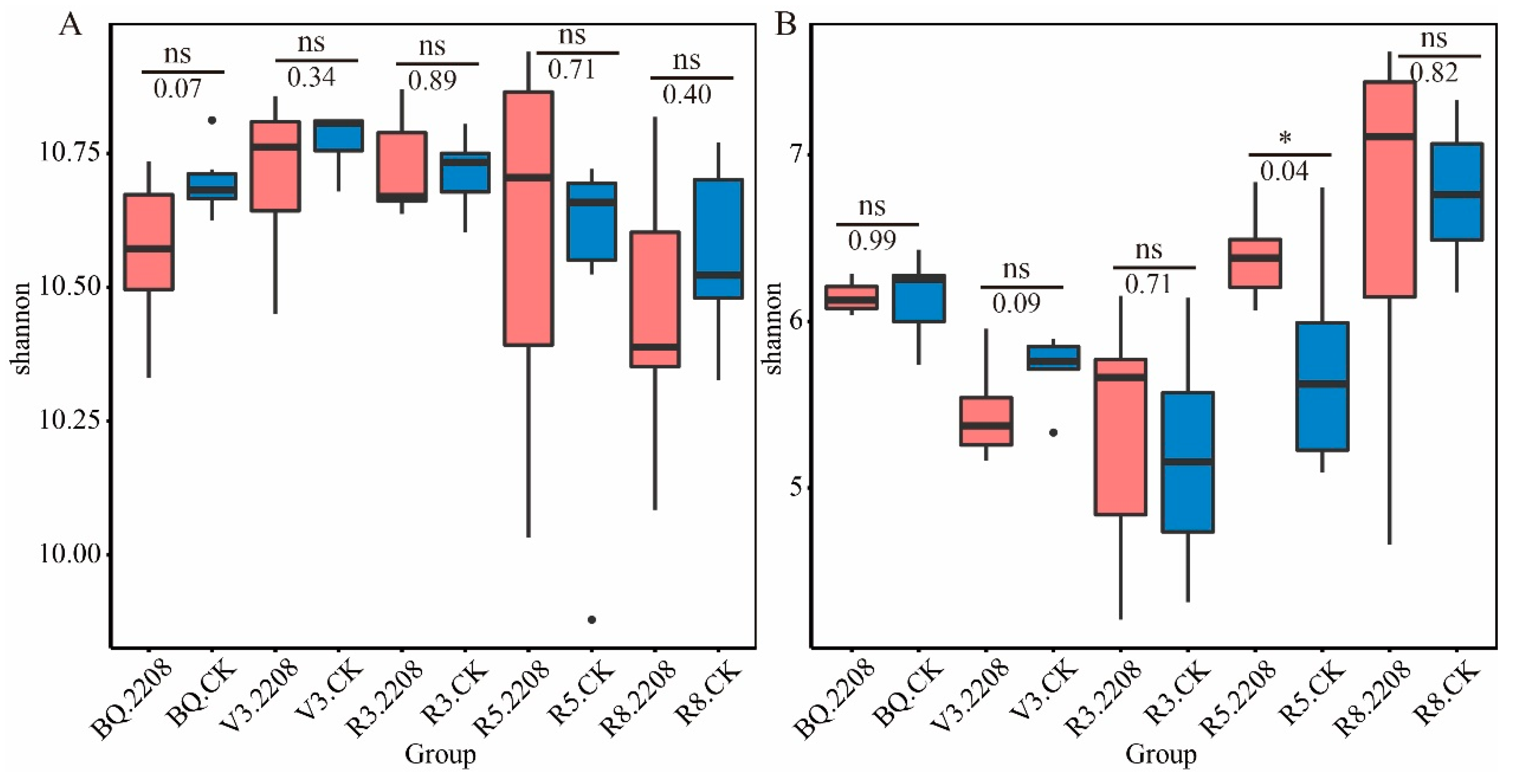
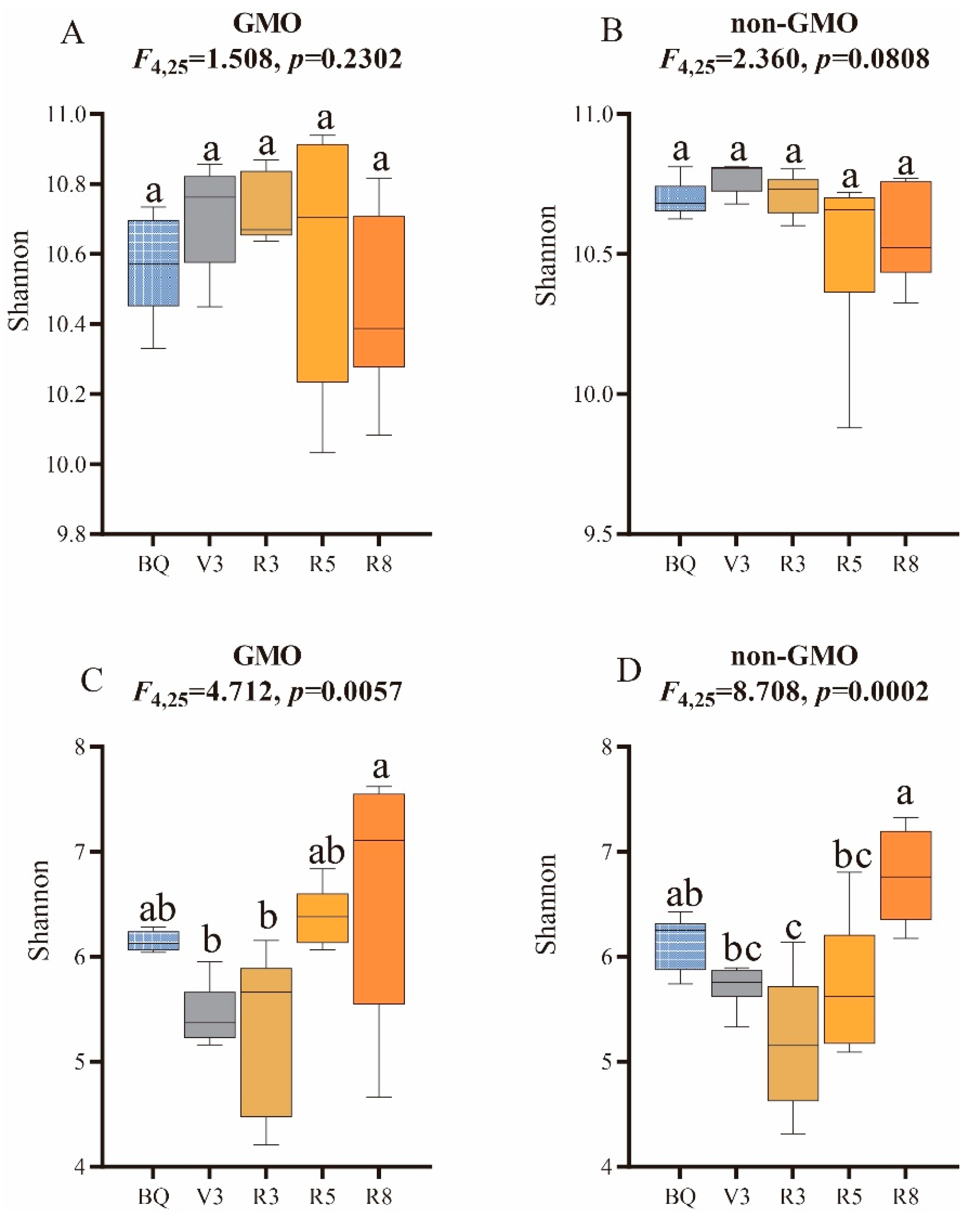
3.3. The Influence of GMO on Microbial Alpha Diversity of Soybean
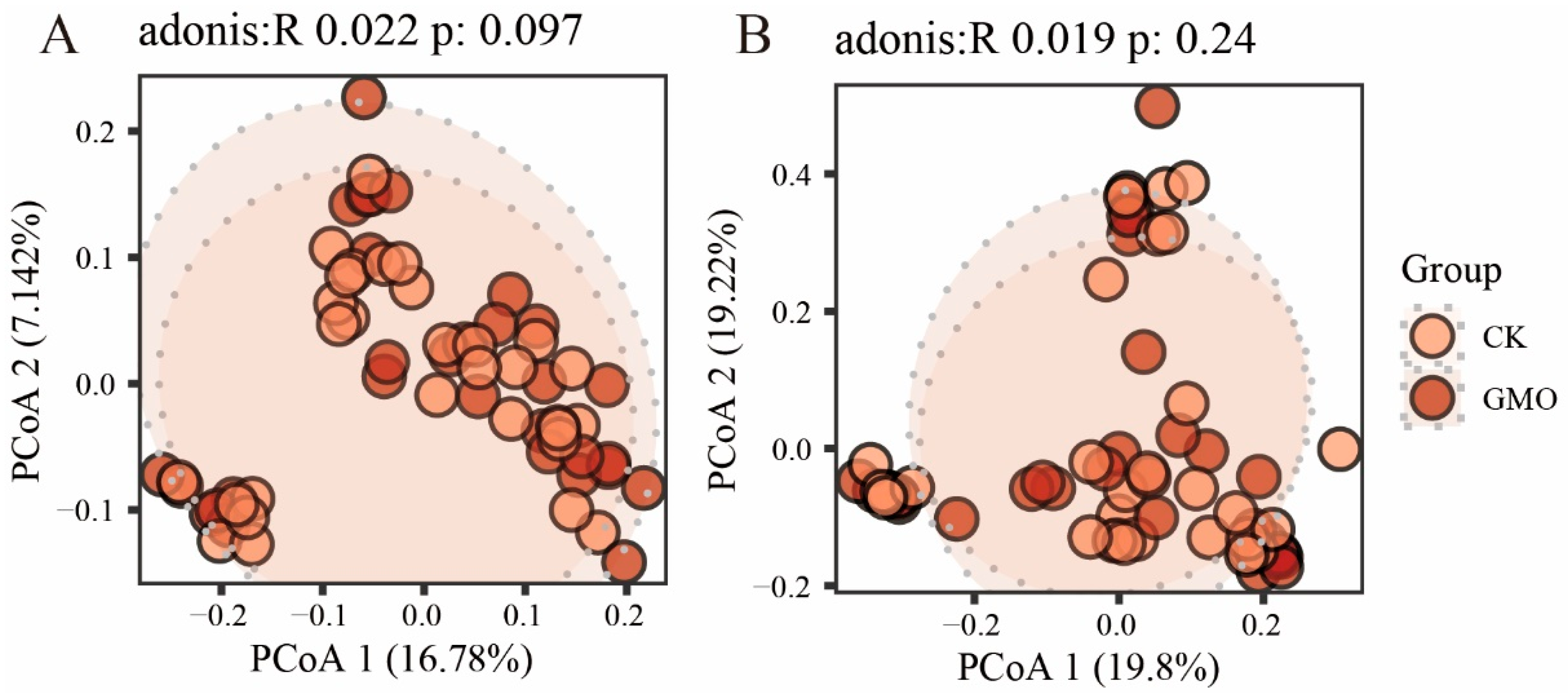
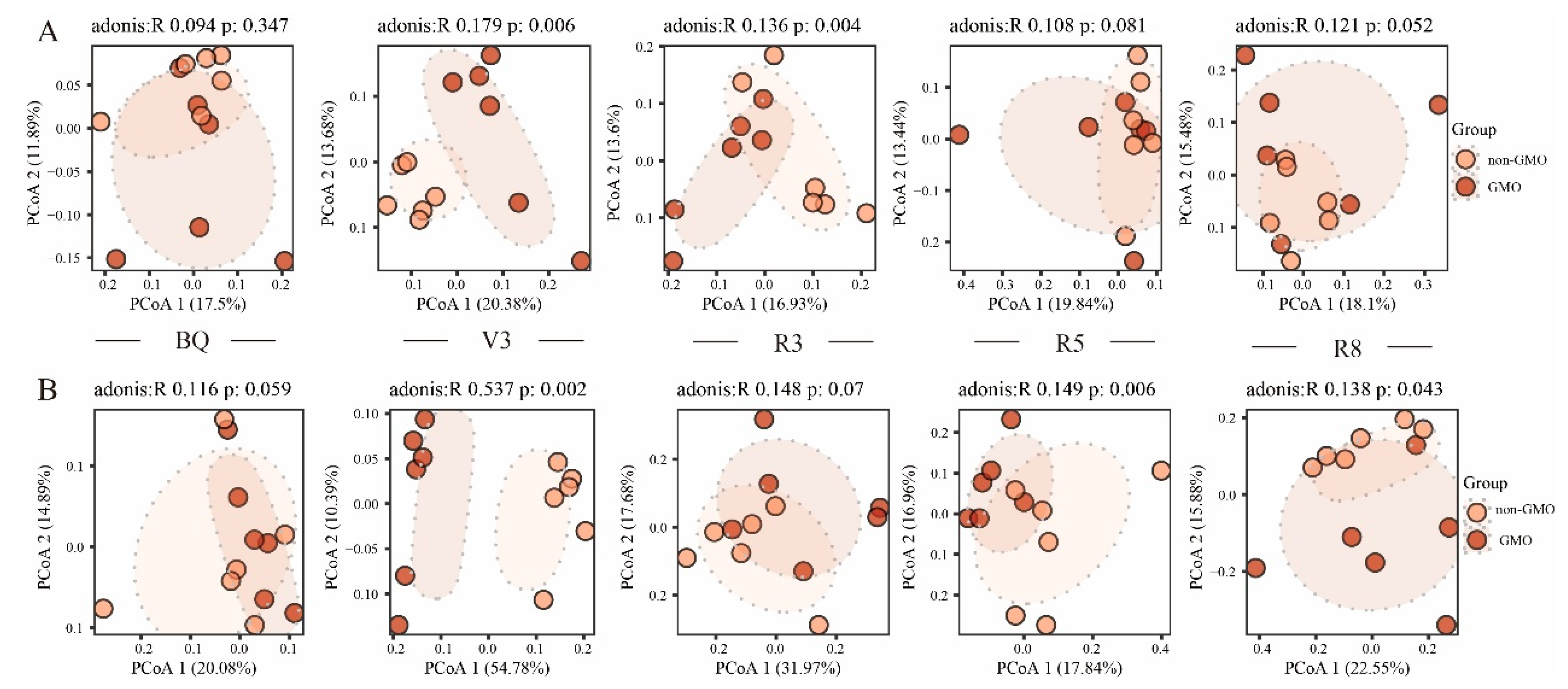
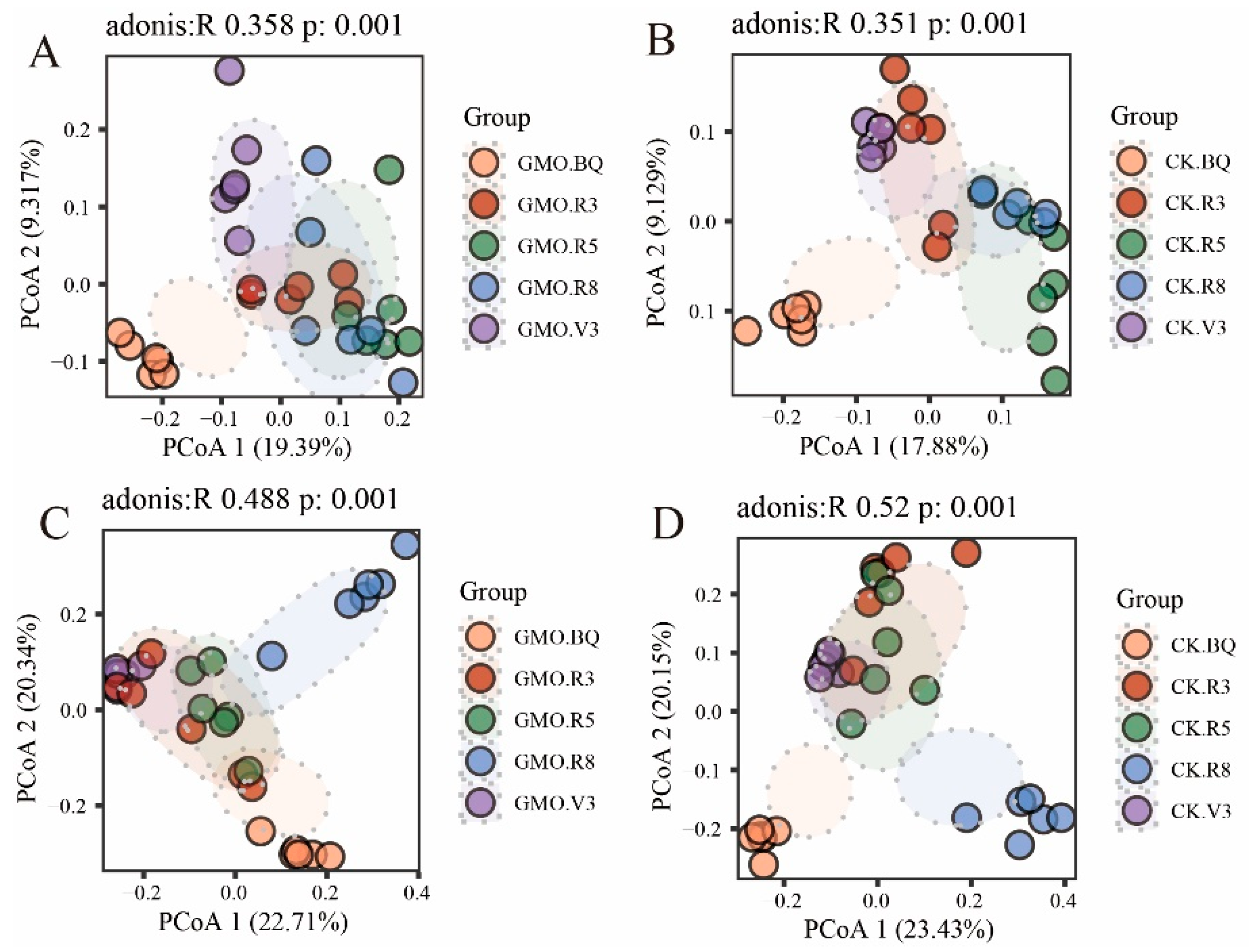
3.4. The Influence of GMO on Microbial Beta Diversity of Soybean
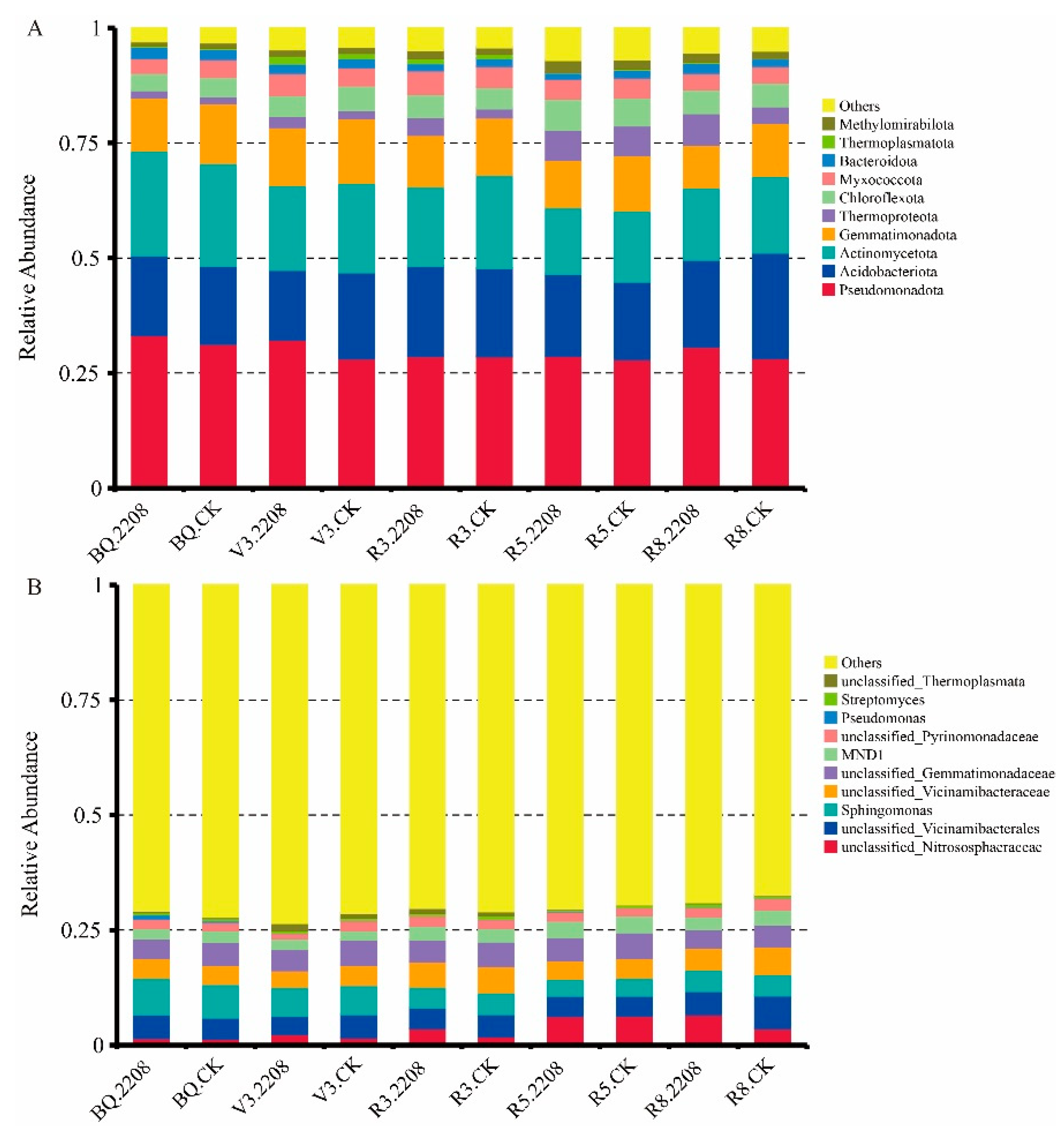
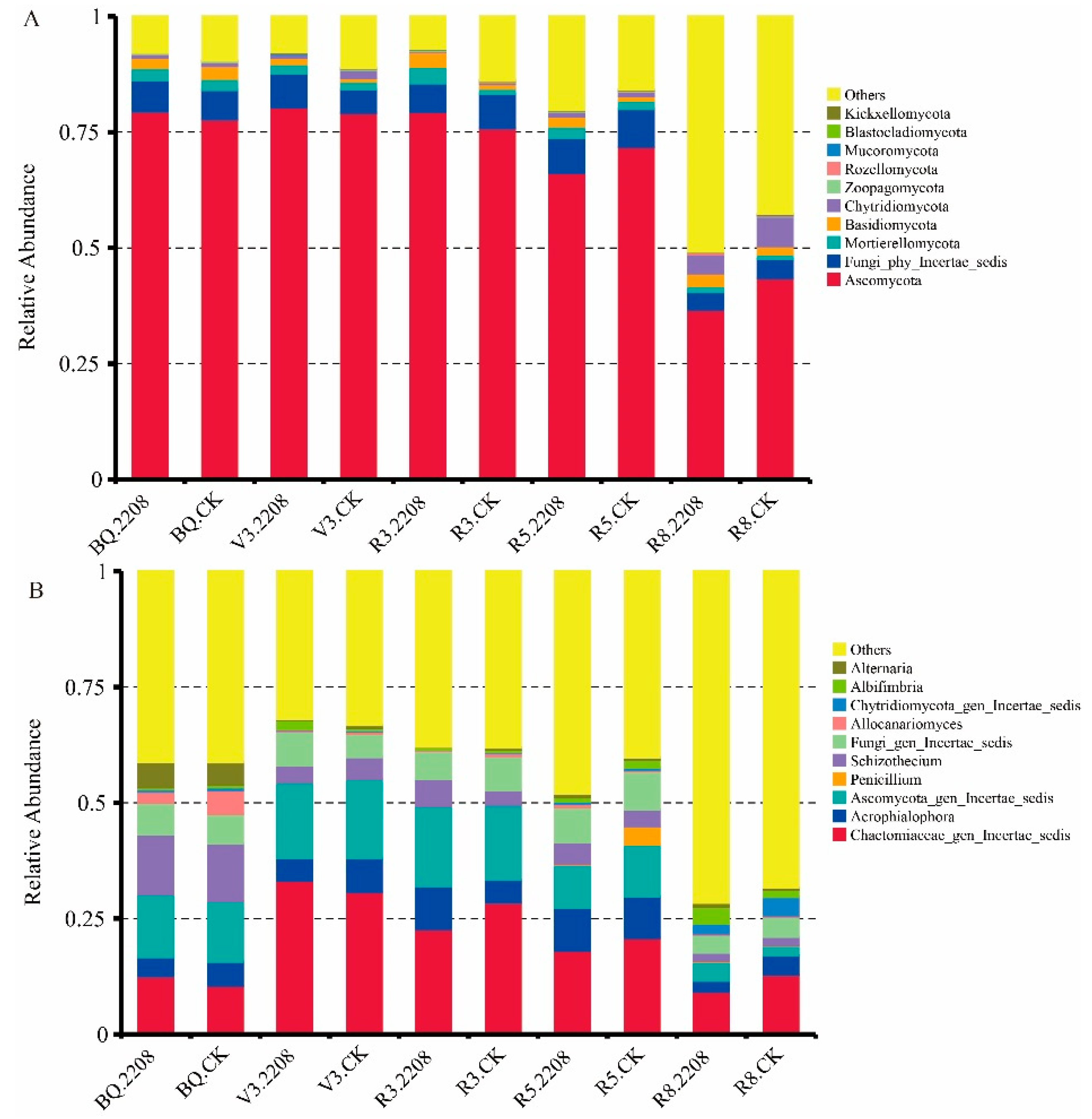
3.5. The Influence of GMO on Microbial Community Structure of Soybean
