Submitted:
16 December 2025
Posted:
17 December 2025
You are already at the latest version
Abstract
Nano Metal–Organic Frameworks (nMOFs) have emerged as a versatile class of porous materials with significant potential in biomedical applications, particularly in cancer treatment. This review explores the pivotal role of nMOFs in facilitating the combina-tion of photodynamic therapy (PDT) and immunotherapy (IMT), focusing on their unique capabilities to synergistically enhance therapeutic outcomes. By serving as effi-cient photosensitizer and immunotherapy drug carriers nMOFs serve as immune re-sponse modulators and enable targeted tumour destruction through reactive oxygen species generation while simultaneously stimulating antitumor immunity. The com-bination of nMOF-based PDT and immunotherapy represents a promising strategy for more effective, personalized cancer treatments. This article highlights recent progress, challenges, and future perspectives in leveraging nMOFs for the synergistic cancer therapy landscape.
Keywords:
photodynamic therapy
; immunotherapy
; immunosuppressant inhibitors
; immunity stimulation
; PDT-IMT
; combination therapy
; metal-organic frameworks
; cancer targeting
; tumor selectivity
1. Introduction
Cancer is a significant global health challenge, characterized by high recurrence and fatality rates, as well as the propensity for metastasis, leading to millions of deaths annually. Traditional treatment modalities such as surgery, radiotherapy (RT), and chemotherapy remain prevalent; however, they are often hindered by issues of poor precision, low efficiency, and severe side effects that diminish their therapeutic effectiveness. In response to these limitations, various innovative treatment strategies have emerged, including PDT, chemodynamic therapy (CDT), photothermal therapy (PT), starvation therapy, and immunotherapy. These methods are favoured for their lower side effects, non-invasive nature, and ease of application. Despite their advantages, these therapies encounter significant challenges. For instance, CDT is constrained by the limited availability of hydrogen peroxide in the tumour microenvironment (TME), while PDT struggles with the instability of photosensitizers and its reliance on oxygen. To enhance the efficacy of cancer treatments, researchers are increasingly turning to advanced nanomaterials. The development of these materials has led to the creation of various innovative therapeutic approaches that aim to overcome the shortcomings associated with conventional cancer therapies [1].
nMOFs have gained attention due to their unique properties that can potentially address the limitations of existing treatment methods. Their potential to improve efficiency, precision, and reduce side effects makes them a promising avenue for advancing cancer therapy. nMOFs consist of organic ligands or linkers and metal clusters or metal ions joined by coordinative bonds connected in one-, two- or three-dimensional networks [2]. nMOFs are exploited in many fields of research, and they are currently explored for their use in biological application as drug/cargo delivery because of their high porosity ranging from 1nm to several nanometres, large surface area ranging from 100 to 1000 m2/g, variable pore size, low cytotoxicity, high biocompatibility, high crystallinity, chemical and thermal stability, stable luminescence and high biodegradability [3,4]. Recent developments have resulted in the creation of biodegradable and non-toxic nMOFs, which help reduce side effects and promote the body’s clearance of these materials following treatment. This property is essential for clinical applications, as it prioritizes patient safety [5].
2. Application of nMOFs in Biomedical Fields
Recently, researchers have devoted significant attention to studying the application of nMOFs in biomedical fields, gaining substantial understanding on how the materials function. Researchers have employed strategies of designing nMOFs for use as delivery systems, as therapeutics or for achieving diagnostics. nMOFs can be designed to deliver therapeutics via passive targeting or active targeting [6]. They can be designed to react to specific stimuli, such as pH levels, redox conditions, or enzymatic activity, enabling the controlled release of therapeutic agents in the tumour microenvironment (TME) [7].
2.1. Metal Organic Frameworks in Drug Delivery
Drug delivery systems (DDSs) that utilize nMOFs have captured the interest of numerous researchers. nMOFs hold significant potential in biomedical applications when compared to traditional materials, as highlighted by several key factors:
- the use of various metal ions or clusters and organic linkers allows for the creation of nMOFs with diverse morphologies, compositions, and sizes, facilitating the loading of a wide range of cargo molecules, from small drugs to larger proteins.
- the tuneable pore sizes and high surface area-to-volume ratios of nMOFs contribute to their ability to achieve substantial drug loading capacities.
- controlled drug release, achieved by adjusting host–guest interactions of nMOFs, can provide controllable drug release, ensuring that the therapeutic effects are directed to tumour sites and controlled.
- due to their biodegradability and safety, most nMOFs are both bio-accessible and biodegradable, which helps mitigate adverse effects on the human body [8].
nMOFs can be modified to specifically target certain cells or tissues, especially in the context of cancer treatment by altering their surface characteristics. Researchers can improve the selective absorption of therapeutic agents by tumour cells, which in turn boosts the effectiveness of the treatment [9]. The structural and chemical properties of nMOFs can be readily adjusted by changing the metal ions and organic ligands utilized in their synthesis. This ability to modify their properties allows for the creation of nMOFs with specific pore sizes and surface functionalities that are customized for particular therapeutic applications [7]. nMOFs can enable the controlled and sustained release of therapeutic agents in response to specific stimuli, such as changes in pH or enzymatic activity. This capability facilitates targeted delivery, reducing side effects on healthy tissues while enhancing therapeutic effects on diseased cells [10]. nMOFs are capable of encapsulating biomacromolecule drugs that are usually unstable under physiological conditions. This encapsulation safeguards the drugs from degradation and aids in their delivery to specific target sites within the body [11].
2.2. Classification of the Efficacy of Combinations Therapies
The anticancer efficacy of combination therapies using MOFs may be classified as synergistic or additive enhancement, or antagonistic depending on whether the efficacy is enhanced or reduced. Synergistic enhancement is achieved when the efficacy of the combination therapy is more than sum of the efficacies of the combined methods. This is shown in equation 1, where EF(nMOF@PDT&IMT) = efficacy of the combination of PDT and IMT, EF(PDT) = efficacy of PDT alone, EF(IMT) = efficacy of the immunotherapy alone.
EF(nMOF@PDT&IMT) > EF(PDT) + EF(IMT)
The efficacy will be classified as additive enhancement if the efficacy of the combination therapy is equal to the sum of the efficacies of the combined methods (equation 2).
EF(nMOF@PDT&IMT) = EF(PDT) + EF(IMT)
The efficacy will be classified as antagonistic if the efficacy of the combination therapy is less than the sum of the efficacies of the combined methods (equation 3).
EF(nMOF@PDT&IMT) < EF(PDT) + EF(IMT)
2.3. PDT Combinations with IMT
Using nMOFs, PDT may be combined with IMT (PDT+IMT) in several ways. Porphyrins and phthalocyanines can be used as linkers in the synthesis of nMOFs to produce PS-based nMOFs (PnMOFs) which function as PSs by generating ROS upon light activation. Non-PS organic linkers can be used to synthesize non-PS-based nMOFs. To facilitate PDT+IMT non-PS-based nMOFs are used as carriers and delivery systems for PSs and IMT drugs. Upon reaching the TME, ROS are generated by the released PSs, the innate immunity is induced by the released immune stimulating drug, and the TME immunosuppression is blocked by the released checkpoint blockade immunotherapy (CBIMT) inhibitors. Conceptually therefore, the combination of PDT with IMT can be achieved as dual or triple combinations of PDT with IMT, PDT with CBIMT or PDT with IMT and CBIMT.
2.4. Applications of Metal Organic Frameworks in Combinations of PDT with IMT
To facilitate the various combinations of PDT with IMT therefore, nMOFs can play passive roles as carrier and delivery systems or active roles as ROS generating PSs. Systemic toxicity can be reduced by encapsulating the nMOFs with biocompatible materials such as carbohydrate or other polymers. TME targeting may be enhanced by functionalization of the biocompatible encapsulation layer with cancer-specific biomolecules such as aptamers, peptides, and cancer cell membrane. Stimulus responsive release of the PS, IMT and CBIMT drugs such as pH-responsive, glutathione-responsive, can enhance TME targeting.
3. Purpose Statement
In this review, we concentrate on some of the recent advancements and accomplishments of notable nMOF-based nanoplatforms designed for effective cancer treatment through the combination of PDT and IMT. The review emphasizes the performance in vitro and in vivo, the significance and necessity of the nanoplatforms in tumour therapy by interpreting key studies in the field. Additionally, we discuss the prospects and challenges associated with the use of such nanoplatforms across various cancer treatment methods. Unlike previous reviews that have addressed the applications of nMOFs in cancer therapy, this review specifically highlights customizable construction strategies and therapeutic mechanisms of the nanoplatforms, transitioning from single therapy to combination therapy. It aims to provide insights into the potential clinical translation of these nanoplatforms for cancer treatment, with the hope of further advancing the development of nMOF-mediated anticancer multiple combination therapeutics involving PDT and IMT.
Figure 8.
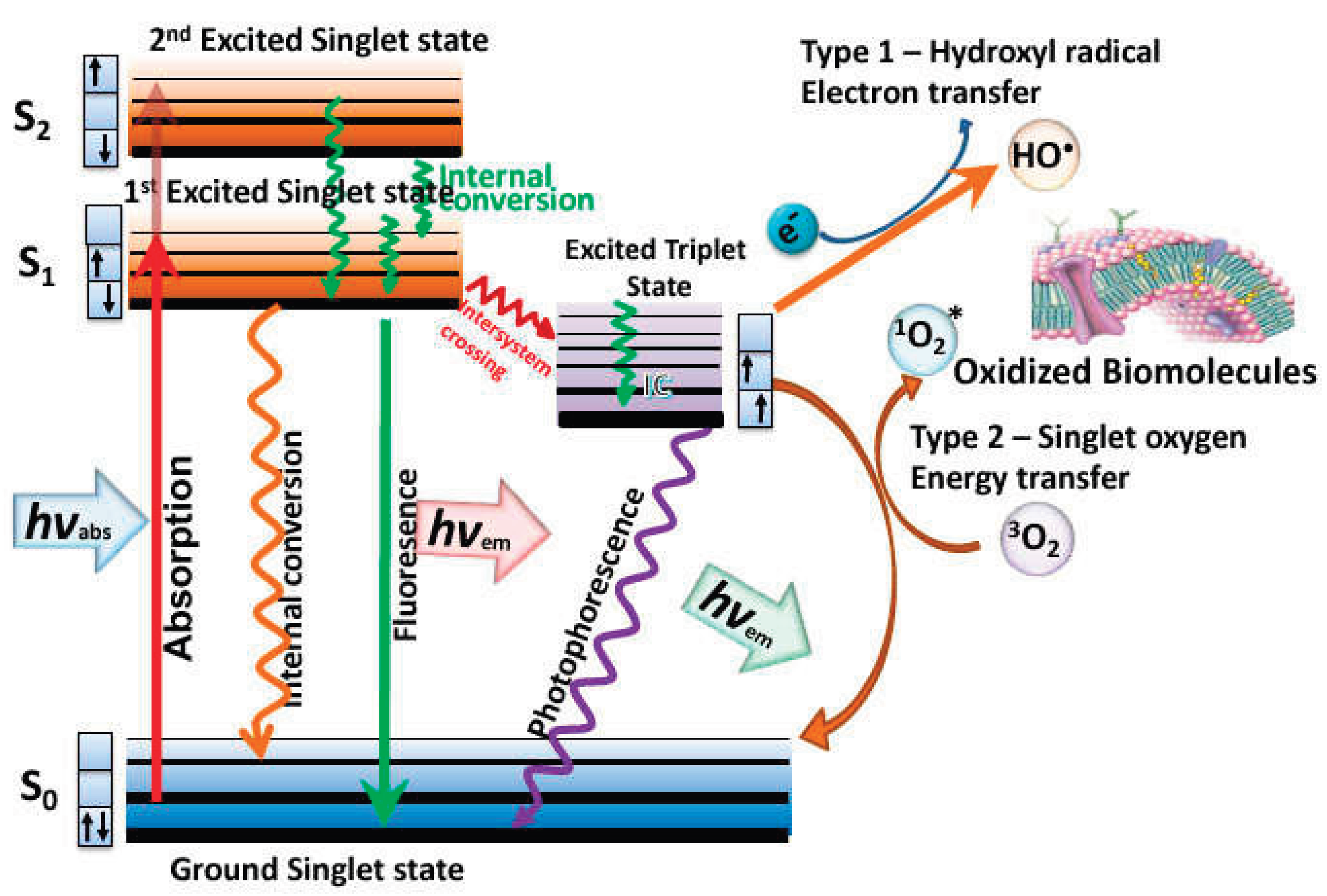
Mechanism of PDT shown by means of a Jablonski diagram [12].
Figure 8.
Mechanism of PDT shown by means of a Jablonski diagram [12].
4. Photodynamic Therapy
PDT is one of the modern and non-invasive forms of therapy that is currently used to treat non-oncological diseases and various types of cancers in different locations on the body. The molecular mechanism of PDT is based on 3 major components; light of appropriate wavelength, oxygen dissolved in intracellular media, and the photosensitizer (PS). These components yield the desired effects within pathological tissues through mutual interactions [13]. The PSs are activated with light of specific wavelength from the ground state reaching a short-live excited singlet state. The excited PS can revert to the ground state by emitting fluorescence. By undergoing intersystem crossing in which the spin of one excited electron is flipped to form a relatively long-lived excited triplet state. The excited triplet state can also revert to the ground state by emitting phosphorescence but significantly it can react with the surrounding substrates (like biomolecules and cell membrane) to produce radicals or react with 3O2 to form reactive oxygen species, like hydroxyl radicals, hydrogen peroxide (type I, reaction) and superoxide anions radicals. Alternatively, the energy that excites the PS can be transferred directly to 3O2 in the ground state to form excited state 1O2 (type II, reaction). The type I and type II reactions can occur simultaneously, with the ratio of these reactions being influenced by the nature of the PS and the concentration of 3O2 and other substrates [14]. A Jablonski diagram is used to illustrate the PDT mechanism (Figure 8). Therefore, there are two mechanisms in which the photodynamic reaction can occur in PDT and both mechanisms are highly dependent on the concentration of oxygen molecules inside the cells.
5. Immunotherapy
5.1. Innate Immunity
IMT it is a type of cancer treatment that enhances the ability of the body’s immune system to identify and attack cancer cells. It operates by stimulating or enhancing the immune system to better recognize and attack cancer. Recently, IMT has emerged as promising therapeutic approach that is effective in treating several types of cancers including breast cancer, pancreatic cancer and colorectal cancer [15]. The IMT mechanism is not only useful in single tumour site but also for patients with metastasizing cancer, where the cancer has spread to other tissues or organs. IMT also displays less side effects and can be combined with other therapies. This enhances its efficacy and reduces the risk of resistance. IMT can also assist the immune system in memorizing the cancer cells so it can attack these types of cells if they show up in the future [16].
Figure 9.
