
Submitted:
13 December 2025
Posted:
15 December 2025
You are already at the latest version
Abstract
Recent advances in neuro-oncology fundamentally challenge the traditional view of cancer as anarchic cellular proliferation, independent of physiological regulation. This review synthesizes the compelling evidence for bidirectional communication between the autonomic nervous system and cancer. Current research reveals that tumors are not isolated masses but complex biological systems integrated with neural networks. They actively recruit nerve fibers through neurotrophin release, generate neurons from cancer stem cells, and establish functional synaptic-like connections with the nervous system. The sympathetic branch consistently exploits this neuro-integration to promote oncogenesis across multiple cancer types. Norepinephrine and epinephrine released from its terminals enhance tumor cell proliferation, angiogenesis, and metastatic dissemination while suppressing anti-tumor immunity. β2-adrenergic receptor signaling, via the activation of cAMP-PKA pathways, serves as a key mediator of these effects. In contrast, the parasympathetic system displays a more nuanced, organ-specific role. Cholinergic signaling through muscarinic receptors can accelerate progression in gastric, pancreatic, and colorectal cancers by promoting stemness and proliferation. Conversely, parasympathetic activity may suppress tumor growth and metastasis in contexts like breast cancer and certain gastrointestinal malignancies, primarily through immunomodulatory mechanisms. This neurobiological framework reconceptualizes tumors as “neuro-integrated organs” that actively shape and exploit host neural architecture. Understanding this interplay repositions the nervous system from a passive bystander to a central and ultimately targetable regulator of oncogenesis. This review provides a narrative synthesis of current literature examining the interplay between the autonomic nervous system and cancer. Our approach involved a targeted survey of recent experimental and clinical studies covering key domains such as tumor innervation, sympathetic and parasympathetic regulation, adrenergic and cholinergic signaling, and neuroimmune interactions. Emphasis was placed on extracting mechanistic insights and identifying translational opportunities for therapeutic modulation. The resulting synthesis provides a conceptual framework for viewing cancer as a neurobiologically integrated disease process.
Keywords:
autonomic nervous system
; sympathetic
; parasympathetic
; adrenergic
; cholinergic
; tumor neurobiology
; cancer
; oncogenesis
; local anesthetics
Introduction
Nerve-Tumor Interaction: A Conceptual Shift
The perception of the nerve-tumor relationship has undergone a profound transformation. For decades, it was viewed through the narrow lens of perineural invasion (PNI), where nerves served as mere structural conduits for migrating cancer cells [1,2]. This simplistic model has given way to a far more complex reality: a dynamic, bidirectional interplay where the nervous system acts as an active collaborator in tumor development. Cancer cells co-opt neural mechanisms, remodel local innervation, and establish feedback loops that fuel their own growth and spread [3,4]. This fundamental reconceptualization, which moves from viewing nerves as passive structures to active collaborators, is summarized in Table 1.
This neuro-neoplastic dialogue is orchestrated through multiple mechanisms:
- Neurotransmitter signaling: A shared language of neuroactive molecules (e.g., norepinephrine, acetylcholine) is used by both nerve and tumor cells to control proliferation, angiogenesis, and immunity [7].
- Synaptic-like communication: Direct, functional contacts between neurons and cancer cells enable rapid signal exchange [8]
Beyond co-opting existing nerves, tumors demonstrate a startling capacity for self-neuralization. Cancer stem cells can differentiate into neuron-like cells, giving rise to de novo intratumoral neurogenesis.[12,13,14,15] This neural mimicry extends to function: tumor cells produce their own neurotransmitters, directly manipulating the neuro-immune interface to forge an immunosuppressive, pro-survival microenvironment. The result is a self-sustaining circuit that integrates neural, immune, and neoplastic signaling to drive immune escape, metabolic reprogramming, and therapeutic resistance.[16,17,18,19,20]
This new understanding reframes tumors as “neuro-integrated organs”—systems that are not just influenced by the host’s neural architecture but actively shape, expand, and exploit it. From this conceptual shift, the field of cancer neuroscience has emerged, viewing the nervous system not as a bystander, but as a central orchestrator of cancer’s evolution.[21,22]
The Tumor as a Neuro-Integrated Organ
We must now view the tumor as a neuro-integrated neo-organ, a system that maintains reciprocal connections with the host nervous system.[21,22,23,27] From its earliest stages, the tumor establishes a continuous dialogue, building its own autonomous neural network through active nerve recruitment and de novo neurogenesis from cancer stem cells. [5‚9‚12‚14‚15‚17‚24]
This innervation is functionally essential. It endows the tumor with the capacity to regulate its own metabolism,[25,26,27] leverage neurotransmitter signals to promote growth and metastasis,[28,29,30,31] and architect an immunosuppressive microenvironment by hijacking neuro-immune pathways.[32,33,34,35,36] The clinical relevance of this architecture is undeniable: higher nerve density consistently correlates with more aggressive disease and poorer patient survival across a range of cancers.[37,38,39,40,41]
The functionality of this network is evidenced by its electrical activity, which integrates the tumor into host autonomic circuits and creates a state of localized dysautonomia that fuels malignancy. The fact that this signaling can be pharmacologically silenced to inhibit tumor progression—for example, with adrenergic blockers—provides definitive proof of a causal, functional link.[42]
Viewing cancer through this neurobiological lens raises profound strategic questions. Effective therapy may now depend on our ability to dismantle this neural scaffolding. Understanding the plasticity of this network is key to overcoming the challenges of recurrence and resistance. Answering these questions is the central mandate of cancer neuroscience and the foundation for the next generation of oncologic therapies.
ANS in Oncogenesis: Re-Envisioning a Central Regulator
We must now re-envision the autonomic nervous system as an active orchestrator of oncogenesis, moving beyond a model where it is a mere background regulator. This shift integrates neurobiology directly into the core of cancer development, alongside genetics and inflammation, with neural inputs shaping virtually every hallmark of cancer, from initiation to therapy resistance (Figure 2).
The canonical mechanisms underpinning this view are now clear: sympathetic hyperactivation fuels the chronic inflammation that lays the groundwork for carcinogenesis, while its neurotransmitters directly promote tumor growth, invasion, and immune evasion.[18,19,20,34,43,44,45,46,47] This established paradigm explains how systemic stressors translate into malignant progression.
However, this view of a one-way influence is incomplete. Emerging evidence reveals a more complex, reciprocal relationship defined by profound neural plasticity. Tumors actively sculpt their own innervation, rewiring local circuits to their advantage.[14,50] Even more remarkably, they can achieve a degree of neural autonomy through de novo neurogenesis from resident cancer stem cells.[12] The culmination of this intricate local dialogue is a systemic autonomic dysregulation, clinically measurable through markers like Heart Rate Variability (HRV). The progressive decline of HRV in cancer patients provides a powerful biomarker of this deep neurobiological integration, cementing the ANS’s role as both a contributor to and a reporter of disease status.[51]
Sympathetic Overdrive: Fueling the Hallmarks of Cancer
The sympathetic nervous system (SNS) is a key accomplice in malignancy, unleashing a sustained barrage of catecholamines into the tumor microenvironment. These mediators, norepinephrine and epinephrine, find their targets in β-adrenergic receptors, which are ubiquitously expressed on cancer cells and the surrounding stromal, endothelial, and immune cells. The ensuing activation of intracellular pathways provides a direct mechanism for orchestrating the fundamental hallmarks of cancer.[6,34,64,64]
The direct innervation of tumor tissues by sympathetic fibers makes this a highly localized and efficient process.[9,21,23,65,66] Through this pathway, β2-adrenergic receptor stimulation via the cAMP-PKA axis can trigger devastating, tumor-specific outcomes. It can drive proliferation and induce a therapy-resistant neuroendocrine phenotype in prostate cancer,[5,67,68,69] or it can fuel tumor growth by promoting the formation of new blood vessels through VEGF upregulation.[19,30,63]
This synergy is perhaps most evident in aggressive cancers like pancreatic adenocarcinoma. Here, adrenergic signaling becomes entangled with neurotrophic pathways, creating a self-amplifying cycle of malignancy and treatment resistance.[70] This is not merely a biological curiosity; its clinical gravity is confirmed by the strong correlation between sympathetic nerve density and reduced patient survival, cementing the role of the SNS as a critical contributor to oncogenesis.[71]
The Vicious Cycle of Adrenergic Dependency
A critical shift in our understanding is the recognition that cancer cells are not just responsive to adrenergic signals, but become functionally dependent on them. This dependency is architected at the molecular level through the upregulation of β2-adrenergic receptors (β2-AR), which are highly expressed in proliferative niches and at the invasive front of tumors. This positions the β2-AR as a requisite axis for tumor evolution and a key therapeutic vulnerability.[72]
This neurochemical dependency is what enables tumors to hijack the autonomic nervous system’s homeostatic machinery. The ANS typically relies on negative feedback to stabilize tissue function. However, in the tumor context, this system is rewired into a positive feedback loop, creating what Makale et al. (2017) term a “runaway” state of self-perpetuating pro-tumorigenic signaling. This runaway loop disrupts tissue equilibrium, accelerating tumor growth and metastasis by upregulating factors like VEGF and IL-6 while simultaneously impairing DNA repair and inhibiting apoptosis.[73]
The power of this dysregulated circuit is evident at the cellular level. In breast cancer models, sustained β2-AR activation enhances proliferation, promotes resistance to treatment via MDR1 expression,[74] and drives invasion by stimulating the formation of matrix-degrading invadopodia through the Src pathway.[75] This detailed mechanistic understanding reveals a clear therapeutic logic: if the loop can be hijacked, it can also be broken. Indeed, preclinical studies show that β-adrenergic blockade with propranolol can reduce tumor burden in pancreatic cancer, and that vagotomy can curb cholinergic-driven growth in gastric models, demonstrating that restoring feedback balance is a viable anti-cancer strategy.⁷³
Sympathetic Modulation of Breast Cancer Bone Metastasis
The bone niche provides a fertile ground for breast cancer metastasis, a process significantly exacerbated by sympathetic nervous system (SNS) activity. Sympathetic neurotransmitters activate β-adrenergic receptors not only directly on breast cancer cells—promoting their proliferation and invasion—but also on the constituent cells of the bone microenvironment.[76] This signaling disrupts normal bone remodeling by enhancing osteoclast activity, which in turn creates osteolytic lesions and establishes a “metastatic vicious cycle.” Simultaneously, sympathetic stimulation fosters a pro-tumorigenic stroma by polarizing tumor-associated macrophages (TAMs) and drives angiogenesis via VEGF upregulation, further facilitating tumor cell survival and dissemination. This localized neurobiological hijacking aligns with broader clinical observations where systemic autonomic dysfunction and chronic stress are correlated with worse patient outcomes, positioning the metastatic bone niche as a critical hub of neuro-neoplastic interaction.