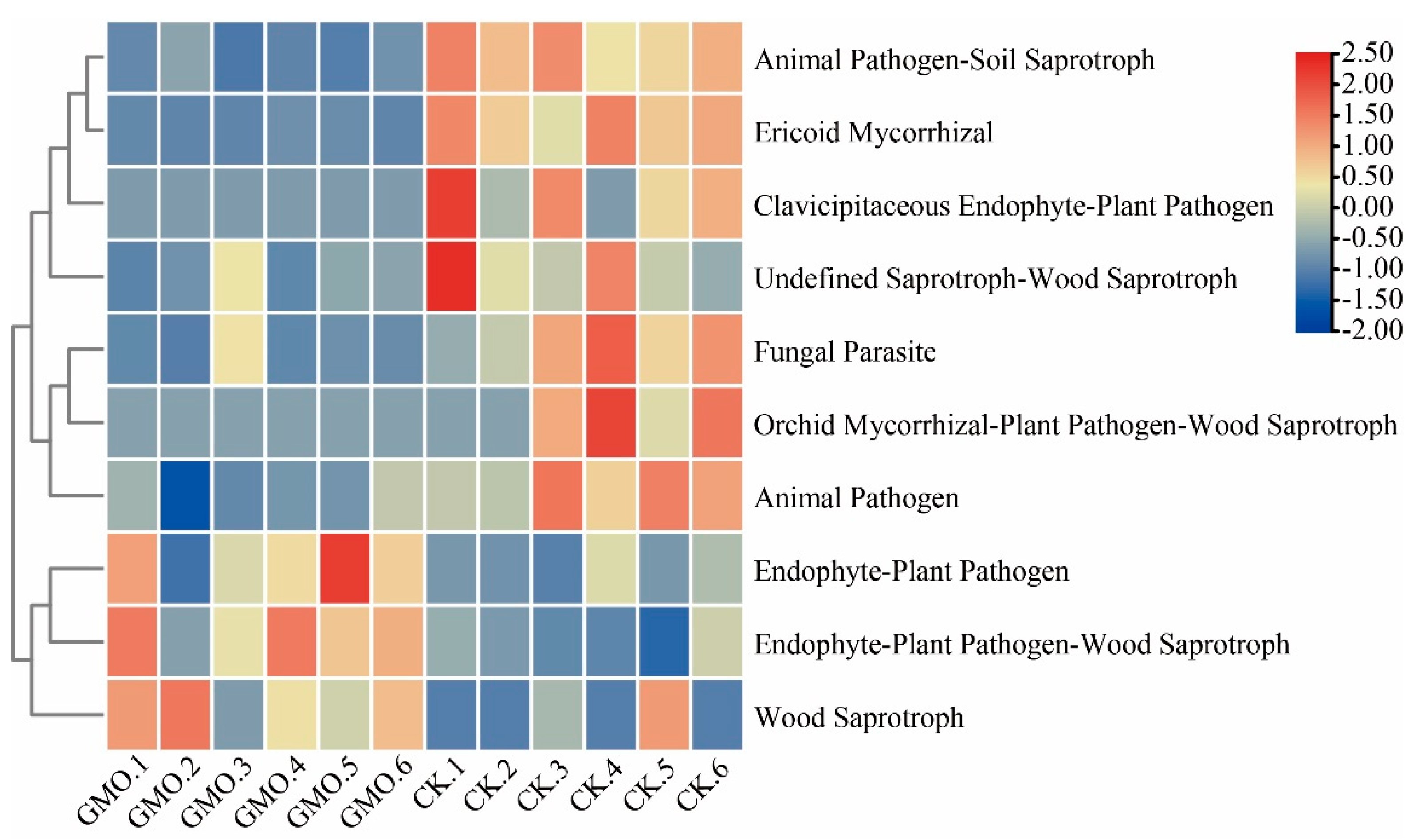
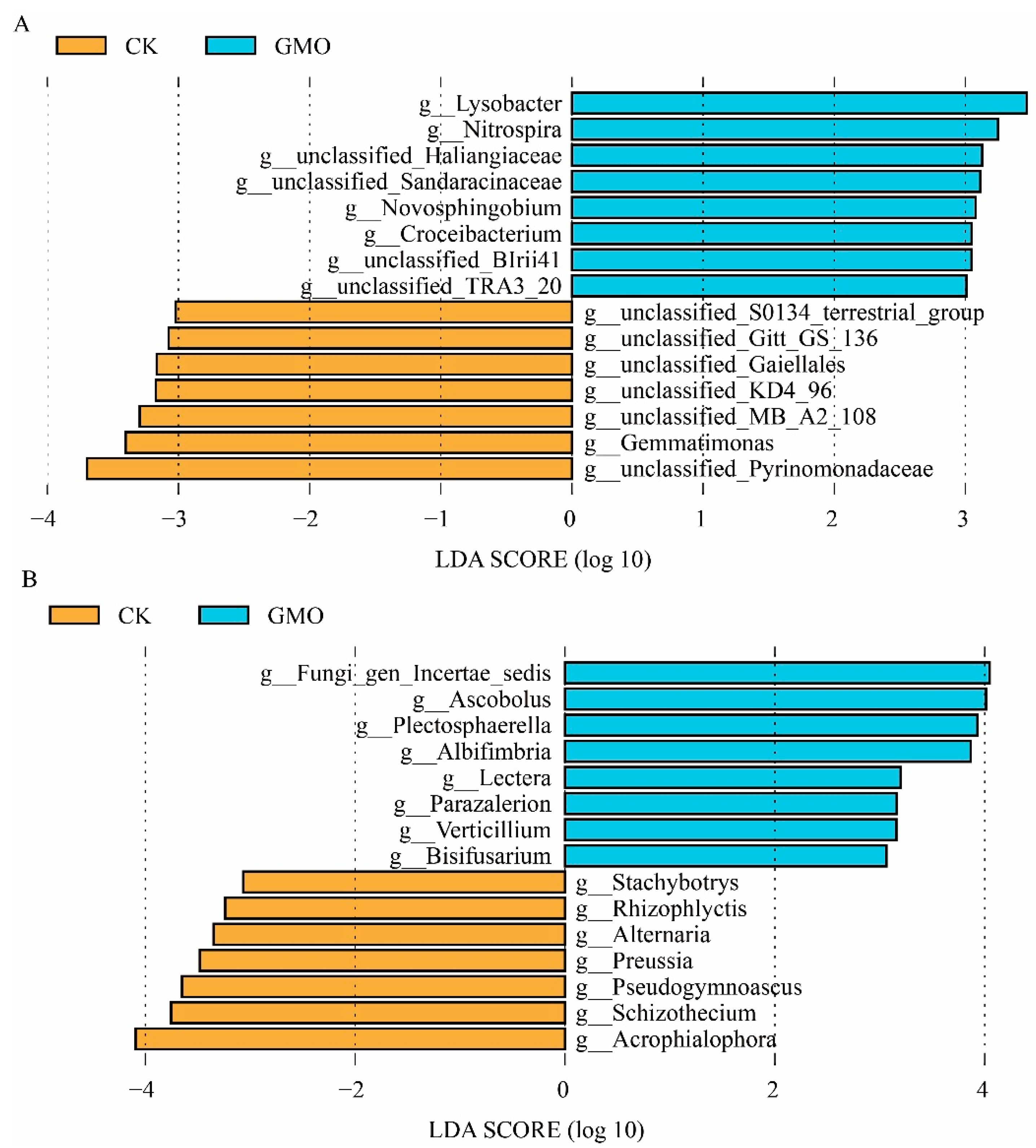
3.6. The Influence of GMO on Microbial Biomarkers of Soybean
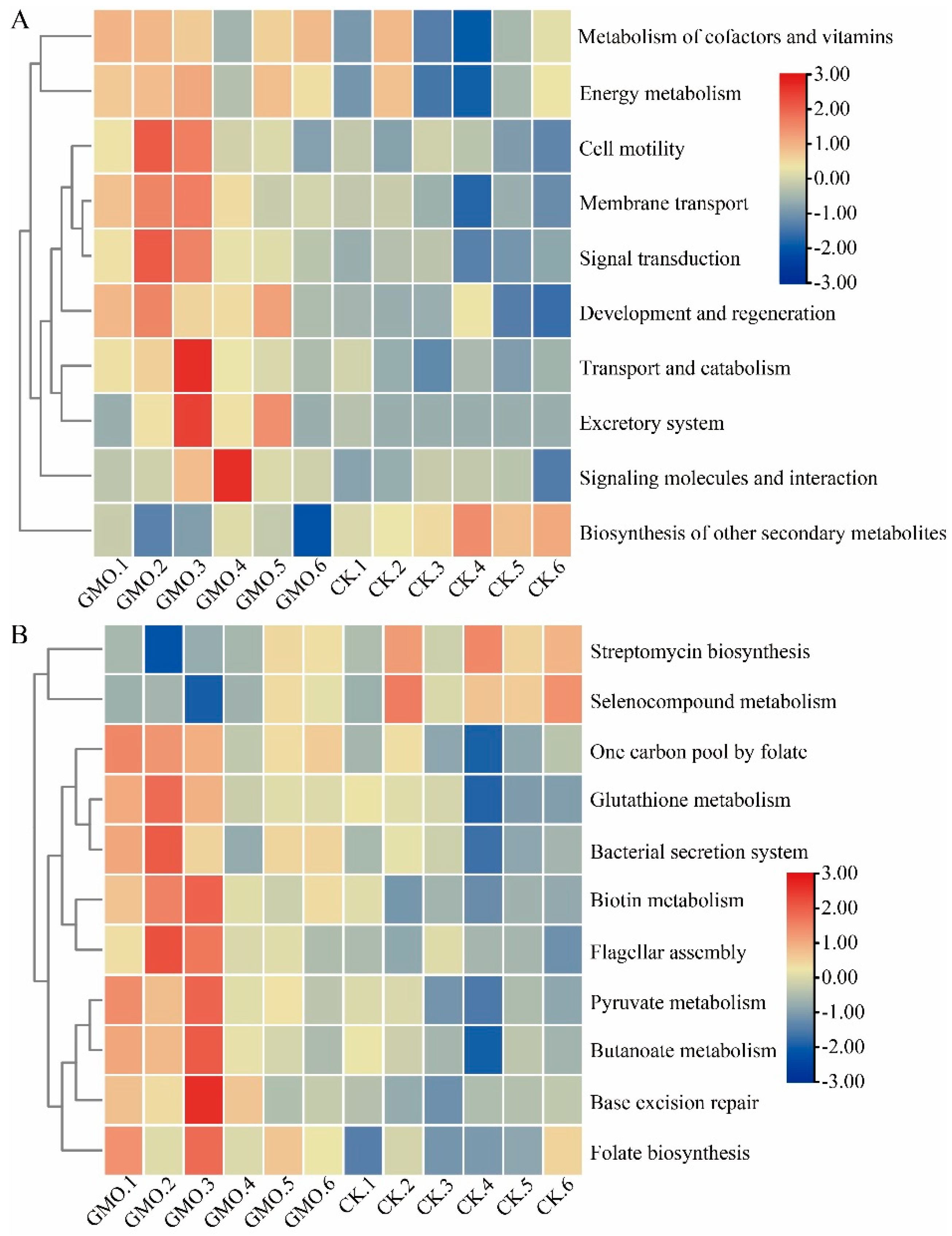
3.7. The Influence of GMO on Microbial Function of Soybean
4. Discussion
4.1. Stage-Specific Modulation of Rhizosphere Environment and Microbiome
4.2. Functional Implications of Microbial Community Shifts
4.3. Ecological Significance and Biosafety Perspective
