
Submitted:
17 December 2025
Posted:
17 December 2025
You are already at the latest version
Abstract
Background: Mammary ductal hyperplasia represents a spectrum of benign proliferative breast lesions, some of which pose elevated risks for malignant transformation into ductal carcinoma in situ and invasive breast cancer. This narrative review explores why only specific types, particularly those with atypia, exhibit higher progression potential, synthesizing epidemiologic, histopathologic, molecular, and environmental insights. Methods: We reviewed key literature from databases, including PubMed, focusing on classification, risk stratification, genetic/epigenetic mechanisms, tumor microenvironment dynamics, and modifiable factors influencing progression. Results: Benign breast lesions are categorized into non-proliferative, proliferative without atypia, and proliferative with atypia, such as atypical ductal hyperplasia and atypical lobular hyperplasia. Atypia represents a morpho-logic continuum toward low-grade ductal carcinoma in situ, driven by genetic alterations, epigenetic reprogramming, and changes in the tumor microenvironment, including stromal remodeling, immune infiltration, hypoxia-induced angiogenesis, and extracellular matrix degradation. Dietary factors, such as high-fat intake and obesity, exacerbate progression through inflammation, insulin resistance, and adipokine imbalance, while environmental toxins, including endocrine disruptors, pesticides, and ionizing radiation, amplify genomic instability. Conclusions: Understanding differential risks and mechanisms underscores the need for stratified surveillance, biomarker-driven interventions, and lifestyle modifications to mitigate progression. Future research should prioritize molecular profiling for personalized prevention in high-risk hyperplasia.
Keywords:
mammary ductal hyperplasia
; malignant transformation
; genetic alterations
; breast cancer
; diet and environmental factors
1. Introduction
Mammary ductal hyperplasia (MDH) is a condition characterized by the abnormal growth of cells within the breast’s milk ducts. This condition can vary in severity, and while it is considered a benign lesion, it is often regarded as a potential precursor or risk factor for the development of breast carcinoma, especially when associated with other pathological changes. The relationship between different types of mammary hyperplasia and the subsequent development of carcinoma is complex, influenced by both genetic and environmental factors.
A comprehensive literature search was conducted between January 2024 and November 2025 using PubMed/MEDLINE as the primary database. The search strategy combined the following key terms: (“ductal hyperplasia” OR “atypical ductal hyperplasia” OR “atypical hyperplasia” OR “flat epithelial atypia” OR “usual ductal hyperplasia” OR “proliferative breast disease”) AND (“breast cancer risk” OR “malignant progression” OR “precursor lesion” OR “genetic alteration*” OR “PIK3CA” OR “tumor microenvironment” OR “stromal remodeling” OR “immune infiltration” OR “angiogenesis” OR “diet*” OR “obesity” OR “endocrine disruptors” OR “environmental factors”). No date restrictions were applied to the electronic search. Still, priority was given to studies published after 2015.
Only peer-reviewed articles published in English were included. Conference abstracts, case reports, and non-English publications were excluded. Studies were selected based on relevance to histologic classification and risk stratification, genetic/epigenetic drivers, tumor microenvironment dynamics, dietary/obesity-related mechanisms, and environmental toxicants influencing progression from hyperplasia to carcinoma. Over 220 references were evaluated; 209 were ultimately cited. The content was synthesized narratively without formal meta-analysis, with emphasis on mechanistic coherence and clinical translation. Figures were created using professional BioRender software with publishing license.
2. Mammary Ductal Hyperplasia (MDH) and Its Types
Breast carcinogenesis is often framed as a multistep evolution from benign epithelial alterations toward ductal carcinoma in situ (DCIS) and eventually invasive breast carcinoma. Yet in practice, most benign epithelial proliferations remain morphologically stable throughout a woman’s lifetime. Discriminating between proliferative lesions that are truly at risk and those that are innocuous is fundamental to guiding surveillance, biopsy decisions, and chemoprevention strategies. A useful conceptual schema subdivides benign mammary epithelial alterations into three broad categories:
- nonproliferative changes;
- proliferative hyperplasia without cytologic atypia
- proliferative hyperplasia with cytologic atypia [1].
In what follows, we review key entities in each category, summarize the epidemiologic evidence on differential cancer risk, and explore plausible mechanistic explanations for why only specific hyperplastic lesions tend to evolve.
2.1. Classification of Benign Breast Lesions and Associated Risks
2.1.1. Nonproliferative Changes
Nonproliferative lesions are those lacking significant epithelial proliferation. Representative lesions include simple cysts, ductal ectasia, apocrine metaplasia, mild epithelial hyperplasia without proliferation, and stromal fibrosis [2]. In large cohorts, women whose benign biopsy reveals only nonproliferative changes do not display a significantly increased risk of subsequent breast cancer relative to the background population. Indeed, nonproliferative disease is used as the baseline reference in risk stratification in many studies [3]. In select analyses, marginal associations between particular nonproliferative features (e.g., microcalcifications, mild epithelial change) and later cancer risk have been reported, although effect sizes are modest and often attenuated with multivariable adjustment. In the classic Hartmann et al. cohort of 9,087 women with benign breast disease, nonproliferative lesions had a relative risk (RR) for breast cancer of 1.27 (95% CI 1.15–1.41) compared to general population, that is lower than in proliferative disease without atypia (RR ~ 1.88) or atypical hyperplasia (RR ~ 4.24) [3].
A recent systematic review emphasized that the breast cancer risk associated with benign breast disease is heterogeneous, largely determined by the extent of epithelial proliferation and the presence or absence of cytologic atypia. While benign breast disease in general correlates with an elevated incidence of breast cancer compared to women without such findings, the magnitude of this increase is substantially lower for nonproliferative lesions than for either proliferative lesions without atypia or atypical hyperplasia [4]. Consequently, nonproliferative disease represents the lowest-risk category within the benign breast disease spectrum.
2.1.2. Proliferative Hyperplasia Without Atypia
This category encompasses lesions with increased epithelial cell numbers but without definite cytologic atypia. Key entities include: usual ductal hyperplasia (UDH), sclerosing adenosis, radial scars, complex sclerosing lesions, intraductal papillomas without atypia, complex proliferative lesions or multifocal proliferative foci.
Epidemiologic evidence indicates that proliferative lesions without atypia confer a modest increase in breast cancer risk—typically in the range of 1.5- to 2.0-fold compared to nonproliferative disease.
For example, in a large case-control nested in benign breast disease cohorts, proliferative lesions without atypia had an adjusted odds ratio (OR) of approximately 1.45 relative to nonproliferative disease [7]. Other classic studies similarly estimate a RR of 1.8–1.9 [3].
With a focus solely on certain lesional entities, UDH is characterized by a haphazard proliferation of heterogeneous epithelial cells forming slit-like spaces, bridging projections, and irregular architecture. Because of its disorganized morphology and lack of monoclonal dominance, UDH is generally considered a marker of increased risk rather than a direct precursor lesion [3,8]. Currently, no reliable prognostic indicators exist to determine which patients with UDH will progress to invasive breast carcinoma. The associated risk level is comparable to that conferred by certain reproductive characteristics, such as early onset of menarche or delayed menopause, and therefore does not justify modification of routine mammographic screening intervals [9].
Radial scars and complex sclerosing lesions often harbor adjacent proliferative changes, and some series report modest independent risk contributions or occasional occult carcinoma [10]. Although once considered potential precursors, current evidence does not confirm a premalignant nature for radial scars. Their modestly increased breast cancer risk (RR of approximately 1.4–1.7) likely reflects coexisting proliferative changes rather than intrinsic malignancy. Larger lesions (>0.6 cm), more common in women over 50, may coexist with atypical hyperplasia or carcinoma [11]. Optimal management of mammographically detected radial scars and complex sclerosing lesions remains debated. Lesions showing epithelial atypia on core biopsy carry a malignancy risk warranting excision [12,13], whereas recent studies suggest that lesions with concordant imaging and pathologic aspects lacking atypia might be managed conservatively [14,15,16].
To sum up, proliferative disease without atypia constitutes an intermediate-risk stratum but, by itself, lacks many of the hallmarks of progression to carcinoma, such as architectural disorder or clonal expansion.
2.1.3. Proliferative Hyperplasia with Atypia
This is the high-risk category, containing principally atypical ductal hyperplasia (ADH) and atypical lobular hyperplasia (ALH), and, in some classification schemes, flat epithelial atypia (FEA). These lesions combine proliferation with cytologic uniformity, partial loss of polarity, and architectural disturbance.
Atypical hyperplasia is identified in 4 to 10% of benign breast biopsies performed for imaging or palpable lesions [5,9]. Multiple long-term follow-up cohorts and meta-analyses demonstrate that proliferative hyperplasia with atypia confers a 3- to 5-fold RR of future breast carcinoma compared to nonproliferative disease [3,17,18,19]. In the Mayo cohort, the estimated cumulative incidence of breast cancer at 25 years in women with atypical hyperplasia was approximately 29% [9]. Recent large-scale analyses using contemporary imaging and biopsy data refined these estimates. In the Breast Cancer Surveillance Consortium cohort, the 10-year incidence of invasive carcinoma after ADH was approximately 5 to 6%, slightly lower than previous figures, with risk modestly higher in excisional than in core needle biopsies (6.7% vs 5%) [20]. These findings suggest that smaller ADH foci, increasingly detected by modern imaging, may carry a lower absolute risk than in earlier cohorts, although ADH remains a significant marker of long-term susceptibility.
The often-quoted approximately 1% annual risk for atypical hyperplasia is a useful heuristic, though actual risk is modulated by age, family history, breast density, and other lesion features [21].
2.1.4. Subtype Nuances: FEA, ADH vs ALH and the Extent of Atypia
FEA represents clonal proliferation of mildly atypical epithelial cells lining terminal duct lobular units, typically associated with microcalcifications and incidentally discovered during mammographic screening [22]. Although limited data exist, evidence indicates that FEA may occasionally precede invasive breast carcinoma [23]. Nonetheless, the probability of progression is very low, and its risk magnitude is considerably lower than that of ADH or ALH [24]. Consequently, FEA should not be managed identically to these higher-risk atypical proliferations.
Several retrospective analyses have reported that up to one-third of FEA lesions identified on core needle biopsy may reveal a more advanced lesion upon surgical excision. However, these findings are inconsistent due to small sample sizes, variable histologic criteria, and differences in radiologic–pathologic correlation, resulting in wide variability in reported upgrade rates. Current evidence suggests that surgical excision is not uniformly required after a diagnosis of pure FEA, particularly when post-biopsy imaging confirms complete removal of the calcifications associated with the lesion. Instead, careful radiologic–pathologic concordance is advised to determine appropriate follow-up and to ensure that management remains individualized and evidence-based [25,26].
ADH is frequently conceptualized as a direct, though non-obligate precursor of low-grade DCIS, whereas ALH is considered primarily a risk indicator or a marker of a susceptible field [27]. In practice, both types are associated with subsequent ductal carcinoma, and their clinical outcomes are often similar [27].
The question of whether the extent (number of foci) of atypia modulates risk has been explored. In the nested case-control study by Collins et al., among women with ADH, increasing the number of foci (≥3) did not significantly increase OR beyond that of 1–2 foci (OR 2.7 vs 3.5, p = 0.58) [28]. In ALH, there was a trend toward higher OR with ≥3 foci (OR 8.0) vs 1–2 foci (OR 5.2), though this was not statistically robust (p = 0.66). Other series suggest some stratification by extent, but the evidence is not consistent enough to uniformly guide management [29]. In another systematic review of determinants of breast cancer risk in women with atypia, use of menopausal hormone therapy, timing of follow-up, and other factors were also modifiers of risk [30].
2.1.5. Upgrade Rates and Interpretive Caution
A recurring complication is that many cases of atypical hyperplasia diagnoses on core-needle biopsy are upgraded to DCIS or invasive carcinoma on subsequent excisional pathology. This upgrade phenomenon clouds the boundary between true progression versus initial under-sampling of coexistent malignancy [21]. Meta-analyses of core-biopsy-diagnosed ADH yield upgrade rates to malignancy in the range of 10–30%, depending on biopsy technique, lesion characteristics, and institutional practice [31]. Disentangling upgrades from genuine evolution remains an ongoing challenge, requiring rigorous pathological and imaging correlation and thoughtful clinical decision-making.
Therefore, the histologic classification of benign breast lesions remains highly relevant to stratified surveillance. For nonproliferative disease, standard screening per population guidelines is generally appropriate. In proliferative disease without atypia, a modest risk elevation may justify closer surveillance in the context of additional risk factors. On the other hand, because atypical hyperplasia confers substantial long-term risk, many centers recommend enhanced surveillance (annual mammography plus supplemental MRI) and consideration of risk-reducing interventions. Excision of the atypical lesion does not eliminate the residual elevated risk conferred by the breast environment; therefore, continued vigilance is required [22,32].
2.2. Mechanistic Frameworks: Why Some Lesions Progress?
Although the above classification and epidemiologic data stratify risk, they do not explain why only particular lesions (especially those with atypia) tend to evolve toward malignancy. Below we synthesize current mechanistic hypotheses, enriched by recent molecular studies, as well as represented graphically in Figure 1
2.2.1. Degree of Atypia
The presence of cytologic and architectural atypia is perhaps the single most important histologic feature distinguishing benign proliferations that merely signify elevated risk from those that embody early steps in the neoplastic continuum. Within atypical hyperplasia, the severity of cytologic atypia may correlate with proximity to malignant features (e.g., higher nuclear grade, more mitotic activity, loss of polarity). Lesions with more pronounced atypia may be closer to the threshold and thus more likely to cross into carcinoma, especially with additional alterations [33].
2.2.2. Morphologic and Histopathologic Continuum: ADH as a “mini-DCIS”
ADH is generally regarded as a transitional lesion within the biological spectrum of breast epithelial proliferations, representing an intermediate step between UDH and low-grade DCIS. One of the most compelling arguments that atypia represents a transitional state is the striking morphologic and structural affinity between ADH and low-grade DCIS. In fact, many pathologists conceptually regard ADH as a limited, partial, or incipient DCIS, fulfilling many of the same architectural and cytologic criteria, but lacking extent or completeness of ductal involvement [34].
Microscopically, ADH is characterized by a relatively uniform (monomorphic) epithelial population showing mild to moderate nuclear enlargement and minimal pleomorphism. The architectural arrangement often mirrors that of low-grade DCIS, displaying cribriform, micropapillary, or solid patterns; however, these features are typically focal, involving only a limited number of ductal spaces. A partial loss of polarity and reorganization of epithelial layers may also be observed, features that distinguish ADH from the irregular, haphazard cell stratification seen in UDH. From an immunophenotypic standpoint, ADH shares several markers with low-grade DCIS [35]. The atypical cells commonly express estrogen and progesterone receptors and demonstrate a relatively low proliferative index (ki-67), though this is higher than that observed in non-atypical proliferations. Molecular studies further reveal overlapping genetic and epigenetic alterations between ADH and DCIS, reinforcing the notion that ADH represents an early, quantitatively limited precursor in the neoplastic progression toward DCIS [36].
Because ADH clones possess many (though not all) features of low-grade DCIS, they are anatomically and biologically poised for further progression. Indeed, the threshold from ADH to low-grade DCIS is sometimes subtle and somewhat arbitrary (extent, degree of involvement, continuity) in diagnostic practice [37,38]. Thus, from a morphologic standpoint, the presence of atypia pushes a lesion from the benign proliferative region toward the in situ neoplastic region on the histologic continuum.
2.2.3. Epidemiologic and Clinical Evidence: Progression Risk Linked to Atypia
From the population and cohort studies, the differential risk conferred by atypical hyperplasia (vs proliferative disease without atypia) is pronounced and consistent. Whereas non-atypical proliferative disease confers a RR of approximately 1.5 to 2, atypical hyperplasia confers approximately 4–5x risk of future carcinoma [9]. Moreover, long-term follow-up data support that atypical hyperplasia is a strong independent predictor of later DCIS and invasive carcinoma, even after adjustment for confounders (age, family history, breast density) [9]. This suggests that atypia is not merely a marker but likely resides on the causal pathway.
Observational cohorts of ADH (particularly “pure ADH” without concurrent lesions) show meaningful rates of later carcinoma, consistent with a subset of these lesions evolving over many years [21]. Additionally, in analyses of biopsy upgrade rates (concurrent, initially occult carcinoma), ADH diagnoses on core biopsy are upgraded to DCIS or invasive cancer in a variable proportion (10–30%) depending on biopsy technique, lesion features, and institutional practice. This suggests that in many cases, ADH is a “step on the ladder” adjacent to more advanced neoplasia [21].
In conclusion, atypia is a powerful discriminator of risk and a clinically actionable histologic marker of progression potential.
2.2.4. Microenvironmental and Epithelial Constraints: How Atypia May Loosen Barriers
The molecular architecture of atypical lesions (discussed below) suggests a “halfway house”, offering enough neoplastic alteration to confer selective advantage, but not yet enough to cross the barrier into invasion. Therefore, while molecular alterations are necessary, they may be insufficient for progression without concomitant loosening of architectural or microenvironmental constraints.
The progression from benign epithelial proliferation to neoplasia also depends on changes within the cellular microenvironment that relax normal growth constraints. Atypical epithelial proliferation may represent a stage at which these regulatory barriers begin to fail, allowing for architectural disorganization and early invasive potential. As epithelial cells acquire atypical features, the structural integrity and functional role of the myoepithelial layer and basement membrane may become compromised. These barriers, normally responsible for maintaining ductal polarity and inhibiting stromal invasion, can appear attenuated, fragmented, or locally absent, diminishing their suppressive influence on epithelial expansion [39].
In parallel, the reciprocal communication between epithelial and stromal compartments becomes altered. Atypical cells may produce or respond abnormally to growth factors, cytokines, and matrix metalloproteinases that remodel the extracellular matrix, increase matrix stiffness, and recruit fibroblasts and immune cells, collectively creating a pro-tumorigenic milieu [40]. Within these altered microenvironments, the spatial organization of cells and local mechanical or metabolic gradients may further relax normal controls such as contact inhibition and nutrient diffusion, favoring the outgrowth of more resilient subclones [41].
Additionally, atypical epithelial populations often display increased resistance to apoptosis and enhanced survival under oxidative or replicative stress, enabling their persistence in settings where non-atypical cells would undergo programmed cell death [42]. Overall, atypia may therefore represent not only a morphological manifestation of genetic instability but also a biologically active phase in which epithelial and stromal interactions evolve to permit escape from architectural suppression—effectively bridging the transition from hyperplasia to neoplasia. As such, atypia may not only reflect accumulated mutations but also actively mediate escape from architectural suppression—providing a bridge from hyperplasia to neoplasia. Once a lesion surpasses the threshold of DCIS, the transition to invasion shares many overlapping features and constraints, and the presence of atypia earlier can be seen as a prelude [43].
Among untreated DCIS, 25–60% are believed to progress to invasive carcinoma over years to decades, depending on grade, molecular subtype, and microenvironment [44]. However, not all DCIS progress to invasive disease. Microenvironmental suppression, immune surveillance, and stromal constraints continue to play a role. In that respect, atypia marks early loosening of barriers, but further events are required to cross into full invasion [45]. Thus, atypia marks the entry point into the neoplastic continuum, with molecular/architectural permissiveness setting the stage for subsequent progression into full in situ and invasive disease [46].
In conclusion, not all atypical lesions are destined to progress. The molecular and morphological heterogeneity within atypical hyperplasia means that some lesions may remain indolent, others may regress, and only a subset evolves. The presence of atypia is necessary but not sufficient, and additional factors (extent, molecular burden, microenvironment, host context) influence the trajectory.
Given the importance of atypia, the pressing translational challenge is to differentiate which atypical lesions will progress versus stay stable. Biomarkers such as gene-expression risk scores, mutational burden, spatial heterogeneity metrics and imaging correlates are vigorously being explored [47,48].
3. Genetic Causes and Mechanisms of Mammary Gland Hyperplasia: From Atypia to Malignant Transformation
The likelihood that a given hyperplastic lesion will progress to cancer correlates with the accumulation of specific genetic mutations and epigenetic modifications. Recent research has revealed how these molecular events, from driver gene mutations (e.g., in PIK3CA, BRCA1/2, TP53) to DNA methylation changes and microRNA dysregulation, can cooperate to drive some hyperplasias along the trajectory to malignancy [49,50].
UDH is a proliferation of ordinary ductal epithelium without atypia. It is generally polyclonal and not considered an actual tumor. Molecular analysis shows that UDH lesions typically lack the consistent clonal genetic alterations seen in atypical lesions [50]. Nevertheless, modern sequencing has detected low-frequency activating mutations in pro-growth pathways even in some UDH: PIK3CA mutations or PI3K-AKT-mTOR pathway alterations [51]. These early mutations may provide a growth advantage but by themselves are insufficient for malignancy, as UDH generally retains normal architecture and growth control.
From a molecular point of view, ADH is a clonal neoplastic proliferation and is considered a genetically advanced precursor lesion [50]. ADH frequently harbors driver mutations commonly seen in luminal-type breast cancers, such as mutations in PIK3CA gene (the catalytic subunit of PI3K), that are present in a large fraction of ADH lesions [53]. Copy number aberrations characteristic of low-grade carcinomas are also observed; classic examples include gains on 1q and losses on 16q. Loss of heterozygosity on chromosome 16q, which harbors the CDH1 tumor suppressor gene, has been detected in some ADH cases. 16q loss is a hallmark of the low-grade pathway and is also frequent in concurrent DCIS or invasive cancers of the luminal A subtype. Overall, the genetic profile of ADH often mirrors that of low-grade DCIS, supporting its role as an immediate precursor [50]. Table 1 summarizes key genetic alterations in ADH and other hyperplasias, while Table 2 provides a more detailed view of key genetic drivers from atypical hyperplasia to cancer.
