Submitted:
09 December 2025
Posted:
14 December 2025
You are already at the latest version
Abstract
Neuromuscular diseases are biologically diverse, clinically heterogeneous, and often difficult to diagnose and treat, creating an urgent need for computational tools that can resolve overlapping phenotypes and enable timely, mechanism-based therapeutics. This narrative review synthesizes recent advances in artificial intelligence as applied to neuromuscular diseases, drawing from peer-reviewed literature from the past five years. Artificial intelligence can augment diagnosis by extracting disease-relevant patterns from imaging, electrophysiology, and multimodal clinical data, improving discrimination between clinically similar entities such as Duchenne and Becker muscular dystrophy. Artificial intelligence can also enhance early detection of amyotrophic lateral sclerosis. Artificial intelligence can utilize digital biomarkers of disease progression data from gait, voice, and wearable sensors for enhanced modeling of disease outcomes. Deep learning–based multi-omics integration, high-throughput phenotypic screening, and artificial intelligence-based protein structure predictive models are accelerating the path from causative mutation, to molecular mechanism, and on to candidate therapy. Despite these advances, significant challenges remain, including data scarcity, heterogeneous acquisition methods, limited external validation, and the need for interpretable models that can win clinician acceptance. Addressing these constraints is essential to moving high-performance research tools from the laboratory to the neuromuscular clinic. Artificial intelligence has the potential to shorten the diagnostic odyssey and accelerate the historically slow development of targeted therapeutics for rare neuromuscular diseases.
Keywords:
artificial intelligence
; neuromuscular diseases
; machine learning
; rare diseases
; neurogenetic diseases
; diagnosis
; prognosis
; digital biomarkers
; multi-omics
1. Introduction: Computational Imperatives for Neuromuscular Diseases
Neuromuscular diseases represent a diverse and biologically complex group of neurological disorders [1,2] that primarily affect the motor neuron, peripheral nerve, neuromuscular junction, or muscle (Table 1). A subset of neuromuscular diseases, particularly immune-mediated conditions such as myasthenia gravis and chronic inflammatory demyelinating polyneuropathy, has already benefited from advances in immunotherapy developed largely through traditional drug discovery and trial methods that did not rely on artificial intelligence [3,4,5,6]. In contrast, many predominantly neurogenetic neuromuscular diseases, including amyotrophic lateral sclerosis, the muscular dystrophies, and Charcot–Marie–Tooth disease, remain difficult to diagnose promptly and to treat effectively. This narrative review focuses on these difficult-to-diagnose and difficult-to-treat neuromuscular diseases, with particular emphasis on how advances in artificial intelligence and machine learning can expedite clinical diagnosis and accelerate the discovery and optimization of mechanism-based therapies.
Most neuromuscular diseases are uncommon, with prevalences well below 1% in the general population. Two notable exceptions—carpal tunnel syndrome and diabetic length-dependent polyneuropathy—affect more than 1% of adults and are among the most common neurological conditions [7,8]. Most neuromuscular diseases are neurogenetic in origin, although some have autoimmune, metabolic, toxic, traumatic, and nutritional etiologies (Table 1).
Despite their relative rarity, neuromuscular diseases account for a disproportionately large share of rare and genetic disorders. OrphaData [9] lists 6,171 rare diseases, of which approximately 522 (8.5%) fall within the neuromuscular domain. The genetic landscape is similarly broad: Online Mendelian Inheritance in Man [10] catalogs 9,326 phenotypes with confirmed or suspected genetic etiologies, and Manjunath et al. [11] identified 747 neuromuscular disease–associated genes and 1,240 neuromuscular phenotypes, suggesting that roughly 12% of Online Mendelian Inheritance in Man phenotypes are neuromuscular. Because pathogenic variants in a single gene may produce multiple clinically distinct diseases or marked phenotypic variability within a single disorder, the true phenotypic space is larger than gene counts alone imply. For example, pathogenic variants in TBK1 may manifest as amyotrophic lateral sclerosis, frontotemporal dementia, or a mixed amyotrophic lateral sclerosis–frontotemporal dementia syndrome, whereas pathogenic variants in PMP22 may cause Charcot–Marie–Tooth disease type 1A (CMT1A) or hereditary neuropathy with liability to pressure palsies (HNPP).
Online databases demonstrate the phenotypic complexity of neuromuscular diseases. The Online Mendelian Inheritance in Man [10] emphasizes phenotypes at the disease level, while the Human Phenotype Ontology [12] encodes granular phenotypic features such as weakness, sensory loss, or spasticity. In clinical practice, both levels matter: clinicians reason in terms of disease entities , yet diagnosis is based on the pattern of individual signs, symptoms, imaging abnormalities, and electrophysiological findings that those entities produce.
Charcot–Marie–Tooth disease illustrates the diagnostic complexity that arises from this multilayered structure (Table 2). Although most Charcot-Marie Tooth disease variants share canonical features such as distal weakness, sensory loss, hyporeflexia, and muscle atrophy, nline Mendelian Inheritance in Man lists more than 100 phenotypically distinct Charcot-Marie Tooth disease entities, including CMT1, CMT2, CMT4, and X-linked CMT. More than 120 genes have been implicated in hereditary neuropathies, including PMP22, MPZ, GJB1, MFN2, NEFL, SH3TC2, and GDAP1. Even single genes may produce multiple phenotypes. For example, NEFL, which encodes the neurofilament light chain protein, is associated with at least three Charcot-Marie-Tooth disease phenotypes (CMT1F, CMT2E, and dominant intermediate CMT). ClinVar [13] lists 786 NEFL variants, 28 of which are reported as associated with Charcot-Marie-Tooth disease, spanning missense, nonsense, frameshift, splice-site, regulatory, and structural alterations. Each variant has the potential to produce distinct effects on neurofilament assembly, axonal transport, protein stability, and protein–protein interactions.
These layers of complexity—multiple diseases per gene, multiple genes per phenotype, and many variants per gene—make accurate diagnosis of neuromuscular diseases intrinsically difficult [14,15,16]. A single disorder may present with strikingly different clinical features, while the same signs or symptoms may arise from dozens of distinct conditions. This crowded phenotypic space yields many plausible explanations for any given presentation and contributes to prolonged diagnostic delays—the so-called “diagnostic odyssey”—that are well documented in neuromuscular disease [17,18].
Until recently, these challenges in diagnosing neuromuscular diseases were matched by parallel challenges in developing effective therapies. Many neuromuscular diseases were considered incurable, and the field was characterized by considerable therapeutic pessimism. For amyotrophic lateral sclerosis alone, more than fifty compounds that showed promise in preclinical models ultimately failed in human trials [19,20,21]. This long-standing gulf—between expanding biological insights and persistent therapeutic failures—reinforced the perception that most neuromuscular diseases were untreatable.
This landscape is now changing rapidly. The emergence of targeted therapies—including antisense oligonucleotides, gene replacement therapies, molecular chaperones, and precision small molecules—is transforming previously untreatable disorders into conditions with potential treatment pathways [22]. These advances demand computationally informed diagnoses that match the precision of emerging therapeutic strategies.
Together, these developments create three imperatives for neuromuscular medicine:
- Computational tools for diagnosis that overcome heterogeneous presentations, integrate multimodal data, resolve phenotypic overlap, and shorten the time to accurate diagnosis.
- Computational tools for disease progression and outcome that measure disease progression more precisely, automate these measurements, and better model clinical outcomes.
- Computational tools for therapeutics that improve patient stratification for clinical trials, connect gene mutations to protein structure and function, predict phenotype from altered protein structure and function, reduce failure rates in clinical trials, and critically assist in drug discovery and design.
Artificial intelligence—including deep learning and large language models—is uniquely positioned to address these computational imperatives (Figure 1). Modern neuromuscular practice generates vast, high-dimensional datasets across modalities that include imaging, electrophysiology, genomics, proteomics, and clinical observation. Traditional statistical approaches can struggle with the nonlinear, longitudinal, and multimodal complexity of these data. In contrast, contemporary artificial intelligence methods integrate heterogeneous data streams and create insightful simulations and models [23]. As a result, artificial intelligence is emerging as an enabling technology that provides computational capabilities that can jumpstart rapid improvements in the diagnosis and treatment of rare neuromuscular diseases [24,25].