Colorectal Cancer: The Paradox of Sympathetic Signaling
Sympathetic innervation in colorectal cancer presents a biological paradox, with its influence appearing to shift dramatically with disease stage. In early-stage disease, a higher density of sympathetic nerve fibers is associated with improved prognosis and reduced nodal metastasis. Yet, in advanced stages, the upregulation of β-adrenergic receptors (β2-AR) on tumor cells becomes a clear driver of progression, invasiveness, and dissemination. This dichotomy highlights a critical distinction between nerve presence and receptor-mediated signaling, suggesting that the ultimate effect of the sympathetic nervous system is highly plastic and context-specific.[15,105]
The Adrenergic Axis as a Driver of Liver Cancer Malignancy
In hepatocellular carcinoma (HCC), the sympathetic nervous system (SNS) acts as a pivotal driver of tumor progression. The liver’s direct sympathetic innervation provides a structural conduit for adrenergic signals, integrating these inputs directly into the pathophysiology of HCC. This axis is further amplified by systemic factors like chronic stress, which, via the hypothalamic-pituitary-adrenal (HPA) axis, elevates catecholamine secretion and compromises immune surveillance.
At the tumor level, this sustained adrenergic signaling promotes malignancy through multiple mechanisms. It enhances tumor cell proliferation, upregulates pro-angiogenic factors like VEGF, and shapes a pro-tumorigenic microenvironment by facilitating the recruitment of tumor-associated macrophages. Aggressive HCC phenotypes capitalize on this pathway by upregulating β2-adrenergic receptors, a molecular adaptation that intensifies their responsiveness to adrenergic stimuli and locks in a state of accelerated growth and metastatic potential.[107]
Neural Colonizers: The Brain as a Source for Tumor Innervation
The traditional boundaries of neuro-oncology have been shattered by the discovery that the brain itself can be a source of tumor-innervating neurons. Groundbreaking work by Mauffrey et al. has shown that neural progenitors from the brain’s subventricular zone (SVZ) can breach the blood-brain barrier, migrate to prostate tumors, and differentiate into adrenergic neo-neurons. These newly formed neurons are not passive bystanders; they actively drive early-stage tumor growth and metastasis. This process represents a pathological hijacking of conserved embryonic pathways, where fundamental programs like neurogenesis and angiogenesis are co-opted to build and sustain a malignant microenvironment.[77]
“Stress,” What Stress? Redefining Stressors in Cancer Biology
The question posed is not whether stress influences cancer, but what we define as “stress.” The conventional focus has been on chronic psychoemotional distress, a recognized trigger of sympathetic overactivation known to promote epithelial-mesenchymal transition (EMT) and metastasis.[78,43,66] However, this definition is critically incomplete. It overlooks a host of powerful biological stressors.
Chronic inflammatory lesions, unresolved pain, and visceral dysfunctions function as persistent physiological insults that hijack the very same sympathetic-autonomic reflex arcs.[16] The outcome is a shared pathological state: a sustained catecholamine release that compromises neuroimmune communication and cultivates a microenvironment ripe for tumorigenesis.[34,63,64] Therefore, the answer to “What stress?” is a unified physiological state of sympathetic hyperactivity, regardless of its trigger. This broader definition is essential, as it implies that mitigating the biological drivers of sympathetic overdrive is as fundamental to cancer management as addressing its psychoemotional components.
Immune Sabotage: The Sympathetic Hijacking of the Tumor Microenvironment
Sympathetic signaling’s most insidious role in cancer may be its ability to orchestrate tumor escape through a comprehensive hijacking of the immune system. This occurs at the neuroimmune interface, where norepinephrine binds to β-adrenergic receptors ubiquitously expressed on immune cells, triggering a cascade that systematically dismantles anti-tumor defenses.
The strategy is twofold: disarming protectors while recruiting collaborators. On one hand, adrenergic signaling directly blunts the effectiveness of innate and adaptive immunity by suppressing the cytotoxic functions of lymphocytes and the antigen-presenting capacity of dendritic cells.[63,64,34] On the other, it actively cultivates a treacherous microenvironment by promoting the influx and pro-tumorigenic activity of myeloid-derived suppressor cells and M2-polarized macrophages. These cells then secrete a cocktail of immunosuppressive factors that further cripple cytotoxic responses and fuel angiogenesis.
This entire subversive campaign is fueled by the systemic effects of chronic stress. The hypothalamic-pituitary-adrenal (HPA) axis provides a continuous supply of catecholamines and glucocorticoids that systemically inhibit natural killer cells and disrupt T-cell function. The outcome is a self-sustaining, permissive neuroimmune environment, engineered by the sympathetic nervous system to allow tumors not only to survive, but to thrive, metastasize, and resist therapy.[35,36,46,79,80]
Vagal Duality: The Context-Dependent Role of Parasympathetic Signaling
In stark contrast to the consistently pro-tumorigenic role of the sympathetic nervous system, the parasympathetic nervous system (PNS) exhibits a paradoxical and context-dependent influence on cancer. Its primary neurotransmitter, acetylcholine (ACh), acting through muscarinic receptors (mAChRs), can either drive or suppress tumor progression. The ultimate outcome of this signaling is dictated by a complex interplay of factors, including cancer type, disease stage, and the specific molecular and cellular composition of the tumor microenvironment. This functional duality necessitates a nuanced understanding of cholinergic pathways to guide the development of future targeted oncological therapies.[18,20]
The Pro-Tumorigenic Face of Parasympathetic Signaling
Despite its broader anti-inflammatory roles, the parasympathetic nervous system can act as a potent driver of malignancy in certain contexts. This pro-tumorigenic capacity is mediated by acetylcholine, which can have profound, context-specific effects on tumor biology. In pancreatic ductal adenocarcinoma, muscarinic receptor activation directly enhances cancer cell proliferation and stemness, two core drivers of tumorigenesis.[82]
This oncogenic potential can also be realized through intricate signaling within the tumor microenvironment. Gastric tumors, for example, can establish a feed-forward loop with cholinergic neurons via nerve growth factor (NGF) to accelerate their own growth.²⁸ Colorectal cancer cells take this a step further, creating their own autocrine cholinergic loops to ensure their survival and proliferation.⁸³ Collectively, these findings reveal a darker side to parasympathetic signaling, necessitating the development of highly selective neuromodulatory therapies capable of distinguishing between its protective systemic roles and its context-specific oncogenic actions.