ALH is the lobular counterpart of ADH, arising in the terminal duct-lobular units. It is characterized by a monomorphic proliferation of discohesive, dyshesive cells (often due to loss of E-cadherin expression) filling part of the lobule. ALH and the related LCIS are collectively referred to as lobular neoplasia, which confers a risk of subsequent carcinoma similar to that of ADH. Genetically, lobular neoplasia is defined by inactivation of the CDH1 gene, which encodes E-cadherin. E-cadherin loss is observed in more than 90% of ALH/LCIS, either through mutations in CDH1 gene or 16q22 loss of heterozygosity [50,53]. This loss abrogates cell-cell adhesion and is a defining early step in the lobular lineage of breast cancer development [50]. In addition to CDH1, lobular neoplastic lesions often harbor mutations in PI3K pathway genes similar to those in ADH. For example, PIK3CA mutations are found in a substantial subset of ALH/LCIS and invasive lobular carcinomas [50]. Recurrent mutations in AKT1 gene (another PI3K pathway oncogene) are also reported in lobular cancers and may be present even at the in situ stage. Remarkably, lobular neoplasia can be multifocal and bilateral; genetically, independent ALH/LCIS foci in the same breast may arise from a field effect of genetically altered but morphologically normal cells distributed through the lobe. The presence of early BRCA2 or TBX3 gene mutations in morphologically normal epithelium has been documented in some cases [50]. This field of altered lobules can give rise to clonally related neoplastic lesions along both ductal and lobular pathways (if additional ductal or lobular lineage-specific hits occur) [50].
While genetic mutations lay the foundation for neoplastic transformation, epigenetic modifications provide additional layers of dysregulation in mammary hyperplasia. Epigenetic changes, heritable alterations in gene expression that do not stem from DNA sequence mutations, are now understood to occur early in breast lesion development [51]. These include DNA CpG island methylation, post-translational histone modifications, and deregulated expression of non-coding RNAs such as microRNAs. In hyperplastic breast tissue, epigenetic alterations can silence tumor suppressor pathways or activate oncogenic programs, thereby complementing genetic drivers in promoting cell proliferation and abnormal survival. Importantly, epigenetic changes are reversible in principle, and their presence in precursor lesions raises the possibility of early intervention to halt or reverse progression (though therapeutic implications are beyond the scope of this review) [66].
Aberrant DNA methylation is one of the earliest and most consistent epigenetic abnormalities observed during breast carcinogenesis. Even at the stage of hyperplasia, especially atypical hyperplasias, specific genes acquire promoter hypermethylation that leads to transcriptional silencing. For example, the promoters of tumor suppressor genes such as RASSF1A and APC are frequently hypermethylated in pre-invasive breast lesions, including ADH and DCIS. RASSF1A is a cell cycle regulator frequently silenced in breast cancer, and studies have detected RASSF1A methylation in a significant fraction of atypical hyperplasias and carcinoma in situ. Similarly, TWIST1, HIN1, p16INK4a and BRCA1 are among other genes reported to undergo methylation-associated silencing early in the progression sequence [67]. In lobular neoplasia, besides global methylation changes, CDH1 promoter methylation can serve as a second hit in cases lacking a CDH1 mutation; in fact, CDH1 promoter hypermethylation is found in a subset of LCIS/ILC, contributing to E-cadherin loss [62]. Overall, the presence of DNA methylation changes in a hyperplastic lesion is a red flag for a biologically advanced state.
MicroRNAs (miRNAs) are 20-22 nucleotide non-coding RNAs that post-transcriptionally regulate sets of target genes. Altered miRNA expression profiles are well documented in breast cancers, and emerging evidence indicates that miRNA dysregulation starts at pre-cancerous stages [68]. In fact, certain miRNA changes in hyperplasias may signal the development of malignancy and serve as early biomarkers. It was noted that oncomiRs such as miR-21 and miR-155, which promote proliferation and inhibit apoptosis, are progressively upregulated from normal epithelium to ADH to DCIS [72]. These miRNAs are key regulators of breast tumorigenesis: for instance, miR-21 suppresses PTEN and other tumor suppressors, while miR-155 modulates TP53 and SOCS1, thereby driving cell growth. On the other hand, some tumor-suppressor miRNAs are found to be downregulated in precursors. A striking recent example is miR-1297, frequently underexpressed in FEA lesions; Scafetta et al. compared miRNA profiles of normal breast epithelium, FEA, and DCIS: miR-1297 was significantly downregulated in FEA and DCIS relative to normal tissue [51]. MiR-1297 normally targets the oncogenic receptor Ephrin-A2 (EPHA2), so its loss may unleash pro-proliferative signaling. Functional experiments confirmed that restoring miR-1297 in breast cells inhibited growth, whereas miR-1297 loss enhanced proliferation and altered 3D acinar morphology. Thus miR-1297 downregulation represents an early event in mammary neoplastic transformation, potentially driving the progression of FEA toward malignancy [51]. This illustrates how even minute non-obligate lesions can harbor significant regulatory changes.
3.1. Mechanism of Malignant Transformation
The process by which ADH transitions into carcinoma involves the accumulation of multiple genetic mutations in key pathways governing cellular growth, survival, and apoptosis [69]. These mutations enable hyperplastic cells to evade normal regulatory controls, fostering an escalating potential for invasive behavior. This lesion bridges benign proliferations and DCIS, with features such as cribriform patterns, micropapillae, and uniform nuclear enlargement, all of which are discernible under microscopy [70]. The existing literature indicates that malignant transformation is not obligatory but is often driven by molecular aberrations, which can manifest as synchronous or metachronous carcinomas [71]. For instance, upgrades from ADH to DCIS or IDC occur in 22% to 65% of cases upon excisional biopsy, highlighting histopathological progression marked by increased extent of atypia and loss of myoepithelial integrity [72].
Additionally, the hypothetical multistep model of breast carcinogenesis proposes progression from normal epithelium to invasive carcinoma through intraductal hyperplasia (IDH), without and with atypia, including ADH and DCIS, with cytogenetic and molecular-genetic analyses revealing accumulation of genetic alterations [52]. Comparative genomic hybridization (CGH) and fluorescence in situ hybridization (FISH) studies reveal DNA amplification in the chromosomal region 20q13 in early IDH stages, indicating early cytogenetic changes in presumptive precursors [73].
3.1.1. Gene Expression Changes
The onset of atypia in ADH is frequently associated with alterations in gene expression of oncogenes (e.g., HER2/ERBB2, Cyclin D1) and tumor suppressor genes (e.g., TP53, BRCA1/2), which histopathologically manifest as proliferative and architectural abnormalities. Transcriptional profiling of matched normal, ADH, and carcinoma samples reveals upregulation of ERBB2, FOXA1, and GATA3 in ADH, correlating with estrogen receptor (ER) positivity and luminal phenotypes observable on IHC [74]. These changes are evident in H&E sections as monotonous cell populations with nuclear enlargement and rare mitoses, distinguishing ADH from usual ductal hyperplasia (UDH) by uniform spacing and polarization. In synchronous ADH, gene expression patterns cluster with those of low-grade DCIS, exhibiting enriched pathways in membrane transport, fatty acid metabolism, and phenylalanine metabolism, which may contribute to the cribriform architecture and micropapillary formations observed microscopically [75].
Matrix metalloproteinase-1 (MMP-1) emerges as a potential progression biomarker, with elevated expression in synchronous versus pure ADH, aligning with basement membrane discontinuities visible on periodic acid-Schiff (PAS) staining [76]. Furthermore, hub gene analyses identify RRM2, TOP2A, PBK, MELK, and NUSAP1 as progressively upregulated from normal mammary epithelium to ADH, DCIS, and IDC, which is associated with cell cycle deregulation and proliferation. Histopathologically, this correlates with increased ki67 positivity in atypical cells, indicating heightened mitotic activity in transitioning lesions [77]. In atypical hyperplasia (AH), including both ductal and lobular variants, gene signatures exhibit shared alterations, such as the downregulation of immune-related genes and the upregulation of proliferation markers, which are evident in multifocal lesions characterized by expanded ducts and acini in HE [78]. ERBB2 overexpression, even without amplification, is noted in ADH, with IHC revealing moderate cytoplasmic and membranous staining in atypical epithelial cells, potentially driving the transition to HER2-positive IDC as seen in 7% of subsequent carcinomas [79].
These expression shifts are more pronounced in premenopausal women, where higher-grade features like punctate necrosis predict malignant upgrade, underscoring hormonal influences on histopathological progression [80]. Cyclin D1 gene amplification and protein overexpression occur in benign breast disease and ADH, with frequencies similar to normal tissue in non-atypical hyperplasias (amplification 15-19%, overexpression 13%) but higher in ADH (amplification 27%, overexpression 57%), approaching DCIS (35% amplification, 50% overexpression) and IDC levels (25% amplification, 64% overexpression). Assessed via differential PCR and IHC, these changes precede histologic alterations but increase in atypical lesions [81]. BigH3 protein expression decreases progressively from benign tissues to DCIS, lobular carcinoma, and IDC, with benign tissue showing a 23-fold increase compared to infiltrating colloid carcinoma, correlating with malignancy in tissue microarray IHC of 192 cases [82].
In vitro hypoxia models show increased HIF-1alpha, GLUT1, and CAIX expression, with in vivo IHC absent in normal, DH, or ADH but present in DCIS (GLUT1 56.8%, CAIX 25.0%) and IDC (GLUT1 44.1%, CAIX 30.5%), higher in high-grade lesions (p=0.001 for GLUT1, p=0.036 for CAIX in DCIS). Notch1 and JAG1 hypomethylation inversely correlate with protein overexpression in IDC (Notch1 88.7%, JAG1 89.9%) versus ADH (36.0%, 45.0%), associating with lymph node metastasis and TNM stages [83]. In postmenopausal macaques, estrogen (E2) increases proliferation, epithelial area, and progesterone receptor expression (p<0.05), with higher columnar cell hyperplasia in E2-treated groups (p<0.05), showing greater ESR1 and Ki67 in lesional tissue. MiR-205-5p downregulation in metastatic 21T series correlates with higher histopathological grades (EG III) and invasion rates (21MT-1: 66.7 cells, p≤0.01) [84]. HER2 amplification and overexpression are absent in atypical ductal hyperplasia (ADH). Still, they are characteristically present in high-grade DCIS, implicating a contributory role in the clonal proliferation underlying neoplastic progression [85].
3.1.2. Loss of Tumor Suppressor Genes
The inactivation of genes such as PTEN, p16, and RB often leads to uncontrolled cell division and contributes to the progression of malignancy, as evidenced by histopathological findings including loss of cellular polarity and increased nuclear pleomorphism. Loss of heterozygosity (LOH) at tumor suppressor loci is an early event in ADH, targeting regions like 16q (harboring CDH1/E-cadherin) and 17p (TP53), observed in 42% of pure ADH cases via allelic imbalance analysis [86]. This manifests histopathologically as disrupted myoepithelial layers, with reduced p63 and smooth muscle actin (SMA) expression on IHC, facilitating epithelial-stromal interactions in atypical ducts. In paired UDH and ADH samples, shared LOH at 11p15.5, 13q14, 16q24.3, and 17p13.1 suggests a continuum, with ADH showing additional losses that correlate with cribriform patterns and partial ductal involvement [87]. RB1 and BRCA1/2 regions are implicated, with 16q deletions being common in low-grade pathways, leading to E-cadherin loss and lobular-like features in some ADH lesions, which are visible as expanded acini with uniform cells on HE [88]. PTEN inactivation, though less frequent in pure ADH, accumulates in synchronous cases, associating with nuclear atypia and mitoses that border on DCIS criteria [89]. Histopathological grading reveals that subsequent carcinomas from ADH exhibit 31% grade 1, 43-53% grade 2, and 16-26% grade 3, with suppressor losses contributing to this spectrum rather than exclusively low-grade disease. p16 alterations, often epigenetic, are noted in AH, correlating with heterogeneous CK5/6 negativity on IHC, which distinguishes it from UDH’s mosaic pattern and indicates suppressor-mediated dedifferentiation [90].
In high-risk families, ADH prevalence reaches 39%, with BRCA1/2 mutations enhancing suppressor loss, as seen in prophylactic mastectomy specimens with multifocal atypia and microcalcifications [91]. These inactivations drive morphological shifts, such as the formation of rigid bridges and solid growth, underscoring their role in malignant evolution. FHIT and WWOX expression decreases from normal to ADH, DCIS, and IDC, with higher detectable rates in benign tissue/ADH (rate ratios 2.95-4.58 for mRNA/protein) versus in situ/invasive stages [92]. Accumulation of chromosomal imbalances has been documented in premalignant breast lesions, including recurrent gains on chromosomes 3p and 8q, as well as losses on 16q, in ADH, which may implicate tumor suppressor genes in early neoplastic changes [93]. LOH at 11q13 has been observed in approximately 9% of ADH cases, 0% of low-grade DCIS, and 35% of high-grade DCIS, indicating a potential role for this alteration in facilitating the transition to invasive carcinoma. In intraductal hyperplasia, mutant p53 and cyclin D1 overexpression are more frequently identified in DCIS than in precursor lesions, with LOH at multiple loci associating suppressor gene inactivation with disease progression [94].
3.1.3. Genetic Instability
Atypical lesions tend to harbor genomic instability, with chromosomal aberrations and mutations in critical loci, particularly those involved in DNA repair and cell cycle regulation (e.g., BRCA1, TP53 mutations), histopathologically evident as aneuploidy and clonal expansions [95]. Aneuploidy is detected in 15-44% of ADH by flow cytometry and in all cases via FISH with multiple probes, higher than in non-atypical lesions but lower than in carcinomas. CGH identifies copy number aberrations like 16q loss and 1q gain in pure ADH, mirroring DCIS/IDC, with histopathological correlates including ductal spaces filled with monotonous cells and pseudo-lumens [96]. Next-generation sequencing in small cohorts reveals shared aneuploidy (e.g., 1q gain) and somatic mutations, but with intra-lesional heterogeneity in 46% of multifocal ADH, manifesting as mixed clonal proliferations on HE. TP53 mutations accumulate in synchronous ADH, associating with nuclear pleomorphism and mitoses, while BRCA1 defects enhance instability, as seen in high-risk lesions with frequent LOH [97].
Centrosomal abnormalities, with increased α- and γ-tubulin expression on IHC, are observed in ADH and carcinoma, indicating mitotic spindle defects that correlate with aneuploidy and invasive foci. Molecular profiles reveal advanced changes, including gross chromosomal rearrangements and epigenetic alterations in AH, with histopathological features such as calcifications and multifocality predicting higher instability and upgrade risk [98]. In progression models, instability drives from diploid ADH to aneuploid high-grade BC, though most ADH-associated cancers are ER-positive with moderate grades, emphasizing a low-grade pathway with occasional escalation. Allelic imbalance (AI) frequencies are <5% in benign tissue and ADH, 20% in DCIS, 25% in invasive carcinomas, with significant differences between ADH and DCIS (p<0.0001), suggesting instability predetermines biology at in situ stage [99].
Genetic alterations are absent in fibroadenomas, even associated with cancer, but LOH in DCIS within fibroadenomas, indicating no role in carcinogenesis [100]. c-erbB-2 amplification is absent in hyperplasia and ADH, but present in high-grade DCIS (10-40% in IDC), highlighting instability in the precursor to invasive transition [101].
3.1.4. Microenvironmental Factors
The surrounding tissue microenvironment, including stromal changes and inflammatory responses, can promote the progression of atypical lesions into invasive cancer, with histopathological evidence of field cancerization and multifocality [102]. Ipsilateral predominance of subsequent BC (2:1 ratio) suggests a microenvironmental field effect, persisting post-excision, as observed in cohorts with 80% ipsilateral events in the first 5 years [103].
Stromal alterations, such as desmoplasia and calcification, are common in ADH, as revealed by HE staining, which shows periductal fibrosis and immune infiltrates that promote progression [104]. Lobular involution extent inversely correlates with risk, with absent involution (SIR 7.66) associating with denser stroma and atypical expansions [105]. Inflammatory responses, including downregulation of immune genes in AH, contribute to evasion, histopathologically evident as reduced lymphocytic aggregates in progressing lesions [106].
Microenvironmental hormones, like in premenopausal women, heighten the risk of higher-grade carcinomas, with features like punctate necrosis on core biopsy predicting upgrade [107]. Field cancerization explains the multifocal appearance of ADH (higher risk if calcified), with non-clonal origins in cancer-prone tissue, as seen in heterogeneous lesions separated by stroma [108]. Tamoxifen’s risk reduction supports microenvironmental modulation, with histopathological reversal of atypia in treated cases [109].
Overall, these factors integrate with genetic changes to drive transformation, advocating for microenvironment-targeted interventions. Extra-tumoral tissue in BC patients shows ductal hyperplasia effects similar to non-neoplastic tissue under oral contraceptive use, indicating stromal-inflammatory influences [110]. In progression, hypoxia shifts HIF-1alpha role from proliferation response in early stages (DH, ADH) to tumor promotion, with GLUT1/CAIX in DCIS/IDC adapting to hypoxia/acidosis for aggressive phenotypes [111]. E2 in macaques induces columnar cell changes/hyperplasia, with greater ESR1/PGR/Ki67 in lesions, suggesting a hyper response in terminal ductal lobular units [112].
4. Epigenetic Modifications in Atypical Hyperplasia and Their Role in Malignant Transformation
4.1. General Features
Epigenetic reprogramming has a key role in mammary tumorigenesis, favouring the evolution of atypical hyperplasia to invasive carcinoma through changing gene expression, stimulating a stem cell-like condition, and allowing epithelial-to-mesenchymal transition (EMT) [113]. The main epigenetic mechanisms comprise alterations in DNA methylation, histone changes, and non-coding RNA expression, loss of genomic imprinting, reactivation of developmental pathways like Wnt/β-catenin signalling and Notch signalling, that could interrupt tumor suppressor genes and promote oncogenes [114,115]. Another important epigenetic mechanism is cell plasticity and stemness [116]. Thus, epigenetic reprogramming may confer stem cell-like properties to tumor cells, enabling limitless self-renewal and supporting tumor heterogeneity. Still, these identical flexible epigenetic changes offer therapeutic opportunities, as preventing epigenetic converters may reestablish normal gene expression, making tumors responsive to therapy and improving the efficiency of immunotherapies [117].
Figure 2.
Epigenetic drivers of malignant transformation if atypical hyperplasia of the breast. Created in BioRender. Elena Mihaela, J. (2025) https://BioRender.com/r65vbpl.
Figure 2.
Epigenetic drivers of malignant transformation if atypical hyperplasia of the breast. Created in BioRender. Elena Mihaela, J. (2025) https://BioRender.com/r65vbpl.
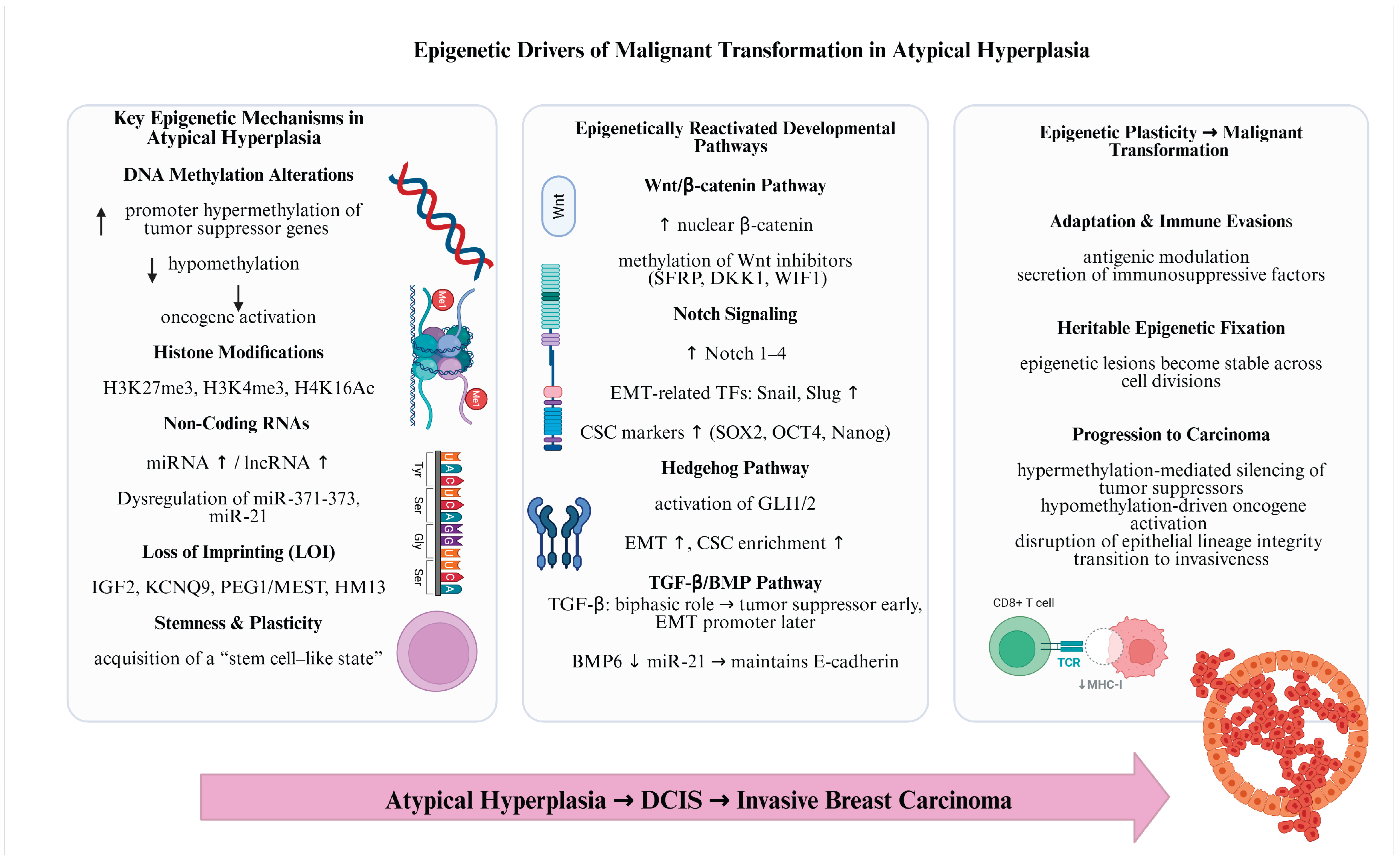
4.2. The Main Epigenetic Mechanisms Involved in Breast Tumorigenesis
Loss of Imprinting
It was found that genomic imprinting has a pivotal role in development and evolution. Loss of imprinting (LOI) represents an initial phase alteration in neoplasia. Imprinting is defined as the monoallelic expression of genes in a parent-of-origin-specific approach [118]. It was shown that in diploid eukaryotic entities, the maternal and paternal clones of main genes are expressed at equal quantities. Concerning imprinted genes, nonetheless, just one allele is transcriptionally dynamic [102]. Imprinting models are variable among tissues [119]. Most imprinted genes are regulated by imprinting control regions, which are generally modulated by DNA methylation, yet H3K27me3 also contributes to this regulation [120,121]. Moreover, as the number of imprinted genes is fundamental, disruption of imprinting is associated with some human imprinting syndromes and could lead to cancer by promoting oncogenic or suppressing antitumour processes [122,123].