2. Methods
This narrative review summarizes recent work in computational neuromuscular medicine, with emphasis on how artificial intelligence is accelerating advances in diagnosis and treatment. We identified primary sources through structured searches of PubMed and Google Scholar, focusing on peer-reviewed studies that examined artificial intelligence or machine learning approaches to diagnosis, disease progression and outcome prediction, or therapeutics in human neuromuscular diseases. We included English-language articles published between 2019 and 2025 and excluded studies restricted to animal models or purely methodological machine-learning work without a direct neuromuscular application. When specific subtopics required more detailed coverage, we performed additional targeted keyword searches and screened the reference lists of key articles. All included papers were reviewed manually for relevance and quality by the multidisciplinary author team, which has expertise in both clinical neurology and artificial intelligence.
3. Artificial Intelligence and Machine Learning for Diagnosis of Rare Neuromuscular Diseases
Artificial intelligence and machine learning are increasingly used to augment neuromuscular disease diagnosis, particularly where disorders occupy a very narrow “diagnostic space” and are difficult to distinguish clinically. By extracting subtle patterns from imaging, electrophysiology, and other modalities, artificial intelligence can improve accuracy and sharpen differentiation between closely overlapping neuromuscular disease subtypes, with particular impact on rare and ultra-rare entities where clinical experience is limited [2,17,18].
3.1. Advanced Imaging and Radiomics
Artificial intelligence-based medical image analysis is transforming the diagnostic evaluation of myopathies and muscular dystrophies. Ultrasound imaging—non-invasive, dynamic, and repeatable—has increasingly been paired with artificial intelligence to support clinical assessment [2]. For example, a lightweight deep learning model, NMD-AssistNet, achieved an accuracy of 0.9502 and an AUC of 0.9776 for automated classification of neuromuscular diseases from muscle ultrasound images [2]. Saliency analyses showed that the model concentrated on clinically meaningful muscle regions, supporting both its face validity and interpretability.
Beyond ultrasound, artificial intelligence enables high-throughput extraction of quantitative features from MRI (radiomics). Machine learning algorithms applied to thigh Dixon-sequence MRI radiomics have shown strong performance in differentiating phenotypically similar neuromuscular diseases in early stages—specifically Duchenne muscular dystrophy and Becker muscular dystrophy [26]. In a cohort of 62 patients, radiomic models using logistic regression, k-nearest neighbors, support vector machines, multilayer perceptrons, and random forests achieved 81.2–90.6% accuracy (versus 69.4% for radiologists), 71.0–86.0% specificity (versus 19.0%), and F1 scores of 85.2–92.6%, while maintaining high sensitivity (85.6–95.0% versus 95.1%) [26].
Importantly, the clinical value of these models is not limited to early detection of abnormal muscle. Their major contribution lies in improving differential diagnosis. In this example, machine learning models markedly increased specificity for Becker muscular dystrophy, reducing misclassification of Becker muscular dystrophy as Duchenne muscular dystrophy—a distinction that carries major implications for prognosis, clinical trial eligibility, and therapeutic planning. These results illustrate how artificial intelligence-enhanced imaging enables more precise phenotypic discrimination and supports earlier, more accurate diagnostic decision-making.
3.2. Electrophysiology and Signal-Based Diagnosis
Electrodiagnostic testing—including nerve conduction studies and electromyography—remains central to neuromuscular diagnosis, but interpretation requires specialized expertise and is subject to inter-rater variability. artificial intelligence and machine learning approaches have been applied to both conventional summary metrics (for example, motor unit duration, amplitude, and recruitment pattern) and raw waveforms, with the goal of automating pattern recognition and supporting non-expert clinicians [25,27].
Early work focused on handcrafted time- and frequency-domain features from needle electromyography or surface electromyography and utilized classical machine learning classifiers. Using features extracted from multi-muscle electromyography recordings and time–frequency transforms such as the Hilbert–Huang transform, Torres-Castillo et al. [28] demonstrated accurate discrimination among normal controls and several neuromuscular disease categories, illustrating the feasibility of automated electromyography-based screening in heterogeneous clinical populations [28]. More recent studies employ deep learning architectures that operate directly on raw or minimally processed electromyography signals, obviating manual feature extraction and capturing subtle waveform characteristics that may be difficult for human readers to appreciate [27]. Diagnostic performance was high in research cohorts. Across studies summarized by Piñeros-Fernández [25] and by Taha and Morren [27], machine learning models trained on electrodiagnostic data achieve accuracies ranging from approximately 67% to 99% on some classification tasks, including the differentiation of amyotrophic lateral sclerosis from other motor neuron and myopathic disorders and discriminating between myopathic and neuropathic patterns [17,27]. In several series, model performance matched or exceeded that of experienced electromyographers, particularly for narrowly defined binary or ternary classification problems.
Beyond categorical diagnosis, artificial intelligence has been used to quantify disease severity and track progression based on electromyography data. Regression models linking motor unit parameters or interference patterns to clinical scales, as well as unsupervised approaches that cluster electromyography recordings into latent disease states, have shown promise for creating more objective electrodiagnostic biomarkers [27]. However, most current electromyography-based artificial intelligence systems are confined to single-center datasets. Data is often collected under tightly controlled acquisition protocols, and validation across different laboratories, machines, and operator techniques is limited. Standardization of acquisition and annotation, together with prospective evaluation in routine electrodiagnostic practice, will be necessary before these tools can reliably be implemented in routine clinical settings.
3.3. Integration of Multimodal Clinical Data
A major advantage of artificial intelligence is its ability to integrate heterogeneous data into unified predictive models. Deep learning has been applied successfully in highly complex settings—for example, classifying pediatric intensive care patients with neuromuscular diseases using combinations of clinical variables, laboratory results, and neurophysiologic measurements [29]. In amyotrophic lateral sclerosis and other rare neuromuscular diseases, multimodal data fusion models that combine clinical rating scales, electrophysiology, imaging-derived features, and genetic markers have demonstrated superior diagnostic accuracy compared to single-modality approaches [23]. These artificial intelligence-augmented methods shift reliance from isolated biomarkers towards integrated, high-resolution phenotypic profiles that represent the full biological and clinical heterogeneity of neuromuscular diseases.
3.4. Phenotype-Driven and Database-Supported Diagnosis
The expansion of structured phenotype resources such as Online Mendelian Inheritance in Man, Orphadata, and the Human Phenotype Ontology has enabled phenotype-driven diagnostic tools that are particularly well suited to rare neuromuscular diseases [9,10,12]. In contrast to traditional “gene-first” approaches, these systems start from the patient’s clinical presentation: signs, symptoms, imaging findings, and ancillary test results are mapped to standardized Human Phenotype Ontology terms and compared against curated gene–phenotype and disease–phenotype profiles. The output is a ranked list of candidate genes or diagnoses that are most compatible with the observed phenotype, guiding targeted genetic testing and interpretation.
Artificial intelligence and machine learning methods substantially enhance this phenotype-driven workflow. Natural language processing models can extract neuromuscular-relevant features from unstructured clinical notes and convert them into Human Phenotype Ontology concepts with high accuracy, reducing the burden of manual encoding and capturing additional detail from longitudinal records [17,30,31,32,33]. Machine-learning algorithms applied to Online Mendelian Inheritance in Man and Orphadata have been used to construct gene–phenotype co-occurrence networks and cluster neuromuscular diseases into phenotypically coherent groups, revealing informative symptom clusters that may not be obvious from single-database queries [34]. These clusters can improve differential diagnosis by highlighting combinations of features that strongly point toward particular genetic etiologies (for example, specific constellations of distal weakness, contractures, cardiomyopathy, and respiratory involvement in muscular dystrophies) [34,35]. Machine learning approaches can help disambiguate and resolve overlapping disease phenotypes by improved visualization of phenotypic features [36].