The Protective Shield: Vagal Tone in Cancer Suppression
The parasympathetic nervous system presents a stark paradox in cancer biology: while cholinergic signaling can sometimes fuel malignancy, vagal activity itself often acts as a powerful brake on tumor progression. This protective role is evident in cancers like breast and pancreas, where intact vagal signaling promotes anti-tumor immunity and suppresses metastasis.[50,47,84] The prognostic implications are profound. Studies consistently link intact vagal integrity with improved cancer outcomes, while vagotomy—the surgical interruption of this pathway—is associated with accelerated tumor growth and diminished survival.[85] Indeed, higher vagal nerve activity has been shown to correlate with significantly longer overall survival across multiple cancer types, firmly establishing the parasympathetic tone as a critical protective factor against oncogenesis.[86,87]
Friend or Foe?
So, is the autonomic nervous system a friend or a foe in the landscape of oncogenesis? The evidence paints a complex and dualistic picture. The sympathetic nervous system consistently emerges as a foe: its overactivation reliably promotes tumor proliferation, angiogenesis, immune evasion, and metastasis. It acts as a clear and unambiguous driver of malignancy.
The true complexity of the “friend or foe” question resides within the parasympathetic nervous system. Far from being a simple counterweight, its influence is profoundly context-dependent, capable of either suppressing or accelerating cancer progression depending on the tumor type, stage, and microenvironment, an organ-specific duality illustrated in Figure 3. Therefore, the ANS is not a monolithic entity but a system with one consistently pro-tumorigenic branch and one highly ambivalent branch. This intricate duality underscores the need for precision neuromodulation—strategies that can inhibit the foe-like sympathetic pathways while selectively harnessing the friendly, protective potential of the parasympathetic system.

Table 2.
The Dualistic Role of the Autonomic Nervous System in Cancer Progression.
| Autonomic branch | Functional Impact on Tumor Biology |
|---|---|
|
Sympathetic (Consistently the "Foe") |
Consistently Pro-Tumorigenic Actions: - Drives tumor cell proliferation, invasion, and metastatic spread. - Orchestrates an immunosuppressive microenvironment and promotes therapy resistance. - Mediates the oncogenic effects of chronic systemic stress. |
|
Parasympathetic (The "Friend and Foe") |
Context-Dependent Duality Pro-Tumorigenic Actions (“Foe”): - Can accelerate proliferation and invasion in specific contexts (e.g., gastric, colon, some pancreatic cancers). Anti-Tumorigenic Actions ("Friend"): - Can inhibit tumor growth and metastasis in other contexts (e.g., breast, certain GI and pancreatic cancers). - Enhances systemic anti-tumor immune responses. |
Neuro-Targeted Therapies: A Paradigm Shift in Cancer Treatment
Viewing cancer through a neurobiological lens does more than add another layer of complexity; it unveils an entirely new class of therapeutic targets within the autonomic nervous system. The core principle of this approach is that the ANS is not merely an influencer of tumor growth but a master regulator of the microenvironment’s adaptive responses to stress, inflammation, and immune attack. Consequently, therapeutically restoring autonomic balance is not an ancillary strategy but a direct intervention into the machinery of oncogenesis. This opens a new frontier in treatment, where manipulating neural circuits could become as fundamental to oncology as targeting genetic mutations or boosting immune responses.[22,63,88]
β-Adrenergic Blockade: Countering Sympathetic Overdrive
Pharmacologic inhibition of sympathetic signaling is one of the most developed neuro-oncologic strategies. Nonselective β-blockers, such as propranolol, directly counteract the effects of chronic sympathetic overdrive by attenuating catecholamine-driven tumor proliferation, angiogenesis, and metastatic dissemination. By reducing vascular endothelial growth factor (VEGF) expression and limiting inflammatory cytokine release, these agents dismantle the pro-tumorigenic microenvironment. Clinical observations consistently suggest that β-blocker exposure correlates with improved survival and reduced recurrence in several malignancies, positioning them as powerful adjuvant modulators of oncologic therapy.[89,90,91]
Vagal Modulation: Restoring the Cholinergic Anti-inflammatory Reflex
Complementing the strategy of sympathetic blockade is the active enhancement of parasympathetic tone. The vagus nerve provides an intrinsic anti-inflammatory and immunoregulatory influence, and its stimulation can re-calibrate the autonomic system. By activating the cholinergic anti-inflammatory reflex, vagal modulation lowers systemic cytokine levels and reactivates cell-mediated immunity, restraining tumor progression and improving host resilience. However, the context-dependent duality of cholinergic signaling warrants caution. In tumors where this pathway is known to be pro-tumorigenic, such as in certain gastric or pancreatic cancers, nonspecific vagal stimulation could theoretically carry risks. This highlights the critical need to move beyond systemic modulation toward receptor-specific or tissue-targeted strategies. A range of techniques, from invasive electrical stimulation to noninvasive methods, are now under investigation to achieve this delicate autonomic rebalancing in cancer patients.[47,55,92,9347,55,92,93]
Surgical and Chemical Denervation: Dismantling Local Neural Circuits
A more direct approach involves the targeted disruption of the tumor’s neural infrastructure. While context-dependent, surgical or chemical sympathectomy has suppressed tumor growth in multiple preclinical models by physically interrupting the local feedback loops between nerves and cancer cells. This denervation starves the tumor of catecholamine availability, effectively diminishing the neural support system that sustains its viability and growth.[19,27,94,95]
Local Anesthetics: Multi-Modal Neuroimmune Modulation
Beyond their analgesic function, local anesthetics (LAs) are emerging as uniquely versatile agents, capable of disrupting the neuro-cancer axis at multiple, synergistic levels. Their most immediate relevance lies in their capacity to dismantle the pro-tumorigenic sympathetic framework. By blunting autonomic reflex pathways or through direct nerve blockade, LAs can shield the patient from the oncogenic consequences of the surgical stress response, a mechanism that may underpin the improved outcomes observed with regional versus general anesthesia in some clinical studies.[96,97,98,99]
This neuro-modulatory function, however, is only the opening chapter of a much deeper biological story. A compelling body of preclinical evidence has repositioned LAs as direct anti-cancer agents and, crucially, as potent chemosensitizers. Their actions constitute a multi-pronged assault on the machinery of malignancy. They can achieve chemosensitization by disabling drug efflux pumps like P-glycoprotein, effectively trapping conventional therapies inside resistant cancer cells. Concurrently, they launch a direct attack by inducing cytotoxicity through apoptosis and by dismantling the cell's metastatic potential via the blockade of voltage-gated sodium channels. Finally, they can orchestrate a profound disruption of oncogenic signaling, interfering with core survival pathways like PI3K/AKT/mTOR and even enacting epigenetic reprogramming through mechanisms such as DNA demethylation.[100,101,102,103,104,105]
The investigation into these multi-modal actions now marks a clinical crossroads where mechanistic promise meets the rigor of prospective evidence. Decades of preclinical research have built this strong case, a foundation that now finds its first crucial clinical support in the pilot RCT by Alexa et al. (2023). This trial demonstrated that a perioperative lidocaine infusion alone could significantly curb disease recurrence in colorectal cancer, validating the systemic anti-cancer potential of LAs in a clinical setting.[109] This study, however, illuminates a path as much as it answers a question. It confirms the drug's efficacy as a standalone agent but leaves the most compelling hypothesis—its ability to synergize with chemotherapy—untested in patients. The field is therefore poised at this crossroads, armed with a powerful biological rationale but still lacking the definitive, large-scale trial evidence required to translate this synergy into standard oncologic practice.
The Common Challenge: From Preclinical Promise to Clinical Reality
The therapeutic paradigm emerging from these neuro-targeted strategies—whether through systemic β-blockade, vagal modulation, targeted denervation, or the multi-modal action of local anesthetics—is as compelling as it is incomplete. These approaches collectively represent the therapeutic vanguard of cancer neuroscience, yet they advance on a path where profound mechanistic insights and promising preclinical data consistently outpace definitive clinical validation. Their integration into standard oncologic care, therefore, awaits the evidence that can only be provided by large-scale, robust randomized controlled trials. Navigating this critical translational journey from biological rationale to proven patient benefit is not just a scientific challenge; it is the defining mandate for the future of this entire field.
Conclusions
This review has charted a paradigm shift in our understanding of cancer, reframing the autonomic nervous system from a passive bystander to a central orchestrator of oncogenesis. The evidence compels us to view tumors as “neuro-integrated organs,” systems whose survival depends on a continuous dialogue with the host. This neurobiological lens clarifies the “Friend or Foe” paradox: the sympathetic system emerges as a consistent “foe,” driving malignancy, while the parasympathetic system’s role is profoundly ambivalent, acting as both “friend” and “foe” depending on the context. Recognizing this intricate reality is not an endpoint, but the starting point for a new chapter in cancer research, one defined by critical unanswered questions and a clear path toward a new class of therapies.