LOI develops in a biallelic expression from the stimulation of the dormant allele. Research in mice revealed that demethylation of imprinted genes leading to LOI made cells more sensitive to cellular transformation and carcinogenesis [102]. Initiation of a growth-promoting allele leads to aberrant cell multiplication, a trigger of cancer [124].
LOI represents an essential abnormality in breast neoplasia growth and evolution, concerning the aberrant expression of imprinted genes linked to epigenetic dysregulation, usually DNA hypomethylation [108]. Reports have discovered imprinted genes such as HM13, KCNQ9, IGF2 and PEG1/MEST that display LOI in breast cancer, directing to modified gene expression responsible for tumor progression, with possible diagnostic and therapeutic values [107,108].
Research highlighted that HM13 shows LOI and expression upregulation in mammary tumors, involving DNA demethylation and possibly related to better prognosis in different types of cancer [125].
LOI at the KCNQ9 locus has been involved in the overexpression of the TASK3 potassium channel and is linked to triple-negative breast cancer [126]. The connection between KCNQ9 hypomethylation and triple-negative breast cancers indicates that targeting the KCNK9 DMR may represent a probable approach for inhibiting this form of breast cancer [110].
Persistent LOI of PEG1/MEST has been detected in invasive breast carcinomas, indicating its role in the evolution from hyperplasia to invasive cancer [108]. LOI of the IGF2 gene could determine its overexpression, supporting the growth of tumor-initiating cells and overall chromatin variability [127].
Reactivation of Developmental Pathways
Several of the best described signaling pathways influencing self-renewal and differentiation in mature stem cells, like Wnt/β-catenin, Notch, Hedgehog and TGF-β/BMP pathways are commonly regulated in cancer via epigenetic processes [97].
Abnormal epigenetic changes in atypical duct hyperplasia could restart developmental paths, for example Notch signaling and Wnt/β-catenin signaling, that are usually suppressed in mature cells but are reactivated through cancer development [97,99].
Notch signaling controls cell multiplication, differentiation and influences cell outcome and programmed cell death [99]. Notch signaling is controlled via Notch receptors (1-4) and ligands for instance Delta-like ligand (DDL 1/3/4) and Jagged 1/2 [128]. Notch signaling collaborates with transcription factors of epithelial-to-mesenchymal transition (Snail and Slug) and stemness (SOX2, Nanog, and OCT4), in addition to accelerating the acquisition of both epithelial-to-mesenchymal transition (EMT) and stemness [129]. Notch, in combination with TGF-β, induces Slug expression to modulate EMT. Furthermore, Notch signaling controls distinct target genes associated to cancer stem cells (CSCs) and facilitate chemoresistance [130]. For example, IL-6, a Notch target gene, sustains the self-renewal capacity of CSCs, and Notch-regulated activation of PKB protects cells from programmed cell death, thereby promoting survival [114]. Inactivation of Notch signaling destroys precursor cells that are like CSCs, which promote the function of Notch in stemness and correlate with chemoresistance. MDR1 is highly expressed in CSCs, during which the Notch signal is linked to NF-κB and related PI3K/Akt activation, controlling MRP2, a carrier that facilitates the preservation of stemness [97].
Wnt signaling pathway, which regulates the expression of target genes via β-catenin, represents a key mechanism of stem cells and CSCs and is abnormally activated through the growth of several human cancers [99]. Acquisition of function mutations of the CTNNB1 gene (encoding β-catenin) and deficit of function mutations of AXIN and genes were observed to be the principal processes responsible for Wnt signaling alteration in cancers [131]. Several genes implicated in the Wnt/β-catenin signaling are methylated and suppressed in mammary gland cancer, involving the Wnt inhibitors SFRP1-5, WIF1, and DKK1, also SRY-box containing gene 17 (SOX17) and APC [115]. Current findings reveal that the Wnt/β-catenin pathway may additionally be controlled by histone mutations in cancer [99]. Over-expression of EZH2 within the mammary gland causes β-catenin nuclear deposit and stimulation of Wnt pathway plus intraductal epithelial hyperplasia [132]. Moreover, the transcriptional suppression of a Wnt antagonist, DACT3, was related to divalent H3K4me3 and H3K27me3 histone changes [133]. Additionally, Dkk-1 suppressed expression was caused by reduced H4K16Ac and amplified H3K27me3, plus the enrolment of Suz12, SirT1, EZH2, and BMI1 to its promoter [134]. Also, miRNAs have a key role in the modulation of various participants of the Wnt/β-catenin pathways. It was shown that β-catenin/Lef1 transactivates the miR-371-373 group associated with CSC self-renewal, and in sequence, these miRNAs control the Wnt/β-catenin signaling via directing DKK1 inhibitor [135].
Hedgehog (Hh) signaling pathway modulates the multiplication of stem and precursor cells in several tissues and modifications in this pathway have been involved in tumor growth [99,136]. Hh signaling pathway cooperates with Wnt-β-catenin signaling, inducing tumorigenesis and cancer severity [137]. This mechanism could regulate both CSC and EMT pathways by increasing key transcription factors (Slug, ZEB1, Snail, ZEB 2, FOXC2 and TWIST2) and stem cell markers (CD44, BM1 and CD133) [121]. Enhancement of abnormal Hh signaling mechanisms in destructive, metastatic and chemoresistant mammary gland cancer types leads to a small amount of E-cadherin and enhanced expression of Twist, FOXC2, SIP1, Snail, vimentin, N-cadherin and fibronectin [113].
BMP/TGF-β signaling pathways control numerous biological processes like proliferation, differentiation and programmed cell death [97]. Deregulating the molecular effectors of TGF-β signaling could lead to cancer [99]. TGF-β functions as a tumor suppressor in tumor induction. Initial phases of tumor development and deactivation of the TGF-β tumor suppressor pathway represent the principal phase in the growth of many tumors [138]. Still, in the final phase it produces tumor progression, EMT, and metastasis [99]. BMP-6 acts as a suppressor of mammary cancer EMT by saving E-cadherin expression. Recent findings propose that this mechanism is facilitated by the BMP-6-stimulated transcriptional suppression of miR-21, which is over-expressed in destructive mammary gland neoplasia [139].
4.3. Epigenetic Plasticity
Epigenetic plasticity enables cells to dynamically alter gene expression patterns in response to the evolving tumor microenvironment, facilitating adaptation and survival under stressful conditions such as hypoxia or nutrient scarcity. This adaptability also allows cancer cells to circumvent intrinsic cellular checkpoints that typically restrict aberrant proliferation, including apoptosis and cell cycle arrest mechanisms [140]. Furthermore, it promotes immune evasion, enabling tumor cells to persist and thrive by modulating surface antigens or secreting immunosuppressive factors. The underlying processes involve key epigenetic modifications, such as histone acetylation or methylation, aberrant DNA methylation at promoter regions, and dysregulation of noncoding RNAs, which collectively enhance this phenotypic flexibility during early neoplastic stages [141].
These epigenetic alterations can become heritably fixed through successive cell divisions, stabilizing a malignant phenotype that supports sustained tumor growth. In the context of breast cancer, such changes drive progression from premalignant lesions like atypical ductal hyperplasia by repressing tumor suppressor genes (e.g., via hypermethylation) or activating oncogenes, thereby disrupting lineage integrity and promoting invasive characteristics [142]. Environmental factors, including dietary influences or endocrine disruptors, can further amplify these epigenetic shifts, underscoring the interplay between extrinsic cues and intrinsic molecular reprogramming in cancer evolution [143].
5. Tumor Microenvironment Changes in Driving Malignant Transformation of Atypical Hyperplasia
The tumor microenvironment (TME) in breast ADH encompasses a multifaceted and evolving landscape involving neoplastic epithelial cells, stromal fibroblasts, immune cells, vascular networks, and extracellular matrix (ECM) elements. These components interact synergistically to modulate the transition from premalignant states like ADH to DCIS and ultimately IDC. Subtle TME modifications in ADH, such as early stromal fibrosis, immune cell recruitment, and alterations in the basement membrane, are detectable through IHC and serve as harbingers of malignant progression. Existing literature highlights that these TME shifts, including disruptions of the myoepithelial layer and activation of fibroblasts, are pivotal in facilitating invasion. For instance, reduced myoepithelial continuity in ADH lesions, as assessed by the co-expression of alpha-smooth muscle actin (α-SMA) and p63 via multiplex IHC, correlates with an increased risk of progression to DCIS. Moreover, gene expression profiling and spatial analyses reveal transcriptional reprogramming in the stroma that supports epithelial invasion. This review synthesizes histopathological evidence from recent studies, highlighting how TME dynamics drive the malignant transformation of ADH, with implications for biomarker development and targeted interventions to halt disease progression.
5.1. Key TME Components and Their Impact on Malignant Transformation
5.1.1. Stromal Remodeling
Remodelling in ADH involves the activation and phenotypic shift of fibroblasts, leading to desmoplasia and a supportive niche for epithelial proliferation and invasion. Histopathologically, early ADH exhibits periductal fibrosis with an increased density of α-SMA-positive cancer-associated fibroblasts (CAFs), which are observable on IHC as spindle-shaped cells encircling atypical ducts [149]. These CAFs secrete cytokines and growth factors, such as hepatocyte growth factor (HGF), which upregulate the MET receptor in epithelial cells, fostering morphological changes indicative of invasive potential. In coculture models mimicking ADH progression, premalignant cells like MCF10DCIS display upregulated HGF/MET signaling when interacting with mammary fibroblasts, resulting in altered acinar structures in three-dimensional (3D) cultures, characterized by delayed cavitation and increased apoptosis upon HGF blockade [150].
This stromal-epithelial crosstalk is evident in vivo, where high HGF signature expression correlates with basal-like subtypes and poorer survival, highlighting stromal contributions to ADH malignant transformation. Furthermore, myoepithelial cells in ADH undergo phenotypic alterations, as indicated by IHC, which reveals reduced continuity—measured as increased distances between α-SMA/p63 co-positive cells—compared to normal tissue. This discontinuity, significantly lower in ADH bordering DCIS, permits greater epithelial-stromal interactions, potentially allowing immune cell access and promoting progression [151]. In obesity-associated models, high-fat diet (HFD)-induced stromal changes accelerate ADH formation, with H&E sections showing expanded atypical ducts and DCIS-like lesions; weight loss reverses this via kinome reprogramming, downregulating PKC-α and MEK3 while upregulating AMPKα, as detected by proteomic arrays in unaffected glands [152]. Additionally, inhibitor of differentiation-1 (Id-1) overexpression in ADH, assessed by IHC, correlates with morphologic progression, driving shifts in carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) from apical to cytoplasmic/membranous patterns, disrupting cell adhesion and facilitating stromal invasion [153]. Stromal fibroblasts in high-grade ADH-adjacent areas lose CD34 expression while gaining α-SMA, as seen in IHC, inversely correlating with microvessel density (MVD) and indicating a desmoplastic response that stiffens the matrix and aids epithelial migration [154]. These histopathological features underscore stromal remodeling as a critical driver, with early interventions targeting CAFs potentially arresting ADH progression.
5.1.2. Immune Cell Infiltration
The immune landscape in ADH’s TME features progressive infiltration of lymphocytes and macrophages, which can either suppress or promote malignant transformation depending on their phenotype and spatial distribution [155]. Histopathologically, HE staining of ADH biopsies reveals periductal lymphocytic aggregates, with higher stromal lymphocyte counts predicting upgrade to DCIS or IDC (p<0.01). In B3 lesions including ADH, elevated stromal lymphocytes, quantified per international guidelines, combine with patient age and lesion type to yield a predictive model for upgrade, suggesting immune surveillance failure in progressing cases [156]. Multiplex IHC further delineates this, showing increased tumor-infiltrating lymphocytes (TILs) in TP53-mutated ADH, with CD3+CD8+ T cells associated with favorable outcomes and CD3+Foxp3+ regulatory T cells (Tregs) linked to recurrence. Spatial proximity analyses via IHC demonstrate that closer T cell-tumor cell interactions correlate with reduced invasive risk, implying immune-mediated containment in non-progressing ADH [157].
In hypoxic conditions, myeloid cells release S100A9, which fosters systemicimmunosuppression, as evidenced by altered CD4+/CD8+ ratios in lymphoid organs and reduced T cell proliferation in cocultures [158]. Histologically, ADH progressing to DCIS shows enhanced stromal immune infiltrates, including macrophages, on H&E, contributing to ECM degradation via protease secretion [159]. In basal-like precursors, immune evasion is marked by increased polymorphonuclear neutrophils (PMNs) in the stroma, as detected by Ly6G+ IHC, which inhibit T cell function and promote atypical hyperplasia [138]. These findings are compounded in obesity, where HFD elevates inflammatory cytokines, leading to denser immune infiltrates in ADH lesions, reversible by weight loss that restores lean phenotypes and reduces pre-neoplastic inflammation. Overall, histopathological evidence points to a shift from anti-tumor to pro-tumor immune profiles in progressing ADH, with TIL density and subtype serving as prognostic indicators [160].
5.1.3. Hypoxia and Angiogenesis
Hypoxic niches in expanding ADH lesions trigger angiogenesis, providing sustenance for proliferation and invasion. Histopathologically, central necrosis in atypical ducts on HE signals hypoxia, with IHC showing nuclear hypoxia-inducible factor-1α (HIF-1α) stabilization in epithelial and stromal cells. This induces vascular endothelial growth factor (VEGF) expression, correlating with elevated MVD counted via CD31 IHC, particularly in high-grade ADH transitioning to DCIS [161]. Dynamic contrast-enhanced MRI (DCE-MRI) quantifies this angiogenic switch, revealing increased vessel permeability and vascular fraction in atypical hyperplasia, persisting through progression and confirmed by microvessel counting [162]. In transgenic models, hypoxia-driven small extracellular vesicles (sEVs) containing HIF-1α promote angiogenesis, with CD31 staining showing higher microvessel density and in vivo probes confirming vascular proliferation [141].
Angiogenesis precedes significant stromagenesis in ADH, as evidenced by VEGF-positive epithelial cells and immature vessels on IHC, fostering a permissive environment for invasion.
In obesity contexts, HFD exacerbates hypoxic signaling, accelerating angiogenic changes in ADH, while weight loss mitigates this via kinase modulation, reducing vascular support for progression [163]. HGF/MET pathways also contribute, with upregulated signaling in ADH inducing tortuous vessels observable in 3D models. These histopathological markers highlight hypoxia and angiogenesis as early drivers, with therapeutic targeting of HIF-1α or VEGF potentially preventing ADH malignant evolution.
5.1.5. Extracellular Matrix (ECM) and Cell-Cell Interactions
ECM remodeling in ADH disrupts structural integrity, enabling epithelial invasion. Histopathologically, Masson’s trichrome staining reveals disorganized collagen in periductal stroma, with IHC showing elevated matrix metalloproteinases (MMPs) from activated fibroblasts degrading basement membranes. Basement membrane discontinuities, visible on periodic acid-Schiff (PAS) staining, coincide with myoepithelial loss, marked by reduced α-SMA/p63 expression, facilitating EMT-like changes with vimentin positivity in atypical cells. RNA sequencing of ADH tissues identifies upregulated ECM-related genes, correlating with increased colony formation and migration in Matrigel assays, indicative of invasive potential [164].
Cell-cell adhesions weaken, with IHC demonstrating E-cadherin loss and CEACAM1 redistribution from apical to cytoplasmic patterns in progressing ADH (p<0.05), driven by Id-1 overexpression. In 3D cultures, HGF/MET activation induces morphological shifts, such as epithelial budding and ECM invasion, reversible by signaling inhibition [165]. Obesity amplifies ECM stiffening via kinase upregulation, promoting ADH to DCIS transition, while weight loss restores matrix homeostasis. These changes, including integrin-mediated signaling enhancements, underscore ECM and adhesion disruptions as key histopathological features in ADH transformation [166].
5.1.4. Exosomes and Tumor Signaling
Exosomes in ADH’s TME mediate intercellular signaling, transferring oncogenic cargo to remodel the niche. Although direct visualization is challenging, indirect evidence from IHC shows exosome-driven alterations, such as HIF-1α nuclear accumulation in recipient cells [137]. Hypoxic sEVs from premalignant cells induce atypical hyperplasia, as evidenced by HE staining, which reveals ductal expansion and an increase in ki67+ proliferation. These vesicles reprogram progenitors, disrupting cytokeratin 8/14 polarity and promoting EMT via RNA-seq-identified pathways [167]. In progression models, exosomal HIF-1α accelerates oncogene-driven tumorigenesis, with plasma detection correlating to recurrence in luminal cancers. Stromal responses, including α-SMA upregulation, suggest exosome-mediated CAF activation, though limited histopathological data exists [168]. Targeting exosomal cargo may offer novel strategies to interrupt ADH signaling networks [169].
In conclusion, the histopathological evolution of the TME in ADH—encompassing stromal activation, immune shifts, hypoxic angiogenesis, ECM degradation, and exosomal communication—critically drives malignant transformation. Integrated analyses from IHC, imaging, and molecular profiling provide robust evidence for these mechanisms, advocating for TME-targeted biomarkers and therapies to mitigate progression risks in high-risk patients.
6. Diet and Environmental Factors Influencing the Transformation of Atypical Ductal Hyperplasia (ADH) into Breast Cancer
6.1. Dietary Factors Influencing ADH Progression
According to the American Cancer Society, about 30% of the breast cancers in postmenopausal patients are related to modifiable risk factors, including diet [170]. Moreover, some research suggests that a healthy diet could prevent breast cancer onset, improve therapeutic outcome and decrease the risk of recurrence among these patients [171].
High-fat diet is an important risk factor for the onset and progression of breast cancer, through multiple pathogenetic mechanisms, involving increased systemic inflammation and oxidative stress [172]. Increased intake of total and saturated animal fat, found in red meat and high-fat dairy, was linked to the risk of breast cancer due to activation of pro-inflammatory mechanisms such as the NF-kB signaling, with downstream expression of cytokines, growth factors, and other molecules capable of supporting tumor progression [173,174]. The association between intake of monounsaturated fatty acids and breast cancer appears to depend on the source (olive oil or margarines) and the food processing methods. Intake of olive oil could have a beneficial effect by reducing insulin resistance, whereas hydrogenated oleic acids found in margarines seem to increase breast cancer risk. Polyunsaturated fatty acids can exert different effects on breast cancer, depending on the position of the double bond [175]. Thus, ω-6 fatty acids, such as linoleic acid and arachidonic acid may promote mammary tumorigenesis by producing eicosanoids and by undergoing oxidation, which results in enhanced cell damage [176]. By contrast, marine ω-3 fatty acids seem to have a protective effect [154].
A high-fat diet can disrupt the gut microbiota, increasing the risk of malignancies, by the activation of the “gut-bone marrow-tumor” axis. Altered microbiota can produce large amounts of leucine, capable of activating the mTORC1 signaling in myeloid progenitor cells, resulting in differentiation of polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs). An increased number of PMN-MDSCs infiltrating the tumor promotes breast cancer growth and metastasis [177].
High-fat diets may promote tumor cell metastasis by preactivating platelets and endothelial cells and by forming premetastatic niches. Within niches, the crosstalk between activated platelets, endothelial cells and cancer cells promotes metastasis [178].
Additionally, high-fat diets are a major driver of obesity, which itself increases breast cancer risk and progression through complex signaling pathways affecting cell growth and angiogenesis [179].
Obesity is a high-risk factor for breast cancer and poor prognosis in women after menopause [180]. Moreover, the risk of triple negative breast cancer and mortality post diagnosis is significantly increased in both pre- and postmenopausal obese women, compared with normal-weight women [181].