Diagnostic decision-support systems for rare diseases increasingly integrate these resources. Phenotype-driven gene-prioritization tools such as Phen2Gene and related knowledge-graph frameworks take Human Phenotype Ontology-encoded patient data and return ranked candidate genes, often outperforming manual literature search or naive Online Mendelian Inheritance in Man look-up in both speed and completeness [37,38] In neuromuscular clinics, such tools can be layered onto exome or genome sequencing pipelines to help resolve cases with multiple variants of uncertain significance by identifying the subset of variants that best explains the observed neuromuscular phenotype [39].
Despite these advances, clinical deployment remains limited. Many phenotype-driven systems are research prototypes, and their performance depends heavily on the completeness and accuracy of both the underlying databases and the clinical phenotype encoding [18,40,41,42]. Neuromuscular phenotypes are often underrepresented or described at varying levels of granularity across sources, and complex patients may have overlapping central and peripheral nervous system features that challenge current algorithms. Moving from proof-of-concept tools to robust clinical decision support will require continued curation of neuromuscular gene–phenotype relationships, better harmonization of ontologies, and prospective evaluation within real-world diagnostic workflows [43,44,45,46,47,48].
3.5. Limitations and Gaps in Applying artificial intelligence to Diagnosis of Neuromuscular Diseases
Despite impressive performance in research settings, diagnostic artificial intelligence for rare neuromuscular diseases faces several limitations. Data are often fragmented across institutions, sample sizes are small, and case mixes are biased toward tertiary centers. Imaging protocols and electromyography acquisition methods are heterogeneous. External validation is uncommon, and few models have been utilized in real-world clinical workflows with prospective evaluation. For rare diseases, where every misclassification may have outsized consequences for a small patient population, robust calibration, explainability, and human oversight are essential [17,18,23].
4. Artificial Intelligence and Machine Learning for Disease Progression and Outcome Prediction
Accurately measuring disease progression and forecasting disease outcomes is central to counseling patients, designing clinical trials, and allocating resources in neuromuscular medicine. For rare neuromuscular diseases, where natural history data are sparse and heterogeneous, artificial intelligence-based progression models and digital biomarkers can be especially valuable.
4.1. Prognostic Modeling in Amyotrophic Lateral Sclerosis and Other Rare Meuromuscular diseases
In amyotrophic lateral sclerosis, deep learning models have demonstrated superior predictive performance using large, harmonized datasets such as PRO-ACT [49]. Optimized models—most notably extreme gradient boosting—achieved a sensitivity of 100%, specificity of 97.44%, accuracy of 97.96%, F1-score of 95.96%, Matthews correlation coefficient of 94.12%, and an AUC of 0.9550 in distinguishing bulbar-onset from limb-onset amyotrophic lateral sclerosis [49]. Tree-based ensemble methods iteratively correct residual errors and are well-suited to capturing nonlinear relationships in heterogeneous clinical data.
Similar approaches have been applied to survival prediction, respiratory decline, and loss of ambulation in other rare neuromuscular diseases, using combinations of clinical scales, pulmonary function tests, and laboratory biomarkers. These models can be used to generate individualized risk estimates, stratify patients into prognostic subgroups, and simulate counterfactual trajectories under different treatment assumptions [50,51].
4.2. Digital Biomarkers and Remote Monitoring
Wearable sensors, smartphone-based assessments, and home-based devices can capture high-frequency, ecologically valid signals—gait, balance, voice, facial expression, and limb movement—that reflect neuromuscular function more sensitively than episodic clinic visits. In several rare neuromuscular diseases, artificial intelligence models applied to these signals have begun to yield candidate “digital biomarkers” that correlate with, and in some cases outperform, traditional rating scales.
Gait analysis is a prominent example. In dystrophinopathies and spinal muscular atrophy, inertial measurement units (IMUs) placed on the lower limbs or trunk, combined with computer-vision–based pose estimation from video, have been used to extract stride length, cadence, joint angles, and symmetry indices [52,53]. Supervised learning models trained on these features can distinguish affected individuals from controls and differentiate between ambulatory stages. Longitudinal analyses can detect subtle changes in gait that precede clinically apparent decline. Because these measures can be collected at home, they offer a more granular view of day-to-day variability than clinic-based six-minute walk tests or timed function tests.
Voice and speech are similarly rich sources of digital biomarkers, particularly in amyotrophic lateral sclerosis and bulbar-onset motor neuron disease. Deep learning models operating on acoustic features (for example, jitter, shimmer, formant trajectories, and spectral envelopes) or on raw audio waveforms have been shown to classify amyotrophic lateral sclerosis versus controls, estimate disease severity, and track progression over time [54,55,56].In some studies, voice-based models detect early bulbar involvement before large changes in Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) scores, suggesting a role in pre-symptomatic or prodromal monitoring in genetically at-risk individuals.
Beyond gait and voice, actigraphy and multi-sensor wearables have been used to quantify overall activity levels, sleep–wake patterns, and limb use asymmetries in hereditary neuropathies and inflammatory myopathies. Machine learning models applied to these longitudinal time series can segment daily routines into activity states, detect deviations associated with exacerbations or treatment responses, and produce composite progression scores that correlate with physician-rated scales. Because these data streams are dense and noisy, artificial intelligence methods are particularly important for feature extraction, anomaly detection, and robust modeling of within-subject trajectories [57,58,59,60,61,62].
For rare neuromuscular diseases, digital biomarkers have several advantages: they reduce the need for frequent in-person visits to specialized centers, increase the effective sample size by capturing more data points per patient, and can support decentralized or hybrid clinical trial designs. However, challenges remain, including device heterogeneity, variable patient adherence, uncertainty about the minimal clinically important difference for digital endpoints, and limited regulatory experience with artificial intelligence-derived biomarkers. Carefully designed validation studies that link digital measures to established clinical and functional outcomes will be necessary before these tools can be widely adopted as primary or surrogate endpoints in rare neuromuscular disease trials [54,63].
4.3. Subtyping, Deep Phenotyping, and Latent Trajectories
Unsupervised learning and clustering methods have been used to identify latent subtypes within clinically defined neuromuscular diseases, revealing groups with distinctive progression profiles and treatment responses. In amyotrophic lateral sclerosis, for example, clustering of longitudinal clinical, imaging, and biomarker data has uncovered subgroups with slower and fast progression, subgroups with bulbar and spinal predominance, and subgroups with cortical and spinal involvement [64]. In muscular dystrophies and hereditary neuropathies, similar approaches may identify patients who are most likely to benefit from specific therapies or who require alternative trial designs.
4.4. Methodological and Practical Challenges
Prognostic artificial intelligence models for rare neuromuscular diseases face several methodological and practical constraints, including small sample sizes, sampling bias, limited generalizability across sites and populations, heterogeneous follow-up intervals, and missing data [18,65]. Models developed from enriched clinical trial cohorts may fail to represent community-based populations. Moreover, the regulatory acceptance of artificial intelligence-derived surrogate endpoints remains limited, underscoring the need for rigorous validation against clinically meaningful outcomes [66,67]. Despite these challenges, the integration of digital phenotyping, multi-omics data, and advanced machine learning architectures offers substantial promise for improving the measurement of disease progression and enhancing the accuracy of prognostic modeling.
5. Artificial Intelligence and Machine Learning for Therapeutics in Neuromuscular Diseases
On the therapeutic side, artificial intelligence and machine learning are increasingly being applied to accelerate new drug discovery, drug repurposing, clinical trial design, and the development of gene- and cell-based therapies for rare neuromuscular diseases. These approaches aim to shorten the path from mutation to mechanism to treatment and to mitigate the historically high failure rates that have characterized neuromuscular drug development [19,20,21,22].