The path forward requires us to venture beyond the established roles of adrenergic and cholinergic signaling. We must now ask: What are the functions of the broader neuro-modulatory landscape, including neuropeptides and other neuroactive molecules? Can we map the specific neuronal subtypes that innervate different tumors to move from broad autonomic modulation to precision targeting? The revolutionary discovery of a brain-tumor axis that seeds prostate cancer with neural progenitors demands investigation in other malignancies—is this a universal mechanism of neural hijacking? Answering these questions will be fundamental to truly understanding the tumor as a systemic, neuro-integrated entity.
Translating this new paradigm into clinical practice is the ultimate goal and the greatest challenge. The immediate priority is the development of robust biomarkers, such as standardized measures of nerve density and Heart Rate Variability, to move the assessment of a tumor’s neural landscape from the laboratory to the clinic. These tools are prerequisites for designing the next generation of clinical trials, which must be sophisticated enough to account for the context-dependent duality of parasympathetic signaling. While repurposing drugs like β-blockers has shown immense promise, the long-term vision must be the creation of novel agents that can selectively dismantle the neural scaffolding supporting the tumor. The emerging, multi-modal role of local anesthetics—capable of not only blunting sympathetic input but also exerting direct anti-tumor effects—perfectly illustrates this new frontier, where single agents can target multiple facets of the neuro-cancer axis.
Ultimately, this journey leads toward a new era of integrated oncology. The neural profile of a tumor may one day become as critical to treatment decisions as its genetic or immunological signature. The therapeutic goal will expand from simply killing cancer cells to actively re-calibrating the physiological environment that allows them to thrive. This is the path forward: to develop therapies that are not only more effective, but that work in concert with the body’s own neural regulatory systems to restore balance.
Author Contributions
M.A. (Michele Acanfora): Conceptualization, Literature search, Writing – original draft, Review & editing. M.E. (Maurizio Episodio): Literature search, Writing – original draft. M.B. (Massimo Bonucci): Supervision, Critical review. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare that they have no conflict of interest. This review received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
- Chen, S.H.; Zhang, B.Y.; Zhou, B.; Zhu, C.Z.; Xu, L.Z.; Huang, T. Perineural invasion of cancer: a complex crosstalk between cells and molecules in the perineural niche. Am J Cancer Res. 2019, 9, 1–21. [Google Scholar]
- Huang, S.; Zhu, J.; Yu, L.; Huang, Y.; Hu, Y. Cancer-nervous system crosstalk: from biological mechanism to therapeutic opportunities. Mol Cancer 2025, 24, 133. [Google Scholar] [CrossRef]
- Zhang, Y.; Liao, Q.; Wen, X.; Fan, J.; Yuan, T.; Tong, X.; et al. Hijacking of the nervous system in cancer: mechanism and therapeutic targets. Mol Cancer 2025, 24, 44. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Liang, X.; Tang, Y. Neuroscience in peripheral cancers: tumors hijacking nerves and neuroimmune crosstalk. MedComm 2024, 5, e784. [Google Scholar] [CrossRef] [PubMed]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; et al. Autonomic Nerve Development Contributes to Prostate Cancer Progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [PubMed]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat Rev Cancer 2020, 20, 143–57. [Google Scholar] [CrossRef]
- Jiang, S.H.; Hu, L.P.; Wang, X.; Li, J.; Zhang, Z.G. Neurotransmitters: emerging targets in cancer. Oncogene 2020, 39, 503–15. [Google Scholar] [CrossRef]
- Wang, H.; Song, X.; Shen, H.; et al. Cancer neuroscience in head and neck: interactions, modulation, and therapeutic strategies. Mol Cancer 2025, 24, 101. [Google Scholar] [CrossRef]
- Silverman, D.A.; Martinez, V.K.; Dougherty, P.M.; Myers, J.N.; Calin, G.A.; Amit, M. Cancer-Associated Neurogenesis and Nerve-Cancer Cross-talk. Cancer Research 2021, 81, 1431–40. [Google Scholar] [CrossRef]
- Ventura, S.; Evans, B.A. Does the autonomic nervous system contribute to the initiation and progression of prostate cancer? Asian J Androl. 2013, 15, 715–6. [Google Scholar] [CrossRef]
- Bautista, M.; Krishnan, A. The autonomic regulation of tumor growth and the missing links. Front Oncol. 2020, 10, 566353. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Fan, C.; Shangguan, W.; Liu, Y.; Li, Y.; Shang, Y.; et al. Neurons generated from carcinoma stem cells support cancer progression. Sig Transduct Target Ther. 2017, 2, 16036. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S. The neural regulation of cancer. Science 2019, 366, 965–965. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Villagrana, R.D.; Albores-García, D.; Cervantes-Villagrana, A.R.; Sánchez-Reyes, J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct Target Ther. 2020, 5, 99. [Google Scholar] [CrossRef]
- Yaman, I.; Ağaç Çobanoğlu, D.; Xie, T.; Ye, Y.; Amit, M. Advances in understanding cancer-associated neurogenesis and its implications on the neuroimmune axis in cancer. Pharmacology & Therapeutics 2022, 239, 108199. [Google Scholar] [CrossRef]
- Hanoun, M.; Maryanovich, M.; Arnal-Estapé, A.; Frenette, P.S. Neural regulation of hematopoiesis, inflammation, and cancer. Neuron 2015, 86, 360–73. [Google Scholar] [CrossRef]
- Arese, M.; Bussolino, F.; Pergolizzi, M.; Bizzozero, L.; Pascale, A. Tumor progression: the neuronal input. Ann Transl Med. 2018, 6, 89. [Google Scholar] [CrossRef]
- Hutchings, C.; Phillips, J.A.; Djamgoz, M.B.A. Nerve input to tumours: Pathophysiological consequences of a dynamic relationship. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2020, 1874, 188411. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Arnal-Estapé, A.; Maryanovich, M.; Nakahara, F.; Cruz, C.D.; Finley, L.W.S.; et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017, 358, 321–6. [Google Scholar] [CrossRef]