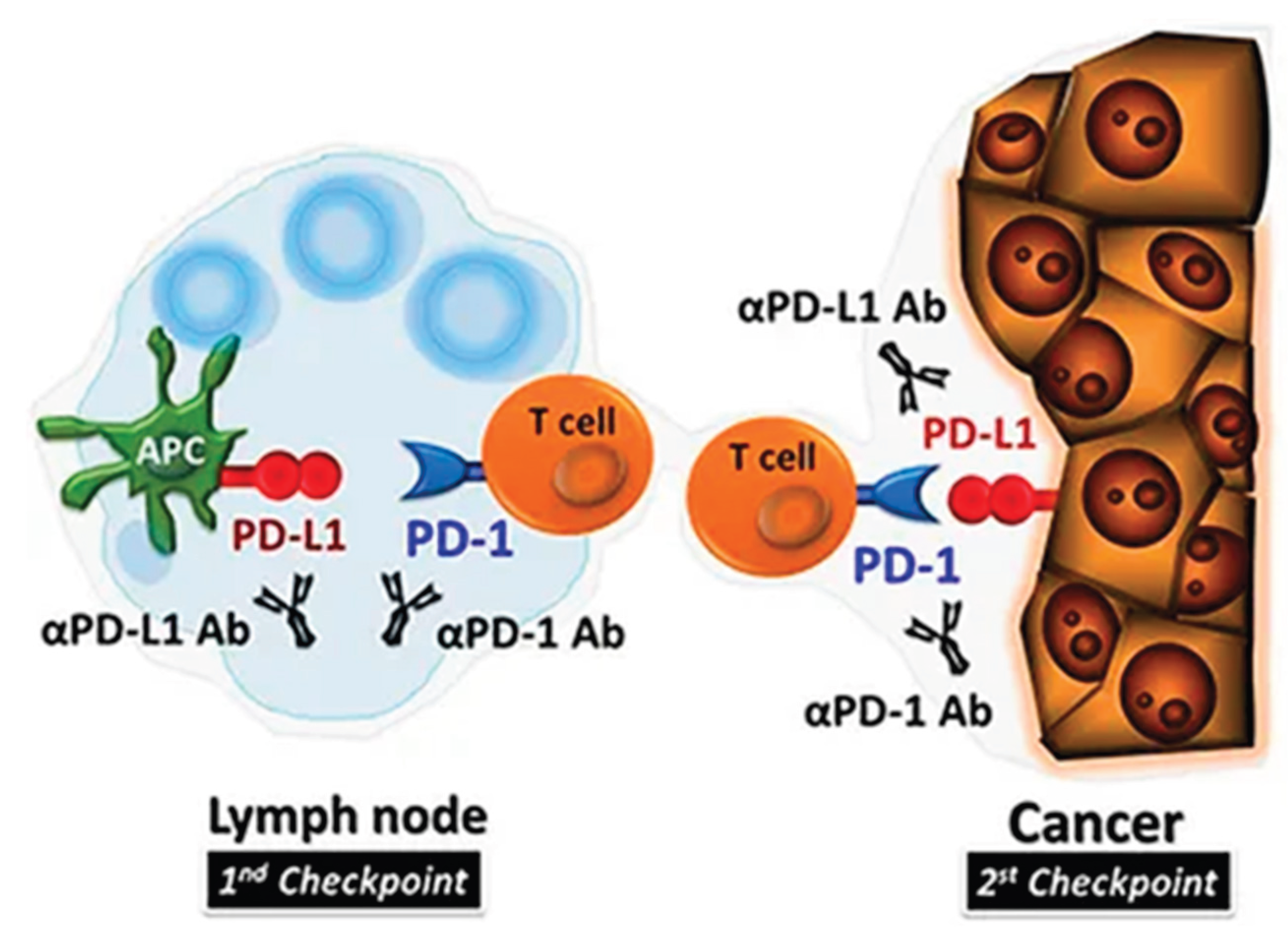
The mechanism illustrating how the PD-1 checkpoint inhibitors of T-cells binds to PDL-1 of tumour cells [17].
Figure 9.
The mechanism illustrating how the PD-1 checkpoint inhibitors of T-cells binds to PDL-1 of tumour cells [17].
5.2. Checkpoint Inhibitors
Negative regulation of the immune system has been shown to have antitumor activity in several solid cancers like advanced melanoma and Non–Small Cell Lung Cancer (NSCLC), where the immune checkpoint inhibitors are the motive force of negative regulation [15]. T-cells exhibit receptors to which the antigens as well as ligands bind, leading to function inhibition or activation, this depends on the type of T-cell receptor that is targeted. The cancer targets and uses the immune checkpoint mechanism to escape the immune system via negative feedback mechanism, which is accomplished by the binding of cancer cells to inhibitory receptors on T-cells like cytotoxic T lymphocyte associated protein 4 and programmed cell death protein 1 (PD 1) inhibitory receptors [18]. The antigens present in cancer cells bind to these inhibitory receptors, and they hinder T-cell proliferation and activation, resulting in weak immune responses against the tumour. The major function of IMT is the application of antibodies that will block checkpoint in the immune system [15].
6. Synergistic Effects of Combining PDT and IMT
The integration of PDT and IMT has emerged as the new promising approach for the cancer treatment. This PDT+IMT combination leverages the strength of both therapies for the enhancement of therapeutic efficacy with minimum side effects. The combination permits the synergistic effect where the PDT can destroy to tumours, followed by the IMT initiated immune response. As a result, this combination leads to increased tumour control compared to therapies being applied alone [17].
Due to targeting of multiple mechanisms and pathways, the combination of therapeutic approaches is likely to decrease the drug resistance which develops in cancerous tumours. The ROS generated by PDT can make cancer cells more susceptible to ensuing treatments by IMT [17,19] . The combination of therapies can be effective in targeting a wide range of cancers. This makes it a flexible option in the oncology field [20]. It not only destroys the cancer cells, but it can also significantly improve the anti-tumour response by recruiting immune cells like macrophages and neutrophils. While tumours create an immunosuppressive microenvironment that significantly limits the effectiveness of IMT, PDT plays an important role in disrupting this environment through promoting maturation of antigen presenting cells and inducing inflammation [21]. The PDT+IMT combination is less invasive compared to traditional surgical or the chemotherapy approach. As a result, this combination results in few side-effects and it enhances the quality of life in patients [22].
6.1. Combination with Innate Immunity Stimulation
PDT can transform the so called “cold” tumours, which typically show low responsiveness to IMT, into “hot” tumours by inducing immunogenic cell death (ICD) and triggering an inflammatory response. This conversion is essential for improving the effectiveness of subsequent immunotherapeutic approaches [23]. The integration of PDT with checkpoint inhibitors, such as anti-PD-1 or anti-PD-L1 antibodies, can greatly improve T-cell infiltration and activation in tumours. Research indicates that PDT enhances the efficacy of R837 by creating a more favourable immune environment, effectively countering the immunosuppressive conditions commonly found in tumours [24].
The combination of PDT with IMT has demonstrated enhanced treatment outcomes in multiple preclinical models. Kleinovink et al.(2016) investigated the efficacy of combining the IMT with PDT for advanced cancer [25]. Thus, the application of PDT using the PS, Bremachlorin significantly delayed tumour growth in established tumours. When PDT was combined with therapeutic synthetic long peptide (SLP) vaccination, one-third of the treated mice were cured. Notably, all cured mice demonstrated complete protection against subsequent tumour challenges, and this combination treatment not only targeted primary tumours but also led to the eradication of distant secondary tumours, indicating that a systemic anti-tumour immune response had been induced. PDT alone triggered a substantial CD8+ T-cell response against the tumour, which was further amplified when combined with SLP vaccination, and this response was essential for the therapeutic effectiveness of the IMT and PDT combination therapy [25].
Hwang et al. (2020) investigated the impact of combining PDT with a TLR5 agonist, specifically flagellin-adjuvanted tumour-specific peptide vaccination (FlaB-Vax), to enhance PD-1 blockade-mediated melanoma suppression using a mouse model with B16-F10 tumour implants [26]. In this bilateral mouse melanoma model, the researchers assessed how the peritumoral delivery of FlaB-Vax and PDT tumour ablation could improve the effectiveness of PD-1 blockade. A photosensitizing agent, pheophorbide A (PhA), was utilized for laser-triggered photodynamic destruction of the primary tumour. The effects of this combination therapy, along with PD-1 blockade, were evaluated on tumour growth and survival rates. Additionally, effector cytokines that promote the activation of CD8+ T cells and antigen-presenting cells within both tumour tissue and tumour-draining lymph nodes (TDLNs) were measured [26]. The combination of PDT and FlaB-Vax also increased the infiltration of tumour antigen-reactive CD8+ T cells and the accumulation of migratory CXCL10-secreting CD103+ dendritic cells (DCs), which presumably contributed to tumour antigen cross-presentation within the tumour microenvironment (TME) [26].

Table 1.
The table representing the impact of combining PDT-IMT in cancer/tumour cells.
| Combination | Photosensitizer | IMT agent | Impact on tumour/cancer cells |
| PDT + checkpoint inhibitors PDT + SLP Vaccination |
mTHPC Bremachlorin |
anti-PD-1 or anti-PD-L1 antibodies Synthetic Long Peptides (SLP) |
Research indicates that PDT enhances the efficacy of R837 by creating a more favourable immune environment [24]. When PDT was combined with therapeutic synthetic long peptide (SLP) vaccination, one-third of the treated mice were cured [25] |
| PDT + Immune Checkpoint Blockade | Several, like Ce6 | Anti-PD-1, Anti-PD-L1 | Improves antitumor immune response while prohibiting the metastasis and recurrence [23]. |
| PDT + Anti-PD-L1 | Photofrin | Anti-PD-L1 | Enhanced activation of CD8+ T-cells and decreased tumour growth observed in preclinical models [27]. |
| PDT + TLR5 agonist | Mono-L-aspartyl chlorin e6 | FlaB-Vax | Increased the infiltration of tumour antigen-reactive CD8+ T cells [26] |
6.2. PDT and Checkpoint Blockade Immunotherapy
Ongoing research has been investigating the potential synergistic effects of combining PDT with CTLA-4 inhibition and other immune checkpoint blockade inhibitors. For instance, in double tumour models of colon carcinoma, specifically using MC38 or CT26 cell lines, the addition of CTLA-4 blockade prior to Bremachlorin-PDT resulted in a significant reduction in tumour burden compared to either treatment alone. It was observed that CTLA-4 blockade was more effective against smaller secondary tumours than against primary tumours. Furthermore, findings from CD8 T cell depletion studies indicated that this synergistic effect relied on the presence of CD8 T cells [28,29].
Yuan et al. (2021) investigated the underlying mechanisms to increase the PD-L1 blockages using the multifunction nanoparticles (NPs) containing mTHPC PSs for anti-tumour efficacy on colorectal cancer[30]. In this study, the mTHPC@VeC/T-RGD NPs under the 660-nm near-infrared (NIR) laser were shown to kill tumour cells by inducing apoptosis and/or necrosis while also stimulating a systemic immune response. This response could be further enhanced by PD-L1 blockade, which inhibited both primary and distant tumour growth and helped establish long-term immunological memory in the host to prevent tumour recurrence. Additionally, it was found that mTHPC@VeC/T-RGD NP-mediated PDT sensitized tumours to PD-L1 blockade therapy primarily because PDT-induced hypoxia activated the hypoxia-inducible factor 1α (HIF-1α) signalling pathway, leading to increased PD-L1 expression in colorectal cancer (CRC). Overall, the findings demonstrated that mTHPC@VeC/T-RGD NP-mediated PDT is a promising strategy that could enhance the efficacy of anti-PD-L1 checkpoint blockade immunotherapies in CRC [30].
Duan et al 92016). demonstrated that Zn-pyrophosphate (ZnP) NPs loaded with the PS pyro lipid (ZnP@pyro) could kill tumour cells upon light irradiation by directly inducing apoptosis and/or necrosis, as well as indirectly disrupting tumour vasculature and enhancing tumour immunogenicity. Furthermore, the immunogenic ZnP@pyro PDT treatment sensitized tumours to checkpoint inhibition mediated by a PD-L1 antibody, resulting not only in the eradication of the primary 4T1 breast tumour but also in a significant reduction of lung metastasis. The abscopal effects observed in both 4T1 and TUBO bilateral syngeneic mouse models further illustrated that the combination of ZnP@pyro PDT treatment with anti-PD-L1 led to the complete eradication of light-irradiated primary tumours and the inhibition of untreated distant tumours by generating a systemic tumour-specific cytotoxic T cell response. These findings indicated that nanoparticle-mediated PDT could enhance the systemic efficacy of checkpoint blockade immunotherapies by activating both the innate and adaptive immune systems within the tumour microenvironment [31].
Huang et al (2019). The study involved conjugating the PS protoporphyrin IX (PpIX) with the IDO inhibitor NLG919 through ester bonds. This coupled drug was incorporated into liposomes and used to treat mice with 4T1 breast cancer, resulting in significant inhibition of both in situ and distant tumours. To control drug release, a prodrug vesicle was designed with a dual-activation mechanism involving matrix metalloproteinase-2 (MMP-2) exfoliation and glutathione (GSH) reduction. These vesicles, known as enzyme-activatable prodrug vesicles (EAPVs), consisted of an enzyme-liable PEG corona, a PS (pyrogalophylchlorophyll a), and an IDO1 inhibitor (NLG919)-based prodrug sensitive to reduction [32]. The EAPVs remained stable in the bloodstream, minimizing drug leakage and phototoxicity to normal tissues. They accumulated at the tumour site via the enhanced permeability and retention (EPR) effect and were activated for fluorescence imaging and PDT upon MMP-2-mediated cleavage of the PEG coating. Simultaneously, the NLG919 prodrugs were reduced by the GSH-rich tumour microenvironment to release the active drug and inhibit IDO1. This approach was effective in treating both CT26 and 4T1 tumours, with significant tumour suppression observed in both models. The combination therapy enhanced dendritic cell (DC) maturation and increased the infiltration of IFN-γ-positive CD4+ and CD8+ T lymphocytes into the tumour. Additionally, the inactivation of IDO-1 by NLG919 led to a decrease in the kynurenine to tryptophan ratio, resulting in an increased ratio of cytotoxic T lymphocytes (CTLs) to regulatory T cells (Tregs). These findings demonstrated that EAPV-mediated photodynamic IMT significantly promoted DC maturation in tumour-draining lymph nodes, enhancing anti-tumour immune responses and creating an immune-promoting environment for synergistic tumour treatment [32].
Table 2.
The table representing the impact of combining PDT-IMT (checkpoint blockade) in cancer/tumour cells.
Table 2.
The table representing the impact of combining PDT-IMT (checkpoint blockade) in cancer/tumour cells.
| Combination | Photosensitizer | IMT agents | Type of cancer | Impact on tumour/cancer cells |
| PDT + checkpoint blockage | mTHPC | PD-L1 blockade | colorectal cancer | Inhibited both primary and distant tumour growth and helped establish long-term immunological memory in the host to prevent tumour recurrence [30]. |
| PDT + checkpoint blockage | ZnP@pyro | PD-L1 antibody | primary 4T1 breast tumour | led to the complete eradication of light-irradiated primary tumours and the inhibition of untreated distant tumours by generating a systemic tumour-specific cytotoxic T cell response [31]. |
| PDT + check-point blockage | protoporphyrin IX (PpIX) | IDO1 inhibitor | 4T1 breast cancer | The combination therapy enhanced dendritic cell (DC) maturation and increased the in-filtration of IFN-γ-positive CD4+ and CD8+ T lymphocytes into the tumour, which demonstrated to be effective in treating both CT26 and 4T1 tumours [32]. |
6.3. Synergized PDT- Immunoadjuvant Therapy
Calreticulin, a protein that binds calcium ions, is a well-known damage-associated molecular patterns (DAMPs) produced in cells undergoing pre-apoptosis. Typically found in the endoplasmic reticulum, it moves to the cell surface during apoptosis and acts as a signal for phagocytosis. Due to its immunogenic properties, calreticulin has been used as a substitute for traditional immunoadjuvants like OVA and CpG [33,34]. Korbelik et al. performed a study to investigate the potential of externally added calreticulin to enhance the antitumor effects of PDT. The results showed that recombinant calreticulin bound to mouse SCCVII tumour cells treated with PDT. In immunocompetent C3H/HeN mice, administering calreticulin (0.4 mg/mouse) peritoumorally after PDT increased cure rates compared to PDT alone. However, this benefit was not seen in immunodeficient NOD-scid mice. Additionally, adding recombinant calreticulin to PDT-treated cells used in a vaccine protocol improved therapeutic outcomes. The study found that calreticulin gene expression decreased in PDT-treated cells but remained unchanged in other tissues. Overall, externally added calreticulin can augment antitumor responses mediated by PDT or PDT-generated vaccines, serving as an effective adjuvant for cancer treatment involving PDT and other stress-inducing therapies [33].