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Council, N.R.; Plants, C.O.G.M.P.-P. enetically modified pest-protected plants: science and regulation, (2000).
- Yang, X.; Chen, S.; Liu, D.; Zhao, S.; Kang, G.; Song, C.; Tian, Z.; Li, H.; Chen, J.; Li, X.; et al. Insect resistant and glyphosate tolerant maize, Bt11 × MIR162 × GA21, can enhance management of fall armyworm and weeds in tropical Asia. Èntomol. Gen. 2024. [Google Scholar] [CrossRef]
- Ricroch, A.E.; Martin-Laffon, J.; Rault, B.; Pallares, V.C.; Kuntz, M. Next biotechnological plants for addressing global challenges: The contribution of transgenesis and new breeding techniques. New Biotechnol. 2022, 66, 25–35. [Google Scholar] [CrossRef]
- Chen, Q.; Song, Y.; An, Y.; Lu, Y.; Zhong, G. Soil Microorganisms: Their Role in Enhancing Crop Nutrition and Health. Diversity 2024, 16, 734. [Google Scholar] [CrossRef]
- Snelders, N.C.; Rovenich, H.; Thomma, B.P.H.J. Microbiota manipulation through the secretion of effector proteins is fundamental to the wealth of lifestyles in the fungal kingdom. FEMS Microbiol. Rev. 2022, 46. [Google Scholar] [CrossRef]