- Erin, N.; Shurin, G.V.; Baraldi, J.H.; Shurin, M.R. Regulation of Carcinogenesis by Sensory Neurons and Neuromediators. Cancers 2022, 14, 2333. [Google Scholar] [CrossRef]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor neurobiology and the war of nerves in cancer. Cancer Discov. 2019, 9, 702–10. [Google Scholar] [CrossRef] [PubMed]
- Magnon, C.; Hondermarck, H. The neural addiction of cancer. Nat Rev Cancer 2023, 23, 143–58. [Google Scholar] [CrossRef]
- Gysler, S.M.; Drapkin, R. Tumor innervation: peripheral nerves take control of the tumor microenvironment. J Clin Invest. 2021, 131, e147276. [Google Scholar] [CrossRef] [PubMed]
- Madeo, M.; Colbert, P.L.; Vermeer, D.W.; Lucido, C.T.; Cain, J.T.; Vichaya, E.G.; et al. Cancer exosomes induce tumor innervation. Nat Commun. 2018, 9, 4284. [Google Scholar] [CrossRef] [PubMed]
- Albo, D.; Akay, C.L.; Marshall, C.L.; Wilks, J.A.; Verstovsek, G.; Liu, H.; et al. Neurogenesis in colorectal cancer is a marker of aggressive tumor behavior and poor outcomes. Cancer 2011, 117, 4834–45. [Google Scholar] [CrossRef]
- Saloman, J.L.; Albers, K.M.; Li, D.; Hartman, D.J.; Crawford, H.C.; Muha, E.A.; et al. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc Natl Acad Sci USA 2016, 113, 3078–83. [Google Scholar] [CrossRef]
- Zhao, C.M.; Hayakawa, Y.; Kodama, Y.; Muthupalani, S.; Westphalen, C.B.; Andersen, G.T.; et al. Denervation suppresses gastric tumorigenesis. Sci Transl Med. 2014, 6, 250ra115. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Sakitani, K.; Konishi, M.; Asfaha, S.; Niikura, R.; Tomita, H.; et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell. 2017, 31, 21–34. [Google Scholar] [CrossRef]
- Allen, J.K.; Armaiz-Pena, G.N.; Nagaraja, A.S.; Sadaoui, N.C.; Ortiz, T.; Dood, R.; et al. Sustained adrenergic signaling promotes intratumoral innervation through BDNF induction. Cancer Res. 2018, 78, 3233–42. [Google Scholar] [CrossRef]
- Tilan, J.; Kitlinska, J. Sympathetic neurotransmitters and tumor angiogenesis—link between stress and cancer progression. J Oncol. 2010, 2010, 539706. [Google Scholar] [CrossRef]
- Yang, E.V.; Kim, S.J.; Donovan, E.L.; Chen, M.; Gross, A.C.; Webster Marketon, J.I.; et al. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: implications for stress-related enhancement of tumor progression. Brain Behav Immun. 2009, 23, 267–75. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Hiyama, T.; Fujimura, A.; Yoshikawa, S. Sympathetic and parasympathetic innervation in cancer: therapeutic implications. Clin Auton Res. 2021, 31, 165–78. [Google Scholar] [CrossRef] [PubMed]
- Amit, M.; Baruch, E.; Nagarajan, P.; Gleber-Netto, F.; Rao, X.; Xie, T.; et al. Inflammation induced by tumor-associated nerves promotes resistance to anti-PD-1 therapy in cancer patients and is targetable by IL-6 blockade. PREPRINT 2023. [Google Scholar]
- Qiao, G.; Chen, M.; Bucsek, M.J.; Repasky, E.A.; Hylander, B.L. Adrenergic Signaling: A Targetable Checkpoint Limiting Development of the Antitumor Immune Response. Front Immunol. 2018, 9, 164. [Google Scholar] [CrossRef]
- Qin, J.F.; Jin, F.J.; Li, N.; Guan, H.T.; Lan, L.; Ni, H.; et al. Adrenergic receptor β2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB Reports 2015, 48, 295–300. [Google Scholar] [CrossRef]
- Kizil, B.; De Virgiliis, F.; Scheiermann, C. Neural control of tumor immunity. The FEBS Journal. 2024, 291, 4670–9. [Google Scholar] [CrossRef]
- Li, X.; Peng, X.; Yang, S.; Wei, S.; Fan, Q.; Liu, J.; et al. Targeting tumor innervation: premises, promises, and challenges. Cell Death Discov. 2022, 8, 131. [Google Scholar] [CrossRef]
- Wang, H.; Huo, R.; He, K.; Li, W.; Gao, Y.; He, W.; et al. Increased nerve density adversely affects outcome in colorectal cancer and denervation suppresses tumor growth. J Transl Med. 2025, 23, 112. [Google Scholar] [CrossRef]
- Perez-Pacheco, C.; Schmitd, L.B.; Furgal, A.; Bellile, E.L.; Liu, M.; Fattah, A.; et al. Increased Nerve Density Adversely Affects Outcome in Oral Cancer. Clinical Cancer Research 2023, 29, 2501–12. [Google Scholar] [CrossRef]
- Ali, S.R.; Jordan, M.; Nagarajan, P.; Amit, M. Nerve Density and Neuronal Biomarkers in Cancer. Cancers 2022, 14, 4817. [Google Scholar] [CrossRef]
- Schmitd, L.B.; Perez-Pacheco, C.; D’Silva, N.J. Nerve density in cancer: Less is better. FASEB BioAdvances 2021, 3, 773–86. [Google Scholar] [CrossRef] [PubMed]
- McCallum, G.A.; Shiralkar, J.; Suciu, D.; Covarrubias, G.; Yu, J.S.; Karathanasis, E.; et al. Chronic neural activity recorded within breast tumors. Sci Rep. 2020, 10, 14824. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; He, Z.; Yin, K.; Li, B.; Zhang, L.; et al. Chronic stress promotes gastric cancer progression and metastasis: an essential role for ADRB2. Cell Death Dis. 2019, 10, 788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Sig Transduct Target Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–52. [Google Scholar] [CrossRef]
- Nevin, J.T.; Moussa, M.; Corwin, W.L.; Mandoiu, I.I.; Srivastava, P.K. Sympathetic nervous tone limits the development of myeloid-derived suppressor cells. Sci Immunol. 2020, 5, eaay9368. [Google Scholar] [CrossRef]
- Brem, S. Vagus nerve stimulation: Novel concept for the treatment of glioblastoma and solid cancers by cytokine (interleukin-6) reduction, attenuating the SASP, enhancing tumor immunity. Brain, Behavior, & Immunity - Health 2024, 42, 100859. [Google Scholar] [CrossRef]
- Muñoz, J.P.; Calaf, G.M. Acetylcholine, Another Factor in Breast Cancer. Biology 2023, 12, 1418. [Google Scholar] [CrossRef]
- Delbro, D.S. Do neuro-humoral signaling molecules participate in colorectal carcinogenesis/cancer progression? Neurogastroenterology & Motility 2012, 24, 96–9. [Google Scholar]
- Zhao, Q.; Yang, Y.; Liang, X.; Du, G.; Liu, L.; Lu, L.; et al. The clinicopathological significance of neurogenesis in breast cancer. BMC Cancer 2014, 14, 484. [Google Scholar] [CrossRef]
- Ben-David, K.; Wittels, H.L.; Wishon, M.J.; Lee, S.J.; McDonald, S.M.; Wittels, Howard S. Tracking Cancer: Exploring Heart Rate Variability Patterns by Cancer Location and Progression. Cancers 2024, 16, 962. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.R.; Jordan, M.; Nagarajan, P.; Amit, M. Nerve density and neuronal biomarkers in cancer. Cancers 2022, 14, 4829. [Google Scholar] [CrossRef] [PubMed]