The maturation of immature dendritic cells (DCs) into mature DCs can be triggered by various factors, including heat shock proteins, inflammatory cytokines like TNFα, IL-1, and IL-6, inflammasomes, and Toll-like receptors (TLRs). Recently, advancements have been made in developing nanomaterials to enhance DC activation, with a focus on designing nanomaterials that incorporate TLR agonists [35]. R837, a TLR7 agonist, facilitates the maturation of dendritic cells (DCs) and enhances their ability to phagocytose tumour-associated antigens (TAAs). This process promotes the activation and proliferation of antigen-specific lymphocytes in the draining lymph nodes, thereby boosting the immune response [36]. Meng et al prepared the light triggered in situ gelation composing PS [37]. The immune adjuvant R837 was incorporated into the system to induce robust antitumor immune responses following PDT. By leveraging this adjuvant, the hydrogel system was able to significantly amplify immune responses through repeated stimulation cycles [37].
Ge at al. (2018) prepared nanodrug carriers based on poly(ethylene glycol)-block-poly(lactic-co-glycolic acid) copolymer-encapsulated Fe3O4 super particles (SPs) were developed as immune adjuvants. These carriers are loaded with imiquimod (R837), a Toll-like receptor 7 agonist, to enhance immune responses [38]. The nanodrug carriers, known as Fe3O4-R837 SPs, can destroy Calreticulin through photothermal therapy (PTT) under near-infrared laser irradiation. This process generates tumour-associated antigens due to the high efficiency of magnetic attraction and photothermal effects. PTT also triggers the release of R837, an immune adjuvant that stimulates a robust antitumor immune response. When combined with checkpoint blockade therapy using PD-L1 antibodies, the Fe3O4-R837 SPs not only eliminate primary tumours but also prevented metastasis to the lungs and liver. Additionally, this synergistic approach exhibits abscopal effects, completely inhibiting the growth of untreated distant tumours by promoting the infiltration of CD45+ leukocytes. These findings indicate that Fe3O4-R837 SPs can significantly enhance the therapeutic efficacy of PD-L1 checkpoint blockade therapy by activating both innate and adaptive immune systems [38].
Chen et al (2016). investigated indocyanine green (ICG), a photothermal agent, and imiquimod (R837), a Toll-like-receptor-7 agonist, were co-encapsulated by poly(lactic-co-glycolic) acid (PLGA). The formed PLGA-ICG-R837 NPs, composed of three clinically approved components, were used for near-infrared laser-triggered photothermal ablation of primary tumours. This process generated tumour-associated antigens, which, in the presence of R837-containing NPs as an adjuvant, exhibited vaccine-like functions. When combined with checkpoint blockade using anti-cytotoxic T-lymphocyte antigen-4 (CTLA4), the generated immunological responses were able to attack remaining tumour cells in mice, inhibiting metastasis and potentially being applicable to various tumour models. This strategy also provided a strong immunological memory effect, offering protection against tumour rechallenge after the initial tumours were eliminated [39].
Table 3.
The table representing synergized PDT- Immunoadjuvant Therapy in cancer/tumour cells.
| Combination | Photosensitizer | IMT agent | Cancer type | Impact on tumour/cancer cells |
| PDT + immune adjuvants | Temoporfin and chlorin e6 (Ce6) | Calreticulin | SCCVII tumour cells | The study found that calreticulin gene expression decreased in PDT-treated cells but remained unchanged in other tissues. Overall, externally added calreticulin can augment antitumor responses mediated by PDT or PDT-generated vaccines, serving as an effective adjuvant for cancer treatment involving PDT and other stress-inducing therapies [33]. |
| photothermal therapy + immune adjuvants | Fe3O4-R837 SPs | Fe3O4 super particles (SPs), imiquimod (R837),PD-L1 antibodies and | 4T1 breast tumor | PTT also triggers the release of R837, an immune adjuvant that stimulates a robust anti-tumour immune response. When combined with checkpoint blockade therapy using PD-L1 antibodies, the Fe3O4-R837 SPs not only eliminate primary tumours but also prevented metastasis to the lungs and liver [38]. |
| Photothermal + Immunoadjuvant | indocyanine green (ICG) | imiquimod (R837) | Various cancers | The combination of photothermal agent with checkpoint blockage generated immunological responses were able to attack remaining tumour cells in mice, inhibiting metastasis and potentially being applicable to various tumour models [39]. |
7. MOFs as Carriers for PDT+IMT Therapeutics
nMOFs can be categorized into active and inactive types. Active nMOFs function not only as carriers but also have intrinsic therapeutic properties, such as antitumor effects or immunostimulatory capabilities, offering added therapeutic advantages. nMOFs can enable the combination of PDT and IMT by co-delivering innate immunity stimulants and immune checkpoint blockade inhibitors with PSs. This dual delivery boosts immune responses while directly targeting tumour cells. The ICD 40t0riggered by PDT activates dendritic cells and enhances T-cell responses, thereby increasing the effectiveness of checkpoint blockade immunotherapies [40]. Liang et al. investigated the effect of the PCN-224 nMOF loaded with programmed death-ligand 1 inhibitor (iPD-L1) against an aggressive triple native breast cancer . The in-vitro studies showed that the irradiation of nanocomplexes with 600nm laser has a robust capacity to generate singlet oxygen. PDT with high immunogenicity resulted in tumour necrosis and apoptosis, triggering ICD that prompts tumour cells to release danger-associated molecular patterns. When used in conjunction with iPD-L1, this combination therapy enhanced dendritic cell maturation, boosting T-cell activation and infiltration within the tumour, and modified the tumour immune microenvironment. This ultimately led to the inhibition of tumour growth and prevented distant tumour progression [40].
In 2016, Lin et al synthesized a photodynamic nano-material-organic framework (PnMOF) made from 5,10,15,20-tetra(p-benzoato)chlorin (H4TBC) and Hf6(μ3-O)4(μ3-OH)4 secondary building units (SBUs), referred to as TBC-Hf. They subsequently encapsulated an indoleamine 2,3-dioxygenase (IDO) inhibitor (IDOi) within the channels of the nMOF. The resulting IDOi@TBC-Hf demonstrated a synergistic effect in combining PDT and IMT against both local and distant tumours in colorectal cancer models [41]. Following this work, Lin and his team reported numerous other nMOFs designed for the combination of PDT and IMT. In 2018, benzoporphyrin-based nMOF (TBP-MOF) was reported, featuring a 10-connected Zr6 cluster and it significantly enhanced photophysical properties compared to traditional porphyrin-based nMOFs. TBP-MOF demonstrates red-shifted absorption bands and strong near-infrared luminescence suitable for bioimaging, while the π-extended benzoporphyrin linkers facilitate the generation of 1O2, thereby improving oxygen-dependent PDT. From the study, it was indicated that poly(ethylene glycol)-modified nanoscale TBP-MOF (TBP-nMOF) serves as an effective PDT agent even in hypoxic TMEs. Furthermore, it was showed that the low oxygen-dependent PDT of TBP-nMOF, when combined with αPD-1 checkpoint blockade therapy, not only inhibits primary tumour growth but also triggers an antitumor immune response that suppresses metastatic tumour development [41].
Table 4.
Table summarizing some of the nMOF-mediated combinations of IMT with PDT.
| nMOF-Mediated Combinations | Therapeutic Agents | Mechanism | Immunologic Modulation | Effectiveness |
|---|---|---|---|---|
| Zr-MOFs with TLR Agonists | TLR agonists | Activates immune pathways next to PDT | Improves immune responses through TLR signalling | Synergistic effect on tumour growth inhibition [23,42]. |
| ZIF-8@PDT + IMT | PS | Synergistic effects of PDT and IMT | Modulates tumour microenvironment to enhance immune responses | Enhanced therapeutic outcomes in resistant tumours [23,43] |
| AuNC@MnO2 NPs | Gold Nanoclusters (AuNCs), MnO2 | Induces ICD and enhances PDT efficacy | Increases tumour-associated antigens (TAAs), dendritic cells (mDC), CD4+, CD8+, NK cells | Enhanced immune response leading to tumour regression [44] |
7.1. Metabolic Pathways in nMOF
With their highly porous and tuneable architectures, nMOFs serve as versatile nanocarriers that can be engineered to interact with cellular metabolism in multiple ways. When delivered to tumour cells, nMOFs may release metal ions such as copper, iron, or zinc in a controlled manner, triggering oxidative stress and DNA damage that lead to cell death. Additionally, nMOFs can modulate intracellular environments by scavenging antioxidants like glutathione (GSH), thereby enhancing reactive oxygen species (ROS)-based therapies such as PDT. Their interaction with metabolic pathways influences the generation of ROS and the activation of PSs, critical for effective PDT [10,45].
They can also alter metabolic processes by regulating intracellular pathways such as mitochondrial function, ATP production, and antioxidant defences. Some nMOFs are designed to catalyse Fenton or Fenton-like reactions within the TME, converting endogenous hydrogen peroxide into highly toxic hydroxyl radicals (·OH), further enhancing cancer cell killing through oxidative damage. Understanding these metabolic interactions, the release kinetics of therapeutic agents and ions, their effects on cellular redox state, metabolic alterations, and the TME response enables the fine-tuning of nMOF design to maximize tumour targeting, efficacy, and minimize systemic toxicity. This detailed elucidation of metabolic pathways is foundational for developing next-generation nMOF-based nanotherapeutics with improved cancer treatment outcomes [46,47].
7.2. In-Vitro Studies for nMOFs as Carries of PDT+IMT Therapeutics
Liang et al (2023). investigated the effect of PDT+IMT using metal-organic frameworks to inhibit metastatic progression of triple-negative breast cancer [40]. In this study, PCN-224, a MOF known for its excellent biocompatibility and biodegradability, was prepared by solvothermal method, and used to deliver the PD-L1 small molecule inhibitor BMS-202, enabling a synergistic anti-tumour approach for combining PDT and IMT. Additionally, hyaluronic acid (HA)-modified polyethylene glycol (HA-PEG) was synthesized and applied as an outer coating for the nanocomplex to extend its circulation time within the body [40].
To study the uptake behaviour of the nanocomplexes, C-6 was used as a fluorescent tracer for PCN-224/HP NPs, and their cellular uptake was examined. C-6 can be excited by a 488 nm laser and detected in the fluorescein isothiocyanate (FITC) channel of flow cytometry. Both the number of green-fluorescent particles within the cells and the fluorescence intensity increased over time, as shown in Figure 10a,b and confirmed by flow cytometry assays. These results demonstrated that the PCN-224/HP NPs are internalized by tumour cells in a time-dependent manner. After 2 hours of incubation, the green fluorescence from the nanocomplex overlapped with the red fluorescence of lysosomes, producing a yellow signal, which intensified after 4 hours. With longer incubation, some nanocomposites were observed to escape from lysosomes (Figure 10c) [40].
The cytotoxicity of the nanocomposite against cancer cells was assessed. Importantly, the viability of L929 cells remained nearly unchanged after treatment with PCN-224/HP and BMS@P/HP across a PCN-224 concentration range of 1.56 to 50.00 μg/ml, indicating good cytocompatibility (Figure 10a). While BMS-202, as a non-peptide PD-1/PD-L1 inhibitor, blocks the PD-L1/PD-1 pathway, it does not possess intrinsic tumour-killing effects. Cell viability significantly decreased when the nanocomplexes were combined with 660 nm laser irradiation (Figure 10b). Calcein-AM, which is non-fluorescent, is hydrolysed by intracellular esterases in live cells to produce membrane-impermeable, green-fluorescent Calcein. In contrast, propidium iodide (PI) stains dead cells with compromised membranes. Compared to the non-irradiated group, the nanocomplex group exposed to 660 nm laser showed stronger red fluorescence, indicating increased 4T1 cell death (Figure 10c), consistent with MTT assay findings. Moreover, PDT inhibited 4T1 cell proliferation and migration in vitro, as demonstrated by a colony formation assay (Figure 10d) [40].
Reactive oxygen species (ROS) are common by-products of various cellular metabolic processes. Elevated ROS levels can cause damage to proteins, nucleic acids, lipids, cell membranes, and organelles, ultimately leading to cell death . To evaluate the ROS-generating capability of the nanocomplexes under 660 nm laser irradiation in 4T1 cells, we used DCFH-DA as a ROS-sensitive probe. The non-fluorescent DCFH-DA is hydrolysed by intracellular esterases into DCFH, which cannot cross the cell membrane and thus accumulates inside the cells. Intracellular ROS oxidize DCFH into green-fluorescent DCF. The group treated with the nanocomposite and irradiated with the 660 nm laser exhibited strong green fluorescence compared to the control (Figure 10e), indicating that PCN-224 nanocarriers were effectively internalized by cells and activated photodynamic therapy. Flow cytometry analysis (Figure 10d) showed only weak green fluorescence in the PCN-224/HP NPs, BMS-202, and BMS@P/HP NPs groups compared to controls, likely reflecting the influence of the drugs on cellular metabolism [40].
Figure 11.
(a) Immunofluorescence imaging of 4T1 cells stained with an anti-HMGB1 antibody. (b) Measurement of the relative mean fluorescence intensity of calreticulin (CRT). (c) Assessment of extracellular ATP levels using an ATP assay kit. (d) Analysis of CD80 and CD86 expression on dendritic cells (DCs) following exposure to various treatments [48].
Figure 11.
(a) Immunofluorescence imaging of 4T1 cells stained with an anti-HMGB1 antibody. (b) Measurement of the relative mean fluorescence intensity of calreticulin (CRT). (c) Assessment of extracellular ATP levels using an ATP assay kit. (d) Analysis of CD80 and CD86 expression on dendritic cells (DCs) following exposure to various treatments [48].