- Sun, K.; Niu, Z.; Xiao, S.; Qi, X.; Li, S.; Chen, M.; Dai, L.; Si, Y. Artificially regulated humification in creating humic-like biostimulators. npj Clean Water 2024, 7, 47. [Google Scholar] [CrossRef]
- Bano, S.; Wu, X.; Zhang, X. Towards sustainable agriculture: rhizosphere microbiome engineering. Appl. Microbiol. Biotechnol. 2021, 105, 7141–7160. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Duan, Y.; Wei, R.; Yuan, Y.; Yan, H.; Tang, T.; Shang, H. Rhizosphere Microbiome and Nutrient Fluxes Reveal Subtle Biosafety Signals in Transgenic Cotton. Microorganisms 2025, 13, 2702. [Google Scholar] [CrossRef]
- Sharma, B.; Sharma, K.; Sharma, P.; Gupta, I.; Kour, J.; Kour, K. Genetically modified maize. In Genetically Modified Crops and Food Security; Routledge, 2022; pp. 85–96. [Google Scholar]
- Lynch, J.M.; Benedetti, A.; Insam, H.; Nuti, M.P.; Smalla, K.; Torsvik, V.; Nannipieri, P. Microbial diversity in soil: ecological theories, the contribution of molecular techniques and the impact of transgenic plants and transgenic microorganisms. Biol. Fertil. Soils 2004, 40, 363–385. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, Y.; Li, X. USEARCH 12: Open-source software for sequencing analysis in bioinformatics and microbiome. iMeta 2024, 3, e236. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- R. Santos, Silva, FNO, Fernandes, AM, Teixeira, FG, Soto, DAS, Brito, CJ, & Miarka, B. 2022.