5.1. Target Discovery and Drug Repurposing
Network-based and multi-omics approaches can integrate gene expression, proteomics, protein–protein interaction networks, and clinical phenotypes to identify candidate therapeutic targets and repurposable drugs [68,69]. In amyotrophic lateral sclerosis, for example, artificial intelligence-driven analyses have highlighted pathways related to RNA metabolism, proteostasis, and neuroinflammation, and have suggested existing compounds that modulate these pathways as candidates for rapid repurposing [69,70,71]. In muscular dystrophies and hereditary neuropathies, similar pipelines have been used to prioritize small molecules and biologics for experimental testing [72,73].
5.2. Multi-Omics Integration and Mechanistic Modeling
artificial intelligence architectures such as graph neural networks and variational autoencoders can integrate multi-omics data to infer disease mechanisms and predict the impact of perturbations. In rare neuromuscular diseases, where single-gene defects may have pleiotropic downstream effects, these methods can help map from genotype to pathway dysregulation to candidate interventions. Coupling these models with patient-derived induced pluripotent stem cell (iPSC) systems and high-content imaging enables closed-loop discovery in which artificial intelligence proposes perturbations, experimental systems test them, and results iteratively refine the model [74,75,76,77,78,79].
5.3. Artificial Intelligence-Enabled Gene and RNA Therapies
Gene replacement, exon skipping, and antisense oligonucleotide therapies have transformed the outlook for several rare neuromuscular diseases, including spinal muscular atrophy and Duchenne muscular dystrophy. artificial intelligence contributes along this therapeutic pipeline by optimizing antisense oligonucleotide sequences, predicting off-target effects, modeling vector tropism and transduction efficiency, and simulating immune responses. Machine learning models trained on large CRISPR and oligonucleotide datasets can prioritize constructs with favorable on-target/off-target profiles and pharmacodynamic properties, while structure-based protein models estimate the effects of missense variants on folding, stability, and protein–protein interactions. In rare neuromuscular diseases driven by a limited set of recurrent variants, these combined approaches can help determine whether genetic correction is likely to restore function, anticipate dominant-negative or gain-of-function mechanisms, and integrate with functional genomics and patient-derived models to begin closing the loop from variant interpretation to therapeutic design and strategy [80,81,82,83,84,85,86].
5.4. Clinical Trial Design and Patient Stratification
artificial intelligence can also enhance clinical trial design by predicting which patients are most likely to benefit from a given therapy, which endpoints are most sensitive to change, and what sample sizes are required to detect clinically meaningful effects [87,88]. Digital biomarkers derived from sensors and imaging can serve as secondary or exploratory endpoints, and disease-progression models can inform enrichment strategies that increase statistical power in small rare-disease trials.
5.5. Ethical, Regulatory, and Practical Considerations
artificial intelligence-driven therapeutics for rare neuromuscular diseases raise ethical and regulatory questions, including how to validate model predictions when patient numbers are small, how to ensure equitable access to bespoke gene-targeted therapies, and how to manage uncertainty about long-term safety. Transparent reporting of model development, external validation, and post-marketing surveillance will be essential, alongside sustained collaboration among clinicians, data scientists, regulators, and patient communities [89,90,91].
6. Discussion: Knowledge Gaps and Future Directions
Across diagnosis, prognosis, and therapeutics, several cross-cutting challenges continue to limit the realization of artificial intelligence’s potential in rare neuromuscular diseases. Data remain fragmented across institutions, and many rare neuromuscular diseases lack even basic natural history cohorts. Standardization of imaging, electrophysiology, and digital phenotyping protocols is incomplete. Most models are trained retrospectively and lack prospective validation or deployment studies, and few efforts have systematically combined multi-omics data, deep phenotyping, and digital biomarkers in ways that are both biologically informative and clinically actionable. Future work should prioritize:
- Multicenter, interoperable datasets that capture the breadth of rare neuromuscular disease phenotypes.
- Harmonized acquisition and annotation standards for imaging, electrophysiology, and digital signals.
- Extensive and harmonized use of standardized terminologies with machine-readable codes across the biomedical literature and electronic health records (e.g., Online Mendelian Inheritance in Man, Human Phenotype Ontology, Orphadata, Gene Ontology).
- Prospective, clinician-in-the-loop deployments of diagnostic and prognostic artificial intelligence tools.
- Integration of biological and clinical modeling across scales, from molecule to motor unit to patient.
- Ethical frameworks that address data governance, algorithmic bias, and equitable access to artificial intelligence-enabled therapies.
7. Conclusions
Rare neuromuscular diseases exemplify both the challenges and the opportunities of precision medicine. Phenotypic complexity, genetic heterogeneity, and small patient populations have historically impeded timely diagnosis and effective treatment. artificial intelligence and machine learning directly target these obstacles by enhancing diagnostic accuracy through multimodal pattern recognition, enabling more precise and continuous monitoring of disease progression, and accelerating the discovery and optimization of mechanism-based therapies.
The field is still in transition from proof-of-concept models to routinely deployed clinical tools. Realizing the full potential of artificial intelligence in neuromuscular medicine will require robust data infrastructures, careful attention to interpretability and equity, and sustained collaboration among clinicians, computational scientists, and patients. If these conditions are met, artificial intelligence has the potential not only to shorten the diagnostic odyssey but also to transform therapeutic development, bringing care to people living with rare neuromuscular diseases that is both effective and personalized. In this sense, artificial intelligence is well positioned to jumpstart the next generation of advances in the diagnosis and treatment of neuromuscular diseases.
Author Contributions
Conceptualization, DCW II and DCW III; Search Methodology and Literature Search, all authors; Original Draft Preparation, DCW III; Revisions, all authors. All authors have read and agreed to the published version of the manuscript.
Funding
DCW II received support from the Missouri S&T Mary K. Finley Endowment and the Kummer Institute for Artificial Intelligence and Autonomous Systems, and was also supported by NSF Award Number 2420248 under the project title EAGER: Solving Representation Learning and Catastrophic Forgetting with Adaptive Resonance Theory. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the National Science Foundation or the U.S. Government. The U.S. Government is authorized to reproduce and distribute reprints for Government purposes notwithstanding any copyright notation herein.
Institutional Review Board Statement
No institutional review was required for this work. No protected health information was used.