Potential pathogenic mechanisms underlying these associations include: increased estrogen levels, dysregulated insulin and Insulin-like growth factor (IGF) signaling, altered adipokines released by adipose cells, and chronic systemic inflammation indicated by high levels of C-reactive protein [161,182].
In postmenopausal women, the link between obesity and high estrogen levels is more significant, since, after menopause, the adipose tissue is the main source of estrogen. Excess estrogen derived from androgen aromatization in the adipose tissue, associated with obesity-related decrease in sex hormone binding globulin causes a hormonal imbalance, which is linked to the high risk of breast cancer [183,184]. A meta-analysis reported that obese or overweight breast cancer patients develop larger and higher-grade malignancy tumors, more frequent positive lymph nodes and distant metastasis, lower survival rate and worse overall prognostic [185]. This could be explained by the estrogen’s ability to fuel the growth of hyperplastic cells and increase the risk of DNA mutations [186].
Imbalance in adipokines due to increased mass of adipose tissue is also responsible for direct activation of breast cancer cells, contributing to tumor growth and invasion. Low adiponectin levels and high leptin levels were linked to poor prognosis of breast cancer. Adiponectin has a protective role by inhibiting cancer growth [187]. High leptin levels secreted by excess adipose tissue activate multiple mechanisms, including Jak2/Stat3, MAPK, PI3K-AKT, thus increasing tumor cell proliferation and invasion [188].
Chronic systemic inflammation maintains a pro-inflammatory state in adipose tissue, leading to increased secretion of inflammatory cytokines that promote tumor progression and metastasis. In obese individuals, adipose tissue is infiltrated by numerous macrophages, which form crown-like structures surrounding degenerating adipocytes. The presence of such structures in the mammary adipose stroma have been associated with increased levels of blood glucose, insulin, triglycerides, C-reactive protein and IL-6 [189]. Moreover, these patients had a higher risk of developing breast cancer, tumor metastasis and a lower survival rate [190,191]. The mechanisms underlying the poor prognosis in obese patients in association with the accumulation of macrophages and crown-like structures in the mammary gland include: the activation of pathways such as: NF-κB, hypoxia-inducible factor 1-alpha (HIF-1α), and PI3K-AKT-mTOR.
NF-κB, expressed in human breast cancer tumor cells, supports cancer cells’ survival and differentiation into an aggressive phenotype, and is also responsible for the ET resistance in ER+ breast tumors [192]. Expression of HIF-1α in adipose tissue macrophages has been detected in patients with insulin resistance and altered glucose metabolism [193].
PI3K-AKT-mTOR pathway controls cell growth and survival and has been demonstrated to cross-talk with estrogen receptor (ER) pathway; thus, alterations of PI3K-AKT-mTOR pathway are present in ER+ breast cancer [194].
Insulin resistance, which often accompanies obesity, leads to elevated levels of insulin and IGF, known as potent mitogens. Insulin and IGF-related tumorigenesis occurs by activation of oncogenic pathways such as PI3K/Ras-Raf in the affected cells, resulting in cell proliferation and ADH progression [195].
According to data published by the World Cancer Research Fund, a higher risk of breast cancer is linked to alcohol consumption, even in low amounts. Pathogenic mechanisms underlying the association between ethanol intake and mammary tumorigenesis include: oxidative stress by accumulation of reactive oxygen species (ROS), systemic inflammation due to increased production of pro-inflammatory cytokines, and insulin resistance [196]. Breast cancer initiation in patients who consume ethanol is the consequence of hormone levels alterations, mainly increasing estrogen by activation of aromatase [197]. Thus, alcohol was found to promote the development of ER+ breast tumors, but not ER- cancers. Additionally, alcohol is converted into acetaldehyde, a carcinogenic metabolite that causes DNA damage and induces epigenetic modifications by abnormal DNA methylation, both of which contribute to cancer initiation and progression [176].
Reducing high-fat foods, especially processed meats, fried goods, and high-fat dairy, and replacing them with unsaturated fats, fruits, vegetables, and the correct supplementation with vitamins and minerals is a key lifestyle recommendation for breast cancer prevention and outcome management [150].
Phytoestrogens are plant-derived compounds found in various foods: fruits, vegetables, grains, legumes such as soybeans, flaxseeds, tofu, and lentils [198]. Due to their molecular structure, these compounds perform a wide range of pharmacologic effects, acting as antioxidants, anti-inflammatories, anti-tumoral agents, and can also be effective as endocrine modulators. Upon binding to the estrogen receptors, phytoestrogens can mimic or regulate the function of estrogen in the body [164]. Phenolic phytoestrogens contain two main groups of active pharmacologic substances: flavonoids (including:flavone, isoflavone, flavonol phytoestrogens, dihydroflavone phytoestrogens) and non-flavonoid compounds (mainly terpenoids) [199]. Some studies suggest that phytoestrogens might have a protective role against breast cancer by acting as antagonists to estrogen in the breast tissue, potentially reducing the risk of malignancy in estrogen-sensitive cancers [164]. This protective role is explained by the ability of phenolic compounds to bind to the estrogen receptor and to prevent the interaction of endogenous estrogen with the G protein-coupled estrogen receptor (GPER) [200]. Moreover, the hydroxyl group in xenoestrogens plays a role in performing estrogen-like effects [201]. However, the effect of phytoestrogens on the progression of ADH is still debated, as some studies suggest that they may stimulate estrogen receptors in certain contexts, promoting cell proliferation in hormonally driven breast tumors.
Vitamins can also have anti-cancer properties. Studies suggest that vitamin D may inhibit tumor cell proliferation in mammary hyperplastic lesions, whereas vitamins C, E, and carotenoids exert antioxidant properties [202]. Dietary fibers are beneficial for breast cancer prevention by reducing glucose absorption and glycemic levels, and by regulating the balance of gastrointestinal microbiome [203].
6.2. Environmental Factors and Toxins Influencing ADH Progression
Although ADH already reflects a significant factor for the malignant transformation, other environmental and toxic factors such as endocrine disruptors chemicals, air pollution, pesticides, radiation exposure, etc. may have an important role in how ADH lesions evolve [204].
6.2.1. Ionizing Radiations
One of the most powerful environmental factors influencing ADH Progression is ionizing radiation, which directly induces double-stranded DNA breaks and also, in mammary epithelial cells, chromosomal instability. Studies show that even low doses of radiation can cause the activation of DNA repair and cell cycle genes (e.g., BRCA1). This leads to a proliferative effect in breast tissue [205].
Studies in mice have suggested that radiation exposure causes the activation of genes that are involved in proliferative and metabolic pathways that play a role in carcinogenesis in the mammary glands [206].
6.2.2. Chemical Endocrine Disruptors
Endocrine disruptors are natural or synthetic substances that are able to affect the physiological functions of the endocrine system [207]. These chemicals (EDCs) represent a class of anthropogenic compounds (e.g., bisphenol A, phthalates, and perfluoroalkyl substances) that mimic or antagonize natural hormones. Low-dose exposure to BPA and DDT significantly increases aromatase (CYP19A1) biosynthesis and 17β-estradiol production in mammary epithelial cells, leading to sustained activation and proliferation of Erα [208].
Bisphenol A (BPA) is used to produce polycarbonate plastics (e.g., plastic bottles), and recent studies discovered that exposure to BPA, due to the ability to mimic estrogen, can increase the growth of cancer cells in the breast [209].
6.2.3. Air Pollution
Airborne pollutants and particulate matter (PM) contain a multitude of compounds such as polycyclic aromatic hydrocarbons (PAHs) and metals which can stimulate the progression of mammary cancer [210].
Exposure to PM2.5 can lead to oxidative damage of the DNA and can also increase the expression level of oxidation markers. Cellular responses to these factors are activation of transcription factors, release of inflammatory mediators, etc. which can lead to cellular apoptosis. Studies reveald that urban residents have higher PM2.5 exposure and present a higher risk of developing breast cancer [211].
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous in the environment and are formed in the organic fuels combustion process. Studies have corelated PAH exposure with developmant of breast cancer [212].
6.2.4. Pesticides
Pesticides such as organophosphates, organochlorines and fungicides have been proven to increase the risk of breast cancer [213]. Organophosphates, a class of chemicals used as pesticides, are proven to act as endocrine disruptors and can increase the risk of mammary tumor development [214].
Dichlorodiphenyltrichloroethane (DDT), a synthetic organochlorine, is found to be in the adipose tissue and studies revealed that higher levels of DDT was found in women with breast cancer compared to women without cancer and a lower DDT level [215].
7. Conclusions
This review has examined the complex spectrum of MDH and its potential role as a precursor to breast cancer, highlighting why certain subtypes, especially those with atypia, pose a higher risk of becoming malignant. By combining epidemiologic, histopathologic, molecular, and environmental data, we have emphasized the multifactorial nature of progression from benign proliferative lesions to DCIS and invasive cancer. Key findings show that benign breast lesions are categorized into nonproliferative changes (low risk), proliferative hyperplasia without atypia (moderate risk), and proliferative hyperplasia with atypia (high risk). Atypical ductal hyperplasia and ALH serve as crucial transitional states, both morphologically and genetically bridging benignity and malignancy. These atypical forms display early cancer-related changes, such as PIK3CA mutations, BRCA1/2 dysfunction, and epigenetic modifications, including miR-21 upregulation, that disturb cellular balance and promote genomic instability. The tumor microenvironment further increases this risk through stromal remodeling, immune dysregulation, hypoxia-driven angiogenesis, and ECM breakdown via MMPs. Modifiable factors worsen these processes: obesity and high-fat diets sustain chronic inflammation through adipokines such as leptin, while environmental exposures to ionizing radiation, endocrine disruptors, air pollutants, and pesticides cause DNA damage and hormonal imbalance, primarily affecting high-risk hyperplasia.
Stratified risk assessments enable targeted monitoring: women with atypical hyperplasia might benefit from increased mammographic screening, chemoprevention, or preventive procedures, while women with non-atypical lesions need routine surveillance. Lifestyle changes, such as weight management and reduced toxin exposure, can help prevent progression. Biomarker-based strategies, including molecular testing for PIK3CA or TP53, could tailor treatment and identify those who may benefit from early intervention. Environmental links, while strong, often rely on epidemiological links rather than direct mechanistic proof. Future research should focus on long-term cohort studies that integrate omics data to understand progression pathways better. Ultimately, this review advocates for a comprehensive approach to MDH management, combining genetic risk, environmental factors, and personalized prevention strategies to lower breast cancer rates.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, ASS and MMO; methodology, BAG; software, TCP; validation, ASS, BAG and LPM; formal analysis, RTS; investigation, BAB, ED; resources, AMC; data curation, MJ; writing—original draft preparation, ASS; writing—review and editing, CC; visualization, LPM; supervision, LPM; project administration, CC; funding acquisition, CC. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.:
Abbreviations
The following abbreviations are used in this manuscript:
MDH - Mammary ductal hyperplasia
DCIS - ductal carcinoma in situ
RR - relative risk
UDH - usual ductal hyperplasia
OR - odds ratio
ADH - atypical ductal hyperplasia
ALH - atypical lobular hyperplasia
FEA - flat epithelial atypia
MRI - Magnetic Resonance Imaging
LCIS - lobular in situ carcinoma
PIK3CA - Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
BRCA1/2 - BReast CAncer gene 1/2
TP53 - Tumor Protein 53
DNA - Deoxyribonucleic Acid
K-AKT-mTOR - Phosphoinositide 3-kinase - AKR mouse thymoma - Mechanistic (mammalian) Target of Rapamycin
CDH1 - Cadherin-1
TBX3 - T-box transcription factor 3
CCLs - Columnar cell lesions
ER - Estrogen Receptor
NF1 - Neurofibromin 1
GATA - Glutamyl aminotransferase subunit A
MAP3K1 - Mitogen - activated protein kinase kinase kinase 1
CBFB - Core-Binding Factor, Beta Subunit
16q LOH – 16 q Loss of Heterozygosity
PTEN - phosphatase and tensin homolog
IBC - invasive breast cancers
ERBB2 - v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2
HR – hormone receptors
RUNX1 - Runt-related transcription factor 1
FOXA1 - Forkhead box protein A1
KMT2C (MLL3)/ KMT2D (MLL2) - type 2 lysine methyltransferases
ARID 1A/ARID 1B - AT-Rich Interaction Domain 1A Gene/ AT-Rich Interaction Domain 1B Gene
NCOR1/ NCOR2 - Nuclear Receptor Co-Repressor 1/ Nuclear Receptor Co-Repressor 2
CREBBP/ EP300 - CAMP Response Element-Binding-Binding Protein/E1A-binding protein P300
NOTCH1 - Neurogenic locus notch homolog protein 1
RNAs – Ribonucleic acid
RASSF1A - Ras Association Domain Family 1, isoform A
APC - Adenomatous Polyposis Coli
TWIST1 - Twist Family BHLH Transcription Factor 1
HIN1 - Hypermethylated in Cancer 1
p16INK4a - Cyclin-Dependent Kinase Inhibitor 2A
TP53 - Tumor Protein p53
SOCS1 - Suppressor of Cytokine Signaling 1
CGH - Comparative genomic hybridization
FISH - Fluorescence in situ hybridization
HER 2 - Human Epidermal Growth Factor Receptor 2
IHC – Immunohistochemistry
MMP-1 - Matrix metalloproteinase-1
PAS - periodic acid-Schiff
RRM2 – Ribonucleotide Reductase Regulatory Subunit M2
TOP2A – Topoisomerase II Alpha
PBK – PDZ-Binding Kinase
MELK – Maternal Embryonic Leucine Zipper Kinase
NUSAP1 - Nucleolar and Spindle-Associated Protein 1
AH - atypical hyperplasia
H&E – haematoxylin and eosin
PCR – polymerase chain reaction
HIF-1alpha - Hypoxia-Inducible Factor 1 alpha
GLUT1 - Glucose Transporter 1
CAIX - Carbonic Anhydrase IX
JAG1 - Jagged 1
RB - Retinoblastoma protein
LOH - Loss of heterozygosity
TNM - Tumor–Node–Metastasis staging system
SMA - and smooth muscle actin
CK5/6 - Cytokeratin 5/6
FHIT - Fragile Histidine Triad
WWOX - WW Domain–Containing Oxidoreductase
CGH - Comparative Genomic Hybridization.
AI – Allelic imbalance
c-erbB-2 - HER2
BC - breast cancers
SIR – Signal-to-Intensity Ratio
ESR1 - Estrogen receptor 1
PGR - Progesterone receptor
Ki-67 - Tumor proliferation
EMT - epithelial-to-mesenchymal transition
Wnt– Wingless Int-1
LOI - Loss of imprinting
H3K27me3 – Histone 3 lysine 27 trimethylation
HM13 - Histocompatibility minor 13
KCNQ9 - Potassium voltage-gated channel subfamily Q member 9
IGF2 - Insulin-like growth factor 2
PEG1/MEST - Paternally expressed gene 1 / Mesoderm-specific transcript
TASK3 – TWIK-related acid-sensitive potassium channel 3
KCNK9 DMR – Potassium channel subfamily K member 9 differentially methylated region
TGF-β - Transforming Growth Factor beta
BMP - Bone Morphogenetic Protein
DDL- Delta-like ligand (DLL?
SOX2 - SRY (Sex-determining Region Y)-Box 2
OCT4 - Octamer-binding transcription factor 4
CSCs - cancer stem cells
IL-6 - Interleukin 6
PKB - Protein Kinase B (also known as AKT)
NF-κB - Nuclear Factor kappa-light-chain-enhancer of activated B cells
MRP2 - Multidrug Resistance-associated Protein 2
CTNNB1 - Catenin Beta 1 (β-catenin)
AXIN - Axis Inhibition Protein
SFRP1-5 - Secreted Frizzled-Related Proteins 1–5
WIF1 - Wnt Inhibitory Factor 1
DKK1 - Dickkopf-related protein 1
SRY - Sex-determining Region Y
APC - Adenomatous Polyposis Coli
EZH2 - Enhancer of Zeste Homolog 2
DACT3 - Dishevelled-binding Antagonist of β-catenin 3
H3K4me3 - Histone 3 lysine 4 trimethylation
H3K27me3 - Histone 3 lysine 27 trimethylation
Suz12 - Suppressor of Zeste 12
SirT1 - Sirtuin 1
BMI1 - B lymphoma Mo-MLV insertion region 1 homolog
Lef1 - Lymphoid Enhancer-binding Factor 1
Slug - Snail family transcriptional repressor 2
ZEB1 - Zinc Finger E-box-binding Homeobox 1
Snail - Snail family transcriptional repressor 1
ZEB2 - Zinc Finger E-box-binding Homeobox 2
FOXC2 - Forkhead Box C2
TWIST2 - Twist family bHLH transcription factor 2
CD44 - Cluster of Differentiation 44
CD133 - Cluster of Differentiation 133
SIP1 - Smad Interacting Protein 1
BMP-6 - Bone Morphogenetic Protein 6
ECM - extracellular matrix
TME - tumor microenvironment
α-SMA - alpha-smooth muscle actin
CAFs - cancer-associated fibroblasts
HGF - hepatocyte growth factor
MET receptor - Mesenchymal-Epithelial Transition receptor
MCF10 - original non-tumorigenic human mammary epithelial cell line
HGF/MET - Hepatocyte Growth Factor/ MET receptor signaling pathway
HFD - high-fat diet
CEACAM1 - in carcinoembryonic antigen-related cell adhesion molecule 1
MVD - microvessel density
PKC-α - Protein Kinase C alpha
MEK3 - Mitogen-activated protein kinase kinase 3
AMPKα - AMP-activated protein kinase alpha subunit
TILs - tumor-infiltrating lymphocytes
Foxp3 - Forkhead box P3
Tregs - regulatory T cells
S100A9 - S100 calcium-binding protein A9
PMN - polymorphonuclear neutrophils
Ly6G+ - Lymphocyte antigen 6 complex, locus G positive
HIF 1α- 1hypoxia-inducible factor-1α
VEGF - vascular endothelial growth factor
MVD - Microvessel Density
DCE – MRI - Dynamic contrast-enhanced MRI
sEVs - small extracellular vesicles
PMN-MDSCs - polymorphonuclear myeloid-derived suppressor cells
IGF - Insulin-like growth factor
MAPK - Mitogen-Activated Protein Kinase
ET - Endocrine Therapy
ROS - reactive oxygen species
GPER - G protein-coupled estrogen receptor
EDCs - Endocrine-disrupting chemicals
BPA - Bisphenol A
DDT – Dichlorodiphenyltrichloroethane
CYP19A1 (Cytochrome P450 Family 19 Subfamily A Member 1
PM- particulate matter
PAHs - polycyclic aromatic hydrocarbons
References
- Kumar, V; Abbas, AK; Aster, JC. Robbins and Cotran pathologic basis of disease, 10th ed.; Philadelphia (PA); Elsevier, 2020. [Google Scholar]
- Klassen, C.L.; Hines, S.L.; Ghosh, K. Common Benign Breast Concerns for the Primary Care Physician. CCJM 2019, 86, 57–65. [Google Scholar] [CrossRef]
- Hartmann, L.C.; Degnim, A.C.; Pankratz, V.S.; Blake, C.; Iii, L.J.M. Benign Breast Disease and the Risk of Breast Cancer. n engl j med 2005. [Google Scholar] [CrossRef]
- Burke, A.; O’Driscoll, J.; Abubakar, M.; Bennett, K.E.; Carmody, E.; Flanagan, F.; Gierach, G.L.; Mullooly, M. A Systematic Review of Determinants of Breast Cancer Risk among Women with Benign Breast Disease. npj Breast Cancer 2025, 11, 16. [Google Scholar] [CrossRef]
- Stachs, A.; Stubert, J.; Reimer, T.; Hartmann, S. Benign Breast Disease in Women. Deutsches Ärzteblatt international 2019. [Google Scholar] [CrossRef]
- Vaidyanathan, L.; Barnard, K.; Elnicki, D.M. Benign Breast Disease: When to Treat, When to Reassure, When to Refer. Cleveland Clinic Journal of Medicine 2002, 69, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Kabat, G.C.; Jones, J.G.; Olson, N.; Negassa, A.; Duggan, C.; Ginsberg, M.; Kandel, R.A.; Glass, A.G.; Rohan, T.E. A Multi-Center Prospective Cohort Study of Benign Breast Disease and Risk of Subsequent Breast Cancer. Cancer Causes Control 2010, 21, 821–828. [Google Scholar] [CrossRef]
- Collins, L.C.; Baer, H.J.; Tamimi, R.M.; Connolly, J.L.; Colditz, G.A.; Schnitt, S.J. The Influence of Family History on Breast Cancer Risk in Women with Biopsy-confirmed Benign Breast Disease: Results from the Nurses’ Health Study. Cancer 2006, 107, 1240–1247. [Google Scholar] [CrossRef]
- Hartmann, L.C.; Degnim, A.C.; Santen, R.J.; Dupont, W.D.; Ghosh, K. Atypical Hyperplasia of the Breast — Risk Assessment and Management Options. N Engl J Med 2015, 372, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.A.; Newell, M.S. Radial Scars of the Breast Encountered at Core Biopsy: Review of Histologic, Imaging, and Management Considerations. American Journal of Roentgenology 2017, 209, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.M.; Cha, J.H.; Shin, H.J.; Chae, E.Y.; Choi, W.J.; Kim, H.H.; Oh, H.-Y. Radial Scars/Complex Sclerosing Lesions of the Breast: Radiologic and Clinicopathologic Correlation. BMC Med Imaging 2018, 18, 39. [Google Scholar] [CrossRef]
- Lavoué, V.; Fritel, X.; Antoine, M.; Beltjens, F.; Bendifallah, S.; Boisserie-Lacroix, M.; Boulanger, L.; Canlorbe, G.; Catteau-Jonard, S.; Chabbert-Buffet, N.; et al. Clinical Practice Guidelines from the French College of Gynecologists and Obstetricians (CNGOF): Benign Breast Tumors – Short Text. European Journal of Obstetrics & Gynecology and Reproductive Biology 2016, 200, 16–23. [Google Scholar] [CrossRef]
- Ferreira, A.I.; Borges, S.; Sousa, A.; Ribeiro, C.; Mesquita, A.; Martins, P.C.; Peyroteo, M.; Coimbra, N.; Leal, C.; Reis, P.; et al. Radial Scar of the Breast: Is It Possible to Avoid Surgery? European Journal of Surgical Oncology (EJSO) 2017, 43, 1265–1272. [Google Scholar] [CrossRef]
- Sohn, V.Y.; Causey, M.W.; Steele, S.R.; Keylock, J.B.; Brown, T.A. The Treatment of Radial Scars in the Modern Era—Surgical Excision Is Not Required. The American SurgeonTM 2010, 76, 522–525. [Google Scholar] [CrossRef]
- Donaldson, A.R.; Sieck, L.; Booth, C.N.; Calhoun, B.C. Radial Scars Diagnosed on Breast Core Biopsy: Frequency of Atypia and Carcinoma on Excision and Implications for Management. The Breast 2016, 30, 201–207. [Google Scholar] [CrossRef]
- Chou, W.Y.Y.; Veis, D.J.; Aft, R. Radial Scar on Image-Guided Breast Biopsy: Is Surgical Excision Necessary? Breast Cancer Res Treat 2018, 170, 313–320. [Google Scholar] [CrossRef]
- Page, D.L.; Dupont, W.D.; Rogers, L.W.; Rados, M.S. Atypical Hyperplastic Lesions of the Female Breast. A Long-Term Follow-up Study. Cancer 1985, 55, 2698–2708. [Google Scholar] [CrossRef]
- Degnim, A.C.; Visscher, D.W.; Berman, H.K.; Frost, M.H.; Sellers, T.A.; Vierkant, R.A.; Maloney, S.D.; Pankratz, V.S.; De Groen, P.C.; Lingle, W.L.; et al. Stratification of Breast Cancer Risk in Women With Atypia: A Mayo Cohort Study. JCO 2007, 25, 2671–2677. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.C.; Baer, H.J.; Tamimi, R.M.; Connolly, J.L.; Colditz, G.A.; Schnitt, S.J. Magnitude and Laterality of Breast Cancer Risk According to Histologic Type of Atypical Hyperplasia: Results from the Nurses’ Health Study. Cancer 2007, 109, 180–187. [Google Scholar] [CrossRef]
- Menes, T.S.; Kerlikowske, K.; Lange, J.; Jaffer, S.; Rosenberg, R.; Miglioretti, D.L. Subsequent Breast Cancer Risk Following Diagnosis of Atypical Ductal Hyperplasia on Needle Biopsy. JAMA Oncol 2017, 3, 36. [Google Scholar] [CrossRef] [PubMed]
- Kader, T.; Hill, P.; Rakha, E.A.; Campbell, I.G.; Gorringe, K.L. Atypical Ductal Hyperplasia: Update on Diagnosis, Management, and Molecular Landscape. Breast Cancer Res 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board Breast Tumours; International Agency for Research on Cancer, 2019.