- Kõljalg, U.; Larsson, K.-H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; et al. UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 2005, 166, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- de Micheaux, P.L.; Drouilhet, R.; Liquet, B. The R software. Fundamentals of programming and statistical analysis 2013, 978–971. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘vegan’, Community ecology package; version 2, 2013, 1-295.
- Wickham, H.; Wickham, M.H. Package tidyverse, Easily install and load the ‘Tidyverse; 2017. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Lehman, R.M.; Cambardella, C.A.; Stott, D.E.; Acosta-Martinez, V.; Manter, D.K.; Buyer, J.S.; Maul, J.E.; Smith, J.L.; Collins, H.P.; Halvorson, J.J.; et al. Understanding and Enhancing Soil Biological Health: The Solution for Reversing Soil Degradation. Sustainability 2015, 7, 988–1027. [Google Scholar] [CrossRef]
- Khan, N. Decoding the long-term impacts of genetic modifications in hormone pathways on plant physiology and ecosystem stability. Environ. Rev. 2025, 33, 1–20. [Google Scholar] [CrossRef]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P.; et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Ajilogba, C.F.; Olanrewaju, O.S.; Babalola, O.O. Plant Growth Stage Drives the Temporal and Spatial Dynamics of the Bacterial Microbiome in the Rhizosphere of Vigna subterranea. Front. Microbiol. 2022, 13, 825377. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Onyino, J.; Gao, X. Current Advances in the Functional Diversity and Mechanisms Underlying Endophyte–Plant Interactions. Microorganisms 2024, 12, 779. [Google Scholar] [CrossRef] [PubMed]
- Baetz, U.; Martinoia, E. Root exudates: the hidden part of plant defense. Trends Plant Sci. 2014, 19, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Icoz, I.; Stotzky, G. Fate and effects of insect-resistant Bt crops in soil ecosystems. Soil Biol. Biochem. 2008, 40, 559–586. [Google Scholar] [CrossRef]
- Dyer, A.R.; Knapp, E.E.; Rice, K.J. Unintentional Selection and Genetic Changes in Native Perennial Grass Populations During Commercial Seed Production. Ecol. Restor. 2016, 34, 39–48. [Google Scholar] [CrossRef]

| Indexes | Groups | BQ | V3 | R3 | R5 | R8 |
|---|---|---|---|---|---|---|
| TK (g/Kg) |
GMO | 22.70±1.68a | 19.27±0.54a | 19.77±0.70a | 19.82±1.07a | 19.68±0.26a |
| CK | 20.08±0.67b | 20.05±0.64a | 20.43±0.63a | 20.20±0.76a | 19.87±0.58a | |
| AK (mg/Kg) |
GMO | 914.00±52.29a | 1012.50±51.72a | 1055.83±58.37a | 1002.00±100.73a | 1013.00±36.3a |
| CK | 946.67±41.97a | 1024.83±62.23a | 1056.00±48.43a | 1064.00±55.17a | 996.17±38.97a | |
| EN (mg/Kg) |
GMO | 162.00±42.09a | 128.43±21.92a | 95.03±9.29a | 103.33±22.82a | 92.77±16.81a |
| CK | 182.33±43.28a | 111.90±15.8b | 114.15±16.8b | 100.75±23.48a | 100.35±18.05a | |
| pH | GMO | 7.35±0.14a | 6.97±0.19a | 7.37±0.27a | 7.00±0.18b | 7.12±0.08b |
| CK | 7.42±0.04a | 7.38±0.26a | 7.62±0.15a | 7.62±0.33a | 7.68±0.19a | |
| OM (g/Kg) |
GMO | 19.98±0.71a | 17.77±0.69b | 16.85±0.78b | 18.38±0.83a | 20.33±1.66a |
| CK | 19.88±0.53a | 18.95±0.32a | 20.28±2.36a | 19.47±1.34a | 19.18±1.99a | |
| TP (g/Kg) |
GMO | 0.72±0.31a | 1.32±0.02a | 1.29±0.06a | 1.32±0.1a | 1.33±0.07a |
| CK | 1.31±0.04a | 1.26±0.07a | 1.38±0.06a | 1.24±0.11a | 1.26±0.15a | |
| AP (mg/Kg) |
GMO | 152.67±7.34a | 150.83±11.63a | 125.00±9.14b | 125.17±14.11a | 130.83±15.16a |
| CK | 144.5±11.84a | 137.83±14.41a | 136.17±5.64a | 115.83±17.99a | 118.33±12.27a | |
| TN (%) |
GMO | 0.15±0.00a | 0.13±0.00b | 0.12±0.00b | 0.13±0.00a | 0.14±0.01a |
| CK | 0.15±0.00a | 0.14±0.00a | 0.14±0.01a | 0.14±0.01a | 0.14±0.01a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