Data Availability Statement
Not applicable. All materials used are available in this submission.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jankovic, J.; Mazziotta, J.C.; Pomeroy, S.L.; Newman, N.J. (Eds.) Bradley and Daroff’s Neurology in Clinical Practice, 8 ed.; Elsevier: Philadelphia, 2022; p. 2400. [Google Scholar]
- Xie, J.; Zhang, Z. Development of a deep learning model for automated diagnosis of neuromuscular diseases using ultrasound imaging. Frontiers in Neurology 2025, 16, 1640428. [Google Scholar] [CrossRef]
- Iorio, R. Myasthenia gravis: The changing treatment landscape in the era of molecular therapies. Nature Reviews Neurology 2024, 20, 84–98. [Google Scholar] [CrossRef]
- Rajabally, Y.A. Chronic inflammatory demyelinating polyradiculoneuropathy: Current therapeutic approaches and future outlooks. ImmunoTargets and Therapy 2024, 99–110. [Google Scholar] [CrossRef]
- Wolfe, G.I.; Hanson, J.E.; Silvestri, N.J. Myasthenia gravis: The evolving therapeutic landscape. eNeurologicalSci 2024, 37, 100541. [Google Scholar] [CrossRef] [PubMed]
- Ripellino, P.; Fleetwood, T.; Cantello, R.; Comi, C. Treatment of chronic inflammatory demyelinating polyneuropathy: From molecular bases to practical considerations. Autoimmune diseases 2014, 2014, 201657. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nature reviews Disease primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Dale, A.M.; Harris-Adamson, C.; Rempel, D.; Gerr, F.; Hegmann, K.; Silverstein, B.; Burt, S.; Garg, A.; Kapellusch, J.; Merlino, L.; et al. Prevalence and incidence of carpal tunnel syndrome in US working populations: Pooled analysis of six prospective studies. Scandinavian journal of work, environment & health 2013, 39, 495. [Google Scholar]
- Rath, A.; Olry, A.; Dhombres, F.; Brandt, M.M.; Urbero, B.; Ayme, S. Representation of rare diseases in health information systems: The Orphanet approach to serve a wide range of end users. Human mutation 2012, 33, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.; Bocchini, C.; Hamosh, A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®). Human mutation 2011, 32, 564–567. [Google Scholar] [CrossRef]
- Manjunath, U.; Kulkarni, S.S.; Reddy, H.N.; Anil, A.; Iyer, G.R.; Mishra, R.K.; et al. NMPhenogen: A comprehensive database for genotype-phenotype correlation in neuromuscular genetic disorders. Frontiers in Neuroscience 2025, 19, 1696899. [Google Scholar] [CrossRef]
- Robinson, P.N. Deep phenotyping for precision medicine. Human mutation 2012, 33, 777–780. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic acids research 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Krenn, M.; Wagner, M.; Zulehner, G.; Weng, R.; Jäger, F.; Keritam, O.; Sener, M.; Brücke, C.; Milenkovic, I.; Langer, A.; et al. Next-generation sequencing and comprehensive data reassessment in 263 adult patients with neuromuscular disorders: Insights into the gray zone of molecular diagnoses. Journal of Neurology 2023, 271, 1937–1946. [Google Scholar] [CrossRef]
- Ng, K.; Chin, H.; Chin, A.; Goh, D. Using gene panels in the diagnosis of neuromuscular disorders: A mini-review. Frontiers in Neurology 2022, 13. [Google Scholar] [CrossRef]
- Rosenberg, A.; Tian, C.; He, H.; Ulm, E.; Collins Ruff, K.; B. Nagaraj, C. An evaluation of clinical presentation and genetic testing approaches for patients with neuromuscular disorders. American Journal of Medical Genetics Part A 2023, 191, 2679–2692. [Google Scholar] [CrossRef]
- Piñeros-Fernández, M.C. Artificial intelligence applications in the diagnosis of neuromuscular diseases: A narrative review. Cureus 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Decherchi, S.; Pedrini, E.; Mordenti, M.; Cavalli, A.; Sangiorgi, L. Opportunities and challenges for machine learning in rare diseases. Frontiers in medicine 2021, 8, 747612. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Brooks, B.R.; Silani, V. Clinical trials in amyotrophic lateral sclerosis: Why so many negative trials and how can trials be improved? The Lancet Neurology 2014, 13, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Frontiers in aging neuroscience 2017, 9, 68. [Google Scholar] [CrossRef]
- Corcia, P.; Lunetta, C.; Vourc’h, P.; Pradat, P.F.; Blasco, H. Time for optimism in amyotrophic lateral sclerosis. European Journal of Neurology 2023, 30, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, N.; Claeys, K.; Schoser, B. Implementing new technologies for neuromuscular disorders. 2024. [Google Scholar]
- Tiwari, S.; Shukla, A. Review on Classification of Amyotrophic Lateral Sclerosis Using Ensemble Classifiers. Engineering Proceedings 2025, 82, 114. [Google Scholar] [CrossRef]
- Scodellaro, R.; Zschüntzsch, J.; Hell, A.K.; Alves, F. A first explainable-AI-based workflow integrating forward-forward and backpropagation-trained networks of label-free multiphoton microscopy images to assess human biopsies of rare neuromuscular disease. Computer methods and programs in biomedicine 2025, 265, 108733. [Google Scholar] [CrossRef]
- Piñeros-Fernández, M.C. Artificial intelligence applications in the diagnosis of neuromuscular diseases: A narrative review. Cureus 2023, 15. [Google Scholar] [CrossRef]
- Chen, T.; Zhu, H.; Hu, Y.; Huang, Y.; He, W.; Luo, Y.; Wu, Z.; Fang, D.; Sun, L.; Zeng, H.; et al. Machine learning-based radiomics using MRI to differentiate early-stage Duchenne and Becker muscular dystrophy in children. BMC Musculoskeletal Disorders 2025, 26, 287. [Google Scholar] [CrossRef]
- Taha, M.A.; Morren, J.A. The role of artificial intelligence in electrodiagnostic and neuromuscular medicine: Current state and future directions. Muscle & nerve 2024, 69, 260–272. [Google Scholar]
- Torres-Castillo, J.R.; Lopez-Lopez, C.O.; Padilla-Castaneda, M.A. Neuromuscular disorders detection through time-frequency analysis and classification of multi-muscular EMG signals using Hilbert-Huang transform. Biomedical Signal Processing and Control 2022, 71, 103037. [Google Scholar] [CrossRef]
- Cooray, G.; Nastasi, L.; Motan, D.; Deeb, J. Evaluating paediatric peripheral neuromuscular disorders using deep neural networks on electrodiagnostic data. Clinical Neurophysiology Practice 2025. [Google Scholar] [CrossRef] [PubMed]
- Hier, D.B.; Munzir, S.I.; Stahlfeld, A.; Obafemi-Ajayi, T.; Carrithers, M.D. High-Throughput Phenotyping of Clinical Text Using Large Language Models. In Proceedings of the 2024 IEEE EMBS International Conference on Biomedical and Health Informatics (BHI); IEEE, 2024; pp. 1–8. [Google Scholar]
- Azizi, S.; Hier, D.B.; Wunsch II, D.C. Enhanced neurologic concept recognition using a named entity recognition model based on transformers. Frontiers in Digital Health 2022, 4, 1065581. [Google Scholar] [CrossRef] [PubMed]
- Hier, D.B.; Carrithers, M.A.; Platt, S.K.; Nguyen, A.; Giannopoulos, I.; Obafemi-Ajayi, T. Preprocessing of Physician Notes by LLMs Improves Clinical Concept Extraction Without Information Loss. Information 2025, 16, 446. [Google Scholar] [CrossRef]
- Munzir, S.I.; Hier, D.B.; Oommen, C.; Carrithers, M.D. A large language model outperforms other computational approaches to the high-throughput phenotyping of physician notes. In Proceedings of the AMIA Annual Symposium Proceedings; 2025; Vol. 2024, p. 838. [Google Scholar]