- Schnitt, S.J. The Diagnosis and Management of Pre-Invasive Breast Disease: Flat Epithelial Atypia – Classification, Pathologic Features and Clinical Significance. Breast Cancer Res 2003, 5, 263. [Google Scholar] [CrossRef]
- Lamb, L.R.; Bahl, M.; Gadd, M.A.; Lehman, C.D. Flat Epithelial Atypia: Upgrade Rates and Risk-Stratification Approach to Support Informed Decision Making. Journal of the American College of Surgeons 2017, 225, 696–701. [Google Scholar] [CrossRef]
- Dialani, V.; Venkataraman, S.; Frieling, G.; Schnitt, S.J.; Mehta, T.S. Does Isolated Flat Epithelial Atypia on Vacuum-Assisted Breast Core Biopsy Require Surgical Excision? Breast J 2014, 20, 606–614. [Google Scholar] [CrossRef] [PubMed]
- DiPasquale, A.; Silverman, S.; Farag, E.; Peiris, L. Flat Epithelial Atypia: Are We Being Too Aggressive? Breast Cancer Res Treat 2020, 179, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.C.; Radisky, D.C.; Frost, M.H.; Santen, R.J.; Vierkant, R.A.; Benetti, L.L.; Tarabishy, Y.; Ghosh, K.; Visscher, D.W.; Degnim, A.C. Understanding the Premalignant Potential of Atypical Hyperplasia through Its Natural History: A Longitudinal Cohort Study. Cancer Prevention Research 2014, 7, 211–217. [Google Scholar] [CrossRef]
- Collins, L.C.; Aroner, S.A.; Connolly, J.L.; Colditz, G.A.; Schnitt, S.J.; Tamimi, R.M. Breast Cancer Risk by Extent and Type of Atypical Hyperplasia: An Update from the N Urses’ H Ealth S Tudies. Cancer 2016, 122, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Degnim, A.C.; Dupont, W.D.; Radisky, D.C.; Vierkant, R.A.; Frank, R.D.; Frost, M.H.; Winham, S.J.; Sanders, M.E.; Smith, J.R.; Page, D.L.; et al. Extent of Atypical Hyperplasia Stratifies Breast Cancer Risk in 2 Independent Cohorts of Women. Cancer 2016, 122, 2971–2978. [Google Scholar] [CrossRef]
- Burke, A.; O’Driscoll, J.; Abubakar, M.; Bennett, K.E.; Carmody, E.; Flanagan, F.; Gierach, G.L.; Mullooly, M. A Systematic Review of Determinants of Breast Cancer Risk among Women with Benign Breast Disease. npj Breast Cancer 2025, 11, 16. [Google Scholar] [CrossRef]
- Amin, A.L.; Wagner, J.L. Contemporary Management of Atypical Breast Lesions Identified on Percutaneous Biopsy: A Narrative Review. Ann Breast Surg 2021, 5, 9–9. [Google Scholar] [CrossRef]
- Klassen, C.L.; Fraker, J.L.; Pruthi, S. Updates on Management of Atypical Hyperplasia of the Breast. Mayo Clinic Proceedings 2025, 100, 1051–1057. [Google Scholar] [CrossRef]
- Chou, AJ; Bing, RS; Ding, DC. Endometrial Atypical Hyperplasia and Risk of Endometrial Cancer. Diagnostics (Basel). 2024 Nov 5;14(22):2471. [CrossRef] [PubMed] [PubMed Central]
- Pinder, S.E.; Ellis, I.O. The Diagnosis and Management of Pre-Invasive Breast Disease: Ductal Carcinoma in Situ (DCIS) and Atypical Ductal Hyperplasia (ADH) – Current Definitions and Classification. Breast Cancer Res 2003, 5, 254. [Google Scholar] [CrossRef] [PubMed]
- Baker, J; Noguchi, N; Marinovich, ML; Sprague, BL; Salisbury, E; Houssami, N. Atypical ductal or lobular hyperplasia, lobular carcinoma in-situ, flat epithelial atypia, and future risk of developing breast cancer: systematic review and meta-analysis. Breast. 2024 Oct;77:103807. [CrossRef]
- Han, LK; Hussain, A; Dodelzon, K; Ginter, PS; Towne, WS; Marti, JL. Active Surveillance of Atypical Ductal Hyperplasia of the Breast. Clin Breast Cancer. 2023 Aug;23(6):649-657. Epub 2023 May 21. [CrossRef] [PubMed]
- Page, D.L.; Dupont, W.D.; Rogers, L.W.; Rados, M.S. Atypical Hyperplastic Lesions of the Female Breast. A Long-Term Follow-up Study. Cancer 1985, 55, 2698–2708. [Google Scholar] [CrossRef]
- Tavassoli, F.A.; Norris, H.J. A Comparison of the Results of Long-Term Follow-up for Atypical Intraductal Hyperplasia and Intraductal Hyperplasia of the Breast. Cancer 1990, 65, 518–529. [Google Scholar] [CrossRef]
- Borowsky, A; Glencer, A; Ramalingam, K; Schindler, N; Mori, H; Ghule, P; Lee, K; Nachmanson, D; Officer, A; Harismendy, O; Stein, J; Stein, G; Evans, M; Weaver, D; Yau, C; Hirst, G; Campbell, M; Esserman, L. Tumor microenvironmental determinants of high-risk DCIS progression. Res Sq [Preprint]. 2024 May 9:rs.3.rs-4126092. [CrossRef]
- Hu, D.; Li, Z.; Zheng, B.; Lin, X.; Pan, Y.; Gong, P.; Zhuo, W.; Hu, Y.; Chen, C.; Chen, L.; et al. Cancer-associated Fibroblasts in Breast Cancer: Challenges and Opportunities. Cancer Communications 2022, 42, 401–434. [Google Scholar] [CrossRef]
- Northey, J.J.; Barrett, A.S.; Acerbi, I.; Hayward, M.-K.; Talamantes, S.; Dean, I.S.; Mouw, J.K.; Ponik, S.M.; Lakins, J.N.; Huang, P.-J.; et al. Stiff Stroma Increases Breast Cancer Risk by Inducing the Oncogene ZNF217. Journal of Clinical Investigation 2020, 130, 5721–5737. [Google Scholar] [CrossRef]
- Westerman, R.; Liu, Z.; Mohamud, R.; Mussa, A. Editorial: Oxidative Stress in Breast Cancer. Front. Oncol. 2025, 15, 1661039. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Yu, W.; Liu, J.; Tang, D.; Yang, L.; Chen, X. Oxidative Cell Death in Cancer: Mechanisms and Therapeutic Opportunities. Cell Death Dis 2024, 15, 556. [Google Scholar] [CrossRef]
- Wang, J.; Li, B.; Luo, M.; Huang, J.; Zhang, K.; Zheng, S.; Zhang, S.; Zhou, J. Progression from Ductal Carcinoma in Situ to Invasive Breast Cancer: Molecular Features and Clinical Significance. Sig Transduct Target Ther 2024, 9, 83. [Google Scholar] [CrossRef]
- Prajzendanc, K. DCIS Progression and the Tumor Microenvironment: Molecular Insights and Prognostic Challenges. Cancers 2025, 17, 1925. [Google Scholar] [CrossRef]
- Sanders, M.E.; Schuyler, P.A.; Dupont, W.D.; Page, D.L. The Natural History of Low-grade Ductal Carcinoma in Situ of the Breast in Women Treated by Biopsy Only Revealed over 30 Years of Long-term Follow-up. Cancer 2005, 103, 2481–2484. [Google Scholar] [CrossRef] [PubMed]
- Bhala, R.; Cando, L.F.; Verma, P.; Adesoye, T.; Bhardwaj, A. Biomarkers Predicting Progression and Prognosis of Ductal Carcinoma In Situ (DCIS). Anticancer Res 2025, 45, 1305–1328. [Google Scholar] [CrossRef]
- Gregory, K.J.; Roberts, A.L.; Conlon, E.M.; Mayfield, J.A.; Hagen, M.J.; Crisi, G.M.; Bentley, B.A.; Kane, J.J.; Makari-Judson, G.; Mason, H.S.; et al. Gene Expression Signature of Atypical Breast Hyperplasia and Regulation by SFRP1. Breast Cancer Res 2019, 21, 76. [Google Scholar] [CrossRef]
- Bombonati, A; Sgroi, DC. The molecular pathology of breast cancer progression. J Pathol. 2011 Jan;223(2):307-17. Epub 2010 Nov 16. [CrossRef] [PubMed]
- Krings, G; Shamir, ER; Shelley Hwang, E; Chen, YY. Genetic Analysis of Early Neoplasia in the Breast: Next-Generation Sequencing of Flat Epithelial Atypia and Associated Ductal and Lobular Lesions. Mod Pathol. 2025 Jun 17;38(11):100820. Epub ahead of print. [CrossRef] [PubMed]
- Keraite, I; Alvarez-Garcia, V; Garcia-Murillas, I; Beaney, M; Turner, NC; Bartos, C; Oikonomidou, O; Kersaudy-Kerhoas, M; Leslie, NR. PIK3CA mutation enrichment and quantitation from blood and tissue. Sci Rep. 2020 Oct 13;10(1):17082. [CrossRef]
- Van Baelen, K; Geukens, T; Maetens, M; Tjan-Heijnen, V C.G.; Lord, C J; Linn, S; et al. Current and future diagnostic and treatment strategies for patients with invasive lobular breast cancer. Ann Oncol. 2022, 33(8), 769–785. [Google Scholar] [CrossRef]
- Sokolova, A; Lakhani, SR. Lobular carcinoma in situ: diagnostic criteria and molecular correlates. Mod Pathol. 2021 Jan;34(Suppl 1):8-14. Epub 2020 Oct 6. [CrossRef]
- Cai, YW; Liu, CC; Zhang, YW; Liu, YM; Chen, L; Xiong, X; Shao, ZM; Yu, KD. MAP3K1 mutations confer tumor immune heterogeneity in hormone receptor-positive HER2-negative breast cancer. J Clin Invest. 2024 Nov 12;135(2):e183656. [CrossRef]
- Mortimer, JE; Lindsey, B; Zukin, M; et al. Prevalence of BRCA1 and BRCA2 variants in an unselected population of women with breast cancer. JAMA Netw Open. 2025, 8(9), e2531577. [Google Scholar] [CrossRef] [PubMed]
- Plesea, RM; Riza, AL; Ahmet, AM; et al. Clinically significant BRCA1 and BRCA2 germline variants in breast cancer—A single-center experience. Cancers (Basel) 2025, 17(1), 39. [Google Scholar] [CrossRef]
- Sandström, J; Bomanson, J; Pérez-Tenorio, G; Jönsson, C; Nordenskjöld, B; Fornander, T; Lindström, LS; Stål, O. GATA3 and markers of epithelial-mesenchymal transition predict long-term benefit from tamoxifen in ER-positive breast cancer. NPJ Breast Cancer. 2024 Sep 6;10(1):78. [CrossRef]
- Dopeso, H; Gazzo, AM; Derakhshan, F; Brown, DN; Selenica, P; Jalali, S; Da Cruz Paula, A; Marra, A; da Silva, EM; Basili, T; Gusain, L; Colon-Cartagena, L; Bhaloo, SI; Green, H; Vanderbilt, C; Oesterreich, S; Grabenstetter, A; Kuba, MG; Ross, D; Giri, D; Wen, HY; Zhang, H; Brogi, E; Weigelt, B; Pareja, F; Reis-Filho, JS. Genomic and epigenomic basis of breast invasive lobular carcinomas lacking CDH1 genetic alterations. NPJ Precis Oncol. 2024 Feb 12;8(1):33. [CrossRef]
- Smyth, LM; Tamura, K; Oliveira, M; Ciruelos, EM; Mayer, IA; Sablin, MP; Biganzoli, L; Ambrose, HJ; Ashton, J; Barnicle, A; Cashell, DD; Corcoran, C; de Bruin, EC; Foxley, A; Hauser, J; Lindemann, JPO; Maudsley, R; McEwen, R; Moschetta, M; Pass, M; Rowlands, V; Schiavon, G; Banerji, U; Scaltriti, M; Taylor, BS; Chandarlapaty, S; Baselga, J; Hyman, DM. Capivasertib, an AKT Kinase Inhibitor, as Monotherapy or in Combination with Fulvestrant in Patients with AKT1E17K-Mutant, ER-Positive Metastatic Breast Cancer. Clin Cancer Res. 2020 Aug 1;26(15):3947-3957. Epub 2020 Apr 20. [CrossRef]
- Chen, KM; Stephen, JK; Raju, U; Worsham, MJ. Delineating an epigenetic continuum for initiation, transformation and progression to breast cancer. Cancers (Basel). 2011 Jun;3(2):1580-92. [CrossRef] [PubMed]
- Herzog, SK; Fuqua, SAW. ESR1 mutations and therapeutic resistance in metastatic breast cancer: progress and remaining challenges. Br J Cancer. 2022 Feb;126(2):174-186. Epub 2021 Oct 7. [CrossRef]
- Arruabarrena-Aristorena, A; Maag, JLV; Kittane, S; Cai, Y; Karthaus, WR; Ladewig, E; Park, J; Kannan, S; Ferrando, L; Cocco, E; Ho, SY; Tan, DS; Sallaku, M; Wu, F; Acevedo, B; Selenica, P; Ross, DS; Witkin, M; Sawyers, CL; Reis-Filho, JS; Verma, CS; Jauch, R; Koche, R; Baselga, J; Razavi, P; Toska, E; Scaltriti, M. FOXA1 Mutations Reveal Distinct Chromatin Profiles and Influence Therapeutic Response in Breast Cancer. Cancer Cell. 2020 Oct 12;38(4):534-550.e9. Epub 2020 Sep 3. [CrossRef]
- Marei, HE. Epigenetic regulators in cancer therapy and progression. NPJ Precis Oncol 2025, 9, 206. [Google Scholar] [CrossRef] [PubMed]
- Maqsood, Q; Khan, MU; Fatima, T; et al. Recent insights into breast cancer: molecular pathways, epigenetic regulation, and emerging targeted therapies. Breast Cancer (Auckl) 2025, 19, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Prajzendanc, K; Domagała, P; Hybiak, J; et al. BRCA1 promoter hypermethylation is not associated with germline variants in Polish breast cancer patients. Hered Cancer Clin Pract. 2025, 23(1), 18. [Google Scholar] [CrossRef] [PubMed]
- Yang, H; Fang, Y; Wang, H; Lu, T; Chen, Q; Liu, H. Progress in epigenetic research of breast cancer: a bibliometric analysis since the 2000s. Front Oncol. 2025 Sep 5;15:1619346. [CrossRef]
- Pu, R; Laitala, L; Alli, P; et al. Methylation profiling of benign and malignant breast lesions and its application to cytopathology. Mod Pathol. 2003, 16(11), 1095–1101. [Google Scholar] [CrossRef]
- Mihai, AM; Ianculescu, LM; Suciu, N. MiRNAs as potential biomarkers in early breast cancer detection: a systematic review. J Med Life 2024, 17(6), 549–554. [Google Scholar] [CrossRef]
- Keohavong, P; Gao, WM; Mady, HH; Kanbour-Shakir, A; Melhem, MF. Analysis of p53 mutations in cells taken from paraffin-embedded tissue sections of ductal carcinoma in situ and atypical ductal hyperplasia of the breast. Cancer Lett. 2004 Aug 20;212(1):121-30. [CrossRef]
- Panda, KM; Naik, R. A Clinicopathological Study of Benign Phyllodes Tumour of Breast with Emphasis on Unusual Features. J Clin Diagn Res. 2016 Jul;10(7):EC14-7. Epub 2016 Jul 1. [CrossRef]
- Kader, T; Hill, P; Rakha, EA; Campbell, IG; Gorringe, KL. Atypical ductal hyperplasia: update on diagnosis, management, and molecular landscape. Breast Cancer Res. 2018 May 2;20(1):39. [CrossRef]
- Laws, A; Leonard, S; Hershey, E; Stokes, S; Vincuilla, J; Sharma, E; Milliron, K; Garber, JE; Merajver, SD; King, TA; Pilewskie, ML. Upgrade Rates and Breast Cancer Development Among Germline Pathogenic Variant Carriers with High-Risk Breast Lesions. Ann Surg Oncol. 2024 May;31(5):3120-3127. Epub 2024 Jan 23. [CrossRef]
- Werner, M; Mattis, A; Aubele, M; Cummings, M; Zitzelsberger, H; Hutzler, P; Höfler, H. 20q13.2 amplification in intraductal hyperplasia adjacent to in situ and invasive ductal carcinoma of the breast. Virchows Arch. 1999 Nov;435(5):469-72. [CrossRef]
- Martin, EM; Orlando, KA; Yokobori, K; Wade, PA. The estrogen receptor/GATA3/FOXA1 transcriptional network: lessons learned from breast cancer. Curr Opin Struct Biol. 2021 Dec;71:65-70. Epub 2021 Jul 2. [CrossRef]
- Lin, Y; Fu, F; Lv, J; Wang, M; Li, Y; Zhang, J; Wang, C. Identification of potential key genes for HER-2 positive breast cancer based on bioinformatics analysis. Medicine (Baltimore). 2020 Jan;99(1):e18445. [CrossRef]
- Kader, T; Hill, P; Rakha, EA; Campbell, IG; Gorringe, KL. Atypical ductal hyperplasia: update on diagnosis, management, and molecular landscape. Breast Cancer Res. 2018 May 2;20(1):39. [CrossRef]
- Santisteban, M; Reynolds, C; Barr Fritcher, EG; Frost, MH; Vierkant, RA; Anderson, SS; Degnim, AC; Visscher, DW; Pankratz, VS; Hartmann, LC. Ki67: a time-varying biomarker of risk of breast cancer in atypical hyperplasia. Breast Cancer Res Treat. 2010 Jun;121(2):431-7. Epub 2009 Sep 23. [CrossRef] [PubMed]
- Gregory, KJ; Roberts, AL; Conlon, EM; Mayfield, JA; Hagen, MJ; Crisi, GM; Bentley, BA; Kane, JJ; Makari-Judson, G; Mason, HS; Yu, J; Zhu, LJ; Simin, K; Johnson, JPS; Khan, A; Schneider, BR; Schneider, SS; Jerry, DJ. Gene expression signature of atypical breast hyperplasia and regulation by SFRP1. Breast Cancer Res. 2019 Jun 27;21(1):76. [CrossRef] [PubMed]
- Grassini, D; Cascardi, E; Sarotto, I; Annaratone, L; Sapino, A; Berrino, E; Marchiò, C. Unusual Patterns of HER2 Expression in Breast Cancer: Insights and Perspectives. Pathobiology Epub 2022 May 2. 2022, 89(5), 278–296. [Google Scholar] [CrossRef] [PubMed]
- Luen, SJ; Viale, G; Nik-Zainal, S; Savas, P; Kammler, R; Dell’Orto, P; Biasi, O; Degasperi, A; Brown, LC; Láng, I; MacGrogan, G; Tondini, C; Bellet, M; Villa, F; Bernardo, A; Ciruelos, E; Karlsson, P; Neven, P; Climent, M; Müller, B; Jochum, W; Bonnefoi, H; Martino, S; Davidson, NE; Geyer, C; Chia, SK; Ingle, JN; Coleman, R; Solbach, C; Thürlimann, B; Colleoni, M; Coates, AS; Goldhirsch, A; Fleming, GF; Francis, PA; Speed, TP; Regan, MM; Loi, S. Genomic characterisation of hormone receptor-positive breast cancer arising in very young women. Ann Oncol. 2023 Apr;34(4):397-409. Epub 2023 Jan 25. [CrossRef]
- Fico, F; Santamaria-Martínez, A. TGFBI modulates tumour hypoxia and promotes breast cancer metastasis. Mol Oncol. 2020 Dec;14(12):3198-3210. Epub 2020 Nov 5. [CrossRef]
- Wang, H; Wang, X; Xu, L. Transforming growth factor-induced gene TGFBI is correlated with the prognosis and immune infiltrations of breast cancer. World J Surg Oncol. 2024 Jan 20;22(1):22. [CrossRef]
- Han, D; Li, Z; Luo, L; Jiang, H. Targeting Hypoxia and HIF1α in Triple-Negative Breast Cancer: New Insights from Gene Expression Profiling and Implications for Therapy. Biology (Basel). 2024 Jul 31;13(8):577. [CrossRef] [PubMed]
- Hobi, G; Cline, JM; Ethun, KF; Simillion, C; Keller, I; Stute, P. Impact of 6 month conjugated equine estrogen versus estradiol-treatment on biomarkers and enriched gene sets in healthy mammary tissue of non-human primates. PLoS One. 2022 Mar 17;17(3):e0264057. [CrossRef] [PubMed]
- Van Bockstal, MR; Wesseling, J; Lips, EH; Smidt, M; Galant, C; van Deurzen, CHM. Systematic assessment of HER2 status in ductal carcinoma in situ of the breast: a perspective on the potential clinical relevance. Breast Cancer Res. 2024 Aug 27;26(1):125. [CrossRef]
- Sapino, A; Marchiò, C; Kulka, J. “Borderline” epithelial lesions of the breast: what have we learned in the past three decades? Pathologica. 2021 Oct;113(5):354-359. [CrossRef] [PubMed]
- Batistatou, A; Stefanou, D; Arkoumani, E; Agnantis, NJ. The usefulness of p63 as a marker of breast myoepithelial cells. In Vivo. 2003 Nov-Dec;17(6):573-6.