- Mravec, B.; Dubravicky, J.; Tibensky, M.; Horvathova, L. Effect of the nervous system on cancer: Analysis of clinical studies. BLL 2019, 120, 119–23. [Google Scholar] [CrossRef] [PubMed]
- Schonkeren, S.L.; Thijssen, M.S.; Vaes, N.; Boesmans, W.; Melotte, V. The Emerging Role of Nerves and Glia in Colorectal Cancer. Cancers 2021, 13, 152. [Google Scholar] [CrossRef]
- Abdullahi, A.; Wong, T.W.L.; Ng, S.S.M. Putative role of non-invasive vagus nerve stimulation in cancer pathology and immunotherapy: Can this be a hidden treasure, especially for the elderly? Cancer Medicine 2023, 12, 19081–90. [Google Scholar] [CrossRef]
- Shurin, M.R.; Shurin, G.V.; Zlotnikov, S.B.; Bunimovich, Y.L. The neuroimmune axis in the tumor microenvironment. J Immunol. 2020, 204, 280–95. [Google Scholar] [CrossRef]
- Moreno-Smith, M.; Lutgendorf, S.K.; Sood, A.K. Impact of stress on cancer metastasis. Future Oncol. 2010, 6, 1863–81. [Google Scholar] [CrossRef]
- Sood, A.K.; Bhatty, R.; Kamat, A.A.; Landen, C.N.; Han, L.; Thaker, P.H.; et al. Stress Hormone–Mediated Invasion of Ovarian Cancer Cells. Clinical Cancer Research 2006, 12, 369–75. [Google Scholar] [CrossRef]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006, 12, 939–44. [Google Scholar] [CrossRef]
- Dai, S.; Mo, Y.; Wang, Y.; Xiang, B.; Liao, Q.; Zhou, M.; et al. Chronic stress promotes cancer development. Front Oncol. 2020, 10, 1492. [Google Scholar] [CrossRef]
- Hassan, S.; Karpova, Y.; Baiz, D.; Yancey, D.; Pullikuth, A.; Flores, A.; et al. Behavioral stress accelerates prostate cancer development in mice. J Clin Invest. 2013, 123, 874–86. [Google Scholar] [CrossRef]
- Valles, S.L.; Benlloch, M.; Rodriguez, M.L.; Mena, S.; Pellicer, J.A.; Asensi, M.; et al. Stress hormones promote growth of B16-F10 melanoma metastases: an interleukin 6- and glutathione-dependent mechanism. J Transl Med. 2013, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.W.; Nagaraja, A.S.; Lutgendorf, S.K.; Green, P.A.; Sood, A.K. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer 2015, 15, 563–72. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.W.L.; Kokolus, K.M.; Reed, C.B.; Hylander, B.L.; Ma, W.W.; Repasky, E.A. A nervous tumor microenvironment: the impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol Immunother. 2014, 63, 1115–28. [Google Scholar] [CrossRef]
- Magnon, C. Role of the autonomic nervous system in tumorigenesis and metastasis. Molecular & Cellular Oncology 2015, 2, e975643. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.E.; Dai, H.; Powell, M.; Li, R.; Ding, Y.; Wheeler, T.M.; et al. Cancer-related axonogenesis and neurogenesis in prostate cancer. Clin Cancer Res. 2008, 14, 7593–603. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular pathways: beta-adrenergic signaling in cancer. Clin Cancer Res. 2012, 18, 1201–6. [Google Scholar] [CrossRef]
- Zhao, Y.; Cao, R.; Ma, D.; Zhang, H.; Zhang, L.; Zhang, Y.; et al. Beta-adrenergic signaling on neuroendocrine differentiation, angiogenesis, and metastasis in prostate cancer progression. Asian J Androl. 2019, 21, 253–9. [Google Scholar]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Taskén, K.A. β-adrenergic receptor signaling in prostate cancer. Front Oncol. 2015, 4, 375. [Google Scholar] [CrossRef]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Datta, M.; et al. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell. 2018, 33, 75–90.e7. [Google Scholar] [CrossRef]
- Ferdoushi, A.; Griffin, N.; Marsland, M.; Xu, X.; Faulkner, S.; Gao, F.; et al. Tumor innervation and clinical outcome in pancreatic cancer. Sci Rep. 2021, 11, 7390. [Google Scholar] [CrossRef]
- Oliveira, A.F.; Bretes, L.; Furtado, I. Correlation of beta-2 adrenergic receptor expression in tumor-free surgical margin and at the invasive front of oral squamous cell carcinoma. J Oncol. 2016, 2016, 1651467. [Google Scholar] [CrossRef] [PubMed]
- Makale, M.T.; Kesari, S.; Wrasidlo, W. The autonomic nervous system and cancer. J Drug Target. 2017, 25, 395–403. [Google Scholar] [CrossRef]
- Lüthy, I.A.; Bruzzone, A.; Piñero, C.P.; Castillo, L.F.; Chiesa, I.J.; Vázquez, S.M.; et al. Adrenoceptors: non conventional target for breast cancer? Curr Med Chem. 2009, 16, 1850–62. [Google Scholar] [CrossRef] [PubMed]
- Creed, S.J.; Le, C.P.; Hassan, M.; Pon, C.K.; Albold, S.; Chan, K.T.; et al. β2-adrenoceptor signaling regulates invadopodia formation to enhance tumor cell invasion. Breast Cancer Res. 2015, 17, 145. [Google Scholar] [CrossRef]
- Conceição, F.; Sousa, D.M.; Loessberg-Zahl, J.; Neto, E.; Porcino, C.; Salgado, A.; et al. Sympathetic activity in breast cancer and metastasis: partners in crime. Bone Res. 2021, 9, 9. [Google Scholar] [CrossRef]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Mallavialle, A.; Castagné, R.; Goulard, M.; et al. Neural progenitors from the central nervous system infiltrate prostate tumors to promote development and metastasis. Nat Cancer 2019, 1, 167–80. [Google Scholar]
- Kim-Fuchs, C.; Le, C.P.; Pimentel, M.A.; Shackleford, D.; Ferrari, D.; Angst, E.; et al. Chronic stress accelerates pancreatic cancer growth and invasion. Gut 2014, 63, 1313–22. [Google Scholar]
- Nissen, M.D.; Sloan, E.K.; Mattarollo, S.R. β-adrenergic signaling impairs antitumor CD8+ T-cell responses to B-cell lymphoma immunotherapy. Cancer Immunol Res. 2018, 6, 98–109. [Google Scholar] [CrossRef]
- Kuol, N.; Stojanovska, V.; Apostolopoulos, V.; Nurgali, K. Crosstalk between cancer and the neuro-immune system. J Neuroimmunol. 2018, 315, 15–23. [Google Scholar] [CrossRef]
- Yu, H.; Xia, H.; Tang, Q.; Xu, H.; Wei, G.; Chen, Y.; et al. Acetylcholine acts through M3 muscarinic receptor to activate the EGFR signaling and promotes gastric cancer cell proliferation. Sci Rep. 2017, 7, 40802. [Google Scholar] [CrossRef] [PubMed]
- Schledwitz, A.; Xie, G.; Raufman, J.P. Differential actions of muscarinic receptor subtypes in gastric, pancreatic, and colon cancer. Int J Mol Sci. 2021, 22, 13153. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Samimi, R.; Xie, G.; Shant, J.; Drachenberg, C.; Wade, M.; et al. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am J Physiol Gastrointest Liver Physiol. 2008, 295, G591–597. [Google Scholar] [CrossRef] [PubMed]
- Renz, B.W.; Tanaka, T.; Sunagawa, M.; Takahashi, R.; Jiang, Z.; Macchini, M.; et al. Cholinergic signaling via muscarinic receptors directly and indirectly suppresses pancreatic tumorigenesis and cancer stemness. Cancer Discov. 2018, 8, 1458–73. [Google Scholar] [CrossRef]