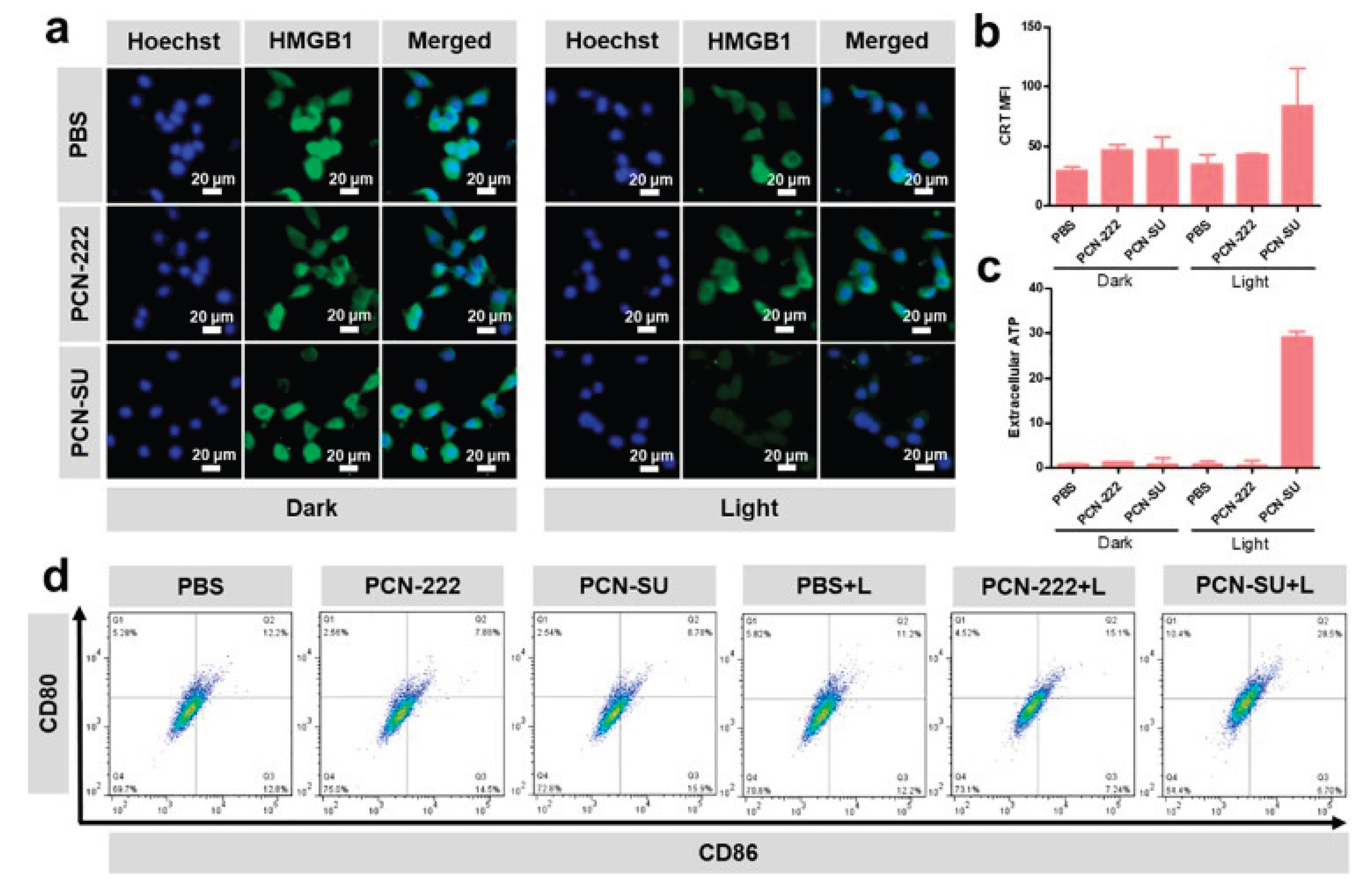
Li at al (2022). synthesized spindle-shaped PCN-SU through a sulfonation reaction for near-infrared (NIR)-enhanced photodynamic IMT targeting 4T1 tumours. It was observed that PDT induces ICD, releasing DAMPs that serve as “eat me” and “find me” signals, which help recruit and activate macrophages and antigen-presenting cells (APCs) to initiate an anti-tumour immune response. To assess the ICD effect induced by PCN-SU on tumour cells under near-infrared (NIR) irradiation, related DAMPs such as calreticulin translocation (CRT), high mobility group box 1 (HMGB1) protein release from the nucleus, and extracellular ATP secretion were examined in vitro [48].
Figure 12.
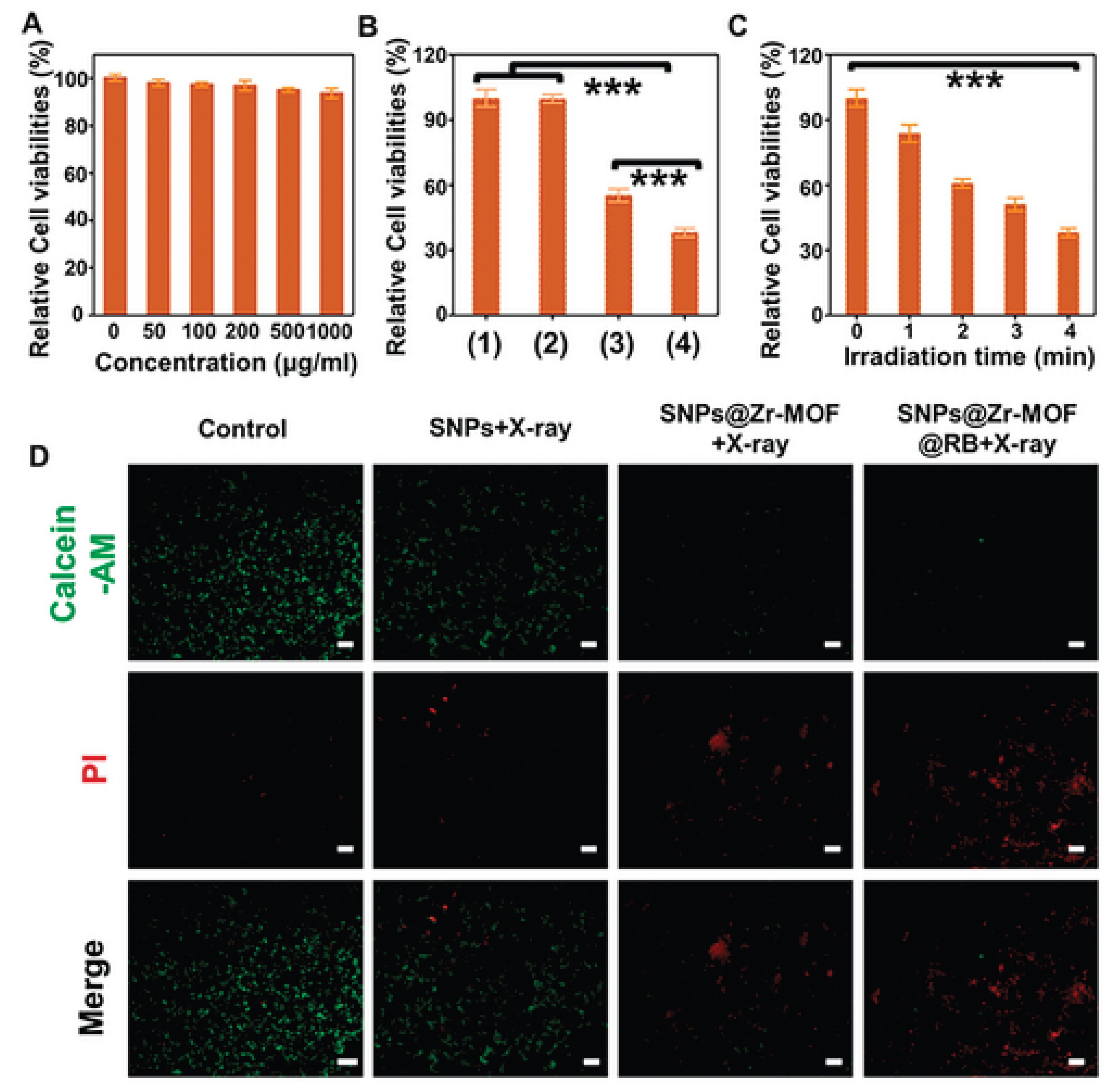
(a) Cell viability of 4T1 tumour cells after treatment with varying concentrations of the SNPs@Zr-MOF@RB nanoprobe. (b) Viability of 4T1 cells exposed to different samples with or without soft X-ray irradiation: (1) Control, (2) SNPs plus X-ray, (3) SNPs@Zr-MOF plus X-ray, and (4) SNPs@Zr-MOF@RB plus X-ray. (c) Survival rates of 4T1 cells treated with the SNPs@Zr-MOF@RB nanoprobe under varying durations of soft X-ray exposure. (d) Fluorescent microscopy images of 4T1 cells stained with Calcein-AM (green indicating live cells) and propidium iodide (PI, red indicating dead cells) following soft X-ray irradiation (tube voltage: 45 kVp). Statistical significance: ***P < 0.001. Scale bars represent 200 µm [49].
Figure 12.
(a) Cell viability of 4T1 tumour cells after treatment with varying concentrations of the SNPs@Zr-MOF@RB nanoprobe. (b) Viability of 4T1 cells exposed to different samples with or without soft X-ray irradiation: (1) Control, (2) SNPs plus X-ray, (3) SNPs@Zr-MOF plus X-ray, and (4) SNPs@Zr-MOF@RB plus X-ray. (c) Survival rates of 4T1 cells treated with the SNPs@Zr-MOF@RB nanoprobe under varying durations of soft X-ray exposure. (d) Fluorescent microscopy images of 4T1 cells stained with Calcein-AM (green indicating live cells) and propidium iodide (PI, red indicating dead cells) following soft X-ray irradiation (tube voltage: 45 kVp). Statistical significance: ***P < 0.001. Scale bars represent 200 µm [49].
Following 730 nm irradiation, immunofluorescence analysis showed HMGB1 protein migrating from the nucleus, indicated by green fluorescence. The PCN-SU + light (L) group displayed weaker green fluorescence compared to the PBS, PBS + L, PCN-222, PCN-222 + L, and PCN-SU groups, indicating rapid HMGB1 release from 4T1 cells. Similarly, confocal laser scanning microscopy (CLSM) revealed stronger CRT translocation in the PCN-SU + L group versus the others. ATP assay results showed extracellular ATP levels in PCN-SU + L were tenfold higher than in other groups.
Furthermore, the supernatant from treated 4T1 cells stimulated maturation of mouse bone marrow-derived dendritic cells (DC2.4), evidenced by increased expression of co-stimulatory molecules CD80 and CD86 measured via flow cytometry. The PCN-SU + L group induced a significant rise in mature dendritic cells (CD80+CD86+, 28.5%) compared to PBS (12.2%), PBS + L (11.2%), PCN-222 (7.88%), and PCN-SU (8.78%) groups, and was nearly twice as high as the PCN-222 + L group (15.1%).
These findings indicate that PCN-SU activated by NIR-triggered PDT induces enhanced ICD and promotes dendritic cell maturation, thereby stimulating systemic immune responses in vitro [48].
Zhao et al (2021). investigated A soft X-ray-activated nanoprobe which was strategically designed by combining a porphyrin zirconium-based PnMOF with lanthanide NaYF4:Gd,Tb@NaYF4 scintillator NPs (SNPs) using a novel in situ growth method, enabling synergistic PDT and IMT for tumour treatment. Before investigating the in vivo therapeutic efficacy of the soft X-ray mediated treatment, the nanoprobe cytotoxicity was evaluated [49]. As illustrated in Figure 12a below, the 4T1 cells exhibited increased viability rate of >90% after exposure to varying concentrations (0–1 mg/mL) of the SNPs@Zr-MOF@RB nanoprobe for 24 hours, indicating its low cytotoxicity and excellent biocompatibility. Subsequently, the in vitro therapeutic effect triggered by soft X-ray irradiation was evaluated. As illustrated in Figure 12b below, the cell survival SNPs@Zr-MOF plus X-ray and SNPs@Zr-MOF@RB nanoprobe plus X-ray groups were remarkably decreased, due to the effective production of reactive oxygen species (ROS) activated by soft X-rays. While the cell survival remained stable in both the control group and the group treated with SNPs plus X-ray. Subsequently, the in vitro therapeutic effects of the SNPs@Zr-MOF@RB nanoprobe under varying durations of X-ray irradiation were investigated. As illustrated in Figure 12C, extending the irradiation time from 1 to 4 minutes led to a gradual decline in cell survival, attributed to increased ROS production. Additionally, a live/dead cell co-staining assay was conducted using Calcein-acetoxymethyl (AM) for live cells (green fluorescence) and propidium iodide (PI) for dead cells (red fluorescence). Figure 12D clearly shows a high rate of cell death in 4T1 cells treated with both SNPs@Zr-MOF plus X-ray and SNPs@Zr-MOF@RB nanoprobe plus X-ray, demonstrating the effective soft X-ray triggered PDT of cancer cells in vitro [49].
Liu(2024) and colleagues prepared a tumour targeted nanomedicine ( designated as PCN@HA) which was engineered to increase PDT against tumour cells through modification with hyaluronic acid (HA), that may prey on CD44 receptor which are expressed in the surface of cancer cells. The PCN@HA is said to produce a singlet oxygen that not only destroys and kills the cancer cells but also eliminate the tumours [50]. The binding of HA in CD44 was verified via monitoring cellular uptake assessments, the prepared PCN@HA was incubated with NIH 3T3 cells (CD44-negative cells) and 4T1 cells (murine mammary carcinoma cell line, CD44-positive cells). After incubation with cancer cells, the targeting capability of PCN@HA was demonstrated by confocal laser scanning microscopy (CLSM). Notably, a clear red fluorescence signal from H2TCPP was detected in 4T1 cells, whereas no fluorescence was observed in NIH 3T3 cells (Figure 13A). These findings clearly demonstrate that PCN@HA specifically targets 4T1 cells, attributed to the HA modification, highlighting its excellent potential for targeted drug delivery [50].
The biocompatibility and phototoxicity of PCN@HA were evaluated using the Cell Counting Kit-8 (CCK-8) assay. 4T1 cells were exposed to varying concentrations of PCN@HA for 24 hours. The nanomedicine demonstrated excellent biocompatibility, with cell viability remaining above 90% even at high concentrations of up to 20 μM without irradiation. This indicated minimal cytotoxicity and strong biocompatibility of PCN@HA in the absence of light exposure (Figure 13B). Conversely, PCN@HA exhibited significant phototoxic effects on 4T1 cells following laser irradiation. As shown in Figure 13C, both PCN-224 and PCN@HA displayed toxicity under laser treatment, with the PCN@HA + laser group showing notably higher phototoxicity compared to the PCN-224 + laser group. Further in vitro cytotoxicity was assessed using Calcein-AM and propidium iodide (PI) double staining observed by fluorescence microscopy. Figure 13D revealed abundant green fluorescence indicating live cells in both the control and PCN@HA without irradiation groups. In contrast, marked red fluorescence signifying dead cells was observed in PCN-224 + laser and PCN@HA + laser groups, with the latter showing significantly greater toxicity. These findings align with the CCK-8 results and further emphasize the crucial role of HA in enhancing targeted PDT efficacy [50].
Table 5.