- Taha, SR; Boulos, F. E-cadherin staining in the diagnosis of lobular versus ductal neoplasms of the breast: the emperor has no clothes. Histopathology. 2025 Feb;86(3):327-340. Epub 2024 Aug 13. [CrossRef]
- Zhan, H; Antony, VM; Tang, H; Theriot, J; Liang, Y; Hui, P; Krishnamurti, U; DiGiovanna, MP. PTEN inactivating mutations are associated with hormone receptor loss during breast cancer recurrence. Breast Cancer Res Treat. 2025 Jun;211(2):441-447. Epub 2025 Mar 10. [CrossRef]
- Moradi, G; Ahmadinejad, N; Zarei, D; Sadighi, N. Sonographic Correlations With Histological Grade and Biomarker Profiles in Breast Invasive Ductal Carcinoma. Cancer Rep (Hoboken). 2025 Aug;8(8):e70288. [CrossRef]
- Boyraz, B; Ly, A. Spectrum of histopathologic findings in risk-reducing bilateral prophylactic mastectomy in patients with and without BRCA mutations. Hum Pathol. 2024 Sep;151:105534. Epub 2023 Nov 22. [CrossRef]
- Dabbs, DJ; Carter, G; Fudge, M; Peng, Y; Swalsky, P; Finkelstein, S. Molecular alterations in columnar cell lesions of the breast. Mod Pathol. 2006 Mar;19(3):344-9. [CrossRef]
- Aubele, MM; Cummings, MC; Mattis, AE; Zitzelsberger, HF; Walch, AK; Kremer, M; Höfler, H; Werner, M. Accumulation of chromosomal imbalances from intraductal proliferative lesions to adjacent in situ and invasive ductal breast cancer. Diagn Mol Pathol. 2000 Mar;9(1):14-9. [CrossRef] [PubMed]
- Chuaqui, RF; Zhuang, Z; Emmert-Buck, MR; Liotta, LA; Merino, MJ. Analysis of loss of heterozygosity on chromosome 11q13 in atypical ductal hyperplasia and in situ carcinoma of the breast. Am J Pathol. 1997 Jan;150(1):297-303. [PubMed]
- Ghaleb, A; Padellan, M; Marchenko, N. Mutant p53 drives the loss of heterozygosity by the upregulation of Nek2 in breast cancer cells. Breast Cancer Res. 2020 Dec 2;22(1):133. [CrossRef]
- Privitera, AP; Barresi, V; Condorelli, DF. Aberrations of Chromosomes 1 and 16 in Breast Cancer: A Framework for Cooperation of Transcriptionally Dysregulated Genes. Cancers (Basel). 2021 Mar 30;13(7):1585. [CrossRef]
- Krings, G; Shamir, ER; Shelley Hwang, E; Chen, YY. Genetic Analysis of Early Neoplasia in the Breast: Next-Generation Sequencing of Flat Epithelial Atypia and Associated Ductal and Lobular Lesions. Mod Pathol. 2025 Jun 17;38(11):100820. [CrossRef] [PubMed]
- Cai, F; Li, Y; Liu, H; Luo, J. Single-cell and Spatial Transcriptomic Analyses Implicate Formation of the Immunosuppressive Microenvironment during Breast Tumor Progression. J Immunol. 2024 Nov 1;213(9):1392-1401. [CrossRef]
- Fasching, PA; Hu, C; Hart, SN; Ruebner, M; Polley, EC; Gnanaolivu, RD; Hartkopf, AD; Huebner, H; Janni, W; Hadji, P; Tesch, H; Uhrig, S; Ettl, J; Lux, MP; Lüftner, D; Wallwiener, M; Wurmthaler, LA; Goossens, C; Müller, V; Beckmann, MW; Hein, A; Anetsberger, D; Belleville, E; Wimberger, P; Untch, M; Ekici, AB; Kolberg, HC; Hartmann, A; Taran, FA; Fehm, TN; Wallwiener, D; Brucker, SY; Schneeweiss, A; Häberle, L; Couch, FJ. Susceptibility gene mutations in germline and tumors of patients with HER2-negative advanced breast cancer. NPJ Breast Cancer. 2024 Jul 13;10(1):57. [CrossRef]
- Walter, C; Hartkopf, A; Koch, A; Klaumünzer, M; Schulze, M; Grischke, EM; Taran, FA; Brucker, S; Battke, F; Biskup, S. Sequencing for an interdisciplinary molecular tumor board in patients with advanced breast cancer: experiences from a case series. Oncotarget. 2020 Sep 1;11(35):3279-3285. [CrossRef]
- Daoud, SA; Ismail, WM; Abdelhamid, MS; Nabil, TM; Daoud, SA. Possible Prognostic Role of HER2/Neu in Ductal Carcinoma In Situ and Atypical Ductal Proliferative Lesions of the Breast. Asian Pac J Cancer Prev. 2016, 17(8), 3733–6. [Google Scholar]
- Niwińska, A; Olszewski, WP. The role of stromal immune microenvironment in the progression of ductal carcinoma in situ (DCIS) to invasive breast cancer. Breast Cancer Res. 2021 Dec 24;23(1):118. [CrossRef] [PubMed]
- Hartmann, LC; Radisky, DC; Frost, MH; Santen, RJ; Vierkant, RA; Benetti, LL; Tarabishy, Y; Ghosh, K; Visscher, DW; Degnim, AC. Understanding the premalignant potential of atypical hyperplasia through its natural history: a longitudinal cohort study. Cancer Prev Res (Phila). 2014 Feb;7(2):211-7. Epub 2014 Jan 30. [CrossRef]
- Mascharak, S; Guo, JL; Foster, DS; Khan, A; Davitt, MF; Nguyen, AT; Burcham, AR; Chinta, MS; Guardino, NJ; Griffin, M; Lopez, DM; Miller, E; Januszyk, M; Raghavan, SS; Longacre, TA; Delitto, DJ; Norton, JA; Longaker, MT. Desmoplastic stromal signatures predict patient outcomes in pancreatic ductal adenocarcinoma. Cell Rep Med. 2023 Nov 21;4(11):101248. Epub 2023 Oct 20. [CrossRef]
- Gierach, GL; Brinton, LA; Sherman, ME. Lobular involution, mammographic density, and breast cancer risk: visualizing the future? J Natl Cancer Inst. 2010 Nov 17;102(22):1685-7. Epub 2010 Oct 29. [CrossRef]
- Zhao, H; Wu, L; Yan, G; Chen, Y; Zhou, M; Wu, Y; Li, Y. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021 Jul 12;6(1):263. [CrossRef]
- Greene, AJE; Davis, J; Moon, J; Dubin, I; Cruz, A; Gupta, M; Moazzez, A; Ozao-Choy, J; Gupta, E; Manchandia, T; Kalantari, BN; Rahbar, G; Dauphine, C. Determination of Factors Associated with Upstage in Atypical Ductal Hyperplasia to Identify Low-Risk Patients Where Active Surveillance May be an Alternative. Ann Surg Oncol. 2024 May;31(5):3177-3185. Epub 2024 Feb 22. [CrossRef] [PubMed]
- Rageth, CJ; Rubenov, R; Bronz, C; Dietrich, D; Tausch, C; Rodewald, AK; Varga, Z. Atypical ductal hyperplasia and the risk of underestimation: tissue sampling method, multifocality, and associated calcification significantly influence the diagnostic upgrade rate based on subsequent surgical specimens. Breast Cancer. 2019 Jul;26(4):452-458. Epub 2018 Dec 27. Erratum in: Breast Cancer. 2021 Jan;28(1):246. doi: 10.1007/s12282-020-01186-w. [CrossRef] [PubMed]
- Cornell, LF; Klassen, CL; Ghosh, K; Ball, C; Advani, P; Pruthi, S. Improved Uptake and Adherence to Risk-Reducing Medication with the Use of Low-Dose Tamoxifen in Patients at High Risk for Breast Cancer. Cancer Prev Res (Phila). 2024 Dec 3;17(12):565-570. [CrossRef]
- Vamre, TB; Stalsberg, H; Thomas, DB. WHO Collaborative Study of Neoplasia and Steroid Contraceptives. Extra-tumoral breast tissue in breast cancer patients: variations with steroid contraceptive use. Int J Cancer. 2006 Jun 1;118(11):2827-31.
- Urrutia, AA; Mesa-Ciller, C; Guajardo-Grence, A; Alkan, HF; Soro-Arnáiz, I; Vandekeere, A; Ferreira Campos, AM; Igelmann, S; Fernández-Arroyo, L; Rinaldi, G; Lorendeau, D; De Bock, K; Fendt, SM; Aragonés, J. HIF1α-dependent uncoupling of glycolysis suppresses tumor cell proliferation. Cell Rep. 2024 Apr 23;43(4):114103. Epub 2024 Apr 11. [CrossRef]
- Wood, CE; Hester, JM; Appt, SE; Geisinger, KR; Cline, JM. Estrogen effects on epithelial proliferation and benign proliferative lesions in the postmenopausal primate mammary gland. Lab Invest. 2008 Sep;88(9):938-48. Epub 2008 Jul 7. [CrossRef]
- Ponnusamy, L; Mahalingaiah, PK S; Chang, YW; Singh, KP. Role of cellular reprogramming and epigenetic dysregulation in acquired chemoresistance in breast cancer. Cancer Drug Resist. 2019, 2, 297–312. [Google Scholar] [CrossRef]
- Darwiche, N. Epigenetic mechanisms and the hallmarks of cancer: an intimate affair. Am J Cancer Res. 2020 Jul 1;10(7):1954-1978.
- Muñoz, P; Iliou, MS; Esteller, M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. 2012 Dec;6(6):620-36. Epub 2012 Oct 26. [CrossRef]
- Daniel Romero-Mujalli, Laura I R Fuchs, Martin Haase, Jan-Peter Hildebrandt, Franz J Weissing, Tomás A Revilla, Emergence of phenotypic plasticity through epigenetic mechanisms, Evolution Letters, Volume 8, Issue 4, August 2024, Pages 561–574. [CrossRef]
- Connolly, R; Stearns, V. Epigenetics as a therapeutic target in breast cancer. J Mammary Gland Biol Neoplasia. 2012 Dec;17(3-4):191-204. [CrossRef]
- Goovaerts, T.; Steyaert, S.; Vandenbussche, C.A.; et al. A comprehensive overview of genomic imprinting in breast and its deregulation in cancer. Nat Commun 9, 4120 (2018). [CrossRef]
- Baran, Y; Subramaniam, M; Biton, A; Tukiainen, T; Tsang, EK; Rivas, MA; Pirinen, M; Gutierrez-Arcelus, M; Smith, KS; Kukurba, KR; Zhang, R; Eng, C; Torgerson, DG; Urbanek, C; Li, JB; Rodriguez-Santana, JR; Burchard, EG; Seibold, MA; MacArthur, DG; Montgomery, SB; Zaitlen, NA; Lappalainen, T.; GTEx Consortium. The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 2015 Jul;25(7):927-36. Epub 2015 May 7. [CrossRef]
- Maupetit-Méhouas, S; Montibus, B; Nury, D; Tayama, C; Wassef, M; Kota, SK; Fogli, A; Cerqueira Campos, F; Hata, K; Feil, R; Margueron, R; Nakabayashi, K; Court, F; Arnaud, P. Imprinting control regions (ICRs) are marked by mono-allelic bivalent chromatin when transcriptionally inactive. Nucleic Acids Res. 2016 Jan 29;44(2):621-35. Epub 2015 Sep 22. [CrossRef] [PubMed]
- Inoue, A; Jiang, L; Lu, F; Suzuki, T; Zhang, Y. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature. 2017 Jul 27;547(7664):419-424. Epub 2017 Jul 19. [CrossRef] [PubMed]
- Horsthemke, B. In brief: genomic imprinting and imprinting diseases. J Pathol. 2014 Apr;232(5):485-7. Epub 2014 Jan 29. [CrossRef] [PubMed]
- Harrison, K; Hoad, G; Scott, P; Simpson, L; Horgan, GW; Smyth, E; Heys, SD; Haggarty, P. Breast cancer risk and imprinting methylation in blood. Clin Epigenetics. 2015 Sep 4;7(1):92. [CrossRef] [PubMed]
- Amin, MT; Coussement, L; De Meyer, T. Characterization of Loss-of-Imprinting in Breast Cancer at the Cellular Level by Integrating Single-Cell Full-Length Transcriptome with Bulk RNA-Seq Data. Biomolecules. 2024 Dec 14;14(12):1598. 1598. [CrossRef]
- Yang, H; Li, Z; Wang, Z; Zhang, X; Dai, X; Zhou, G; Ding, Q. Histocompatibility Minor 13 (HM13), targeted by miR-760, exerts oncogenic role in breast cancer by suppressing autophagy and activating PI3K-AKT-mTOR pathway. Cell Death Dis. 2022 Sep 25;13(8):728. [CrossRef]
- Skaar, DA; Dietze, EC; Alva-Ornelas, JA; Ann, D; Schones, DE; Hyslop, T; Sistrunk, C; Zalles, C; Ambrose, A; Kennedy, K; Idassi, O; Miranda Carboni, G; Gould, MN; Jirtle, RL; Seewaldt, VL. Epigenetic Dysregulation of KCNK9 Imprinting and Triple-Negative Breast Cancer. Cancers (Basel). 2021 Nov 30;13(23):6031. 6031. [CrossRef]
- Cui, H. Loss of imprinting of IGF2 as an epigenetic marker for the risk of human cancer. Dis Markers 2007, 23(1-2), 105–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z; Li, Y; Kong, D; Sarkar, FH. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets. 2010 Jun;11(6):745-51. [CrossRef]
- Liu, X; Fan, D. The epithelial-mesenchymal transition and cancer stem cells: functional and mechanistic links. Curr Pharm Des. 2015;21(10):1279-91. [CrossRef] [PubMed]
- Chen, W; Qin, Y; Liu, S. Cytokines, breast cancer stem cells (BCSCs) and chemoresistance. Clin Transl Med. 2018 Sep 3;7(1):27. [CrossRef]
- Zhang, W; Glöckner, SC; Guo, M; Machida, EO; Wang, DH; Easwaran, H; Van Neste, L; Herman, JG; Schuebel, KE; Watkins, DN; Ahuja, N; Baylin, SB. Epigenetic inactivation of the canonical Wnt antagonist SRY-box containing gene 17 in colorectal cancer. Cancer Res. 2008 Apr 15;68(8):2764-72. Erratum in: Cancer Res. 2008 Jul 15;68(14):6030. [CrossRef]
- Li, X; Gonzalez, ME; Toy, K; Filzen, T; Merajver, SD; Kleer, CG. Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. Am J Pathol. 2009 Sep;175(3):1246-54. Epub 2009 Aug 6. [CrossRef]
- Jiang, X; Tan, J; Li, J; Kivimäe, S; Yang, X; Zhuang, L; Lee, PL; Chan, MT; Stanton, LW; Liu, ET; Cheyette, BN; Yu, Q. DACT3 is an epigenetic regulator of Wnt/beta-catenin signaling in colorectal cancer and is a therapeutic target of histone modifications. 2008 Jun;13(6):529-41. [CrossRef]
- Hussain, M; Rao, M; Humphries, AE; Hong, JA; Liu, F; Yang, M; Caragacianu, D; Schrump, DS. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009 Apr 15;69(8):3570-8. Epub 2009 Apr 7. [CrossRef]
- Inui, M; Martello, G; Piccolo, S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010 Apr;11(4):252-63. Epub 2010 Mar 10. [CrossRef]
- Katoh, Y; Katoh, M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009 Sep;9(7):873-86. [CrossRef]
- Jiang, J; Hui, CC. Hedgehog signaling in development and cancer. Dev Cell. 2008 Dec;15(6):801-12. [CrossRef]
- Sakaki-Yumoto, M; Katsuno, Y; Derynck, R. TGF-β family signaling in stem cells. Biochim Biophys Acta. 2013 Feb;1830(2):2280-96. Epub 2012 Aug 16. [CrossRef]
- Du, J; Yang, S; An, D; Hu, F; Yuan, W; Zhai, C; Zhu, T. BMP-6 inhibits microRNA-21 expression in breast cancer through repressing deltaEF1 and AP-1. Cell Res. 2009 Apr;19(4):487-96. [CrossRef]
- Tufail, M; Hu, JJ; Liang, J; He, CY; Wan, WD; Huang, YQ; Jiang, CH; Wu, H; Li, N. Hallmarks of cancer resistance. iScience. 2024 May 15;27(6):109979. [CrossRef]
- Wainwright, EN; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer. 2017 May;3(5):372-386. Epub 2017 May 5. [CrossRef]
- Shi, ZD; Pang, K; Wu, ZX; Dong, Y; Hao, L; Qin, JX; Wang, W; Chen, ZS; Han, CH. Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies. Signal Transduct Target Ther. 2023 Mar 11;8(1):113. [CrossRef]
- Pang, L; Zhou, F; Liu, Y; Ali, H; Khan, F; Heimberger, AB; Chen, P. Epigenetic regulation of tumor immunity. J Clin Invest. 2024 Jun 17;134(12):e178540. [CrossRef]
- Hoque, MO; Prencipe, M; Poeta, ML; Barbano, R; Valori, VM; Copetti, M; Gallo, AP; Brait, M; Maiello, E; Apicella, A; Rossiello, R; Zito, F; Stefania, T; Paradiso, A; Carella, M; Dallapiccola, B; Murgo, R; Carosi, I; Bisceglia, M; Fazio, VM; Sidransky, D; Parrella, P. Changes in CpG islands promoter methylation patterns during ductal breast carcinoma progression. Cancer Epidemiol Biomarkers Prev. 2009 Oct;18(10):2694-700. Epub 2009 Sep 29. [CrossRef] [PubMed]
- Harrison BT, D’Alfonso TM, Schnitt SJ. Columnar cell lesions and flat epithelial atypia. In: Shin SJ, Chen YY, Ginter PS, editors. A Comprehensive Guide to Core Needle Biopsies of the Breast. Cham: Springer; 2022. p. 331-350. [CrossRef]
- Otterlei Fjørtoft, M; Huse, K; Rye, IH. The Tumor Immune Microenvironment in Breast Cancer Progression. Acta Oncol. 2024 May 23;63:359-367. [CrossRef]
- Kim, A; Mo, K; Kwon, H; Choe, S; Park, M; Kwak, W; Yoon, H. Epigenetic Regulation in Breast Cancer: Insights on Epidrugs. Epigenomes. 2023 Feb 18;7(1):6. 6. [CrossRef]
- Brena, D; Huang, MB; Bond, V. Extracellular vesicle-mediated transport: Reprogramming a tumor microenvironment conducive with breast cancer progression and metastasis. Transl Oncol. 2022 Jan;15(1):101286. Epub 2021 Nov 25. [CrossRef]
- Sun, Y; Yang, D; Xi, L; Chen, Y; Fu, L; Sun, K; Yin, J; Li, X; Liu, S; Qin, Y; Liu, M; Hou, Y. Primed atypical ductal hyperplasia-associated fibroblasts promote cell growth and polarity changes of transformed epithelium-like breast cancer MCF-7 cells via miR-200b/c-IKKβ signaling. Cell Death Dis. 2018 Jan 26;9(2):122. [CrossRef]
- Casbas-Hernandez, P; D’Arcy, M; Roman-Perez, E; Brauer, HA; McNaughton, K; Miller, SM; Chhetri, RK; Oldenburg, AL; Fleming, JM; Amos, KD; Makowski, L; Troester, MA. Role of HGF in epithelial-stromal cell interactions during progression from benign breast disease to ductal carcinoma in situ. Breast Cancer Res. 2013;15(5):R82. [CrossRef]
- Borowsky, A; Glencer, A; Ramalingam, K; Schindler, N; Mori, H; Ghule, P; Lee, K; Nachmanson, D; Officer, A; Harismendy, O; Stein, J; Stein, G; Evans, M; Weaver, D; Yau, C; Hirst, G; Campbell, M; Esserman, L. Tumor microenvironmental determinants of high-risk DCIS progression. Res Sq [Preprint]. 2024 May 9:rs.3.rs-4126092. [CrossRef]
- Qin, Y; Sundaram, S; Essaid, L; Chen, X; Miller, SM; Yan, F; Darr, DB; Galanko, JA; Montgomery, SA; Major, MB; Johnson, GL; Troester, MA; Makowski, L. Weight loss reduces basal-like breast cancer through kinome reprogramming. Cancer Cell Int. 2016 Apr 1;16:26. [CrossRef]
- Liu, Q; Yang, YM; Zhang, QH; Zhang, TG; Zhou, Q; Zhou, CJ. Inhibitor of differentiation is overexpressed with progression of benign to malignant lesions and related with carcinoembryonic antigen-related cell adhesion molecule 1 distribution in mammary glands. Ann Diagn Pathol. 2011 Feb;15(1):30-6. Epub 2010 Nov 24. [CrossRef]