- Partecke, L.I.; Käding, A.; Trung, D.N.; Diedrich, S.; Sendler, M.; Weiss, F.; et al. Subdiaphragmatic vagotomy promotes tumor growth and reduces survival via TNFα in a murine pancreatic cancer model. Oncotarget 2017, 8, 22501–12. [Google Scholar] [CrossRef]
- Huang WB, Lai H zhou, Long J, Ma Q, Fu X, You FM; et al. Vagal nerve activity and cancer prognosis: a systematic review and meta-analysis. BMC Cancer 2025, 25, 579. [Google Scholar] [CrossRef]
- Erin, N.; Barkan, T.; Akcan, G.; Yorukoglu, K. Activation of vagus nerve by semapimod alters substance P levels and decreases breast cancer metastasis. Regul Pept. 2012, 179(1-3), 101–8. [Google Scholar] [CrossRef]
- Kazi, R.; Bunimovich, Y.L. The nervous system: a new target in the fight against cancer. Anti-Cancer Drugs 2018, 29, 929. [Google Scholar] [CrossRef]
- Fæstad, A.A.; Galván, J.A.; Schürch, C.M.; Seifert, B.; Zlobec, I.; Krebs, P.; et al. Blockade of beta-adrenergic receptors reduces cancer growth and enhances the response to anti-CTLA4 therapy by modulating the tumor microenvironment. Oncogene 2022, 41, 1364–75. [Google Scholar] [CrossRef]
- Hering, D.; Winklewski, P.J.; Gruszecka, A.; Rudzinska, M.; Kucharska, A.; Markuszewski, L.; et al. Blockage of cholinergic signaling via muscarinic acetylcholine receptor 3 inhibits tumor growth in human colorectal adenocarcinoma. Int J Mol Sci. 2021, 22, 12358. [Google Scholar] [CrossRef]
- Coelho, M.; Moz, M.; Correia, G.; Teixeira, A.; Medeiros, R.; Ribeiro, L. β-adrenergic modulation of cancer cell proliferation: available evidence and clinical perspectives. J Cancer Res Clin Oncol. 2017, 143, 275–91. [Google Scholar] [CrossRef] [PubMed]
- Kumaria, A.; Ashkan, K.; Powell, M.P.; Solo, A.; Hanrahan, J.G.; Bhangoo, R.; et al. Neuromodulation as an anticancer strategy. Front Oncol. 2023, 13, 1264619. [Google Scholar]
- Wu, J.; Lu, X.; Yan, C. Neuro-immune-cancer interactions: Mechanisms and therapeutic implications for tumor modulation. Brain Behavior and Immunity Integrative 2025, 10, 100119. [Google Scholar] [CrossRef]
- Saloman, J.L.; Albers, K.M.; Rhim, A.D.; Davis, B.M. Can stopping nerves, stop cancer? Trends Neurosci. 2016, 39, 880–9. [Google Scholar] [CrossRef]
- Mitsou, G.; Burke, O.; Ashok, D.; Hewage, S.; Jobling, P.; Chuah, P.; et al. Radical tumor denervation activates potent local and global cancer treatment. Cancers 2023, 9, eadi8236. [Google Scholar]
- Piegeler, T.; Votta-Velis, E.G.; Bakhshi, F.R.; Mao, M.; Carnegie, G.; Bonini, M.G.; et al. Do amide local anesthetics play a therapeutic role in the perioperative management of cancer patients? Int Anesth Res Soc. 2016, 122, 1994–2004. [Google Scholar]
- Xie, M.; Huang, Y.; Lin, X.; Yang, L.; Fan, Q.; Wang, Z.; et al. Regional anesthesia might reduce recurrence and metastasis rates in adult patients with cancers after surgery: a meta-analysis. BMC Anesthesiol. 2024, 24, 124. [Google Scholar] [CrossRef]
- Nunez-Rodriguez, S.; Dono, A.; Bonnin, D.; Nunez-Castilla, J.; Sheth, S.A.; Esquenazi, Y. Local anesthetics, regional anesthesia and cancer biology. J Neurooncol 2025, 166, 209–23. [Google Scholar]
- Cassinello, F.; Prieto, I.; del Olmo, M.; Rivas, S.; Strichartz, G.R. Cancer surgery: how may anesthesia influence outcome? J Clin Anesth. 2015, 27, 262–72. [Google Scholar]
- Lynch, C. Local Anesthetics as…Cancer Therapy? Anesthesia & Analgesia 2018, 127, 601–2. [Google Scholar] [CrossRef]
- Krömer, A.; Farag, M.; Birkenhauer, U.; Stadler, A.; Mueller, J.E.; Zeman, F.; et al. Local anesthetics affect tumor biology in an ex vivo tissue model of non-small cell lung cancer. Cancers 2022, 14, 5594. [Google Scholar]
- Zhang, Y.; Jing, Y.; Pan, R.; Ding, K.; Chen, R.; Meng, Q. Mechanisms of Cancer Inhibition by Local Anesthetics. Front Pharmacol 2021. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Kepp, O.; Kroemer, G. Immunogenic stress induced by local anesthetics injected into neoplastic lesions. Oncoimmunology 2022, 11, 2043672. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Kepp, O.; Kroemer, G. Local anesthetics elicit immune-dependent anticancer effects. J Immunother Cancer 2022, 10, e004607. [Google Scholar] [CrossRef]
- Bezu, L.; Kepp, O.; Kroemer, G. Impact of local anesthetics on epigenetics in cancer. Int J Mol Sci. 2022, 23, 15583. [Google Scholar] [CrossRef]
- Zhou, H.; Shi, B.; Jia, Y.; Qiu, G.; Yang, W.; Li, J.; et al. Expression and significance of autonomic nerves and α9 nicotinic acetylcholine receptor in colorectal cancer. Molecular Medicine Reports 2018, 17, 8423–31. [Google Scholar] [CrossRef]
- Li, W.; Zhang, J.; Gao, Y.; Kong, X.; Sun, X. Nervous system in hepatocellular carcinoma: correlation, mechanisms, therapeutic implications, and future perspectives. BBA Rev Cancer 2025, 1880, 189345. [Google Scholar] [CrossRef]
- Abdelaatti, A.; Buggy, D.J.; Wall, T.P. Local anaesthetics and chemotherapeutic agents: a systematic review of preclinical evidence of interactions and cancer biology. BJA Open. 2024, 10, 100284. [Google Scholar] [CrossRef]
- Alexa, A.L.; Ciocan, A.; Zaharie, F.; Valean, D.; Sargarovschi, S.; Breazu, C.; Al Hajjar, N.; Ionescu, D. The Influence of Intravenous Lidocaine Infusion on Postoperative Outcome and Neutrophil-to-Lymphocyte Ratio in Colorectal Cancer Patients. A Pilot Study. JGLD [Internet] cited. 2023, 32, 156–61. [Google Scholar]
Figure 1.
In the tumor microenvironment, NGF promotes sympathetic fiber growth, which releases norepinephrine, enhancing tumor proliferation, angiogenesis, and invasiveness. This neurotrophic-autonomic loop supports cancer progression.
Figure 1.
In the tumor microenvironment, NGF promotes sympathetic fiber growth, which releases norepinephrine, enhancing tumor proliferation, angiogenesis, and invasiveness. This neurotrophic-autonomic loop supports cancer progression.
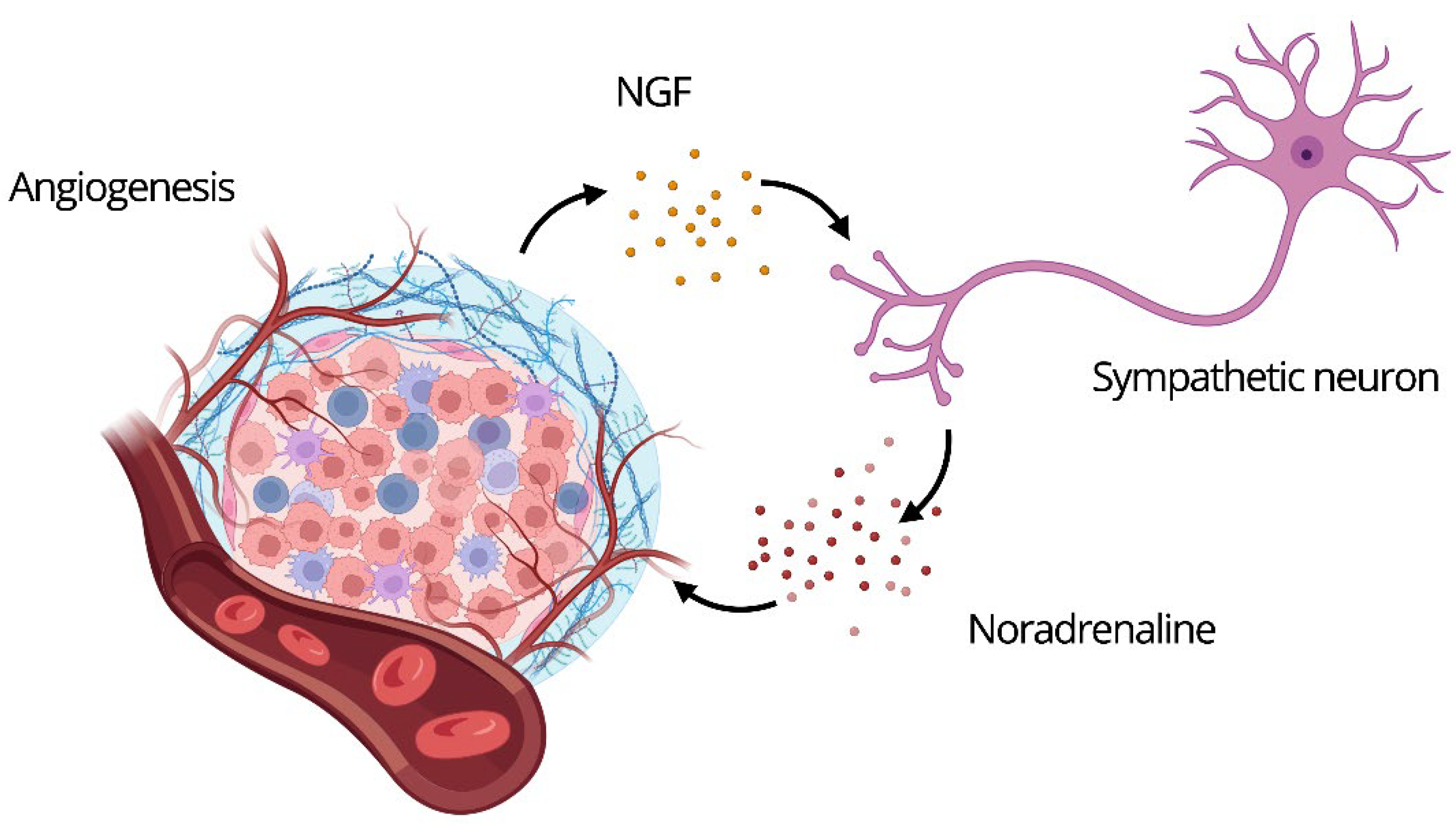
Figure 2.
The interplay between the nervous system and cancer: neural input shapes tumor development by directly stimulating cancer cells and promoting their survival.
Figure 2.
The interplay between the nervous system and cancer: neural input shapes tumor development by directly stimulating cancer cells and promoting their survival.