- Díaz-Santiago, E.; Claros, M.G.; Yahyaoui, R.; de Diego-Otero, Y.; Calvo, R.; Hoenicka, J.; Palau, F.; Ranea, J.A.; Perkins, J.R. Decoding neuromuscular disorders using phenotypic clusters obtained from co-occurrence networks. Frontiers in Molecular Biosciences 2021, 8, 635074. [Google Scholar] [CrossRef]
- Wong, A.K.; Sealfon, R.S.; Theesfeld, C.L.; Troyanskaya, O.G. Decoding disease: From genomes to networks to phenotypes. Nature Reviews Genetics 2021, 22, 774–790. [Google Scholar] [CrossRef]
- Hier, D.B.; Yelugam, R.; Carrithers, M.D.; Wunsch III, D.C. The visualization of Orphadata neurology phenotypes. Frontiers in Digital Health 2023, 5, 1064936. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Havrilla, J.M.; Fang, L.; Chen, Y.; Peng, J.; Liu, C.; Wu, C.; Sarmady, M.; Botas, P.; Isla, J.; et al. Phen2Gene: Rapid phenotype-driven gene prioritization for rare diseases. NAR genomics and Bioinformatics 2020, 2, lqaa032. [Google Scholar] [CrossRef]
- Gnanaolivu, R.; Oliver, G.; Jenkinson, G.; Blake, E.; Chen, W.; Chia, N.; Klee, E.W.; Wang, C. A clinical knowledge graph-based framework to prioritize candidate genes for facilitating diagnosis of Mendelian diseases and rare genetic conditions. BMC bioinformatics 2025, 26, 82. [Google Scholar] [CrossRef]
- Nuredini, A.; Savarese, M.; Santorelli, F.M.; Tupler, R.G. Empowering clinicians with artificial intelligence in hereditary neuromuscular disorders: Training the Next Generation through the CoMPaSS-NMD Young Investigator Training program and the CoMPaSS-NMD Autumn School Workshop Report. Acta Myologica 2025, 44, 62. [Google Scholar]
- Schaefer, J.; Lehne, M.; Schepers, J.; Prasser, F.; Thun, S. The use of machine learning in rare diseases: A scoping review. Orphanet journal of rare diseases 2020, 15, 145. [Google Scholar] [CrossRef]
- Banerjee, J.; Taroni, J.N.; Allaway, R.J.; Prasad, D.V.; Guinney, J.; Greene, C. Machine learning in rare disease. Nature Methods 2023, 20, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Faviez, C.; Chen, X.; Garcelon, N.; Neuraz, A.; Knebelmann, B.; Salomon, R.; Lyonnet, S.; Saunier, S.; Burgun, A. Diagnosis support systems for rare diseases: A scoping review. Orphanet journal of rare diseases 2020, 15, 94. [Google Scholar] [CrossRef]
- Groza, T.; Köhler, S.; Doelken, S.; Collier, N.; Oellrich, A.; Smedley, D.; Couto, F.M.; Baynam, G.; Zankl, A.; Robinson, P.N. Automatic concept recognition using the Human Phenotype Ontology reference and test suite corpora. Database: The Journal of Biological Databases and Curation 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Groza, T.; Gration, D.; Baynam, G.; Robinson, P.N. FastHPOCR: Pragmatic, fast, and accurate concept recognition using the human phenotype ontology. Bioinformatics 2024, 40. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Wang, L.; Liu, H. Phenotypic Analysis of Clinical Narratives Using Human Phenotype Ontology. Studies in health technology and informatics 2020, 245, 581–585. [Google Scholar] [CrossRef]
- Liu, C.; Kury, F.; Li, Z.; Ta, C.N.; Wang, K.; Weng, C. Doc2Hpo: A web application for efficient and accurate HPO concept curation. Nucleic Acids Research 2019, 47, W566–W570. [Google Scholar] [CrossRef]
- Köhler, S.; Doelken, S.; Mungall, C.; Bauer, S.; Firth, H.; Bailleul-Forestier, I.; Black, G.; Brown, D.L.; Brudno, M.; Campbell, J.; et al. The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic acids research 2014, 42 Database issue, D966–D974. [Google Scholar] [CrossRef]
- Gao, J.; Liu, L.; Yao, S.; Huang, X.; Mamitsuka, H.; Zhu, S. HPOAnnotator: Improving large-scale prediction of HPO annotations by low-rank approximation with HPO semantic similarities and multiple PPI networks. BMC Medical Genomics 2019, 12. [Google Scholar] [CrossRef]
- Qin, H.; Hussain, L.; Liu, Z.; Yan, X.; Awwad, F.A.; Butt, F.M.; Salaria, U.A.; Ismail, E.A. Optimizing deep learning models to combat amyotrophic lateral sclerosis (ALS) disease progression. Digital health 2025, 11, 20552076251349719. [Google Scholar] [CrossRef] [PubMed]
- Blemker, S.S.; Riem, L.; DuCharme, O.; Pinette, M.; Costanzo, K.E.; Weatherley, E.; Statland, J.; Tapscott, S.J.; Wang, L.H.; Shaw, D.W.; et al. Multi-scale machine learning model predicts muscle and functional disease progression. Scientific Reports 2025, 15, 25339. [Google Scholar] [CrossRef]
- Edwards, A.S.; Kaplan, B.; Jie, T. A primer on machine learning. Transplantation 2021, 105, 699–703. [Google Scholar] [CrossRef]
- Calderone, A.; Latella, D.; Bonanno, M.; Quartarone, A.; Mojdehdehbaher, S.; Celesti, A.; Calabrò, R.S. Towards transforming neurorehabilitation: The impact of artificial intelligence on diagnosis and treatment of neurological disorders. Biomedicines 2024, 12, 2415. [Google Scholar] [CrossRef]
- Belić, M.; Bobić, V.; Badža, M.; Šolaja, N.; Đurić-Jovičić, M.; Kostić, V.S. Artificial intelligence for assisting diagnostics and assessment of Parkinson’s disease—A review. Clinical neurology and neurosurgery 2019, 184, 105442. [Google Scholar] [CrossRef] [PubMed]
- Youn, B.Y.; Ko, Y.; Moon, S.; Lee, J.; Ko, S.G.; Kim, J.Y. Digital biomarkers for neuromuscular disorders: A systematic scoping review. Diagnostics 2021, 11, 1275. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Kothare, H.; Ramanarayanan, V. Multimodal speech biomarkers for remote monitoring of ALS disease progression. Computers in Biology and Medicine 2024, 180, 108949. [Google Scholar] [CrossRef]
- Dubbioso, R.; Spisto, M.; Verde, L.; Iuzzolino, V.V.; Senerchia, G.; Salvatore, E.; De Pietro, G.; De Falco, I.; Sannino, G. Voice signals database of ALS patients with different dysarthria severity and healthy controls. Scientific Data 2024, 11, 800. [Google Scholar] [CrossRef]
- Bortolani, S.; Brusa, C.; Rolle, E.; Monforte, M.; De Arcangelis, V.; Ricci, E.; Mongini, T.E.; Tasca, G. Technology outcome measures in neuromuscular disorders: A systematic review. European Journal of Neurology 2022, 29, 1266–1278. [Google Scholar] [CrossRef] [PubMed]
- Dabnichki, P.; Pang, T.Y. Wearable Sensors and Motion Analysis for Neurological Patient Support. Biosensors 2024, 14, 628. [Google Scholar] [CrossRef]
- Gupta, A.S.; Patel, S.; Premasiri, A.; Vieira, F. At-home wearables and machine learning sensitively capture disease progression in amyotrophic lateral sclerosis. Nature communications 2023, 14, 5080. [Google Scholar] [CrossRef]
- van Unnik, J.W.; Meyjes, M.; van Mantgem, M.R.J.; van den Berg, L.H.; van Eijk, R.P. Remote monitoring of amyotrophic lateral sclerosis using wearable sensors detects differences in disease progression and survival: A prospective cohort study. EBioMedicine 2024, 103. [Google Scholar] [CrossRef]
- Izuka, S.; Sen, P.; Komai, T.; Fujio, K.; Knitza, J.; Gupta, L. Digital approaches in myositis. 2024. [Google Scholar]
- Torri, F.; Vadi, G.; Meli, A.; Loprieno, S.; Schirinzi, E.; Lopriore, P.; Ricci, G.; Siciliano, G.; Mancuso, M. The use of digital tools in rare neurological diseases towards a new care model: A narrative review. Neurological Sciences 2024, 45, 4657–4668. [Google Scholar] [CrossRef]
- Izmailova, E.S.; Demanuele, C.; McCarthy, M. Digital health technology derived measures: Biomarkers or clinical outcome assessments? Clinical and Translational Science 2023, 16, 1113–1120. [Google Scholar] [CrossRef]
- Tiwari, S.; Shukla, A. Review on Classification of Amyotrophic Lateral Sclerosis Using Ensemble Classifiers. Engineering Proceedings 2025, 82, 114. [Google Scholar] [CrossRef]