- Pavlakis, K; Messini, I; Vrekoussis, T; Yiannou, P; Keramopoullos, D; Louvrou, N; Liakakos, T; Stathopoulos, EN. The assessment of angiogenesis and fibroblastic stromagenesis in hyperplastic and pre-invasive breast lesions. BMC Cancer. 2008 Apr 2;8:88. [CrossRef] [PubMed]
- Degnim, AC; Hoskin, TL; Arshad, M; Frost, MH; Winham, SJ; Brahmbhatt, RA; Pena, A; Carter, JM; Stallings-Mann, ML; Murphy, LM; Miller, EE; Denison, LA; Vachon, CM; Knutson, KL; Radisky, DC; Visscher, DW. Alterations in the Immune Cell Composition in Premalignant Breast Tissue that Precede Breast Cancer Development. Clin Cancer Res. 2017 Jul 15;23(14):3945-3952. Epub 2017 Jan 26. [CrossRef]
- Kader, T; Provenzano, E; Jayawardana, MW; Hendry, S; Pang, JM; Elder, K; Byrne, DJ; Tjoeka, L; Frazer, HM; House, E; Jayasinghe, SI; Keane, H; Murugasu, A; Rajan, N; Miligy, IM; Toss, M; Green, AR; Rakha, EA; Fox, SB; Mann, GB; Campbell, IG; Gorringe, KL. Stromal lymphocytes are associated with upgrade of B3 breast lesions. Breast Cancer Res. 2024 Jul 8;26(1):115. [CrossRef]
- Borowsky, A; Glencer, A; Ramalingam, K; Schindler, N; Mori, H; Ghule, P; Lee, K; Nachmanson, D; Officer, A; Harismendy, O; Stein, J; Stein, G; Evans, M; Weaver, D; Yau, C; Hirst, G; Campbell, M; Esserman, L. Tumor microenvironmental determinants of high-risk DCIS progression. Res Sq [Preprint]. 2024 May 9:rs.3.rs-4126092. [CrossRef]
- Bertolini, I; Perego, M; Nefedova, Y; Lin, C; Milcarek, A; Vogel, P; Ghosh, JC; Kossenkov, AV; Altieri, DC. Intercellular hif1α reprograms mammary progenitors and myeloid immune evasion to drive high-risk breast lesions. J Clin Invest. 2023 Apr 17;133(8):e164348. [CrossRef]
- Cichon, MA; Degnim, AC; Visscher, DW; Radisky, DC. Microenvironmental influences that drive progression from benign breast disease to invasive breast cancer. J Mammary Gland Biol Neoplasia. 2010 Dec;15(4):389-97. [CrossRef]
- Kuziel, G; Moore, BN; Haugstad, GP; Xiong, Y; Williams, AE; Arendt, LM. Alterations in the mammary gland and tumor microenvironment of formerly obese mice. BMC Cancer. 2023 Dec 1;23(1):1183. [CrossRef]
- Bluff, JE; Menakuru, SR; Cross, SS; Higham, SE; Balasubramanian, SP; Brown, NJ; Reed, MW; Staton, CA. Angiogenesis is associated with the onset of hyperplasia in human ductal breast disease. Br J Cancer. 2009 Aug 18;101(4):666-72. Epub 2009 Jul 21. [CrossRef]
- Ikeda, O; Nishimura, R; Miyayama, H; Yasunaga, T; Ozaki, Y; Tuji, A; Yamashita, Y. Evaluation of tumor angiogenesis using dynamic enhanced magnetic resonance imaging: comparison of plasma vascular endothelial growth factor, hemodynamic, and pharmacokinetic parameters. Acta Radiol. 2004 Jul;45(4):446-52. [CrossRef]
- Silva, P; Hernández, N; Tapia, H; Gaete-Ramírez, B; Torres, P; Flores, T; Herrera, D; Cáceres-Verschae, A; Acuña, RA; Varas-Godoy, M; Torres, VA. Tumor-derived hypoxic small extracellular vesicles promote endothelial cell migration and tube formation via ALS2/Rab5/β-catenin signaling. FASEB J. 2024 Jun 15;38(11):e23716. [CrossRef]
- Winkler, J; Abisoye-Ogunniyan, A; Metcalf, KJ; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020 Oct 9;11(1):5120. [CrossRef]
- Wang, JL; Sun, SZ; Qu, X; Liu, WJ; Wang, YY; Lv, CX; Sun, JZ; Ma, R. Clinicopathological significance of CEACAM1 gene expression in breast cancer. Chin J Physiol. 2011 Oct 31;54(5):332-8.
- Seo, BR; Bhardwaj, P; Choi, S; Gonzalez, J; Andresen Eguiluz, RC; Wang, K; Mohanan, S; Morris, PG; Du, B; Zhou, XK; Vahdat, LT; Verma, A; Elemento, O; Hudis, CA; Williams, RM; Gourdon, D; Dannenberg, AJ; Fischbach, C. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci Transl Med. 2015 Aug 19;7(301):301ra130. [CrossRef] [PubMed]
- Kang, Y; Meng, L; Bai, S; Li, S. Extracellular vesicles: the “Trojan Horse” within breast cancer host microenvironments. Mol Cancer. 2025 Jun 23;24(1):183. [CrossRef] [PubMed]
- Brena, D; Huang, MB; Bond, V. Extracellular vesicle-mediated transport: Reprogramming a tumor microenvironment conducive with breast cancer progression and metastasis. Transl Oncol. 2022 Jan;15(1):101286. Epub 2021 Nov 25. [CrossRef]
- Sneider, A; Liu, Y; Starich, B; Du, W; Nair, PR; Marar, C; Faqih, N; Ciotti, GE; Kim, JH; Krishnan, S; Ibrahim, S; Igboko, M; Locke, A; Lewis, DM; Hong, H; Karl, MN; Vij, R; Russo, GC; Gómez-de-Mariscal, E; Habibi, M; Muñoz-Barrutia, A; Gu, L; Eisinger-Mathason, TSK; Wirtz, D. Small Extracellular Vesicles Promote Stiffness-mediated Metastasis. Cancer Res Commun. 2024 May 9;4(5):1240-1252. [CrossRef]
- Tamimi, RM; Spiegelman, D; Smith-Warner, SA; Wang, M; Pazaris, M; Willett, WC; Eliassen, AH; Hunter, DJ. Population Attributable Risk of Modifiable and Nonmodifiable Breast Cancer Risk Factors in Postmenopausal Breast Cancer. Am J Epidemiol. 2016 Dec 15;184(12):884-8930. Epub 2016 Dec 6. [CrossRef]
- Seiler, A.; Chen, M. A.; Brown, R. L.; Fagundes, C. P. (2018). Obesity, Dietary Factors, Nutrition, and Breast Cancer Risk. Current breast cancer reports, 10(1), 14–27. [CrossRef]
- Uhomoibhi, T. O.; Okobi, T. J.; Okobi, O. E.; Koko, J. O.; Uhomoibhi, O.; Igbinosun, O. E.; Ehibor, U. D.; Boms, M. G.; Abdulgaffar, R. A.; Hammed, B. L.; Ibeanu, C.; Segun, E. O.; Adeosun, A. A.; Evbayekha, E. O.; Alex, K. B. (2022). High-Fat Diet as a Risk Factor for Breast Cancer: A Meta-Analysis. Cureus, 14(12), e32309. [CrossRef]
- Boyd, NF; Stone, J; Vogt, KN; Connelly, BS; Martin, LJ; Minkin, S. Dietary fat and breast cancer risk revisited: a meta-analysis of the published literature. Br J Cancer. 2003 Nov 3;89(9):1672-85. [CrossRef]
- Khodarahmi, M; Azadbakht, L. The association between different kinds of fat intake and breast cancer risk in women. Int J Prev Med. 2014 Jan;5(1):6-15. [PubMed]
- Fay, MP; Freedman, LS; Clifford, CK; Midthune, DN. Effect of different types and amounts of fat on the development of mammary tumors in rodents: a review. Cancer Res. 1997 Sep 15;57(18):3979-88.
- Wirfält, E; Mattisson, I; Gullberg, B; Johansson, U; Olsson, H; Berglund, G. Postmenopausal breast cancer is associated with high intakes of omega6 fatty acids (Sweden). Cancer Causes Control. 2002 Dec;13(10):883-93. [CrossRef]
- Chen, J; Liu, X; Zou, Y; Gong, J; Ge, Z; Lin, X; Zhang, W; Huang, H; Zhao, J; Saw, PE; Lu, Y; Hu, H; Song, E. A high-fat diet promotes cancer progression by inducing gut microbiota-mediated leucine production and PMN-MDSC differentiation. Proc Natl Acad Sci U S A. 2024 May 14;121(20):e230677612. Epub 2024 May 6. [CrossRef]
- Hergueta-Redondo, M; Sánchez-Redondo, S; Hurtado, B; Santos, V; Pérez-Martínez, M; Ximénez-Embún, P; McDowell, SAC; Mazariegos, MS; Mata, G; Torres-Ruiz, R; Rodríguez-Perales, S; Martínez, L; Graña-Castro, O; Megias, D; Quail, D; Quintela-Fandino, M; Peinado, H. The impact of a high fat diet and platelet activation on pre-metastatic niche formation. Nat Commun. 2025 Apr 2;16(1):2897. [CrossRef]
- Rose, DP; Gracheck, PJ; Vona-Davis, L. The Interactions of Obesity, Inflammation and Insulin Resistance in Breast Cancer. Cancers (Basel). 2015 Oct 26;7(4):2147-68. [CrossRef]
- Goodwin, PJ; Stambolic, V. Obesity and insulin resistance in breast cancer--chemoprevention strategies with a focus on metformin. Breast. 2011 Oct;20 Suppl 3:S31-5. Erratum in: Breast. 2012 Apr;21(2):224. [CrossRef]
- Phipps, AI; Chlebowski, RT; Prentice, R; McTiernan, A; Stefanick, ML; Wactawski-Wende, J; Kuller, LH; Adams-Campbell, LL; Lane, D; Vitolins, M; Kabat, GC; Rohan, TE; Li, CI. Body size, physical activity, and risk of triple-negative and estrogen receptor-positive breast cancer. Cancer Epidemiol Biomarkers Prev. 2011 Mar;20(3):454-63. Epub 2011 Mar 1. [CrossRef] [PubMed]
- Protani, M; Coory, M; Martin, JH. Effect of obesity on survival of women with breast cancer: systematic review and meta-analysis. Breast Cancer Res Treat. 2010 Oct;123(3):627-35. Epub 2010 Jun 23. [CrossRef]
- Javed, SR; Skolariki, A; Zameer, MZ; Lord, SR. Implications of obesity and insulin resistance for the treatment of oestrogen receptor-positive breast cancer. Br J Cancer. 2024 Dec;131(11):1724-1736. Epub 2024 Sep 9. [CrossRef]
- Dehesh, T; Fadaghi, S; Seyedi, M; Abolhadi, E; Ilaghi, M; Shams, P; Ajam, F; Mosleh-Shirazi, MA; Dehesh, P. The relation between obesity and breast cancer risk in women by considering menstruation status and geographical variations: a systematic review and meta-analysis. BMC Womens Health. 2023 Jul 26;23(1):392. [CrossRef]
- Chan, DSM; Vieira, AR; Aune, D; Bandera, EV; Greenwood, DC; McTiernan, A; Navarro Rosenblatt, D; Thune, I; Vieira, R; Norat, T. Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Ann Oncol. 2014 Oct;25(10):1901-1914. [CrossRef] [PubMed]
- Al-Shami, K; Awadi, S; Khamees, A; Alsheikh, AM; Al-Sharif, S; Ala’ Bereshy, R; Al-Eitan, SF; Banikhaled, SH; Al-Qudimat, AR; Al-Zoubi, RM; Al Zoubi, MS. Estrogens and the risk of breast cancer: A narrative review of literature. Heliyon. 2023 Sep 17;9(9):e20224. [CrossRef] [PubMed]
- Grossmann, ME; Nkhata, KJ; Mizuno, NK; Ray, A; Cleary, MP. Effects of adiponectin on breast cancer cell growth and signaling. Br J Cancer. 2008 Jan 29;98(2):370-9. Epub 2008 Jan 8. [CrossRef]
- Caffa, I; Spagnolo, V; Vernieri, C; Valdemarin, F; Becherini, P; Wei, M; Brandhorst, S; Zucal, C; Driehuis, E; Ferrando, L; Piacente, F; Tagliafico, A; Cilli, M; Mastracci, L; Vellone, VG; Piazza, S; Cremonini, AL; Gradaschi, R; Mantero, C; Passalacqua, M; Ballestrero, A; Zoppoli, G; Cea, M; Arrighi, A; Odetti, P; Monacelli, F; Salvadori, G; Cortellino, S; Clevers, H; De Braud, F; Sukkar, SG; Provenzani, A; Longo, VD; Nencioni, A. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature. 2020 Jul;583(7817):620-624. Epub 2020 Jul 15. Erratum in: Nature. 2020 Dec;588(7839):E33. doi: 10.1038/s41586-020-2957-6. [CrossRef]
- Iyengar, NM; Zhou, XK; Gucalp, A; Morris, PG; Howe, LR; Giri, DD; Morrow, M; Wang, H; Pollak, M; Jones, LW; Hudis, CA; Dannenberg, AJ. Systemic Correlates of White Adipose Tissue Inflammation in Early-Stage Breast Cancer. Clin Cancer Res. 2016 May 1;22(9):2283-9. Epub 2015 Dec 28. [CrossRef]
- Chang, MC; Eslami, Z; Ennis, M; Goodwin, PJ. Crown-like structures in breast adipose tissue of breast cancer patients: associations with CD68 expression, obesity, metabolic factors and prognosis. [CrossRef]
- Birts, CN; Savva, C; Laversin, SA; Lefas, A; Krishnan, J; Schapira, A; Ashton-Key, M; Crispin, M; Johnson, PWM; Blaydes, JP; Copson, E; Cutress, RI; Beers, SA. Prognostic significance of crown-like structures to trastuzumab response in patients with primary invasive HER2 + breast carcinoma. Sci Rep. 2022 May 24;12(1):7802. [CrossRef]
- Hill, AA; Anderson-Baucum, EK; Kennedy, AJ; Webb, CD; Yull, FE; Hasty, AH. Activation of NF-κB drives the enhanced survival of adipose tissue macrophages in an obesogenic environment. Mol Metab. 2015 Jul 28;4(10):665-77. [CrossRef]
- Takikawa, A; Mahmood, A; Nawaz, A; Kado, T; Okabe, K; Yamamoto, S; Aminuddin, A; Senda, S; Tsuneyama, K; Ikutani, M; Watanabe, Y; Igarashi, Y; Nagai, Y; Takatsu, K; Koizumi, K; Imura, J; Goda, N; Sasahara, M; Matsumoto, M; Saeki, K; Nakagawa, T; Fujisaka, S; Usui, I; Tobe, K. HIF-1α in Myeloid Cells Promotes Adipose Tissue Remodeling Toward Insulin Resistance. Diabetes. 2016 Dec;65(12):3649-3659. Epub 2016 Sep 13. [CrossRef]
- Campbell, RA; Bhat-Nakshatri, P; Patel, NM; Constantinidou, D; Ali, S; Nakshatri, H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001 Mar 30;276(13):9817-24. Epub 2001 Jan 3. [CrossRef]
- Denduluri, SK; Idowu, O; Wang, Z; Liao, Z; Yan, Z; Mohammed, MK; Ye, J; Wei, Q; Wang, J; Zhao, L; Luu, HH. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015 Mar 1;2(1):13-25. [CrossRef]
- Sohi, I; Rehm, J; Saab, M; Virmani, L; Franklin, A; Sánchez, G; Jhumi, M; Irshad, A; Shah, H; Correia, D; Ferrari, P; Ferreira-Borges, C; Lauby-Secretan, B; Galea, G; Gapstur, S; Neufeld, M; Rumgay, H; Soerjomataram, I; Shield, K. Alcoholic beverage consumption and female breast cancer risk: A systematic review and meta-analysis of prospective cohort studies. Alcohol Clin Exp Res (Hoboken). 2024 Dec;48(12):2222-2241. Epub 2024 Nov 24. [CrossRef]
- Fanfarillo, F; Caronti, B; Lucarelli, M; Francati, S; Tarani, L; Ceccanti, M; Piccioni, MG; Verdone, L; Caserta, M; Venditti, S; Ferraguti, G; Fiore, M. Alcohol Consumption and Breast and Ovarian Cancer Development: Molecular Pathways and Mechanisms. Curr Issues Mol Biol. 2024 Dec 20;46(12):14438-14452. [CrossRef]
- Dixon, RA. Phytoestrogens. Annu Rev Plant Biol. 2004;55:225-61. Annu Rev Plant Biol. 2004, 55, 225–61. [Google Scholar] [CrossRef]
- Liu, ZQ. What about the progress in the synthesis of flavonoid from 2020? Eur J Med Chem. 2022 Dec 5;243:114671. Epub 2022 Aug 27. [CrossRef]
- Jordan, VC; Mittal, S; Gosden, B; Koch, R; Lieberman, ME. Structure-activity relationships of estrogens. Environ Health Perspect. 1985 Sep;61:97-110. [CrossRef]
- Hamblen, EL; Cronin, MT; Schultz, TW. Estrogenicity and acute toxicity of selected anilines using a recombinant yeast assay. Chemosphere. 2003 Aug;52(7):1173-81. [CrossRef] [PubMed]
- Teegarden, D; Romieu, I; Lelièvre, SA. Redefining the impact of nutrition on breast cancer incidence: is epigenetics involved? Nutr Res Rev. 2012 Jun;25(1):68-95. [CrossRef]
- Zengul, AG; Demark-Wahnefried, W; Barnes, S; Morrow, CD; Bertrand, B; Berryhill, TF; Frugé, AD. Associations between Dietary Fiber, the Fecal Microbiota and Estrogen Metabolism in Postmenopausal Women with Breast Cancer. Nutr Cancer. 2021;73(7):1108-1117. Epub 2020 Jun 26. [CrossRef]
- Brody, JG; Rudel, RA. Environmental pollutants and breast cancer. Environ Health Perspect. 2003 Jun;111(8):1007-19. [CrossRef]
- Datta, K; Hyduke, DR; Suman, S; Moon, BH; Johnson, MD; Fornace, AJ, Jr. Exposure to ionizing radiation induced persistent gene expression changes in mouse mammary gland. Radiat Oncol. 2012 Dec 5;7:205. [CrossRef] [PubMed]
- Delgado, O; Batten, KG; Richardson, JA; Xie, XJ; Gazdar, AF; Kaisani, AA; Girard, L; Behrens, C; Suraokar, M; Fasciani, G; Wright, WE; Story, MD; Wistuba, II; Minna, JD; Shay, JW. Radiation-enhanced lung cancer progression in a transgenic mouse model of lung cancer is predictive of outcomes in human lung and breast cancer. Clin Cancer Res. 2014 Mar 15;20(6):1610-22. Epub 2014 Jan 31. [CrossRef]
- Rocha, PRS; Oliveira, VD; Vasques, CI; Dos Reis, PED; Amato, AA. Exposure to endocrine disruptors and risk of breast cancer: A systematic review. Crit Rev Oncol Hematol. 2021 May;161:103330. Epub 2021 Apr 20. [CrossRef]
- Williams, GP; Darbre, PD. Low-dose environmental endocrine disruptors, increase aromatase activity, estradiol biosynthesis and cell proliferation in human breast cells. Mol Cell Endocrinol. 2019 Apr 15;486:55-64. Epub 2019 Feb 26. [CrossRef]
- Kwon, Y. Potential Pro-Tumorigenic Effect of Bisphenol A in Breast Cancer via Altering the Tumor Microenvironment. Cancers (Basel). 2022 Jun 19;14(12):3021. [CrossRef]
- Sweeney, C; Lazennec, G; Vogel, CFA. Environmental exposure and the role of AhR in the tumor microenvironment of breast cancer. Front Pharmacol. 2022 Dec 15;13:1095289. [CrossRef]
- Guo, Q; Wang, X; Gao, Y; Zhou, J; Huang, C; Zhang, Z; Chu, H. Relationship between particulate matter exposure and female breast cancer incidence and mortality: a systematic review and meta-analysis. Int Arch Occup Environ Health. 2021 Feb;94(2):191-201. Epub 2020 Sep 10. [CrossRef]
- Lichtiger, L; Rivera, J; Sahay, D; Miller, RL. Polycyclic Aromatic Hydrocarbons and Mammary Cancer Risk: Does Obesity Matter too? J Cancer Immunol (Wilmington). 2021;3(3):154-162. J Cancer Immunol (Wilmington) 2021, 3(3), 154–162. [Google Scholar]
- Panis, C; Candiotto, LZP; Gaboardi, SC; Teixeira, GT; Alves, FM; da Silva, JC; Scandolara, TB; Rech, D; Gurzenda, S; Ponmattam, J; Ohm, J; Castro, MC; Lemos, B. Exposure to Pesticides and Breast Cancer in an Agricultural Region in Brazil. Environ Sci Technol. 2024 Jun 18;58(24):10470-10481. Epub 2024 Jun 6. [CrossRef]
- Tayour, C; Ritz, B; Langholz, B; Mills, PK; Wu, A; Wilson, JP; Shahabi, K; Cockburn, M. A case-control study of breast cancer risk and ambient exposure to pesticides. Environ Epidemiol. 2019 Oct 14;3(5):e070. [CrossRef]
- Snedeker, SM. Pesticides and breast cancer risk: a review of DDT, DDE, and dieldrin. Environ Health Perspect. 2001 Mar;109 Suppl 1(Suppl 1):35-47. [CrossRef]
Figure 1.