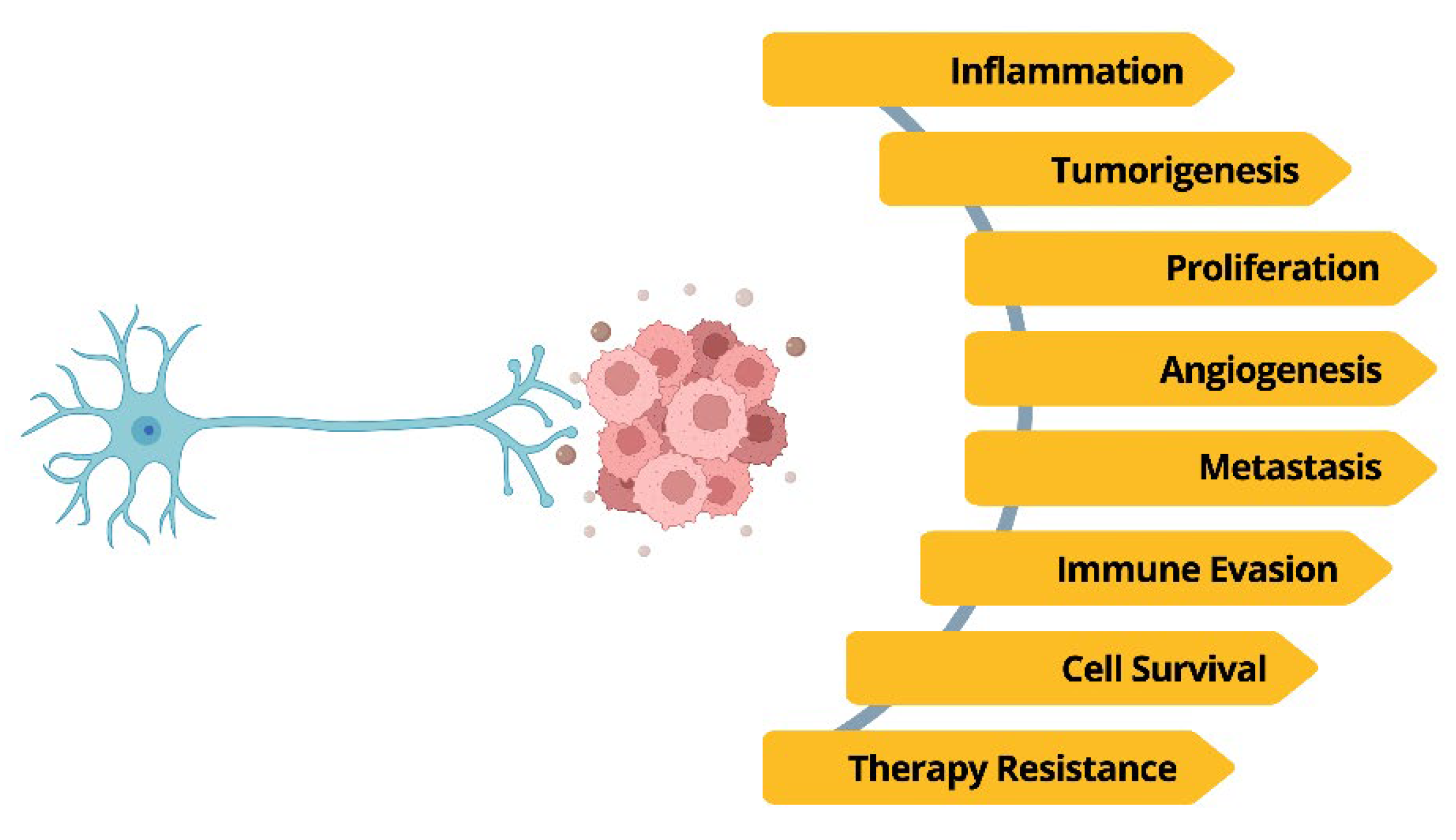
Figure 3.
Organ-specific effects of the Autonomic Nervous System in cancer.
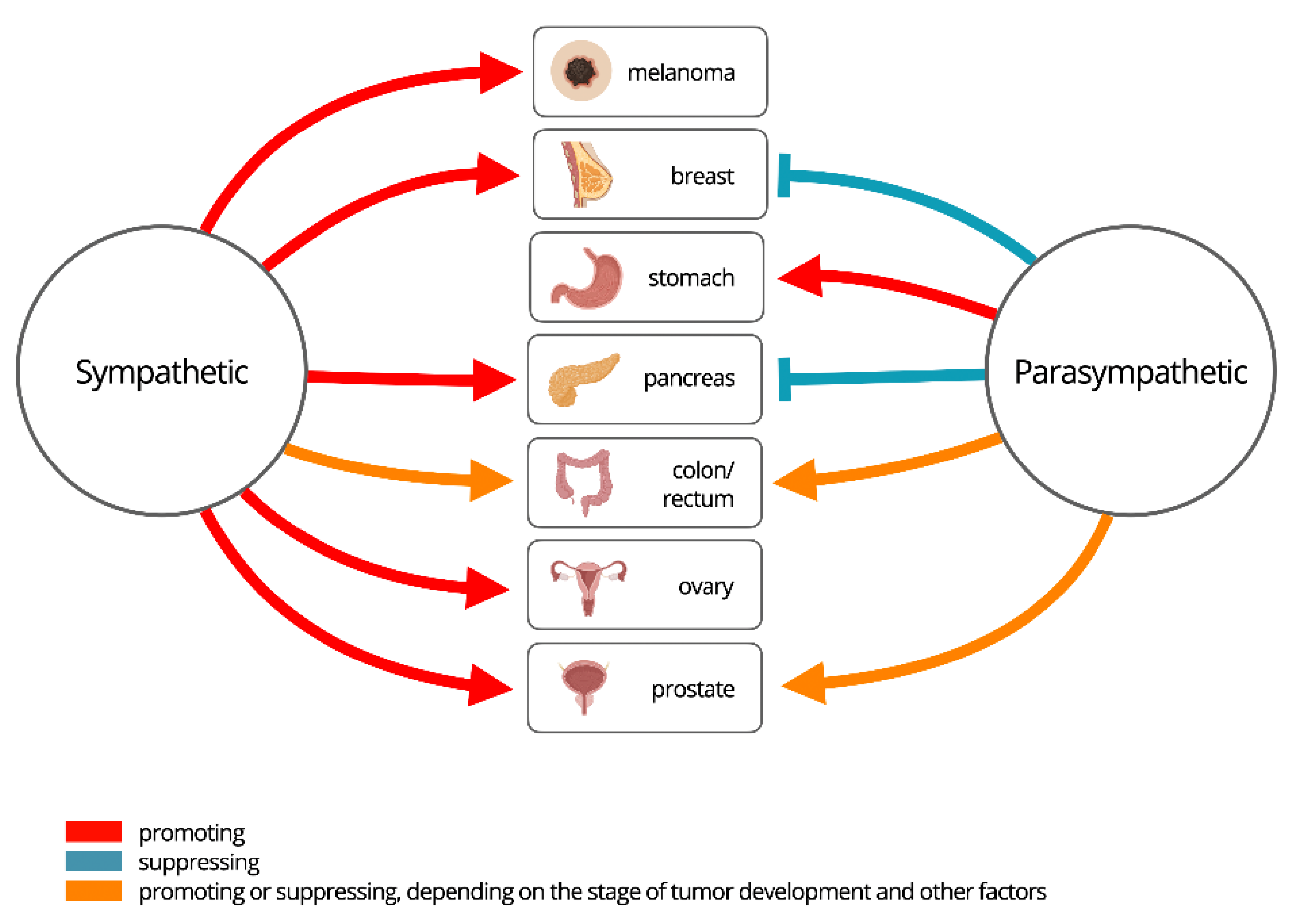
Table 1.
Comparison between traditional and emerging views on ANS roles in oncogenesis.
| Topic | Traditional View | New Evidence |
|---|---|---|
| Role of the ANS in oncogenesis | The ANS was considered irrelevant to oncogenesis; cancer was thought to result solely from genetic mutations and environmental factors. | Modern techniques reveal dense sympathetic and parasympathetic innervation in various tumors, influencing tumor initiation and progression.[25,52,29,33,17,53,54] |
| Role of nerve fibers in tumors | Nerve fibers were seen as passive targets of tumor invasion (perineural invasion). | PNI is now recognized as an active mechanism of tumor dissemination, with nerve-derived signals enhancing cancer cell migration.[55,25,52,29,33] |
| Neurogenesis in the tumor context | Neurogenesis was thought to occur exclusively in the CNS and was unrelated to tumors. | Tumor stem cells can differentiate into functional neurons and contribute to neoneurogenesis within the tumor microenvironment.[12] |
| Origin of tumor-associated nerve fibers | Nerve fibers in tumors were believed to originate only from adjacent tissues. | Human CSCs produce functional autonomic neurons that integrate into the tumor stroma and modulate cancer progression.[12] |
| Impact of the ANS on antitumor immunity | Immunity was believed to be regulated exclusively by the immune system, with no neuronal contribution. | The vagus nerve modulates immune response via cholinergic anti-inflammatory pathways; adrenergic signaling affects myeloid-derived cells.[55,29,33,46,56] |
| Neurotransmitters and cancer | Neurotransmitters were viewed solely as synaptic messengers with no role in cancer biology. | Dopamine, GABA, glutamate, and serotonin directly influence tumor proliferation, immune evasion, and vascular remodeling.[7] |
| Psycho-emotional influence on cancer | Stress and emotional states were considered psychosocial factors without biological relevance in cancer progression. | Chronic stress activates the sympathetic system, increasing norepinephrine and promoting tumor growth and metastasis.[43,57,58,59,60,61,35,62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