Table summarizing in-vitro studies for nMOFs as carries of PDT+IMT therapeutics.
| Study | Type of MOF | PDT + IMT Modality | Cancer model | Key Findings / Outcomes | Cytotoxicity assessment |
| Investigated the effect of PDT+IMT using metal-organic frameworks to inhibit metastatic progression of triple-negative breast cancer | PCN-224 | PDT + PD-L1 small molecule inhibitor BMS-202 | triple-negative breast cancer | synergistic anti-tumour approach for combining PDT and IMT | The cytotoxicity of the nanocomposite against cancer cells was assessed. Importantly, the viability of L929 cells remained nearly unchanged after treatment with PCN-224/HP and BMS@P/HP across a PCN-224 concentration range of 1.56 to 50.00 μg/ml, indicating good cytocompatibility [40]. |
| synthesized spindle-shaped PCN-SU through a sulfonation reaction for near-infrared (NIR)-enhanced photodynamic IMT targeting 4T1 tumours | PCN-222 |
PCN-SU + IMT agent | 4T1 tumours | PDT induces ICD, releasing DAMPs that serve as signals, which help recruit and activate macrophages and antigen-presenting cells (APCs) to initiate an anti-tumour immune response | Cytotoxicity was assessed indirectly by measuring HMGB1 release, CRT translocation, and extracellular ATP release after 730 nm light irradiation. The PCN-SU + light (L) group showed rapid HMGB1 release, stronger CRT translocation, and significantly increased extracellular ATP levels (10x higher), indicating enhanced immunogenic cell death (ICD) and cytotoxicity against 4T1 cells [48]. |
| investigated A soft X-ray-activated nanoprobe which was strategically designed by combining a porphyrin zirconium-based PnMOF with lanthanide NaYF4:Gd,Tb@NaYF4 scintillator NPs (SNPs) using a novel in situ growth method | Zr-MOF | SNPs@Zr-MOF@RB nano-probe | 4T1 cancer cells. | The in vitro therapeutic effects of the SNPs@Zr-MOF@RB nanoprobe under varying durations of X-ray irradiation time from 1 to 4 minutes led to a gradual decline in cell survival, attributed to increased ROS production. | The 4T1 cells exhibited increased viability rate of >90% after exposure to varying concentrations (0–1 mg/mL) of the SNPs@Zr-MOF@RB nanoprobe for 24 hours, indicating its low cytotoxicity and excellent biocompatibility [49]. |
| prepared a tumour targeted nanomedicine ( designated as PCN@HA) which was engineered to increase PDT against tumour cells through modification with hyaluronic acid (HA) | PCN-MOF | PCN@HA + IMT Adjuvant | 4T1 cells | The PCN@HA is said to produce a singlet oxygen that not only destroys and kills the cancer cells but also eliminate the tumours. ). These findings clearly demonstrate that PCN@HA specifically targets 4T1 cells, attributed to the HA modification, highlighting its excellent potential for targeted drug delivery | The biocompatibility and phototoxicity of PCN@HA were evaluated using the Cell Counting Kit-8 (CCK-8) assay. 4T1 cells were exposed to varying concentrations of PCN@HA for 24 hours. Minimal cytotoxicity was observed even at high concentrations of up to 20 μM without irradiation [50]. |
7.3. In-Vivo Studies of nMOFs as Carries of PDT+IMT Therapeutics
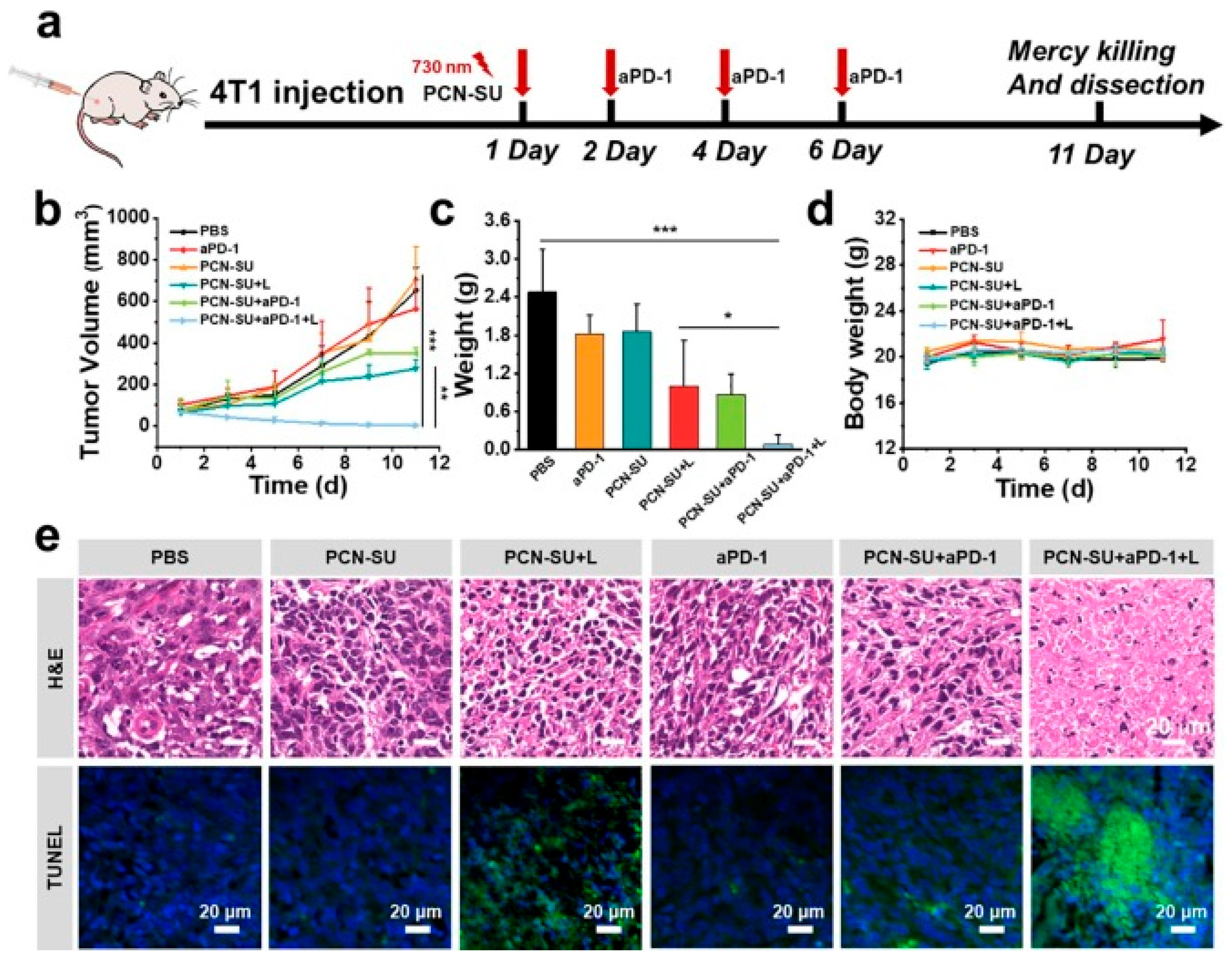
Following the excellent in vitro results of the PCN-SU synthesized through a sulfonation reaction for near-infrared (NIR)-enhanced PDT+IMT, antitumor efficacy in 4T1 tumour-bearing BALB/c mice was evaluated. Since the interaction between programmed cell death ligand 1 (PD-L1) on cancer cells and programmed cell death 1 (PD-1) on T cells suppresses T cell activation and their ability to kill tumour cells, the checkpoint inhibitor anti-PD-1 (aPD-1) was used to block this binding and restore T cell function. To assess the antitumor effect of PDT-IMT in vivo, 4T1 tumour models were established by subcutaneously injecting 2 × 10^6 cells into female BALB/c mice. The mice were divided into six groups receiving different treatments: PBS, aPD-1, PCN-SU, PCN-SU + light, PCN-SU+aPD-1, and PCN-SU + aPD-1 + light. Results showed no significant tumour growth inhibition in the PBS, aPD-1, and PCN-SU groups. The PCN-SU+aPD-1+L (PDT+IMT) group, however, achieved a remarkable 99.6% tumour growth inhibition, higher than the 57.6% inhibition from PCN-SU+L (PDT alone), demonstrating a synergistic effect between PDT and aPD-1 IMT. Tumour tissue weight was also smallest in the PCN-SU + aPD-1 + L group by day 11. Throughout treatment, the mice’s body weights remained stable, indicating minimal side effects. Even at a PCN-SU concentration of 250 μg/mL, there was no significant blood cell damage or haemolysis. Histological analysis (H&E staining) of major organs showed no organ damage from any treatment. Additionally, H&E and TUNEL assays confirmed that PCN-SU with 730 nm light promoted tumour cell apoptosis, and this effect was significantly enhanced when combined with anti-PD-1 [48].
Figure 14.
In vivo therapeutic effectiveness of PCN-SU in activating anti-tumour immune responses was evaluated in 4T1 tumour-bearing mice. a) A schematic diagram depicts the experimental setup for tumour IMT conducted in vivo. b) Tumour growth curves were recorded for 4T1 breast tumour-bearing mice receiving treatments including PBS, aPD-1, PCN-SU, PCN-SU combined with light (L), PCN-SU with aPD-1, and PCN-SU with both aPD-1 and light. c) Weights of the excised 4T1 breast tumours were measured. d) Changes in body weight of the tumour-bearing mice were monitored. e) Histological examination included photomicrographs of tumour tissues stained with haematoxylin and eosin (H&E) and immunocytochemistry images showing TUNEL staining. Statistical significance was assessed using two-tailed Student’s t-test or one-way ANOVA, with *P < 0.05, **P < 0.01, and ***P < 0.001 indicating levels of significance [48].
Figure 14.
In vivo therapeutic effectiveness of PCN-SU in activating anti-tumour immune responses was evaluated in 4T1 tumour-bearing mice. a) A schematic diagram depicts the experimental setup for tumour IMT conducted in vivo. b) Tumour growth curves were recorded for 4T1 breast tumour-bearing mice receiving treatments including PBS, aPD-1, PCN-SU, PCN-SU combined with light (L), PCN-SU with aPD-1, and PCN-SU with both aPD-1 and light. c) Weights of the excised 4T1 breast tumours were measured. d) Changes in body weight of the tumour-bearing mice were monitored. e) Histological examination included photomicrographs of tumour tissues stained with haematoxylin and eosin (H&E) and immunocytochemistry images showing TUNEL staining. Statistical significance was assessed using two-tailed Student’s t-test or one-way ANOVA, with *P < 0.05, **P < 0.01, and ***P < 0.001 indicating levels of significance [48].
To validate that the PDT induced the antitumor immunity through the ICD in-vivo, the immunocytochemistry staining assay was utilised to analyse the exposure of CRT and release of HMGB1. As illustrated in Figure 15(a) the immunohistochemical staining images showed more notable expression of HMGB1 and CRT in the PCN-SU + L group when compared to both PCN-SU and PBS groups, this implies PCN-SU could trigger ICD after NIR light in in-vivo. Additionally, the lymph nodes were extracted from the sacrificed mice for the detection of dendritic cells (DC) maturation level via flow cytometry. The PCN-SU + L group displayed higher CD80 and CD86 expression when compared to other groups. DCs present tumour antigens to antigen-specific T cells, triggering CD8+ cytotoxic T lymphocyte (CTL) responses and promoting their infiltration into tumours. To assess T cell infiltration in tumour tissues, the proportions of activated CD4+ and CD8+ T cells were measured. The PCN-SU + L + aPD-1 group exhibited a notably higher percentage of activated CD8+ T cells (19.1%) compared to PBS (1.92%), aPD-1 (1.21%), PCN-SU (1.91%), PCN-SU + L (6.40%), and PCN-SU + aPD-1 (6.70%) groups, while the levels of activated CD4+ T cells remained largely unchanged (as illustrated in Figure 15b) [48].
Motivated by the outstanding in vitro therapeutic results, soft X-ray activated PDT was conducted on xenografted tumours in mice (Figure 16A). The 4T1 tumour-bearing mice were randomly divided into four groups (n = 5 per group) and treated with PBS, SNPs + X-ray, SNPs@Zr-MOF + X-ray, and SNPs@Zr-MOF@RB + X-ray. With no significant body weight loss observed across groups, tumour growth rapidly progressed in the control and SNPs + X-ray groups, while the SNPs@Zr-MOF + X-ray group demonstrated effective tumour growth inhibition (Figure 15D, E). Notably, the SNPs@Zr-MOF@RB + X-ray group, which generated enhanced ROS levels, further improved tumour growth suppression, indicating superior soft X-ray induced PDT (Figure 16E). Tumour cell death induced by PDT was assessed through haematoxylin and eosin (H&E) and staining and TUNEL assays; the SNPs@Zr-MOF@RB plus X-ray group exhibited the most pronounced apoptosis compared to the SNPs@Zr-MOF plus X-ray group, while minimal apoptosis was found in control and SNPs plus X-ray groups (Figure 16F, G). Additionally, H&E analysis of major organs from normal mice injected intravenously with SNPs@Zr-MOF@RB nanoprobe for 6 and 12 days showed no evident tissue damage or necrosis when compared to untreated controls (Figure S13, Supporting Information). These findings confirm that SNPs@Zr-MOF@RB nanoprobes possess excellent biocompatibility and effectively facilitate soft X-ray triggered PDT in vivo [49].
In a study conducted by Liu et al, to evaluate combining PDT with anti-PD-L1 checkpoint blockade to eliminate residual cancer cells and inhibit tumour metastasis, a bilateral tumour model was established by implanting 4T1 cells into the flanks of mice, where the tumours on the left side served as primary tumours subjected to direct PDT, and the tumours on the right side were used as metastatic tumours without direct application of PDT. The tumour-bearing mice were randomly divided into four groups (n = 5): PBS control, anti-PD-L1, PCN@HA + laser, and PCN@HA + laser + anti-PD-L1. For PDT groups, PCN@HA was intravenously administered on days 0, 2, 4, and 6, followed by 660 nm laser irradiation of the primary tumours the next day. Anti-PD-L1 was injected intraperitoneally 24 hours post-irradiation.
As shown in Figure 17A, neither PBS nor anti-PD-L1 alone significantly affected the growth of primary or metastatic tumours. In contrast, both PCN@HA + laser and PCN@HA + laser + anti-PD-L1 treatments suppressed primary tumour growth and inhibited metastatic tumour progression, indicating activation of the immune system [50].
Flow cytometry analysis of CD4+ and CD8+ T cells in tumours and spleens demonstrated that the PCN@HA + laser + anti-PD-L1 group had the highest proportions of tumour-infiltrating cytotoxic CD8+ T lymphocytes and helper CD4+ T lymphocytes compared to other groups, confirming effective activation of tumour immunity by PCN@HA-mediated PDT. Moreover, PDT markedly increased secretion of interferon gamma (IFN-γ) and tumour necrosis factor alpha (TNF-α), with the highest levels observed in the PCN@HA + laser + anti-PD-L1 group, further highlighting the enhanced antitumor immune response. Collectively, these results suggest that combining PCN@HA-mediated PDT with anti-PD-L1 checkpoint blockade elicits robust immune responses and holds great promise for future the clinical anticancer PDT + IMT combination therapy [50].
Table 6.