- Jing Yeo, C.J.; Ramasamy, S.; Joel Leong, F.; Nag, S.; Simmons, Z. A neuromuscular clinician’s primer on machine learning. Journal of Neuromuscular Diseases 2025, 22143602251329240. [Google Scholar] [CrossRef]
- Jadoenathmisier, K.D.; Gardarsdottir, H.; Mol, P.G.; Pasmooij, A.M. Insights from the European Medicines Agency on digital health technology derived endpoints. Drug Discovery Today 2025, 104388. [Google Scholar] [CrossRef]
- Bakker, J.P.; Izmailova, E.S.; Clement, A.; Hoffmann, S.; Leptak, C.; Menetski, J.P.; Wagner, J.A. Regulatory pathways for qualification and acceptance of digital health technology-derived clinical trial endpoints: Considerations for sponsors. Clinical Pharmacology & Therapeutics 2025, 117, 56–72. [Google Scholar]
- Yu, M.; Xu, J.; Dutta, R.; Trapp, B.; Pieper, A.A.; Cheng, F. Network medicine informed multiomics integration identifies drug targets and repurposable medicines for Amyotrophic Lateral Sclerosis. NPJ Systems Biology and Applications 2024, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Pun, F.W.; Liu, B.H.M.; Long, X.; Leung, H.W.; Leung, G.H.D.; Mewborne, Q.T.; Gao, J.; Shneyderman, A.; Ozerov, I.V.; Wang, J.; et al. Identification of therapeutic targets for amyotrophic lateral sclerosis using PandaOmics–an AI-enabled biological target discovery platform. Frontiers in aging neuroscience 2022, 14, 914017. [Google Scholar] [CrossRef]
- Sunildutt, N.; Ahmed, F.; Chethikkattuveli Salih, A.R.; Lim, J.H.; Choi, K.H. Integrating transcriptomic and structural insights: Revealing drug repurposing opportunities for sporadic ALS. ACS omega 2024, 9, 3793–3806. [Google Scholar] [CrossRef]
- Gerring, Z.F.; Bhalala, O.G.; Fearnley, L.G.; Oikari, L.E.; White, A.R.; Derks, E.M.; Watson, R.; Yassi, N.; Bahlo, M.; Reay, W.R. Drug repurposing candidates for amyotrophic lateral sclerosis using common and rare genetic variants. Brain Communications 2025, 7, fcaf184. [Google Scholar] [CrossRef]
- Hoolachan, J.M.; McCallion, E.; Sutton, E.R.; Çetin, Ö.; Pacheco-Torres, P.; Dimitriadi, M.; Sari, S.; Miller, G.J.; Okoh, M.; Walter, L.M.; et al. A transcriptomics-based drug repositioning approach to identify drugs with similar activities for the treatment of muscle pathologies in spinal muscular atrophy (SMA) models. Human molecular genetics 2024, 33, 400–425. [Google Scholar] [CrossRef]
- Lombardo, S.D.; Basile, M.S.; Ciurleo, R.; Bramanti, A.; Arcidiacono, A.; Mangano, K.; Bramanti, P.; Nicoletti, F.; Fagone, P. A network medicine approach for drug repurposing in duchenne muscular dystrophy. Genes 2021, 12, 543. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, J.; Xiao, Y.; Zhang, S.; Li, L. CoupleVAE: Coupled variational autoencoders for predicting perturbational single-cell RNA sequencing data. Briefings in Bioinformatics 2025, 26. [Google Scholar] [CrossRef]
- Baxi, E.G.; Thompson, T.; Li, J.; Kaye, J.A.; Lim, R.G.; Wu, J.; Ramamoorthy, D.; Lima, L.; Vaibhav, V.; Matlock, A.; et al. Answer ALS, a large-scale resource for sporadic and familial ALS combining clinical and multi-omics data from induced pluripotent cell lines. Nature neuroscience 2022, 25, 226–237. [Google Scholar] [CrossRef]
- Benkirane, H.; Pradat, Y.; Michiels, S.; Cournède, P.H. CustOmics: A versatile deep-learning based strategy for multi-omics integration. PLOS Computational Biology 2023, 19, e1010921. [Google Scholar] [CrossRef]
- Lee, G.B.; Mazli, W.N.A.b.; Hao, L. Multiomics evaluation of human iPSCs and iPSC-derived neurons. Journal of proteome research 2024, 23, 3149–3160. [Google Scholar] [CrossRef]
- Doncevic, D.; Herrmann, C. Biologically informed variational autoencoders allow predictive modeling of genetic and drug-induced perturbations. Bioinformatics 2023, 39, btad387. [Google Scholar] [CrossRef] [PubMed]
- Phatnani, H.; Kwan, J.; Sareen, D.; Broach, J.R.; Simmons, Z.; Arcila-Londono, X.; Lee, E.B.; Van Deerlin, V.M.; Shneider, N.A.; Fraenkel, E.; et al. An integrated multi-omic analysis of iPSC-derived motor neurons from C9ORF72 ALS patients. Iscience 2021, 24. [Google Scholar] [CrossRef]
- Leckie, J.; Yokota, T. Integrating Machine Learning-Based Approaches into the Design of ASO Therapies. Genes 2025, 16, 185. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.; Kwon, M.; Seo, D.; Kim, D.H.; Lee, D.; Lee, K.; Kim, E.; Kang, M.; Ryu, J.H. ASOptimizer: Optimizing antisense oligonucleotides through deep learning for IDO1 gene regulation. Molecular Therapy Nucleic Acids 2024, 35. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Lee, D.; Hwang, G.; Lee, K.; Kang, M. ASOptimizer: Optimizing chemical diversity of antisense oligonucleotides through deep learning. Nucleic Acids Research 2025, gkaf392. [Google Scholar] [CrossRef]
- Lin, J.; Wong, K.C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics 2018, 34, i656–i663. [Google Scholar] [CrossRef]
- Erdoğan, S. Integration of artificial intelligence and genome editing system for determining the treatment of genetic disorders. Balkan Medical Journal 2024, 41, 419. [Google Scholar] [CrossRef]
- Kim, M.g.; Go, M.j.; Kang, S.H.; Jeong, S.h.; Lim, K. Revolutionizing CRISPR technology with artificial intelligence. Experimental & Molecular Medicine 2025, 57, 1419–1431. [Google Scholar] [CrossRef]
- Koutsoni, E.; Konstantakos, V.; Nentidis, A.; Krithara, A.; Paliouras, G. Modeling the off-target effects of CRISPR-Cas9 experiments for the treatment of Duchenne Muscular Dystrophy. In Proceedings of the Proceedings of the 12th Hellenic Conference on Artificial Intelligence; 2022; pp. 1–7. [Google Scholar]
- Zhang, B.; Zhang, L.; Chen, Q.; Jin, Z.; Liu, S.; Zhang, S. Harnessing artificial intelligence to improve clinical trial design. Communications Medicine 2023, 3, 191. [Google Scholar] [CrossRef] [PubMed]
- Askin, S.; Burkhalter, D.; Calado, G.; El Dakrouni, S. Artificial intelligence applied to clinical trials: Opportunities and challenges. Health and technology 2023, 13, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Refolo, P.; Raimondi, C.; Astratinei, V.; Battaglia, L.; Borràs, J.M.; Closa, P.; Lo Scalzo, A.; Marchetti, M.; Muñoz-López, S.; Sampietro-Colom, L.; et al. Ethical, Legal, and Social Assessment of AI-Based Technologies for Prevention and Diagnosis of Rare Diseases in Health Technology Assessment Processes. Healthcare 2025, 13, 829. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.L.; Sawai, T. Navigating equity in global access to genome therapy expanding access to potentially transformative therapies and benefiting those in need requires global policy changes. Frontiers in Genetics 2024, 15, 1381172. [Google Scholar] [CrossRef]
- Ibrahim, H.; Liu, X.; Rivera, S.C.; Moher, D.; Chan, A.W.; Sydes, M.R.; Calvert, M.J.; Denniston, A.K. Reporting guidelines for clinical trials of artificial intelligence interventions: The SPIRIT-AI and CONSORT-AI guidelines. Trials 2021, 22, 11. [Google Scholar] [CrossRef]
Figure 1.
Accelerated diagnosis and treatment for a hypothetical genetic neuromuscular disease. Opportunities include improved genomic diagnosis of diseases (1), enhanced prediction of protein amino acid sequence changes based on genetic variants (2), prediction of abnormal protein folding (3), correlation of protein conformation with protein function (4), prediction of phenotype from altered protein function (5), prediction of disease diagnosis from phenotype (6), improved prediction of disease progression (7), accelerated drug discovery (8), and prediction of disease based on altered protein function (9).
Figure 1.