Progression of ductal hyperplasia. Created in BioRender. Elena Mihaela, J. (2025) https://BioRender.com/innlvj8.
Figure 1.
Progression of ductal hyperplasia. Created in BioRender. Elena Mihaela, J. (2025) https://BioRender.com/innlvj8.
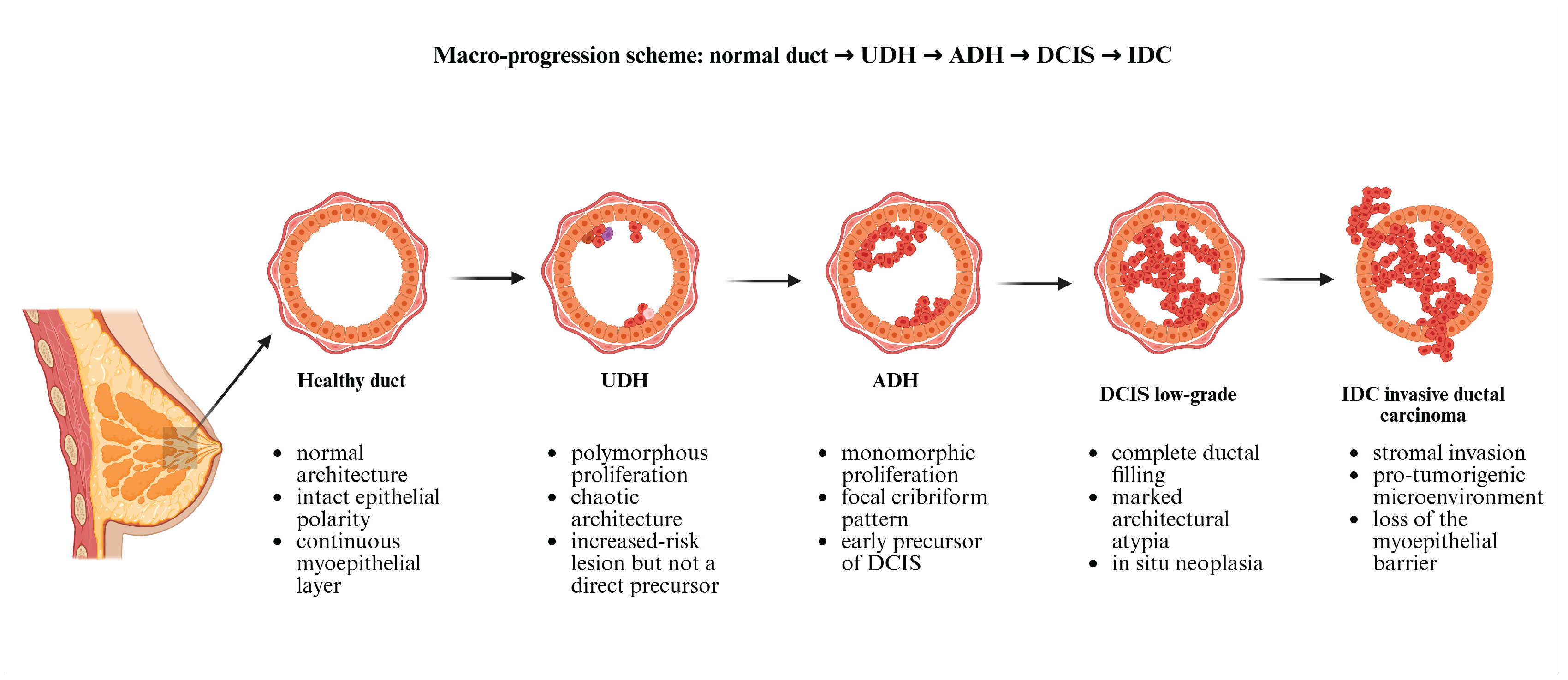

Table 1.
Common genetic alterations in mammary hyperplasia subtypes.
| Lesion Type | Characteristic genetic alterations (mutations and copy number changes) |
|---|---|
| Usual Ductal Hyperplasia (UDH) | Polyclonal proliferation with minimal or no clonal driver mutations. Occasional activating PI3K-AKT pathway mutations (e.g., PIK3CA hotspot mutations) have been detected. It lacks consistent chromosomal aberrations; considered a benign hyperplastic response rather than true neoplasia. [50] |
| Atypical Ductal Hyperplasia (ADH) | Clonal neoplastic lesion sharing many alterations with low-grade DCIS. Frequent PIK3CA mutations (on the order of 30-40% of cases). Loss of chromosome 16q (harboring CDH1 and other genes) is common. Other recurrent mutations in luminal breast cancer genes (e.g., GATA3, MAP3K1, CBFB genes) may be present. Overall, ADH exhibits a luminal A-like genomic profile indicative of early cancerous change. [53] |
| Atypical Lobular Hyperplasia (ALH) | Almost uniformly associated with E-cadherin loss due to CDH1 gene inactivation. This occurs via CDH1 gene mutation or 16q LOH in the majority of cases. Often accompanied by PIK3CA gene mutations similar to those in invasive lobular carcinoma. Occasional mutations in AKT1 or PTEN genes are reported. The genetic pattern overlaps with ER-positive lobular carcinoma, minus additional alterations required for invasion [52]. |
| Columnar Cell Lesions (CCL) and Flat epithelial atypia (FEA) |
High frequency of early oncogenic mutations despite bland histology. PIK3CA gene activating mutations are present in 50-60% of cases, making this a hallmark of FEA/CCLs. Some cases show modest copy number changes (e.g., gains on 1q; losses on 16q or 17p). Clonal relationship to adjacent ADH/DCIS is evidenced by shared mutations, supporting CCL/FEA as the earliest morphologic precursor in low-grade tumor evolution. [53]. |
Legend Table 1: PI3K-AKT = Phosphoinositide 3-kinase-- Protein kinase B signaling pathway; PIK3CA = Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha;CDH1=Cadherin-1 ; DNA-binding protein GATA3 (GATA3) = DNA binding protein 3; mitogen-activated protein kinase kinase kinase 1 (MAP3K1) = Mitogen-Activated Protein Kinase 1;core-binding factor subunit beta (CBFB) = Core-Binding Factor Subunit Beta; AKT1 = Serine/Threonine Ki-nase 1; phosphatase and tensin homolog (PTEN) = Phosphatase and Tensin Homolog; ER = Estrogen Recep-tor.
Table 2.
Key molecular drivers in malignant progression of atypical mammary hyperplasia.
| Gene/locus | Pathway/function | Frequent alterations in ADH/FEA | Pathological implication | Reference |
|---|---|---|---|---|
| PIK3CA | PI3K/AKT signaling (oncogene) | H1047R, E545K, E542K (missense mutations) | Frequent in FEA/ADH and luminal DCIS/IBC; early driver; sustained proliferation; defines low-grade pathway | [54] |
| TP53 | Genome integrity/ apoptosis (tumor suppressor) |
R175H, R248Q/W, R273H/C (missense mutations) |
Subset of atypia/DCIS on progression trajectory; common in high-grade disease; often with ERBB2 amplification | [52] |
| BRCA1 | Homologous recombination DNA repair (tumor suppressor) |
c.68_69delAG (185delAG), c.5266dupC (5382insC); numerous truncations | High-risk hereditary context; genomic instability in early clones | [55] |
| BRCA2 | Homologous recombination DNA repair (tumor suppressor) |
Multiple frameshifts/nonsense variants across exons | High-risk hereditary context; facilitates mutation accumulation | [56] |
| GATA3 | Luminal lineage transcription Factor | Frameshift/truncations; splice-site alterations | Supports luminal differentiation program in HR+ precursors | [57] |
| CDH1 | E-cadherin; cell–cell adhesion (tumor suppressor) |
Bi-allelic inactivation/truncating mutations | Marks the lobular pathway (atypical lobular hyperplasia to lobular carcinoma) | [58] |
| AKT1 | Serine/threonine kinase downstream of PI3K (oncogene) |
E17K (PH domain hotspot) | Activates AKT independent of PI3K, specific to HR+ lesions | [59] |
| PTEN | PI3K negative regulator; lipid phosphatase (tumor suppressor) |
Loss-of-function mutations/deletions; promoter silencing | Cooperates with PIK3CA activation in atypia/DCIS | [50] |
| ERBB2 (HER2) | Receptor tyrosine kinase (oncogene) |
Amplification; S310F/Y; Y772_A775dup (kinase insertion) | Low-level expression in some early lesions; targetable alterations | [60] |
| ERBB3 (HER3) | Receptor tyrosine kinase; dimerizes with HER2 | E928G and kinase-domain substitutions | Co-occurs with ERBB2 mutations; luminal contexts | [62] |
| RUNX1/ CBFB | Core-binding factor complex; lineage and differentiation | RUNX1 Runt domain loss-of-function; CBFB alterations | Luminal tumors/precursors; tumor-suppressor roles | [50] |
| MAP3K1 | MAPK signaling; luminal A-associated | Multiple truncations/missense; pathway-inactivating | Co-mutation patterns with PIK3CA/CBFB in luminal A | [58] |
| ESR1 | Estrogen receptor alpha (nuclear receptor) |
Ligand-binding domain: Y537S, D538G (rare in primary) | Rare in early lesions; prevalent in endocrine-resistant metastases | [61] |
| FOXA1 | Pioneer transcription factor for ER program | Wing2/DBD hotspots (e.g., R219); indels affecting DNA binding | Luminal program maintenance in HR+ tumors; early presence reported | [62] |
| KMT2C (MLL3)/ KMT2D (MLL2) | Histone H3K4 methyltransferases (chromatin modifier) | Truncating mutations across coding exons | Epigenetic deregulation in luminal tumors/precursors | [63] |
| ARID1A/ ARID1B | SWI/SNF chromatin remodeling complex (tumor suppressor) | Frameshift/nonsense mutations; loss of protein expression | Subset of HR+/basal lesions; chromatin accessibility shifts | [64] |
| NCOR1/ NCOR2 | Nuclear receptor co-repressors; ER signaling modulation | Truncations and splice variants | Associated with endocrine response and luminal programs | [65] |
| CREBBP/ EP300 | Histone acetyltransferases; transcriptional co-activators | Loss-of-function mutations | Transcriptional reprogramming in early neoplasia | [68] |
| PIK3R1 | PI3K regulatory subunit p85α (tumor suppressor-like) |
Truncations/indels disrupting SH2 domains | Cooperates with PIK3CA in PI3K activation | [54] |
| NOTCH1 | NOTCH signaling (context-dependent) | PEST-domain truncations; activating mutations | Subset of breast lesions; cross-talk with ER and PI3K | [52] |
Legend Table 2: ADH = Atypical Ductal Hyperplasia; FEA = Flat Epithelial Atypia; CIS = Ductal Carcinoma In Situ; IBC = Invasive Breast Carcinoma; HR+ = Hormone Receptor Positive; ER = Estrogen Receptor; PIK3CA = Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha;TP53 = Tumor Protein p53;BRCA1 = Breast Cancer Gene 1; BRCA2 = Breast Cancer Gene 2,GATA3 =- DNA Binding Protein 3; CDH1 = Cadherin 1; AKT1 = Serine/Threonine Kinase 1; PTEN = Phosphatase and Tensin Homolog;erb-b2 receptor tyrosine kinase 2 (ERBB2) (hematoxylin–eosin (HE)R2) = Erb-B2 Receptor Tyrosine Kinase 2 / Human Epi-dermal Growth Factor Receptor 2; erb-b2 receptor tyrosine kinase 3 (ERBB3) (human epidermal growth factor receptor 3 (HER3)) = Erb-B2 Receptor Tyrosine Kinase 3 / Human Epidermal Growth Factor Receptor 3;RUNX1 =- Runt-Related Transcription Factor 1; CBFB = Core-Binding Factor Subunit Beta;MAP3K1 = Mi-togen-Activated Protein Kinase 1;estrogen receptor 1 (ESR1) = Estrogen Receptor 1;forkhead box A1 (FOXA1) = Forkhead Box A1; KMT2C (MLL3) = Lysine Methyltransferase 2C / Mixed-Lineage Leukemia 3; KMT2D (MLL2 = Lysine Methyltransferase 2D / Mixed-Lineage Leukemia 2;ARID1A = AT-Rich Interaction Domain 1A; ARID1B = AT-Rich Interaction Domain 1B;NCOR1=Nuclear Receptor Corepressor 1;NCOR2 = Nuclear Receptor Corepressor 2;CREBBP = CREB Binding Protein;EP300 = E1A Binding Protein p300;PIK3R1 = Phosphatidylinositol-3-Kinase Regulatory Subunit 1; NOTCH1 = Notch Receptor 1;MAPK = Mitogen-Activated Protein Kinase; SWI/SNF = Switch/Sucrose Non-Fermentable;SH2 = Src Homology 2; PEST = Proline–Glutamate–Serine–Threonine; DBD = DNA-Binding Domain; PH = Pleckstrin Homolo-gy;PI3K = Phosphoinositide 3-Kinase.
Table 3.
Notable epigenetic changes in mammary hyperplasia subtypes.
| Lesion type/stage | Epigenetic alterations |
|---|---|
| Early proliferative lesions (UDH, mild hyperplasia) |
Subtle epigenetic deviations may begin. Global DNA methylation levels start to increase in proliferative breast tissue. Promoter methylation is generally low in UDH, but isolated foci can show initial methylation of genes like RASSF1A. Histones in UDH largely retain normal patterns, although minor shifts toward a more closed chromatin conformation have been noted in high-risk patients. MiRNA expression is near-normal, aside from slight upregulation of proliferative miRNAs (e.g., miR-21) in some cases. Overall, UDH lacks the pronounced epigenetic silencing seen in atypia [144]. |
| Atypical hyperplasias (ADH, ALH, FEA) |
Promoter hypermethylation becomes common and non-random. Frequent methylation of tumor suppressor genes (e.g., RASSF1A, APC) is documented in ADH and FEA lesions. In lobular neoplasia, CDH1 promoter methylation is an alternative mechanism to gene mutation for turning off E-cadherin [62]. These methylation marks often coexist with repressive histone modifications; for instance, hypermethylated genes show enriched H3K27me3 and decreased acetylation, reinforcing chromatin compaction. Genome-wide, atypical lesions exhibit a shift toward a cancer-like epigenome with dozens of genes epigenetically silenced. MicroRNA profiles are significantly altered: oncomiRs such as miR-21 and miR-155 are elevated in ADH, correlating with suppressed PTEN and TP53 pathways [145]. Conversely, certain anti-proliferative miRNAs (e.g., miR-1297, miR-125b) are downregulated in FEA/ADH [62]. These changes act in concert to disable cell-cycle checkpoints and promote survival, thereby potentiating progression to carcinoma in situ. |
| Carcinoma in situ (DCIS, LCIS) |
Widespread epigenetic reprogramming is present. DCIS lesions show hypermethylation at numerous loci, e.g., more than 40% of DCIS cases have RASSF1A gene methylation [71], and many harbor methylation of p16, cyclin D2, and others. LCIS (especially pleomorphic LCIS) likewise accumulates methylation events, though classical LCIS may rely more on CDH1 loss by mutation. Global DNA hypomethylation begins to be evident in carcinoma in situ, contributing to genomic instability. Histone modification patterns are clearly abnormal: high levels of HDACs lead to histone hypoacetylation, and Polycomb complexes maintain silencing of differentiation genes. MicroRNAs in CIS lesions often mirror invasive cancer profiles; for example, miR-21 and miR-210 are strongly up, while miR-205 (an anti-Epithelial–mesenchymal transition (EMT) miRNA is down. Such miRNA dysregulation in CIS has been linked with aggressive features and may predict which in situ lesions are likely to invade [72]. |
Legend Table 3: UDH = Usual Ductal Hyperplasia; AHs = Atypical Hyperplasias; ADH = Atypical Ductal Hyperplasia; ALH = Atypical Lobular Hyperplasia ; Flat Epithelial Atypia; DCIS = Ductal Carcinoma In Situ; LCIS = Lobular Carcinoma In Situ ;LN = Lobular Neoplasia; CIS = Carcinoma In Situ;RASSF1A = Ras Association Domain Family Member 1;APC = Adenomatous Polyposis Coli;CDH1 = Cadherin 1;PTEN = Phosphatase and Tensin Homolog;TP53 = Tumor Protein p53;DNA = Deoxyribonucleic Acid;H3K27me3 = Trimethylation of Lysine 27 on Histone H3;HDACs = Histone Deacetylases; miRNA = MicroRNA ;miR-21 = MicroRNA 21 ;miR-155 = MicroRNA 155 ;miR-1297 = MicroRNA 1297 ;miR-125b = MicroRNA 125b ;miR-210 = MicroRNA 210 ;miR-205 = MicroRNA 205 ;EMT = Epithelial–Mesenchymal Transition; OncomiRs = Oncogenic MicroRNAs.
Table 4.
Molecular and cellular effects of epigenetic mechanisms driving malignancy in atypical hyperplasia.
Table 4.
Molecular and cellular effects of epigenetic mechanisms driving malignancy in atypical hyperplasia.
| Mechanism | Gene targets/factor | Molecular results in ADH/DCIS | Functional consequences |
|---|---|---|---|
| Promoter hypermethylation (DNA) | RASSF1, CDKN2A (p16INK4a), BRCA1, PTEN | Stable silencing of tumor suppressors | Loss of cell cycle arrest; early event in monoclonal progression [146] |
| Oncogene hypomethylation (DNA) | HER2, CCND1 (Cyclin D1) | Transcriptional increase | Overexpression of proliferation drivers [147] |
| Non-coding RNA dysregulation | miR-21 (Upregulation) | Post-transcriptional inhibition of PTEN | Constitutive activation of the PI3K/AKT pathway; promotes survival [148] |
| Histone modifications/remodeling | KMT2C/D, EZH2 | Aberrant acetylation/methylation patterns | Transcriptional reprogramming for malignant phenotype [49] |
Legend Table 4: ADH = Atypical Ductal Hyperplasia; DCIS = Ductal Carcinoma In Situ; DNA = Deoxyri-bonucleic Acid; RASSF1 = Ras Association Domain Family Member 1; CDKN2A = Cyclin-Dependent Ki-nase Inhibitor 2A;p16INK4a = p16 Inhibitor of Cyclin-Dependent Kinase 4a; BRCA1 = Breast Cancer Gene 1 ;PTEN = Phosphatase and Tensin Homolog; HER2 = Human Epidermal Growth Factor Receptor 2; CCND1 = Cyclin D1;miR-21 = MicroRNA 21; PI3K = Phosphoinositide 3-Kinase; AKT = AKT Ser-ine/Threonine Kinase; KMT2C = Lysine Methyltransferase 2C; KMT2D = Lysine Methyltransferase 2D; EZH2 = Enhancer of Zeste Homolog 2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