Table summarizing in-vivo studies for nMOFs as carries of PDT+IMT therapeutics.
| Study | PDT + IMT Modality | Cancer / Mouse Mode | Key Findings / Outcomes | Immune Mechanisms Identified |
|---|---|---|---|---|
| PCN-SU synthesized through a sulfonation reaction for near-infrared (NIR)-enhanced PDT+IMT, antitumor efficacy in 4T1 tumour-bearing BALB/c mice | PCN-SU + PD-1 and aPD-1 | 4T1 tumour-bearing BALB/c mice | The PCN-SU+aPD-1+L (PDT+IMT) group, however, achieved a remarkable 99.6% tumour growth inhibition, higher than the 57.6% inhibition from PCN-SU+L (PDT alone), demonstrating a synergistic effect between PDT and aPD-1 IMT | The PCN-SU + L group displayed higher CD80 and CD86 expression when compared to other groups. The PCN-SU + L + aPD-1 group exhibited a notably higher percentage of activated CD8+ T cells (19.1%) compared to PBS (1.92%), aPD-1 (1.21%), PCN-SU (1.91%), PCN-SU + L (6.40%), and PCN-SU + aPD-1 (6.70%) groups, while the levels of activated CD4+ T cells remained largely un-change [48]. |
| Soft X-ray triggered PDT utilizing the SNPs@Zr-MOF and SNPs@Zr-MOF@RB nanoprobes in 4T1 tumor-bearing mice. | Photodynamic therapy (PDT) activated by soft X-rays, employing silver nanoparticles (SNPs) incorporated within a zirconium-based metal-organic framework (Zr-MOF), with or without the inclusion of Rose Bengal (RB) as the photosensitizing agent. | 4T1 xenografted breast tumours in mice | The combination of SNPs@Zr-MOF and X-ray treatment markedly suppressed tumor growth compared to the control groups (PBS and SNPs with X-ray). The group treated with SNPs@Zr-MOF@RB and X-ray exhibited the most effective tumor suppression, attributed to increased production of reactive oxygen species (ROS). No significant loss in body weight or major organ toxicity was detected, indicating favorable biocompatibility. | The study primarily investigates the effects of photodynamic therapy (PDT); however, immune-related mechanisms such as dendritic cell activation or T cell responses were not specifically addressed in this excerpt [49]. |
| evaluated combining PCN@HA-mediated PDT with anti-PD-L1 checkpoint blockade in a bilateral 4T1 tumour mouse model | PCN@HA nanoparticles + Immune Checkpoint Blockade (anti-PD-L1 antibody) | Bilateral mammary carcinoma tumours derived from 4T1 cells were implanted in mice. | Treatment with either PBS or anti-PD-L1 alone did not significantly impact tumor growth. Both PCN@HA combined with laser-induced photodynamic therapy (PDT) and PCN@HA with laser plus anti-PD-L1 effectively suppressed the growth of primary tumors and prevented metastasis. The combined treatment of PCN@HA, laser, and anti-PD-L1 demonstrated greater efficacy than PDT alone. | Tumor immunity was activated through an increase in tumor-infiltrating CD8+ and CD4+ T cells. The antitumor immune response was further strengthened by enhanced production of pro-inflammatory cytokines such as IFN-γ and TNF-α [50]. |
8. Challenges
Despite the advances and advantages of nMOFs in the synergistic PDT + IMT combination for cancer treatment, it faces several challenges and limitations including biological challenges that present significant limitations in its clinical translation. While many studies focus on the uptake of these nanoplatforms, there is insufficient understanding of the mechanisms of the breakdown of nMOFs within tumour cells and their clearance from the body. A detailed elucidation of the metabolic pathways for nMOF materials is essential for optimizing their therapeutic efficacy. Additionally, tumour heterogeneity among patients, along with varying TME conditions such as blood flow, pH fluctuations, and oxygen levels complicates targeted drug delivery and diminishes therapeutic effectiveness. Therefore, it is necessary to investigate the specific conditions of each tumour to apply the most suitable nMOF composites [1,10]. Understanding heterogeneity has driven the creation of innovative clinical trial models like umbrella trials, basket trials, and master protocols. Umbrella trials test various treatments tailored to distinct molecular subtypes within a single type of cancer. Basket trials evaluate a single therapy across different cancer types that share the same molecular characteristics. Master protocols encompass multiple sub-studies under one comprehensive trial framework. These designs enable more efficient assessment of targeted therapies that are matched to the specific molecular profiles of patients [51].
In vivo toxicity remains another critical barrier to the clinical application of nanomaterials. Although preliminary results suggest that nMOFs exhibit minimal cytotoxicity, comprehensive in vivo toxicity studies across various nMOF types are still lacking. Initial investigations indicate that factors such as size, shape, functionalization, and solvent systems can significantly affect the biodistribution and toxicity of nMOFs. The presence of toxic metals like lead, arsenic, cadmium, and chromium in some nMOFs may also pose serious health risks, these metals may be toxic because they can bind to enzymes and proteins, disrupting their normal function and leading to harmful biological effects. As a result, all nMOF-based drug development studies always conduct cytotoxicity studies (after in-vitro and before in-vivo) before embarking on anticancer efficacy. Any material that fails toxicity studies is discarded. Consequently, extensive research into the long-term impacts of nMOFs and the combination therapies that they facilitate is necessary to better assess their biosafety [52,53].
Preclinical research aims to clarify mechanisms of action, drug delivery systems, treatment safety, efficacy, and stability of nanomedicines in cancer therapy. This understanding is vital to avoid costly limitations that could impede future investments and developments. Selecting appropriate preclinical models is critical for assessing the effects of nanomedicine therapies on the immune system and minimizing risks associated with clinical applications. Although rodent models are commonly used to study immune responses to treatments, significant differences exist between rodent and human immune responses regarding cell development, activation, complexity, proliferation, and function. Furthermore, many widely used mouse xenograft models may overestimate the effectiveness of nanomedicine due to an exaggerated enhanced permeability and retention effect [53,54].
9. Future Perspectives and Innovations
The methodologies for synthesis and characterization of nMOFs are likely to lead to new nMOFs with enhanced features. For applications in combinations of PDT with other non-invasive technologies, nMOFs with large hydrophobic pores are desired. This field has been attracting rising research attention. While new and innovative methods for synthesis have emerged recently, newer ones are still appearing in the literature. The vast array of nMOFs has given rise to many novel technological applications, notably in biomedical areas such as drug delivery and therapeutics. However, trends show new technological applications fuelled by enhanced surface area and porosity. The challenges that have emerged in the biomedical applications of nMOFs point to a future of numerous research studies aimed at ameliorating some of them, notably breakdown and toxicity.
Current knowledge suggests that a triple combination involving PDT and immune modulation therapy (IMT), which effectively merges immune stimulation with the inhibition of immunosuppression alongside PDT, has yet to be established using nMOFs. Although research has shown that PDT can bolster immune responses and enhance tumour control when paired with certain immunotherapies, a comprehensive synergistic effect that integrates all three elements—immune activation, immunosuppression inhibition, and PDT—remains elusive. This underscores the necessity for further investigation into these combinations to develop more effective cancer treatment approaches.
10. Conclusions
The combination of PDT with SLP therapeutics cured one-third of mice in in-vivo studies and thus prevented metastasis and eradicated the secondary cancer tumour. Furthermore, the PDT+IMT combination not only improved the antitumor immune response but also prohibited metastasis. PDT transforms cold tumours that show less response to IMT therapeutics, into hot tumours and this increases the infiltration and activation of CAR T-cells and CD8+ T-cells which decrease tumour growth. The synergistic potential of using nMOFs in combining PDT with IMT not only enhances tumour targeting but also promotes a more robust immune response against cancer cells. By facilitating the localized delivery of PSs and immune-modulating agents, nMOFs can significantly improve therapeutic outcomes and address challenges associated with traditional cancer therapies, such as drug resistance and systemic toxicity.
Looking ahead, the continued exploration of nMOF-based nanoplatforms is essential for realizing their full potential in clinical applications. Future research should focus on optimizing the design and functionalization of nMOFs to enhance their therapeutic efficacy, biocompatibility and cancer cell targeting. Furthermore, more clinical trials should be carried out for validating these innovative strategies and determining their effectiveness in diverse cancer types.
Author Contributions
MS = Musawenkosi Shange, SM = Samson Mohomane, SP = Sandile Phinda Songca. Conceptualization, methodology, SP and MS. Validation, formal analysis, investigation, resources, data curation, visualization, review writing, and editing, supervision, MS, SM and SP. Writing, original draft preparation, MS. Project administration, SP and SM. Funding acquisition, SP. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Research Foundation (South Africa), under the Competitive Support Program for Rated and Unrated Researchers (SRUG220323502, SRUG200210502723, CSRP170424228543), and The APC was funded by the University of Zululand.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript
| AA | Acetic acid |
| BDC | benzene dicarboxylic acid |
| BTC | 1,3,5- benzenetricarboxylate |
| CDT | chemodynamic therapy |
| DDQ | dichlorodicyanoquinone |
| DDS | drug delivery systems |
| DEF | diethyl formamide |
| DMA | dimethyl acetamide |
| DMF | dimethyl formamide |
| DMSO | dimethyl sulphoxide |
| EPR | enhanced permeability and retention |
| EtOH | ethanol |
| ICD | immunogenic cell death |
| ICIs | immune checkpoint inhibitors |
| IMT | immunotherapy |
| LAG | Liquid assisted griding |
| ILAG | Ion and Liquid assisted griding |
| MeOH | methanol |
| MOF | Metal organic framework |
| NDC | naphthalene dicarboxylic acid |
| PDT | Photodynamic therapy |
| PS | photosensitizer |
| PT | Photothermal therapy |
| RME | reverse microemulsion |
| ROS | Reactive oxygen species |
| STS | solvothermal synthesis |
| TME | Tumour microenvironment |
| CAR | chimeric antigen receptors |
| TAM | tumour-associated macrophages |
References
- Yang, J.; Dai, D.; Zhang, X.; Teng, L.; Ma, L.; Yang, Y.-W. Multifunctional metal-organic framework (MOF)-based nanoplatforms for cancer therapy: from single to combination therapy. Theranostics 2023, 13, 295.
- He, S.; Wu, L.; Li, X.; Sun, H.; Xiong, T.; Liu, J.; Huang, C.; Xu, H.; Sun, H.; Chen, W. Metal-organic frameworks for advanced drug delivery. Acta Pharmaceutica Sinica B 2021, 11, 2362-2395.
- Ye, Y.; Zhao, Y.; Sun, Y.; Cao, J. Recent progress of metal-organic framework-based photodynamic therapy for cancer treatment. International Journal of Nanomedicine 2022, 2367-2395.
- Mallakpour, S.; Nikkhoo, E.; Hussain, C.M. Application of MOF materials as drug delivery systems for cancer therapy and dermal treatment. Coordination Chemistry Reviews 2022, 451, 214262.
- Pattnaik, A.K.; Priyadarshini, N.; Priyadarshini, P.; Behera, G.C.; Parida, K. Recent advancements in metal organic framework-modified multifunctional materials for photodynamic therapy. Materials Advances 2024.
- Li, B.; Ashrafizadeh, M.; Jiao, T. Biomedical application of metal-organic frameworks (MOFs) in cancer therapy: stimuli-responsive and biomimetic nanocomposites in targeted delivery, phototherapy and diagnosis. International Journal of Biological Macromolecules 2024, 260, 129391.
- Wang, A.; Walden, M.; Ettlinger, R.; Kiessling, F.; Gassensmith, J.J.; Lammers, T.; Wuttke, S.; Peña, Q. Biomedical metal–organic framework materials: perspectives and challenges. Advanced functional materials 2024, 34, 2308589.
- Dong, X.; Mu, Y.; Shen, L.; Wang, H.; Huang, C.; Meng, C.; Zhang, Y. Structure engineering of Mn2SiO4/C architecture improving the potential window boosting aqueous Li-ion storage. Chemical Engineering Journal 2023, 456, 141031.
- Nirosha Yalamandala, B.; Shen, W.T.; Min, S.H.; Chiang, W.H.; Chang, S.J.; Hu, S.H. Advances in functional metal-organic frameworks based on-demand drug delivery systems for tumor therapeutics. Advanced NanoBiomed Research 2021, 1, 2100014.
- Shano, L.B.; Karthikeyan, S.; Kennedy, L.J.; Chinnathambi, S.; Pandian, G.N. MOFs for next-generation cancer therapeutics through a biophysical approach—a review. Frontiers in Bioengineering and Biotechnology 2024, 12, 1397804.
- Tong, P.-H.; Zhu, L.; Zang, Y.; Li, J.; He, X.-P.; James, T.D. Metal–organic frameworks (MOFs) as host materials for the enhanced delivery of biomacromolecular therapeutics. Chemical Communications 2021, 57, 12098-12110.
- Hamblin, M.R.; Abrahamse, H. Inorganic salts and antimicrobial photodynamic therapy: mechanistic conundrums? Molecules 2018, 23, 3190.
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy–mechanisms, photosensitizers and combinations. Biomedicine & pharmacotherapy 2018, 106, 1098-1107.
- Zhang, J.; Jiang, C.; Longo, J.P.F.; Azevedo, R.B.; Zhang, H.; Muehlmann, L.A. An updated overview on the development of new photosensitizers for anticancer photodynamic therapy. Acta pharmaceutica sinica B 2018, 8, 137-146.
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. British journal of cancer 2019, 120, 6-15.
- Bayer, V.; Amaya, B.; Baniewicz, D.; Callahan, C.; Marsh, L.; McCoy, A.S. Cancer Immunotherapy. Clinical journal of oncology nursing 2017, 21.
- Yang, S.; Wu, G.-l.; Li, N.; Wang, M.; Wu, P.; He, Y.; Zhou, W.; Xiao, H.; Tan, X.; Tang, L. A mitochondria-targeted molecular phototheranostic platform for NIR-II imaging-guided synergistic photothermal/photodynamic/immune therapy. Journal of Nanobiotechnology 2022, 20, 475.
- Herzberg, B.; Campo, M.J.; Gainor, J.F. Immune checkpoint inhibitors in non-small cell lung cancer. The oncologist 2017, 22, 81-88.
- Kim, K.; Park, M.-H. Advancing cancer treatment: enhanced combination therapy through functionalized porous nanoparticles. Biomedicines 2024, 12, 326.
- Bienia, A.; Wiecheć-Cudak, O.; Murzyn, A.A.; Krzykawska-Serda, M. Photodynamic therapy and hyperthermia in combination treatment—Neglected forces in the fight against cancer. Pharmaceutics 2021, 13, 1147.
- Dudzik, T.; Domański, I.; Makuch, S. The impact of photodynamic therapy on immune system in cancer–an update. Frontiers in immunology 2024, 15, 1335920.
- Liu, Z.; Xie, Z.; Li, W.; Wu, X.; Jiang, X.; Li, G.; Cao, L.; Zhang, D.; Wang, Q.; Xue, P. Photodynamic immunotherapy of cancers based on nanotechnology: recent advances and future challenges. Journal of Nanobiotechnology 2021, 19, 160.
- Thiruppathi, J.; Vijayan, V.; Park, I.-K.; Lee, S.E.; Rhee, J.H. Enhancing cancer immunotherapy with photodynamic therapy and nanoparticle: making tumor microenvironment hotter to make immunotherapeutic work better. Frontiers in Immunology 2024, 15, 1375767.
- Alvarez, N.; Sevilla, A. Current advances in photodynamic therapy (PDT) and the future potential of PDT-combinatorial cancer therapies. International Journal of Molecular Sciences 2024, 25, 1023.
- Kleinovink, J.W.; van Driel, P.B.; Snoeks, T.J.; Prokopi, N.; Fransen, M.F.; Cruz, L.J.; Mezzanotte, L.; Chan, A.; Löwik, C.W.; Ossendorp, F. Combination of photodynamic therapy and specific immunotherapy efficiently eradicates established tumors. Clinical Cancer Research 2016, 22, 1459-1468.
- Hwang, H.S.; Cherukula, K.; Bang, Y.J.; Vijayan, V.; Moon, M.J.; Thiruppathi, J.; Puth, S.; Jeong, Y.Y.; Park, I.-K.; Lee, S.E. Combination of photodynamic therapy and a flagellin-adjuvanted cancer vaccine potentiated the anti-PD-1-mediated melanoma suppression. Cells 2020, 9, 2432.