Accelerated diagnosis and treatment for a hypothetical genetic neuromuscular disease. Opportunities include improved genomic diagnosis of diseases (1), enhanced prediction of protein amino acid sequence changes based on genetic variants (2), prediction of abnormal protein folding (3), correlation of protein conformation with protein function (4), prediction of phenotype from altered protein function (5), prediction of disease diagnosis from phenotype (6), improved prediction of disease progression (7), accelerated drug discovery (8), and prediction of disease based on altered protein function (9).
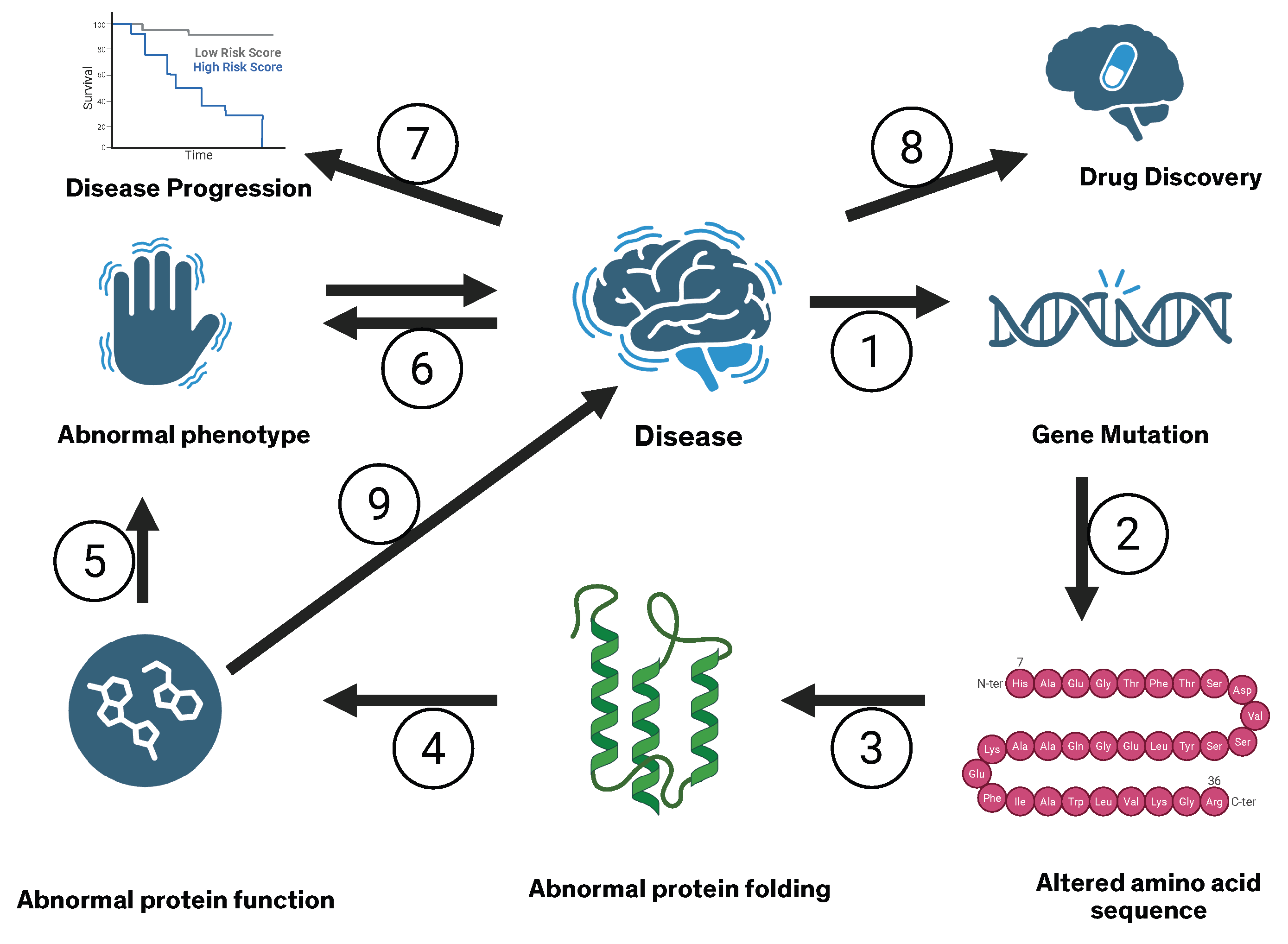

Table 1.
Example Neuromuscular Diseases by Locus of Pathology.
| Disease locus | Abbrev. | Brief description | Prevalence |
|---|---|---|---|
| Motor neuron | |||
| Amyotrophic lateral sclerosis | ALS | Progressive degeneration of upper and lower motor neurons; usually sporadic, with a minority of familial cases involving C9orf72, SOD1, TARDBP, or FUS. | Rare |
| Proximal spinal muscular atrophy | SMA | Childhood-onset hereditary lower motor neuron disease most often caused by SMN1. | Rare |
| Axon | |||
| Charcot–Marie–Tooth disease (axonal) | CMT2 | Axonal neuropathy due to pathogenic variants in multiple genes. | Rare |
| Diabetic distal symmetric polyneuropathy | DSPN | Length-dependent axonal polyneuropathy due to chronic hyperglycemia and metabolic and vascular factors. | Common |
| Myelin | |||
| Charcot–Marie–Tooth disease (demyelinating) | CMT1A | Hereditary demyelinating neuropathy caused by PMP22 gene duplication. | Uncommon |
| Chronic inflammatory demyelinating polyneuropathy | CIDP | Immune-mediated demyelinating neuropathy affecting peripheral nerves and roots. | Rare |
| Neuromuscular junction | |||
| Myasthenia gravis | MG | Autoimmune postsynaptic neuromuscular junction disorder, most commonly mediated by antibodies to acetylcholine receptors. | Uncommon |
| Lambert–Eaton myasthenic syndrome | LEMS | Autoimmune presynaptic neuromuscular junction disorder caused by antibodies to P/Q-type voltage-gated calcium channels, often paraneoplastic. | Ultra-rare |
| Muscle | |||
| Duchenne muscular dystrophy | DMD | X-linked recessive dystrophinopathy due to pathogenic variants in DMD. | Rare |
| Myotonic dystrophy type 1 | DM1 | Autosomal dominant CTG-repeat expansion in DMPK, causing a multisystem distal myopathy with myotonia. | Rare |
Notes. Prevalence categories follow approximate Orphanet-style bands: Common (>1,000 per 1,000,000), Uncommon (≈100–900 per 1,000,000), Rare (≈1–90 per 1,000,000), and Ultra-rare (<1 per 1,000,000).[web:39] The disorders listed here are representative examples for each anatomical locus of pathology rather than an exhaustive catalog. Apparent prevalence may differ from incidence depending on disease duration and survival (for example, MG has relatively low incidence but higher prevalence, whereas amyotrophic lateral sclerosis has higher incidence but lower prevalence due to short survival). When relevant, illustrative gene loci are shown in italics (for example, SOD1).
Table 2.
Genetic Complexity of Charcot–Marie–Tooth Disease (CMT).
| Context | CMT is a neurogenetic neuropathy with both motor and sensory features. |
|---|---|
| Canonical symptoms | Distal weakness, sensory loss, hyporeflexia, and muscle atrophy. |
| Online Mendelian Inheritance in Man phenotypes | Over 100 phenotypically distinct CMT entities in Online Mendelian Inheritance in Man, including CMT1, CMT2, CMT4, dominant intermediate CMT (DI-CMT), X-linked CMT, and intermediate forms. |
| Genes implicated in CMT | Over 120 genes associated with hereditary neuropathies. Examples include PMP22, MPZ, GJB1, MFN2, NEFL, SH3TC2, and GDAP1. |
| Example gene: NEFL | |
| Gene-level complexity | NEFL is associated with three CMT phenotypes: CMT1F, CMT2E, and DI-CMT. ClinVar lists 786 NEFL variants, of which 28 are associated with CMT. |
| Protein-level complexity | These 786 NEFL variants correspond to many possible amino acid substitutions or truncations, each potentially altering neurofilament assembly, axonal transport, protein stability, and molecular interactions in distinct ways. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