- Mušković, M.; Pokrajac, R.; Malatesti, N. Combination of two photosensitisers in anticancer, antimicrobial and upconversion photodynamic therapy. Pharmaceuticals 2023, 16, 613.
- Cramer, G.M.; Moon, E.K.; Cengel, K.A.; Busch, T.M. Photodynamic therapy and immune checkpoint blockade. Photochemistry and Photobiology 2020, 96, 954-961.
- Kleinovink, J.W.; Ossendorp, F. Combination of Photodynamic Therapy and Immune Checkpoint Blockade. In Photodynamic Therapy: Methods and Protocols; Springer: 2022; pp. 589-596.
- Yuan, Z.; Fan, G.; Wu, H.; Liu, C.; Zhan, Y.; Qiu, Y.; Shou, C.; Gao, F.; Zhang, J.; Yin, P. Photodynamic therapy synergizes with PD-L1 checkpoint blockade for immunotherapy of CRC by multifunctional nanoparticles. Molecular Therapy 2021, 29, 2931-2948.
- Duan, X.; Chan, C.; Guo, N.; Han, W.; Weichselbaum, R.R.; Lin, W. Photodynamic therapy mediated by nontoxic core–shell nanoparticles synergizes with immune checkpoint blockade to elicit antitumor immunity and antimetastatic effect on breast cancer. Journal of the American Chemical Society 2016, 138, 16686-16695.
- Huang, Z.; Wei, G.; Zeng, Z.; Huang, Y.; Huang, L.; Shen, Y.; Sun, X.; Xu, C.; Zhao, C. Enhanced cancer therapy through synergetic photodynamic/immune checkpoint blockade mediated by a liposomal conjugate comprised of porphyrin and IDO inhibitor. Theranostics 2019, 9, 5542.
- Korbelik, M.; Banath, J.; Saw, K.M.; Zhang, W.; Čiplys, E. Calreticulin as cancer treatment adjuvant: combination with photodynamic therapy and photodynamic therapy-generated vaccines. Frontiers in oncology 2015, 5, 15.
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nature medicine 2007, 13, 54-61.
- Guo, R.; Wang, S.; Zhao, L.; Zong, Q.; Li, T.; Ling, G.; Zhang, P. Engineered nanomaterials for synergistic photo-immunotherapy. Biomaterials 2022, 282, 121425.
- Zhao, Y.; Liu, X.; Liu, X.; Yu, J.; Bai, X.; Wu, X.; Guo, X.; Liu, Z.; Liu, X. Combination of phototherapy with immune checkpoint blockade: Theory and practice in cancer. Frontiers in immunology 2022, 13, 955920.
- Meng, Z.; Zhou, X.; Xu, J.; Han, X.; Dong, Z.; Wang, H.; Zhang, Y.; She, J.; Xu, L.; Wang, C. Light-triggered in situ gelation to enable robust photodynamic-immunotherapy by repeated stimulations. Advanced Materials 2019, 31, 1900927.
- Ge, R.; Liu, C.; Zhang, X.; Wang, W.; Li, B.; Liu, J.; Liu, Y.; Sun, H.; Zhang, D.; Hou, Y. Photothermal-activatable Fe3O4 superparticle nanodrug carriers with PD-L1 immune checkpoint blockade for anti-metastatic cancer immunotherapy. ACS Applied Materials & Interfaces 2018, 10, 20342-20355.
- Chen, Q.; Xu, L.; Liang, C.; Wang, C.; Peng, R.; Liu, Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nature communications 2016, 7, 13193.
- Liang, X.; Mu, M.; Chen, B.; Fan, R.; Chen, H.; Zou, B.; Han, B.; Guo, G. Metal-organic framework-based photodynamic combined immunotherapy against the distant development of triple-negative breast cancer. Biomaterials Research 2023, 27, 120.
- Zeng, J.-Y.; Zou, M.-Z.; Zhang, M.; Wang, X.-S.; Zeng, X.; Cong, H.; Zhang, X.-Z. π-extended benzoporphyrin-based metal–organic framework for inhibition of tumor metastasis. ACS nano 2018, 12, 4630-4640.
- Hua, J.; Wu, P.; Gan, L.; Zhang, Z.; He, J.; Zhong, L.; Zhao, Y.; Huang, Y. Current strategies for tumor photodynamic therapy combined with immunotherapy. Frontiers in Oncology 2021, 11, 738323.
- Song, Y.; Wang, L.; Xie, Z. Metal–organic frameworks for photodynamic therapy: emerging synergistic cancer therapy. Biotechnology Journal 2021, 16, 1900382.
- Ji, B.; Wei, M.; Yang, B. Recent advances in nanomedicines for photodynamic therapy (PDT)-driven cancer immunotherapy. Theranostics 2022, 12, 434.
- Prasad, S.B.; Shinde, A.; Srinivasrao, D.A.; Famta, P.; Shah, S.; Kolipaka, T.; Pandey, G.; Gaonker, D.; Vambhurkar, G.; Khairnar, P. Metal-Organic Frameworks as Therapeutic Chameleons: Revolutionizing the Cancer Therapy Employing Novel Nanoarchitectonics. Asian Journal of Pharmaceutical Sciences 2025, 101054.
- Wang, N.; Song, W.; Ji, J.; Guo, W.; Du, Q. Metal-organic framework nanomaterials alter cellular metabolism in bladder cancer. Ecotoxicology and Environmental Safety 2025, 298, 118292.
- Soman, S.; Kulkarni, S.; Kulkarni, J.; Dhas, N.; Roy, A.A.; Pokale, R.; Mukharya, A.; Mutalik, S. Metal–organic frameworks: a biomimetic odyssey in cancer theranostics. Nanoscale 2025, 17, 12620-12647.
- Li, Y.; Zhou, J.; Chen, Y.; Pei, Q.; Li, Y.; Wang, L.; Xie, Z. Near-infrared light-boosted photodynamic-immunotherapy based on sulfonated metal-organic framework nanospindle. Chemical Engineering Journal 2022, 437, 135370.
- Zhao, X.; Li, Y.; Du, L.; Deng, Z.; Jiang, M.; Zeng, S. Soft x-ray stimulated lanthanide@ MOF nanoprobe for amplifying deep tissue synergistic photodynamic and antitumor immunotherapy. Advanced Healthcare Materials 2021, 10, 2101174.
- Liu, Y.; Zou, B.; Yang, K.; Jiao, L.; Zhao, H.; Bai, P.; Tian, Y.; Zhang, R. Tumor targeted porphyrin-based metal–organic framework for photodynamic and checkpoint blockade immunotherapy. Colloids and Surfaces B: Biointerfaces 2024, 239, 113965.
- Hirakawa, A.; Asano, J.; Sato, H.; Teramukai, S. Master protocol trials in oncology: review and new trial designs. Contemporary clinical trials communications 2018, 12, 1-8.
- Harvey, P.D.; Plé, J. Recent advances in nanoscale metal–organic frameworks towards cancer cell cytotoxicity: an overview. Journal of inorganic and organometallic polymers and materials 2021, 31, 2715-2756.
- Fernandes, P.D.; Magalhães, F.D.; Pereira, R.F.; Pinto, A.M. Metal-organic frameworks applications in synergistic cancer photo-immunotherapy. Polymers 2023, 15, 1490.
- Nguyen, N.T.T.; Nguyen, T.T.T.; Ge, S.; Liew, R.K.; Nguyen, D.T.C.; Van Tran, T. Recent progress and challenges of MOF-based nanocomposites in bioimaging, biosensing and biocarriers for drug delivery. Nanoscale Advances 2024, 6, 1800-1821.
Figure 10.
In vitro evaluation of antitumor effects of the nanocomplexes. (a) MTT assay assessed the cytotoxicity of BMS-202, PCN-224/HP, and BMS@P/HP NPs on normal L929 cells. (b) The cytotoxicity of these agents was evaluated on 4T1 tumor cells using the MTT assay (n = 3, significance levels: P < 0.05, P < 0.01, P < 0.001, P < 0.001, ***** < 0.0001). © LIVE/DEAD staining was performed on 4T1 cells treated with different drugs; live cells fluoresced green with Calcein-AM, and dead cells fluoresced red with propidium iodide (PI). Scale bar: 100 μm. (d) ROS levels in 4T1 cells were detected by flow cytometry, and (e) fluorescence microscopy was used to visualize ROS. Scale bar: 100 μm [40].
Figure 10.
In vitro evaluation of antitumor effects of the nanocomplexes. (a) MTT assay assessed the cytotoxicity of BMS-202, PCN-224/HP, and BMS@P/HP NPs on normal L929 cells. (b) The cytotoxicity of these agents was evaluated on 4T1 tumor cells using the MTT assay (n = 3, significance levels: P < 0.05, P < 0.01, P < 0.001, P < 0.001, ***** < 0.0001). © LIVE/DEAD staining was performed on 4T1 cells treated with different drugs; live cells fluoresced green with Calcein-AM, and dead cells fluoresced red with propidium iodide (PI). Scale bar: 100 μm. (d) ROS levels in 4T1 cells were detected by flow cytometry, and (e) fluorescence microscopy was used to visualize ROS. Scale bar: 100 μm [40].
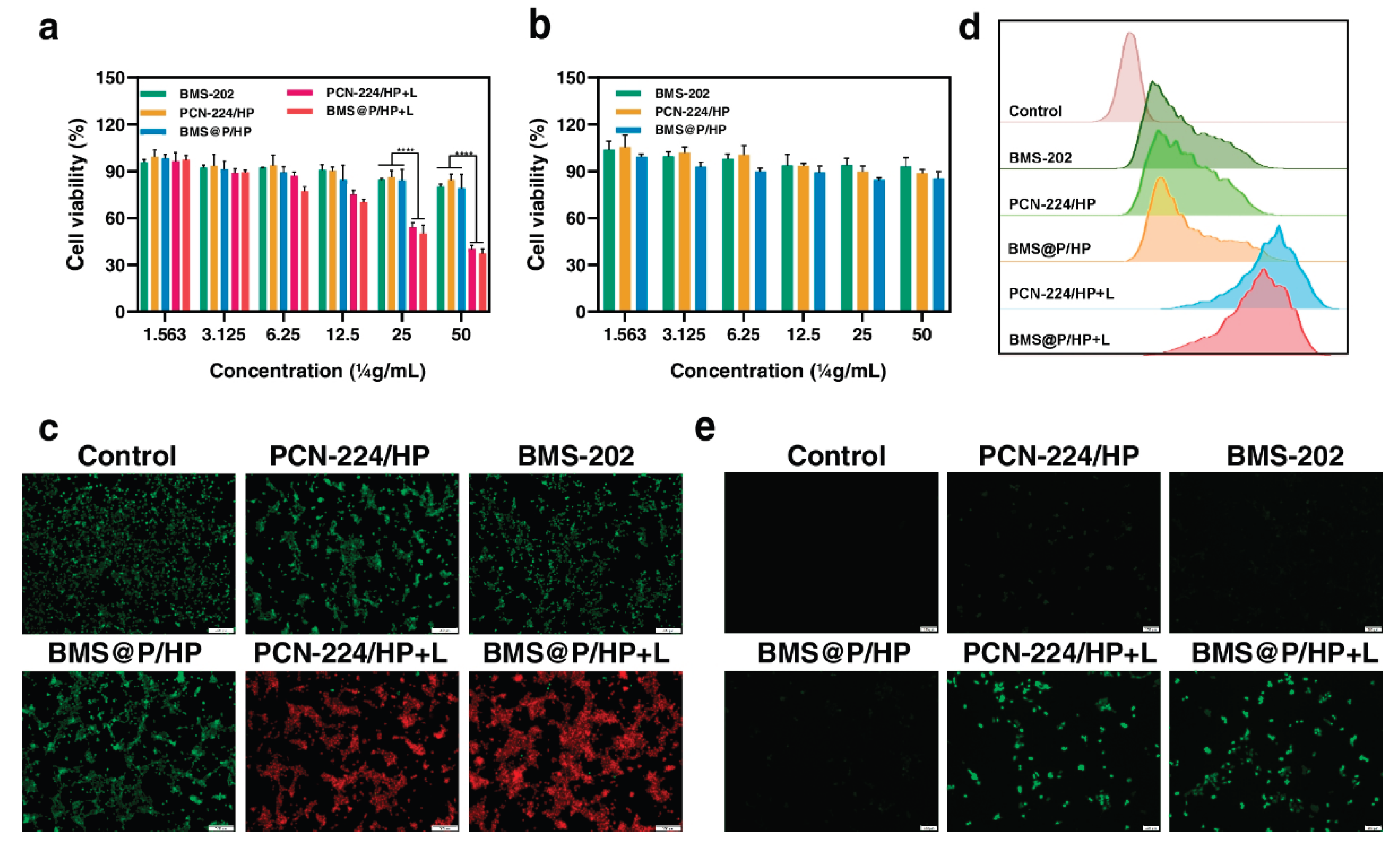
Figure 13.
(A) Confocal laser scanning microscopy (CLSM) images of 4T1 and HK-2 cells following treatment with PCN@HA, scale bar: 30 μm. (B, C) Viability of 4T1 cells incubated with PCN-224 and PCN@HA, both without and with irradiation. (D) Assessment of live and dead cells after different treatments, with live cells stained by calcein-AM (green) and dead cells stained by propidium iodide (PI, red), scale bar: 100 μm.
Figure 13.
(A) Confocal laser scanning microscopy (CLSM) images of 4T1 and HK-2 cells following treatment with PCN@HA, scale bar: 30 μm. (B, C) Viability of 4T1 cells incubated with PCN-224 and PCN@HA, both without and with irradiation. (D) Assessment of live and dead cells after different treatments, with live cells stained by calcein-AM (green) and dead cells stained by propidium iodide (PI, red), scale bar: 100 μm.
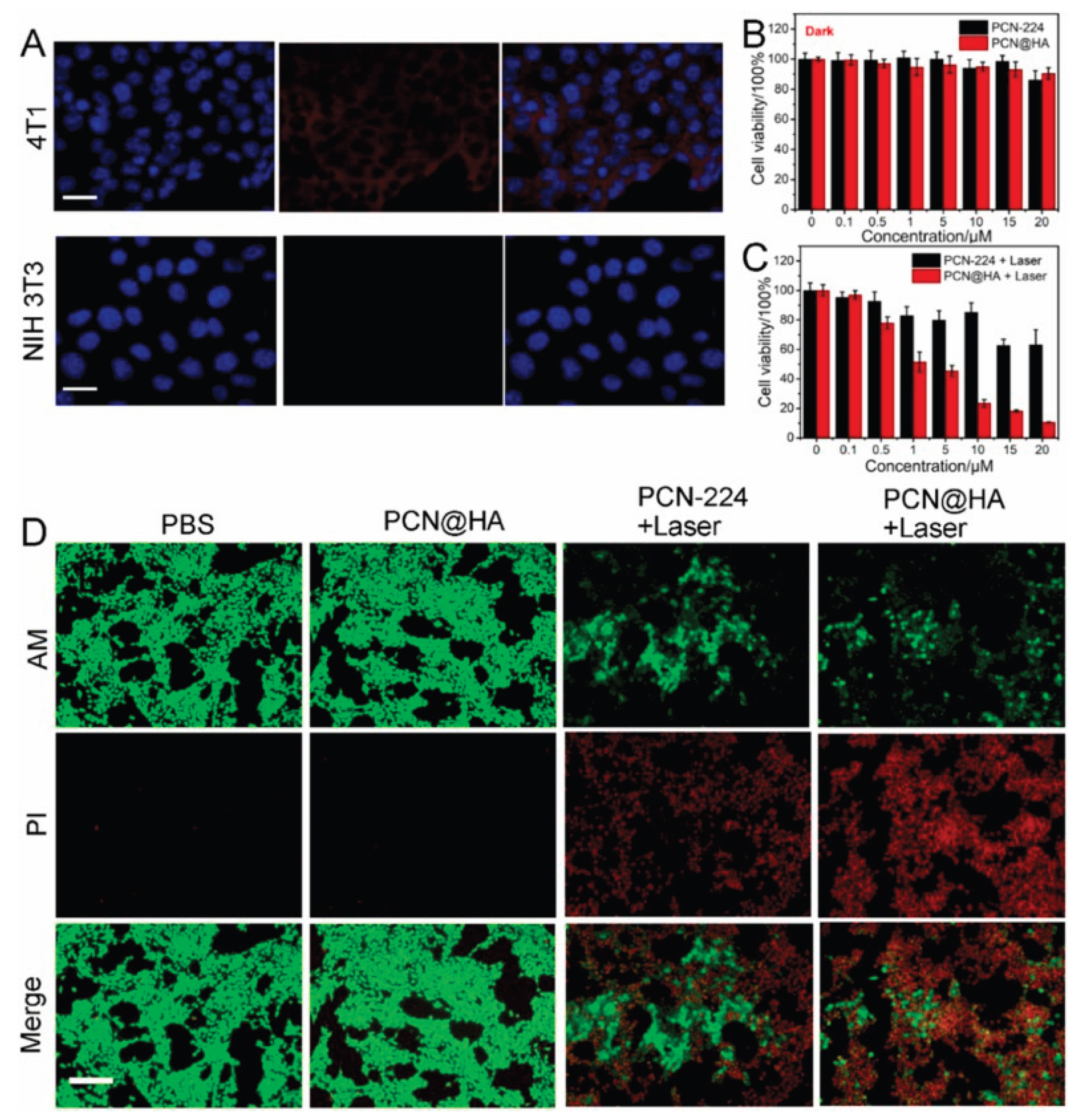
Figure 15.
a) Immunohistochemical analysis of calreticulin (CRT) and high mobility group box 1 (HMGB1) expression in tumour tissue samples. b) Quantification of the average proportion of tumour-infiltrating CD8+ cytotoxic T cells within the tumour microenvironment.
Figure 15.
a) Immunohistochemical analysis of calreticulin (CRT) and high mobility group box 1 (HMGB1) expression in tumour tissue samples. b) Quantification of the average proportion of tumour-infiltrating CD8+ cytotoxic T cells within the tumour microenvironment.
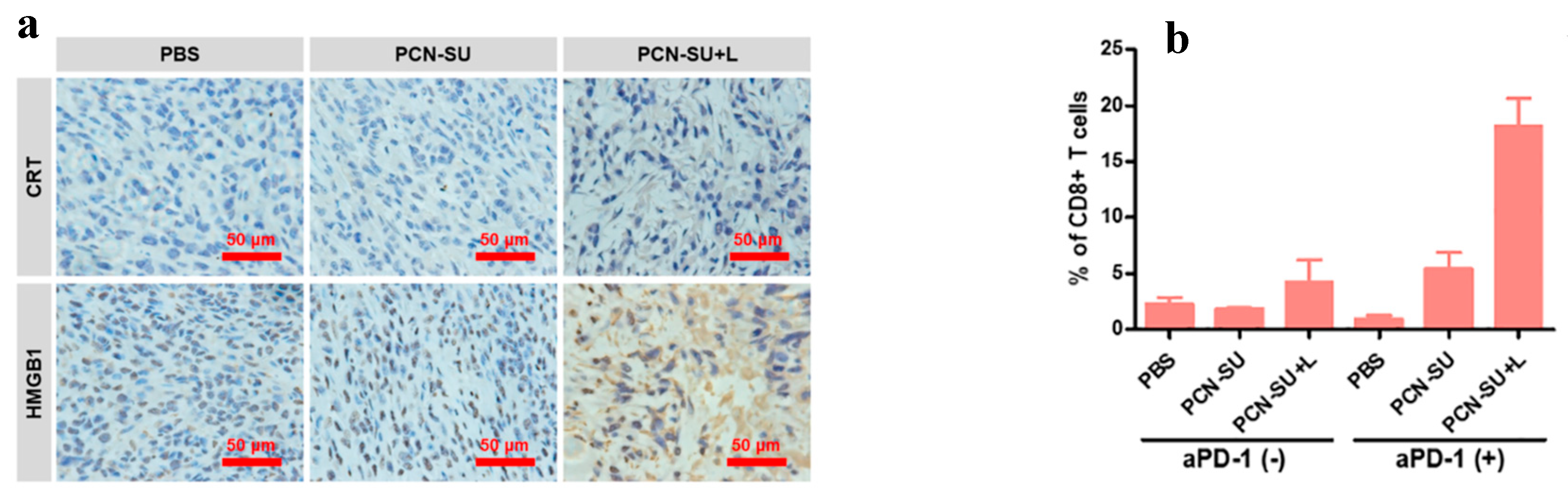
Figure 16.
(A) Diagram illustrating the soft X-ray activated PDT of tumours in vivo (tube voltage: 45 kVp). (B) Quantitative analysis of yttrium (Y) distribution in major organs of 4T1 tumour-bearing mice at 3, 6, 12, and 24 hours following intravenous injection of the SNPs@Zr-MOF@RB nanoprobe. (C) Body weight change curves of tumour-bearing mice over 12 days of treatment. (D) Tumour growth curves in 4T1 tumour-bearing mice subjected to various treatments. (E) Photographs of tumours excised from 4T1 tumour-bearing mice after 12 days of. (F,G) Representative tumour tissue sections stained with haematoxylin and eosin (H&E) and analysed by TUNEL assay following different treatments (scale bars: 50 µm). (H) Immunofluorescence imaging showing HMGB1 translocation from the tumour cell nucleus to the cytoplasm after various treatments (scale bar: 20 µm). (I) Quantitative analysis of fluorescence distribution patterns over distance across different treatment groups. Statistical significance: *P < 0.05, ***P < 0.001.
Figure 16.
(A) Diagram illustrating the soft X-ray activated PDT of tumours in vivo (tube voltage: 45 kVp). (B) Quantitative analysis of yttrium (Y) distribution in major organs of 4T1 tumour-bearing mice at 3, 6, 12, and 24 hours following intravenous injection of the SNPs@Zr-MOF@RB nanoprobe. (C) Body weight change curves of tumour-bearing mice over 12 days of treatment. (D) Tumour growth curves in 4T1 tumour-bearing mice subjected to various treatments. (E) Photographs of tumours excised from 4T1 tumour-bearing mice after 12 days of. (F,G) Representative tumour tissue sections stained with haematoxylin and eosin (H&E) and analysed by TUNEL assay following different treatments (scale bars: 50 µm). (H) Immunofluorescence imaging showing HMGB1 translocation from the tumour cell nucleus to the cytoplasm after various treatments (scale bar: 20 µm). (I) Quantitative analysis of fluorescence distribution patterns over distance across different treatment groups. Statistical significance: *P < 0.05, ***P < 0.001.
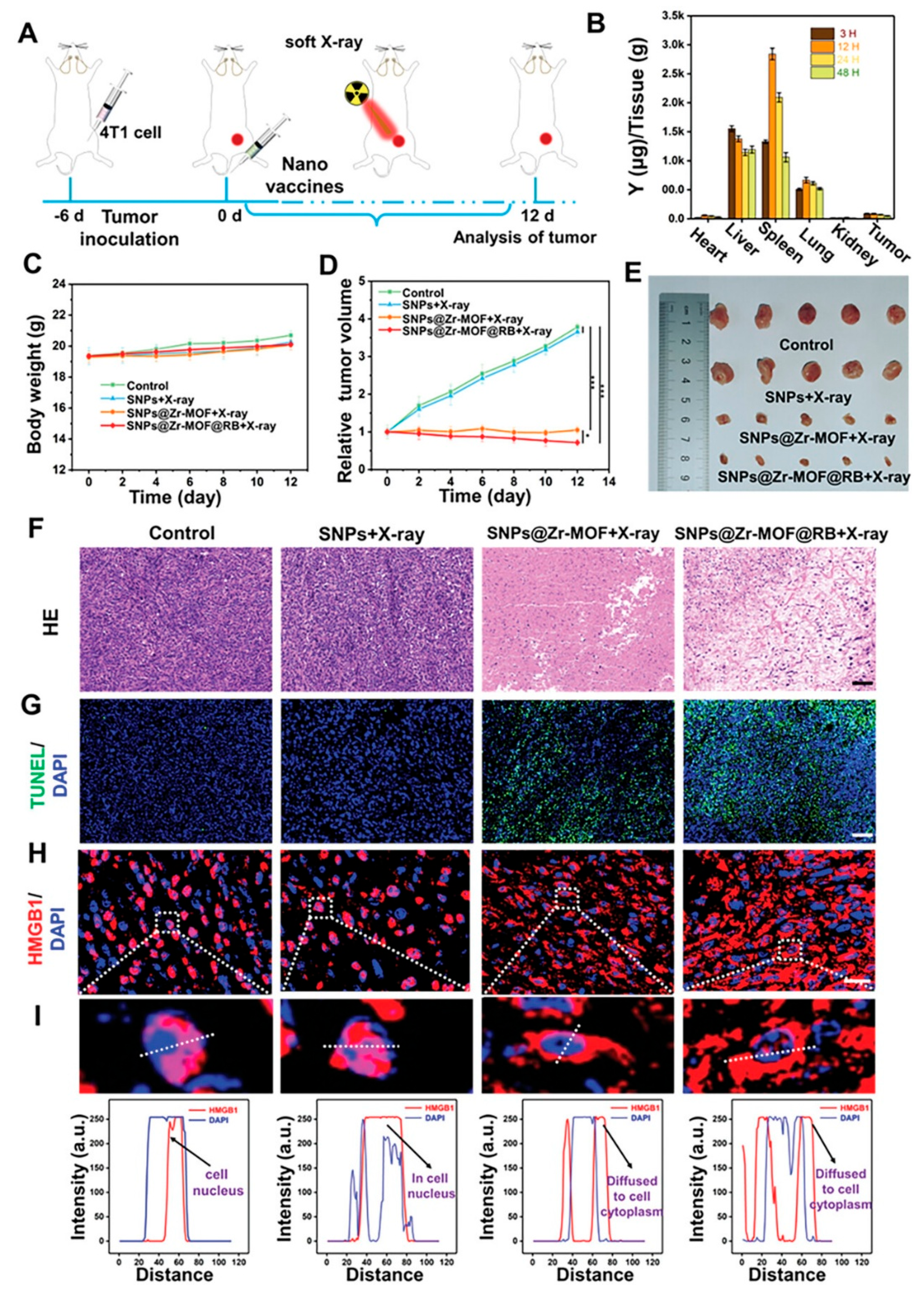
Figure 17.
(a) Volume of primary tumours and (b) volume of metastatic tumours in Balb/c mice subjected to various treatments. (c) Levels of interferon-gamma (IFN-γ) secretion and (d) tumour necrosis factor-alpha (TNF-α) secretion by splenocytes isolated from immunized mice [50].
Figure 17.
(a) Volume of primary tumours and (b) volume of metastatic tumours in Balb/c mice subjected to various treatments. (c) Levels of interferon-gamma (IFN-γ) secretion and (d) tumour necrosis factor-alpha (TNF-α) secretion by splenocytes isolated from immunized mice [50].
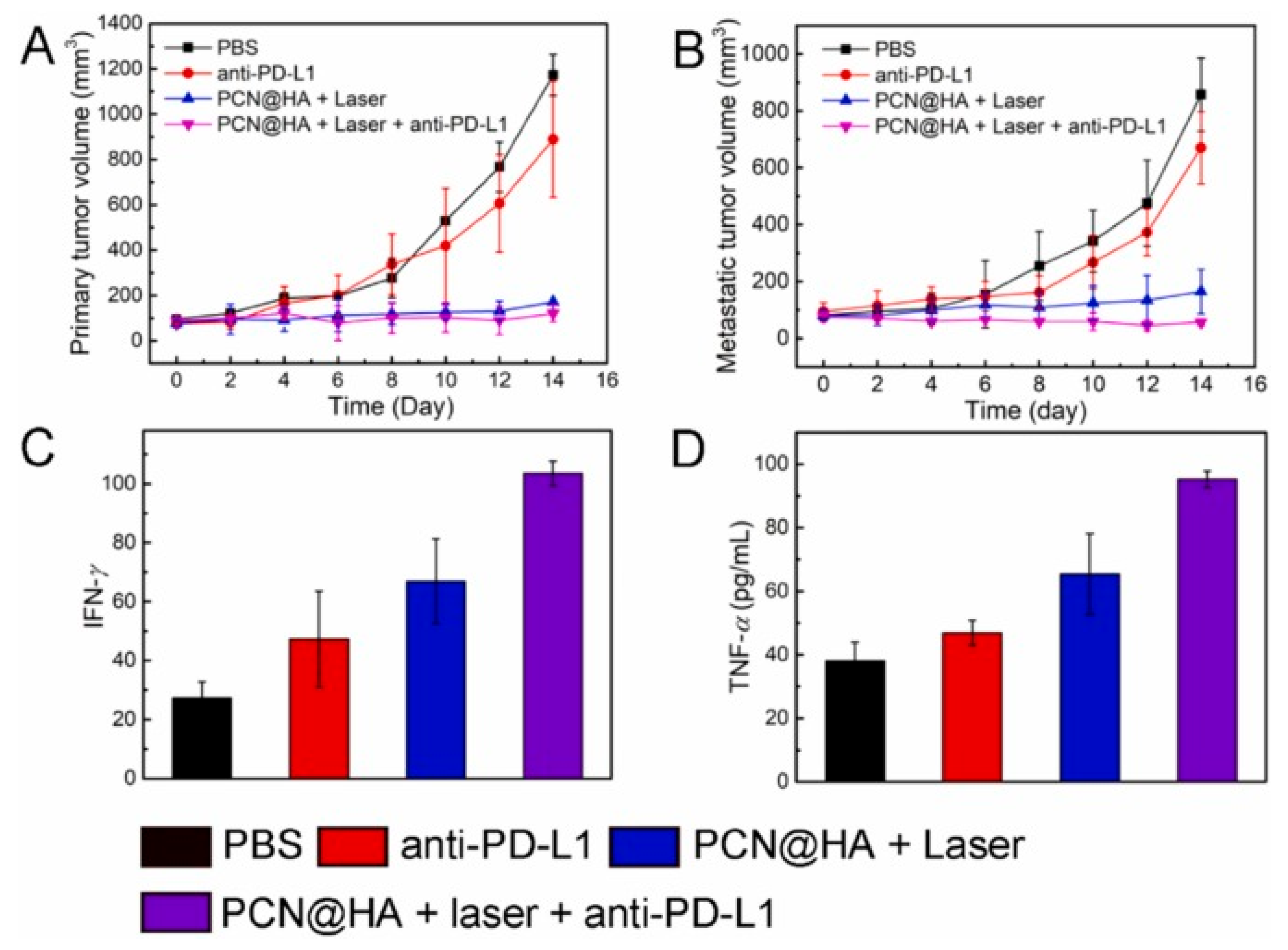
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



